

Abstract
Lung adenocarcinomas exhibit various patterns of genomic alterations. During the development of this cancer, KRAS serves as a driver oncogene with a relatively high mutational frequency. Emerging data suggest that lung adenocarcinomas with KRAS mutations can show enhanced PD-L1 expression and additional somatic mutations, thus linking the prospect of applying immune checkpoint blockade therapy to this disease. However, the responses of KRAS-mutant lung adenocarcinomas to this therapy are distinct, which is largely attributed to the heterogeneity in the tumoral immune milieus. Recently, it was revealed that KRAS-mutant lung adenocarcinomas simultaneously expressing either a LKB1 or TP53 mutation typically have different immune profiles of their tumours: tumours with a KRAS/TP53 co-mutation generally present with a significant upregulation of PD-L1 expression and tumoricidal T-cell accumulation, and those with a KRAS/LKB1 co-mutation are frequently negative for PD-L1 expression and have few tumoricidal immune infiltrates. In this regard, interrogating TP53 or LKB1 mutation in addition to PD-L1 expression will be promising in guiding clinical use of immune checkpoint blockade therapy for KRAS-mutant lung adenocarcinomas.
Keywords: cancer immune milieus, KRAS gene, LKB1 gene, Lung adenocarcinoma, TP53 gene
Introduction
Currently, non-small cell lung cancer (NSCLC) is uniquely prevalent worldwide. KRAS serves as a critical driver gene for lung carcinogenesis. Notably, the frequency of KRAS mutation in lung adenocarcinomas (LUADs) varies among human populations,1 with a KRAS mutation frequency of 26.1% in Western patients and 11.2% in East-Asian patients.1 KRAS mutations are rare in lung squamous cell carcinoma.1 Mechanistically, KRAS mutation leads to the robust activation of MAPK and PI3K cascades, independent of their corresponding upstream signals,2 ultimately increasing the aggressive behaviour of these cancer cells.
Compared with KRAS wild-type cases, KRAS mutation cases generally correlate with shortened survival times for NSCLC patients, even those receiving conventional therapies.3,4 For example, a retrospective study reported that Chinese patients with metastatic NSCLC with a KRAS mutation have significantly lower progression-free survival (PFS) after first-line chemotherapy than those without a KRAS and EGFR mutation and ALK/ROS1 fusion.5 In addition, tyrosine kinase inhibitors against mutated EGFR exert negligible therapeutic effects on KRAS-mutant LUAD patients.6 In this regard, alternative strategies should be developed to treat lung cancers with KRAS mutations.
To our knowledge, therapies by using immune checkpoint inhibitor (ICI) alone (e.g. KEYNOTE-0247 and KEYNOTE-0428) or in combination with chemotherapy (e.g. KEYNOTE-1899 or KEYNOTE-40710) with or without antiangiogenics (e.g. IMpower-15011) are the mainstay of treatment in first-line therapy for inoperable NSCLC. In a second or later line for advanced NSCLC patients with KRAS mutation, the objective response rate (ORR) of ICI monotherapy was reported as 26%.12 Moreover, the KRAS mutation was found to be positively related to the upregulation of PD-L1 in LUAD,13,14 thus indicating a potential of KRAS mutation in predicting the effectiveness of ICI therapy.
In fact, carcinogenic mutations shape the immune landscape of tumours.15 However, several studies revealed the heterogeneity of the tumour and immune milieus in KRAS-mutant LUAD.13,14,16 Typically, the immune milieu in KRAS/TP53 co-mutant tumours is profoundly different from that in tumours with KRAS/LKB1 co-mutations, indicating that KRAS/TP53-mutant tumours are ‘immune-hot’, whereas KRAS/LKB1-mutant tumours are ‘immune-cold’.14,16 (Figure 1 and Table 1) This distinction suggests that KRAS-mutant LUAD is not a unique disease. In this review, we will focus on the KRAS co-mutation with LKB1 or TP53 which forms the immune nature in LUAD, aiming to guide the ICI therapy for LUAD patients.
Figure 1.
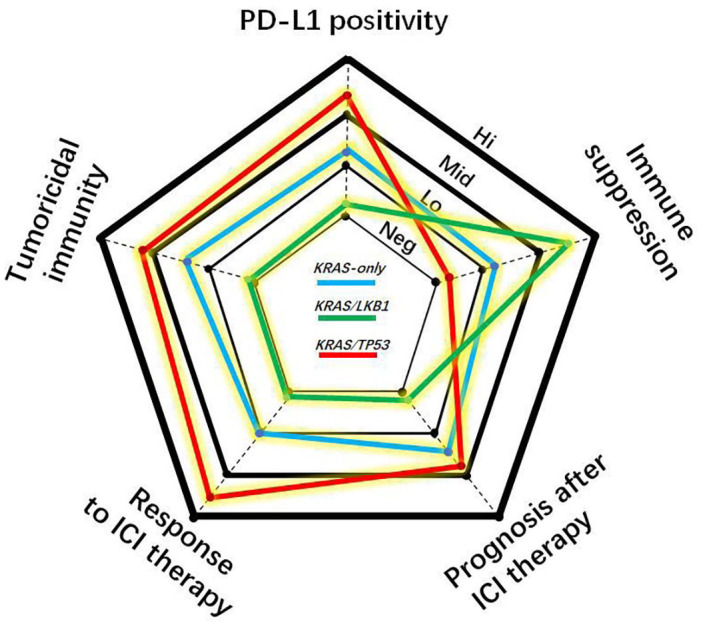
Radar plot ranking the cancer immunity and response to immune checkpoint blockade therapy of lung adenocarcinomas with KRAS-only, KRAS/LKB1 or KRAS/TP53 mutational pattern into four degrees including negative (Neg), low (Lo), middle (Mid) and high (Hi).
ICI, immune checkpoint inhibitor; PD-L1, programmed death-ligand 1.
Table 1.
