

Abstract
Droplet digital polymerase chain reaction (ddPCR) is a highly sensitive and accurate method for quantification of nucleic acid sequences. We used absolute quantification of mutated v‐Ki‐ras2 Kirsten rat sarcoma viral oncogene homology gene (KRAS) by ddPCR to investigate the prognostic role of mutated KRAS in patients with KRAS‐mutated lung adenocarcinomas. Pre‐treatment plasma samples from 60 patients with stages I–IV KRAS‐mutated lung adenocarcinomas were analysed for KRAS mutations. The associations between survival, detectable KRAS mutations in plasma, and the plasma concentration of mutated KRAS were assessed. Overall, 23 of 60 (38%) patients had detectable KRAS mutation in plasma. The percentage of patients with detectable mutation was 8% in stage I, 30% in stage II, 71% in stage III, and 73% in stage IV. Estimated overall median progression‐free survival (PFS) and overall survival (OS) were 26.2 months [95% confidence interval (CI) 12.5–39.9] and 50.8 months (95% CI 0–107.3), respectively. Patients with detectable mutations in plasma had significantly worse median PFS compared to patients with undetectable mutation (13.1 versus 70.1 months) and shorter median OS (20.7 versus not reached). High circulating tumour DNA (ctDNA) concentrations of mutated KRAS were significantly associated with shorter PFS [hazard ratio (HR) 1.008, 95% CI 1.004–1.012] and OS (HR 1.007, 95% CI 1.003–1.011). All associations remained statistically significant in multivariable analyses. In conclusion, ddPCR is an accurate and easily feasible technique for quantification of KRAS mutations in ctDNA. The presence of detectable KRAS mutation in plasma at baseline was associated with worse PFS and OS. High concentration of mutated KRAS in ctDNA was an independent negative prognostic factor for both PFS and OS.
Keywords: non‐small cell lung cancer, liquid biopsy, plasma analyses, droplet digital PCR, ctDNA levels
Introduction
Despite recent advances in the treatment of non‐small cell lung cancer (NSCLC) with the introduction of tyrosine kinase inhibitors and immune checkpoint inhibitors, lung cancer is still the leading cause of death worldwide. Next‐generation sequencing (NGS) technologies have become widely accessible and have improved the knowledge of druggable genetic aberrations in tumour DNA, as well as genetic aberrations interfering with targeted treatment strategies. However, there is still a need for easily assessable biomarkers that can be used for better prognostication of individual patients. Analyses of circulating cell‐free DNA (cfDNA) and circulating tumour DNA (ctDNA) are promising approaches. cfDNA comprises DNA released from non‐malignant cells and a smaller ctDNA fraction derived from apoptotic and necrotic tumour cells [1] and possibly by active release from tumour cells [2]. ctDNA may include information from genetically heterogenous areas of the primary tumour and metastatic sites, possibly reflecting the whole tumour genome [3]. ctDNA analyses have a growing number of clinical applications. In lung adenocarcinoma patients, ctDNA analyses for the detection of druggable genetic aberrations, or mutations associated with treatment resistance mechanisms, have become an important alternative when tissue is not available for analysis [4, 5, 6, 7, 8, 9]. Studies also indicate that detectable ctDNA and high pre‐treatment ctDNA concentrations are associated with a negative impact on survival in various cancer types [10, 11, 12, 13]. Detectable ctDNA after treatment for localised cancer may predict recurrence [14], while an increase in ctDNA concentration during follow‐up may indicate relapse or progression, enabling disease monitoring and detection of progression in advance of image detection [15, 16, 17].
Hot‐spot mutations in codons 12 and 13 in exon 2 of the v‐Ki‐ras2 Kirsten rat sarcoma viral oncogene homology gene (KRAS) are the most common driver mutations in lung adenocarcinomas and are found in approximately 25–30% of non‐Asian patients [18, 19]. Thus, we considered KRAS a relevant biomarker for investigating the prognostic role of ctDNA in patients with lung adenocarcinomas harbouring KRAS mutations. Using droplet digital polymerase chain reaction (ddPCR) technology for accurate quantification of mutated KRAS, we aimed to explore whether there was a difference in progression‐free survival (PFS) and overall survival (OS) between patients with and without detectable KRAS mutations in plasma at baseline. We also explored whether there was an association between the plasma concentration of mutated KRAS and PFS and OS.
Materials and methods
Ethics
This study was approved by the Regional Committee for Medical and Health Research Ethics (REC) in Central Norway. Both tissue and plasma samples were collected from Biobank1®, the regional research biobank in Central Norway. The biobank is approved by the REC in Central Norway, the Ministry of Health and Care Services, and the Norwegian Data Protection Authority. Patients enrolled in the biobank are over 18 years old and have given written informed consent.
Patient inclusion and tumour specimens
A total of 553 patients with NSCLC were included in the biobank from 2007 to 2018. All tumour specimens in the biobank were reviewed and classified according to the World Health Organization (WHO) 2015 classification of lung tumours [20] by two lung pathologists (SGFW and VGD). Clinical and pathological disease stages were restaged according to the Eighth Edition of the TNM Classification for lung cancer. Patients fulfilling the following criteria were included: treatment‐naïve KRAS‐mutated lung adenocarcinoma, available blood sample collected prior to first treatment, and a time interval of ≥5 years between the final control of any previous cancer and the lung cancer. Sixty patients fulfilled all criteria and were eligible for this study (Figure 1). Clinicopathological baseline characteristics were collected from the hospital medical records. Tumour specimens were tested for KRAS mutations using either the KRAS mutation analysis protocol implemented for routine diagnostics at the Department of Pathology, St. Olavs Hospital, or targeted NGS using the Trusight Tumour 26 panel (Illumina®, San Diego, CA, USA).
Figure 1.
