

Abstract
Oseltamivir represents one of the most successful neuraminidase (NA) inhibitors in the current anti-influenza therapy. The 150-cavity of NA was identified as an additional binding pocket, and novel NA inhibitors have been designed to occupy the 150-cavity based on the structure information of oseltamivir carboxylate (OC) in complex with NA. In this study, a series of C-5-NH2-acyl derivatives of OC containing the pyrazole moiety were synthesized. Several derivatives exhibited substantial inhibitory activity against NA. Moreover, in silico ADME evaluation indicated that the derivatives were drug-like with higher oral absorption rates and greater cell permeability than OC. Additionally, molecular docking studies revealed that the derivatives interacted with both the NA enzyme active site and 150-cavity as expected. The results provided useful information for further structural optimization of OC.
Keywords: influenza virus, neuraminidase inhibitor, 150-cavity, oseltamivir derivatives, pyrazole
1. Introduction
Seasonal flu epidemics and pandemics caused substantial mortality and economic loss. Reports showed that up to 0.65 million respiratory deaths per annum are attributed to seasonal flu globally [1]. According to the antigenic differences in their nucleoprotein (NP) and matrix protein (M1), influenza viruses are classified as types A, B, and C [2]. Different subtypes of influenza A virus were named according to two important surface antigenic glycoproteins, hemagglutinin (HA) and neuraminidase (NA) [3]. So far, 18 HA subtypes (H1–18) and 11 NA subtypes (N1–N11) were identified [4], in which several different subtypes caused severe pandemics in the last century, including the 1918 Spanish flu (H1N1), 1957 Asian flu (H2N2), 1968 Hong Kong flu (H3N2) [5], and 2009 swine flu (H1N1 pdm09) [6].
Presently, there are three classes of anti-influenza drugs, consisting of the M2 channel inhibitors, the neuraminidase inhibitors (NAIs) [7], and the recently approved cap-dependent endonuclease inhibitor baloxavir marboxil [8] (Figure 1), although the viral resistance to all the drugs has been reported [9]. NAIs have been approved as antivirals in clinical use for about 20 years, and they have a mode of action of disrupting the release of nascent viruses from host cells. Amongst all the NAIs, oseltamivir phosphate (OP, ethyl ester prodrug) has become a first-line therapy since its approval in 1999 [10].
Figure 1.

Representative anti-influenza drugs. M2 channel inhibitors: (i,ii); NAIs: (iii–vi) and cap-dependent endonuclease inhibitor (vii).
Phylogenetically, NAs can be divided into two groups: group 1 contains N1, N4, N5, and N8, and group 2 contains N2, N3, N6, N7, and N9 [11]. Based on the structural observations, Russel et al. disclosed that group-1 neuraminidases contain a so-called 150-cavity adjacent to their active sites, which suggests a new binding pocket for drug design [12]. Besides, it was also reported that N2 NA could be induced to an open position by the binding of oseltamivir carboxylate to NA [13]. Crystal structures of OC in complex with NAs indicated that the C-5-NH2 group of OC close to the 150-cavity of NA might be a possible modification site. On this basis, several C-5-NH2 modified OC derivatives have been reported as 150-cavity binders, including second amine derivatives (viii–xiii) [14,15,16,17]. These derivatives inhibited NA at the nM level, and most of them possessed the selectivity against group 1 NAs. Other derivatives, such as triazolylated analogue (xiv) [18], pyridyl analogue (xv) [19], and C-5-NH2-acyl analogue (xvi) (Figure 2) [20], showed moderate activity against NA.
Figure 2.

Representative C-5-NH2 modified OC derivatives designed as 150-cavity binders: secondary amine derivatives (viii–xiii), triazolylated analogue (xiv), pyridyl analogue (xv) and acyl analogue (xvi).
Previously, we designed and synthesized a class of heterocycle-bearing secondary amine derivatives of OC for probing the 150-cavity [17]. Results indicated that the introduction of heterocyclic side chains could maintain the antiviral activity of OC with appropriate ADME properties. In this study, in an attempt to develop novel NA 150-cavity binders, we intended to modify the C-5-NH2 group of OC by incorporating pyrazole moiety. A series of pyrazole-containing C-5-NH2-acyl OC analogues were prepared, and their inhibitory activities to NA of a clinically isolated A/H3N2 influenza virus strain from Hong Kong were evaluated.
2. Results and Discussion
2.1. Design of Novel Oseltamivir Analogues
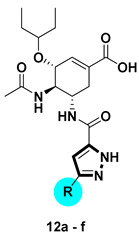
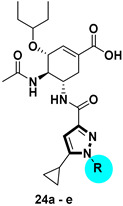
The reported co-crystal structure of OC in complex with NA (PDB: 2HU0) showed that C-5-NH2 of OC is adjacent to the 150-cavity, which suggests that the C-5-NH2 may become a modification site towards the discovery of novel NA inhibitors. Based on this information, attempts on the development of 150-cavity binders as OC inhibitors with increased activity or overcome drug resistance were carried out. Herein, we designed, synthesized, and tested a series of C-5-NH2-acyl analogues of OC containing the pyrazole moiety through an amide bond with different substitutions on pyrazole (Figure 3).
Figure 3.

Design of the pyrazole-based C-5-NH2-acyl oseltamivir analogues.
2.2. Chemistry
The pyrazole-based C-5-NH2-acyl OC derivatives were synthesized following the synthetic routes described in Scheme 1, Scheme 2, Scheme 3 and Scheme 4.
Scheme 1.

Synthetic route of compounds 6a–k. Reagents and conditions: (a) for 3a–g: 1a–g, K2CO3, acetone, reflux, overnight, 60–90%; for 3h–k: 1h–k, CuI, trans-N,N′-dimethyl-1,2-cyclohexanediamine, K2CO3, Tol., 110 °C, 24 h, 30–55%. (b) NaOH, MeOH/H2O, 50 °C; (c) oseltamivir phosphate, HATU, DIPEA, DCM, rt, 60–90%; (d) NaOH, MeOH/H2O, 50 °C, 40–70%.
Scheme 2.

Synthetic route of compounds 12a–f. Reagents and conditions: (a) Dimethyl oxalate, t-BuOK, Tol, rt; (b) NH2NH2⋅H2O, AcOH, 90 °C, 4 h, 70–80%; (c) NaOH, MeOH/H2O, 50 °C, overnight, 80–90%; (d) oseltamivir phosphate, HATU, DIPEA, DCM, rt, 40–80%; (e) NaOH, MeOH/H2O, 50 °C, 50–70%.
Scheme 3.

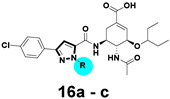
Synthetic route of compounds 16a-c. Reagents and conditions: (a) R3Br, K2CO3, acetone, reflux, 60–70%; (b) NaOH, MeOH/H2O, 50 °C, 80–90%; (c) oseltamivir phosphate, HATU, DIPEA, DCM, rt, 40–90%; (d) NaOH, MeOH/H2O, 50 °C, 40–60%.
Scheme 4.

Synthetic route of compounds 24a–e and 26a–d. Reagents and conditions: (a) Dimethyl oxalate, t-BuOK, Tol, rt; (b) R4NHNH2⋅HCl, EtOH, reflux, 5 h, 19a–e: 40–80%, 20a–d: 15–30%; (c) NaOH, MeOH/H2O, 50 °C, 80–90%; (d) HATU, DIPEA, DCM, rt, 50–80%; (e) NaOH, MeOH/H2O, 50 °C, 40–70%.
As shown in Scheme 1, we first prepared a series of 1H-pyrazole-4-carboxamide derivatives 6a–k. Ethyl 1H-pyrazole-4-carboxylate 2 reacted with various aryl bromides in the presence of K2CO3 [21] to furnish 3a–g. The arylated derivatives 3h–k were prepared by the coupling of aryl iodides with compound 2 catalyzed by the CuI-diamine system with K2CO3 as the base [21]. Hydrolysis of the esters 3a–k occurred under basic conditions to afford 1H-pyrazole-4-carboxylic acid derivatives 4a–k. The condensation of oseltamivir phosphate with substituted pyrazolecarboxylic acids 4a–k using the HATU/DIPEA methodology provided 5a–k. Finally, hydrolysis of ethyl ester gave compounds 6a–k.
The 1H-pyrazole-5-carboxamide derivatives 12a–f were synthesized as illustrated in Scheme 2. Treatment of methyl ketones 7a–f with dimethyl oxalate in the presence of potassium tert-butoxide afforded intermediates 8a–f, which were subjected to cyclization with hydrazine at 50 °C in AcOH to give methyl 1H-pyrazole-5-carboxylate derivatives 9a–f [22], followed by hydrolysis to furnish the 1H-pyrazole-5-carboxylic acid derivatives 10a–f. The condensation of 10a–f with oseltamivir phosphate under the HATU/DIPEA condition provided 11a–f. After hydrolysis of ethyl ester, compounds 12a–f were obtained.
Alkylation at the pyrazole N-1 position of methyl 3-(4-chlorophenyl)-1H-pyrazole-5-carboxylate (9b) was accomplished as outlined in Scheme 3. Compound 9b was respectively alkylated with several alkyl bromides with diverse ring sizes in the presence of K2CO3, followed by hydrolysis of the esters to afford carboxylic acids 14a–c. Subsequently, condensation of acids 14a–c with oseltamivir phosphate gave compounds 15a–c, which underwent facile hydrolysis to provide compounds 16a–c.
1H-pyrazole-3-carboxamide derivatives 24a–e and 1H-pyrazole-5-carboxamide derivatives 26a–d were prepared as shown in Scheme 4. Claisen condensation of commercially available 1-cyclopropylethan-1-one 17 with dimethyl oxalate afforded diketoester 18, which underwent cyclization with substituted hydrazines to give both the corresponding 1,5-disubstituted pyrazole-3-carboxylic acid esters 19a–e, and the isomeric derivatives, 1,3-disubstituted pyrazole-5-carboxylic acid esters 20a–d. The latter could be isolated and purified by column chromatography [23,24]. The esters were hydrolyzed by aqueous sodium hydroxide solution to afford the corresponding carboxylic acids 21a–e and 22a–d. These carboxylic acids reacted with oseltamivir phosphate to furnish compounds 23a–e and 25a–d, respectively. After hydrolysis, compounds 24a–e and 26a–d were obtained.