A characteristic comparison of lung adenocarcinomas with different patterns of KRAS mutation.
| Groups | ‘KRAS-only’ | KRAS/LKB1 co-mutation | KRAS/TP53 co-mutation | KRAS/LKB1/TP53 tri-mutation |
|---|---|---|---|---|
| Characteristics | ||||
| Mutation frequency in KRAS groupRef. | 37–50%14,16,17 | 8–31%14,16,17 | 31–46%14,16,17 | 11–19%18,19 |
| Sensitivity to glucose restriction (versus wild-type KRAS)Ref. | ↑20,21 | ↑↑21 | ↑↑20 | NR |
| Aggression of cancer cell (versus wild-type KRAS)22 | ↑ | ↑↑↑ | ↑↑ | NR |
| Percent of tumour positive for PD-L1 expression23 | 37.5% | 10% | 68.8% | 25% |
| Median TMB value16 | 8.1–11.7 mutations/Megabase in these three groups | NR | ||
| Tumour immune milieuRef. | 1. Certain numbers of CD4+ T,24 CD8+ T,25,26 Treg,16,26 γδT,26 and myeloid cells (including neutrophils and macrophages)26
2. CXCL-9 and CXCL-10 upregulation26 3. CXCL-2, IL-6 and TGF-β downregulation26 |
1. Massive neutrophils with immune-suppressive function24,27
2. Few CD3+ T,14,16,24,27 CD4+ T,24 CD8+ T,14,16,24,27 CD45RO+ T,14,16 CD68+ macrophages24,27 and matured DC;27 3. Low proliferation activity of CD4+ T and CD8+ T24 4. Low potency of immune surveillance28 5. Low production of IFN-γ by CD4+ T, or by CD8+ T24 6. G-CSF, IL-1α, IL-6 and CXCL-724 7. STING inactivation29 8. HLA-DR, CD28, ICOS, CD80 and CD86 downregulation14 9. PD-1, CTLA-4, TIM-3 and LAG-3 downregulation in tumors14,18 10. CD4+ or CD8+ T-cell exhaustion24 |
1. Certain numbers of NK,26 B cell,26 matured DC,27 Treg16,26 and macrophages27
2. Massive CD3+ T, CD8+ T, and CD45RO+ T13,14,16,27 3. CCL-5, CXCL-9, CXCL-10, CXCL-11 and CXCL-13 upregulation in tumours27 4. HLA-DR, CD28, ICOS, CD80 and CD86 upregulation in tumours14,18 5. PD-1, CTLA-4, TIM-3 and LAG-3 upregulation in tumours14,18 6. Antigen presentation↑16,18 7. Antigen recognition↑14 8. T-cell-driven cytotoxicity↑14,27 |
1. CXCL-7, G-CSF and IL-6 upregulation24- Increased amounts of neutrophils with immune-suppressive function 2. CD28, CD86, CTLA-4, TIM-3 and HLA-DR downregulation in tumours18- Reduced amounts of T cells in tumours 3. PD-L1 upregulation18,23- T-cell exhaustion |
| Prognosis after first-line therapy Single-centre data18,19 |
• Upenn data*
Median PFS: NM Median OS: NM • CSU data& Median PFS: 22.29 weeks Median OS: 28.57 weeks |
• Upenn data*
Median PFS: 2.4 months Median OS: 7.1 months • CSU data& Median PFS: 12.43 weeks Median OS: 19.36 weeks |
• Upenn data*
Median PFS: NM Median OS: NM • CSU data& Median PFS: 12.86 weeks Median OS: 16.29 weeks |
Upenn data*
Median PFS: 13 months Median OS: 22 months • CSU data& Median PFS: 12.29 weeks Median OS: 20.22 weeks |
| ORR to immune checkpoint blockade therapy • SU2C cohort16 (MSKCC+MDACC+MGH) |
28.6% | 7.4% | 35.7% | NR |
| CheckMate-057 cohort16 | 18.2% | 0% | 57.1% | NR |
| • KEYNOTE-04230
(PD-L1 ⩾ 1%) |
31% (LKB1 mutation: n = 16 patients) versus 29% (LKB1 wide-type: n = 214 patients) | |||
| Prognosis after immune checkpoint blockade therapy • SU2C cohort16 (MSKCC+MDACC+MGH) • CheckMate-057 cohort16 |
Median PFS: 2.7 months Median OS: 16.1 months Median PFS: 2.1 months Median OS: ~7 months |
Median PFS: 1.8 months Median OS: 6.4 months Median PFS: 2.0 months Median OS: ~27 months |
Median PFS: 3.0 months Median OS: 16.0 months Median PFS: 5.1 months Median OS: ~15 months |
Median PFS: NR
Median OS: NR Median PFS: NR Median OS: NR |
| • KEYNOTE-04230
(PD-L1 ⩾ 1%) |
LKB1 mutation Median PFS: 5 months Median OS: 18 months |
LKB1 wide-type Median PFS: 6 months Median OS: 17 months |
||
| PD-L1/TIL paradigm31 | Positive/Positive | Negative/Negative | Positive/Positive | Positive/Negative |
| Precision of mutational pattern in predicting ICI response as tumoral PD-L1 expression increasing Ref. | Increase16,32,33 | Increase30 | Increase16,32 | NR |
Over 80% of enrolled patients receiving chemotherapy during first-line management.
Chemotherapy by using pemetrexed plus platinum regimen.
CCL-5, chemokine C-C motif ligand-5; CSU, XiangYa School of Medicine; CTLA-4, cytotoxic T-lymphocyte associated protein-4; CXCL, chemokine C-X-C motif ligand; G-CSF, granulocyte-colony stimulating factor; HLA-DR, human leukocyte antigen (locus) DR; ICI, immune checkpoint inhibition; ICOS, inducible T-cell co-stimulator; IFN-γ, interferon-gamma; KRAS, Kirsten rats sarcoma viral oncogene homolog; LAG-3, lymphocyte-activation gene-3; LKB1, liver kinase B1 gene; MDACC, MD Anderson Cancer Centre; MGH, Massachusetts General Hospital; MSKCC, Memorial Sloan-Kettering Cancer Centre; NR, not reported; OS, overall survival; PD-1, programmed death-1; PD-L1, programmed death-ligand 1; PFS, progression-free survival; STING, stimulator of interferon genes; TGF-β, transforming growth factor-beta; TIL, tumour-infiltrating lymphocyte; TIM-3, T-cell immunoglobulin and mucin domain-containing molecule-3; TMB, tumour mutation burden; TP53, tumour protein-53 gene; UPenn, University of Pennsylvania.