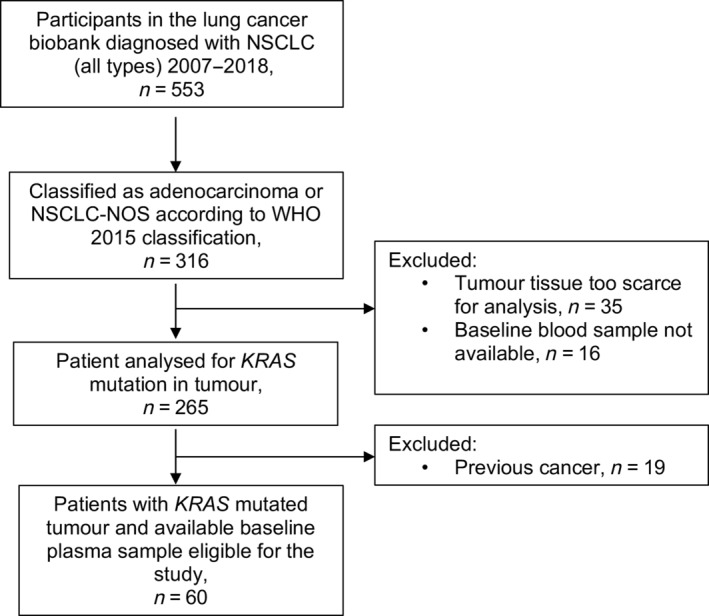
Outline of patient selection. NOS, not otherwise specified.
Blood samples and DNA extraction from plasma
A total of 10–18 ml peripheral whole blood was collected in ethylenediaminetetraacetic (EDTA) acid tubes. Plasma was prepared within 1 h by centrifugation at 4 °C, either at 2500 × g for 10 min or at 1500 × g for 15 min. Samples collected after 2016 had a second centrifugation step at 10 000 × g for 10 min at 4 °C. The plasma samples were then aliquoted, transferred to cryotubes (Nalgene™ Cryotubes, Thermo Scientific™; Thermo Fisher Scientific Inc., Waltham, MA, USA), and stored at −80 °C until DNA extraction. The plasma samples were thawed at room temperature. cfDNA was extracted from 2 to 4 ml plasma using the QIAamp® Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany) according to the manufacturer's manual and was finally eluted in 50 μl of elution buffer. The DNA concentration was quantified by a Qubit fluorometer (Thermo Fisher Scientific Inc.).
Plasma samples used as KRAS mutation‐negative controls were prepared according to the same protocol from deidentified healthy donor blood obtained from the Department of Immunology and Transfusion Medicine at St. Olavs Hospital and from patients included in the biobank.
Validation of ddPCR assays
Before running the patient samples, we performed mutation serial dilutions for each assay to test the ability to detect and quantify specific mutant alleles in a background of excess wild‐type (wt) DNA. Mutated KRAS plasmid templates were serially diluted to 1:2 from a stock solution of 250 copies/μl down to 2 copies/μl for all target KRAS mutations and further down to 0.5 copies/μl for KRAS G12A. A 2‐μl plasmid solution was spiked into a background of 4 ng of genomic wt DNA (approximately 1212 copies) extracted from blood samples from healthy individuals. The plasmid titre samples and PCR reagents were loaded onto the QX200 Droplet Digital PCR System (Bio‐Rad Laboratories Inc., Hercules, CA, USA) in triplicate or quadruplicate. In addition to each serial dilution assay, we ran the following controls: KRAS wt reference standards (Horizon Discovery, Cambridge, UK) for assessment of assay performance; wt genomic DNA controls extracted from blood samples from healthy individuals (the same DNA used in the KRAS plasmid dilutions) for the detection of false‐positive mutant KRAS; and DNA‐free, non‐template controls (NTCs) for contamination. The PCR and data analyses were performed as described for the KRAS analyses of plasma samples. The wt reference standards, NTCs, and cfDNA from healthy donors were used as controls when running the patient samples. The wt genomic DNA from healthy donors was also used as a control in some of the set‐ups. The performance of the ddPCR assays was investigated using regression analysis to assess the linearity of the serial dilutions.
For determination of the limit of detection (LOD), the distribution of false‐positive mutant KRAS copies in the wt standards and wt controls per reaction for each assay in both the mutation titration series and patient sample set‐ups was recorded. A 95% confidence interval (CI) for false positives per assay was calculated, and the upper limit of the CI was defined as the LOD.
KRAS analysis of plasma
KRAS analysis of plasma DNA was performed using the QX200 Droplet Digital PCR System and Bio‐Rad's ddPCR KRAS mutation assays (Bio‐Rad Laboratories). In brief, 5 μl of plasma DNA (2–30 ng) was mixed in a total reaction volume of 20 μl with ddPCR Supermix and primers and Taqman probes (Bio‐Rad Laboratories) for both wt and mutated KRAS alleles (p.G12D, p.G12A, p.G12V, p.G12C, p.G12S, and p.G13D). The reaction mixture was partitioned into aqueous droplets in oil by the QX200 Droplet Generator and then transferred to a PCR plate. The PCR was carried out with a two‐step thermal cycling protocol: 10 min at 95 °C, 40 cycles of 30 s at 94 °C and 60 s at 56 °C, and 10 min at 96 °C. The reaction was held at 4 °C until the plate was transferred to the Bio‐Rad's QX200 Droplet Reader to determine the level of fluorescence in each droplet. The data were processed and analysed using QuantaSoft version 1.74 software (Bio‐Rad Laboratories). The number of mutated and wt KRAS‐positive droplets was converted to the copy number/μl of PCR reaction mixture by the software, and the concentrations of mutated and wt KRAS copies/ml plasma were then calculated. Three or four replicas of each patient sample were run in each experimental set‐up. In addition, each set‐up also included a mutation‐positive control, a mutation‐negative control, and an NTC. Plasmid DNA containing fragments of the KRAS exon 2 target mutations or DNA isolated from KRAS mutation‐positive tumour samples was used as the mutation‐positive control. As mutation‐negative controls, either cfDNA from healthy blood donors or KRAS wt reference standards (Horizon) were used.
