

Abstract
Acute myeloid leukemia (AML) is a heterogeneous disease with variable presentation, molecular phenotype, and cytogenetic abnormalities and has seen very little improvement in patient survival over the last few decades. This heterogeneity supports poor prognosis partially through the variability in response to the standard chemotherapy. Further understanding of molecular heterogeneity has promoted the development of novel treatments, some of which target mitochondrial metabolism and function. This review discusses the relative dependency that AML cells have on mitochondrial function, and the ability to pivot this reliance to target important subsets of AML cells, including leukemia stem cells (LSCs). LSCs are tumor-initiating cells that are resistant to standard chemotherapy and promote the persistence and relapse of AML. Historically, LSCs have been targeted based on immunophenotype, but recent developments in the understanding of LSC metabolism has demonstrated unique abilities to target LSCs while sparing normal hematopoietic stem cells (HSCs) through inhibition of mitochondrial function. Here we highlight the use of small molecules that have been demonstrated to effectively target mitochondrial function. IACS-010759 and ME-344 target the electron transport chain (ETC) to inhibit oxidative phosphorylation (OXPHOS). The imipridone family (ONC201, ONC206, ONC212) of inhibitors target mitochondria through activation of ClpP mitochondrial protease and reduce function of essential pathways. These molecules offer a new mechanism for developing clinical therapies in AML and support novel strategies to target LSCs in parallel with conventional therapies.
Keywords: Acute myeloid leukemia, Mitochondria, Oxidative phosphorylation, IACS-010759, ME-344, ONC201
1. Introduction
Acute myeloid leukemia (AML) is a rapidly progressing hematopoietic malignancy that is characterized by the accumulation of clonal myeloid progenitor cells which are unable to differentiate into mature blood cells [1]. There has been little improvement in prognosis and long-term outcomes for AML patients in the last few decades. The 5-year overall survival rates for adult patients remain low (~27%) and therapy involving cytotoxic drugs, cytarabine and anthracycline, has remained the standard of care for medically fit patients with AML for more than 45 years [2]. The treatment of relapsed/refractory (R/R) AML has become increasingly important as only 30–40% of patients will have long-term disease-free survival following conventional therapy and 3 year survival is less than 10% for R/R AML [3]. Another consideration of the complications of intensive induction therapies is the limitation of age and comorbidities, as the majority of AML patients are elderly and cannot tolerate complications associated with intensive chemotherapy.
There has been progress in the development of more targeted therapies that focus on genetic and phenotypic characteristics to better address the heterogeneity of AML. This includes the FDA approval of FLT3 inhibitors (midostaurin and gilteritinib), isocitrate dehydrogenase (IDH) mutant inhibitors (enasidenib and ivosidenib), BCL-2 inhibitor (venetoclax), Hedgehog pathway inhibitor (glasdegib), antibody-drug conjugate (gemtuzumab ozogamicin), and liposomal cytarabine and daunorubicin at a ratio of 5:1 (CPX-3510). More recent studies have been focusing on targeting biological function of bulk AML cells and leukemia stem cells (LSCs) for potential therapeutic development. This has included better understanding of cellular metabolism and mitochondrial function in AML and efforts to target pathways that are common throughout AML, despite its heterogeneity. Importantly, there has been increasing focus on the understanding of how LSCs support disease persistence and progression. This review covers the important role that mitochondria have in maintenance of AML and as potential drug targets.
2. Mitochondria in AML
Alterations of mitochondrial metabolism in cancer have been well established over the last sixty years. This includes utilizing aerobic glycolysis, upregulating glutamine metabolism, induction of fatty acid oxidation and amino acid metabolism, and regulating oxidative phosphorylation (OXPHOS) [4]. The mitochondria are home to many of the core metabolic pathways that play a role in metabolizing carbohydrates, lipids, and amino acids, which are essential for cell and cancer proliferation. In normal postmitotic cells mitochondria generate over 90% of cellular energy in the form of ATP and they also produce important intermediates such as NADH, NADPH, and FADH2. They are also the main site of reactive oxygen species (ROS) production and the intrinsic apoptotic pathway, both of which can be altered to support cancer cell progression and survival [5]. One of the main functions of mitochondria is utilizing pyruvate from glycolysis to support the production of intermediates (NADH, NADPH, FADH2) that fuel the electron transport chain (ETC) and OXPHOS to produce ATP [5]. Though many cancer cells utilize aerobic glycolysis to meet the anabolic demands of cell growth and proliferation, also known as the ‘Warburg effect’, some subsets of AML cells have been found to be uniquely reliant on mitochondrial function to support cell survival. This phenomenon has been increasingly studied as a potential therapeutic target [5].
AML cells have been found to have a higher mitochondrial mass in comparison with normal hematopoietic cell counterparts and this is also true when comparing LSCs to normal hematopoietic stem cells (HSCs). Though there is an increase in mitochondrial mass, there is not a representative increase in respiratory chain activity and AML cells have a lower spare reserve capacity (maximal respiratory capacity when the mitochondrial membrane potential is uncoupled) [6]. This apparent mitochondrial dysfunction leads to increased susceptibility to mitochondrial oxidative stress induction in AML cells. Induction of oxidative stress increases ROS levels in both AML cell lines and primary patient samples and reduces tumor volume in in vivo studies [6]. Additionally, induction of oxidative stress in AML patient derived xenograft models reduced leukemia burden and dampens secondary engraftment, showing efficacy against LSCs and leukemia progenitor cells [6]. Some AML cells also display an upregulation of mitochondrial DNA (mtDNA) biosynthesis pathways compared to normal hematopoietic cells and inhibition of these pathways has been correlated with inhibition of mitochondrial function, and specifically OXPHOS [7], because all of the 13 mitochondrial DNA encoded proteins are part of the OXPHOS machinery. Inhibiting mitochondrial translation can impair normal function of OXPHOS through downregulation of ETC complexes and can selectively target both blasts and LSCs in AML [8]. Many AML cells, including LSCs, overexpress the antiapoptotic protein, BCL-2, which can function indirectly to regulate mitochondrial OXPHOS [9]. These studies have demonstrated the unique reliance that AML has on mitochondrial function in comparison to normal hematopoietic cells.
Mutations in IDH1/2 genes are found in nearly 20% of AML patients and are enriched in patients with normal cytogenetics, though they can be found along with FLT3-ITD and NPM1 mutations [10,11]. IDH1 and IDH2 enzymes catalyze the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG) with NADP+ as a cofactor. IDH1/2 mutations are heterozygous point mutations that result in an inability to convert isocitrate to α-KG. This is also a gain-of-function mutation that catalyzes the NADPH dependent reduction of α-KG to 2-hydroxyglutarate (2-HG), an oncometabolite [12]. The presence of α-KG is essential for adequate function of many metabolic and epigenetic processes and the combination of α-KG loss and inhibition of dioxygenases by 2-HG supports leukemogenesis [13,14]. IDH enzymes are important in generating NADPH from NADP+, and loss of NADPH with mutations in IDH is related to decreased cellular ability to regulate the redox state of the cell [4]. IDH enzymes are extremely important for normal cellular metabolism, and mutations in these enzymes lead to changes in the metabolic profile of these cells. Earlier studies have demonstrated that colorectal cancer cells with mutations in the IDH1 gene are unable to utilize glutamine metabolism under hypoxic conditions, which results in an increased reliance on OXPHOS [15]. The authors also demonstrated that mutations in IDH1/2 lead to increased sensitivity to compounds that induce mitochondrial stress and inhibit OXPHOS [15]. Supporting their reliance on OXPHOS, IDH mutant cells have been found to depend on BCL-2 for survival [16]. Cell death can be induced in these cells by targeting BCL-2 via shRNA knockdown or BCL-2 specific inhibition with the BH3 mimetic venetoclax. This study also found an overlap in targeting IDH1 mutated cells and LSCs as inhibition of BCL-2 in IDH1 mutated patient samples was able to eradicate LSCs and prevent secondary engraftment in murine models [16]. Studies have demonstrated that the production of 2-HG in IDH1/2 mutated cells results in the inhibition of ETC complex IV (cytochrome c oxidase, COX) by binding to the enzyme and inhibiting the reduction of oxygen. This inhibition of COX was found to be essential for sensitivity to BCL-2 inhibition and IDH1/2 mutations promote leukemia cell survival through this dysregulation of mitochondrial function [16].