Carcinogenic KRAS mutation
KRAS, the Kristen rat sarcoma viral oncogene, is a member of the RAS family of genes, which includes HRAS and NRAS.34 At a steady state, RAS genes encode intracellular guanine nucleotide-binding proteins, which have GTPase activity. When conjugated with GTP, RAS proteins are activated to increase cell proliferation and survival. However, the RAS protein bound with GDP is inactive.34
Nearly four decades ago, the KRAS mutation was revealed to drive carcinogenesis in the lung.4 Mutant KRAS is characterized by a single nucleotide base missense mutation, which frequently occurs in codon 12 and codon 13 of exon 2 or exon 3.4,34 Nucleotide base substitutions in KRAS hot codons are presented as G12A, G12C, G12D, G12S, G12V, G13C or G13D.12 Among these mutations, G12C most frequently occurs in KRAS-mutant LUAD.35 Functionally, the missense mutation leads to the subsequent activation of the KRAS protein due to an impairment in its GTPase function, thus activating cellular processes that are critical in cancer invasion and metastasis.4,34
Notably, heterogeneous missense mutations of KRAS variably enable cancer cells to gain a growth advantage. To explain this notion, an in vitro experiment demonstrated that PI3K/Akt and MAPK were preferentially activated in NSCLC cells with the G12D mutation but not with the G12C or G12V mutation.36 Notably, refractory NSCLC patients with a G12C or G12V mutation generally had a shortened PFS compared with patients harbouring other missense mutations of KRAS.36 Intriguingly, another study indicated that first-line chemotherapy was superior in prolonging the PFS of NSCLC patients, especially those with the missense mutation of G12C.17 Similarly, second-line or subsequent therapies based on ICI drugs provided a survival benefit to patients in the G12C subgroup, who showed no significant difference in PFS compared with the patients with other missense mutations.12 However, in NSCLC, the KRAS mutation is still regarded as a negative regulator of chemotherapy.3,4 Nevertheless, several lines of data suggested that patients with KRAS-mutant LUAD generally had a better PFS upon ICI therapy than those with wild-type KRAS.32,33 Hereafter, we will illustrate the impact of KRAS mutation on tumoral immune profiles in LUAD.
KRAS mutation and the immune nature of lung adenocarcinoma
Although accumulative evidence suggests the feasibility of using the KRAS mutation for selecting candidates for ICI therapy among LUAD patients,12,16,32,33 this criterion is not necessarily applicable for all KRAS-mutant patients because many patients carry mutations in other genes, such as LKB1, which seems to nearly abolish the effectiveness of ICI therapy.16 In contrast, a TP53 mutation was associated with an enhanced survival benefit from ICI therapy for KRAS-mutant LUAD patients compared with its effectiveness for ‘KRAS-only’ patients.16,32 In fact, the ‘KRAS-only’ group included patients who likely had simultaneous mutations with other genes, such as CDKN2A/B,2,14 KEAP12,14 or PIK3CA.2 In this regard, functional aberrations in these genes were confirmed to synergize with Kras mutations in lung carcinogenesis of mice.37–39 However, in LUAD, frequencies of these mutations were not as high as those of the KRAS/TP53 or KRAS/LKB1 co-mutation.2 In addition, the value of these mutations in guiding ICI therapy for LUAD patients is still uncertain. For example, in contrast to TP53- or LKB1-mutant carriers, LUAD patients with the KRAS/CDKN2A/B co-mutation exhibited mixed immune profiles in their tumours,14 possibly distorting the assessment of ICI therapeutic effectiveness for patients with the KRAS/CDKN2A/B co-mutation. In view of these issues, patients with these co-mutational patterns have generally been classified in the ‘KRAS-only’ group. Notably, the mutational frequencies of ‘KRAS-only’, KRAS/TP53 and KRAS/LKB1 in LUAD tumours remain consistent regardless of disease stage or the administration of platinum-based chemotherapy,14 thus indicating that these are the three major patterns of gene mutations in KRAS-mutant LUAD despite cancer cell clone evolution during disease progression or the level of chemotherapy received.
‘KRAS-only’ mutation
A certain number of LUAD cases present with ‘KRAS-only’ mutations. The frequency of the ‘KRAS-only’ mutation is reported to vary from 37% to 50% in the whole KRAS-mutant group.14,16,17 (Table 1). Such a high rate of KRAS mutation can be attributed to carcinogen exposure or heredity. For example, NSCLC patients with a history of smoking have a higher frequency of KRAS mutations than patients who never smoked.25 In addition, among KRAS-mutant LUAD patients, smokers have a significantly higher somatic mutation load than non-smokers, implying divergent routes to carcinogenesis despite a KRAS mutation, as was also confirmed in a mouse model of Kras mutation.40 Theoretically, a high mutation load correlating with the excessive production of tumoral neoantigens induces recruitment tumoricidal T cells from the periphery to recognize and subsequently kill clones positive for the antigens processed by dendritic cells (DCs).32 Compared with an EGFR mutation, a KRAS mutation can significantly increase the number of CD8+ T subsets in LUAD tumours25,26 (Table 1). In Kras-mutant mice, regulatory T cells, γδ T cells and myeloid cells were found in increased numbers in lung tumours26 (Table 1). In addition, genes encoding CXCL-9 and CXCL-10 showed upregulated expression in the lung tumours of these mice, and genes encoding immune-suppressive factors, such as TGF-β or CXCL-2, showed downregulated expression in chemokine-recruiting myeloid-derived suppressive cells (MDSCs)26 (Table 1). However, to counteract this inhibition, the KRAS mutation induces PD-L1 upregulation in an ERK-phosphorylation-dependent manner, ultimately enabling cancer cells to escape from immune attacks by causing PD-1/PD-L1 axis-induced T-cell exhaustion.41 However, despite this theory, not all LUAD patients with a ‘KRAS-only’ mutation, even those are highly positive for PD-L1 expression in their tumours, will respond to ICI therapy, whereas a few patients in the ‘KRAS-only’ group but negative for PD-L1 still benefit from ICI therapy,16 indicating that PD-L1 is not a reliable marker in these situations. What are the mechanisms driving these outcomes?
As mentioned above, carcinogen-induced and inherited KRAS mutations associated with LUAD are genetically heterogeneous.40 In addition to showing a significant reduction in somatic mutations, a significantly higher frequency of genomic copy-number aberrations (GCAs) was found in lung tumours of germline KrasG12D-mutant mouse models than was found in carcinogen-induced Kras-mutant tumours.40 To exemplify the role of GCAs in lung carcinogenesis, a recent study revealed that lung tumour cells in mice with homozygous Kras mutations (KrasG12D/G12D) showed significantly enhanced glucose consumption, acquisition of the glycolytic phenotype, higher levels of glucose-derived tricarboxylic acid cycle metabolites, and greater resistance to oxidative stress than cells with heterozygous Kras mutations (KrasG12D/− mice) or with wild-type Kras (Kraswt/wt mice), thus confirming a relationship between mutant Kras copy gain and cell metabolic reprogramming.20 More strikingly, mutant Kras copy gain during lung cancer progression from an early to a late stage drives a low- to high-grade switch in tumour pathology, indicating that mutant Kras copy gain predisposes lung cancer cells to acquire a more aggressive phenotype.20 Nevertheless, these tumour cells are more sensitive to a low-glucose environment than KrasG12D/− or Kraswt/wt cells.20 Besides, chronic glucose restriction can reduce interferon-γ production in tumoricidal T cells.42 Hence, glucose competition between cancer cells and T cells will induce T-cell exhaustion.43 In this case, a strategy against aberrant glucose metabolism in cancer cells will become a potential way to improve the effectiveness of ICI therapy. In fact, as cancer progresses, metabolic reprogramming in immune infiltrates, such as macrophages, DCs, MDSCs, neutrophils, natural killer cells (NK), T cells, and B cells, serves as a critical mechanism for regulating their pro- or anticancer properties.44 In this regard, an alternative strategy is urgent to determine the anticancer properties of immune infiltrates. For example, acetate can increase histone-3 acetylation of PD-1+ T cells in an acetyl-CoA synthetase-dependent manner, thus restoring their production of IFN-γ.43 If combining with ICI therapy, the tumoricidal activity of T cells should be improved.