Statistics
Chi‐square test for independence was used to explore the association between the fraction of patients with detectable KRAS and clinical stage. The independent‐samples t‐test was used to compare the DNA yield from plasma between patients with and without detectable KRAS mutations in plasma. The Kruskal–Wallis H‐test was used to compare the DNA yield from plasma, and the plasma concentration of mutated KRAS, across disease stages. PFS was defined as the time from diagnosis until objective progression or death by any cause, and OS was defined as the time from diagnosis until death by any cause. Using the reversed Kaplan–Meier method, median follow‐up time for PFS and OS was calculated for event‐free patients from the date of diagnosis to the date the collection of survival data was completed (July 2019). Survival was estimated using the Kaplan–Meier method and compared using the log‐rank test. The Cox proportional hazard model was used for multivariable analyses, adjusting for the following baseline characteristics: age, sex, disease stage, WHO performance status (PS), and treatment. The significance level was defined as a two‐sided p < 0.05. All analyses were performed using the IBM SPSS Statistics for Windows version 25.0. (IBM Corp., Armonk, NY, USA).
Results
Patient characteristics
An overview of patient characteristics is presented in Table 1. Median age was 69 (range 47–83) years; all patients were former or current smokers; 37 (62%) were women; and 25 (42%) had stage I disease, 10 (17%) stage II, 14 (23%) stage III, and 11 (18%) had stage IV disease. Thirty‐three patients (55%) had WHO PS 0, 23 (38%) PS 1, and 4 (7%) PS 2. The primary treatment was surgery in 42 patients (70%), curative chemoradiotherapy in 3 (5%), and palliative treatment in 15 (25%). Of the latter, 12 (80%) received platinum‐doublet chemotherapy, 1 (7%) received whole‐brain radiotherapy only, and 2 (13%) received palliative radiotherapy. The distribution of KRAS mutations was as follows: G12C in 31 (52%) patients, G12V in 13 (22%), G12D in 9 (15%), G12A in 3 (5%), G12S in 2 (3%), and G13D in 2 (3%) patients. Detailed clinical data and tumour characteristics are listed in supplementary material, Table S1.
Table 1.
Baseline characteristics.
| Characteristic | Total (N = 60) | KRAS mutation status in plasma | |
|---|---|---|---|
| Not detected (n = 37) | Detected (n = 23) | ||
| Age | 69 (47–83) | 71 (54–79) | 64 (47–83) |
| Sex | |||
| Female | 37 (62) | 21 (57) | 16 (70) |
| Male | 23 (38) | 16 (43) | 7 (30) |
| Smoking history | |||
| Non‐smoker | 0 (0) | 0 (0) | 0 (0) |
| Smoker/former smoker | 60 (100) | 37 (100) | 23 (100) |
| WHO performance status | |||
| 0 | 33 (55) | 22 (60) | 11 (48) |
| 1 | 23 (38) | 12 (32) | 11 (48) |
| 2 | 4 (7) | 3 (8) | 1 (4) |
| Clinical disease stage | |||
| IA | 17 (28) | 15 (41) | 2 (9) |
| IB | 8 (13) | 8 (22) | 0 (0) |
| IIA | 4 (7) | 2 (5) | 2 (9) |
| IIB | 6 (10) | 5 (14) | 1 (4) |
| IIIA | 9 (15) | 3 (8) | 6 (26) |
| IIIB | 5 (8) | 1 (3) | 4 (17) |
| IVA | 8 (13) | 2 (5) | 6 (26) |
| IVB | 3 (5) | 1 (3) | 2 (9) |
| Histology | |||
| Adenocarcinoma | 60 (100) | 37 (62) | 23 (38) |
| Therapy | |||
| Complete resection | 42 (70) | 32 (87) | 10 (44) |
| Curative chemoradiotherapy | 3 (5) | 0 (0) | 3 (13) |
| Palliative | 15 (25) | 5 (14) | 10 (44) |
| KRAS mutation | |||
| G12C | 31 (52) | 19 (51) | 12 (52) |
| G12V | 13 (22) | 7 (19) | 6 (26) |
| G12D | 9 (15) | 7 (19) | 2 (9) |
| G12S | 2 (3) | 1 (3) | 1 (4) |
| G12A | 3 (5) | 2 (5) | 1 (4) |
| G13D | 2 (3) | 1 (3) | 1 (4) |
Data are presented as median (range) or n (%).
Assay validation and determination of detection limits
Regression analyses revealed good linearity over the range of dilutions with the following R 2 values: 0.9957 for G12V, 0.9906 for G12C, 0.9963 for G12D, 0.9979 for G12S, 0.9884 for G12A, and 0.9933 for G13D (see supplementary material, Figure S1). The average number of false‐positive mutated KRAS copies detected per reaction was 0 for G12V, 0.0326 for G12C, 0.392 for G12D, 0.093 for G12S, 0 for G12A, and 0.063 for G13D. The upper limit of the 95% CI for each assay was defined as the LOD and was as follows: 0 for G12V and G12A, 0.69 mutated copies for G12D, 0 for G12A, 0.10 mutated copies for G12C, 0.29 mutated copies for G12S, and 0.19 mutated copies for G13D.