3. Leukemia stem cells (LSCs)
LSCs play a central role in AML and are linked to relapse and disease persistence [17]. Indeed, LSCs are defined to have stem cell characteristics of self-renewal and proliferative capacity and differentiate into AML bulk cells that have no self-renewal capacity [18,19]. Patients with a high proportion of LSCs at the time of diagnosis are associated with high residual disease after therapy and poor survival, demonstrating that targeting these cells is important for improved remission and prognosis [20]. Originally, LSCs were thought to have a specific phenotype of CD34+/CD38−, but recent studies have found that this population of cells is more heterogeneous than previously understood [21,22]. Other markers that have been tied to LSCs include CD123, CD32, CD33, CD45RA, CD47, CD96, CD99, ILIRAP, CD44, CD25, GPR56, CLL-1, and TIM-3, demonstrating the heterogeneity that exists [19,23–32]. While some of these markers can be found on normal hematopoietic stem cells, some are unique to LSCs (CD90, CD117, CD123) [18]. In addition to existing heterogeneity between patients and within AML, LSCs have been found to be more frequent and have more diverse phenotypes following relapse in AML patients [21]. This persistence that leads to relapse and divergence of disease makes targeting them during initial treatment even more important. Though there are some immunotherapies currently undergoing clinical trials to target the immunophenotype of LSCs [33], efficacy in eliminating this population has yet to be seen.
Though finding a reliable LSC phenotype has been difficult, targeting LSCs based on their unique metabolic profiles has raised some promising options for therapies in recent years [34]. LSCs are quiescent and resistant to cytotoxic therapies, making them difficult to target with conventional AML therapies [25,35,36]. Earlier studies demonstrated that LSCs can be identified and isolated based on low levels of ROS (ROS-low) in comparison to more mature leukemia blasts which demonstrate higher levels of ROS (ROS-high) [37]. The authors further observed that this population of ROS-low LSCs demonstrated a higher dependence on OXPHOS over glycolysis to meet cellular energy demand and were impaired in their reserve glycolytic capacity, distinguishing them from normal hematopoietic stem cells that rely more heavily on glycolysis (Fig. 1) [37,38]. This apparent reliance on OXPHOS in ROS-low LSCs was related to BCL-2 overexpression, as BCL-2 inhibitors caused decreases in oxygen consumption and a drop in ATP production, further demonstrating the inability of these cells to effectively utilize glycolysis [37]. In parallel, LSCs have been found to be uniquely reliant on amino acid metabolism to support OXPHOS and cell survival (Fig. 1) [39]. Interestingly, LSCs do not exhibit the same reliance on glucose supplied by lipid metabolism to support OXPHOS and thus amino acid metabolism has become an interesting possibility for targeting LSCs. As mentioned previously, BCL-2 inhibitors (venetoclax) inhibit OXPHOS in LSCs and lead to cell death [37]. Further studies have found that this inhibition is likely through venetoclax modifying amino acid metabolism availability in LSCs, leading to the reduction in OXPHOS and cell death [39]. Most recently, Roca-Portoles et al. demonstrated that venetoclax inhibition of OXPHOS and the TCA cycle in solid tumor cancer cells is independent of its BCL-2 inhibition and proposed that its therapeutic effect is instead due to the induction of an integrated stress response (ISR) [40]. This distinction supports the concept that venetoclax might be a good therapy option for targeting LSCs as it has two independent mechanisms of targeting them – through BCL-2 inhibition and separately through suppression of metabolism and OXPHOS.
Fig. 1.
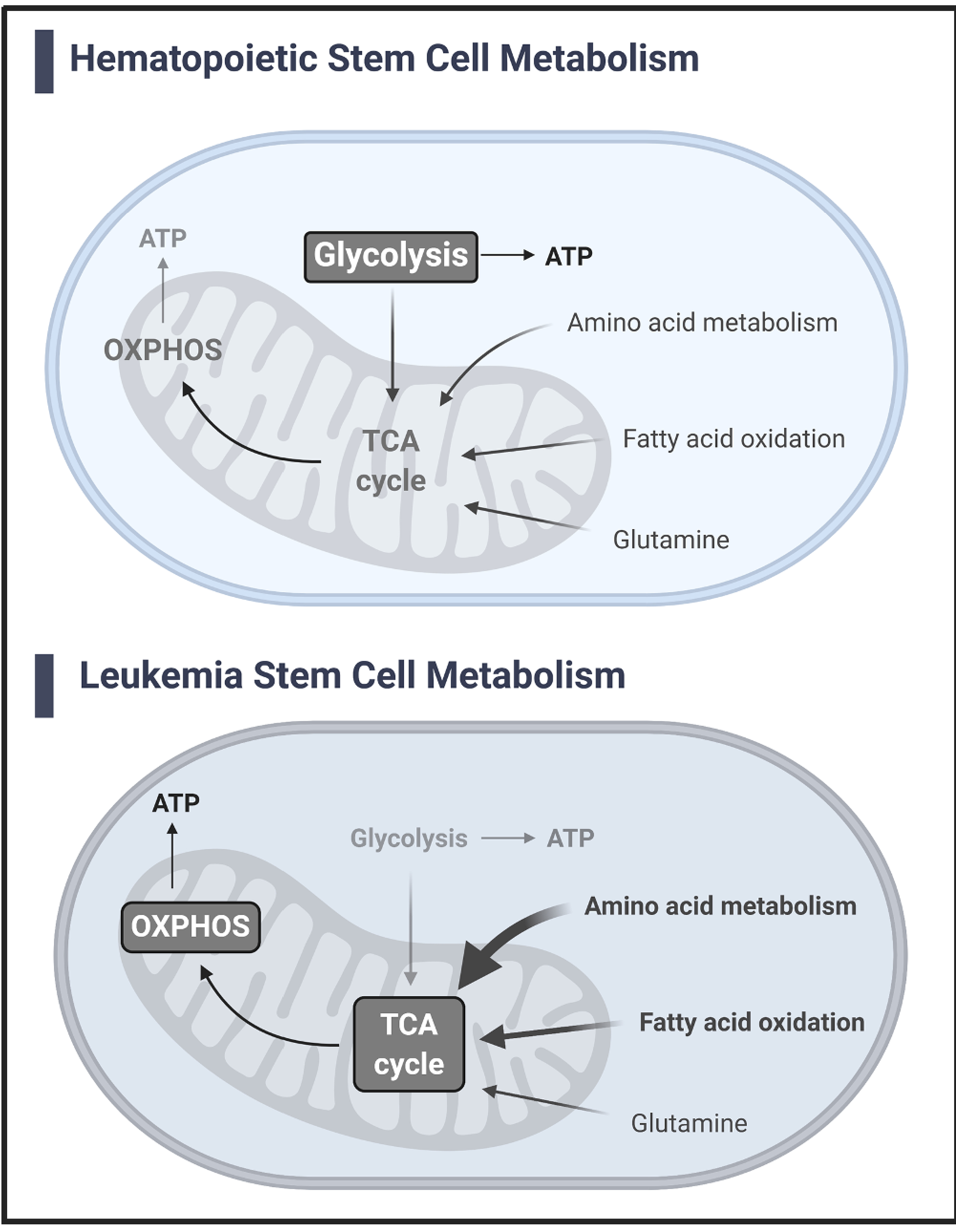
Hematopoietic stem cell and leukemia stem cell metabolism [38,39]. Normal hematopoietic stem cells (HSCs) are more dependent on glycolysis over OXPHOS to support cellular metabolism and survival. Leukemia stem cells (LSCs) are uniquely dependent on OXPHOS to support cellular metabolism and survival and are glycolytically deficient. LSCs rely on amino acid metabolism over fatty acid oxidation or glutamine to fuel the TCA cycle in most cases. In relapse LSCs however, fatty acid oxidation is upregulated under amino acid depletion and targeting multiple pathways is necessary.