2.3. Neuraminidase Enzyme Inhibitory Assay
A clinically isolated A/H3N2 strain from Hong Kong was cultivated with host cell cultures followed by lysis. All of the newly synthesized 29 compounds were evaluated for their inhibitory activity to A/H3N2 NA with OC as the reference drug [17]. The results were shown in Table 1.
Table 1.
Inhibitory rates of designed compounds against A/H3N2 influenza NA a.
| Derivatives | Cpd. | R | 10 μM | Cpd. | R | 10 μM |
|---|---|---|---|---|---|---|
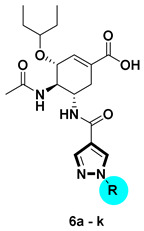
|
OC | \ | 97.46% | 6f |
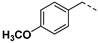
|
26.79% |
| 6a |
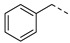
|
32.02% | 6g |
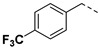
|
21.72% | |
| 6b |
|
18.88% | 6h |
|
38.37% | |
| 6c |
|
23.88% | 6i |
|
52.31% | |
| 6d |
|
12.26% | 6j |
|
34.15% | |
| 6e |
|
23.37% | 6k |
|
25.67% | |
|
12a |
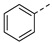
|
42.81% | 12d |
|
46.7% |
| 12b |
|
50.43% | 12e |
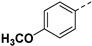
|
42.02% | |
| 12c |
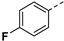
|
56.45% | 12f |
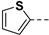
|
40.02% | |
|
16a |
|
31.50% | 16c |
|
48.50% |
| 16b |
|
39.81% | ||||
|
24a |
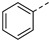
|
30.57% | 24d |
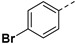
|
42.68% |
| 24b |
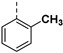
|
60.91% | 24e |
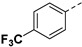
|
44.47% | |
| 24c |
|
34.75% | ||||
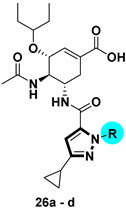
|
26a |
|
30.41% | 26c |
|
31.53% |
| 26b |
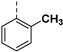
|
72.80% | 26d |
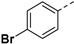
|
37.17% |
a, Ao is the absorbance of the control (no drug control), Abg is the absorbance of the background, Ax is the absorbance of the sample or control compound (OC).
Regarding the first series 6a–k with a 1H-pyrazole-4-carboxamide moiety, most of the compounds displayed mild NA inhibitory activity. Overall, compounds 6a–g containing benzyl or substituted benzyl groups showed less activity than the derivatives 6h–k with substituted phenyls. Compound 6i with a 4-methylphenyl group substituted at pyrazole N-1 position exhibited the highest NA inhibitory activity of 52.31%. Interestingly, compound 6j with a strong electron-donating group (OCH3) and 6k with a weak electron-withdrawing group (Cl) at the 4-position of the phenyl ring showed inferior inhibitory activities to 6i. These results suggest that both steric and electronic properties need to be further investigated to clarify the properties for binding to the 150-cavity of NA.
For 1H-pyrazole-5-carboxamide derivatives 12a–f, all the derivatives showed interesting inhibitory activity at 10 μM. The inhibitory activities of the compounds increased with the electron-withdrawing capability of the halide groups at 4-position of the phenyl ring (12c > 12b > 12a; 12d >12e). Compound 12c with the 4-fluorophenyl group was the most active compound in this series, with the percentages of inhibition of 56.45%.
The alkylation of the N-1 position of the pyrazole moiety of compound 12b provided compounds 16a–c. Results demonstrated that the cyclohexylmethyl group at N-1 of pyzarole (16c) was preferred. 16c showed similar inhibitory activity to 12b, and the decreased ring sizes in 16a and 16b caused inferior activities.
1H-pyrazole-3-carboxamide derivatives 24a–e and their isomers 1H-pyrazole-5-carboxamide derivatives 26a–d displayed diverse NA inhibitory activities from 30% to 72%. When the substituents at para- or meta- positions (e.g., 24c–e, 26c, and 26d), the inhibitory potencies were similar to the non-substituted benzene derivatives 24a or 26a. Compounds 24b and its isomer 26b with 2-methylphenyl substituted on the pyrazole ring exhibited the most potent inhibitory activity of 60.91% and 72.80% at 10 μM, respectively. The outstanding inhibitory activity of both compounds suggests that the ortho effect of the substitutions might be beneficial to the activity.
Briefly, the structure-activity relationship (SAR) discussed above showed that 1H-pyrazole-5-carboxamide or 1H-pyrazole-3-carboxamide were the optimal linker fragments, and they provided different electronic properties from the substitutions to the N atom of the pyrazole. Together with our previous study [17], the SAR obtained here will guide the further design of more potent NA inhibitors.
Overall, most of the pyrazole-based C-5-NH2-acyl analogues of OC inhibited the enzyme activity of A/H3N2 NA. Compounds 6i, 12c, 24b, and 26b represented the most active compounds in each series. Their IC50 values against NA were then determined. As shown in Table 2, compounds 6i, 12c, 24b, and 26b exhibited moderate NA inhibitory activity, among which compound 12c and 26b displayed the best NA inhibitory activity with an IC50 value of 6.98 ± 0.08 μM and 12.27 ± 0.14 μM, respectively. Interestingly, 24b and 26b against NA at 10 μM exhibited to be more potent than 12c, but compound 12c inhibited NA with a lower IC50 value, probably because 12c may elicit stronger inhibitory activity at lower concentrations.
Table 2.
Inhibitory activity of 6i, 12c, 24b, and 26b against clinically isolated A/H3N2 NA (μM) a.
| No. | OC b | 6i | 12c | 24b | 26b |
|---|---|---|---|---|---|
| IC50 (μM) a | 1.04 ± 0.01 | 27.56 ± 0.55 | 6.98 ± 0.08 | 26.99 ± 0.31 | 12.27 ± 0.14 |
a IC50 is the compound concentration producing 50% inhibition of NA, the values are the mean of three independent experiments. b IC50 unit of OC is nM.
2.4. In Silico ADME Prediction
To further explore the druggability of these OC derivatives, the ADME properties were predicted by Qikprop (Maestro 10.2) [17]. The ADME descriptors of compounds 6i, 12c, 24b, and 26b were calculated, including octanol-water partitioning coefficient (QPlogPo/w), aqueous solubility (QPlogS), binding to human serum albumin (QPlogKhsa), brain/blood partition coefficient (QPlogBB), number of likely metabolic reactions (#metab), central nervous system activity (CNS), predicted IC50 value for the blockage of HERG K+ channels (QPloghERG), apparent Caco-2 cell permeability (QPPCaco), apparent MDCK cell permeability (QPPMDCK), and human oral absorption.
As shown in Table 3, the four C5-NH-acyl derivatives were predicted to be significantly more lipophilic with lower aqueous solubility in comparison with OC. Moreover, the percentages of human oral absorption of the new derivatives were 60~70%, higher than 32.7% for OC. Meanwhile, QPPCaco and QPPMDCK of the derivatives were predicted to be 3- or 7-fold higher than that of OC. These data suggest that our derivatives could be well absorbed by the intestine and thereof could be orally administrated directly instead of converting to the ester prodrug as OC. Other parameters such as QPlogBB, CNS, QPloghERG, and #metab remained within the standard ranges which suggests these derivatives should be metabolically stable and nontoxic to both the nervous system and heart.
Table 3.
Comparison of computational pharmacokinetic parameters (ADME) of OC derivatives.
| Principal Descriptors | OC | 6i | 12c | 24b | 26b | Standard Range * |
|---|---|---|---|---|---|---|
| QPlogPo/w | −1.654 | 3.361 | 3.337 | 4.106 | 4.380 | −2.0–6.5 |
| QPlogS | −1.164 | −5.741 | −5.931 | −6.323 | −6.648 | −6.5–0.5 |
| QPlogKhsa | −0.873 | −0.043 | −0.036 | 0.277 | 0.291 | −1.5–1.5 |
| QPlogBB | −1.05 | −2.186 | −2.186 | −2.086 | −1.714 | −3.0–1.2 |
| #metab | 3 | 3 | 2 | 3 | 3 | 1–8 |
| CNS | −2 | −2 | −2 | −2 | −2 | −2 (inactivity), +2 (activity) |
| QPloghERG | −1.322 | −3.348 | −2.975 | −3.006 | −3.085 | <−5 |
| QPPCaco | 7.34 | 20.787 | 17.378 | 22.727 | 54.32 | <25 poor, >500 great |
| QPPMDCK | 6.146 | 17.413 | 19.348 | 20.759 | 42.816 | <25 poor, >500 great |
|
(%) Human Oral
Absorption |
32.756 | 70.213 | 68.675 | 62.305 | 70.687 | <25% is poor, >80% is high |
|
Human Oral
Absorption |
Middle | Middle | Middle | Middle | Middle |
* Statistics of 95% of known drugs according to Qikprop (Maestro 10.2).
2.5. Molecular Docking Studies
As described previously, molecular docking studies were performed to reveal the potential interactions between NA and the newly designed OC derivatives. All the final compounds were docked with the crystal structure of NA (PDB: 2HU0) using Glide of Maestro 10.2 [17]. Results showed that most of the derivatives fitted into the NA active site with their OC motif and occupied the 150-cavity with the substituted pyrazole moieties as designed (Figure S1 and Figure 4A).
Figure 4.

(A) Binding positions of compounds 12c (green) and 26b (cyan) to NA (PDB: 2HU0); Detailed interactions of 12c (B) and 26b (C) with NA: Hydrogen/ionic bonding interactions (green) and hydrophobic interactions (magenta).
Details of the interactions between compound 12c and NA are shown in Figure 4B and the key H-bonds were summarized in Table 4. The OC part of 12c formed hydrogen bonding and hydrophobic interactions with the NA active site, including hydrogen bonds between the carboxylic acid group and residues Asn294 and Ser246, and hydrophobic interactions between the pentan-3-yloxy group and residues Arg152 and Trp178. In addition, the 5-(3-(4-fluorophenyl)-1H-pyrazole-5-carboxamide side chain formed hydrogen bonds with residues Arg371 and Arg430, and hydrophobic interaction with Pro431.
Table 4.