KRAS/LKB1 co-mutation
LKB1, also known as STK11, is a cancer-suppressive gene that encodes a protein that regulates cell metabolism and growth through the AMP-activated protein kinase (AMPK) cascade.45 LKB1 inactivation is common in LUAD,2 and subsequent AMPK inactivation is a hallmark of LUAD with the KRAS/LKB1 co-mutation.14 According to published data, in KRAS-mutant groups from different cohorts, the frequency of KRAS/LKB1 co-mutation ranged from 8% to 31%14,16,17 (Table 1). Mechanistically, LKB1 gene inactivation in LUAD is mainly driven by genomic copy-number deletion, inactive mutation and downregulating its own expression, ultimately resulting in depletion of LKB1 protein.2 Significantly, Lkb1 deletion in KrasG12D-mutant mice was found to accelerate lung carcinogenesis, exhibiting a specific correlation with adenocarcinoma metastasis.22 Consistent with this finding, integrative profiling of LUAD genome revealed that the ‘proximal-proliferative’ subgroup harbours the highest frequency of LKB1 inactivation and/or KRAS mutation and/or KEAP1 mutation.2 In addition to the KEAP1 mutation, genomic copy-number depletion of LKB1 can give rise to KEAP1 depletion because both genes reside on the short arm of chromosome 19, with their loci next to each other.14 Functionally, KEAP1-coded protein causes the degradation of NF-E2-related factor 2 (NRF2).38 If stabilized when KEAP1 is inactivated, then NRF2 activates the antioxidant programme against cellular oxidative stress.38 An in vitro model revealed that the KrasG12D mutation can elevate mouse Nrf2 transcription via Jun and Myc proteins.46 Notably, Nrf2 expression can upregulate the expression of glutathione utilization genes in cancer cells with the KrasG12D mutation.20 These cells also exhibit reduced proliferation because of inhibited glutathione synthesis.46 These data suggest that Nrf2 affects cancer progression through a mechanism of metabolic reprogramming. To exemplify this notion, the metabolism of a mouse model with Lkb1-loss-induced oxidative stress was rescued by the presence of glutamine.47 Consistent with this finding, another mouse model showed that the Keap1-loss-induced progression of Kras-mutant lung tumours depended on glutaminolysis.48 However, intriguingly, LKB1 inactivation synergized with KRAS mutation to increase the sensitivity of human lung cancer cells to glucose restriction, reflecting higher glucose consumption to sustain their growth.21 Glucose competition between cancer cells and T cells to suppress anticancer immunity has been previously described.43 Therefore, the metabolic reprogramming of glucose by KEAP1 inactivation or by NRF2 activation will further accelerate lung carcinogenesis in the context of KRAS/LKB1 co-mutation.
In contrast to the ‘KRAS-only’ mutation, tumoral immune suppression has been identified in LUAD with the KRAS/LKB1 co-mutation, suggesting that these tumours are naturally refractory to ICI therapy.14 In addition, data from several cohorts revealed that a large portion of LUAD cases with the KRAS/LKB1 co-mutation were negative for PD-L1 expression14,16 (Table 1). Furthermore, the LKB1 mutation was revealed to be mostly enriched in cases negative for PD-L1 expression.16,27 Most likely, LKB1 inactivation antagonizes KRAS mutation-induced PD-L1 expression because overexpressing LKB1 in KRAS-mutant lung cancer cells gave rise to an upregulation of PD-L1 expression in these cells.49 To our knowledge, testing PD-L1 expression is currently recommended for guiding anti-PD-1 therapy for LUAD patients without EGFR mutation and ALK/ROS1 fusion.7,8 However, LUAD patients with LKB1 mutation that exhibit highly positive PD-L1 expression in their tumours have significantly shorter PFS and overall survival (OS) after anti-PD-1 or -PD-L1 monotherapy than those with wide-type of LKB1,16 indicating that the LKB1 mutation negatively impacts on the prognosis of LUAD patients after ICI therapy. Besides, in contrast to PD-L1 expression, an intermediate to high tumour mutation burden (TMB) was found in LUAD patients with the KRAS/LKB1 co-mutation.16 In fact, the TMB values in the ‘KRAS-only’, KRAS/LKB1 and KRAS/TP53 groups were comparable, ranging from 8.1 to 11.7 muts/Mb16 (Table 1). However, no correlation between TMB values with PD-L1 expressing levels was found.50 Moreover, TMB is not a valuable biomarker for predicting the effectiveness of ICI therapy in the context of LKB1 mutation because evidence has suggested that patients with metastatic NSCLC with the LKB1 mutation but high plasma TMB were unlikely to respond to anti-PD-1 therapy.51 In this regard, TMB appears to be of little value for guiding the clinical use of ICI therapy for LKB1-mutant LUAD. What are the mechanisms underlying these outcomes?
