

Summary
CpG islands often exhibit low DNA methylation, high histone H3 lysine 4 trimethylation, low nucleosome density, and high DNase I hypersensitivity, yet the rules by which CpG islands are sensed remain poorly understood. In this study, we first evaluated the relationships between the DNA and the chromatin properties of CpG islands in embryonic stem cells using modified bacterial artificial chromosomes. Then, using a bioinformatic approach, we identified strict CpG-island density and length thresholds in mouse embryonic stem and differentiated cells that consistently specify low DNA methylation levels. Surprisingly, the human genome exhibited a dramatically different relationship between DNA properties and DNA methylation levels of CpG islands. Further analysis allowed speculation that this difference is accommodated in part by evolutionary changes in the nucleotide composition of orthologous promoters. Thus, a change in the rules by which CpG-island properties are sensed may have co-evolved with compensatory genome adaptation events during mammalian evolution.
Keywords: CpG islands, DNA methylation, promoters, chromatin, nucleosome, epigenetics, embryonic stem cells, evolution
Graphical abstract
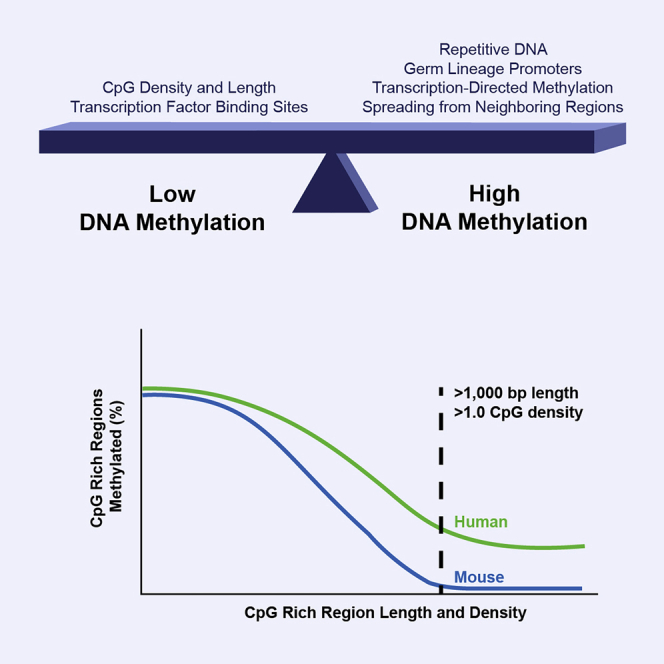
Highlights
-
•
Strict DNA property rules can predict low DNA methylation at murine CpG islands
-
•
Mice and humans differ greatly in how DNA properties influence DNA methylation
-
•
Evolutionary adaptation of CpG islands compensates for the species differences
Langerman and colleagues examined relationships between DNA and chromatin properties of CpG islands. They identified rules by which the length of a CpG-rich region and CpG density influence DNA methylation. These rules differ between humans and mice, with the differences permitted during evolution by adaptations at the DNA and chromatin levels.
Introduction
Two fundamental goals of the eukaryotic gene regulation field are to understand how specific chromatin features are acquired at defined regions of the genome and to determine how these features help orchestrate the proper regulation of gene expression. Many chromatin features, including histone and DNA modifications, are regulated by enzymes recruited by transcription factors that bind DNA in a sequence-specific manner. However, chromatin structure can also be influenced by the nucleotide composition of a DNA region, or by molecular processes such as DNA replication, transcription, and DNA repair (Segal et al., 2006; Deaton and Bird, 2011; Morrison and Shen, 2009).
In mammals, CpG islands serve as an example of the impact of intrinsic nucleotide composition on chromatin structure (Deaton and Bird, 2011). CpG islands are found at approximately 70% of mammalian promoters and have been defined using different CpG dinucleotide prevalence criteria (Irizarry et al., 2009; Wu et al., 2010; Yu et al., 2017). Most definitions consider the length of the CpG-rich region (CGR), GC content, and the ratio of the observed density of CpG dinucleotides to the density expected if CpGs were distributed randomly (obs/exp ratio). Regardless of the criteria used, CpG islands often exhibit low DNA methylation, low nucleosome density, high DNase I hypersensitivity, and detectable histone H3 lysine 4 trimethylation (H3K4me3) (Bock et al., 2007). When associated with poised or silent genes, CpG islands can also exhibit high H3K27me3 (Bernstein et al., 2006).
Experimental evidence supports the hypothesis that the intrinsic DNA properties of CpG islands play an intimate role in specifying their characteristic chromatin properties (Lövkvist et al., 2016). The discovery of a protein motif that recognizes unmethylated CpG dinucleotides, termed the CXXC motif, uncovered one strategy by which the presence of several unmethylated CpGs in a CpG island might be sensed (Clouaire et al., 2012; Xu et al., 2018). CXXC domains can bind directly to and protect unmethylated CpG dinucleotides (Cierpicki et al., 2010). Studies have suggested that the sensing of intrinsic CpG island properties has functional impacts on transcription and development (Agarwal and Shendure, 2020; Hartl et al., 2019). However, the precise rules by which the DNA properties of CpG islands are sensed by CXXC motif-containing proteins or other chromatin regulators remain unknown. For example, is a defined density of CpG dinucleotides within a CGR of defined length essential for an impact on chromatin structure?
The intrinsic instability of nucleosomes assembled at CpG islands may also contribute to their characteristic chromatin properties. Early biochemical experiments demonstrated that GC base pairs help stabilize nucleosomes, but nucleosome stability is compromised in the absence of properly positioned AT dinucleotides, which provide the flexibility needed for DNA to wrap around the histone octamer (Drew and Travers, 1985; Lowary and Widom, 1998). In vitro biochemical studies and genome-wide nucleosome mapping studies have confirmed that CpG islands often exhibit intrinsic nucleosome instability (Fenouil et al., 2012; Ramirez-Carrozzi et al., 2009), raising the possibility that this instability contributes to the acquisition of the other chromatin properties that characterize CpG islands.
Although the DNA properties of CpG islands have the potential to contribute to their characteristic chromatin features, elucidation of this relationship has been confounded by the presence of transcription factor binding sites in CpG-island promoters. In fact, transcription factors have been shown to play a major role in dictating the chromatin properties of CpG islands through their ability to help maintain low CpG methylation levels and recruit enzymes involved in H3K4me3 deposition (Krebs et al., 2014). Is it possible that transcription factor binding is fully responsible for the chromatin properties of CpG islands, or do their DNA properties make an additional contribution?
To increase our understanding of CpG islands, we first evaluated DNA methylation at representative CpG islands and CpG-island variants in the context of bacterial artificial chromosomes (BACs) stably integrated into mouse embryonic stem cells (ESCs). We then carried out a systematic bioinformatic analysis of the relationships between the various DNA properties of CGRs and their characteristic chromatin properties. We also asked whether we could identify CpG-density and CGR-length thresholds that consistently coincided with low DNA methylation levels throughout the genome. This analysis led to a number of insights, the most striking of which was the existence of a dramatically different relationship between DNA properties and the DNA methylation state of the mouse and human genomes. Further analysis led to speculation that the two species adapted to these differences through alterations in the nucleotide composition of orthologous promoters and through the use of distinct mechanisms to mark silent genes.