Hydrogen bonding interactions between NA (PDB: 2HU0) and compounds 12c and 26b.
| H-Bonding between NA and 12c | H-Bonding between NA and 26b | ||
|---|---|---|---|
| Hydrogen Bonds | Distance (Å) | Hydrogen Bonds | Distance (Å) |
| SER246: HG/12c: O9 | 2.10 | ARG156: HH11/26b: O19 | 1.82 |
| ASN294: HD21/12c: O8 | 3.15 | ARG156: HH21/26b: O19 | 2.67 |
| ARG371: HH11/12c: O23 | 2.24 | ARG292: HH21/26b: O9 | 1.92 |
| ARG371: HH21/12c: O23 | 1.95 | ARG371: HH11/26b: O9 | 1.57 |
| ARG371: HH21/12c: N27 | 2.67 | ARG371: HH21/26b: O9 | 3.01 |
| ARG430: HH22/12c: F34 | 2.93 | 26b: H59/ASP151: OD1 | 2.71 |
26b interacts with some different residues in the NA active site when compared with compound 12c (Figure 4C, Table 4). The carboxylic group formed ionic bonding interactions with residue Arg292 and Arg371, and hydrogen bonding with Tyr406. Additionally, the C-4 amide group contacted Arg156 via two hydrogen bonds. These changes might enhance the stability of the 12c-NA complex. Finally, hydrophobic interactions were formed between the phenyl ring and residues Pro431 and Arg430. Overall, the additional interactions of OC derivatives with NA may improve the binding affinity.
3. Materials and Methods
3.1. Biology
NA Enzyme Inhibitory Assay
The NA inhibitory assay was performed using the commercially available NA-Fluor™ Influenza Neuraminidase Assay Kit as described previously [17].
3.2. Chemistry
3.2.1. General Methods
Starting materials and reagents, unless otherwise stated, were commercial grade and used without further purification. Thin-layer chromatography (TLC) on glass sheets (Silica gel F254) was visualized under UV light and used to monitor all the reactions. Column chromatography was carried out using silica gel (200–300 mesh). Compound structures were confirmed by 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra which were acquired using a BRUKER AVANCE-III spectrometer. NMR chemical shifts were given in δ (ppm) and coupling constants (J) in Hz. The solvents for NMR were DMSO-d6 (δ 2.50 for 1H) or chloroform (δ 7.26 for 1H). High-resolution MS spectra were measured using a Micromass® Q-ToF2 mass spectrometer.
3.2.2. General Procedures for the Synthesis of Compounds 3a–k
For compounds 3a–g: A mixture of ethyl-4-pyrazole carboxylate (280 mg, 2 mmol), K2CO3 (1.38 g, 10 mmol), and a bromomethyl arene (2.4 mmol) in acetone (10 mL) was heated to reflux for 12 h. After cooling to room temperature, K2CO3 was filtered off and the filtrate was concentrated, purified by column chromatography, eluting with a gradient of hexane/ethyl acetate (15:1 to 6:1) to provide compounds 3a–g.
For compounds 3h–3k: To a Schlenk tube were added ethyl-4-pyrazole carboxylate (280 mg, 2 mmol), an aryl iodide (2.2 mmol), CuI (76 mg, 0.4 mmol), K2CO3 (580 mg, 4.2 mmol) and trans-N,N′-dimethyl-1,2-cyclohexanediamine (113 mg, 0.8 mmol) and toluene (5 mL). The reaction tube was charged with N2 and immersed in a preheated oil bath for stirring at 110 °C for 24 h. The reaction mixture was concentrated, and the resulting residue was purified by column chromatography, eluting with a gradient of hexane/ethyl acetate (20:1 to 10:1) to provide the desired products.
3.2.3. General Procedure for the Synthesis of Compounds 4a–k
Each ethyl ester of 3a–k was dissolved in MeOH (10 mL), 1 M NaOH aqueous solution (2 mL) was added. The mixture was stirred at 50 °C overnight. The solvent was removed under reduced pressure and the residue was diluted with 10 mL water and washed with DCM. The aqueous phase was acidified by 1 M HCl to precipitate the product which was collected by filtration and dried in vacuo. The products were used directly for the next step of the reaction.
3.2.4. General Procedure for the Synthesis of Compounds 5a–k
A mixture of oseltamivir phosphate (82 mg, 0.2 mmol), each pyrazole-4-carboxylic acid derivative of 4a–k (0.24 mmol), HATU (115 mg, 0.3 mmol) and DIPEA (83 μL, 0.5 mmol) in DCM (10 mL) was stirred at room temperature overnight. The reaction was quenched by saturated NaHCO3 aqueous solution and the resulting mixture was extracted with DCM (20 mL × 3). The combined organic phases were washed with brine, dried over Na2SO4, concentrated, and purified by silica gel column chromatography, eluting with a gradient of DCM/MeOH (50:1 to 30:1) to afford the titled compound.
3.2.5. General Procedure for the Synthesis of Compounds 6a–k
Each compound of 5a–k (0.1 mmol) was dissolved in MeOH (10 mL), 1 M NaOH aqueous solution (2 mL) was added. The mixture was heated to 50 °C overnight. The solvent was removed under reduced pressure and the residue was diluted with 10 mL water and washed with DCM (10 mL × 2). The aqueous phase was acidified by 1 M HCl to precipitate the product which was collected by filtration and dried in vacuo.
3.2.6. (3R,4R,5S)-4-acetamido-5-(1-benzyl-1H-pyrazole-4-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (6a)
Yield 59%; white solid, mp 273–275 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.60 (s, 1H), 8.18 (s, 1H), 7.97–7.80 (m, 2H), 7.79 (s, 1H), 7.42–7.28 (m, 3H), 7.25 (d, J = 6.7 Hz, 2H), 6.63 (s, 1H), 5.35 (s, 2H), 4.08 (ddd, J = 19.4, 17.3, 9.0 Hz, 2H), 3.83 (dd, J = 19.9, 8.9 Hz, 1H), 3.45–3.30 (m, 1H), 2.55 (d, J = 5.1 Hz, 1H), 2.38–2.24 (m, 1H), 1.68 (s, 3H), 1.53–1.30 (m, 4H), 0.84 (t, J = 7.3 Hz, 3H), 0.77 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.3, 167.9, 162.0, 139.2, 138.3, 137.4, 131.8, 129.8, 129.0, 128.3, 128.2, 119.1, 81.6, 75.5, 55.5, 54.6, 48.3, 30.8, 26.3, 25.8, 23.2, 9.8, 9.5. HRMS (ESI): calcd for C25H31N4O5, [M-H]− 467.2300, found 467.2309.
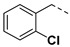
3.2.7. (3R,4R,5S)-4-acetamido-5-(1-(2-chlorobenzyl)-1H-pyrazole-4-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (6b)
Yield 52%; white solid, mp 265–267 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.16 (s, 1H), 7.94–7.83 (m, 2H), 7.82 (s, 1H), 7.50 (dd, J = 7.6, 1.4 Hz, 1H), 7.42–7.31 (m, 2H), 7.12 (dd, J = 7.3, 1.8 Hz, 1H), 6.63 (s, 1H), 5.46 (s, 2H), 4.14 (d, J = 8.1 Hz, 1H), 4.11–4.02 (m, 1H), 3.84 (dd, J = 20.0, 9.0 Hz, 1H), 3.45–3.30 (m, 1H), 2.57–2.51 (m, 1H), 2.32 (dd, J = 17.5, 10.4 Hz, 1H), 1.68 (s, 3H), 1.52–1.30 (m, 4H), 0.84 (t, J = 7.3 Hz, 3H), 0.77 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.3, 167.7, 161.9, 139.4, 138.2, 134.6, 132.9, 132.2, 130.8, 130.4, 130.0, 129.8, 128.0, 119.1, 81.6, 75.6, 54.6, 53.2, 48.4, 30.8, 26.3, 25.8, 23.2, 9.8, 9.5. HRMS (ESI): calcd for C25H30ClN4O5, [M-H]− 501.1910, found 501.1912.
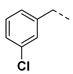
3.2.8. (3R,4R,5S)-4-acetamido-5-(1-(3-chlorobenzyl)-1H-pyrazole-4-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (6c)
Yield 63%; white solid, mp 245–247 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.62 (s, 1H), 8.21 (s, 1H), 7.84 (dd, J = 12.9, 9.2 Hz, 2H), 7.80 (s, 1H), 7.44–7.34 (m, 2H), 7.29 (s, 1H), 7.25–7.17 (m, 1H), 6.63 (s, 1H), 5.38 (s, 2H), 4.17–4.01 (m, 2H), 3.83 (dd, J = 19.9, 9.1 Hz, 1H), 3.45–3.30 (m, 1H), 2.55–2.51 (m, 1H), 2.32 (dd, J = 17.6, 10.4 Hz, 1H), 1.67 (s, 3H), 1.53–1.34 (m, 4H), 0.84 (t, J = 7.3 Hz, 3H), 0.76 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.2, 167.7, 161.9, 139.9, 139.4, 138.2, 133.6, 132.0, 131.0, 129.8, 128.2, 128.0, 126.0, 119.3, 81.6, 75.6, 54.7, 54.6, 48.4, 30.8, 26.3, 25.8, 23.2, 9.8, 9.5. HRMS (ESI): calcd for C25H30ClN4O5, [M-H]− 501.1910, found 501.1912.
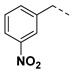
3.2.9. (3R,4R,5S)-4-acetamido-5-(1-(3-nitrobenzyl)-1H-pyrazole-4-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (6d)
Yield 59%; white solid, mp 236–238 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.28 (s, 1H), 8.22–8.15 (m, 1H), 8.12 (s, 1H), 7.94–7.79 (m, 3H), 7.71 (d, J = 7.6 Hz, 1H), 7.67 (t, J = 7.8 Hz, 1H), 6.63 (s, 1H), 5.54 (s, 2H), 4.17–4.01 (m, 2H), 3.84 (dd, J = 19.9, 9.0 Hz, 1H), 3.45–3.30 (m, 1H), 2.60–2.51 (m, 1H), 2.32 (dd, J = 17.5, 10.4 Hz, 1H), 1.68 (s, 3H), 1.51–1.31 (m, 4H), 0.84 (t, J = 7.3 Hz, 3H), 0.76 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.3, 167.8, 161.9, 148.3, 139.7, 139.6, 138.1, 134.9, 132.3, 130.7, 129.9, 123.3, 122.8, 119.4, 81.6, 75.6, 54.6, 54.4, 48.4, 30.8, 26.3, 25.7, 23.1, 9.8, 9.5. HRMS (ESI): calcd for C25H30N5O7, [M-H]− 512.2151, found 512.2156.