Clinical investigations of venetoclax therapy in combination with hypomethylating agents (HMAs), decitabine or azacitidine, was carried out in conjunction with the isolation of LSC enriched populations from patient samples before and after starting therapy [41,42]. The oxygen consumption rate (OCR) in these cells was quantified as an indicator of OXPHOS. They found that LSCs from patients treated with venetoclax had decreased OCR compared to those from patients treated with azacitidine or decitabine alone and these findings were replicated in vitro [41,42]. Amino acid metabolism was significantly altered in LSCs isolated from patients following treatment with venetoclax and azacitidine and these changes reflect a likely inhibition of complex I of the ETC. Combination therapy was found to inhibit complex II activity through reduction of glutathione, which is crucial for post translational modification of the succinate dehydrogenase A (sdhA) component of complex II [41]. In addition to the finding that LSCs are reliant on amino acid metabolism to support OXPHOS, Jones et al. found that ROS-low LSCs isolated from relapsed patients can compensate for amino acid loss by increasing fatty acid metabolism into the TCA cycle and rescue reductions in OXPHOS [39]. This is in line with other studies that depict adipocyte support of LSCs in the BM microenvironment by supplying lipids for fatty acid oxidation (FAO) [43]. Jones et al. further demonstrated that inhibiting fatty acid uptake in combination with venetoclax and azacytidine significantly reduced LSC viability, suggesting that targeting both fatty acid and amino acid metabolism offers a unique therapeutic strategy to eradicate relapse LSCs (Fig. 1) [44].
4. Promising drugs for targeting LSCs
As discussed above, some AML cells, including IDH mutated cells and LSCs have a reliance on OXPHOS that offers a unique target for new therapies. Studies in both LSCs and IDH mutated cells show that BCL-2 inhibitors are effective in decreasing OXPHOS and ATP production and inducing cell death, demonstrating a possible mechanism for targeting mitochondrial respiration [16,37]. Other promising inhibitors of OXPHOS include IACS-010759, ME-344, and the imipridone family (ONC201/ONC212), most of which are currently in clinical trials.
IACS-010759 is a small molecule inhibitor of complex I of the ETC that specifically binds to the ND1 subunit to inhibit electron transfer through the ETC (Fig. 2) [45]. It has been demonstrated to inhibit OXPHOS in cells and can effectively target cells that are glycolysis-deficient [46,47]. As previous studies demonstrated that AML cells with glycolytic deficiencies are more reliant on OXPHOS, Molina et al. demonstrated that AML cell lines were sensitive to the effects of IACS-010759 both in in vitro and in vivo studies in a manner that was dependent on cellular availability of glycolysis induction [46]. In addition to extending survival in patient derived xenograft (PDX) murine models, evaluation of blasts isolated from in vivo studies demonstrated that IACS-010759 effectively inhibits OXPHOS in vivo [46] As AML cells are particularly sensitive to drugs that target mitochondria, IACS-010759 used in combination with vinorelbine (a microtubule inhibitor) resulted in synergistic induction of cell death in AML cell lines and primary patient samples [48]. Importantly, normal hematopoietic cells were largely spared from cell death induction, likely due to their dependence on glycolysis to support cell survival. In cells that upregulate glycolysis in response to OXPHOS inhibition by IACS-010759, cell death could be induced by using 2-deoxy-D-glucose to prevent glycolysis [47]. Additionally, studies in mantle cell lymphoma found that cells resistant to ibrutinib were more reliant on OXPHOS and IACS-010759 was successful in sensitizing resistant cells to cell death [49]. Some studies have suggested that a complex I deficiency may offer resistance to IACS-010759 in AML and inhibit induction of the intrinsic apoptosis pathway [50]. In a recent study, we have demonstrated that the combination of IACS-010759 with the selective BCL-2 inhibitor venetoclax, is synergistic and effective in inducing cell death in OXPHOS-reliant AML cells [51]. This combination significantly extended survival in cell line derived xenograft studies and targeted leukemia progenitor cells in primary AML patient samples [51]. These initial studies demonstrated the ability to target AML cells via their reliance on OXPHOS and the potential to synergistically pair it with drugs that also target mitochondria or glycolysis. IACS-010759 is currently in phase I clinical trials in R/R AML and other advanced solid tumors (NCT02882321, NCT03291938).
Fig. 2.
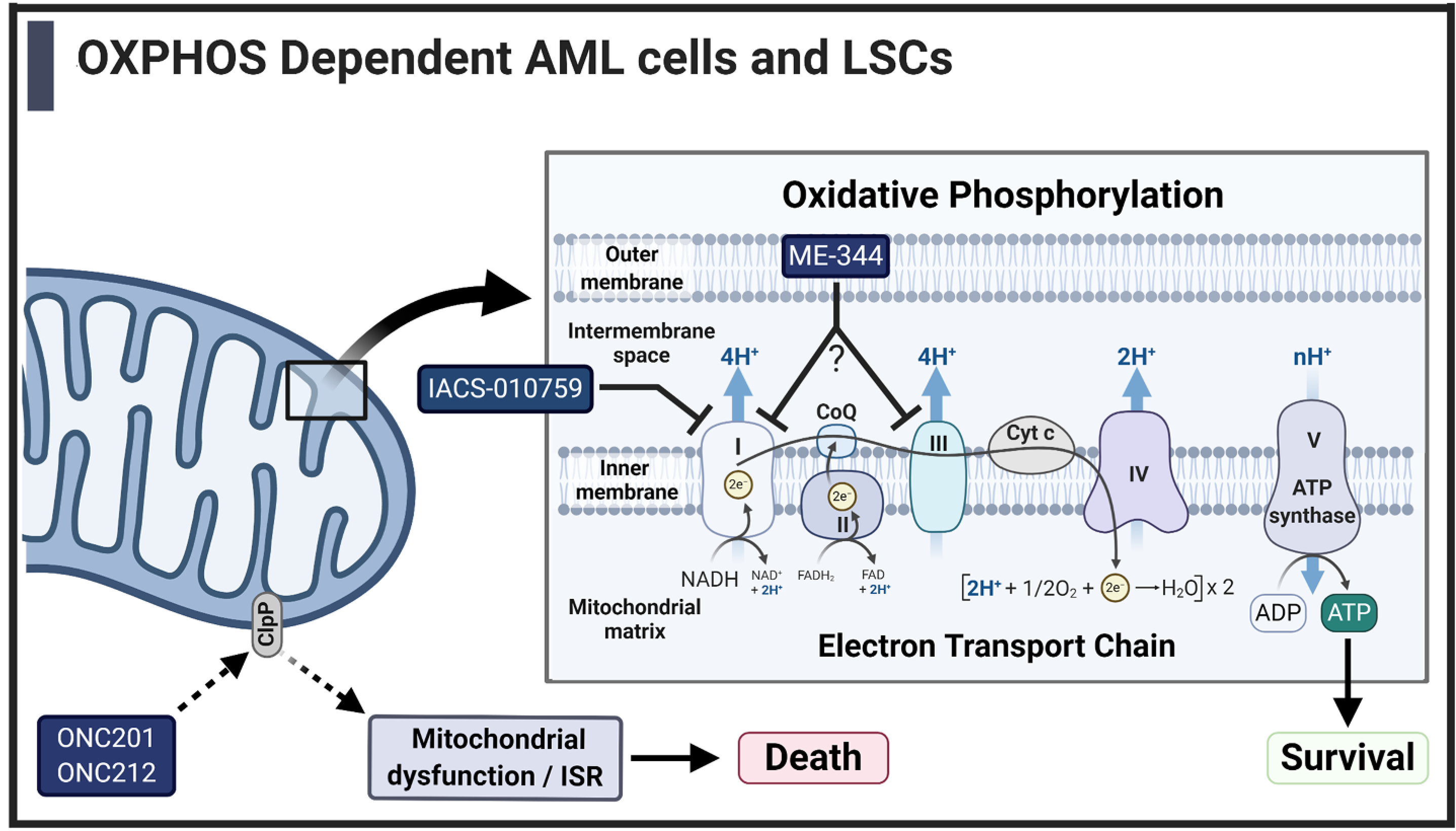
Promising Drugs for targeting OXPHOS dependent AML cells and LSCs [46,48,55–57,67,71,73,75,76]. Some AML cells and LSCs rely on OXPHOS to supply ATP for basic cellular functions and survival. This is a unique target for novel therapies such as ONC201, ONC212, IACS-010759 and ME-344. IACS-010759 and ME-344 suppress OXPHOS by directly targeting complexes of the ETC. While IACS-010759 has been shown to inhibit the activity of complex I, ME-344 can repress the contribution of both complex I and complex III to OXPHOS. On the other hand, ONC201 and ONC212 target the general functionality of mitochondria in AML cells through activation of the mitochondrial protease ClpP, ultimately leading to mitochondrial dysfunction and cell death.
