

Abstract
目的
探讨一个遗传性凝血因子Ⅴ(FⅤ)缺陷症家系的分子致病机制。
方法
DNA直接测序法分析先证者F5的全部外显子、侧翼序列、5′和3′端非翻译区及家系成员(共3代11人)相应的突变位点区域。通过CAT法检测凝血酶生成量;用ClustalX软件分析突变位点的保守性;用MutationTaster、PolyPhen-2、PROVEAN、LRT和SIFT等在线生物信息学软件预测突变位点对蛋白质功能的影响;用Swiss-PdbViewer软件分析氨基酸突变前后蛋白模型及分子间作用力的变化。
结果
先证者F5第8外显子存在c.1258G>T杂合错义突变(p.Gly392Cys)及第14外显子存在c.4797delG杂合缺失突变,导致框移并产生截断蛋白(p.Glu1572Lys fsX19);其祖父和父亲存在p.Gly392Cys杂合突变;其外祖母、母亲、小姨母和表妹均存在p.Glu1572Lys fsX19杂合突变。先证者凝血酶生成延迟和达峰时间比值明显增高。保守性分析结果表明,p.Gly392在10种同源物种中位于保守区域。五个在线生物信息学软件对p.Gly392Cys预测均显示为致病的突变,Mutation Taster对p.Glu1572Lys fsX19预测也显示为致病突变。蛋白模型分析显示,Gly392突变为Cys392后可导致原有氢键延长,并形成新的空间位阻,影响蛋白结构的稳定性。
结论
该家系F5第8外显子c.1258G>T杂合错义突变及第14外显子c.4797delG杂合缺失突变可能与该家系FⅤ水平降低有关。
Keywords: 凝血因子Ⅴ缺陷症, 遗传性, 生物信息学
Abstract
Objective
To explore the molecular pathogenesis of a family with hereditary factor Ⅴ(FⅤ)deficiency.
Methods
All the exons, flanking sequences, 5′ and 3′ untranslated regions of the F5 of the proband, and the corresponding mutation sites of the family members were analyzed via direct DNA sequencing. The CAT measurement was used to detect the amount of thrombin produced. The ClustalX software was used to analyze the conservation of mutation sites. The online bioinformatics software, Mutation Taster, PolyPhen-2, PROVEAN, LRT, and SIFT were applied to predict the effects of mutation sites on protein function. The Swiss-PdbViewer software was used to analyze the changes in the protein model and intermolecular force before and after amino acid variation.
Results
The proband had a heterozygous missense mutation c.1258G>T(p.Gly392Cys)in exon 8 of the F5, and a heterozygous deletion mutation c.4797delG(p.Glu1572Lys fsX19)in exon 14, which results in a frameshift and produces a truncated protein. Her grandfather and father had p.Gly392Cys heterozygous variation, whereas her maternal grandmother, mother, little aunt, and cousin all had p.Glu1572LysfsX19 heterozygous variation. The ratio of proband's thrombin generation delay to peak time was significantly increased. Conservation analysis results showed that p.Gly392 was located in a conserved region among the 10 homologous species. Five online bioinformatics software predicted that p.Gly392Cys was pathogenic, and Mutation Taster also predicted p.Glu1572Lys fsX19 as a pathogenic variant. Protein model analysis showed that the replacement of Gly392 by Cys392 can lead to the extension of the original hydrogen bond and the formation of a new steric hindrance, which affected the stability of the protein structure.
Conclusion
The c.1258G>T heterozygous missense mutation in exon 8 and the c.4797delG heterozygous deletion mutation in exon 14 of the F5 may be responsible for the decrease of FⅤ levels in this family.