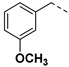
3.2.10. (3R,4R,5S)-4-acetamido-5-(1-(3-methoxybenzyl)-1H-pyrazole-4-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (6e)
Yield 62%; white solid, mp 259–261 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.17 (s, 1H), 7.88 (d, J = 9.1 Hz, 1H), 7.82 (d, J = 8.9 Hz, 1H), 7.79 (s, 1H), 7.27 (t, J = 7.8 Hz, 1H), 6.88 (dd, J = 8.1, 2.1 Hz, 1H), 6.84–6.74 (m, 2H), 6.63 (s, 1H), 5.32 (s, 2H), 4.18–4.00 (m, 2H), 3.83 (dd, J = 20.0, 9.0 Hz, 1H), 3.73 (s, 3H), 3.45–3.30 (m, 1H), 2.59–2.50 (m, 1H), 2.37–2.27 (m, 1H), 1.67 (s, 3H), 1.51–1.28 (m, 4H), 0.84 (t, J = 7.3 Hz, 3H), 0.77 (t, J = 7.4 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.2, 167.7, 162.0, 159.8, 139.2, 138.9, 138.3, 131.8, 130.2, 129.8, 120.3, 119.1, 114.0, 113.6, 81.6, 75.6, 55.5, 55.4, 54.6, 48.3, 30.8, 26.3, 25.7, 23.1, 9.8, 9.5. HRMS (ESI): calcd for C26H33N4O6, [M-H]− 497.2406, found 497.2410.
3.2.11. (3R,4R,5S)-4-acetamido-5-(1-(4-methoxybenzyl)-1H-pyrazole-4-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (6f)
Yield 45%; white solid, mp 269–271 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.11 (s, 1H), 7.87 (d, J = 9.1 Hz, 1H), 7.79 (d, J = 8.9 Hz, 1H), 7.76 (s, 1H), 7.24 (d, J = 8.5 Hz, 2H), 6.92 (d, J = 8.5 Hz, 2H), 6.63 (s, 1H), 5.26 (s, 2H), 4.14 (d, J = 8.0 Hz, 1H), 4.10–4.00 (m, 1H), 3.83 (dd, J = 19.9, 9.0 Hz, 1H), 3.74 (s, 3H), 3.45–3.30 (m, 1H), 2.63–2.45 (m, 1H), 2.34–2.20 (m, 1H), 1.68 (s, 3H), 1.53–1.30 (m, 4H), 0.84 (t, J = 7.3 Hz, 3H), 0.77 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.3, 167.7, 162.0, 159.4, 139.0, 138.2, 131.4, 129.9, 129.8, 129.2, 119.0, 114.4, 81.6, 75.6, 55.6, 55.0, 54.6, 48.3, 30.8, 26.3, 25.7, 23.2, 9.8, 9.5. HRMS (ESI): calcd for C26H33N4O6, [M-H]− 497.2408, found 497.2410.
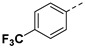
3.2.12. (3R,4R,5S)-4-acetamido-3-(pentan-3-yloxy)-5-(1-(4-(trifluoromethyl) benzyl)-1H-pyrazole-4-carboxamido) cyclohex-1-ene-1-carboxylic acid (6g)
Yield 51%; white solid, mp 259–261 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.24 (s, 1H), 7.86 (dd, J = 15.2, 10.8 Hz, 3H), 7.73 (d, J = 8.1 Hz, 2H), 7.44 (d, J = 8.0 Hz, 2H), 6.64 (s, 1H), 5.48 (s, 2H), 4.22–4.01 (m, 2H), 3.84 (dd, J = 19.8, 9.1 Hz, 1H), 3.40–3.30 (m, 1H), 2.60–2.51 (m, 1H), 2.32 (dd, J = 17.5, 10.4 Hz, 1H), 1.68 (s, 3H), 1.53–1.31 (m, 4H), 0.84 (t, J = 7.3 Hz, 3H), 0.77 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.3, 167.7, 161.9, 142.2, 139.5, 138.3, 132.3, 129.7, 128.85 (d, J = 31.7 Hz), 128.83, 126.0 (d, J = 3.2 Hz), 125.9 (d, J = 3.2 Hz), 124.6 (q, J = 260.3 Hz), 119.3, 81.6, 75.6, 54.8, 54.56, 48.4, 30.8, 26.3, 25.7, 23.2, 9.8, 9.5. HRMS (ESI): calcd for C26H30F3N4O5, [M-H]− 535.2174, found 535.2178.
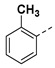
3.2.13. (3R,4R,5S)-4-acetamido-3-(pentan-3-yloxy)-5-(1-(o-tolyl)-1H-pyrazole-4-carboxamido) cyclohex-1-ene-1-carboxylic acid (6h)
Yield 61%; white solid, mp 253–255 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.34 (s, 1H), 8.02 (s, 1H), 7.93 (dd, J = 9.0, 4.2 Hz, 2H), 7.47–7.32 (m, 4H), 6.65 (s, 1H), 4.22–4.05 (m, 2H), 3.87 (dd, J = 20.0, 9.0 Hz, 1H), 3.45–3.30 (s, 1H), 2.58 (dd, J = 17.6, 5.0 Hz, 1H), 2.34 (dd, J = 17.4, 10.4 Hz, 1H), 2.21 (s, 3H), 1.71 (s, 3H), 1.52–1.34 (m, 4H), 0.85 (t, J = 7.3 Hz, 3H), 0.78 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.4, 167.7, 161.9, 140.0, 139.6, 138.3, 133.2, 132.8, 131.8, 129.8, 129.2, 127.3, 126.2, 119.8, 81.6, 75.6, 54.7, 48.5, 30.8, 26.3, 25.8, 23.2, 18.2, 9.9, 9.5. HRMS (ESI): calcd for C25H31N4O5, [M-H]− 467.2300, found 467.2304.
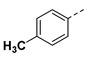
3.2.14. (3R,4R,5S)-4-acetamido-3-(pentan-3-yloxy)-5-(1-(p-tolyl)-1H-pyrazole-4-carboxamido) cyclohex-1-ene-1-carboxylic acid (6i)
Yield 45%; white solid, mp 282–284 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.79 (s, 1H), 8.03 (s, 1H), 7.98 (d, J = 8.9 Hz, 1H), 7.92 (d, J = 9.1 Hz, 1H), 7.72 (d, J = 8.2 Hz, 2H), 7.33 (d, J = 8.1 Hz, 2H), 6.66 (s, 1H), 4.25–4.07 (m, 2H), 3.87 (dd, J = 19.8, 9.1 Hz, 1H), 3.45–3.30 (m, 1H), 2.58 (dd, J = 17.5, 4.8 Hz, 1H), 2.45–2.30 (m, 1H), 2.35 (s, 3H), 1.70 (s, 3H), 1.53–1.33 (m, 4H), 0.85 (t, J = 7.3 Hz, 3H), 0.78 (t, J = 7.3 Hz, 3H), 0.67–0.66 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ 170.3, 167.7, 161.7, 140.4, 138.3, 137.4, 136.9, 130.5, 129.8, 129.0, 120.8, 119.1, 81.6, 75.6, 54.7, 48.4, 30.8, 26.3, 25.8, 23.2, 20.9, 9.9, 9.5. HRMS (ESI): calcd for C25H31N4O5, [M-H]− 467.2300, found 467.2306.
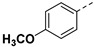
3.2.15. (3R,4R,5S)-4-acetamido-5-(1-(4-methoxyphenyl)-1H-pyrazole-4-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (6j)
Yield 47%; white solid, mp 277–279 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.72 (s, 1H), 8.01 (s, 1H), 7.94 (dd, J = 14.9, 9.2 Hz, 2H), 7.74 (d, J = 8.8 Hz, 2H), 7.08 (d, J = 8.7 Hz, 2H), 6.65 (s, 1H), 4.20–4.08 (m, 2H), 3.93–3.85 (m, 1H), 3.80 (s, 3H), 3.45–3.30 (m, 1H), 2.57 (dd, J = 17.4, 4.3 Hz, 1H), 2.41–2.30 (m, 1H), 1.70 (s, 3H), 1.51–1.33 (m, 4H), 0.85 (t, J = 7.2 Hz, 3H), 0.78 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.3, 167.8, 161.8, 158.6, 140.2, 138.1, 133.2, 130.0, 128.9, 120.8, 120.6, 115.2, 81.6, 75.6, 55.9, 54.7, 48.4, 30.9, 26.3, 25.8, 23.2, 9.9, 9.5. HRMS (ESI): calcd for C25H31N4O6, [M-H]− 483.2249, found 483.2252.
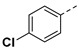
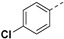
3.2.16. (3R,4R,5S)-4-acetamido-5-(1-(4-chlorophenyl)-1H-pyrazole-4-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (6k)
Yield 55%; white solid, mp 295–297 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.87 (s, 1H), 8.08 (s, 1H), 8.02 (d, J = 8.9 Hz, 1H), 7.92 (d, J = 9.3 Hz, 1H), 7.88 (d, J = 8.6 Hz, 2H), 7.60 (d, J = 8.6 Hz, 2H), 6.65 (s, 1H), 4.22–4.07 (m, 2H), 3.87 (dd, J = 19.7, 9.3 Hz, 1H), 3.45–3.30 (m, 1H), 2.57 (dd, J = 17.5, 4.7 Hz, 1H), 2.41–2.30 (m, 1H), 1.70 (s, 3H), 1.52–1.33 (m, 4H), 0.85 (t, J = 7.2 Hz, 3H), 0.77 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.3, 167.7, 161.5, 140.8, 138.4, 138.3, 131.6, 130.1, 129.8, 129.5, 121.3, 120.9, 81.6, 75.6, 54.7, 48.5, 30.8, 26.3, 25.7, 23.2, 9.9, 9.5. HRMS (ESI): calcd for C24H28ClN4O5, [M-H]− 487.1754, found 487.1759.
3.2.17. General Procedure for the Synthesis of Compounds 9a–f
To a stirred solution of dimethyl oxalate (1.30 g, 11 mmol) and an aryl methyl ketone (10 mmol) in toluene (60 mL) was added a solution of potassium tert-butoxide (1.35 g, 12 mmol) in THF (60 mL). The resulting solution was stirred at room temperature overnight. The reaction was quenched with 1 N HCl solution and extracted with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to provide the crude products 8a–f.