Another molecule that has recently been discovered to target mitochondria is ME-344, a second generation isoflavone. It is a demethylated metabolite of NV-128, which is known to disrupt mitochondria and induce both caspase-independent and -dependent cell death [52–54]. Early studies on ME-344 have demonstrated that it inhibits the ETC through inhibiting complexes I and III (Fig. 2), modifies redox signaling, and inhibits heme oxygenase 1 (HO-1) in solid tumor cells [55–57]. These preclinical models have demonstrated that ME-344 is able to suppress OXPHOS in numerous cancer cells and, similar to IACS-010759, some cells respond with increasing metabolism through glycolytic pathways, highlighting that cells which are glycolytic deficient might be more sensitive to inhibition by ME-344. More recently, in lung cancer cells, ME-344 has been shown to bind to VDAC1/2, which are essential for the translocation of Bax to the mitochondria and transport of ATP to the cytosol. Silencing of VDAC1/2 rescued ME-344-induced apoptosis and reduced the translocation of Bax to the mitochondria [58]. However, this study did not determine if silencing of VDAC had any effect on OXPHOS or ATP transport. So far, only one study has investigated the use of ME-344 in hematologic malignancies. Jeyaraju et al. found that ME-344 reduced cell growth and viability of numerous AML cell lines and was cytotoxic to primary AML patient samples, but normal hematopoietic cells remained viable [59]. ME-344 was also successful in targeting AML cells in in vivo murine models [59]. Cell death in AML cells was not dependent on the generation of ROS, as treatment with N-acetylcysteine, an antioxidant, did not rescue cells. Instead, the authors found that ME-344 interacts with tubulin to prevent polymerization and induce cytoskeletal changes and cell cycle arrest, leading to cell death [59]. This novel understanding of the mechanism of ME-344 in AML demonstrates its potential drug promiscuity, a known characteristic of flavone molecules, and it is important to note that evaluation of mitochondrial respiration has not been evaluated yet in AML cells. Its success in preclinical studies has pushed ME-344 into several clinical trials in solid tumors, both as a single agent therapy and in combination with a topoisomerase inhibitor [60–62]. Data from these clinical trials indicate that ME-344 has some promise as a therapy for solid tumors that should be further evaluated. Additionally, patients with HER-2 negative breast cancer treated with ME-344 had increased levels of succinate dehydrogenase, a component of complex II of the ETC, which indirectly suggests inhibition of complex I in vivo [62]. There are no current clinical trials in hematologic malignancies.
The imipridone class of compounds represent a unique group of mitotoxic drugs discovered by Oncoceutics, Inc. ONC201 is the founding member of the impipridone family and was shown to induce TNF-related apoptosis-inducing ligand (TRAIL) gene transcription [63,64]. ONC201 is a selective antagonist of the G protein-coupled receptor (GPCR), dopamine D2-like receptor (DRD2), and antagonism of DRD2 results in the induction of an ISR in cancer cells [65]. Second generation imipridones, ONC206 and ONC212, have highly increased anti-tumor activity and activate similar pathways compared to ONC201 [66]. ONC201 was found to be effective in inducing cell death in hematologic malignancies independent of TRAIL induction, and relied on induction of ISR (Fig. 2) [67]. Other studies in solid tumors also demonstrated a role of ISR induction in response to ONC201 [68,69]. Further investigating into the mechanism of ONC201, Greer et al. found that ONC201 reduces cellular ATP in breast cancer cells and indirectly inhibits mitochondrial respiration [70]. In addition to inhibiting mitochondrial function, ONC201 decreased mitochondrial DNA and inhibited translation of important functional enzymes. This study demonstrated that ONC201 targets mitochondria of cancer cells and can be used in cells, such as AML, that demonstrate a reliance on mitochondrial function for survival (Fig. 2) [70]. ONC212 similarly binds to a GPCR, GPR132, which is a novel target in hematologic malignancies. ONC212 activation of GPR132 induces mitochondrial cell death via ISR (Fig. 2) and is much more potent than ONC201 in AML cell lines and in vivo models [71]. These authors further demonstrated therapeutic potential of the combination of ONC212 and the FDA approved BCL-2 inhibitor, venetoclax, as BCL-2 has been implicated as a protective factor for cells under ISR [71].
Important to the understanding of the ONC201/ONC212 mechanism of action, Cole and colleagues found that ClpP, a mitochondrial protease that is overexpressed in up to 45% of primary AML samples, is important for leukemia cell survival, and was found on both AML bulk and LSC populations [72]. Importantly, ClpP expression was not upregulated in normal hematopoietic cells and genetic knockdown of Clpp in murine models demonstrated normal hematopoiesis. Additionally, they found that ClpP largely interacted with enzymes important in mitochondrial respiration and mitochondrial metabolism, and knockdown resulted in impaired OXPHOS and increases in ROS [72]. Through use of small molecule inhibitor of ClpP, this study demonstrated that inhibiting ClpP was effective in both in vitro and in mouse xenograft studies, showing promising results as a potential AML therapy. Recent studies have found that ONC201 targets mitochondria, specifically detailing that it activates ClpP (Fig. 2) [72–75]. In a screen to find small molecules that activate ClpP, Ishizawa et al. found that ONC201 and ONC212 were strong activators of ClpP and further demonstrated that ClpP expression was essential for ONC201/ONC212 induction of AML cell death (Fig. 2). Additionally, ONC201 and ONC212 reduced OXPHOS in AML cells and AML sensitivity to these imipridones was correlated to their reliance on OXPHOS and low spare reserve respiratory capacity [73]. Effectively, these studies have found that activation of ClpP results in the destruction of mitochondrial proteins, including complex I and II, resulting in the loss of OXPHOS and induction of ISR and cell death (Fig. 2) [76]. These preclinical studies support the concept that the compounds of the imipridone family mediate a mitochondrial stress response in cancer cells including AML cells which are uniquely sensitive due to their reliance on mitochondrial respiration and function.