Results
Experimental Examination of Relationship between CpG island and DNA Methylation in Mouse ESCs
To extend our understanding of the relationship between CpG islands and chromatin properties, we first took advantage of a previously described strategy (Mendenhall et al., 2010) in which defined CGRs corresponding to known or artificial CpG islands were inserted into a 136-kb gene-desert BAC. The modified BACs were then pre-methylated (to help ensure broad methylation in the absence of activities promoting selective loss of methylation) and stably introduced into ESCs, followed by an analysis of DNA methylation in individual stably transfected clones by bisulfite sequencing; the goal was to examine the impact of DNA features on DNA methylation in a BAC system in which the DNA sequences could be readily manipulated. The properties of the 15 sequences inserted into the BAC for the current analysis are summarized in Figure 1A.
Figure 1.

Experimental Analysis of CpG-Rich Sequences in a Gene-Desert BAC
(A) The table displays the basic DNA properties of CpG-rich sequences inserted into a gene-desert BAC, which was then pre-methylated and introduced into mouse ESCs.
(B) The diagrams represent the DNA methylation status in ESCs at inserts of the Il12b CpG island and deletion variants. Each circle represents a particular CpG position, the color of which represents the average ratio of methylated to unmethylated CpGs determined by bisulfite sequencing across multiple clones. The data shown for each insert are an aggregate of three or four clones. The key shows the color that corresponds to each DNA methylation level.
(C) The graph shows qPCR results from H3K4me3 ChIP DNA with primers specific to select inserts from (B). The signal is relative to the input fraction and normalized to a native unmethylated downstream CpG island on the transgenic BAC. The signal at a control region 2 kb upstream of the insertion site is also shown. The black bars represent the standard error from two immunoprecipitation replicates of at least two clones.
(D) The diagrams show the DNA methylation status at transgenic inserts of the 601 sequence and variants, similar to (B).
(E) The graph shows qPCR results from ChIP for H3K4me3 with primers specific to the 601, “601 reduced CpG,” and “601 plus CpG” variant inserts in ESCs. Normalization and controls are as in (C).
(F) The diagrams show DNA methylation levels determined by bisulfite sequencing at transgenic inserts of either 601 or “601 reduced CpG” fused to the Il12b CpG island 207–277 bp sequence from (B).
(G) The graph shows qPCR results from H3K4me3 ChIP DNA with primers specific to the inserts of 601 and “601 reduced CpG” fusion variant fusions to the Il12b CpG island 207–277 bp fragment.
(H) The diagrams show DNA methylation levels at transgenic inserts of tandem 601 (601 (×2)) or Il12b 1–140 bp fragments (Il12b 1–140 bp (×2)). Also shown are DNA methylation levels at an insert composed of 601 plus the adaptor used to adjoin tandem sequences.
(I) The graph shows qPCR results from H3K4me3 ChIP DNA with primers specific to the tandem insertions in (H).
We first used this assay to analyze a small, 277-bp CGR of unknown function located 1 kb upstream of the mouse Il12b gene; this sequence is representative of a non-promoter CGR at the low end of the length and CpG density spectrum (see below) while exhibiting low DNA methylation and a significant H3K4me3 chromatin immunoprecipitation sequencing (ChIP-seq) peak in mouse ESCs (Vincent et al., 2013). When analyzed by bisulfite sequencing and H3K4me3 ChIP in the context of the gene-desert BAC stably integrated into the mouse ESC genome, this Il12b fragment was found to lack DNA methylation and to exhibit an H3K4me3 ChIP signal (Figures 1B and 1C). CpGs flanking the Il12b insert exhibited high DNA methylation and low H3K4me3 (data not shown).
We then analyzed six sub-fragments of this Il12b sequence to determine whether its DNA methylation and H3K4me3 properties are dictated by its overall DNA properties or by a specific DNA element. The results revealed that a 36-bp fragment (207–242) was necessary and sufficient to establish the unmethylated DNA state (Figure 1B), consistent with a model in which a transcription factor binding site rather than the DNA properties of the 277-bp fragment was responsible for the low DNA methylation. The identity of the factor responsible for the low DNA methylation remains unknown, but a near-consensus binding site for CTCF, a known mediator of low DNA methylation, is present within this 36-bp sequence (Feldmann et al., 2013). Consistent with a possible role for CTCF, deletions of 9 or 15 bp within the CTCF binding site resulted in a substantial increase in DNA methylation in the context of the full-length CpG island (data not shown). Notably, only the intact, full-length 277-bp fragment acquired high H3K4me3 (Figure 1C), suggesting that additional transcription factors and/or specific DNA properties are needed for acquisition of this modification.
To further explore the possibility that DNA properties of CGRs might influence acquisition of the characteristic chromatin properties of CpG islands, we focused on a synthetic 150-bp CGR known as 601, which was originally identified as a sequence capable of assembling into a highly stable nucleosome (Lowary and Widom, 1998); this property is attributable to the fact that it combines a high GC content with properly spaced AT dinucleotides to allow optimal wrapping around the histone octamer. When analyzed in the context of the gene-desert BAC, 601 DNA remained heavily methylated and lacked H3K4me3 (Figures 1D and 1E), indicating that 13 CpG dinucleotides in a 150-bp fragment are insufficient for acquisition of low DNA methylation and high H3K4me3. We also tested two variants of 601; in one variant, “601 reduced CpG,” 10 of the 13 CpGs were converted to other dinucleotides, GpC or GpG, thereby retaining only 3 CpGs. In the other variant, “601 plus CpG,” 7 GpCs were converted to CpGs, thereby increasing the CpG density to an obs/exp ratio of 2.13, which exceeds the density of almost all CGRs in the human genome. Interestingly, the CpGs in both constructs remained heavily methylated when tested in the context of the gene-desert BAC, and H3K4me3 levels remained low (Figures 1D and 1E). Thus, altering CpG numbers and CpG density in a short DNA fragment appears insufficient to drive the loss of DNA methylation and acquisition of high H3K4me3.
Importantly, two different types of alterations to the 601 insert led to loss of DNA methylation. First, insertion of the 70-bp Il12b fragment that supports loss of DNA methylation adjacent to 601 promoted the efficient loss of methylation throughout the 601 sequence (Figure 1F), suggesting that the presence of an appropriate transcription factor binding site adjacent to a CpG-rich fragment is sufficient to promote loss of methylation. This spreading activity was not dependent on the high CpG density in 601, as similar loss of methylation was observed when the Il12b sequence was inserted adjacent to the “601 reduced CpG” variant (Figure 1F). The spreading activity also did not require placement of the Il12b fragment immediately adjacent to 601, as reduced 601 methylation was also observed when a 375-bp CpG-deficient BAC spacer fragment was inserted between 601 and the 70-bp Il12b fragment (data not shown). Interestingly, these constructs did not acquire appreciable H3K4me3 levels (Figure 1G), despite having CpG densities and lengths similar to those of the full-length Il12b CpG island.
