

Abstract
Background -
The hypothesis of the Dilated Cardiomyopathy (DCM) Precision Medicine Study is that most DCM has a genetic basis. The study returns results to probands, and when indicated, to relatives. While both the American College of Medical Genetics and Genomics/Association for Molecular Pathology and ClinGen’s MYH7-cardiomyopathy specifications provide relevant guidance for variant interpretation, further gene and disease specific considerations were required for DCM. To this end, we tailored the ClinGen MYH7-cardiomyopathy variant interpretation framework; the specifications implemented for the study are presented here.
Methods -
Modifications were created and approved by an external Variant Adjudication Oversight Committee. After a pilot using 81 probands, further adjustments were made, resulting in 27 criteria (9 modifications of the ClinGen MYH7 framework and reintroduction of two ACMG/AMP criteria that were deemed not applicable by the ClinGen MYH7 working group).
Results -
These criteria were applied to 2,059 variants in a test set of 97 probands. Variants were classified as benign (n=1,702), likely benign (n=33), uncertain significance (n=71), likely pathogenic (LP; n=12), and pathogenic (P; n=3). Only 2/15 LP/P variants were identified in Non-Hispanic African ancestry probands.
Conclusions –
We tailored the ClinGen MYH7 criteria for our study. Our preliminary data show that 15/97 (15.5%) probands have LP/P variants, most of which were identified in probands of Non-Hispanic European ancestry. We anticipate continued evolution of our approach, one that will be informed by new insights on variant interpretation and a greater understanding of the genetic architecture of DCM.
Clinical Trial Registration -
Journal Subject Terms: Genetics, Clinical Studies, Cardiomyopathy
Keywords: genetic testing, genetics, dilated cardiomyopathy
Introduction
The Dilated Cardiomyopathy (DCM) Precision Medicine Study was launched in 2015 to test the hypothesis that both familial and non-familial DCM have a genetic basis. For this study, DCM is defined as non-ischemic disease and idiopathic, after excluding non-genetic etiologies.1 The study, which aims to recruit 1,300 individuals with DCM (600 non-Hispanic ethnicity [NHE] African Ancestry, 600 NHE-European Ancestry, and 100 HE; 50% female) and 2,600 relatives, includes a randomized controlled trial to evaluate the effectiveness of an educational guide (Family Heart Talk) on the uptake of and adherence to cardiovascular screening in at-risk relatives. Families are recruited at DCM Consortium sites, by telephone, and mail.2
After exome sequencing in probands and affected relatives, results from analysis of 35 DCM genes (see Supplemental Material) are returned to probands and relatives of probands who are found to carry likely pathogenic or pathogenic (LP/P) variants. As part of the study, periodic screening is recommended for unaffected relatives with LP/P variants. Unaffected relatives who do not carry a known LP or P variant identified in the proband are discharged from surveillance. For at-risk relatives of probands in whom a LP/P variant is not identified, surveillance is recommended. These screening recommendations follow Heart Failure Society of America and the American College of Medical Genetics and Genomics (ACMG)3, 4 guidelines and depend on accurate variant interpretation.
The ability to conduct large scale molecular genetic testing has resulted in an increased volume of sequence variants, and, consequently, new challenges in interpretation. This prompted ACMG to revise prior guidance,5 resulting in updated standards, jointly with the Association for Molecular Pathology (AMP).6 Recognizing that the ACMG/AMP standards serve a broad spectrum of single-gene conditions, the ClinGen7 Cardiovascular Domain Working Group (CDWG) tailored ACMG/AMP guidance for cardiovascular disease.8 Incorporating the genetic features of cardiomyopathy (phenotypic, locus and allelic heterogeneity, reduced and adult onset penetrance, multiple variants interacting to cause disease), the ClinGen Inherited Cardiomyopathy Expert Panel (CMP-EP), which included two members of this author group (A.M., R.E.H.), published specifications for hypertrophic and dilated cardiomyopathies focused on MYH7.9 The MYH7 specifications provided a foundation for the variant interpretation criteria needed to conduct our study; however, further specifications were required to incorporate considerations that are unique to DCM. To this end, we tailored the ClinGen MYH7 variant interpretation framework. The resulting criteria, along with preliminary data, are presented here.
Methods
Please see the Supplemental Material for complete methods. The relevant variant curation criteria are summarized (Table 1). The non-pilot variant data used for this analysis, excluding 1,500 unique variants unlikely to change protein function based on predicted Sequence Ontology10 impacts, are provided in the Supplemental Material. Source code and complete variant and adjudication data are available upon request from the authors. All research participants provided signed informed consent. Institutional Review Board approval was obtained for this study, initially locally at each of the participating clinical sites of the DCM Consortium, and later with study oversight provided by a single Institutional Review Board at the University of Pennsylvania.
Table 1.