Plasma
The plasma samples were obtained as part of the primary investigation, with a median of 19 (range 0–307) days prior to first treatment. In 38 patients, the plasma samples were collected <38 days before first treatment; in 18 patients, between 30 and 89 days before first treatment; and in 4 patients, >90 days prior to first treatment (see supplementary material, Table S1). The median cfDNA yield from plasma was 1.04 (range 0.40–6.24) ng/μl for disease stage I, 0.93 (range 0.46–5.03) ng/μl for stage II, 0.75 (range 0.40–1.58) ng/μl for stage III, and 0.76 (range 0.37–1.28) ng/μl for stage IV. There was no difference in the cfDNA yield across the disease stages (p = 0.119). The median DNA yield in patients with detectable KRAS mutations in plasma was 0.89 (range 0.41–2.04) ng/μl and 0.90 (range 0.37–0.6.24) ng/μl in patients with non‐detectable KRAS mutation. The difference in DNA yield between these groups was not significant (p = 0.67). Overall, 23 of 60 (38%) patients had detectable KRAS mutation in plasma. The proportion of patients with detectable KRAS mutation in plasma increased significantly with increasing disease stage: in stage I, 2 of 25 (8%) had detectable mutation; in stage II, 3 of 10 (30%); in stage III, 10 of 14 (71%), and in stage IV, 8 of 11 (73%) (p < 0.001). The concentration of mutated KRAS increased significantly with disease stage, with a median of 0 (range 0–3.3) copies/ml for stage I, 0 (range 0–92.4) for stage II, 11.26 (range 0–217.8) for stage III, and 84.1 (range 0–1208.0) for stage IV (p < 0.001). Within the group of patients with detectable mutations, the median concentration was 37.0 (range 1.1–1208) mutated KRAS copies/ml plasma. Results of the ctDNA analyses are summarised in supplementary material, Table S1.
Progression‐free and overall survival
The median follow up for PFS was 38.7 (range 9.44–115.5) months; 27 patients were alive and progression free when the data collection was completed. The median follow‐up for OS was 40.9 (range 9.4–142.0) months; 32 patients were alive at the time of data collection completion.
For all patients, the estimated median PFS was 26.2 months (95% CI 12.5–39.9), and the estimated median OS was 50.8 months (95% CI 0–107.3). Patients with detectable plasma KRAS mutations had significantly worse median PFS compared with the patients with non‐detectable KRAS mutation (13.1 versus 70.1 months, p < 0.001) and significantly shorter median OS (20.7 versus not reached, p = 0.002) (Figure 2). These associations remained statistically significant in the multivariable analyses, adjusting for baseline characteristics for both PFS [hazard ratio (HR) 2.76, 95% CI 1.064–7.162, p = 0.037] and OS (HR 3.609, 95% CI 1.261–10.328, p = 0.017).
Figure 2.
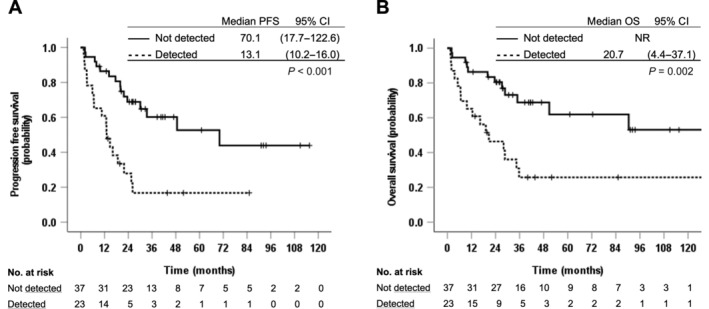
Association between detectable KRAS mutation in plasma and PFS (A) and OS (B). NR, not reached.
The concentration of mutated KRAS was significantly associated with both shorter PFS (HR 1.008, 95% CI 1.004–1.012, p < 0.001) and OS (HR 1.007, 95% CI 1.003–1.011, p = 0.001). The associations remained statistically significant in the multivariable analyses (Table 2) for both PFS (HR 1.008, 95% CI 1.002–1.014, p = 0.005) and OS (HR 1.009, 95% CI 1.003–1.016, p = 0.004).
Table 2.
Associations between mutant KRAS plasma concentration, PFS, and OS.
| PFS | OS | ||||||
|---|---|---|---|---|---|---|---|
| HR | 95% CI | p | HR | 95% CI | p | ||
| Age (years) | 1.01 | 0.96–1.07 | 0.65 | 1.05 | 1.00–1.11 | 0.06 | |
| Sex | Female | 1 (ref) | |||||
| Male | 0.66 | 0.26–1.70 | 0.39 | 0.55 | 0.19–1.55 | 0.26 | |
| Clinical stage | I | 1 (ref) | 1 (ref) | ||||
| II | 2.11 | 0.54–8.25 | 0.28 | 1.82 | 0.41–8.14 | 0.43 | |
| III | 2.70 | 0.71–10.30 | 0.15 | 2.20 | 0.47–10.40 | 0.32 | |
| IV | 2.18 | 0.22–21.94 | 0.51 | 3.58 | 0.28–45.46 | 0.33 | |
| WHO PS | 0 | 1 (ref) | 1 (ref) | ||||
| 1 | 0.72 | 0.30–1.73 | 0.47 | 0.84 | 0.33–2.10 | 0.70 | |
| 2 | 4.45 | 0.74–26.68 | 0.10 | 8.31 | 1.24–55.62 | 0.03 | |
| Treatment intention | Complete resection | 1 (ref) | 1 (ref) | ||||
| Curative radiochemotherapy | 1.02 | 0.18–5.72 | 0.99 | 0.61 | 0.06–6.02 | 0.68 | |
| Palliative chemotherapy and/or radiotherapy | 9.76 | 1.55–61.49 | 0.02 | 5.48 | 0.86–34.79 | 0.07 | |
| Mutant KRAS concentration | (mutated copies/ml plasma) | 1.008 | 1.002–1.014 | 0.005 | 1.009 | 1.003–1.016 | 0.004 |
To illustrate the association between mutated KRAS plasma concentration and survival, the patients were split into three groups: (1) those with plasma concentrations of mutated KRAS above the median of 37.0 mutated copies/ml, (2) those with concentrations below 37.0 mutated copies/ml, and (3) those without detectable KRAS mutations. For the three groups, the median PFS was 6.7 versus 16.1 versus 70.1 months, respectively (p < 0.001), and the median OS was 6.7 months, 28.1 months, and not reached, respectively (p = 0.002) (Figure 3).