The second strategy that resulted in loss of DNA methylation at 601 was duplication of the sequence. Despite the inability of a single copy of 601 to support loss of DNA methylation, we observed efficient loss of methylation at the tandem sequence termed “601 (×2),” as well as acquisition of high H3K4me3 (Figures 1H and 1I). An insert containing only one copy of 601 adjoining the linker region used in the tandem version (“601 + adaptor”) retained high DNA methylation, demonstrating that the linker DNA does not contain a cryptic transcription factor binding site to promote methylation loss (Figure 1H). Although nucleosome density was difficult to accurately measure with these short sequences, these results also show that a CGR that is capable of assembling into stable nucleosomes can still readily acquire low DNA methylation and high H3K4me3.
A similar result was obtained when the Il12b CpG island sub-fragment, 1–140 bp, was duplicated; unlike the single insertion of this sequence, the duplicated version efficiently lost its methylation, with moderately increased H3K4me3 (Figures 1H and 1I). Together, the data in this figure support a hypothesis in which both transcription factor binding sites and the DNA properties of CpG islands can contribute to the loss of DNA methylation and acquisition of H3K4me3.
Relationships between the DNA Properties of CpG-Rich Regions
To define further the relationship between the DNA properties of CpG islands and their characteristic chromatin properties, we developed a bioinformatic approach. We first defined a collection of CGRs within the human genome using low-stringency criteria. Most previous studies used higher stringency criteria to capture primarily those DNA segments that function as CpG islands (Irizarry et al., 2009; Wu et al., 2010; Yu et al., 2017). In contrast, a large fraction of the DNA segments within our collection lack CpG-island properties and functions, thereby facilitating a detailed examination of the relationships between DNA properties and chromatin properties.
Specifically, we selected all non-repetitive DNA segments of at least 150 bp that possess an obs/exp ratio for CpG dinucleotides (i.e., CpG density) of at least 0.55. If two or more overlapping 150-bp segments met this density criterion, they were merged into one longer segment. We did not normalize for GC percentage when calculating CpG density; normalization would be based on an uncertain assumption that GC content does not contribute to regulatory functions. Using these criteria, 173,307 CGRs were identified, comprising approximately 5% of the non-repetitive portion of the human genome. As expected, broad ranges of CpG numbers, densities, CGR lengths, and GC percentages were observed within the pool of CGRs (Figure 2A).
Figure 2.

Nucleotide Properties of Human CGRs
(A) The bar graph shows the distribution of the frequency of all 173,307 human CGRs within bins for total CpG number, total length, average CpG density, or average GC content.
(B) A frequency histogram shows the CGR distribution among total length bins and CpG density bins. Red denotes high occurrence. For each length bin, the highest co-occurring CpG density bin is indicated (black box).
From an analysis of the relationships between the various DNA properties of the CGRs, the most striking finding was a dramatic difference in the CpG density distribution as a function of CGR length (Figure 2B). CGRs between 150 and 600 bp in length frequently possess CpG densities at the low end of the range used in the analysis (0.55–0.60). However, CGRs greater than 800 bp in length were found to be distributed around a much higher CpG density peak of 1.00–1.25, with surprisingly few CGRs near the density minimum (Figure 2B). The infrequent occurrence of long CGRs with low average CpG densities is consistent with a hypothesis in which long CGRs have remained unmethylated in germ cell lineages with such high levels of stability that they have had little opportunity for loss of CpG dinucleotides during evolution via 5-methylcytosine deamination (Cohen et al., 2011).
Relationships between Chromatin Properties of CGRs
As an initial step toward evaluating chromatin properties, published datasets from human ESCs (see Experimental Procedures) were analyzed, revealing the expected distribution of each chromatin property among the set of CGRs (Figure S1). Of greatest relevance for the current analysis, a bimodal distribution of DNA methylation was observed in the ESCs, with 17% and 77% of CGRs exhibiting either low (0%–30%) or high (70%–100%) DNA methylation levels, and only 6% exhibiting intermediate levels.
The datasets were then used to examine relationships between the chromatin properties. As expected, these results revealed close correlations between low CpG methylation, high or medium H3K4me3, and high or medium DNase HS (Figure S2). In contrast, nucleosome density and H3K27me3 did not correlate with these chromatin properties (Figure S2). Overall, the results support a model in which the acquisition of histone H3K4me3, DNase HS, and low DNA methylation at CGRs relies on closely linked mechanisms. In contrast, distinct mechanisms may be responsible for the broad acquisition of low nucleosome density and measurable histone H3K27me3.
The modest correlation between DNase HS and nucleosome density (derived from micrococcal nuclease sequencing [MNase-seq] data) was initially surprising, given that both assays provide a measure of the physical accessibility of chromatin to nuclease cleavage. However, this lack of correlation is consistent with a model in which the DNase HS assay monitors the eviction of one or more nucleosomes from a specific portion of a CGR (leading to hypersensitivity to one or more focused DNase I cleavage events), whereas the MNase assay provides an average nucleosome density throughout the entire CGR. In fact, subsequent analyses showed that nucleosome density correlates most closely with GC percentage rather than the chromatin properties associated with active transcription or with transcription itself (data not shown). Thus, we speculate that, although GC percentage may be a major driver of overall nucleosome density within a CGR, this property may have little relevance to transcriptional control.
Further analysis of genomic location revealed that CGRs at promoters exhibited much lower DNA methylation levels, higher H3K4me3, and higher DNase HS than non-promoter CGRs (Figure S3). Promoter CGRs also exhibited higher CpG densities and were generally longer than non-promoter CGRs (Figure S3). Thus, the three properties associated with active chromatin correlate with DNA properties, but also with regions (i.e., promoters) that are known to be transcriptionally active. Subsequent analyses, in which promoter and non-promoter CGRs were separated into bins based on their DNA properties, with the chromatin properties of each bin then examined, yielded results consistent with the notion that both DNA properties and transcription factor binding influence the chromatin properties of CGRs (data not shown). Importantly, repetition of the above analysis with mouse ESCs yielded similar results (data not shown).
DNA Properties in Mouse ESCs that Consistently Coincide with Low DNA Methylation
The data presented above are consistent with models in which the chromatin properties of CpG islands are influenced by both intrinsic DNA properties and transcription factor binding. However, these results consist largely of statistical trends rather than precise rules. We therefore asked whether specific DNA properties could be identified that consistently predict a defined chromatin state, with the analysis first performed with mouse CGRs. We focused on DNA methylation because of the bimodal distribution of this chromatin property, which allowed reliable quantitation and greater consistency among datasets.
When considering CpG density and CGR length simultaneously in a mouse ESC dataset, we first noted that high DNA methylation was not observed at any of the 5,154 promoter CGRs, and at only 1 of the 714 non-promoter CGRs, exhibiting a CpG density >1.0 and a CGR length >1 kb (Figure 3A, mouse ESC v.6.5 data, >1 CpG density/>1,000 bp length rows). Notably, the 5,154 promoter CGRs that fulfill these criteria represent approximately 21% of all RefSeq promoters. When the CGR length stringency was arbitrarily reduced to 600 bp, 2,591 additional promoter and 677 non-promoter CGRs were added to the pool, yet none of the additional promoters and only 2 of the non-promoter CGRs exhibited high DNA methylation (Figure 3A, mouse ESC v.6.5 data, >1 CpG density/600–1,000 bp rows). An arbitrary reduction in the CpG density stringency to 0.8, while maintaining the CGR length stringency of 1 kb, or simultaneous reductions in the length and density stringencies to 600 bp and 0.8, respectively, resulted in only gradual increases in the percentage of methylated CGRs (Figure 3A, mouse ESC v.6.5 data). Examination of additional bins showed that the prevalence of high DNA methylation increases as the CpG density and CGR length stringencies are reduced (data not shown). Importantly, highly similar profiles were observed with a second independent mouse ESC DNA methylation dataset (Figure 3A, mouse ESC E14 data [please note that the total number of CGRs stated differs slightly among datasets due to differences in CGRs exhibiting sequencing reads]).