DCM Precision Medicine Study Variant Interpretation Criteria
| Strength | Specification type | Rule abbreviation | Rule description |
|---|---|---|---|
| Pathogenic criteria | |||
| Very strong | Reintroduced ACMG criterion for DCM gene specification | PVS1 | Null variant in LMNA or SCN5A |
| Strong | none | PS1 | Same amino acid change as a previously established pathogenic variant |
| none | PS2 | De novo (paternity confirmed) in a patient with disease and no family history | |
| none | PS3 | Well-established in vitro or in vivo functional studies supportive of a damaging effect | |
| none | PS4 | Prevalence of variant in affected individuals is significantly increased compared to controls OR variant identified in ≥15 probands with consistent phenotypes OR variant identified in ≥10 confirmed unlreated probands with consistent phenotypes | |
| none | PP1_Strong | Variant segregates in ≥7 meioses in DCM genes with established evidence | |
| Strength modification, DCM gene specification | PVS1_Strong | Null variant in FLNC, BAG3,and TTN A-band | |
| Moderate | DCM gene specification | PM1 | Located in hotspot (For DCM, RBM20 exon 9, amino acids 634, 636, 637, 638) |
| DCM gene specification, study specific | PM2 | Absent from gnomAD or at extremely low frequency in all gnomAD non-founder populations with available data | |
| Reintroduced ACMG criterion, DCM gene specification | PM3 | For recessive disorders, detected in transwith a pathogenic variant (rarely observed with TNNI3 and TNNT2 variants) | |
| none | PM4 | In-frame deletions/insertions of any size in a non-repeat region or stop-loss variants | |
| none | PM5 | Different missense change at same amino acid residue previously established as pathogenic | |
| DCM specific | PM6 | De novo without confirmation of paternity (parental clinical data required) | |
| DCM gene specification | PVS1_Moderate | Null variant in gene with evidence supporting loss of function as disease mechanism (VCL, PLN, DSP) | |
| none | PS4_Moderate | Variant identified in ≥6 probands with consistent phenotypes | |
| none | PP1_Moderate | Variant segregates in ≥5 meioses in DCM genes with established evidence | |
| Supporting | none | PP1 | Variant segregates in ≥3 meioses in DCM genes with established evidence |
| Method specification | PP3 | Computational evidence support a deleterious effect (REVEL score >0.7) | |
| none | PS4_Supporting | Variant identified in ≥2 probands with consistent phenotypes | |
| Benign | |||
| Stand alone | none | BA1 | Variant allele frequency is >0.1% in any gnomaAD non-founder population |
| DCM gene specification | BP1_Stand_Alone | Missense variant in TTN | |
| Strong | DCM gene specification, study specific | BS1 | Variant allele frequency is >0.05% in any gnomAD non-founder population |
| none | BS3 | Well established in vitroor in vivo functional studies show no damaging effect | |
| none | BS4 | Non-segregation in affected members of a family | |
| Supporting | none | BP2 | Observed as a compound heterozygote (in trans) or double heterozygote in genes with overlapping function |
| Method specification | BP4 | Multiple lines of computational evidence suggest no impact on gene or gene product (REVEL score <0.15) | |
| Study specific | BP7 | Unlikely to affect protein function based on calculated Sequence Ontology terms for the transcript(s) of interest. | |
Results
Pilot Study
A pilot study was conducted with the first 81 probands enrolled in the study. Of these, 40 (49%) carried only likely benign (LB) or benign (B) variants. The rest carried VUS (n=30, 37%) and LP variants (n=11, 14%). Three challenges were identified from this exercise: limited gene-specifications for predicted loss-of-function (LOF) variants, evaluation of a variant’s absence or rarity in reference populations, and use of computational data. Further adjustments, as described below, were approved by the Variant Adjudication Oversight Committee (VAOC; Chair, GJ; Members, W.B, M.D., J.G-F, H.R).
To address the first challenge, a very strong criterion for pathogenicity for LOF variants in LMNA and SCN5A (PVS1) and a strong criterion for pathogenicity for LOF variants in FLNC, BAG3, and TTN A-band (PVS1_Strong) were added. The moderate criterion (PVS1_Moderate) was applied to LOF variants in VCL, PLN, and DSP. These modified criteria were applied considering available evidence associated with DCM. LOF has been established as a disease mechanism for LMNA-DCM and other LMNA-associated diseases.11 Supporting this are large multicenter studies demonstrating a substantially increased odd ratio (OR) in DCM cases versus controls (OR = 99.7).12 Furthermore, in the latest release of gnomAD (v2.1.1), the LMNA pLI (probability of being loss-of-function intolerant) was established to be 1.00. For SCN5A, LOF variants are pathogenic for Brugada syndrome.13, 14 Also, a large multicenter study demonstrated a significantly increased odds ratio of 16.5 for LOF variants in SCN5A in DCM cases versus controls.12 In addition, the p.Arg219His variant, identified in DCM and arrhythmias,15 was studied in Xenopus oocytes, leading to a proton leak but not sodium current alterations. While no segregation has been demonstrated for LOF in primary DCM, supporting segregation data has been published in a family with sick sinus syndrome, and studies in the human cell line tsA-201 with heterologously expressed mutant sodium channels showed LOF properties of reduced or no sodium current density in conjunction with gating modulations.16 For this gene, the gnomAD pLI score is 0.91. We also note that when a truncating SCN5A variant is identified in a proband with DCM a search for an arrhythmia phenotype is also warranted.