Another study has demonstrated that ONC201 inhibits OXPHOS in glioblastoma cells and is accompanied by an increase in glycolysis. This survival mechanism can be inhibited with the combined use of 2-deoxy-D-glucose leading to subsequent drop in ATP levels and cell death [77]. This drug combination was proposed to be synergistic by targeting multiple metabolic pathways of cancer cells, but also demonstrated that ONC201 might be especially effective in cells that are glycolytically deficient, such as LSCs. Indeed, previous studies by Ishizawa et al. found that ONC201 was able to target leukemia progenitor cells and LSCs, preventing secondary engraftment of primary patient AML samples [67]. ONC201 is currently in numerous clinical trials for advanced solid tumors and in hematologic malignancies such as multiple myeloma and AML (NCT02392572, NCT03932643), and thus far has been well tolerated with favorable bioavailability as an oral therapy. ONC201 has been reported to have promising preliminary clinical results in solid tumors, though there have been no updates on success in hematologic malignancies yet [78–81].
5. Concluding remarks
Here we have discussed the increasing understanding of how mitochondrial dependence and reliance on OXPHOS in LSCs and other subsets of AML cells presents an opportunity for novel therapeutic development. The crucial role that LSCs play in progression and relapse of AML has made investigations into the LSC unique metabolic profile essential for progressing treatment options and approaches. The discovery of drugs that target mitochondrial function, especially through inhibition of OXPHOS, holds promising potential as novel AML therapies for the reduction of LSCs and improving long-term outcomes for AML patients.
Acknowledgements
Figures and graphical abstract were created with biorender.com.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Longo DL, Dohner HDJ. Weisdorf CD. Bloomfield, Acute myeloid leukemia, N. Engl. J. Med. 373 (12) (2015) 1136–1152. [DOI] [PubMed] [Google Scholar]
- [2].Zeidan AM, Podoltsev NA, Wang X, Zhang C, Bewersdorf JP, Shallis RM, Huntington SF, Neparidze N, Giri S, Gore SD, Davidoff AJ, Ma X, Wang R, Patterns of care and clinical outcomes with cytarabine-anthracycline induction chemotherapy for AML patients in the United States, Blood Adv. 4 (2020) 1615–1623, doi: 10.1182/bloodadvances.2020001728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bose P, Vachhani P, Cortes JE, Treatment of relapsed/refractory acute myeloid leukemia, Curr. Treat. Options in Oncol. 18 (3) (2017), 10.1007/s11864-017-0456-2. [DOI] [PubMed] [Google Scholar]
- [4].Wallace DC, Mitochondria and cancer, Nat. Rev. Cancer 12 (10) (2012) 685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Basak NP, Banerjee S, Mitochondrial dependency in progression of acute myeloid leukemia, Mitochondrion 21 (2015) 41–48. [DOI] [PubMed] [Google Scholar]
- [6].Sriskanthadevan S, Jeyaraju DV, Chung TE, Prabha S, Xu W, Skrtic M, Jhas B, Hurren R, Gronda M, Wang X, Jitkova Y, Sukhai MA, Lin F-H, Maclean N, Laister R, Goard CA, Mullen PJ, Xie S, Penn LZ, Rogers IM, Dick JE, Minden MD, Schimmer AD, AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress, Blood 125 (2015) 2120–2130, doi: 10.1182/blood-2014-08-594408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Liyanage SU, Hurren R, Voisin V, Bridon G, Wang X, Xu C, et al. Leveraging increased cytoplasmic nucleoside kinase activity to target mtDNA and oxidative phosphorylation in AML, Blood 129 (2017) 2657–2666, doi: 10.1182/blood-2016-10-741207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Škrtić M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, Hurren R, Jitkova Y, Gronda M, Maclean N, Lai C, Eberhard Y, Bartoszko J, Spagnuolo P, Rutledge A, Datti A, Ketela T, Moffat J, Robinson B, Cameron J, Wrana J, Eaves C, Minden M, Wang JY, Dick J, Humphries K, Nislow C, Giaever G, Schimmer A, Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia, Cancer Cell 20 (5) (2011) 674–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chen ZX, Pervaiz S, Bcl-2 induces pro-oxidant state by engaging mitochondrial respiration in tumor cells, Cell Death Differ. 14 (9) (2007) 1617–1627. [DOI] [PubMed] [Google Scholar]
- [10].Marcucci G, Maharry K, Wu Y-Z, Radmacher MD, Mrózek K, Margeson D, Holland KB, Whitman SP, Becker H, Schwind S, Metzeler KH, Powell BL, Carter TH, Kolitz JE, Wetzler M, Carroll AJ, Baer MR, Caligiuri MA, Larson RA, Bloomfield CD, IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study, JCO 28 (14) (2010) 2348–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].DiNardo CD, Ravandi F, Agresta S, Konopleva M, Takahashi K, Kadia T, Routbort M, Patel KP, Mark Brandt, Pierce S, Garcia-Manero G, Cortes J, Kantarjian H, Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML: IDH mutations in AML, Am. J. Hematol. 90 (8) (2015) 732–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. , The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate, Cancer Cell 17 (2010) 225–234, 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Montalban-Bravo G, DiNardo CD, The role of IDH mutations in acute myeloid leukemia, Fut. Oncol. 14 (10) (2018) 979–993. [DOI] [PubMed] [Google Scholar]
- [14].Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, Yang H, Gross S, Artin E, Saada V, Mylonas E, Quivoron C, Popovici-Muller J, Saunders JO, Salituro FG, Yan S, Murray S, Wei W, Gao Y, Dang L, Dorsch M, Agresta S, Schenkein DP, Biller SA, Su SM, de Botton S, Yen KE, Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation, Science 340 (6132) (2013) 622–626. [DOI] [PubMed] [Google Scholar]
- [15].Grassian AR, Parker SJ, Davidson SM, Divakaruni AS, Green CR, Zhang X, Slocum KL, Pu M, Lin F, Vickers C, Joud-Caldwell C, Chung F, Yin H, Handly ED, Straub C, Growney JD, Vander Heiden MG, Murphy AN, Pagliarini R, Metallo CM, IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism, Cancer Res. 74 (12) (2014) 3317–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chan SM, Thomas D, Corces-Zimmerman MR, Xavy S, Rastogi S, Hong W-J, Zhao F, Medeiros BC, Tyvoll DA, Majeti R, Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia, Nat. Med. 21 (2) (2015) 178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Shlush LI, Mitchell A, Heisler L, Abelson S, Ng SWK, Trotman-Grant A, Medeiros JJF, Rao-Bhatia A, Jaciw-Zurakowsky I, Marke R, McLeod JL, Doedens M, Bader G, Voisin V, ChangJiang Xu, McPherson JD, Hudson TJ, Wang JCY, Minden MD, Dick JE, Tracing the origins of relapse in acute myeloid leukaemia to stem cells, Nature 547 (7661) (2017) 104–108. [DOI] [PubMed] [Google Scholar]