Keywords: Coagulation factor Ⅴ deficiency, Hereditary, Bioinformatics
遗传性凝血因子Ⅴ(FⅤ)缺陷症是一种罕见的常染色体隐性遗传性出血性疾病,发病率约为1/100万[1]–[2]。主要表现为FⅤ活性(FⅤ∶C)降低以及轻重不一的出血或血栓形成,临床表现严重程度与F5基因突变的类型、数目、位点有关。轻至中度FⅤ缺陷症一般由杂合突变引起,通常无临床症状;重度FⅤ缺陷症一般由纯合子或复合杂合突变引起,临床表现为程度不等的出血(皮肤瘀斑、鼻出血、月经量过多、术后出血等)[3]。因此,分析FⅤ基因突变对遗传性FⅤ缺陷症具有重要意义。本文对1例遗传性FⅤ缺陷症患者及家系成员进行实验室表型和基因分析,初步探讨其分子发病机制。
对象与方法
1. 家系资料:先证者,女,27岁,汉族,浙江省瑞安市人,因先兆流产于2020年4月入住于我院中医妇科病房行保胎治疗。凝血常规检查发现血浆凝血酶原时间(PT)34.6 s(参考值12.5~14.5 s)、活化部分凝血活酶时间(APTT)103.9 s(参考值27.0~41.0 s),FⅤ∶C 3%(参考值86%~114%)、FⅤ抗原(FⅤ∶Ag)5%(参考值70%~140%),其他凝血指标无异常。先证者肝、肾功能正常,无明显自发性出血症状。家系其他成员均无自发性出血症状。家系图见图1。
图1. 遗传性凝血因子Ⅴ缺陷症家系图.

2. 健康对照组:选取110名体检健康者作为凝血表型指标对照,其中男76名,女34名,年龄21~48岁,无肝、肾功能异常,无出血及血栓史,无抗凝剂用药史,女性无口服避孕药。本研究通过本院伦理委员会批准(伦理2012-17),所有受试者均签署知情同意书。
3. 标本采集与处理:采集研究对象外周静脉血2.7 ml,用0.109 mol/L枸橼酸钠溶液1∶9抗凝,3 000 r/min离心15 min后,取上层乏血小板血浆用于凝血指标检测,下层血细胞用于基因组DNA抽提。
4. 凝血指标检测:PT、APTT、纤维蛋白原(FIB)、FⅤ∶C、凝血因子Ⅱ活性(FⅡ∶C)、凝血因子Ⅶ活性(FⅦ∶C)和凝血因子Ⅹ活性(FⅩ∶C)采用凝固法在法国Stago STA-R全自动血凝仪上测定(配套试剂由法国STAGO公司提供);FⅤ∶Ag采用酶联免疫双抗体夹心法(纯化的sheep anti-human FⅤIgG由加拿大Cedarlane Laboratories Limited公司提供)。
5. 外周血基因组DNA提取:选用酚-氯仿法提取先证者及家系成员的外周血基因组DNA,并用DU800核酸蛋白分析仪检测所提取基因组DNA的浓度和纯度(基因组DNA提取试剂盒由北京天根生化公司提供)。
6. 引物及PCR扩增:根据F5基因序列(GenBank:AY364535),用Primer Premier 5.0软件分别设计31对引物以覆盖F5基因的所有外显子、5′和3′非翻译区序列、侧翼,引物序列及PCR扩增条件参见文献[4],引物由上海桑尼生物公司合成。
7. PCR产物测序:PCR产物割胶后送上海桑尼生物工程公司纯化后用ABI3730XL型测序仪(美国Applera公司产品)测序。通过Chromas软件与美国NCBI基因库所公布的FⅤ基因序列(GenBank:AY364535)进行比对,查找基因突变的位点,发现错义突变位点后用反向测序予以证实,缺失突变用克隆测序证实。明确先证者基因突变的位点后,再扩增其他家系成员相应突变位点区域并进行测序分析。
8. 凝血酶生成试验:参照文献[5]方法。
9. 氨基酸突变位点保守性分析:用ClustalⅩ-2.1-win软件对人类和NCBI数据库中提供的其他9种同源性物种:原鸡(Gallusgallus)、黑猩猩(Pantroglodytes)、猕猴(Macaca mulatta)、家犬(Canislupus familiaris)、牛(Bos Taurus)、小家鼠(Musmusculus)、热带爪蟾(Xenopus tropicalis)、斑马鱼(Danio rerio)和褐家鼠(Rattus norvegicus)的氨基酸序列(https://www.ncbi.nih.gov/homologene/104)进行比对,分析突变氨基酸的保守性。
10. 生物信息学技术分析:采用MutationTaster、PolyPhen-2、PROVEAN、LRT和SIFT在线软件分析氨基酸突变前后生物信息学特性;用Swiss-pdbViewer软件分析FⅤ蛋白模型野生型和突变型局部空间构型及分子间作用力的变化。
结果
1. 先证者及家系成员表型检测结果:先证者PT、APTT明显延长;FⅤ∶C和FⅤ∶Ag较正常对照显著降低。其祖父、外祖母、母亲、父亲、小姨母和表妹的PT、APTT均轻度延长;除表妹外,FⅤ∶C和FⅤ∶Ag均下降至正常对照的一半左右,家系成员的其他凝血指标均无明显异常(表1)。
表1. 先证者及家系成员主要凝血指标及基因检测结果.