LOF variants in FLNC, BAG3, and the A-band of TTN were assigned strong criteria for pathogenicity. First, for FLNC, a LOF mechanism has been reported17 and the gnomAD pLI = 1.00. Due to the relative gene size of FLNC and LMNA, the pLI calculation appears to be reliable. However, in smaller genes, where variants are less prevalent in the DCM population although absent from controls, the pLI may not reflect the actual contribution of LOF variants to disease. For BAG3, a gene comprising 4 coding exons, despite LOF been known as a disease mechanism,18, 19 the gnomAD pLI = 0.62. In the case of TTN, the coding sequence leads to the generation of alternative splicing isoforms,20 with the heart expressing two major isoforms (N2B from transcript NM_003319.4 and N2BA from transcript NM_001256850.1) that incorporate the Z-line, I-band, A-band, and M-line.20 The N2B and N2BA isoforms diverge mainly for the incorporation in the long cardiac isoform N2BA of a large portion of I-band coding exons, similar to the skeletal muscle isoform N2A derived from transcript NM_133378.4.20 It is known that LOF variants affecting the TTN A-band (defined as chr2:179483218–179400709, NM_001267550.1, GRCh37/hg19) are overrepresented in DCM,21, 22 establishing LOF as a mechanism of disease.12 However, due its large size, potential LOF variants are differently distributed across the various functional domains between DCM cases and controls.23 The high prevalence in the control population of LOF variants in TTN regions other than the A-band led to a calculated gnomAD pLI = 0.00; however, the calculation does not discriminate between the various transcripts and protein domains and thus does not accurately reflect the deleterious role of LOF variants in the A-band.24
LOF variants in VCL, PLN, and DSP were assigned a moderate degree of pathogenicity, although for a DSP variant in this category, the variant must have been observed in at least one unrelated DCM case. We implemented this criterion for VCL and DSP LOF variants based on their enrichment in DCM cases versus controls (ORVCL = 21.3; ORDSP = 41.0).12 In addition, the constraint metrics for DSP support a LOF mechanism (pLI = 1.00). For PLN, a trend for enrichment of LOF variants was observed in DCM cases versus controls (ORPLN = 13.8).12 In addition, PLN LOF variants have been shown to be pathogenic and described in both DCM and HCM).25–29 For both PLN and VCL, the constraint metrics from gnomAD are below threshold (PLN, pLI = 0.45; VCL pLI = 0.05), however, the PLN coding sequence has only one exon, possibly being too small a target to reliably detect population levels of LOF variants. Haploinsufficiency of VCL, although enriched in DCM, may represent a low penetrant event. However, we suggest that additional data are needed to confirm our preliminary VCL observations for LOF.
When evaluating PM2, Wilson’s score confidence intervals could not be calculated if there were no genotype calls at the variant position in both exomes and genomes, which could occur due to either the absence of the variant in all populations or lack of coverage. To allow using PM2 in the former scenario, the criterion was adjusted so it was met if the allele count was sufficient (120,000) in neighboring nucleotides, coverage was sufficient, and a mix of exome and genome data was available in gnomAD.