- [18].Jordan CT, Unique molecular and cellular features of acute myelogenous leukemia stem cells, Leukemia 16 (4) (2002) 559–562. [DOI] [PubMed] [Google Scholar]
- [19].Thomas D, Majeti R, Biology and relevance of human acute myeloid leukemia stem cells, Blood 129 (2017) 1577–1585, doi: 10.1182/blood-2016-10-696054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].van Rhenen A, Feller N, Kelder A, Westra AH, Rombouts E, Zweegman S, et al. , High stem cell frequency in acute myeloid leukemia at diagnosis predicts high minimal residual disease and poor survival, Clin. Cancer Res. 11 (2005) 6520–6527, 10.1158/1078-0432.Ccr-05-0468. [DOI] [PubMed] [Google Scholar]
- [21].Ho TC, LaMere M, Stevens BM, Ashton JM, Myers JR, O’Dwyer KM, et al. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression, Blood 128 (2016) 1671–1678, doi: 10.1182/blood-2016-02-695312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE, A cell initiating human acute myeloid leukaemia after transplantation into SCID mice, Nature 367 (6464) (1994) 645–648. [DOI] [PubMed] [Google Scholar]
- [23].Jordan CT, Upchurch D, Szilvassy SJ, Guzman ML, Howard DS, Pettigrew AL, Meyerrose T, Rossi R, Grimes B, Rizzieri DA, Luger SM, Phillips GL, The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells, Leukemia 14 (10) (2000) 1777–1784. [DOI] [PubMed] [Google Scholar]
- [24].Jin L, Lee EM, Ramshaw HS, Busfield SJ, Peoppl AG, Wilkinson L, et al. , Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells, Cell Stem Cell 5 (2009) 31–42, 10.1016/j.stem.2009.04.018. [DOI] [PubMed] [Google Scholar]
- [25].Saito Y, Kitamura H, Hijikata A, Tomizawa-Murasawa M, Tanaka S, Takagi S, Uchida N, Suzuki N, Sone A, Najima Y, Ozawa H, Wake A, Taniguchi S, Shultz LD, Ohara O, Ishikawa F, Identification of therapeutic targets for quiescent, chemotherapy-resistant human leukemia stem cells, Sci. Transl. Med. 2 (17) (2010), 17ra9–17ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Taussig DC, Pearce DJ, Simpson C, Rohatiner AZ, Lister TA, Kelly G, et al. , Hematopoietic stem cells express multiple myeloid markers: implications for the origin and targeted therapy of acute myeloid leukemia, Blood 106 (2005) 4086–4092, 10.1182/blood-2005-03-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Goardon N, Marchi E, Atzberger A, Quek L, Schuh A, Soneji S, Woll P, Mead A, Alford KA, Rout R, Chaudhury S, Gilkes A, Knapper S, Beldjord K, Begum S, Rose S, Geddes N, Griffiths M, Standen G, Sternberg A, Cavenagh J, Hunter H, Bowen D, Killick S, Robinson L, Price A, Macintyre E, Virgo P, Burnett A, Craddock C, Enver T, Jacobsen SEW, Porcher C, Vyas P, Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia, Cancer Cell 19 (1) (2011) 138–152. [DOI] [PubMed] [Google Scholar]
- [28].Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD Jr., van Rooijen N, Weissman IL, CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells, Cell 138 (2) (2009) 286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hosen N, Park CY, Tatsumi N, Oji Y, Sugiyama H, Gramatzki M, Krensky AM, Weissman IL, CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia, Proc. Natl. Acad. Sci. 104 (26) (2007) 11008–11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Chung SS, Eng WS, Hu W, Khalaj M, Garrett-Bakelman FE, Tavakkoli M, Levine RL, Carroll M, Klimek VM, Melnick AM, Park CY, CD99 is a therapeutic target on disease stem cells in myeloid malignancies, Sci. Transl. Med. 9 (374) (2017) eaaj2025, 10.1126/scitranslmed.aaj2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Barreyro L, Will B, Bartholdy B, Zhou L, Todorova TI, Stanley RF, et al. , Overexpression of IL-1 receptor accessory protein in stem and progenitor cells and outcome correlation in AML and MDS. Blood 120 (2012) 1290–1298, doi: 10.1182/blood-2012-01-404699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kikushige Y, Shima T, Takayanagi S, Urata S, Miyamoto T, Iwasaki H, et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell 7 (2010) 708–717, doi: 10.1016/j.stem.2010.11.014. [DOI] [PubMed] [Google Scholar]
- [33].Valent P, Sadovnik I, Eisenwort G, Bauer K, Herrmann H, Gleixner KV, et al. , Immunotherapy-based targeting and elimination of leukemic stem cells in AML and CML, Int. J. Mol. Sci. 20 (2019) doi: 10.3390/ijms20174233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Jordan CT, Can we selectively target AML stem cells? Best Pract. Res. Clin. Haematol. 32 (4) (2019) 101100, 10.1016/j.beha.2019.101100. [DOI] [PubMed] [Google Scholar]
- [35].Zhang Y i, Chen HX, Zhou SY, Wang SX, Zheng K, Xu DD, Liu YT, Wang XY, Wang X, Yan HZ, Zhang L i, Liu QY, Chen WQ, Wang YF, Sp1 and c-Myc modulate drug resistance of leukemia stem cells by regulating survivin expression through the ERK-MSK MAPK signaling pathway, Mol. Cancer 14 (1) (2015) 56, 10.1186/s12943-015-0326-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bencomo-Alvarez AE, Rubio AJ, Gonzalez MA, Eiring AM, Energy metabolism and drug response in myeloid leukaemic stem cells, Br. J. Haematol. 186 (4) (2019) 524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lagadinou E, Sach A, Callahan K, Rossi R, Neering S, Minhajuddin M, Ashton J, Pei S, Grose V, O’Dwyer K, Liesveld J, Brookes P, Becker M, Jordan C, BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells, Cell Stem Cell 12 (3) (2013) 329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Simsek T, Kocabas F, Zheng J, DeBerardinis RJ, Mahmoud AI, Olson EN, Schneider JW, Zhang CC, Sadek HA, The Distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche, Cell Stem Cell 7 (3) (2010) 380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, Pei S, Khan N, Adane B, Ye H, Krug A, Reinhold D, Smith C, DeGregori J, Pollyea DA, Jordan CT, Inhibition of amino acid metabolism selectively targets human leukemia stem cells, Cancer Cell 35 (2) (2019) 333–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Roca-Portoles A, Rodriguez-Blanco G, Sumpton D, Cloix C, Mullin M, Mackay GM, O’Neill K, Lemgruber L, Luo X u, Tait SWG, Venetoclax causes metabolic reprogramming independent of BCL-2 inhibition, Cell Death Dis. 11 (8) (2020), 10.1038/s41419-020-02867-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Pollyea DA, Stevens BM, Jones CL, Winters A, Pei S, Minhajuddin M, D’Alessandro A, Culp-Hill R, Riemondy KA, Gillen AE, Hesselberth JR, Abbott D, Schatz D, Gutman JA, Purev E, Smith C, Jordan CT, Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia, Nat. Med. 24 (12) (2018) 1859–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, et al. , Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia, Blood 133 (2019) 7–17, doi: 10.1182/blood-2018-08-868752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Tabe Y, Konopleva M, Andreeff M, Fatty acid metabolism, bone marrow adipocytes, and AML, Front. Oncol. 