By profiling the immune infiltrates, several studies have revealed that the numbers of CD3+, CD8+, CD45RO+ T subsets, CD68+ macrophages and mature DCs are significantly decreased, whereas the number of neutrophils is increased in KRAS/LKB1-mutant lung adenocarcinomas compared with the numbers in tumours without LKB1 mutations14,16,27,24 (Table 1). In this situation, genes encoding T-cell costimulatory molecules (e.g. CD28, ICOS, CD80 and CD86), immune checkpoint molecules (e.g. PD-1, PD-L1, LAG-3, and CTLA-4), type I IFN signalling signatures (e.g. STING),29 tumour necrosis factor superfamily members (e.g. TNFSF4 and TNFSF9) and tumour necrosis factor receptor superfamily members (e.g. TNFRSF4, TNFRSF9, TNFRSF14 and TNFRSF18) showed significantly downregulated expression in LKB1-mutant LUAD patients14 (Table 1). In this regard, tumoricidal immunity is dampened due to a lack of T-cell engagement. This explanation is reasonable, as an evidence showed that Kras/Lkb1-mutant mice had significantly higher mRNA and protein levels of G-CSF, CXCL-7, IL-1α and IL-6 and greater STAT3 activation in lung tumours than mice with ‘Kras-only’ mutations, thus profoundly recruiting immune-suppressive neutrophils to lung tumours24 (Table 1). In addition, Kras/Lkb1-mutant mice lacked macrophages and CD4+ and CD8+ T-cell subsets in lung tumours, and either the proliferation or the IFN-γ production by CD4+ or CD8+ subsets were limited24 (Table 1). Instead, these T cells show increased expression of PD-1, CTLA-4, TIM-3 and LAG-3, indicating that they were exhausted24 (Table 1). Thus, the deficiency of immune surveillance in LUAD with KRAS/LKB1 co-mutation has been observed.28
Throughout the above analysis, it is found that LKB1 inactivation generally generates a suppressive immune milieu in KRAS-mutant tumours, which can be characterized by their reduced tumoricidal T-cell number and immune-related gene expression. Together with PD-L1 negativity in most cases, the lung tumour of KRAS/LKB1 co-mutation serves as a paradigm that is negative for T infiltration and PD-L1 expression, in which cancer cells negligibly respond to only a PD-1/PD-L1 blockade.31 In this case, combinational approaches should be considered to treat these tumours. For example, either antiangiogenics52 or AMG510, a KRAS (G12C) mutation-specific inhibitor,53 can enhance the anticancer immunity, thus enabling their combination with ICI therapy for these refractory tumours to be feasible (Figure 2). Other strategies, such as MEK inhibition plus chemoradiation,54 epigenetic regulation using inhibitors against methyltransferase (e.g. DNA methyltransferase 1, DNMT1 or enhancer of zeste homologue 2, EZH2) for restoring STING activation,29 dual inhibition of MEK plus fibroblast growth factor receptor 1 (FGFR1),55 triple inhibition of SRC family kinases, PI3K-mTOR and MEK,56 or normalizing glucose metabolism57 in cancer cells have shown effectiveness against tumours of KRAS/LKB1 co-mutations in preclinical models.
Figure 2.

The flow chart of guiding clinical application of immune checkpoint blockade therapy for KRAS-mutant lung adenocarcinomas in combination with TP53 and LKB1.
CT, chemotherapy; IHC, immunohistochemistry; LUAD, lung adenocarcinoma; NGS, next-generation sequencing; PD-L1, programmed death-l.
Moreover, a few LUAD cases of KRAS/LKB1 co-mutations were found to still respond to ICI therapy.16 This finding can be supported by newly published data based on the KEYNOTE-042 study,30 presenting that LUAD patients with somatic mutation of LKB1 in their tumours could have an ORR, median PFS and OS similar to those with wide-type LKB1 after pembrolizumab monotherapy.30 To our knowledge, the KEYNOTE-042 study was designed to recruit patients that had a positive PD-L1 expression in their tumours.8 From the results of KEYNOTE-042, a positive PD-L1 expression is still valuable in predicting the effectiveness of pembrolizumab independent of the LKB1 status in LUAD tumours. Concerning the clinical significance of LKB1 inactivation in LUAD, although tumours having the LKB1 inactivation and negative PD-L1 expression exhibited poor responses to ICI therapy,16,27,51 a real-world retrospective cohort found that the LKB1 mutation could perform its prognostic value in inoperable LUAD patients irrespective of their receiving ICI therapy or not.58 But intriguingly, a previous study reported that NSCLC patients with LKB1 mutations in exon 1 through exon 2 may have a worse prognosis than those with mutations in exon 3 through exon 9 after radical surgery.59 This indicates that the mutational pattern in LKB1 exons can impact the cancer cell biology as well. In this regard, although whole-exome sequencing (WES) technology was extensively used to detect the LKB1 mutation in the aforementioned studies,16,24,27,30 one limitation is whether or not the mutational pattern of LKB1 exons will affect the response of KRAS-mutant NSCLC to ICI therapy; this answer remains elusive. Besides, the limitation of WES lies in that only an extremely small portion of genomic aberration occurring in cancer cells can be obtained using this technology. In this situation, albeit the LKB1 exons are sequenced to be wide-type, matters distorting the expression of the LKB1 gene cannot be anticipated by solely sequencing the exons. Alternatively, a confirmation of functional LKB1 protein in LUAD cells and subsequent analysis of their network of interactions with other molecules will be useful.60
KRAS/TP53 co-mutation
TP53 is a core cancer-suppressing gene, which encodes the p53 protein in humans and mice to protect against genetic mutations at a steady state.61 Notably, TP53 mutation drives lung carcinogenesis.1,2 In LUAD, TP53 mutation has a higher frequency than EGFR mutation or KRAS mutation.2 In contrast, the frequencies of TP53 mutations are not significantly different among Western and Asian patients with LUAD, as both populations present a nearly 30% mutational frequency among all LUAD cases.1 TP53 mutation can be manifested by genetic or epigenetic errors, including missense, nonsense, frameshift, in-frame indel or splice site alterations.2 Changes caused by TP53 mutations will lead to deficiencies in base-pair proofreading during DNA replication or DNA damage repair, thus giving rise to increased burdens of somatic mutations in cancer cells.62 In fact, similar to the KRAS mutation, the TP53 mutation positively correlates with the somatic mutation burden in LUAD.63 In this regard, KRAS/TP53 co-mutation synergistically increases the production of neoantigens released by lung cancer cells, theoretically inflaming the immune milieu of the tumour.