Evaluation of computational data (PP3 and BP4) was performed manually for this pilot using a holistic approach. To harmonize computational data evaluation, a method based on the REVEL score30 was introduced. Cutoffs were developed for this study on the basis of the publication (which estimated the sensitivity and specificity of each potential cutoff using out-of-bag predictions based on 6,182 HGMD disease mutations and 123,706 rare exome sequence variants)30 and made these data publicly available (https://sites.google.com/site/revelgenomics/). On the basis of these data, variants meeting BP4 must have a REVEL score below 0.15, a cutoff estimated to falsely predict no deleterious effect for ~5% of pathogenic variants while correctly identifying ~55% of neutral variants (Figure 1). Variants meeting PP3 must have a REVEL score greater than 0.7, which would correctly identify ~58% of pathogenic variants and falsely predict a deleterious effect for only ~5% of neutral variants. When comparing calls made before REVEL implementation to those that would have been made after, we found that 5/6 variants below the 0.15 cutoff had been manually classified as meeting BP4. Among the 13 variants that had a REVEL score greater than 0.7, 12 had been previously classified as meeting PP3. Of 26 variants with a REVEL score between 0.15 and 0.7, four had been manually classified as meeting BP4, 14 met PP3, and eight met neither. Although variants with a score between 0.15 and 0.7 would not receive an automatic PP3 or BP4 evaluation using the REVEL method, manual review of the data and clinical judgment can be applied if application of the PP3 or BP4 criteria would lead to a clinically significant difference in variant classification.
Figure 1.
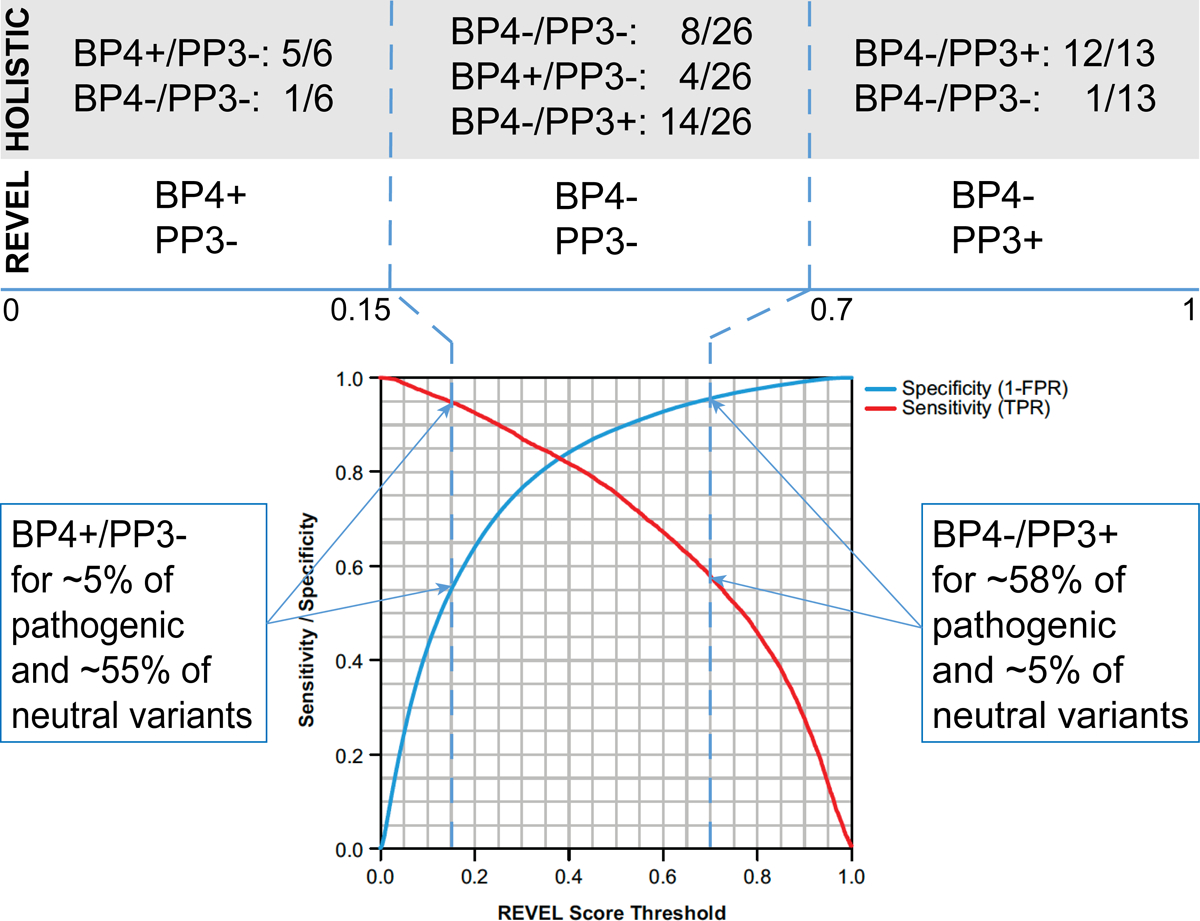
Automated REVEL score Utilization. Cutoffs for BP4/PP3 were based on the estimated sensitivity and specificity reported in Figure S1 of the original publication reproduced here. Comparison of holistic evaluations of variants in the pilot phase with automatic evaluations for the same variants based on the selected REVEL score cutoffs are shown above the blue line.
Results Using DCM Variant Interpretation Criteria Adaptations
Our modifications resulted in 27 criteria for variant interpretation in DCM as summarized (Table 1); full documentation is also provided (Supplemental Material). Using this framework, curation was performed between July 2018-April 2019 for 2,059 variants in an additional set of 97 probands (Supplemental Material). Variants were classified as B (n=1,702), LB (n=33), VUS (n=71), LP (n=12), and P (n=3). 216 (11%) were not adjudicated (low impact variants for which BP7 was met); 22 (1%) were classified as Low Quality.
Among the 97 probands, P variants were identified in 3%, LP in 12%, and VUS in 46%, (Table 2). Most LP/P variants were in TTN (n=8; Table 3). Variants reached LP/P classifications based on case (n=1, PS4; n=3, PS4_Moderate; n=3, PS4_Supporting) and segregation data (n=2, PP1_Strong; n=2, PP1), allele frequency (n=15, PM2), predicted loss of function (n=1, PVS1; n=11, PVS1_Strong), protein length change (n=1, PM4), hotspot location (n=1, PM1), and computational evidence (n=2, PP3). LP/P variant types included stop gained (n=6/15, 40%), frameshift (n=6/15, 40%), in-frame deletion (n=1/15, 7%), and missense (n=2/15, 13%). Of the 15 LP/P variants, 13 were found in individuals of non-Hispanic European ancestry. More than one VUS and above variant were identified in 20 (21%) probands; however, combinations including more than one LP/P variant were not observed in this preliminary sample.
Table 2.
Most Severe Variant Among 97 Probands with DCM, by Ethnicity and Ancestry
| Benign/Likely Benign | Uncertain Signficance | Likely Pathogenic | Pathogenic | Total | |
|---|---|---|---|---|---|
| N (Row %) | |||||
| Non-Hispanic, White | 14 (29.2%) | 21 (43.8%) | 10 (20.8%) | 3 (6.3%) | 48 |
| Hispanic, White | 0 (0%) | 2 (100%) | 0 (0%) | 0 (0%) | 2 |
| Non-Hispanic, African Ancestry | 23 (48.9%) | 22 (46.8%) | 2 (4.3%) | 0 (0%) | 47 |
| Hispanic, African Ancestry | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 |
| TOTAL | 37 (38.1%) | 45 (46.4%) | 12 (12.4%) | 3 (3.1%) | 97 |
Row percentages may not add to exactly 100% due to rounding
Table 3.