Figure 3.
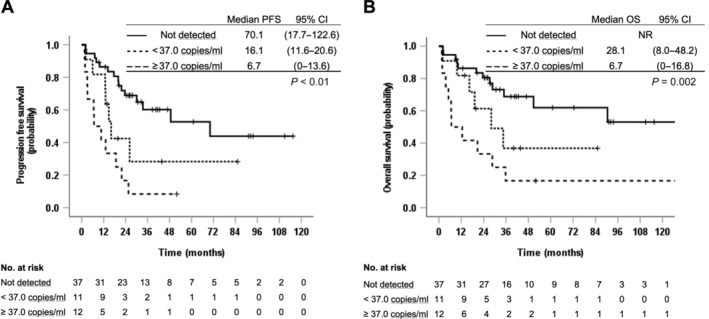
Association between ctDNA concentration of mutated KRAS (copies/ml) and (A) PFS and (B) OS. NR, not reached; 37.0 copies/ml was the median concentration of mutated KRAS among those with detectable KRAS mutations in plasma.
Discussion
In summary, 23 (38%) of the 60 patients had detectable KRAS mutation in their baseline plasma sample. There was no difference in DNA yield across disease stages or between patients with or without detectable KRAS mutations in plasma. The fraction of patients with detectable KRAS mutations and the concentration of mutated KRAS increased with increasing disease stage. Patients with detectable plasma KRAS mutations had significantly worse PFS and OS than the patients with undetectable mutations. There was also a negative association between high concentrations of mutated KRAS and PFS and OS.
The increase in ctDNA concentration with increasing disease stage is well known and is consistent with the results of other studies [3, 10, 14]. In addition, the fraction of patients with detectable KRAS mutations across disease stages in our study is consistent with other studies on NSCLC using ddPCR for the KRAS analyses of plasma [21, 22, 23].
A negative association between detectable KRAS mutations in baseline plasma and survival has been demonstrated in studies on patients with pancreatic and colorectal cancer [24, 25, 26]. Qualitative studies of patients with NSCLC have also demonstrated an association between detectable KRAS mutations in serum or plasma at baseline and worse PFS and/or OS [23, 27, 28, 29, 30, 31, 32, 33]. A recent study, where plasma was analysed for known genetic aberrations in tumour DNA, also showed that detectable baseline ctDNA was associated with shorter OS [34]. However, in three other studies on the detection of KRAS mutations in serum or plasma, no associations with survival were found [35, 36, 37]. The results of two of these studies might be explained by heterogeneity of the study populations (some included non‐adenocarcinoma patients), no prior analyses of corresponding tumour DNA, and a limited number of hot‐spot KRAS mutations targeted in the ctDNA analyses [35, 36]. In the third study [37], only patients with advanced KRAS mutated NSCLC were included, but the proportion of patients with detectable KRAS mutation in plasma was lower (48%) compared to the detection rate in other, similar studies [21, 22]. ctDNA studies of lung adenocarcinoma patients harbouring mutations of the epidermal growth factor receptor (EGFR) gene have also shown diverging results, but these might be explained by differences in treatment history [38, 39, 40].
Quantitative studies of ctDNA levels, assessing either the concentration of ctDNA or the mutant allele frequency (MAF), have shown a negative correlation between high baseline ctDNA concentration and survival in colorectal cancer [10, 41], pancreatic cancer [13, 42], breast cancer [11, 43], oesophageal cancer [44], and malignant melanoma [12, 45]. While many studies on NSCLC have shown a negative association between the levels of cfDNA, as summarised in the meta‐analyses by Ai et al and Cargnin et al [46, 47], few have assessed the impact of mutant ctDNA levels before the first treatment. Two recent studies have suggested that high MAFs of detected alterations are associated with worse PFS and OS also in NSCLC [48, 49].
Our work has some limitations. It is a retrospective study with a limited number of patients, especially with respect to patients with stages I and II disease. We detected mutated KRAS in only a small proportion of the patients with stages I and II disease, which may be explained by the low MAF of ctDNA in early‐stage disease [3, 50]. More comprehensive NGS panels with capturing and sequencing approaches optimised for ctDNA, such as Cancer Personalised Profiling by deep Sequencing (CAPP‐Seq) [15], Tracking Cancer Evolution Through Therapy (TRACERx) [3, 50], and Targeted Error Correction Sequencing (TEC‐Seq) [41], may increase the sensitivity of ctDNA analyses, but detection of ctDNA in early‐stage disease remains a challenge.
As aforementioned, baseline cfDNA levels in plasma or serum may have prognostic value [46, 47] and may also be used in disease monitoring [51]. We did not conduct any additional analyses of cfDNA beyond measuring the DNA yield from plasma prior to ddPCR, although further analyses of the cfDNA levels using reference genes as markers for cfDNA would have been of interest. However, standardised methods for analyses of cfDNA are needed [52, 53].
Another potential limitation of our study is the range of days between blood sample collection and commencing treatment (0–307 days). In four patients, the blood samples were collected >90 days before treatment due to intercurrent diseases or difficulties in obtaining tissue specimens (patient IDs 30, 31, 52, and 57; see supplementary material, Table S1). During the diagnostic work‐up, one of these patients had radiological progression from stage IB to IIB, while there was no radiological progression in the other three patients. The results of the survival analyses were not altered when running sensitivity analyses excluding these four patients (data not shown).