The crude product was dissolved in AcOH (10 mL) and hydrazine hydrate (534 μL, 10 mmol) was added. The solution was heated to 90 °C for 4 h. The solvent was removed under reduced pressure and the residue was dispersed in EtOAc. The precipitate was collected via filtration and washed by a small amount of EtOAc to afford compounds 9a–f without further purification.
3.2.18. General Procedure for the Synthesis of Compounds 10a–f
The preparation of 10a–f was performed similarly as described for compounds 4a–k.
3.2.19. General Procedure for the Synthesis of Compounds 11a–f
The condensation of 10a–f with oseltamivir phosphate to afford 11a–f was performed in a similar manner as described for compounds 5a–k.
3.2.20. General Procedure for the Synthesis of Compounds 12a–f
Each methyl ester of 11a–f was dissolved in MeOH (10 mL), 1 M NaOH aqueous solution (2 mL) was added. The mixture was stirred at 50 °C overnight. The solvent was removed under reduced pressure and the residue was diluted with 10 mL water and washed with DCM (10 mL × 2). The aqueous phase was acidified by 1 M HCl to precipitate the product which was collected by filtration and dried in vacuo.
3.2.21. (3R,4R,5S)-4-acetamido-3-(pentan-3-yloxy)-5-(3-phenyl-1H-pyrazole-5-carboxamido) cyclohex-1-ene-1-carboxylic acid (12a)
Yield 69%; white solid, mp 234–236 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.00 (d, J = 8.6 Hz, 2H), 7.80 (d, J = 7.5 Hz, 2H), 7.46 (t, J = 7.4 Hz, 2H), 7.36 (t, J = 7.2 Hz, 1H), 7.08 (s, 1H), 6.64 (s, 1H), 4.18–4.08 (m, 2H), 4.00–3.85 (m, 1H), 3.46–3.30 (m, 1H), 2.67 (d, J = 15.7 Hz, 1H), 2.35 (dd, J = 16.9, 8.8 Hz, 1H), 1.75 (s, 3H), 1.54–1.34 (m, 4H), 0.86 (t, J = 7.2 Hz, 3H), 0.80 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.4, 168.3, 161.3, 147.4, 145.4, 136.6, 130.7, 129.4, 128.7, 125.7, 102.8, 81.6, 75.3, 53.5, 48.4, 30.7, 26.3, 25.8, 23.2, 9.8, 9.6. HRMS (ESI): calcd for C24H29N4O5, [M-H]− 453.2143, found 453.2144.
3.2.22. (3R,4R,5S)-4-acetamido-5-(3-(4-chlorophenyl)-1H-pyrazole-5-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (12b)
Yield 62%; white solid, mp 266–268 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.74 (s, 1H), 8.05 (s, 1H), 7.95 (d, J = 8.7 Hz, 1H), 7.81 (d, J = 8.1 Hz, 2H), 7.52 (d, J = 8.1 Hz, 2H), 7.12 (s, 1H), 6.67 (s, 1H), 4.25–4.06 (m, 2H), 3.94 (dd, J = 17.6, 8.7 Hz, 1H), 3.45–3.40 (m, 1H), 2.65 (d, J = 14.0 Hz, 1H), 2.38 (dd, J = 16.9, 9.2 Hz, 1H), 1.75 (s, 3H), 1.56–1.31 (m, 4H), 0.86 (t, J = 7.2 Hz, 3H), 0.79 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.4, 167.9, 160.8, 137.4, 133.2, 130.0, 129.5, 127.4, 103.1, 81.6, 75.2, 53.6, 48.4, 30.5, 26.3, 25.8, 23.1, 9.8, 9.6, 9.3. HRMS (ESI): calcd for C24H28ClN4O5, [M-H]− 487.1754, found 487.1761.
3.2.23. (3R,4R,5S)-4-acetamido-5-(3-(4-fluorophenyl)-1H-pyrazole-5-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (12c)
Yield 55%; white solid, mp 259–261 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.46 (s, 1H), 8.02 (s, 1H), 7.96 (d, J = 8.8 Hz, 1H), 7.83 (dd, J = 8.3, 5.5 Hz, 2H), 7.30 (t, J = 8.8 Hz, 2H), 7.07 (s, 1H), 6.68 (s, 1H), 4.22–4.07 (m, 2H), 3.95 (dd, J = 18.2, 8.8 Hz, 1H), 3.45–3.40 (m, 1H), 2.65 (dd, J = 17.3, 4.1 Hz, 1H), 2.38 (dd, J = 17.5, 9.3 Hz, 1H), 1.75 (s, 3H), 1.54–1.34 (m, 4H), 0.86 (t, J = 7.3 Hz, 3H), 0.79 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.4, 167.8, 162.4 (d, J = 243.9 Hz), 160.9, 145.3, 137.6, 129.8, 127.8, 127.7 (d, J = 8.1 Hz), 116.4 (d, J = 21.6 Hz), 102.8, 81.6, 75.2, 53.5, 48.3, 30.4, 26.3, 25.8, 23.1, 9.8, 9.6. HRMS (ESI): calcd for C24H28FN4O5, [M-H]− 471.2049, found 471.2051.
3.2.24. (3R,4R,5S)-4-acetamido-3-(pentan-3-yloxy)-5-(3-(4-(trifluoromethyl) phenyl)-1H-pyrazole-5-carboxamido) cyclohex-1-ene-1-carboxylic acid (12d)
Yield 50%; white solid, mp 270–272 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.86 (s, 1H), 8.13 (s, 1H), 8.02 (s, 1H), 8.00 (s, 1H), 7.96 (d, J = 8.9 Hz, 1H), 7.81 (d, J = 8.1 Hz, 2H), 7.24 (s, 1H), 6.68 (s, 1H), 4.21–4.09 (m, 2H), 3.95 (dd, J = 18.0, 8.9 Hz, 1H), 3.45–3.30 (m, 1H), 2.65 (dd, J = 17.2, 3.6 Hz, 1H), 2.40 (dd, J = 17.3, 9.4 Hz, 1H), 1.74 (s, 3H), 1.52–1.35 (m, 4H), 0.85 (t, J = 7.3 Hz, 3H), 0.79 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.4, 167.8, 160.7, 137.7, 129.8, 128.71, 128.68 (q, J = 31.7 Hz), 126.4 (q, J = 3.6 Hz), 126.2, 126.0, 123.3, 120.6 (q, J = 270.2 Hz), 103.8, 81.6, 75.3, 53.7, 48.4, 30.4, 26.3, 25.8, 23.1, 9.8, 9.5. HRMS (ESI): calcd for C25H28F3N4O5, [M-H]− 521.2017, found 521.2023.
3.2.25. (3R,4R,5S)-4-acetamido-5-(3-(4-methoxyphenyl)-1H-pyrazole-5-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (12e)
Yield 59%; white solid, mp 273–275 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.96 (d, J = 8.6 Hz, 2H), 7.72 (d, J = 8.7 Hz, 2H), 7.02 (d, J = 8.7 Hz, 2H), 6.97 (s, 1H), 6.68 (s, 1H), 4.10 (dd, J = 17.4, 11.7 Hz, 2H), 3.96 (dd, J = 18.2, 8.7 Hz, 1H), 3.79 (s, 3H), 3.45–3.42 (m, 1H), 2.66 (dd, J = 17.5, 4.4 Hz, 1H), 2.36 (dd, J = 17.5, 9.1 Hz, 1H), 1.76 (s, 3H), 1.54–1.35 (m, 4H), 0.86 (t, J = 7.3 Hz, 3H), 0.80 (t, J = 7.3 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.4, 167.8, 161.4, 159.8, 145.4, 137.5, 129.9, 127.1, 122.8, 114.8, 102.0, 81.6, 75.2, 55.7, 53.4, 48.3, 30.5, 26.3, 25.8, 23.1, 9.8, 9.6. HRMS (ESI): calcd for C25H31N4O6, [M-H]− 483.2249, found 483.2253.
3.2.26. (3R,4R,5S)-4-acetamido-3-(pentan-3-yloxy)-5-(3-(thiophen-2-yl)-1H-pyrazole-5-carboxamido) cyclohex-1-ene-1-carboxylic acid (12f)
Yield 65%; white solid, mp 225–227 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.64 (s, 1H), 8.24 (s, 1H), 7.92 (d, J = 8.9 Hz, 1H), 7.54 (s, 1H), 7.41 (s, 1H), 7.13 (s, 1H), 6.94 (s, 1H), 6.65 (s, 1H), 4.19–4.05 (m, 2H), 3.95–3.90 (m, 1H), 3.45–3.30 (m, 1H), 2.60 (d, J = 14.5 Hz, 1H), 2.46–2.33 (m, 1H), 1.73 (s, 3H), 1.56–1.35 (m, 4H), 0.86 (t, J = 7.3 Hz, 3H), 0.79 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.3, 167.9, 160.0, 137.4, 130.1, 128.4, 126.2, 124.6, 102.6, 81.6, 75.3, 53.9, 48.4, 30.5, 26.3, 25.8, 23.6, 9.9, 9.6. HRMS (ESI): calcd for C22H27N4O5S, [M-H]− 459.1708, found 459.1712.
3.2.27. General Procedure for the Synthesis of Compounds 13a–c
A mixture of methyl 3-(4-chlorophenyl)-1H-pyrazole-5-carboxylate (9b, 237 mg, 1 mmol), K2CO3 (276 g, 2 mmol), and alkyl bromide (3 mmol) in acetone (10 mL) was heated to reflux for 12 h. After cooling to room temperature, K2CO3 was filtered off and the filtrate was concentrated and purified by column chromatography, eluting with hexane/ethyl acetate (6:1) to provide the titled compound.
3.2.28. General Procedure for the Synthesis of Compounds 14a–c
The hydrolysis of 13a–c was performed in a similar manner as described for compounds 4a–k.
3.2.29. General Procedure for the Synthesis of Compounds 15a–c
The condensation of 13a–c with oseltamivir phosphate was performed in a similar manner as described for compounds 5a–k.
3.2.30. General Procedure for the Synthesis of Compounds 16a–c
Each ester of 15a–c was dissolved in MeOH (10 mL), 1 M NaOH aqueous solution (2 mL) was added. The mixture was stirred at 50 °C overnight. The solvent was removed under reduced pressure and the residue was diluted with 10 mL water and washed with DCM (10 mL × 2). The aqueous phase was acidified by 1 M HCl to precipitate the product which was collected by filtration and dried in vacuo.