Variant Details for Likely Pathogenic and Pathogenic Variants, by Ethnicity and Ancestry
| Adjudication | Ethnicity, Ancestry | Gene | HGVSc | HGVSp | Impacts |
|---|---|---|---|---|---|
| Pathogenic | Non-Hispanic, White | LMNA | NM_170707.3:c.673C>T | NP_733821.1:p.Arg225Ter | Stop Gained |
| TNNT2 |
NM_000364.3:c.650_652delAGA NM_001001430.2:c.629_631delAGA |
NP_000355.2:p.Lys217del NP_001001430.1:p.Lys210del |
Inframe Deletion | ||
| TTN |
NM_001267550.1:c.61876C>T NM_133378.4:c.54172C>T |
NP_001254479.1:p.Arg20626Ter NP_596869.4:p.Arg18058Ter |
Stop Gained | ||
| Likely Pathogenic | Non-Hispanic, White | BAG3 | NM_004281.3:c.1417C>T | NP_004272.2:p.Arg473Ter | Stop Gained |
| FLNC | NM_001458.4:c.4926_4927insACGTCACA | NP_001449.3:p.Val1643ThrfsTer26 | Frameshift Variant | ||
| FLNC | NM_001458.4:c.1948C>T | NP_001449.3:p.Arg650Ter | Stop Gained | ||
| LMNA | NM_170707.3:c.949G>A | NP_733821.1:p.Glu317Lys | Missense Variant | ||
| RBM20 | NM_001134363.1:c.1906C>T | NP_001127835.1:p.Arg636Cys | Missense Variant | ||
| TTN |
NM_001267550.1:c.90844delA NM_133378.4:c.83140delA |
NP_001254479.1:p.Thr30282ArgfsTer8 NP_596869.4:p.Thr27714ArgfsTer8 |
Frameshift Variant | ||
| TTN |
NM_001267550.1:c.88703_88704delAC NM_133378.4:c.80999_81000delAC |
NP_001254479.1:p.His29568LeufsTer7 NP_596869.4:p.His27000LeufsTer7 |
Frameshift Variant | ||
| TTN |
NM_001267550.1:c.66804_66807delGAAG NM_133378.4:c.59100_59103delGAAG |
NP_001254479.1:p.Lys22269HisfsTer10 NP_596869.4:p.Lys19701HisfsTer10 |
Frameshift Variant | ||
| TTN |
NM_001267550.1:c.64915C>T NM_133378.4:c.57211C>T |
NP_001254479.1:p.Arg21639Ter NP_596869.4:p.Arg19071Ter |
Stop Gained | ||
| TTN |
NM_001267550.1:c.71307_71310dupTGAC NM_133378.4:c.63603_63606dupTGAC |
NP_001254479.1:p.Ser23771Ter NP_596869.4:p.Ser21203Ter |
Frameshift Variant | ||
| Non-Hispanic, African Ancestry | TTN |
NM_001267550.1:c.83515C>T NM_133378.4:c.75811C>T |
NP_001254479.1:p.Arg27839Ter NP_596869.4:p.Arg25271Ter |
Stop Gained | |
| TTN |
NM_001267550.1:c.66931_66932delAA NM_133378.4:c.59227_59228delAA |
NP_001254479.1:p.Lys22311ValfsTer3 NP_596869.4:p.Lys19743ValfsTer3 |
Frameshift Variant |
Discussion
We tailored the ACMG and ClinGen MYH7 variant interpretation criteria to DCM. Previous publications report a ~27% detection rate for LP/P variants in DCM,31 however, the detection rate for LP/P variants using our criteria so far has only been ~16%; a significant proportion of these variants are predicted LOF. Significant due diligence was done when applying PVS1 rules for this study. Variants affecting the initiation codon are not automatically assumed to cause haploinsufficiency due to the possible rescue mechanism of a downstream methionine. Also, variants predicted to lead to premature termination codon or predicted to affect mRNA stability occurring in the last exon and last 55nt of the second-last exon possibly escaping nonsense mediated decay were excluded from PVS1 application. Similarly, invariant splice site variants would be considered as leading to a null variant if a new in-frame splice junction is not predicted and the variant affects all biologically functional transcripts known to be active in the heart and altered in DCM. These principles are consistent with published guidance for interpretation of LOF variants.32
Compared to a decade ago, concluding pathogenicity in Mendelian disorders requires a higher level of evidence.33 This approach strengthens the degree of certainty when reporting variants. While our approach adds gene-specificity, a limitation not unique to this study is that variant adjudication is a probabilistic process. Achieving precision medicine in DCM requires large data sets and functional studies that demonstrate pathogenicity. Further evaluation of these criteria by an independent group, such as the ClinGen CMP-EP, could serve the community by generating international consensus on variant interpretation for DCM.
With continued use of our criteria and an ever-expanding literature body on variant interpretation, cases will emerge that provide opportunities to refine this framework. For example, we recognize that restricting pathogenicity to the A-band of TTN excludes potentially relevant regions that can be identified with more granular analyses.34, 35 We have the infrastructure to support ongoing, thoughtful reanalysis and refinement of our criteria. At study completion, a re-evaluation will be undertaken, incorporating best practices and any new data. In addition, there is potential for reassessment of low-quality variants and those that were not adjudicated. Variant reclassifications may result from this process, and, following published principles for researchers returning results,36 should this occur while the study is active, our protocol has provisions for re-contacting participants.
DCM genetic testing results almost always impact families. In probands, LP/P results may directly affect treatment. For example, DCM patients with LP/P variants in arrhythmogenic genes (e.g., LMNA, FLNC) should have an implantable cardioverter defibrillator placed earlier.3 Negative testing results neither exclude the possibility that the proband’s condition has a genetic etiology, nor relieves relatives from risk.