| 家系成员 | 年龄(岁) | PT(s) | APTT(s) | FIB(g/L) | FⅡ∶C(%) | FⅦ∶C(%) | FⅩ∶C(%) | FⅤ∶C(%) | FⅤ∶Ag(%) | 第8外显子c.1258G>T | 第14外显子c.4797delG |
| 先证者(Ⅲ1) | 33 | 34.6 | 103.9 | 4.20 | 97 | 93 | 89 | 3 | 5 | 杂合子 | 杂合子 |
| 祖父(Ⅰ1) | 79 | 15.3 | 42.7 | 3.91 | 95 | 113 | 98 | 57 | 58 | 杂合子 | 野生型 |
| 祖母(Ⅰ2) | 76 | 13.1 | 35.1 | 3.06 | 100 | 104 | 95 | 107 | 110 | 野生型 | 野生型 |
| 外祖父(Ⅰ3) | 78 | 12.9 | 33.3 | 3.65 | 98 | 114 | 111 | 110 | 124 | 野生型 | 野生型 |
| 外祖母(Ⅰ4) | 76 | 14.1 | 41.9 | 2.98 | 90 | 106 | 94 | 51 | 55 | 野生型 | 杂合子 |
| 父亲(Ⅱ1) | 56 | 14.9 | 45.4 | 2.73 | 102 | 112 | 97 | 49 | 54 | 杂合子 | 野生型 |
| 母亲(Ⅱ2) | 55 | 15.4 | 47.2 | 3.26 | 101 | 98 | 98 | 48 | 46 | 野生型 | 杂合子 |
| 二姨母(Ⅱ3) | 52 | 13.2 | 35.3 | 3.14 | 97 | 91 | 85 | 94 | 101 | 野生型 | 野生型 |
| 小姨母(Ⅱ4) | 50 | 15.1 | 45.6 | 2.84 | 87 | 104 | 89 | 50 | 54 | 野生型 | 杂合子 |
| 小姨夫(Ⅱ5) | 53 | 13.4 | 36.1 | 2.46 | 90 | 97 | 96 | 101 | 107 | 野生型 | 野生型 |