In the RAS-mutant LUAD, several published reports have presented data showing that the frequencies of KRAS/TP53 co-mutation ranged from 31% to 46%14,16,17 (Table 1). Upon ICI therapy, these patients generally had a higher ORR and better prognosis than those with the ‘KRAS-only’ mutation.15 To a certain extent, this outcome can be attributed to a significant upregulation of PD-L1 expression in tumour.13,16,27,64 More intriguingly, it was reported that 30% of patients with KRAS/TP53 mutations but testing negative for PD-L1 expression still responded to ICI therapy.16 Inherently, immune infiltrates contribute to this process. In models of mice with Kras/Trp53 co-mutations, lung tumours were characterized by massive infiltrates, including neutrophils, macrophages, and NK, T and B cells26 (Table 1). In LUAD models with KRAS/TP53 co-mutations, tumours could have significantly higher amounts of CD8+ T-cell subsets than models with ‘KRAS-only’ or ‘TP53-only’ mutations.13 Despite inducing massive immune infiltrates, mutant Trp53 enabled lung tumours in Kras-mutant mice to grow more efficiently than those in mice of the wild-type Trp53.61,65 Consistent with this finding, it was revealed that TP53 mutation further increased cell proliferative activity in KRAS-mutant LUAD models.28 In this regard, it is presumed that some deficiencies probably occur in the processes of immune recognition and/or immune activation of tumoricidal T cells. After investigating the impact of the KRAS/TP53 co-mutation on tumoral immune signatures, it was determined that significantly upregulated genes were mostly enriched in biological processes including antigen presentation, dendritic cell maturation, communication between innate and adaptive immune cells, and antigen recognition by pattern recognition receptors14 (Table 1). These results suggest that T-cell exhaustion largely contributes to lung tumour progression in the presence of the KRAS/TP53 co-mutation, because genes encoding immune checkpoint molecules were revealed to upregulate their expression14 (Table 1). Hence, LUAD with the KRAS/TP53 co-mutation serves as a paradigm of the tumour positive for PD-L1 expression and immune cell infiltration.31 In theory, these tumours proficiently respond to PD-1/PD-L1 blockade.31 However, not all LUAD patients with the KRAS/TP53 co-mutation can benefit from ICI therapy alone.16 Hence, factors that suppress the immune milieu in LUAD tumours with the KRAS/TP53 co-mutation deserve further investigation.
KRAS/LKB1/TP53 tri-mutation
In addition to the KRAS/LKB1 or KRAS/TP53 co-mutation, KRAS/LKB1/TP53 is an alternative mutation pattern in LUAD. As an intrinsic factor in cancer, the genomic landscape impacts the effectiveness of anticancer therapy and the accuracy of patient prognosis.66 However, the prognostic value of the KRAS/LKB1/TP53 tri-mutation in LUAD patients remains undetermined. A retrospective study from a single Chinese centre revealed that patients with metastatic NSCLC of the KRAS/LKB1/TP53 tri-mutation exhibited a shortened PFS after first-line chemotherapy compared with those with KRAS/LKB1 or KRAS/TP53 co-mutations, while a finding of The Cancer Genome Atlas (TCGA) data confirmed that patients in tri-mutation group had lower OS than those either with KRAS/LKB1 or with KRAS/TP53 co-mutations18 (Table 1). In contrast, another retrospective single-centre study indicated that NSCLC patients with KRAS/LKB1/TP53 tri-mutations had higher PFS and OS after first-line therapy than those with KRAS/LKB1 co-mutations67 (Table 1). Probably, inconsistent frequencies of KRAS/LKB1/TP53 tri-mutations in the enrolled patients of these two studies bias the prognostic value of this mutational pattern. Or else, the missense type of KRAS was reported to impact NSCLC patient prognosis.18 This may account for the distinct prognosis of patients with the KRAS/LKB1/TP53 tri-mutation between these two studies.
As in chemotherapy, data concerning the effectiveness of ICI therapy on LUAD patients with the KRAS/LKB1/TP53 tri-mutation have rarely been reported to date. Although a retrospective study from a single centre reported that, after ICI therapy, LUAD patients with the KRAS/LKB1/TP53 tri-mutation had a PFS similar to those with the KRAS/TP53 co-mutation,32 such a tri-mutation pattern should not be expected to enable LUAD patients to respond to ICI therapy in the same way as the KRAS/TP53 co-mutation does. Intrinsically, wild-type p53 has been found to maintain the expression of LKB1 gene at a basal level in the absence of acute cell stress.67 However, most mutant TP53 genes have errors in encoding DNA-binding regions, including the region that can bind with LKB1 promoter.19,67 As a result, mutation of TP53 leads to a reduced expression of LKB1, which has been revealed in tumour cells.67 In this case, LUAD patients with the KRAS/TP53 co-mutation do not completely lose functional proteins encoded by LKB1 gene in their tumours, a remarkable difference from those harbouring the LKB1 gene inactivation, which results in depletion of LKB1 protein in LUAD cells.14,16 In fact, the TCGA data revealed that in addition to a significant upregulation of PD-L1 expression, mRNA levels of genes encoding HLA-DR, CD28, CD86, CTLA-4 and TIM-3 in the KRAS/LKB1/TP53 tri-mutation group showed no differences from those in the KRAS/LKB1 co-mutation group18 (Table 1). But in fact, LUAD with the KRAS/LKB1/TP53 tri-mutation had a lower amount of cancer cells positive for PD-L1 expression than those with the KRAS/TP53 co-mutation23 (Table 1). From these aspects, we can speculate that the absolute number of T cells will be reduced in tumours with the KRAS/LKB1/TP53 tri-mutation. On one hand, molecules including HLA-DR, CD28, CD86, CTLA-4 and TIM-3 are mainly expressed by T cells. On the other hand, genes encoding G-CSF and CXCL-7 were revealed to significantly increase their expression in lung tumours of mice bearing Kras/Lkb1/Trp53 tri-mutations, thus causing the accumulation of immunosuppressive neutrophils in tumours.24 In this regard, it suggests that the immune milieu in tumours with the KRAS/LKB1/TP53 tri-mutation should be suppressive.
In addition, it was found that LUAD cells with the KRAS/LKB1/TP53 tri-mutation exhibited different responses to MEK or JAK inhibition, presenting with increased sensitivity to JAK inhibition with the elevated production of IL-6 by tumour cells but showing resistance to MEK-inhibition-induced cell death.68 LUAD cells with the KRAS/LKB1 co-mutation were tested to have these biological responses occurring to a similar degree with those bearing the KRAS/LKB1/TP53 tri-mutation.68 In contrast, LUAD cells with the KRAS/TP53 co-mutation were found to be sensitive to MEK inhibition but resistant to JAK-inhibition-induced cell death.68 These results indicate that LUAD cells with the KRAS/LKB1/TP53 tri-mutation may share common features in terms of their biological activity and fate with LUAD cells with the KRAS/LKB1 co-mutation, which is, at least in part, manifested by IL-6 elevation and apoptosis in response to JAK inhibition. As a pro-tumorigenic cytokine, IL-6 functions in inducing angiogenesis and immunosuppression in tumours in addition to increasing cancer cell survival, proliferation and invasiveness.69 In this regard, JAK inhibition-induced IL-6 production by apoptotic cancer cells probably assists in improving the resistance of living cancer cells to anticancer therapies. Therefore, cancer immunity commitment after prior anticancer therapies will be critical in predicting whether LUAD patients carrying the KRAS/TP53/LKB1 tri-mutation can benefit from the coming ICI therapy.