Variant reclassifications also have an impact. The highest-impact reclassifications involve downgrading of LP/P variants. In this scenario, where at-risk relatives would have been offered cascade testing, those found not to carry the variant would have been informed in error that no additional cardiac surveillance is recommended. We note, however, that in the case of LP variants a 10% error rate is assumed,6 participants in this category are informed that no additional surveillance is needed beyond what would routinely be recommended for the general population. The counseling conversation also includes discussion of symptoms that should be reported to their physician. Despite these caveats, relatives who do not carry a familial LP or P variant may be discharged from surveillance. When instead of downgraded, variants are upgraded to LP/P, cascade testing is made possible, without which at-risk relatives would have had only the option of cardiac screening. Reclassifications involving changes to and from LB/B and VUS have less clinical impact: cardiac screening should continue in at-risk relatives in every case.3, 4
Although we prefer a conservative approach for return of results in this study, understanding the genetic architecture of DCM will require substantial further research. Indeed, a proportion of DCM may be oligogenic,37 but the proportion following such a mechanism remains uncertain. We note that a high rate of VUSs in BRCA genes were reclassified as LB or B, although in that study only 6.4% (2868 of 44,777) of unique variants were reclassified;38 whether these data predict a similar eventual resolution of VUSs observed in DCM also remains uncertain. Augmented methods for variant interpretation allowing the assessment of criteria consistent with oligogenic inheritance will be needed for disorders with complex genetics, including DCM. Such methods may provide a pathway to evaluate variants that currently do not exceed a VUS classification.
Cohort diversity is essential to understanding the genetic architecture of complex inherited disorders39, including DCM. Our study aims to evaluate the genetic foundation of DCM, regardless of family history, racial, or ethnic background. Our sample of 97 probands included 47 individuals of Non-Hispanic, African Ancestry; actionable variants were identified in only 2 of these participants (4.3% vs. 27.1% in those of Non-Hispanic European ancestry). Although a variant may reach a pathogenic classification based on data from a single family, in reality most pathogenic variants, particularly missense variants, reach such classification only after combining evidence from multiple families and resources. Our study is contributing to the evidence base needed to adjudicate variants in non-European ancestry populations, but at the same time depends on currently existing evidence to classify variants in individuals of diverse backgrounds. Although we present these preliminary results for the purpose of detailing the development and performance of our variant adjudication and return of results framework, our findings echo the call for ongoing attention and inclusion of non-European ancestry families in genetics research.40, 41
Conclusion
The DCM Precision Medicine Study has developed an approach to variant classification by adapting ACMG/AMP and ClinGen guidelines. Following modifications that were deemed necessary after a pilot study of 81 probands, the new framework was applied to a test set of 97 probands, and locked for continued use in this DCM research study. Our preliminary data shows that 15.5% of DCM probands had LP/P variants, and most of these were identified in individuals of Non-Hispanic European ancestry. Our results highlight the value of ethnically and racially diverse family units in research, but also illustrate the challenges in variant interpretation for genetically complex disorders.
Supplementary Material
Acknowledgments:
We thank the DCM families who have participated in this study, without whom this effort would not be possible.
Sources of Funding: Research reported in this publication was supported by a parent award from the National Heart, Lung, And Blood Institute of the National Institutes of Health under Award Number R01HL128857 (Dr. Hershberger), which included a supplement from the National Human Genome Research Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Nonstandard Abbreviations and Acronyms:
- ACMG
American College of Medical Genetics and Genomics
- AMP
Association of Molecular Pathology
- CDWG (of ClinGen)
Cardiovascular Domain Working Group
- ClinGen
Clinical Genome Resource
- CMP-EP (of ClinGen)
Cardiomyopathy Expert Panel
- DCM
dilated cardiomyopathy
- HE
Hispanic ethnicity
- LOF
loss of function
- LP
likely pathogenic
- NHE
non-Hispanic ethnicity
- OR
odds ratio
- P
pathogenic
- VUS
variant of uncertain significance
Appendix: DCM Consortium institutions and personnel participating in this study.
-
Study Principal Investigator and Co-Investigators:
The Ohio State University: Ray E. Hershberger (Principal Investigator), Daniel D. Kinnamon, Ana Morales, Elizabeth Jordan
Nationwide Children’s Hospital: Julie M. Gastier-Foster
University of Washington: Wylie Burke, Deborah J. Bowen, Deborah A. Nickerson, Michael O. Dorschner.
-
DCM Consortium Clinical Site Principal Investigators and Clinical Site Other Significant Contributors (OSC). The following clinical sites and individuals contributed to the submission of R01HL128857 as Site Principal Investigators (Site PI) or as Other Significant Contributors (OSC):
The Ohio State University: Garrie Haas (Site PI), William Abraham (OSC), Philip Binkley (OSC), Ayesha Hasan (OSC), Jennifer Host (OSC), Brent Lampert (OSC), Sakima Smith (OSC)
Tufts University School of Medicine: Gordon Huggins (Site PI), Dr. Huggins also served as study co-principal investigator; David DeNofrio (OSC), Michael Kiernan (OSC)
University of Washington: Daniel Fishbein (Site PI), Richard Cheng (OSC), Todd Dardas (OSC), Wayne Levy (OSC), Claudius Mahr (OSC), Sofia Masri (OSC), April Stempien-Otero (OSC)
University of Maryland: Stephen Gottlieb (Site PI)
Stanford University Medical Center: Matthew Wheeler (Site PI), Euan Ashley (OSC)
Medstar Health Research Institute, Washington Hospital Center: Mark Hofmeyer (Site PI)
Cleveland Clinic Lerner College of Medicine: Wilson Tang (Site PI), Randall Starling (OSC), Rocio Moran (OSC)
University of Pennsylvania: Anjali Owens (Site PI), Kenneth Marguilies (OSC), Thomas Cappola (OSC), Lee Goldberg (OSC), Susan Brozena (OSC), J. Rame (OSC), Rhondalyn McLean (OSC)
University of Mississippi Medical Center: Charles Moore (Site PI), Matthew deShazo (OSC), Robert Long (OSC)
South Miami Hospital: Francisco Jimenez Carcamo (Site PI), Hakop Hrachian-Haftevani (OSC)
The Methodist Hospital Research Institute: Barry Trachtenberg (Site PI), Guhu Ashrith (OSC), Arvind Bhimarahj (OSC), Jerry Estep (OSC)
-
The following clinical site was added following approval of NHGRI supplemental funding but prior to initiation of enrollment:
University of Arizona: Nancy Sweitzer (Site PI)
-
The following clinical sites were added following study activation:
Inova Heart and Vascular Institute: Palak Shah (Site PI)
University of Nebraska Medical Center: Brian Lowes (Site PI)
Louisiana State University: Frank Smart (Site PI)
Emory University: Alanna Morris (Site PI)
Northwestern University: Jane Wilcox (Site PI)
New York University Langone Medical Center: Stephan Pan (Site PI)
Washington University: Gregory Ewald (Site PI)
University of Michigan: Keith Aaronson (Site PI)
University of California – Los Angeles: Jessica Wang (Site PI)
University of Alabama – Birmingham: Salpy Pamboukian (Site PI)
Medical University of South Carolina: Daniel Judge (Site PI)
Cedars-Sinai Medical Center: Evan Kransdorf (Site PI)
University of Texas Southwestern Medical Center: Sonia Garg (Site PI)
-
Study Consultants
Gail P. Jarvik, University of Washington
Eden R. Martin, University of Miami Miller School of Medicine
Heidi Rehm, Harvard Medical School
-
National Institutes of Health Program Personnel
National Heart, Lung, and Blood Institute: Patrice Desvigne-Nickens, James Troendle, Yi-Ping Fu.