Figure 3.

Prevalence of Highly Methylated CGRs Identified Using Defined CpG Density/CGR Length Criteria in Mouse and Human ESC and Differentiated Cells
(A and B) The tables show the counts of CGRs in mouse and human DNA methylation datasets that meet the DNA property criteria labeled at left. CGRs are separated by genomic location and by non-overlapping CpG density and length ranges. Cell line names are in parentheses, with all other datasets derived from primary tissues. For all six mouse (A) and six human (B) datasets, the number of CGRs with high DNA methylation (>70%) in each criteria range is shown under “# Meth” next to the number of regions that qualify for the criteria, “Total.” The last column for each methylome group is the percentage of CGRs methylated for each criterion, “% Meth.” The tables are colored by frequency; increased gray indicates higher CGR numbers, while increased red indicates a higher percentage in the “% Meth” column. CGRs with insufficient bisulfite sequencing reads were discarded, which accounts for the slight variation in qualifying CGRs between methylomes. In addition, promoter CGR classifications were confirmed manually for all methylated mouse CGRs shown and all methylated human CGRs >1.0 CpG density and >1 kb CGR length (see Figure 4). PBL, peripheral blood leukocytes; FB, fibroblasts.
(C) Averaged and rounded values for the mouse and human ESC and somatic cell data from (A) and (B) are shown.
Thus, a strict rule appears to exist in mouse ESCs by which, with only one exception out of 5,868 CGRs, a CpG density >1.0 combined with a CGR length >1 kb coincides with an absence of high methylation. This strict rule breaks down gradually as density and length stringencies are reduced. Notably, the small number of CGRs that were methylated in both of the mouse ESC lines are biased toward exons and introns (Figure 3A). This finding is consistent with evidence that Dnmt3b is recruited to gene bodies during active transcription to promote DNA methylation (Baubec et al., 2015; Morselli et al., 2015).
A Competition Model May Explain the DNA Methylation State of CGRs
An examination of the mouse ESC profiles in Figure 3A seems most consistent with a model in which the DNA methylation state of a CGR is dictated by competition between mechanisms that promote an unmethylated state and opposing mechanisms that promote a methylated state. In this speculative model, CpG density and CGR length help promote an unmethylated state, perhaps through their ability to bind Cfp1 or other unmethylated CpG binding proteins (Cierpicki et al., 2010; Clouaire et al., 2012). At CGRs that exceed a CpG density of 1.0 and a length of 1 kb, such a mechanism may be sufficient to ensure an unmethylated state. Transcription factors may also help promote the unmethylated state at CGRs that exceed these density and length thresholds.
In opposition to these two mechanisms, transcription-directed Dnmt3b recruitment is likely to promote DNA methylation at a subset of transcriptionally active gene bodies. Methylation is also likely to be promoted by other mechanisms, such as spreading from flanking regions of low CpG density. According to this model, as the CpG density and CGR length stringencies are reduced, a larger percentage of CGRs are found to be methylated, with the highest percentage of methylated CGRs in gene bodies (due to transcription-coupled Dnmt3b recruitment), the lowest percentage of methylated CGRs at promoters (due to transcription factor-mediated protection from methylation), and an intermediate percentage of methylated CGRs at intergenic regions.
An Altered Competitive Balance in Differentiated Cells
To extend our analysis, we analyzed DNA methylation datasets from four independent differentiated cell types from mice. Interestingly, in all of these datasets, a larger number of CGRs with high methylation were observed in all genomic locations and in all CGR density/length bins in comparison with the two independent mouse ESC datasets (Figure 3A, promoter data summarized in Figure 4A). For example, although no promoters with a CpG density >1.0 and CGR length >1 kb were methylated in ESCs, five promoters in this stringency bin were methylated in all four differentiated cell types (Figure 4A).
Figure 4.

The Relationships between DNA Properties and DNA Methylation Differ in Mice and Humans and during Development
(A) The pie charts show the gene types associated with the highly methylated mouse promoter CGRs identified using the DNA property criteria at the left (see also Figure 3). Each graph shows the number of CGRs for each mouse cell type that have an associated gene that is either germline related, pseudogene/non-coding, or non-germ related (skin, neuronal, etc.). The five CGRs with CpG density >1.0 and length >1 kb and that have high methylation are from the same five genes in all four differentiated cell types; the names of these five genes are displayed to the right.
(B) The pie charts show the characterization of the confirmed promoter human CGRs with CpG density >1.0 and length >1 kb that are methylated from each cell type, as in (A).
(C) The pie charts show a comparison of the methylated mouse and human CGRs with CpG density >1.0 and length >1 kb, at non-promoter locations in frontal cortex. Each graph shows the number of methylated CGRs at exons, introns, or intergenic regions, colored by the key at the bottom.
(D) The dot plot shows the distribution of average DNA methylation in frontal cortex at all CGRs compared with CpG density, for both mouse (red) and human (black).
Interestingly, the same five promoters were methylated in all four cell types and all were found to be associated with sperm-specific genes (Figure 4A), providing initial evidence that somatic cells contain a mechanism to promote the methylation of germ-lineage-specific promoters possessing CpG criteria that typically would promote an unmethylated state. When the CpG density and CGR length stringencies were reduced, additional methylated promoter CGRs were detected in the four datasets from mouse differentiated cells, with a strong bias toward promoters of germ-lineage genes (Figures 3A and 4A). The acquisition of DNA methylation at the promoters of germ-lineage genes appears to act during the maturation of ESCs to epiblasts, as these same promoters were found to be highly methylated in a mouse epiblast reduced-representation bisulfite sequencing dataset (data not shown). These findings are consistent with a study that documented increased methylation at the CpG-island promoters of germ-lineage genes following ESC maturation (Auclair et al., 2014). More generally, the increased DNA methylation observed in somatic cells in comparison with ESCs is consistent with abundant evidence that chromatin structure is in a more open state in ESCs.
Shifting our attention to non-promoter CGRs in the four differentiated cell types, between 31 and 39 non-promoter CGRs exceeding a density of 1.0 and length of 1 kb were found to be highly methylated in comparison with only one in the ESC datasets, with the vast majority of these methylated non-promoter CGRs found in exons (Figure 3A). The prevalence of methylated CGRs in exons in comparison with introns appears to be due to a bias toward methylation of CGRs near the 3′ end of genes, which tends to coincide with exons (i.e., 3′ untranslated regions) rather than introns (data not shown). Similar to the observations in ESCs, the percentage of methylated CGRs increased gradually with reduced CpG stringencies (Figure 3A).