Conclusions
The nature of the tumoral immune milieu in KRAS-mutant LUAD is not unique. Typically, KRAS/LKB1 and KRAS/TP53 co-mutations generate opposite immune milieus in tumours and responses to ICI therapy. Investigating LKB1 and TP53 mutations probably leads to predictions with higher precision for the effectiveness of ICI therapy in LUAD than those based on PD-L1 or the KRAS mutation alone. However, in LUAD cases negative for EGFR mutation and ALK/ROS1 fusion, current clinical practice guidelines strongly recommend tumoral PD-L1 expression as the conventional test, not detecting the KRAS mutation. Because KRAS mutations occur most frequently in LUAD, testing mutations in LKB1, TP53 and KRAS in addition to PD-L1 expression may be more effective for guiding the clinical use of ICI therapy (Figure 2).
Footnotes
Conflict of interest statement: The authors declare that there is no conflict of interest.
Contributions: GM and CP jointly wrote this paper. XT prepared the figure and table. CP conceived the topic of this paper.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Natural Science Foundation of China [Grant No. 81874254], by Scientific and Technological Developing Scheme Foundation of Jilin Province [Grant No. 20200201400JC], and by Foundation of Scientific Research Planning Project of the 13th Five-year Plan of Jilin Provincial Department of Education [Grant No. JJKH20201043KJ].
ORCID iD: Meichen Gu  https://orcid.org/0000-0002-3236-7785
https://orcid.org/0000-0002-3236-7785
Contributor Information
Meichen Gu, Department of Radiation Oncology & Therapy, The First Hospital of Jilin University, Changchun, P.R. China.
Tiankai Xu, Department of Radiation Oncology & Therapy, The First Hospital of Jilin University, Changchun, P.R. China.
Pengyu Chang, Department of Radiation Oncology & Therapy, The First Hospital of Jilin University, No.71, Xinmin Str, Changchun, 130021, P.R. China.
References
- 1. Dearden S, Stevens J, Wu Y-L, et al. Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap). Ann Oncol 2013; 24: 2371–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014; 511: 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Martin P, Leighl NB, Tsao M-S, et al. KRAS mutations as prognostic and predictive markers in non-small cell lung cancer. J Thorac Oncol 2013; 8: 530–542. [DOI] [PubMed] [Google Scholar]
- 4. Román M, Baraibar I, López I, et al. KRAS oncogene in non-small cell lung cancer: clinical perspectives on the treatment of an old target. Mol Cancer 2018; 17: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jia Y, Jiang T, Li X, et al. Characterization of distinct types of KRAS mutation and its impact on first-line platinum-based chemotherapy in Chinese patients with advanced non-small cell lung cancer. Oncol Lett 2017; 14: 6525–6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roberts PJ, Stinchcombe TE, Der CJ, et al. Personalized medicine in non-small-cell lung cancer: is KRAS a useful marker in selecting patients for epidermal growth factor receptor-targeted therapy? J Clin Oncol 2010; 28: 4769–4777. [DOI] [PubMed] [Google Scholar]
- 7. Reck M, Rodríguez-Abreu D, Robinson AG, et al. Updated analysis of KEYNOTE-024: pembrolizumab versus platinum-based chemotherapy for advanced non-small-cell lung cancer with PD-L1 tumor proportion score of 50% or greater. J Clin Oncol 2019; 37: 537–546. [DOI] [PubMed] [Google Scholar]
- 8. Mok TSK, Wu Y-L, Kudaba I, et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet 2019; 393: 1819–1830. [DOI] [PubMed] [Google Scholar]
- 9. Gadgeel S, Rodríguez-Abreu D, Speranza G, et al. Updated analysis from KEYNOTE-189: pembrolizumab or placebo plus pemetrexed and platinum for previously untreated metastatic nonsquamous non-small-cell lung cancer. J Clin Oncol 2020; 38: 1505–1517. [DOI] [PubMed] [Google Scholar]
- 10. Paz-Ares L, Luft A, Vicente D, et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N Engl J Med 2018; 379: 2040–2051. [DOI] [PubMed] [Google Scholar]
- 11. Socinski MA, Jotte RM, Cappuzzo F, et al. Atelizumab for first-line treatment of metastatic nonsquamous NSCLC. N Engl J Med 2018; 378: 2288–2301. [DOI] [PubMed] [Google Scholar]
- 12. Mazieres J, Drilon A, Lusque A, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann Oncol 2019; 30: 1321–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dong ZY, Zhong WZ, Zhang XC, et al. Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin Cancer Res 2017; 23: 3012–3024. [DOI] [PubMed] [Google Scholar]
- 14. Skoulidis F, Byers LA, Diao L, et al. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov 2015; 5: 860–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wellenstein MD, de Visser KE. Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity 2018; 48: 399–416. [DOI] [PubMed] [Google Scholar]
- 16. Skoulidis F, Goldberg ME, Greenawalt DM, et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov 2018; 8: 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lei L, Wang WX, Yu ZY, et al. A real-world study in advanced non-small cell lung cancer with KRAS mutations. Transl Oncol 2020; 13: 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cai D, Hu C, Li L, et al. The prevalence and prognostic value of KRAS co-mutation subtypes in Chinese advanced non-small cell lung cancer patients. Cancer Med 2020; 9: 84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bange E, Marmarelis ME, Hwang W-T, et al. Impact of KRAS and TP53 co-mutations on outcomes after first-line systemic therapy among patients with STK11-mutated advanced non-small-cell lung cancer. JCO Precis Oncol 2019; 3: PO.18.00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kerr EM, Gaude E, Turrell FK, et al. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature 2016; 531: 110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Caiola E, Falcetta F, Giordano S, et al. Co-occurring KRAS mutation/LKB1 loss in non-small cell lung cancer cells results in enhanced metabolic activity susceptible to caloric restriction: an in vitro integrated multilevel approach. J Exp Clin Cancer Res 2018; 37: 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ji H, Ramsey MR, Hayes DN, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 2007; 448: 807–810. [DOI] [PubMed] [Google Scholar]
- 23. Scheel AH, Ansén S, Schultheis AM, et al. PD-L1 expression in non-small cell lung cancer: correlations with genetic alterations. Oncoimmunology 2016; 5: e1131379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koyama S, Akbay EA, Li YY, et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res 2016; 76: 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ng TL, Liu Y, Dimou A, et al. Predictive value of oncogenic driver subtype, programmed death-1 ligand (PD-L1) score, and smoking status on the efficacy of PD-1/PD-L1 inhibitors in patients with oncogene-driven non-small cell lung cancer. Cancer 2019; 125: 1038–1049. [DOI] [PubMed] [Google Scholar]
- 26. Busch SE, Hanke ML, Kargl J, et al. Lung cancer subtypes generate unique immune responses. J Immunol 2016; 197: 4493–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Biton J, Mansuet-Lupo A, Pécuchet N, et al. TP53, STK11, and EGFR mutations predict tumor immune profile and the response to anti-PD-1 in lung adenocarcinoma. Clin Cancer Res 2018; 24: 5710–5723. [DOI] [PubMed] [Google Scholar]