10 (2020) 155, 10.3389/fonc.2020.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, Pei S, Khan N, Adane B, Ye H, Krug A, Reinhold D, Smith C, DeGregori J, Pollyea DA, Jordan CT, Inhibition of amino acid metabolism selectively targets human leukemia stem cells, Cancer Cell 34 (5) (2018) 724–740.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Tsuji A, Akao T, Masuya T, Murai M, Miyoshi H, IACS-010759, a potent inhibitor of glycolysis-deficient hypoxic tumor cells, inhibits mitochondrial respiratory complex I through a unique mechanism, J. Biol. Chem. 295 (21) (2020) 7481–7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, McAfoos T, Morlacchi P, Ackroyd J, Agip A-N, Al-Atrash G, Asara J, Bardenhagen J, Carrillo CC, Carroll C, Chang E, Ciurea S, Cross JB, Czako B, Deem A, Daver N, de Groot JF, Dong J-W, Feng N, Gao G, Gay J, Do MG, Greer J, Giuliani V, Han J, Han L, Henry VK, Hirst J, Huang S, Jiang Y, Kang Z, Khor T, Konoplev S, Lin Y-H, Liu G, Lodi A, Lofton T, Ma H, Mahendra M, Matre P, Mullinax R, Peoples M, Petrocchi A, Rodriguez-Canale J, Serreli R, Shi T, Smith M, Tabe Y, Theroff J, Tiziani S, Xu Q, Zhang Q i, Muller F, DePinho RA, Toniatti C, Draetta GF, Heffernan TP, Konopleva M, Jones P, Di Francesco ME, Marszalek JR, An inhibitor of oxidative phosphorylation exploits cancer vulnerability, Nat. Med. 24 (7) (2018) 1036–1046. [DOI] [PubMed] [Google Scholar]
- [47].Vangapandu HV, Alston B, Morse J, Ayres ML, Wierda WG, Keating MJ, Marszalek JR, Gandhi V, Biological and metabolic effects of IACS-010759, an OxPhos inhibitor, on chronic lymphocytic leukemia cells, Oncotarget 9 (38) (2018) 24980–24991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Panina SB, Pei J, Baran N, Konopleva M, Kirienko NV, Utilizing synergistic potential of mitochondria-targeting drugs for leukemia therapy, Front. Oncol. 10 (2020) 435, doi: 10.3389/fonc.2020.00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhang L, Yao Y, Zhang S, Liu Y, Guo H, Ahmed M, Bell T, Zhang H, Han G, Lorence E, Badillo M, Zhou S, Sun Y, Di Francesco ME, Feng N, Haun R, Lan R, Mackintosh SG, Mao X, Song X, Zhang J, Pham LV, Lorenzi PL, Marszalek J, Heffernan T, Draetta G, Jones P, Futreal A, Nomie K, Wang L, Wang M, Metabolic reprogramming toward oxidative phosphorylation identifies a therapeutic target for mantle cell lymphoma, Sci. Transl. Med. 11 (491) (2019) eaau1167, 10.1126/scitranslmed.aau1167. [DOI] [PubMed] [Google Scholar]
- [50].Yadav N, Kumar S, Marlowe T, Chaudhary AK, Kumar R, Wang J, O’Malley J, Boland PM, Jayanthi S, Kumar TKS, Yadava N, Chandra D, Oxidative phosphorylation-dependent regulation of cancer cell apoptosis in response to anticancer agents, Cell Death Dis. 6 (11) (2015) e1969 e1969 http://www.nature.com/articles/cddis2015305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Liu F, Kalpage HA, Wang D, Edwards H, Hüttemann M, Ma J, et al. , Cotargeting of mitochondrial complex I and Bcl-2 shows antileukemic activity against acute myeloid leukemia cells reliant on oxidative phosphorylation, Cancers 12 (2020) doi: 10.3390/cancers12092400(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Alvero AB, Montagna MK, Holmberg JC, Craveiro V, Brown D, Mor G, Targeting the mitochondria activates two independent cell death pathways in ovarian cancer stem cells, Mol. Cancer Ther. 10 (8) (2011) 1385–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zhang L, Zhang J, Ye Z, Townsend DM, Tew KD, Pharmacology of ME-344, a novel cytotoxic isoflavone, Adv. Cancer Res. 142 (2019) 187–207, 10.1016/bs.acr.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Alvero AB, Montagna MK, Chen R, Kim KH, Kyungjin K, Visintin I, Fu H-H, Brown D, Mor G, NV-128, a novel isoflavone derivative, induces caspase-independent cell death through the Akt/mammalian target of rapamycin pathway, Cancer 115 (14) (2009) 3204–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lim SC, Carey KT, McKenzie M, Anti-cancer analogues ME-143 and ME-344 exert toxicity by directly inhibiting mitochondrial NADH: ubiquinone oxidoreductase (Complex I), Am. J. Cancer Res. 5 (2015) 689–701. [PMC free article] [PubMed] [Google Scholar]
- [56].Manevich Y, Reyes L, Britten CD, Townsend DM, Tew KD, Redox signaling and bioenergetics influence lung cancer cell line sensitivity to the isoflavone ME-344, J. Pharmacol. Exp. Ther. 358 (2) (2016) 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zhang L, Zhang J, Ye Z, Manevich Y, Ball LE, Bethard JR, Jiang Y-L, Broome A-M, Dalton AC, Wang GY, Townsend DM, Tew KD, Isoflavone ME-344 disrupts redox homeostasis and mitochondrial function by targeting heme oxygenase 1, Cancer Res. 79 (16) (2019) 4072–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhang L, Townsend DM, Morris M, Maldonado EN, Jiang Y-L, Broome A-M, Bethard JR, Ball LE, Tew KD, Voltage-dependent anion channels influence cytotoxicity of ME-344, a therapeutic isoflavone, J. Pharmacol. Exp. Ther. 374 (2) (2020) 308–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Jeyaraju DV, Hurren R, Wang X, MacLean N, Gronda M, Shamas-Din A, Minden MD, Giaever G, Schimmer AD, A novel isoflavone, ME-344, targets the cytoskeleton in acute myeloid leukemia, Oncotarget 7 (31) (2016) 49777–49785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Bendell JC, Patel MR, Infante JR, Kurkjian CD, Jones SF, Pant S, Burris III HA, Moreno O, Esquibel V, Levin W, Moore KN, Phase 1, open-label, dose escalation, safety, and pharmacokinetics study of ME-344 as a single agent in patients with refractory solid tumors: phase 1 ME-344 for refractory solid tumors, Cancer 121 (7) (2015) 1056–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Diamond JR, Goff B, Forster MD, Bendell JC, Britten CD, Gordon MS, Gabra H, Waterhouse DM, Poole M, Ross Camidge D, Hamilton E, Moore KM, Phase Ib study of the mitochondrial inhibitor ME-344 plus topotecan in patients with previously treated, locally advanced or metastatic small cell lung, ovarian and cervical cancers, Invest. New Drugs 35 (5) (2017) 627–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Quintela-Fandino M, Morales S, Cortés-Salgado A, Manso L, Apala JV, Muñoz M, Gasol Cudos A, Salla Fortuny J, Gion M, Lopez-Alonso A, Cortés J, Guerra J, Malón D, Caleiras E, Mulero F, Mouron S, Randomized phase 0/I trial of the mitochondrial inhibitor ME-344 or placebo added to bevacizumab in early HER2-negative breast cancer, Clin. Cancer Res. 26 (1) (2020) 35–45. [DOI] [PubMed] [Google Scholar]