| 表妹(Ⅲ2) | 24 | 16.1 | 46.8 | 2.21 | 93 | 90 | 85 | 89 | 95 | 野生型 | 杂合子 |
| 参考值 | 12.5~14.5 | 27.0~41.0 | 2.00~4.00 | 86~116 | 86~120 | 80~120 | 86~115 | 70~130 | |||
注:PT:凝血酶原时间;APTT:活化部分凝血活酶时间;FIB:纤维蛋白原;FⅤ∶Ag:凝血因子Ⅴ抗原;FⅡ∶C、FⅦ∶C、FⅩ∶C、FⅤ∶C分别为凝血因子Ⅱ、Ⅶ、Ⅹ、Ⅴ活性
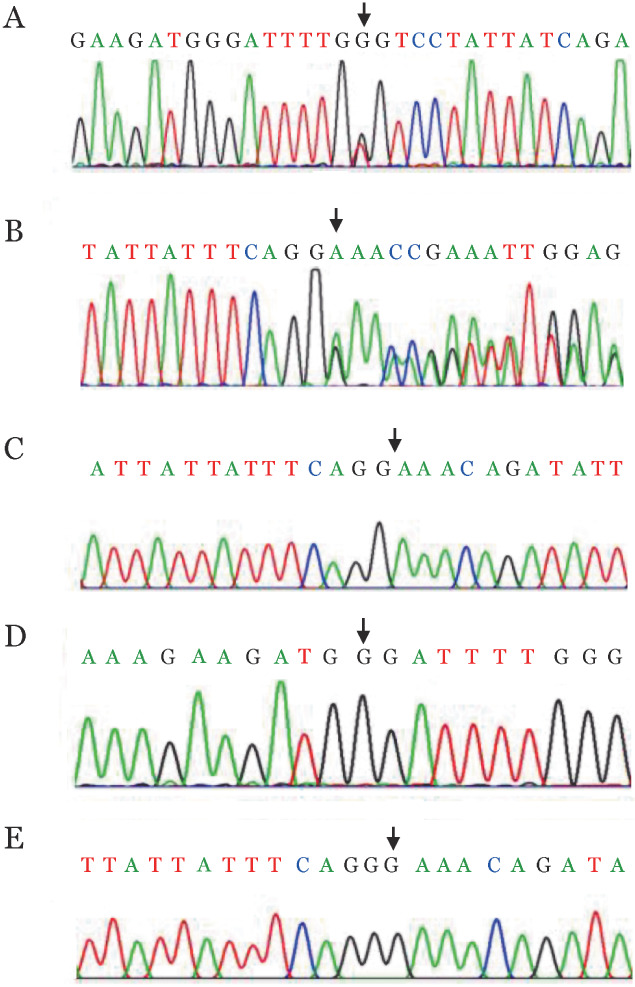
2. 先证者及家系成员基因分析结果:先证者检出F5基因第8外显子c.1258G>T杂合错义突变(p.Gly392Cys)、第14外显子c.4797delG杂合缺失突变(p.Glu1572Lys fsX19),导致1572位的谷氨酸变成赖氨酸后框移19个氨基酸并在1591位提前出现终止翻译,最终产生截断蛋白。其祖父和父亲检出c.1258G>T杂合错义突变,其外祖母、母亲、小姨母和表妹均检出c.4797delG杂合缺失突变,其余家系成员均为野生型(表1、图2)。
图2. F5基因第8和第14外显子测序结果A:先证者c.1258G>T杂合错义突变;B:先证者c.4797delG缺失突变正向测序;C:c.4797delG缺失突变克隆测序;D:第8外显子c.1258野生型;E:第14外显子野生型.
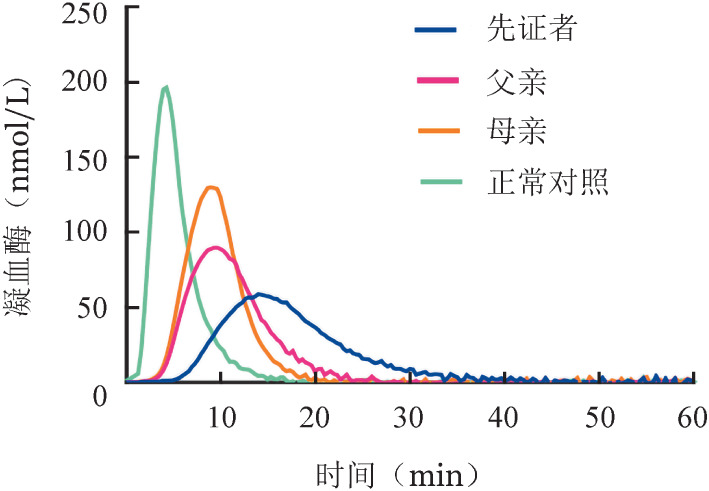
3. 凝血酶生成试验结果:先证者的峰高明显降低,凝血酶生成潜力略微降低,峰高比值为29.6%,显著降低;而其延迟时间(lag time)和达峰时间(tt peak)比值分别延长为3.01和2.92;其父母上述指标也均发生不同程度变化(表2、图3)。
表2. 遗传性凝血因子Ⅴ缺陷症先证者及其父母凝血酶生成试验结果.