- 28. Schabath MB, Welsh EA, Fulp WJ, et al. Differential association of STK11 and TP53 with KRAS mutation-associated gene expression, proliferation and immune surveillance in lung adenocarcinoma. Oncogene 2016; 35: 3209–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kitajima S, Ivanova E, Guo S, et al. Suppression of STING associated with LKB1 loss in KRAS-driven lung cancer. Cancer Discov 2019; 9: 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cho BC, Lopes G, Kowalski DM, et al. Abstract CT084: Relationship between STK11 and KEAP1 mutational status and efficacy in KEYNOTE-042: pembrolizumab monotherapy versus platinum-based chemotherapy as first-line therapy for PD-L1-positive advanced NSCLC. Cancer Res 2020; 80: CT084. [Google Scholar]
- 31. Sanmamed MF, Chen L. A paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell 2018; 175: 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Torralvo J, Friedlaender A, Achard V, et al. The activity of immune checkpoint inhibition in KRAS mutated non-small cell lung cancer: a single centre experience. Cancer Genomics Proteomics 2019; 16: 577–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jeanson A, Tomasini P, Souquet-Bressand M, et al. Efficacy of immune checkpoint inhibitors in KRAS-mutant Non-Small Cell Lung Cancer (NSCLC). J Thorac Oncol 2019; 14: 1095–1101. [DOI] [PubMed] [Google Scholar]
- 34. Westcott PM, To MD. The genetics and biology of KRAS in lung cancer. Chin J Cancer 2013; 32: 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cox AD, Fesik SW, Kimmelman AC, et al. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov 2014; 13: 828–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ihle NT, Byers LA, Kim ES, et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst 2012; 104: 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Green S, Trejo CL, McMahon M. PIK3CA(H1047R) accelerates and enhances KRAS(G12D)-driven lung tumorigenesis. Cancer Res 2015; 75: 5378–5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Best SA, Ding S, Kersbergen A, et al. Distinct initiating events underpin the immune and metabolic heterogeneity of KRAS-mutant lung adenocarcinoma. Nat Commun 2019; 10: 4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schuster K, Venkateswaran N, Rabellino A, et al. Nullifying the CDKN2AB locus promotes mutant K-ras lung tumorigenesis. Mol Cancer Res 2014; 12: 912–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Westcott PM, Halliwill KD, To MD, et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature 2015; 517: 489–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen N, Fang W, Lin Z, et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol Immunother 2017; 66: 1175–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Qiu J, Villa M, Sanin DE, et al. Acetate promotes T cell effector function during glucose restriction. Cell Rep 2019; 27: 2063–2074.e2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chang CH, Qiu J, O’Sullivan D, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015; 162: 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity 2015; 43: 435–449. [DOI] [PubMed] [Google Scholar]
- 45. Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer 2009; 9: 563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. DeNicola GM, Karreth FA, Humpton TJ, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011; 475: 106–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Galan-Cobo A, Sitthideatphaiboon P, Qu X, et al. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res 2019; 79: 3251–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Romero R, Sayin VI, Davidson SM, et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med 2017; 23: 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shen X, Zhao Y, Liu G, et al. Upregulation of programmed death ligand 1 by liver kinase B1 and its implication in programmed death 1 blockade therapy in non-small cell lung cancer. Life Sci 2020; 256: 117923. [DOI] [PubMed] [Google Scholar]
- 50. Rizvi H, Sanchez-Vega F, La K, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-Programmed Death-Ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol 2018; 36: 633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Aggarwal C, Thompson JC, Chien AL, et al. Baseline plasma tumor mutation burden predicts response to pembrolizumab-based therapy in patients with metastatic non-small cell lung cancer. Clin Cancer Res 2020; 26: 2354–2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fukumura D, Kloepper J, Amoozgar Z, et al. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol 2018; 15: 325–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019; 575: 217–223. [DOI] [PubMed] [Google Scholar]
- 54. Wang Y, Li N, Jiang W, et al. Mutant LKB1 confers enhanced radiosensitization in combination with trametinib in KRAS-mutant non-small cell lung cancer. Clin Cancer Res 2018; 24: 5744–5756. [DOI] [PubMed] [Google Scholar]
- 55. Manchado E, Weissmueller S, Morris JP, IV, et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature 2016; 534: 647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Carretero J, Shimamura T, Rikova K, et al. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell 2010; 17: 547–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shackelford DB, Abt E, Gerken L, et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 2013; 23: 143–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Papillon-Cavanagh S, Doshi P, Dobrin R, et al. STK11 and KEAP1 mutations as prognostic biomarkers in an observational real-world lung adenocarcinoma cohort. ESMO Open 2020; 5: e000706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pécuchet N, Laurent-Puig P, Mansuet-Lupo A, et al. Different prognostic impact of STK11 mutations in non-squamous non-small-cell lung cancer. Oncotarget 2017; 8: 23831–23840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Xu JY, Zhang C, Wang X, et al. Integrative proteomic characterization of human lung adenocarcinoma. Cell 2020; 182: 245–261. [DOI] [PubMed] [Google Scholar]
- 61. Junttila MR, Karnezis AN, Garcia D, et al. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature 2010; 468: 567–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shajani-Yi Z, de Abreu FB, Peterson JD, et al. Frequency of somatic TP53 mutations in combination with known pathogenic mutations in colon adenocarcinoma, non-small cell lung carcinoma, and gliomas as identified by next-generation sequencing. Neoplasia 2018; 20: 256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008; 455: 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Assoun S, Theou-Anton N, Nguenang M, et al. Association of TP53 mutations with response and longer survival under immune checkpoint inhibitors in advanced non-small-cell lung cancer. Lung Cancer 2019; 132: 65–71. [DOI] [PubMed] [Google Scholar]
- 65. Meylan E, Dooley AL, Feldser DM, et al. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature 2009; 462: 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science 2013; 339: 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pappas K, Xu J, Zairis S, et al. p53 maintains baseline expression of multiple tumor suppressor genes. Mol Cancer Res 2017; 15: 1051–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kitajima S, Asahina H, Chen T, et al. Overcoming resistance to dual innate immune and MEK inhibition downstream of KRAS. Cancer Cell 2018; 34: 439–452.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jones SA, Jenkins BJ. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat Rev Immunol 2018; 18: 773–789. [DOI] [PubMed] [Google Scholar]