National Human Genome Research Institute: Lucia Hindorff
Footnotes
Disclosures: none
References:
- 1.Hershberger RE, Morales A, Siegfried JD. Clinical and genetic issues in dilated cardiomyopathy: a review for genetics professionals. Genet Med. 2010;12:655–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kinnamon DD, Morales A, Bowen DJ, Burke W, Hershberger RE, DCM Consortium. Toward Genetics-Driven Early Intervention in Dilated Cardiomyopathy: Design and Implementation of the DCM Precision Medicine Study. Circ Cardiovasc Genet. 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hershberger RE, Givertz M, Ho CY, Judge DP, Kantor P, McBride KL, Morales A, Taylor MRG, Vatta M, Ware SM. Genetic Evaluation of Cardiomyopathy - a Heart Failure Society of America Practice Guideline. J Card Fail. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hershberger RE, Givertz MM, Ho CY, Judge DP, Kantor PF, McBride KL, Morales A, Taylor MRG, Vatta M, Ware SM, et al. Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2018;20:899–909. [DOI] [PubMed] [Google Scholar]
- 5.Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, Lyon E, Ward BE. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med. 2008;10:294–300. [DOI] [PubMed] [Google Scholar]
- 6.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, Ledbetter DH, Maglott DR, Martin CL, Nussbaum RL, et al. ClinGen--the Clinical Genome Resource. N Engl J Med. 2015;372:2235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Milko LV, Funke BH, Hershberger RE, Azzariti DR, Lee K, Riggs ER, Rivera-Munoz EA, Weaver MA, Niehaus A, Currey EL, et al. Development of Clinical Domain Working Groups for the Clinical Genome Resource (ClinGen): lessons learned and plans for the future. Genet Med. 2019;21:987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelly MA, Caleshu C, Morales A, Buchan J, Wolf Z, Harrison SM, Cook S, Dillon MW, Garcia J, Haverfield E, et al. Adaptation and validation of the ACMG/AMP variant classification framework for MYH7-associated inherited cardiomyopathies: recommendations by ClinGen’s Inherited Cardiomyopathy Expert Panel. Genet Med. 2018;20:351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eilbeck K, Lewis SE, Mungall CJ, Yandell M, Stein L, Durbin R, Ashburner M. The Sequence Ontology: a tool for the unification of genome annotations. Genome Biol. 2005;6:R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Rijsingen IA, Nannenberg EA, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, van der Kooi AJ, van Tintelen JP, van den Berg MP, Grasso M, et al. Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. Eur J Heart Fail. 2013;15:376–84. [DOI] [PubMed] [Google Scholar]
- 12.Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, et al. Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2016;19:192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berne P, Brugada J. Brugada syndrome 2012. Circ J. 2012;76:1563–71. [DOI] [PubMed] [Google Scholar]
- 14.Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J, Brugada P, Fressart V, Guerchicoff A, Harris-Kerr C, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010;7:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gosselin-Badaroudine P, Keller DI, Huang H, Pouliot V, Chatelier A, Osswald S, Brink M, Chahine M. A proton leak current through the cardiac sodium channel is linked to mixed arrhythmia and the dilated cardiomyopathy phenotype. PLoS One. 2012;7:e38331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abe K, Machida T, Sumitomo N, Yamamoto H, Ohkubo K, Watanabe I, Makiyama T, Fukae S, Kohno M, Harrell DT, et al. Sodium channelopathy underlying familial sick sinus syndrome with early onset and predominantly male characteristics. Circ Arrhythm Electrophysiol. 2014;7:511–7. [DOI] [PubMed] [Google Scholar]
- 17.Ortiz-Genga MF, Cuenca S, Dal Ferro M, Zorio E, Salgado-Aranda R, Climent V, Padron-Barthe L, Duro-Aguado I, Jimenez-Jaimez J, Hidalgo-Olivares VM, et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J Am Coll Cardiol. 2016;68:2440–2451. [DOI] [PubMed] [Google Scholar]
- 18.Franaszczyk M, Bilinska ZT, Sobieszczanska-Malek M, Michalak E, Sleszycka J, Sioma A, Malek LA, Kaczmarska D, Walczak E, Wlodarski P, et al. The BAG3 gene variants in Polish patients with dilated cardiomyopathy: four novel mutations and a genotype-phenotype correlation. J Transl Med. 2014;12:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Norton N, Li D, Reider MJ, Siegfried JD, Rampersaud E, Zuchner S, Mangos S, Gonzalez Quintana J, Wang L, McGee S, et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am J Hum Genet. 