- [63].Allen JE, Kline CLB, Prabhu VV, Wagner J, Ishizawa J.o, Madhukar N, Lev A, Baumeister M, Zhou L, Lulla A, Stogniew M, Schalop L, Benes C, Kaufman HL, Pottorf RS, Nallaganchu BR, Olson GL, Al-Mulla F, Duvic M, Wu GS, Dicker DT, Talekar MK, Lim B, Elemento O, Oster W, Bertino J, Flaherty K, Wang ML, Borthakur G, Andreeff M, Stein M, El-Deiry WS, Discovery and clinical introduction of first-in-class imipridone ONC201, Oncotarget 7 (45) (2016) 74380–74392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Allen JE, Krigsfeld G, Mayes PA, Patel L, Dicker DT, Patel AS, Dolloff NG, Messaris E, Scata KA, Wang W, Zhou J-Y, Wu GS, El-Deiry WS, Dual Inactivation of Akt and ERK by TIC10 signals foxo3a Nuclear translocation, TRAIL gene induction, and potent antitumor effects, Sci. Transl. Med. 5 (171) (2013), 171ra17–171ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kline CLB, Ralff MD, Lulla AR, Wagner JM, Abbosh PH, Dicker DT, Allen JE, El-Deiry WS, Role of dopamine receptors in the anticancer activity of ONC201, Neoplasia 20 (1) (2018) 80–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wagner J, Kline CL, Ralff MD, Lev A, Lulla A, Zhou L, Olson GL, Nallaganchu BR, Benes CH, Allen JE, Prabhu VV, Stogniew M, Oster W, El-Deiry WS, Preclinical evaluation of the imipridone family, analogs of clinical stage anti-cancer small molecule ONC201, reveals potent anti-cancer effects of ONC212, Cell Cycle 16 (19) (2017) 1790–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ishizawa J.o, Kojima K, Chachad D, Ruvolo P, Ruvolo V, Jacamo RO, Borthakur G, Mu H, Zeng Z, Tabe Y, Allen JE, Wang Z, Ma W, Lee HC, Orlowski R, Sarbassov DD, Lorenzi PL, Huang X, Neelapu SS, McDonnell T, Miranda RN, Wang M, Kantarjian H, Konopleva M, Davis RE, Andreeff M, ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies, Sci. Signal. 9 (415) (2016) ra17–ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kline CL, Van den Heuvel AP, Allen JE, Prabhu VV, Dicker DT, El-Deiry WS, ONC201 kills solid tumor cells by triggering an integrated stress response dependent on ATF4 activation by specific eIF2alpha kinases, Sci. Signal. 9 (2016) ra18, 10.1126/scisignal.aac4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yuan X, Kho D, Xu J, Gajan A, Wu K, Wu GS, ONC201 activates ER stress to inhibit the growth of triple-negative breast cancer cells, Oncotarget 8 (13) (2017) 21626–21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Greer YE, Porat-Shliom N, Nagashima K, Stuelten C, Crooks D, Koparde VN, Gilbert SF, Islam C, Ubaldini A, Ji Y, Gattinoni L, Soheilian F, Wang X, Hafner M, Shetty J, Tran B, Jailwala P, Cam M, Lang M, Voeller D, Reinhold WC, Rajapakse V, Pommier Y, Weigert R, Linehan WM, Lipkowitz S, ONC201 kills breast cancer cells in vitro by targeting mitochondria, Oncotarget 9 (26) (2018) 18454–18479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Nii T, Prabhu VV, Ruvolo V, Madhukar N, Zhao R, Mu H, Heese L, Nishida Y, Kojima K, Garnett MJ, McDermott U, Benes CH, Charter N, Deacon S, Elemento O, Allen JE, Oster W, Stogniew M, Ishizawa J.o, Andreeff M, Imipridone ONC212 activates orphan G protein-coupled receptor GPR132 and integrated stress response in acute myeloid leukemia, Leukemia 33 (12) (2019) 2805–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Cole A, Wang Z, Coyaud E, Voisin V, Gronda M, Jitkova Y, Mattson R, Hurren R, Babovic S, Maclean N, Restall I, Wang X, Jeyaraju D, Sukhai M, Prabha S, Bashir S, Ramakrishnan A, Leung E, Qia Y, Zhang N, Combes K, Ketela T, Lin F, Houry W, Aman A, Al-awar R, Zheng W, Wienholds E, Xu C, Dick J, Wang JY, Moffat J, Minden M, Eaves C, Bader G, Hao Z, Kornblau S, Raught B, Schimmer A, Inhibition of the mitochondrial protease ClpP as a therapeutic strategy for human acute myeloid leukemia, Cancer Cell 27 (6) (2015) 864–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Ishizawa J.o, Zarabi SF, Davis RE, Halgas O, Nii T, Jitkova Y, Zhao R, St-Germain J, Heese LE, Egan G, Ruvolo VR, Barghout SH, Nishida Y, Hurren R, Ma W, Gronda M, Link T, Wong K, Mabanglo M, Kojima K, Borthakur G, MacLean N, Ma MCJ, Leber AB, Minden MD, Houry W, Kantarjian H, Stogniew M, Raught B, Pai EF, Schimmer AD, Andreeff M, Mitochondrial ClpP-mediated proteolysis induces selective cancer cell lethality, Cancer Cell 35 (5) (2019) 721–737.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Jacques S, van der Sloot AM, Huard CC, Coulombe-Huntington J, Tsao S, Tollis S, Bertomeu T, Culp EJ, Pallant D, Cook MA, Bonneil E, Thibault P, Wright GD, Tyers M, Imipridone anticancer compounds ectopically activate the ClpP protease and represent a new scaffold for antibiotic development, Genetics 214 (4) (2020) 1103–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Graves PR, Aponte-Collazo LJ, Fennell EMJ, Graves AC, Hale AE, Dicheva N, Herring LE, Gilbert TSK, East MP, McDonald IM, Lockett MR, Ashamalla H, Moorman NJ, Karanewsky DS, Iwanowicz EJ, Holmuhamedov E, Graves LM, Mitochondrial protease ClpP is a target for the anticancer compounds ONC201 and related analogues, ACS Chem. Biol. 14 (5) (2019) 1020–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Wang S, Dougan DA, The direct molecular target for imipridone ONC201 is finally established, Cancer Cell 35 (5) (2019) 707–708. [DOI] [PubMed] [Google Scholar]
- [77].Pruss M, Dwucet A, Tanriover M, Hlavac M, Kast RE, Debatin K-M, Wirtz CR, Halatsch M-E, Siegelin MD, Westhoff M-A, Karpel-Massler G, Dual metabolic reprogramming by ONC201/TIC10 and 2-Deoxyglucose induces energy depletion and synergistic anti-cancer activity in glioblastoma, Br. J. Cancer 122 (8) (2020) 1146–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Arrillaga-Romany I, Chi AS, Allen JE, Oster W, Wen PY, Batchelor TT, A phase 2 study of the first imipridone ONC201, a selective DRD2 antagonist for oncology, administered every three weeks in recurrent glioblastoma, Oncotarget 8 (45) (2017) 79298–79304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Edwards H, Ge Y, ONC201 shows promise in AML treatment, Cell Cycle 17 (3) (2018), 277 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Stein MN, Bertino JR, Kaufman HL, Mayer T, Moss R, Silk A, Chan N, Malhotra J, Rodriguez L, Aisner J, Aiken RD, Haffty BG, DiPaola RS, Saunders T, Zloza A, Damare S, Beckett Y, Yu B, Najmi S, Gabel C, Dickerson S, Zheng L, El-Deiry WS, Allen JE, Stogniew M, Oster W, Mehnert JM, First-in-human clinical trial of oral ONC201 in PATIENTS WITH REFRACTORY SOLID TUMORS, Clin. Cancer Res. 23 (15) (2017) 4163–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Arrillaga-Romany I, Odia Y, Prabhu VV, Tarapore RS, Merdinger K, Stogniew M, et al. , Biological activity of weekly ONC201 in adult recurrent glioblastoma patients. Neuro-oncology 22 (2020) 94–102, doi: 10.1093/neuonc/noz16. [DOI] [PMC free article] [PubMed] [Google Scholar]