The storage time of the plasma samples may also be a potential limitation. The amount of cfDNA in a plasma sample may decrease during storage [54, 55] and could have an impact on the detection rate. We did not, however, find any significant difference in the total KRAS concentration between plasma samples with different storage times (data not shown).
Using a single gene or few genes approach targeting known tumour‐specific mutations in ctDNA by ddPCR has some advantages over NGS. It may minimise the risk of falsely misinterpreting somatic mutations in haematopoietic stem cells associated with clonal haematopoiesis as tumour derived [56, 57]. In addition, for monitoring treatment and disease course, accurate quantification of nucleic acids combined with simple workflow, low cost, and less complex data analysis make ddPCR a feasible technique in the daily routine of a diagnostic laboratory.
Although the assays we used have been validated by the manufacturer, we performed in‐house validation with tests for linearity and establishment of LODs for each assay. In our experience, comparing detection rates of ctDNA across different studies is challenging, even if the same platforms for ctDNA analyses have been used. Data on false‐positive rates and how the LOD was determined is important information that should be available for cross‐study comparison. Lack of concordance between validated commercial kits for KRAS mutation detection, which have the same LOD according to the manufacturers, has been reported [58]. Therefore, the false‐positive rate and LOD should be established in house for the assays used, even if the assays are pre‐validated by the manufacturer.
In conclusion, patients with detectable KRAS mutation in their baseline plasma sample had worse PFS and OS than patients who did not have detectable KRAS mutations. Our findings also suggest that the baseline plasma concentration of mutated KRAS before the first treatment may be an important, independent prognostic factor in KRAS‐positive lung adenocarcinomas. Hence, a quantitative analysis assessing the plasma concentration of mutated KRAS improves the prognostic value compared to qualitative analysis only. These findings are confirmed in other studies, but the value of ctDNA baseline levels in treatment decision‐making must be evaluated in larger prospective clinical trials. Finally, ddPCR for ctDNA analyses is a highly sensitive and accurate technique that can be implemented in a daily diagnostic routine.
Author contributions statement
SGFW participated in planning and designing the study, evaluated all histological material, collected the clinical data, performed statistical analyses, participated in interpretation of laboratory and statistical analyses, and drafted and wrote the manuscript. HYD designed and planned the study and participated in the laboratory set‐up, performed molecular analyses of tissue and plasma, interpreted the molecular analyses, and contributed to manuscript preparation. EFE optimised the laboratory protocol, participated in the laboratory set‐up, performed some of the tissue analyses and all the plasma analyses, interpreted the plasma analyses, and contributed to manuscript preparation. ALO performed some of the tissue analyses and contributed to manuscript preparation. VGD evaluated all histological material and contributed to manuscript preparation. ER contributed to planning the study, gave statistical advice, and contributed to interpretation of statistical analyses and to manuscript preparation. TOH gave statistical advice, participated in performance and interpretation of statistical analyses, and contributed to manuscript preparation. BHG was the group leader and coordinated the study, and contributed to the study design, to patient recruitment, interpretation of statistical analyses, and manuscript drafting and preparation. All authors have read and approved the final manuscript.
Supporting information
Figure S1. Linear regression analyses of serial dilution series of mutated KRAS plasmid templates
Table S1. Patient characteristics and results of ctDNA analyses
Acknowledgements
The authors thank all the patients who have agreed to be included in the biobank, Espen D Jensen and Line MM Skarsvåg for supplying the plasma samples obtained from the biobank, and Hilde JT Lien for technical assistance. This study was funded by the Liaison Committee for Education, Research and Innovation in Central Norway; the Clinic of Laboratory Medicine; and the Cancer Fund at St. Olavs Hospital.
No conflicts of interest were declared.
References
- 1. Stroun M, Lyautey J, Lederrey C, et al. About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin Chim Acta 2001; 313: 139–142. [DOI] [PubMed] [Google Scholar]
- 2. Bronkhorst AJ, Wentzel JF, Aucamp J, et al. Characterization of the cell‐free DNA released by cultured cancer cells. Biochim Biophys Acta 2016; 1863: 157–165. [DOI] [PubMed] [Google Scholar]
- 3. Abbosh C, Birkbak NJ, Wilson GA, et al. Phylogenetic ctDNA analysis depicts early‐stage lung cancer evolution. Nature 2017; 545: 446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marchetti A, Palma JF, Felicioni L, et al. Early prediction of response to tyrosine kinase inhibitors by quantification of EGFR mutations in plasma of NSCLC patients. J Thorac Oncol 2015; 10: 1437–1443. [DOI] [PubMed] [Google Scholar]
- 5. Mok T, Wu Y‐L, Lee JS, et al. Detection and dynamic changes of EGFR mutations from circulating tumor DNA as a predictor of survival outcomes in NSCLC patients treated with first‐line intercalated erlotinib and chemotherapy. Clin Cancer Res 2015; 21: 3196–3203. [DOI] [PubMed] [Google Scholar]