Most notably, increases in the percentages of methylated CGRs increased concordantly at promoters, exons, introns, and intergenic regions in the reduced stringency bins, when the ESC profiles were compared with the somatic cell profiles. This finding is consistent with the competition model, as the balance appears to have shifted in these differentiated cells toward mechanisms that promote methylation and/or away from mechanisms that protect against methylation. Because the altered balance was observed at all genomic locations, we speculate that it is due to reduced potency of a putative mechanism that “measures” CpG density and CGR length to support an unmethylated state. If the altered balance were primarily due to enhanced recruitment of Dnmt3b to gene bodies or to changes in transcription factor binding to promoters, one would expect changes to be observed primarily at gene bodies or promoters.
A Greatly Altered Competitive Balance in Human Cells
Most surprisingly, in human cells in comparison with mouse cells, the balance appears to be tilted much further in favor of the mechanisms promoting DNA methylation. This finding is apparent in two datasets from human ESCs and four datasets from human differentiated cell types (Figures 3B and 4B). Notably, the prevalence of methylated CGRs in human ESCs was comparable to that in human differentiated cell types (Figures 3B and 4B), consistent with the notion that human ESCs are thought to be more analogous to mouse post-epiblast cells than to mouse ESCs (see below). However, much higher percentages of CGRs in all stringency bins and at all genomic locations exhibited high methylation in the six human cell datasets compared with the four datasets derived from differentiated mouse cells (compare Figure 3A to Figure 3B and note averages in Figure 4C).
At human promoters within the highest stringency bin (CpG density >1.0 and CGR length >1.0), methylation was biased toward germ-lineage-specific genes, as in mice (Figure 4B). However, the number of methylated promoters in humans was much larger than in mice and was not limited to germ-lineage genes (compare Figures 4A and 4B). Also similar to the observations in mice, high methylation at human non-promoter locations was most prevalent within exons (Figures 3 and 4C). However, once again, the number of methylated exons was consistently much higher in humans than in mice. For example, mouse and human frontal cortex contained 31 and 629 methylated CGRs, respectively, in the highest stringency bin (Figure 4C). The scatterplot in Figure 4D, which compares DNA methylation and CpG density in mouse and human frontal cortex, further emphasizes the fact that high DNA methylation is observed at CGRs with high CpG densities in humans but not in mice.
As mentioned above, typical human ESC lines, unlike mouse ESC lines, possess molecular characteristics that are most similar to the post-epiblast stage of development, including higher global DNA methylation (Nichols and Smith, 2009). Recently it has been shown that by using certain culture conditions, ESCs can be converted from this primed phenotype to a naive, blastocyst-like stage (Guo et al., 2017; Takashima et al., 2014; Theunissen et al., 2014). By analyzing a DNA methylome generated by Guo et al. (2017) from these naive human ESCs, with a direct comparison with a methylome from the more mature primed cells, we observed that DNA methylation in the high-stringency CGR bins in the naive cells was lower than in the human primed cells (Figure S4), but remained substantially higher than the methylation levels in mouse ESCs (and more comparable to mouse differentiated tissues). Therefore, even at the most blastocyst-like stage with reduced overall DNA methylation, human ESCs maintain a skewed balance toward DNA methylation at CGRs in comparison with mouse ESCs.
Species-Specific Adaptation of Promoter Sequences
The striking difference between humans and mice with respect to the relationship between DNA properties and DNA methylation was unexpected and difficult to rationalize. In particular, one would expect such fundamental relationships to be well conserved through mammalian evolution. Instead, on the basis of the differences described above, many orthologous promoters are predicted to be highly methylated when silent in human cells, but not when silent in mouse cells.
To examine how this fundamental difference might be tolerated, we analyzed the properties of orthologous promoters. Of the human genes whose promoters exhibited a CpG density >1.0, a CGR length >1 kb, and high methylation in human ESCs, 112 had mouse orthologs that could be clearly identified. Strikingly, 25 of these 112 mouse promoters exhibited high methylation in mouse ESCs. On the basis of the data shown in Figure 3, high methylation would not be expected in mice if the mouse promoters possessed the same DNA properties as their human orthologs. Interestingly, each of these mouse promoters exhibited a CpG density that was much lower than that observed in the human ortholog (Figure 5A, right). Furthermore, the two methylated mouse promoters with the highest CpG densities (between 0.75 and 1.00) were shorter than their human orthologs (data not shown). Thus, all of these mouse promoters exhibited properties that are compatible with DNA methylation in mice. In other words, the CpG densities and CGR lengths of the mouse and human promoters appear to have adapted to their distinct, species-specific relationships between DNA properties and DNA methylation; the mouse orthologs possess reduced CpG densities and CGR lengths, allowing them to acquire the same high levels of DNA methylation when silent as their human counterparts. (To determine whether these results can be explained by frequent differences in CpG densities of orthologous promoters, we examined the human orthologs of mouse promoter CGRs in the highest CpG-density bin; 82% of the human orthologs exhibited a high CpG density [>1.0], demonstrating that differences in CpG density are relatively infrequent [data not shown].)
Figure 5.

Species-Specific Adaptation of Promoters to Accommodate the Different Relationships between DNA Properties and DNA Methylation
(A) For this chart, 112 mouse promoter CGRs were selected that were orthologous to methylated human CGRs from ESCs with length >1 kb and CpG density >1.0. The graph to the left shows the corresponding 87 orthologous mouse promoters that were unmethylated in mouse ESCs, numbered and colored by CpG density as described in the key at right; 52 of these mouse ortholog promoters retain high CpG densities that are resistant to DNA methylation in mice. The right graph shows the 25 mouse promoters that were methylated in mouse ESCs similarly labeled and colored; the CpG densities of these promoters are consistently lower than those of their human orthologs, providing an explanation for their ability to be methylated in mouse cells. The very low (<0.55) CpG density group reflects genes for which a mouse ortholog exists, but which have no DNA region that fulfills the minimum criteria of a CGR within 500 bp of the transcription start site (TSS). DNA methylation at very low regions was calculated by averaging all CpGs within 500 bp of the TSS.
(B) Of the 52 mouse promoters from (A) that retain the high CpG densities observed with their human orthologs (and therefore are unmethylated in mice because CGRs with these DNA properties are almost never methylated), 27 were found to be silent in mouse ESCs (<0.5 RPKM). H3K27me3 levels are shown for these 27 promoters, suggesting that H3K27me3 may contribute to the silencing of these “methylation-resistant” mouse promoters.
The mouse orthologs of the remaining 87 methylated human promoters were found to be unmethylated in mouse ESCs (Figure 5A, left). Thirty-five of these 87 mouse promoters exhibited reduced CpG densities, which may allow them to be silenced by DNA methylation in other cell types. However, the other 52 promoters exhibited high CpG densities in mice, just as in humans (Figure 5A, left). Most of these 52 promoter CGRs were also longer than 1 kb, suggesting that they may never be susceptible to DNA methylation in mice, even when silent, in contrast to the high methylation observed at their human orthologs. Twenty-seven of these 52 promoters were expressed below 0.5 RPKM in mouse ESCs. An examination of histone modifications at these 27 promoters revealed that all but one exhibited high or medium levels of H3K27me3 (Figure 5B). These results suggest that, despite CpG properties that render these mouse promoters resistant to DNA methylation (in contrast to their human orthologs), they remain susceptible to silencing via an H3K27me3-dependent mechanism. Thus, we speculate that different rules governing the relationship between DNA properties and DNA methylation in mice and humans can be tolerated because some orthologous promoters have altered their nucleotide compositions in a species-specific manner to maintain susceptibility to DNA methylation in both species; other promoters may rely exclusively on H3K27me3 to maintain a silent state in mice, with DNA methylation contributing to silencing of the human ortholog.