2011;88:273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gigli M, Begay RL, Morea G, Graw SL, Sinagra G, Taylor MR, Granzier H, Mestroni L. A Review of the Giant Protein Titin in Clinical Molecular Diagnostics of Cardiomyopathies. Front Cardiovasc Med. 2016;3:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, Buchan RJ, Walsh R, John S, Wilkinson S, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015;7:270ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schafer S, de Marvao A, Adami E, Fiedler LR, Ng B, Khin E, Rackham OJ, van Heesch S, Pua CJ, Kui M, et al. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet. 2017;49:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huttner IG, Wang LW, Santiago CF, Horvat C, Johnson R, Cheng D, von Frieling-Salewsky M, Hillcoat K, Bemand TJ, Trivedi G, et al. A-Band Titin Truncation in Zebrafish Causes Dilated Cardiomyopathy and Hemodynamic Stress Intolerance. Circ Genom Precis Med. 2018;11:e002135. [DOI] [PubMed] [Google Scholar]
- 25.Haghighi K, Kolokathis F, Pater L, Lynch RA, Asahi M, Gramolini AO, Fan GC, Tsiapras D, Hahn HS, Adamopoulos S, et al. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest. 2003;111:869–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiu C, Tebo M, Ingles J, Yeates L, Arthur JW, Lind JM, Semsarian C. Genetic screening of calcium regulation genes in familial hypertrophic cardiomyopathy. J Mol Cell Cardiol. 2007;43:337–43. [DOI] [PubMed] [Google Scholar]
- 27.Landstrom AP, Adekola BA, Bos JM, Ommen SR, Ackerman MJ. PLN-encoded phospholamban mutation in a large cohort of hypertrophic cardiomyopathy cases: summary of the literature and implications for genetic testing. Am Heart J. 2011;161:165–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ceholski DK, Trieber CA, Holmes CF, Young HS. Lethal, hereditary mutants of phospholamban elude phosphorylation by protein kinase A. J Biol Chem. 2012;287:26596–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Rijsingen IA, van der Zwaag PA, Groeneweg JA, Nannenberg EA, Jongbloed JD, Zwinderman AH, Pinto YM, Dit Deprez RH, Post JG, Tan HL, et al. Outcome in phospholamban R14del carriers: results of a large multicentre cohort study. Circ Cardiovasc Genet. 2014;7:455–65. [DOI] [PubMed] [Google Scholar]
- 30.Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D, et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am J Hum Genet. 2016;99:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, Bowser M, Harrison B, Aaron D, Mahanta LM, et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. 2014;16:601–8. [DOI] [PubMed] [Google Scholar]
- 32.Abou Tayoun AN, Pesaran T, DiStefano MT, Oza A, Rehm HL, Biesecker LG, Harrison SM, ClinGen Sequence Variant Interpretation Working G. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum Mutat. 2018;39:1517–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amendola LM, Jarvik GP, Leo MC, McLaughlin HM, Akkari Y, Amaral MD, Berg JS, Biswas S, Bowling KM, Conlin LK, et al. Performance of ACMG-AMP Variant-Interpretation Guidelines among Nine Laboratories in the Clinical Sequencing Exploratory Research Consortium. Am J Hum Genet. 2016;98:1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deo RC. Alternative Splicing, Internal Promoter, Nonsense-Mediated Decay, or All Three: Explaining the Distribution of Truncation Variants in Titin. Circ Cardiovasc Genet. 2016;9:419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akinrinade O, Alastalo TP, Koskenvuo JW. Relevance of truncating titin mutations in dilated cardiomyopathy. Clin Genet. 2016;90:49–54. [DOI] [PubMed] [Google Scholar]
- 36.Jarvik GP, Amendola LM, Berg JS, Brothers K, Clayton EW, Chung W, Evans BJ, Evans JP, Fullerton SM, Gallego CJ, et al. Return of genomic results to research participants: the floor, the ceiling, and the choices in between. Am J Hum Genet. 2014;94:818–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cowan JR, Kinnamon DD, Morales A, Salyer L, Nickerson DA, Hershberger RE. Multigenic Disease and Bilineal Inheritance in Dilated Cardiomyopathy Is Illustrated in Nonsegregating LMNA Pedigrees. Circ Genom Precis Med. 2018;11:e002038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mersch J, Brown N, Pirzadeh-Miller S, Mundt E, Cox HC, Brown K, Aston M, Esterling L, Manley S, Ross T. Prevalence of Variant Reclassification Following Hereditary Cancer Genetic Testing. JAMA. 2018;320:1266–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hindorff LA, Bonham VL, Ohno-Machado L. Enhancing diversity to reduce health information disparities and build an evidence base for genomic medicine. Per Med. 2018;15:403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Petrovski S, Goldstein DB. Unequal representation of genetic variation across ancestry groups creates healthcare inequality in the application of precision medicine. Genome Biol. 2016;17:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Landry LG, Rehm HL. Association of Racial/Ethnic Categories With the Ability of Genetic Tests to Detect a Cause of Cardiomyopathy. JAMA Cardiol. 2018;3:341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.