| 受检者 | 凝血酶生成潜力比值(%) | 峰值比值(%) | 延迟时间比值 | 达峰时间比值 |
| 先证者(Ⅲ1) | 90.9 | 29.6 | 3.01 | 2.92 |
| 父亲(Ⅱ1) | 92.0 | 45.8 | 2.43 | 2.09 |
| 母亲(Ⅱ2) | 98.4 | 67.0 | 2.34 | 1.98 |
| 参考值 | 86.9~113.1 | 85.3~114.7 | 0.88~1.28 | 0.87~1.15 |
图3. 先证者及其父母的凝血酶生成试验曲线图.
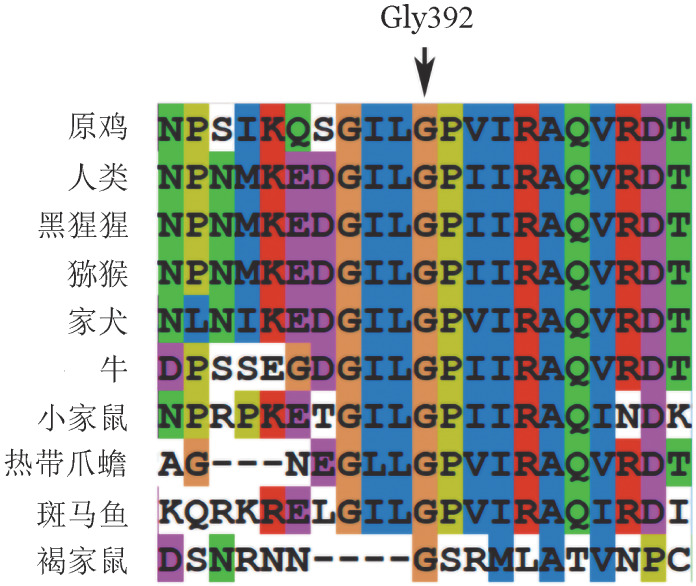
4. 保守性分析结果:ClustalX软件的保守性分析结果表明,p.Gly392在同源物种间高度保守(图4)。
图4. F5基因p.Gly392在同源性物种多重序列比对结果.
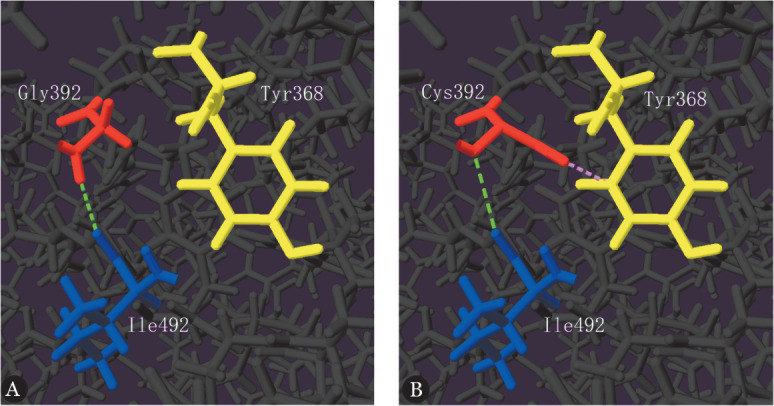
5. 在线生物信息学软件和蛋白结构模型分析结果:MutationTaster、PolyPhen-2、PROVEAN、LRT和SIFT五个在线生物信息学软件对p.Gly392Cys预测结果分别为1.00分、1.00分、−7.67分、0.00分和0.00分,均显示为可影响蛋白质功能的致病突变;MutationTaster对p.Glu1572Lys fsX19预测结果为1.00,显示为可影响蛋白质功能的致病突变。突变蛋白质空间结构模型分析显示:野生型FⅤ蛋白的分子模型结构中Gly392与I1e492之间存在1条氢链,当Gly392突变为Cys392时,其分子之间的氢键明显延长,并且Cys392与Tyr368之间产生了空间位阻,使蛋白质结构发生改变,可能导致蛋白质的稳定性下降(图5)。
图5. 野生型(A)、Gly392Cys突变型(B)凝血因子Ⅴ蛋白质模型图(绿色虚线表示氢键,粉色虚线表示空间位阻).