Discussion
Through a systematic analysis of the relationships between the DNA and the chromatin properties of CGRs in the human and mouse genomes, we provide evidence in support of models in which both transcription factor binding and intrinsic DNA properties contribute to the regulation of the chromatin properties at CpG islands. Of greatest importance, an effort to uncover rules by which DNA properties influence DNA methylation revealed striking differences between mouse ESCs and mouse somatic cells, and between mice and humans, with respect to the intrinsic relationship between DNA properties and DNA methylation. We speculate that the genomes of the two species accommodated these differences through adaptation of their promoter sequences and of the modes of silencing employed at orthologous genes.
The profiles described throughout this analysis support a model in which mechanisms promoting the loss of DNA methylation at CpG islands (transcription factor binding and the recognition of CpG-rich DNA regions) compete with mechanisms that promote DNA methylation (transcription-coupled Dnmt3b recruitment and perhaps spreading from CpG-poor regions). The balance between these opposing mechanisms appears to shift during development and to differ in humans and mice. We propose that the shift in this balance is due to a change in the potency of the unknown mechanism by which CpG density and CGR length properties are sensed, since a change in this one property could explain the fact that the competitive balance is shifted at all genomic regions.
It is important to note that, although a surprisingly firm rule can be defined in mouse ESCs, in that only 1 of 5,868 CGRs with a CpG density >1.0 and CGR length >1 kb exhibits high methylation, the breakdown of this rule in reduced stringency bins is gradual. This observation suggests that more refined rules remain to be elucidated to explain why, for example, 30 exonic CGRs in frontal cortex (CpG density >1.0, length >1 kb) are methylated, whereas the remaining 292 exonic CGRs with similar density and length properties remain unmethylated. Interestingly, 3 of these 30 methylated exonic CGRs contain highly repetitive sequences (data not shown), which may promote the acquisition of DNA methylation. The remaining 27 also exhibit a tendency toward repetitive sequence content that differs substantially from that observed at the CGRs that remain unmethylated (data not shown). Additional studies are also needed to gain an understanding of the mechanism by which CpG density and CGR length are measured. As suggested above, variations in this mechanism may be responsible for the species-specific relationships between DNA properties and DNA methylation, thereby increasing the importance of future studies in this direction.
Experimental procedures
Identification and Analysis of CGRs
A script (see below) was written to scan the repeat-masked hg19 human genome and the mm9 mouse genome for CpG dinucleotide occurrence. Sliding 150-bp windows were scanned and selected if the obs/exp CpG ratio was greater than 0.55:
Obs/exp ratio = CpG number ÷(size of window in bp × probability of random CpG [1/16]). CGRs were defined by specific genomic coordinates and overlapping regions were combined.
Distribution analyses were performed using frequency histograms bins in Microsoft Excel. Genomic locations of non-promoter regions (including genic regions) were defined by the UCSC Refseq database. Promoter locations were determined as overlap ± 500 bp of any transcription start site defined in the UCSC RefSeq database. Overlapping locations were called by the following hierarchy: promoter, UTR (omitted in Figure 3), exon, intron, intergenic.
Analysis of CGR Chromatin Properties
ChIP-seq datasets for H3K4me3 and H3K27me3 for H1 human ESCs and E14/Bruce4 mouse ESCs, and H3K4me3 data from post-mortem human frontal cortex, were obtained from the ENCODE database on the UCSC Genome Browser (Bernstein et al., 2010; Ernst et al., 2011; The Mouse ENCODE Consortium et al., 2014). The histone modification signal at CGRs was calculated by averaging of the ChIP sequencing signal across the CGR position interval. DNase HS data for H1, E14 human ESCs, and human frontal cortex were from publicly available datasets (Thurman et al., 2012; Vierstra et al., 2014). DNase HS signals at CGRs were calculated as the maximal peak score overlapped by the CGR interval. MNase nucleosome mapping data for human and mouse ESCs were downloaded from the GEO database (West et al., 2014). Nucleosome density was calculated by averaging the MNase sequencing signal over the CGR interval. DNA methylation levels were acquired from published datasets for the cell types in Figure 3 (Bernstein et al., 2010; dos Santos et al., 2015; Guo et al., 2017; Hon et al., 2013; Kunde-Ramamoorthy et al., 2014; Lister et al., 2009; Lu et al., 2014; Vincent et al., 2013; Ziller et al., 2013). DNA methylation scores were obtained by averaging all individual CpG methylation scores within the CGR. The hg19 reference genome was used for this analysis because all of the published datasets were mapped in hg19.
BAC Modification and Preparation
The human gene-desert RP11-722D BAC was purchased from CHORI-BACPAC. Exogenous sequences were inserted into the BAC as described (Gong and Yang, 2005). BACs were electroporated into SW102 RecA-expressing bacteria and selected for targeted recombination of GalK and replacement of GalK by minimal galactose medium or deoxygalactose, respectively (Warming et al., 2005). A PGK-neomycin-expressing cassette was introduced into the BAC as described (Wang et al., 2001). Successful recombineering was confirmed by restriction enzyme fingerprinting and sequencing of the insert region.
BAC DNA was isolated using the Large Construct Kit (Qiagen) and linearized with the restriction enzyme PI-SceI (New England Biolabs). BACs were pre-methylated as described (Xu et al., 2009) and BAC integrity was verified on a large pulsed-field gel (Bio-Rad CHEF Mapper XA).
Cell Culture
The R1 male mouse ESC line was grown in Knockout DMEM supplemented with 15% fetal bovine serum (Omega), 0.1 mM non-essential amino acids, 2 mM L-glutamine, 1% penicillin/streptomycin, 0.05 mM β-mercaptoethanol, and 1,000 U/mL LIF (ESGRO, Millipore). All culture products were purchased from Gibco unless otherwise noted. ESCs were maintained in gelatin (STEMCELL Technologies)-coated Petri dishes and on a layer of mouse embryonic fibroblasts mitotically inactivated with mitomycin C. ESCs were removed from plates using trypsin-EDTA (STEMCELL Technologies).
ESCs were grown to confluency on a 10-cm plate prior to transduction with 5–20 μg of BAC DNA by electroporation at 0.27 kV, 500 μF. After a short recovery, the ESCs were replated 1:2. G418/neomycin at 255 μg/μL was added for approximately 10 days to select transfected clones. Single colonies were picked and expanded in G418. Genomic DNA was isolated from stable ESC clones with the DNeasy kit (Qiagen). Integration of BAC DNA was confirmed by genotyping PCR.