- 6. Thompson JC, Yee SS, Troxel AB, et al. Detection of therapeutically targetable driver and resistance mutations in lung cancer patients by next‐generation sequencing of cell‐free circulating tumor DNA. Clin Cancer Res 2016; 22: 5772–5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Oxnard GR, Thress KS, Alden RS, et al. Association between plasma genotyping and outcomes of treatment with osimertinib (AZD9291) in advanced non‐small‐cell lung cancer. J Clin Oncol 2016; 34: 3375–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jenkins S, Yang JCH, Ramalingam SS, et al. Plasma ctDNA analysis for detection of the EGFR T790M mutation in patients with advanced non–small cell lung cancer. J Thorac Oncol 2017; 12: 1061–1070. [DOI] [PubMed] [Google Scholar]
- 9. Rolfo C, Mack PC, Scagliotti GV, et al. Liquid biopsy for advanced non‐small cell lung cancer (NSCLC): a statement paper from the IASLC. J Thorac Oncol 2018; 13: 1248–1268. [DOI] [PubMed] [Google Scholar]
- 10. Bettegowda C, Sausen M, Leary R, et al. Detection of circulating tumor DNA in early‐ and late‐stage human malignancies. Sci Transl Med 2014; 6: 224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Olsson E, Winter C, George A, et al. Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO Mol Med 2015; 7: 1034–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sanmamed MF, Fernández‐Landázuri S, Rodríguez C, et al. Quantitative cell‐free circulating BRAF V600E mutation analysis by use of droplet digital PCR in the Follow‐up of patients with melanoma being treated with BRAF inhibitors. Clin Chem 2015; 61: 297–304. [DOI] [PubMed] [Google Scholar]
- 13. Tjensvoll K, Lapin M, Buhl T, et al. Clinical relevance of circulating KRAS mutated DNA in plasma from patients with advanced pancreatic cancer. Mol Oncol 2016; 10: 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chaudhuri AA, Chabon JJ, Lovejoy AF, et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov 2017; 7: 1394–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Newman AM, Bratman SV, To J, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage HHS Public Access. Nat Med 2014; 20: 548–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sorensen BS, Wu L, Wei W, et al. Monitoring of epidermal growth factor receptor tyrosine kinase inhibitor‐sensitizing and resistance mutations in the plasma DNA of patients with advanced non‐small cell lung cancer during treatment with erlotinib. Cancer 2014; 120: 3896–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gao F, Pfeifer E, Farah H, et al. Microdroplet digital PCR: detection and quantitation of biomarkers in archived tissue and serial plasma samples in patients with lung cancer. J Thorac Oncol 2015; 10: 212–217. [DOI] [PubMed] [Google Scholar]
- 18. Dogan S, Shen R, Ang DC, et al. Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: higher susceptibility of women to smoking‐related KRAS‐mutant cancers. Clin Cancer Res 2012; 18: 6169–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. El Osta B, Behera M, Kim S, et al. Characteristics and outcomes of patients with metastatic KRAS‐mutant lung adenocarcinomas: the Lung Cancer Mutation Consortium experience. J Thorac Oncol 2019; 14: 876–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Travis WD, Brambilla E, Burke AP, et al. WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart. WHO Classification of Tumours, Volume 7 (4th edn). International Agency for Research on Cancer: Lyon, 2015. [Google Scholar]
- 21. Guibert N, Pradines A, Farella M, et al. Monitoring KRAS mutations in circulating DNA and tumor cells using digital droplet PCR during treatment of KRAS‐mutated lung adenocarcinoma. Lung Cancer 2016; 100: 1–4. [DOI] [PubMed] [Google Scholar]
- 22. Sacher AG, Paweletz C, Dahlberg SE, et al. Prospective validation of rapid plasma genotyping for the detection of EGFR and KRAS mutations in advanced lung cancer. JAMA Oncol 2016; 2: 1014–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Isaksson S, George AM, Jönsson M, et al. Pre‐operative plasma cell‐free circulating tumor DNA and serum protein tumor markers as predictors of lung adenocarcinoma recurrence. Acta Oncol 2019; 58: 1079–1086. [DOI] [PubMed] [Google Scholar]
- 24. Spindler KG, Appelt AL, Pallisgaard N, et al. KRAS‐mutated plasma DNA as predictor of outcome from irinotecan monotherapy in metastatic colorectal cancer. Br J Cancer 2013; 109: 3067–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Normanno N, Esposito Abate R, Lambiase M, et al. RAS testing of liquid biopsy correlates with the outcome of metastatic colorectal cancer patients treated with first‐line FOLFIRI plus cetuximab in the CAPRI‐GOIM trial. Ann Oncol 2018; 29: 112–118. [DOI] [PubMed] [Google Scholar]
- 26. Guo S, Shi X, Shen J, et al. Preoperative detection of KRAS G12D mutation in ctDNA is a powerful predictor for early recurrence of resectable PDAC patients. Br J Cancer 2020; 122: 857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ramirez JL, Sarries C, de Castro PL, et al. Methylation patterns and K‐ras mutations in tumor and paired serum of resected non‐small‐cell lung cancer patients. Cancer Lett 2003; 193: 207–216. [DOI] [PubMed] [Google Scholar]
- 28. Kimura T, Holland WS, Kawaguchi T, et al. Mutant DNA in plasma of lung cancer patients: potential for monitoring response to therapy. Ann N Y Acad Sci 2004; 1022: 55–60. [DOI] [PubMed] [Google Scholar]
- 29. Gautschi O, Huegli B, Ziegler A, et al. Origin and prognostic value of circulating KRAS mutations in lung cancer patients. Cancer Lett 2007; 254: 265–273. [DOI] [PubMed] [Google Scholar]
- 30. Wang S, An T, Wang J, et al. Potential clinical significance of a plasma‐based KRAS mutation analysis in patients with advanced non–small cell lung cancer. Clin Cancer Res 2010; 16: 1324–1330. [DOI] [PubMed] [Google Scholar]
- 31. Nygaard AD, Garm Spindler K‐L, Pallisgaard N, et al. The prognostic value of KRAS mutated plasma DNA in advanced non‐small cell lung cancer. Lung Cancer 2013; 79: 312–317. [DOI] [PubMed] [Google Scholar]
- 32. Dowler Nygaard A, Spindler K‐LG, Pallisgaard N, et al. Levels of cell‐free DNA and plasma KRAS during treatment of advanced NSCLC. Oncol Rep 2014; 31: 969–974. [DOI] [PubMed] [Google Scholar]