3.2.31. (3R,4R,5S)-4-acetamido-5-(3-(4-chlorophenyl)-1-(cyclopropylmethyl)-1H-pyrazole-5-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (16a)
Yield 65%; white solid, mp 269–271 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.42 (d, J = 8.9 Hz, 1H), 8.02 (d, J = 9.0 Hz, 1H), 7.76 (d, J = 8.2 Hz, 2H), 7.51 (d, J = 8.2 Hz, 2H), 7.14 (s, 1H), 6.56 (s, 1H), 4.37 (ddd, J = 45.8, 13.6, 7.2 Hz, 2H), 4.17–4.08 (m, 2H), 3.90–3.80 (m, 1H), 3.45–3.32 (m, 1H), 2.59 (d, J = 13.2 Hz, 1H), 2.44–2.33 (m, 1H), 1.73 (s, 3H), 1.54–1.36 (m, 4H), 1.34–1.28 (m, 1H), 0.85 (t, J = 7.3 Hz, 3H), 0.78 (t, J = 7.2 Hz, 3H), 0.46 (d, J = 7.6 Hz, 2H), 0.33–0.40 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 170.3, 169.3, 159.5, 147.7, 137.4, 135.6, 132.7, 132.0, 129.4, 127.1, 104.9, 81.5, 75.8, 55.2, 54.7, 48.8, 31.1, 26.4, 25.7, 23.2, 12.5, 9.9, 9.5, 3.9, 3.8. HRMS (ESI): calcd for C28H34ClN4O5, [M-H]− 541.2223, found 541.2229.
3.2.32. (3R,4R,5S)-4-acetamido-5-(3-(4-chlorophenyl)-1-(cyclobutylmethyl)-1H-pyrazole-5-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (16b)
Yield 51%; white solid, mp 265–267 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.39 (d, J = 8.8 Hz, 1H), 8.02 (d, J = 8.9 Hz, 1H), 7.75 (d, J = 8.0 Hz, 2H), 7.51 (d, J = 8.0 Hz, 2H), 7.12 (s, 1H), 6.63 (s, 1H), 4.54 (ddd, J = 40.1, 13.1, 7.5 Hz, 2H), 4.20–4.10 (s, 2H), 3.88 (dd, J = 19.3, 9.3 Hz, 1H), 3.45–3.30 (m, 1H), 2.82–2.71 (m, 1H), 2.56 (d, J = 13.6 Hz, 1H), 2.46–2.34 (m, 1H), 1.96–1.85 (m, 2H), 1.85–1.65 (m, 7H), 1.54–1.33 (m, 4H), 0.85 (t, J = 7.0 Hz, 3H), 0.78 (t, J = 7.0 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.2, 168.2, 159.5, 147.6, 137.7, 137.5, 132.7, 132.0, 129.4, 127.0, 104.8, 81.6, 75.6, 55.3, 54.5, 48.6, 36.2, 30.7, 26.3, 25.7, 25.5, 25.4, 23.2, 18.2, 9.9, 9.5. HRMS (ESI): calcd for C29H36ClN4O5, [M-H]− 555.2380, found 555.2382.
3.2.33. (3R,4R,5S)-4-acetamido-5-(3-(4-chlorophenyl)-1-(cyclohexylmethyl)-1H-pyrazole-5-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (16c)
Yield 40%; white solid, mp 232–234 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.36 (d, J = 8.9 Hz, 1H), 8.02 (d, J = 9.1 Hz, 1H), 7.75 (d, J = 8.3 Hz, 2H), 7.50 (d, J = 8.3 Hz, 2H), 7.15 (s, 1H), 6.64 (s, 1H), 4.37 (ddd, J = 51.7, 13.0, 7.3 Hz, 2H), 4.22–4.07 (m, 2H), 3.88 (dd, J = 19.5, 9.3 Hz, 1H), 3.45–3.30 (m, 1H), 2.56 (dd, J = 17.6, 4.7 Hz, 1H), 2.48–2.37 (m, 1H), 1.85 (s, 1H), 1.73 (s, 3H), 1.61 (d, J = 16.6 Hz, 3H), 1.54–1.29 (m, 7H), 1.18–0.89 (m, 6H), 0.85 (t, J = 7.2 Hz, 3H), 0.78 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6) δ 170.1, 168.0, 159.4, 147.5, 138.0, 137.8, 132.7, 132.0, 130.1, 129.4, 127.0, 104.7, 81.6, 75.6, 56.6, 54.5, 48.6, 39.0, 30.6, 30.4, 26.34, 26.31, 25.7, 25.6, 23.2, 9.9, 9.5. HRMS (ESI): calcd for C31H40ClN4O5, [M-H]− 583.2693, found 583.2699.
3.2.34. General Procedure for the Synthesis of Compounds 19a–e and 20a–d
To a stirred solution of dimethyl oxalate (1.30 g, 11 mmol) and 1-cyclopropylethan-1-one (991 μL, 10 mmol) in toluene (60 mL) was added a solution of potassium tert-butoxide (1.35 g, 12 mmol) in THF (60 mmol). The resulting solution was stirred at room temperature overnight. The reaction was quenched with 1 N HCl and extracted with ethyl acetate. The combined organic layers were dried over Na2SO4, filtered, and concentrated under reduced pressure to provide crude product 18.
Compound 18 was dissolved in EtOH (15 mL) and aryl hydrazine (12 mmol) was added. The mixture was heated to 85 °C for 4 h. The solvent was removed under reduced pressure and the residue was purified by column chromatography, eluting with a gradient of hexane/ethyl acetate (15:1 to 8:1) to provide the desired product from 19a–e to 20a–d (for 19a–e, the methyl esters were transformed to ethyl esters in EtOH).
3.2.35. General Procedure for the Synthesis of Compounds 21a–e and 22a–d
The hydrolysis of 19a–e and 20a–d was performed in a similar manner as described for compounds 4a–k.
3.2.36. General Procedure for the Synthesis of Compounds 23a–e and 25a–d
The condensation of 21a–e and 22a–d with oseltamivir phosphate was performed in a similar manner as described for compounds 5a–k.
3.2.37. General Procedure for the Synthesis of Compounds 24a–e and 26a–d
Each compound of 23a–e and 25a–d (0.2 mmol) was dissolved in MeOH (10 mL), and 1 M NaOH aqueous solution (2 mL) was added. The mixture was stirred at 50 °C overnight. The solvent was removed in vacuo, the residue was diluted with 10 mL water and washed with DCM (10 mL × 2). The aqueous phase was acidified by 1 M HCl to precipitate the product which was collected by filtration and dried in vacuo.
3.2.38. (3R,4R,5S)-4-acetamido-5-(5-cyclopropyl-1-phenyl-1H-pyrazole-3-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (24a)
Yield 57%; white solid, mp 273–275 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.96 (d, J = 8.2 Hz, 1H), 7.90 (d, J = 8.5 Hz, 1H), 7.66 (d, J = 7.8 Hz, 2H), 7.57 (t, J = 7.7 Hz, 2H), 7.48 (t, J = 7.2 Hz, 1H), 6.69 (s, 1H), 6.48 (s, 1H), 4.19–4.02 (m, 2H), 3.95 (dd, J = 15.9, 8.6 Hz, 1H), 3.45–3.30 (m, 1H), 2.59 (dd, J = 17.8, 4.5 Hz, 1H), 2.39 (dd, J = 17.8, 7.8 Hz, 1H), 1.83 (td, J = 8.2, 4.2 Hz, 1H), 1.78 (d, J = 10.4 Hz, 3H), 1.55–1.33 (m, 4H), 1.00–0.90 (m, 2H), 0.85–0.73 (m, 8H). 13C NMR (100 MHz, DMSO-d6) δ 170.3, 167.9, 161.5, 148.0, 146.7, 139.6, 136.9, 129.9, 129.6, 128.6, 125.2, 103.7, 81.6, 74.6, 52.5, 47.6, 29.8, 26.2, 25.8, 23.1, 9.8, 9.6, 9.2, 9.0, 7.8. HRMS (ESI): calcd for C27H33N4O5, [M-H]− 493.2456, found 493.2461.
3.2.39. (3R,4R,5S)-4-acetamido-5-(5-cyclopropyl-1-(o-tolyl)-1H-pyrazole-3-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (24b)
Yield 61%; white solid, mp 169–171 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.89 (dd, J = 8.1, 3.5 Hz, 2H), 7.45 (s, 2H), 7.39 (s, 2H), 6.67 (s, 1H), 6.41 (s, 1H), 4.16–4.04 (m, 2H), 3.92 (dd, J = 16.3, 8.5 Hz, 1H), 3.45–3.30 (M, 1H), 2.58 (dd, J = 17.7, 4.3 Hz, 1H), 2.35 (dd, J = 17.7, 7.8 Hz, 1H), 2.02 (s, 3H), 1.74 (s, 3H), 1.48–1.30 (m, 5H), 0.85–0.80 (m, 2H), 0.80–0.70 (m, 8H). 13C NMR (100 MHz, DMSO-d6) δ 170.2, 167.8, 161.6, 148.9, 146.5, 138.4, 136.9, 136.1, 131.4, 130.0, 129.9, 128.1, 127.1, 101.5, 81.5, 74.6, 52.6, 47.6, 29.9, 26.1, 25.7, 23.1, 17.3, 9.8, 9.5, 8.8, 8.6, 7.0. HRMS (ESI): calcd for C28H35N4O5, [M-H]− 507.2613, found 507.2617.
3.2.40. (3R,4R,5S)-4-acetamido-5-(5-cyclopropyl-1-(m-tolyl)-1H-pyrazole-3-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (24c)
Yield 53%; white solid, mp 220–222 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.97 (d, J = 8.2 Hz, 1H), 7.90 (d, J = 8.4 Hz, 1H), 7.45 (s, 3H), 7.30 (s, 1H), 6.68 (s, 1H), 6.46 (s, 1H), 4.16–4.06 (m, 2H), 3.95 (dd, J = 15.6, 8.3 Hz, 1H), 3.45–3.40 (m, 1H), 2.58 (dd, J = 17.6, 3.7 Hz, 1H), 2.40 (s, 4H), 1.82 (d, J = 4.6 Hz, 1H), 1.77 (s, 3H), 1.53–1.33 (m, 4H), 0.95 (d, J = 6.9 Hz, 2H), 0.85–0.72 (m, 8H). 13C NMR (100 MHz, DMSO-d6) δ 170.3, 167.9, 161.5, 147.9, 146.6, 139.6, 139.3, 136.6, 130.0, 129.4, 129.2, 125.7, 122.2, 103.6, 81.7, 74.6, 52.4, 47.4, 29.7, 26.2, 25.8, 23.1, 21.3, 9.8, 9.6, 9.2, 9.0, 7.9. HRMS (ESI): calcd for C28H35N4O5, [M-H]− 507.2613, found 507.2618.