讨论
FⅤ是一种单链糖蛋白,在凝血途径和活化蛋白C介导的抗凝血途径中均起到重要的作用。遗传性FⅤ缺陷症,多数患者血浆FⅤ∶Ag水平和FⅤ∶C平行下降,称为Ⅰ型;少数患者血浆FⅤ∶Ag水平正常或接近正常,而FⅤ∶C明显下降,即为Ⅱ型[6]。本例患者血浆FⅤ∶C和FⅤ∶Ag平行下降,属于Ⅰ型。F5基因位于染色体1q21-25,全长80 kb,包含25个外显子和24个内含子。其中,第1~12外显子编码长28个氨基酸的信号肽和A1-A2区,第13外显子编码完整的B区,第14~25外显子编码A3-C1-C2区。成熟FⅤ蛋白含有2196个氨基酸,FⅤ分子由3个A区、1个B区及2个C区组成,其排列顺序为A1-A2-B-A3-C1-C2[7],F5基因改变将影响其结构和功能,从而导致FⅤ缺陷症。
目前已发现了约160多种与FⅤ缺陷症有关的基因突变,包括错义突变、无义突变及缺失、插入、剪切位点突变等。错义突变通常引起单个氨基酸置换,影响FⅤ的折叠和构象改变,分泌途径的质量控制系统将其滞留在细胞内,导致细胞内降解和分泌障碍[8]。本研究发现先证者F5第8外显子存在p.Gly392Cys杂合错义突变及第14外显子存在p.Glu1572Lys fsX19杂合缺失突变的复合杂合突变;其祖父和父亲存在p.Gly392Cys杂合错义突变,其外祖母、母亲、小姨母和表妹均存在p.Glu1572LysfsX19杂合缺失突变。存在不同位点单个杂合突变的家系成员,其FⅤ∶C和FⅤ∶Ag均下降至正常对照的一半左右。从凝血酶生成试验显示,携带单个杂合基因突变位点的家系成员,其凝血酶生成量和凝血酶生成速度均较正常人下降,尤其是同时携带这两个突变位点的先证者,表现为更明显,患者的凝血功能受到较大影响,一定程度上评估了出血风险。本研究中F5基因的p.Gly392Cys突变由傅启华等[9]首次报道,该突变使FⅤ蛋白A2结构域的第392位甘氨酸突变为半胱氨酸。Chen等[10]曾报道,p.Gly392Cys突变会引起半胱氨酸残基的暴露,破坏A2结构域支架,干扰了A2结构域上其他半胱氨酸之间二硫键的形成,从而影响蛋白质结构的稳定性,导致FⅤ蛋白质分泌或稳定性受损。
本研究通过Swiss-Pdb Viewer version软件对p.Gly392Cys突变前后的蛋白结构进行模型分析显示:野生型FⅤ蛋白的分子模型结构中Gly392与I1e492之间存在1条氢链,当Gly392突变为Cys392时,其分子之间的氢键明显延长,并且Cys392与Tyr368之间产生了空间位阻,导致蛋白质的稳定性下降,与上述的报道符合。本研究中的p.Glu1572Lys fsX19杂合缺失突变,使1572位的谷氨酸变成赖氨酸后框移19个氨基酸并在1591位提前出现终止翻译,产生截断蛋白,导致18个氨基酸(1572~1590)被替换和606个氨基酸(1591~2197)丢失,最终使完整的FⅤ蛋白质组装受阻,功能障碍。有研究显示,基因突变后形成结构异常的蛋白质,将会被内质网或蛋白酶体降解途径提前降解,导致血浆中含量降低或活性异常,从而影响其生物学功能[11]。本研究还分析了p.Gly392Cys突变的生物学特性,发现p.Gly392Cys在同源物种间高度保守,说明该位点是FⅤ分子的重要位点,在FⅤ蛋白正常功能的发挥中起重要作用。五个在线生物信息学软件对p.Gly392Cys预测均显示为致病的突变,MutationTaster对p.Glu1572Lys fsX19预测也显示为致病的突变,均可影响蛋白质功能,进一步证实了这两个位点野生型氨基酸在FⅤ蛋白中的重要性。
综上所述,本FⅤ缺陷症家系FⅤ水平降低与p.Gly392Cys杂合错义突变和p.Glu1572Lys fsX19杂合缺失突变有关,其具体的致病机制有待体外表达等试验进一步的探讨。
Funding Statement
基金项目:温州市卫生健康委员会医药卫生科学研究项目计划(2015B06)
Fund program: Medical and Health Science Research Project Plan of Health Commission of Wenzhou(2015B06)
References
- 1.Kanaji S, Kanaji T, Honda M, et al. Identification of four novel mutations in F5 associated with congenital factor V deficiency[J] Int J Hematol. 2009;89(1):71–75. doi: 10.1007/s12185-008-0210-4. [DOI] [PubMed] [Google Scholar]