- 33. Mardinian K, Okamura R, Kato S, et al. Temporal and spatial effects and survival outcomes associated with concordance between tissue and blood KRAS alterations in the pan‐cancer setting. Int J Cancer 2020; 146: 566–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Corradetti MN, Torok JA, Hatch AJ, et al. Dynamic changes in circulating tumor DNA during chemoradiation for locally advanced lung cancer. Adv Radiat Oncol 2019; 4: 748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Camps C, Sirera R, Bremnes R, et al. Is there a prognostic role of K‐ras point mutations in the serum of patients with advanced non‐small cell lung cancer? Lung Cancer 2005; 50: 339–346. [DOI] [PubMed] [Google Scholar]
- 36. Camps C, Jantus‐Lewintre E, Cabrera A, et al. The identification of KRAS mutations at codon 12 in plasma DNA is not a prognostic factor in advanced non‐small cell lung cancer patients. Lung Cancer 2011; 72: 365–369. [DOI] [PubMed] [Google Scholar]
- 37. Zulato E, Attili I, Pavan A, et al. Early assessment of KRAS mutation in cfDNA correlates with risk of progression and death in advanced non‐small‐cell lung cancer. Br J Cancer 2020; 123: 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang X, Zhuo M, Ye X, et al. Quantification of mutant alleles in circulating tumor DNA can predict survival in lung cancer. Oncotarget 2016; 7: 20810–20824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee Y, Park S, Kim WS, et al. Correlation between progression‐free survival, tumor burden, and circulating tumor DNA in the initial diagnosis of advanced‐stage EGFR‐mutated non‐small cell lung cancer. Thorac Cancer 2018; 9: 1104–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim T, Kim EY, Lee SH, et al. Presence of mEGFR ctDNA predicts a poor clinical outcome in lung adenocarcinoma. Thorac Cancer 2019; 10: 2267–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Phallen J, Sausen M, Adleff V, et al. Direct detection of early‐stage cancers using circulating tumor DNA. Sci Transl Med 2017; 9: eaan2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Patel H, Okamura R, Fanta P, et al. Clinical correlates of blood‐derived circulating tumor DNA in pancreatic cancer. J Hematol Oncol 2019; 12: 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liang DH, Ensor JE, Liu Z‐B, et al. Cell‐free DNA as a molecular tool for monitoring disease progression and response to therapy in breast cancer patients. Breast Cancer Res Treat 2016; 155: 139–149. [DOI] [PubMed] [Google Scholar]
- 44. Pasternack H, Fassunke J, Plum PS, et al. Somatic alterations in circulating cell‐free DNA of oesophageal carcinoma patients during primary staging are indicative for post‐surgical tumour recurrence. Sci Rep 2018; 8: 14941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gray ES, Rizos H, Reid AL, et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget 2015; 6: 42008–42018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ai B, Liu H, Huang Y, et al. Circulating cell‐free DNA as a prognostic and predictive biomarker in non‐small cell lung cancer. Oncotarget 2016; 7: 44583–44595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cargnin S, Canonico PL, Genazzani AA, et al. Quantitative analysis of circulating cell‐free DNA for correlation with lung cancer survival: a systematic review and meta‐analysis. J Thorac Oncol 2017; 12: 43–53. [DOI] [PubMed] [Google Scholar]
- 48. Schwaederle M, Husain H, Fanta PT, et al. Use of liquid biopsies in clinical oncology: pilot experience in 168 patients. Clin Cancer Res 2016; 22: 5497–5505. [DOI] [PubMed] [Google Scholar]
- 49. Schwaederlé MC, Patel SP, Husain H, et al. Utility of genomic assessment of blood‐derived circulating tumor DNA (ctDNA) in patients with advanced lung adenocarcinoma. Clin Cancer Res 2017; 23: 5101–5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Abbosh C, Birkbak NJ, Swanton C. Early stage NSCLC — challenges to implementing ctDNA‐based screening and MRD detection. Nat Rev Clin Oncol 2018; 15: 577–586. [DOI] [PubMed] [Google Scholar]
- 51. Wei L, Wu W, Han L, et al. A quantitative analysis of the potential biomarkers of non‐small cell lung cancer by circulating cell‐free DNA. Oncol Lett 2018; 16: 4353–4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Trigg RM, Martinson LJ, Parpart‐Li S, et al. Factors that influence quality and yield of circulating‐free DNA: a systematic review of the methodology literature. Heliyon 2018; 4: e00699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bronkhorst AJ, Ungerer V, Holdenrieder S. The emerging role of cell‐free DNA as a molecular marker for cancer management. Biomol Detect Quantif 2019; 17: 100087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. El Messaoudi S, Rolet F, Mouliere F, et al. Circulating cell free DNA: preanalytical considerations. Clin Chim Acta 2013; 424: 222–230. [DOI] [PubMed] [Google Scholar]
- 55. Guyard A, Boyez A, Pujals A, et al. DNA degrades during storage in formalin‐fixed and paraffin‐embedded tissue blocks. Virchows Arch 2017; 471: 491–500. [DOI] [PubMed] [Google Scholar]
- 56. Genovese G, Kähler AK, Handsaker RE, et al. Clonal hematopoiesis and blood‐cancer risk inferred from blood DNA sequence. N Engl J Med 2014; 371: 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Guo Q, Wang J, Xiao J, et al. Heterogeneous mutation pattern in tumor tissue and circulating tumor DNA warrants parallel NGS panel testing. Mol Cancer 2018; 17: 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dong L, Wang S, Fu B, et al. Evaluation of droplet digital PCR and next generation sequencing for characterizing DNA reference material for KRAS mutation detection. Sci Rep 2018; 8: 9650. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Linear regression analyses of serial dilution series of mutated KRAS plasmid templates
Table S1. Patient characteristics and results of ctDNA analyses