Bisulfite Sequencing and ChIP
Bisulfite treatment of 2.5 μg of genomic DNA was performed as described (Millar et al., 2002). Sequence-specific PCR of the bisulfite-treated DNA was performed using primers specific to BAC regions. The PCR fragments were cloned into the pCRII vector (Invitrogen, K2070-20) and transformed into DH5α E. coli cells. Miniprep plasmid DNA was sequenced using M13 reverse primers (5′-AGGAAACAGCTATGACCAT-3′).
Nuclei from approximately 30 million ESCs were isolated as previously described (Ramirez-Carrozzi et al., 2006). The nuclei were then supplemented with protease inhibitors (α-Complete, Roche) and sonicated in a Diagenode Bioruptor Twin sonicator for 15 min with 30-s cycles. One hundred micrograms of chromatin was incubated overnight at 4°C with 5 μg of an antibody to H3K4me3 (Millipore, 07-473). Chromatin complexes were recovered by binding to Protein A Dynabeads (Invitrogen, 100-02D) and were released from the beads by elution with NaCHO3 1% SDS buffer, and cross-linking was reversed by incubation at 65°C overnight. DNA was purified using the PCR purification kit (Qiagen). The quantity of immunoprecipitated DNA was measured by qPCR on an iCycler (Bio-Rad). The amount of DNA for each primer set was calculated relative to a 5% input chromatin control sample. To control for variable BAC integrants, the percentage inputs for all BAC regions were normalized to the percentage input at a downstream BAC CpG island with consistent enrichment.
Data and Code Availability
The code used to find CGRs is available at https://github.com/teneth/findCGR/.
Author contributions
J.L. and S.T.S. designed the experiments; J.L. performed the experiments; J.L., D.L., M.P., and S.T.S. analyzed the data; and J.L. and S.T.S wrote the manuscript.
Conflicts of interests
The authors declare no competing interests.
Acknowledgments
We thank J. Ernst, K. Plath, A. Smith, and K. Zaret, for helpful discussions and/or critical reading of the manuscript, and L. Smale for preparation of the graphical abstract. This work was supported by NIH grants R01 GM086372 (S.T.S.), P01 GM099134 (S.T.S.), and T32 GM007185 (J.L.).
Published: March 25, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.stemcr.2021.02.016.
Supplemental information
References
- Agarwal V., Shendure J. Predicting mRNA abundance directly from genomic sequence using deep convolutional neural networks. Cell Rep. 2020;31:107663. doi: 10.1016/j.celrep.2020.107663. [DOI] [PubMed] [Google Scholar]
- Auclair G., Guibert S., Bender A., Weber M. Ontogeny of CpG island methylation and specificity of DNMT3 methyltransferases during embryonic development in the mouse. Genome Biol. 2014;15:545. doi: 10.1186/s13059-014-0545-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baubec T., Colombo D.F., Wirbelauer C., Schmidt J., Burger L., Krebs A.R., Akalin A., Schübeler D. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature. 2015;520:243–247. doi: 10.1038/nature14176. [DOI] [PubMed] [Google Scholar]
- Bernstein B.E., Mikkelsen T.S., Xie X., Kamal M., Huebert D.J., Cuff J., Fry B., Meissner A., Wernig M., Plath K. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Bernstein B.E., Stamatoyannopoulos J.A., Costello J.F., Ren B., Milosavljevic A., Meissner A., Kellis M., Marra M.A., Beaudet A.L., Ecker J.R. The NIH roadmap epigenomics mapping consortium. Nat. Biotechnol. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock C., Walter J., Paulsen M., Lengauer T. CpG island mapping by epigenome Prediction. PLoS Comput. Biol. 2007;3:e110. doi: 10.1371/journal.pcbi.0030110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cierpicki T., Risner L.E., Grembecka J., Lukasik S.M., Popovic R., Omonkowska M., Shultis D.D., Zeleznik-Le N.J., Bushweller J.H. Structure of the MLL CXXC domain–DNA complex and its functional role in MLL-AF9 leukemia. Nat. Struct. Mol. Biol. 2010;17:62–68. doi: 10.1038/nsmb.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clouaire T., Webb S., Skene P., Illingworth R., Kerr A., Andrews R., Lee J.-H., Skalnik D., Bird A. Cfp1 integrates both CpG content and gene activity for accurate H3K4me3 deposition in embryonic stem cells. Genes Dev. 2012;26:1714–1728. doi: 10.1101/gad.194209.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen N.M., Kenigsberg E., Tanay A. Primate CpG islands are maintained by heterogeneous evolutionary regimes involving minimal selection. Cell. 2011;145:773–786. doi: 10.1016/j.cell.2011.04.024. [DOI] [PubMed] [Google Scholar]
- Deaton A.M., Bird A. CpG islands and the regulation of transcription. Genes Dev. 2011;25:1010–1022. doi: 10.1101/gad.2037511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Santos C.O., Dolzhenko E., Hodges E., Smith A.D., Hannon G.J. An epigenetic memory of pregnancy in the mouse mammary gland. Cell Rep. 2015;11:1102–1109. doi: 10.1016/j.celrep.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew H.R., Travers A.A. DNA bending and its relation to nucleosome positioning. J. Mol. Biol. 1985;186:773–790. doi: 10.1016/0022-2836(85)90396-1. [DOI] [PubMed] [Google Scholar]
- Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X., Wang L., Issner R., Coyne M. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann A., Ivanek R., Murr R., Gaidatzis D., Burger L., Schübeler D. Transcription factor occupancy can mediate active turnover of DNA methylation at regulatory regions. PLoS Genet. 2013;9:e1003994. doi: 10.1371/journal.pgen.1003994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenouil R., Cauchy P., Koch F., Descostes N., Cabeza J.Z., Innocenti C., Ferrier P., Spicuglia S., Gut M., Gut I. CpG islands and GC content dictate nucleosome depletion in a transcription-independent manner at mammalian promoters. Genome Res. 2012;22:2399–2408. doi: 10.1101/gr.138776.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S., Yang X.W. Modification of bacterial artificial chromosomes (BACs) and preparation of intact BAC DNA for generation of transgenic mice. Curr. Protoc. Neurosci. 2005;31:5.21.1–5.21.13. doi: 10.1002/0471142301.ns0521s31. [DOI] [PubMed] [Google Scholar]