- 2.Asselta R, Peyvandi F. Factor V deficiency[J] Semin Thromb Hemost. 2009;35(4):382–389. doi: 10.1055/s-0029-1225760. [DOI] [PubMed] [Google Scholar]
- 3.Liu HC, Shen MC, Eng HL, et al. Asp68His mutation in the A1 domain of human factor V causes impaired secretion and ineffective translocation[J] Haemophilia. 2014;20(4):e318–e326. doi: 10.1111/hae.12476. [DOI] [PubMed] [Google Scholar]
- 4.谢 耀胜, 张 扬, 朱 丽青, et al. 一个近亲结婚导致的遗传性凝血因子V缺陷症家系分析[J] 中华医学遗传学杂志. 2013;30(2):161–164. doi: 10.3760/cma.j.issn.1003-9406.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 5.金 艳慧, 郝 秀萍, 程 晓丽, et al. 一个遗传性凝血因子Ⅶ缺陷症家系的临床特征和基因分析[J] 中华血液学杂志. 2015;36(5):427–430. doi: 10.3760/cma.j.issn.0253-2727.2015.05.016. [DOI] [Google Scholar]
- 6.曹 丽娟, 王 兆钺, 李 红, et al. 两例遗传性凝血因子Ⅴ缺乏症的产前诊断[J] 中华医学遗传学杂志. 2011;28(6):679–682. doi: 10.3760/cma.j.issn.1003-9406.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 7.Jamila Hirbawi, Michael Kalafatis. Spellbinding Effects of the Acidic COOH-Terminus of Factor Va Heavy Chain on Prothrombinase Activity and Function[J] ACS Omega. 2017;2(9):5529–5537. doi: 10.1021/acsomega.7b00769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu HC, Lin TM, Eng HL, et al. Functional characterization of a novel missense mutation, His147Arg, in A1 domain of FV protein causing type II deficiency[J] Thromb Res. 2014;134(1):153–159. doi: 10.1016/j.thromres.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 9.傅 启华, 王 鸿利, 王 明山, et al. 2种新的凝血因子V基因突变导致的遗传性凝血因子V缺乏症[J] 中华医学杂志. 2003;83(4):312–315. [PubMed] [Google Scholar]
- 10.Chen TY, Lin TM, Chen HY, et al. Gly392Cys missense mutation in the A2 domain of factor V causing severe factor V deficiency: molecular characterization by expression of the recombinant protein[J] Thromb Haemost. 2005;93(3):614–615. [PubMed] [Google Scholar]
- 11.杨 丽红, 金 艳慧, 杨 婷, et al. 两个遗传性蛋白C缺陷症家系表型与基因突变分析[J] 中华医学遗传学杂志. 2017;34(1):10–14. doi: 10.3760/cma.j.issn.1003-9406.2017.01.003. [DOI] [Google Scholar]