3.2.41. (3R,4R,5S)-4-acetamido-5-(1-(4-bromophenyl)-5-cyclopropyl-1H-pyrazole-3-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (24d)
Yield 55%; white solid, mp 166–168 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.98 (d, J = 8.2 Hz, 1H), 7.92 (d, J = 8.4 Hz, 1H), 7.77 (d, J = 8.4 Hz, 2H), 7.64 (d, J = 8.5 Hz, 2H), 6.67 (s, 1H), 6.50 (s, 1H), 4.10 (d, J = 5.4 Hz, 2H), 3.98–3.91 (m, 1H), 3.45–3.30 (m, 1H), 2.64–2.54 (m, 1H), 2.38 (dd, J = 17.7, 7.8 Hz, 1H), 1.89–1.80 (m, 1H), 1.76 (s, 3H), 1.52–1.33 (m, 4H), 0.96 (d, J = 7.8 Hz, 2H), 0.85–0.70 (m, 8H). 13C NMR (100 MHz, DMSO-d6) δ 170.4, 168.0, 161.4, 148.1, 147.0, 138.9, 136.7, 132.6, 130.1, 127.0, 121.4, 104.3, 81.6, 74.7, 52.6, 47.7, 29.9, 26.2, 25.8, 23.1, 9.8, 9.6, 9.1, 8.9, 7.8. HRMS (ESI): calcd for C27H32BrN4O5, [M-H]− 571.1562, found 571.1562.
3.2.42. (3R,4R,5S)-4-acetamido-5-(5-cyclopropyl-1-(4-(trifluoromethyl) phenyl)-1H-pyrazole-3-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (24e)
Yield 60%; white solid, mp 220–222 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.03 (d, J = 8.2 Hz, 1H), 8.00–7. 90 (m, 5H), 6.69 (s, 1H), 6.56 (s, 1H), 4.11 (d, J = 5.3 Hz, 2H), 3.96 (dd, J = 16.3, 8.5 Hz, 1H), 3.45–3.30 (m, 1H), 2.60 (dd, J = 17.6, 4.2 Hz, 1H), 2.40 (dd, J = 17.7, 7.9 Hz, 1H), 1.99–1.89 (m, 1H), 1.77 (s, 3H), 1.51–1.34 (m, 4H), 1.00 (d, J = 7.6 Hz, 2H), 0.72–0.85 (m, 8H). 13C NMR (100 MHz, DMSO-d6) δ 170.4, 167.8, 161.2, 148.3, 147.5, 142.8, 137.1, 129.8, 128.5 (d, J = 32.0 Hz), 126.9 (d, J = 3.1 Hz), 126.9 (d, J = 3.9 Hz), 125.3, 124.4 (q, J = 270.6 Hz), 104.9, 81.6, 74.7, 52.7, 47.8, 29.8, 26.2, 25.8, 23.1, 9.8, 9.6, 9.2, 9.0, 8.0. HRMS (ESI): calcd for C28H32F3N4O5, [M-H]− 561.2330, found 561.2335.
3.2.43. (3R,4R,5S)-4-acetamido-5-(3-cyclopropyl-1-phenyl-1H-pyrazole-5-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (26a)
Yield 51%; white solid, mp 267–269 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.96 (d, J = 8.3 Hz, 1H), 7.90 (d, J = 8.5 Hz, 1H), 7.66 (d, J = 7.8 Hz, 2H), 7.57 (t, J = 7.7 Hz, 2H), 7.49 (t, J = 7.3 Hz, 1H), 6.67 (s, 1H), 6.48 (s, 1H), 4.16–4.06 (m, 2H), 3.94 (dd, J = 15.8, 8.7 Hz, 1H), 3.45–3.30 (m, 1H), 2.59 (dd, J = 17.7, 4.7 Hz, 1H), 2.38 (dd, J = 17.8, 7.9 Hz, 1H), 1.84 (td, J = 8.3, 4.1 Hz, 1H), 1.78 (d, J = 11.8 Hz, 3H), 1.43 (qt, J = 13.8, 7.1 Hz, 4H), 1.04–0.89 (m, 2H), 0.86–0.71 (m, 8H). 13C NMR (100 MHz, DMSO-d6) δ 170.3, 168.0, 161.5, 148.0, 146.7, 139.6, 136.6, 130.1, 129.6, 128.6, 125.2, 103.7, 81.6, 74.7, 52.5, 47.6, 29.8, 26.2, 25.8, 23.2, 9.8, 9.6, 9.2, 9.0, 7.8. HRMS (ESI): calcd for C27H33N4O5, [M-H]− 493.2456, found 493.2461.
3.2.44. (3R,4R,5S)-4-acetamido-5-(3-cyclopropyl-1-(o-tolyl)-1H-pyrazole-5-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (26b)
Yield 45%; white solid, mp 168–170 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.88 (d, J = 8.1 Hz, 2H), 7.42 (d, J = 26.3 Hz, 4H), 6.67 (s, 1H), 6.41 (s, 1H), 4.20–4.10 (m, 2H), 3.92 (dd, J = 16.2, 8.3 Hz, 1H), 3.45–3.30 (m, 1H), 2.58 (dd, J = 17.6, 3.6 Hz, 1H), 2.36 (dd, J = 17.5, 7.6 Hz, 1H), 2.02 (s, 3H), 1.74 (s, 3H), 1.49–1.31 (m, 5H), 0.90–0.85 (m, 2H), 0.80–0.70 (m, 8H). 13C NMR (100 MHz, DMSO-d6) δ 170.2, 167.8, 161.6, 148.9, 146.5, 138.4, 137.0, 136.1, 131.4, 130.0, 129.9, 128.1, 127.1, 101.5, 81.5, 74.7, 52.6, 47.6, 29.9, 26.1, 25.7, 23.1, 17.3, 9.8, 9.5, 8.8, 8.6, 7.0. HRMS (ESI): calcd for C28H35N4O5, [M-H]− 507.2613, found 507.2614.
3.2.45. (3R,4R,5S)-4-acetamido-5-(3-cyclopropyl-1-(m-tolyl)-1H-pyrazole-5-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (26c)
Yield 48%; white solid, mp 159–161 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.97 (d, J = 8.2 Hz, 1H), 7.91 (d, J = 8.4 Hz, 1H), 7.45 (s, 3H), 7.29 (s, 1H), 6.69 (s, 1H), 6.46 (s, 1H), 4.17–4.04 (m, 2H), 3.95 (dd, J = 15.6, 8.2 Hz, 1H), 3.45–3.30 (m, 1H), 2.58 (dd, J = 17.8, 4.0 Hz, 1H), 2.40 (s, 4H), 1.82 (d, J = 4.8 Hz, 1H), 1.77 (s, 3H), 1.50–1.35 (m, 4H), 0.95 (d, J = 7.0 Hz, 2H), 0.85–0.70 (m, 8H). 13C NMR (100 MHz, DMSO-d6) δ 170.3, 167.9, 161.5, 147.9, 146.6, 139.5, 139.3, 136.8, 129.8, 129.4, 129.2, 125.7, 122.1, 103.6, 81.7, 74.6, 52.4, 47.4, 29.7, 26.2, 25.8, 23.1, 21.3, 9.8, 9.6, 9.2, 9.0, 7.9. HRMS (ESI): calcd for C28H35N4O5, [M-H]− 507.2613, found 507.2614.
3.2.46. (3R,4R,5S)-4-acetamido-5-(1-(4-bromophenyl)-3-cyclopropyl-1H-pyrazole-5-carboxamido)-3-(pentan-3-yloxy) cyclohex-1-ene-1-carboxylic acid (26d)
Yield 40%; white solid, mp 169–171 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.97 (d, J = 8.0 Hz, 1H), 7.90 (d, J = 8.3 Hz, 1H), 7.77 (d, J = 8.3 Hz, 2H), 7.64 (d, J = 8.2 Hz, 2H), 6.68 (s, 1H), 6.50 (s, 1H), 4.30–4.05 (m, 2H), 3.94 (dd, J = 15.9, 8.2 Hz, 1H), 3.45–3.30 (m, 1H), 2.58 (d, J = 13.8 Hz, 1H), 2.39 (dd, J = 17.3, 7.5 Hz, 1H), 1.91–1.80 (m, 1H), 1.76 (s, 3H), 1.50–1.35 (m, 4H), 0.96 (d, J = 5.9 Hz, 2H), 0.85–0.70 (m, 8H). 13C NMR (100 MHz, DMSO-d6) δ 170.3, 167.9, 161.3, 148.1, 147.0, 138.9, 136.9, 132.6, 129.9, 127.0, 121.4, 104.3, 81.6, 74.7, 52.6, 47.7, 29.8, 26.2, 25.8, 23.1, 9.8, 9.6, 9.1, 8.9, 7.8. HRMS (ESI): calcd for C27H32BrN4O5, [M-H]− 571.1562, found 571.1565.
3.3. In Silico Studies
The structure of NA was extracted from PDB 2HU0. Structures of the compounds and the protein for docking were imported to Maestro 10.2, and the conformations were generated with the panel “Protein Preparation Wizard” and “LigPrep”, respectively. Molecular docking was performed using the Glide in Maestro 10.2, and ADME properties were predicted by Qikprop (Maestro 10.2). The docking and ADME calculation processes were conducted with the default parameters unless otherwise mentioned.
4. Conclusions
The 150-cavity of NA represents a potential binding site for novel NA inhibitors. In the present study, we designed and synthesized a series of pyrazole-based C-5-NH2-acyl derivatives of OC. Most of the derivatives exhibited moderate to high inhibitory activity against NA at the μM level, in which compounds 12c and 26b demonstrated the greatest activity. Moreover, in silico ADME prediction suggest that the four compounds have drug-like pharmacokinetic properties. Especially, parameters for oral absorption and cell permeability indicated the prospect of direct oral administration of the derivatives, such as 6i, 12c, 24b, and 26b. Further molecular docking studies revealed that the derivatives could dock into the active site and the 150-cavity as expected. However, calculated binding energy showed that these derivatives possessed higher binding energy than OC (Table S1), indicating these modifications would reduce the binding affinity to NA. Considering the inhibitory activity of compounds 12c and 26b, as well as their docking models, it is expected that further optimized NA inhibitors could be identified based on the work described in this study.