- Guo G., von Meyenn F., Rostovskaya M., Clarke J., Dietmann S., Baker D., Sahakyan A., Myers S., Bertone P., Reik W. Epigenetic resetting of human pluripotency. Development. 2017;144:2748–2763. doi: 10.1242/dev.146811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl D., Krebs A.R., Grand R.S., Baubec T., Isbel L., Wirbelauer C., Burger L., Schübeler D. CG dinucleotides enhance promoter activity independent of DNA methylation. Genome Res. 2019;29:554–563. doi: 10.1101/gr.241653.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon G.C., Rajagopal N., Shen Y., McCleary D.F., Yue F., Dang M.D., Ren B. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat. Genet. 2013;45:1198–1206. doi: 10.1038/ng.2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry R.A., Wu H., Feinberg A.P. A species-generalized probabilistic model-based definition of CpG islands. Mamm. Genome. 2009;20:674–680. doi: 10.1007/s00335-009-9222-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs A.R., Dessus-Babus S., Burger L., Schübeler D. High-throughput engineering of a mammalian genome reveals building principles of methylation states at CG rich regions. ELife. 2014;3:e04094. doi: 10.7554/eLife.04094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunde-Ramamoorthy G., Coarfa C., Laritsky E., Kessler N.J., Harris R.A., Xu M., Chen R., Shen L., Milosavljevic A., Waterland R.A. Comparison and quantitative verification of mapping algorithms for whole-genome bisulfite sequencing. Nucleic Acids Res. 2014;42:e43. doi: 10.1093/nar/gkt1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R., Pelizzola M., Dowen R.H., Hawkins R.D., Hon G., Tonti-Filippini J., Nery J.R., Lee L., Ye Z., Ngo Q.-M. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lövkvist C., Dodd I.B., Sneppen K., Haerter J.O. DNA methylation in human epigenomes depends on local topology of CpG sites. Nucleic Acids Res. 2016;44:5123–5132. doi: 10.1093/nar/gkw124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowary P., Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- Lu F., Liu Y., Jiang L., Yamaguchi S., Zhang Y. Role of Tet proteins in enhancer activity and telomere elongation. Genes Dev. 2014;28:2103–2119. doi: 10.1101/gad.248005.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendenhall E.M., Koche R.P., Truong T., Zhou V.W., Issac B., Chi A.S., Ku M., Bernstein B.E. GC-rich sequence elements recruit PRC2 in mammalian ES cells. PLoS Genet. 2010;6:e1001244. doi: 10.1371/journal.pgen.1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar D.S., Warnecke P.M., Melki J.R., Clark S.J. Methylation sequencing from limiting DNA: embryonic, fixed, and microdissected cells. Methods. 2002;27:108–113. doi: 10.1016/s1046-2023(02)00061-0. [DOI] [PubMed] [Google Scholar]
- Morrison A.J., Shen X. Chromatin remodelling beyond transcription: the INO80 and SWR1 complexes. Nat. Rev. Mol. Cell Biol. 2009;10:373–384. doi: 10.1038/nrm2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morselli M., Pastor W.A., Montanini B., Nee K., Ferrari R., Fu K., Bonora G., Rubbi L., Clark A.T., Ottonello S. In vivo targeting of de novo DNA methylation by histone modifications in yeast and mouse. ELife. 2015;4:e06205. doi: 10.7554/eLife.06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J., Smith A. Naive and primed pluripotent states. Cell Stem Cell. 2009;4:487–492. doi: 10.1016/j.stem.2009.05.015. [DOI] [PubMed] [Google Scholar]
- Ramirez-Carrozzi V.R., Nazarian A.A., Li C.C., Gore S.L., Sridharan R., Imbalzano A.N., Smale S.T. Selective and antagonistic functions of SWI/SNF and Mi-2beta nucleosome remodeling complexes during an inflammatory response. Genes Dev. 2006;20:282–296. doi: 10.1101/gad.1383206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Carrozzi V.R., Braas D., Bhatt D.M., Cheng C.S., Hong C., Doty K.R., Black J.C., Hoffmann A., Carey M., Smale S.T. A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell. 2009;138:114–128. doi: 10.1016/j.cell.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E., Fondufe-Mittendorf Y., Chen L., Thåström A., Field Y., Moore I.K., Wang J.-P.Z., Widom J. A genomic code for nucleosome positioning. Nature. 2006;442:772–778. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima Y., Guo G., Loos R., Nichols J., Ficz G., Krueger F., Oxley D., Santos F., Clarke J., Mansfield W. Resetting transcription factor control circuitry toward ground-state pluripotency in human. Cell. 2014;158:1254–1269. doi: 10.1016/j.cell.2014.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Mouse ENCODE Consortium, Yue F., Cheng Y., Breschi A., Vierstra J., Wu W., Ryba T., Sandstrom R., Ma Z., Davis C. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515:355–364. doi: 10.1038/nature13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theunissen T.W., Powell B.E., Wang H., Mitalipova M., Faddah D.A., Reddy J., Fan Z.P., Maetzel D., Ganz K., Shi L. Systematic identification of culture conditions for induction and maintenance of naïve human pluripotency. Cell Stem Cell. 2014;15:524–526. doi: 10.1016/j.stem.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman R.E., Rynes E., Humbert R., Vierstra J., Maurano M.T., Haugen E., Sheffield N.C., Stergachis A.B., Wang H., Vernot B. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierstra J., Rynes E., Sandstrom R., Zhang M., Canfield T., Hansen R.S., Stehling-Sun S., Sabo P.J., Byron R., Humbert R. Mouse regulatory DNA landscapes reveal global principles of cis-regulatory evolution. Science. 2014;346:1007–1012. doi: 10.1126/science.1246426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent J.J., Huang Y., Chen P.-Y., Feng S., Calvopiña J.H., Nee K., Lee S.A., Le T., Yoon A.J., Faull K. Stage-specific roles for Tet1 and Tet2 in DNA demethylation in primordial germ cells. Cell Stem Cell. 2013;12:470–478. doi: 10.1016/j.stem.2013.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Engler P., Longacre A., Storb U. An efficient method for high-fidelity BAC/PAC retrofitting with a selectable marker for mammalian cell transfection. Genome Res. 2001;11:137–142. doi: 10.1101/gr.159001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warming S., Constantino N., Court D., Jenkins N., Copeland N. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 2005;33:e36. doi: 10.1093/nar/gni035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West J.A., Cook A., Alver B.H., Stadtfeld M., Deaton A.M., Hochedlinger K., Park P.J., Tolstorukov M.Y., Kingston R.E. Nucleosomal occupancy changes locally over key regulatory regions during cell differentiation and reprogramming. Nat. Commun. 2014;5:4719. doi: 10.1038/ncomms5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Caffo B., Jaffee H.A., Irizarry R.A., Feinberg A.P. Redefining CpG islands using hidden Markov models. Biostatistics. 2010;11:499–514. doi: 10.1093/biostatistics/kxq005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C., Liu K., Lei M., Yang A., Li Y., Hughes T.R., Min J. DNA sequence recognition of human CXXC domains and their structural determinants. Structure. 2018;26:85–95. doi: 10.1016/j.str.2017.11.022. [DOI] [PubMed] [Google Scholar]
- Xu J., Watts J.A., Pope S.D., Gadue P., Kamps M., Plath K., Zaret K.S., Smale S.T. Transcriptional competence and the active marking of tissue-specific enhancers by defined transcription factors in embryonic and induced pluripotent stem cells. Genes Dev. 2009;23:2824–2838. doi: 10.1101/gad.1861209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu N., Guo X., Zelikovsky A., Pan Y. GaussianCpG: a Gaussian model for detection of CpG island in human genome sequences. BMC Genomics. 2017;18(Suppl 4):392. doi: 10.1186/s12864-017-3731-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziller M.J., Gu H., Müller F., Donaghey J., Tsai L.T.-Y., Kohlbacher O., De Jager P.L., Rosen E.D., Bennett D.A., Bernstein B.E. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500:477–481. doi: 10.1038/nature12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code used to find CGRs is available at https://github.com/teneth/findCGR/.