C-5-NH2 modification of oseltamivir represents one of the most attractive research interests in the development of novel NA 150-cavity binders. Previously, we initiated the strategy of using heterocycles to link oseltamivir and the 150-binding fragments [17]. Inspired by the fact that pyrazole is one of the most widely used heterocycles in drug discovery, we validated our hypothesis that modifying the oseltamivir C-5-NH2 group by incorporating the pyrazole moiety has the potential to develop novel NA inhibitors. This work together with our previous discovery on the development of NA inhibitors containing other heterocycles will pave a solid basis for the design of novel 150-binders as NA inhibitors in the future.
Abbreviations
| OC | oseltamivir carboxylate |
| OP | oseltamivir phosphate |
| HA | Hemagglutinin |
| NA | Neuraminidase |
| NAIs | Neuraminidase inhibitors |
| ADME | absorption, distribution, metabolism and excretion |
| DCM | Dichloromethane |
| DIPEA | N,N-Diisopropylethylamine |
| HATU | 2-(7-Azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate |
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/ph14040371/s1, docking models of the products, binding free energy of representative products, experimental data of the intermediates; 1H and 13C NMR Spectra of the products.
Author Contributions
Conceptualization, X.Y. and C.M.; methodology, X.Y. and C.M.; validation, J.Y. and L.L.; formal analysis, J.Y. and L.L.; writing—original draft preparation, J.Y.; writing—review and editing, J.Y., L.L., J.X., P.K.-s.C., X.Y. and C.M.; supervision, X.Y. and C.M.; project administration, X.Y. and C.M.; funding acquisition, X.Y. and C.M. All authors have read and agreed to the published version of the manuscript.
Funding
The research was supported by Hong Kong Research Grants Council (PolyU 151000/19M and C5008-19G (C.M.), CUHK 141079/19M (X.Y.)), Hong Kong Polytechnic University (State Key Laboratory of Chemical Biology and Drug Discovery, Internal grants 1-ZVPS and G-YBYY, and large equipment fund), and the Chinese University of Hong Kong (Faculty of Medicine Faculty Innovation Award No. FIA2018/A/03 (X.Y.)).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are provided within the article and the Supplementary Materials.
Conflicts of Interest
The authors declare no competing financial interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Word Health Organization Report. [(accessed on 4 December 2020)]; Available online: https://www.who.int/news/item/13-12-2017-up-to-650-000-people-die-of-respiratory-diseases-linked-to-seasonal-flu-each-year.
- 2.Cox N.J., Bender C.A. The molecular epidemiology of influenza viruses. Semin. Virol. 1995;6:359–370. doi: 10.1016/S1044-5773(05)80013-7. [DOI] [Google Scholar]
- 3.Bouvier N.M., Palese P. The biology of influenza viruses. Vaccine. 2008;26(Suppl. S4):D49–D53. doi: 10.1016/j.vaccine.2008.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhuang Q., Wang S., Liu S., Hou G., Li J., Jiang W., Wang K., Peng C., Liu D., Guo A., et al. Diversity and distribution of type A influenza viruses: An updated panorama analysis based on protein sequences. Virol. J. 2019;16:85. doi: 10.1186/s12985-019-1188-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kilbourne E.D. Influenza pandemics of the 20th century. Emerg. Infect. Dis. 2006;12:9–14. doi: 10.3201/eid1201.051254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dawood F.S., Iuliano A.D., Reed C., Meltzer M.I., Shay D.K., Cheng P.-Y., Bandaranayake D., Breiman R.F., Brooks W.A., Buchy P., et al. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: A modelling study. Lancet Infect. Dis. 2012;12:687–695. doi: 10.1016/S1473-3099(12)70121-4. [DOI] [PubMed] [Google Scholar]
- 7.Jefferson T., Demicheli V., Rivetti D., Jones M., Di Pietrantonj C., Rivetti A. Antivirals for influenza in healthy adults: Systematic review. Lancet. 2006;367:303–313. doi: 10.1016/S0140-6736(06)67970-1. [DOI] [PubMed] [Google Scholar]
- 8.Noshi T., Kitano M., Taniguchi K., Yamamoto A., Omoto S., Baba K., Hashimoto T., Ishida K., Kushima Y., Hattori K., et al. In vitro characterization of baloxavir acid, a first-in-class cap-dependent endonuclease inhibitor of the influenza virus polymerase PA subunit. Antivir. Res. 2018;160:109–117. doi: 10.1016/j.antiviral.2018.10.008. [DOI] [PubMed] [Google Scholar]
- 9.Gubareva L.V., Fry A.M. Baloxavir and Treatment-Emergent Resistance: Public Health Insights and Next Steps. J. Infect. Dis. 2020;221:337–339. doi: 10.1093/infdis/jiz245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greener M. Using oseltamivir (Tamiflu) as a first line treatment for seasonal or pandemic flu. Nurs. Times. 2009;105:22. [PubMed] [Google Scholar]
- 11.Gamblin S.J., Skehel J.J. Influenza hemagglutinin and neuraminidase membrane glycoproteins. J. Biol. Chem. 2010;285:28403–28409. doi: 10.1074/jbc.R110.129809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Russell R.J., Haire L.F., Stevens D.J., Collins P.J., Lin Y.P., Blackburn G.M., Hay A.J., Gamblin S.J., Skehel J.J. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature. 2006;443:45–49. doi: 10.1038/nature05114. [DOI] [PubMed] [Google Scholar]
- 13.Wu Y., Qin G., Gao F., Liu Y., Vavricka C.J., Qi J., Jiang H., Yu K., Gao G.F. Induced opening of influenza virus neuraminidase N2 150-loop suggests an important role in inhibitor binding. Sci. Rep. 2013;3:1551. doi: 10.1038/srep01551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie Y., Xu D., Huang B., Ma X., Qi W., Shi F., Liu X., Zhang Y., Xu W. Discovery of N-substituted oseltamivir derivatives as potent and selective inhibitors of H5N1 influenza neuraminidase. J. Med. Chem. 2014;57:8445–8458. doi: 10.1021/jm500892k. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J., Poongavanam V., Kang D., Bertagnin C., Lu H., Kong X., Ju H., Lu X., Gao P., Tian Y., et al. Optimization of N-Substituted Oseltamivir Derivatives as Potent Inhibitors of Group-1 and -2 Influenza A Neuraminidases, Including a Drug-Resistant Variant. J. Med. Chem. 2018;61:6379–6397. doi: 10.1021/acs.jmedchem.8b00929. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J., Murugan N.A., Tian Y., Bertagnin C., Fang Z., Kang D., Kong X., Jia H., Sun Z., Jia R., et al. Structure-Based Optimization of N-Substituted Oseltamivir Derivatives as Potent Anti-Influenza A Virus Agents with Significantly Improved Potency against Oseltamivir-Resistant N1-H274Y Variant. J. Med. Chem. 2018;61:9976–9999. doi: 10.1021/acs.jmedchem.8b01065. [DOI] [PubMed] [Google Scholar]
- 17.Ye J., Yang X., Xu M., Chan P.K., Ma C. Novel N-Substituted oseltamivir derivatives as potent influenza neuraminidase inhibitors: Design, synthesis, biological evaluation, ADME prediction and molecular docking studies. Eur. J. Med. Chem. 2019;182:111635. doi: 10.1016/j.ejmech.2019.111635. [DOI] [PubMed] [Google Scholar]
- 18.Wang P., Oladejo B.O., Li C., Fu L., Zhang S., Qi J., Lv X., Li X. Structure-based design of 5′-substituted 1,2,3-triazolylated oseltamivir derivatives as potent influenza neuraminidase inhibitors. RSC Adv. 2021;11:9528–9541. doi: 10.1039/D1RA00472G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang K., Lei Z., Zhao L., Chen B., Yang F., Liu K., Zhu H., Zhao H., Cao R., Zhang K., et al. Design, synthesis and biological evaluation of oseltamivir derivatives containing pyridyl group as potent inhibitors of neuraminidase for influenza A. Eur. J. Med. Chem. 2020;185:111841. doi: 10.1016/j.ejmech.2019.111841. [DOI] [PubMed] [Google Scholar]
- 20.Wang K., Yang F., Wang L., Liu K., Sun L., Lin B., Hu Y., Wang B., Cheng M., Tian Y. Synthesis and biological evaluation of NH2-acyl oseltamivir analogues as potent neuraminidase inhibitors. Eur. J. Med. Chem. 2017;141:648–656. doi: 10.1016/j.ejmech.2017.10.004. [DOI] [PubMed] [Google Scholar]
- 21.Antilla J.C., Baskin J.M., Barder T.E., Buchwald S.L. Copper-diamine-catalyzed N-arylation of pyrroles, pyrazoles, indazoles, imidazoles, and triazoles. J. Org. Chem. 2004;69:5578–5587. doi: 10.1021/jo049658b. [DOI] [PubMed] [Google Scholar]
- 22.Dawood K.M., Abdel-Gawad H., Mohamed H.A., Badria F.A. Synthesis, anti-HSV-1, and cytotoxic activities of some new pyrazole- and isoxazole-based heterocycles. Med. Chem. Res. 2010;20:912–919. doi: 10.1007/s00044-010-9420-4. [DOI] [Google Scholar]
- 23.Levent S., Caliskan B., Ciftci M., Ozkan Y., Yenicesu I., Unver H., Banoglu E. Pyrazole derivatives as inhibitors of arachidonic acid-induced platelet aggregation. Eur. J. Med. Chem. 2013;64:42–53. doi: 10.1016/j.ejmech.2013.03.048. [DOI] [PubMed] [Google Scholar]
- 24.Aghazadeh Tabrizi M., Baraldi P.G., Baraldi S., Ruggiero E., De Stefano L., Rizzolio F., Di Cesare Mannelli L., Ghelardini C., Chicca A., Lapillo M., et al. Discovery of 1,5-Diphenylpyrazole-3-Carboxamide Derivatives as Potent, Reversible, and Selective Monoacylglycerol Lipase (MAGL) Inhibitors. J. Med. Chem. 2018;61:1340–1354. doi: 10.1021/acs.jmedchem.7b01845. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are provided within the article and the Supplementary Materials.