

Abstract
Cancer immunotherapy has revolutionized the way that we think about treating cancer. Although checkpoint blockade therapy, including anti-PD-1/PD-L1 and anti-CTLA-4, has shown remarkable success, the responses are limited to only a subset of patients. This discrepancy highlights the many overlapping avenues for immune evasion or suppression that can be employed by a tumor. One such mechanism of immunosuppression is adenosinergic signaling within the tumor microenvironment. We provide an overview of the current status of clinical trials targeting the adenosine pathway, including CD73, CD39, and adenosine receptors. Additionally, we highlight several avenues that may be explored to further potentiate responses in the clinic by combining adenosine-targeting agents to target multiple arms of the pathway or by using conventional immunotherapy agents.
Keywords: adenosine, immunotherapy, adenosine receptor, CD73, CD39
INTRODUCTION
Immunotherapy has revolutionized the way that we think about treating cancer. Although immune checkpoint blockade (ICB) therapy, including anti-PD-1/PD-L1 and anti-CTLA-4, has shown remarkable success, the responses are limited to a subset of patients. This discrepancy highlights the many overlapping avenues for immune evasion or suppression that can be employed by a tumor. In order to bring the success of immunotherapy to a wider patient cohort, it will be critical to clinically understand the roles of the diverse array of immunosuppressive mechanisms. One such mechanism is the adenosinergic pathway, whereby extracellular ATP is converted into immunosuppressive adenosine.
At steady state, levels of extracellular ATP are exceedingly low; however, upon cell death or cellular stress, ATP is released extracellularly. The levels of extracellular ATP can rapidly and dramatically increase, and this is often seen in the tumor microenvironment (TME) due to hypoxia, inflammation, and necrotic cell death (1, 2). While ATP itself can be immunostimulatory, it can undergo a stepwise process where it is ultimately converted into the nucleoside adenosine (Figure 1). Canonically, ATP is first degraded into AMP via the ecto-nucleotidase CD39. AMP is then dephosphorylated and converted into adenosine by CD73. Adenosine can subsequently bind to purinergic receptors, including A1, A2a, A2b, and A3 (3). The A2a receptor (A2aR) and A2b receptor (A2bR) are primarily responsible for downstream immunosuppressive signaling following accumulation of intracellular cAMP (4).
Figure 1.
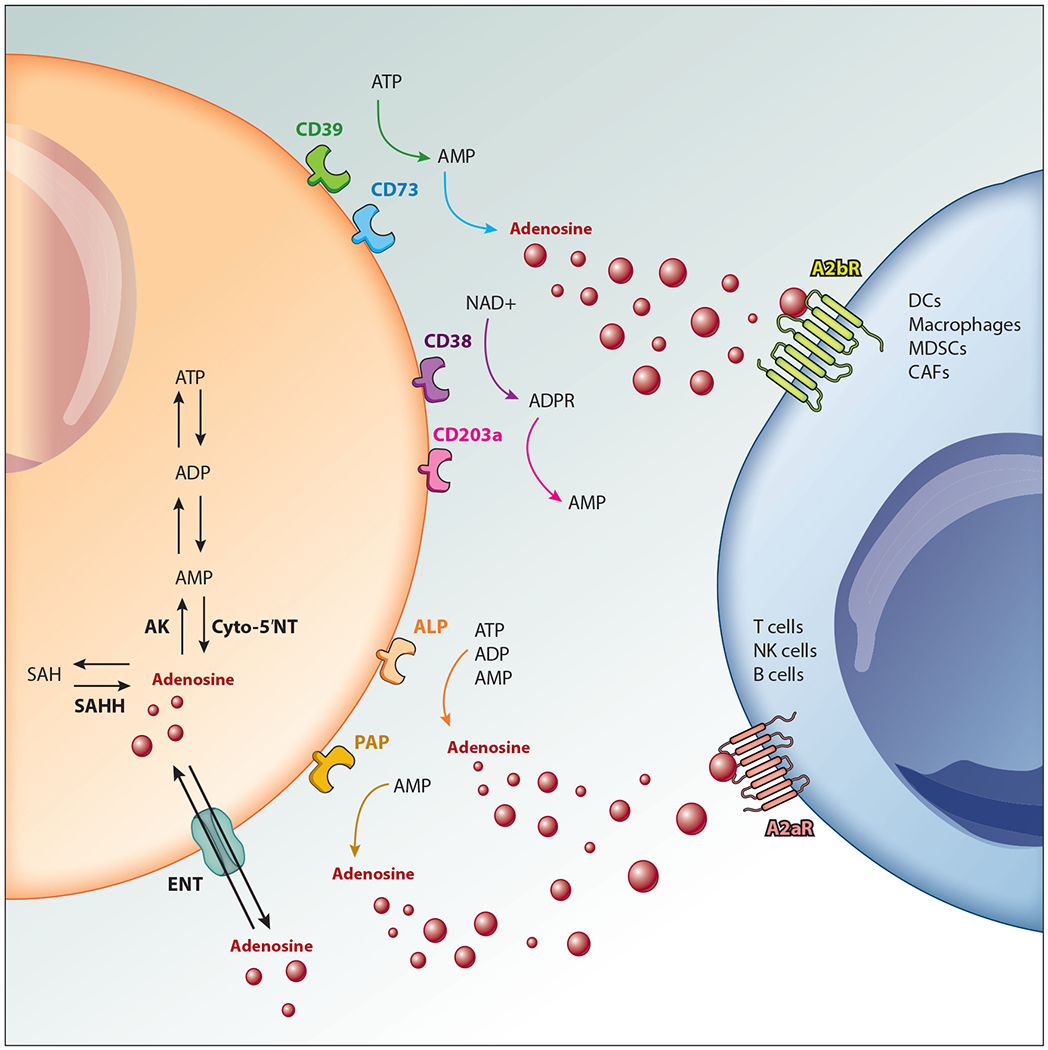
Adenosine production and signaling. Extracellular adenosine is generated in a stepwise process via multiple molecules. CD39 dephosphorylates ATP into AMP, which is then converted into adenosine via CD73. AMP can also be generated via sequential action of CD38 and CD203a. Alternative sources of extracellular adenosine production include alkaline phosphatase (ALP) and prostatic acid peptidase (PAP). Intracellular adenosine is regulated by the balance of the activity of adenosine kinase (AK) and cyto-5′NT/intracellular CD73 or the direct metabolism of S-adenosylhomocysteine (SAH) by the enzyme S-adenosyl-homocysteine hydrolase (SAHH). Adenosine is transported into and out of the cell by equilibrative nucleoside transporters (ENTs) and signals predominantly via A2aR and A2bR on cells within the tumor microenvironment. Other abbreviations: CAF, cancer-associated fibroblast; DC, dendritic cell; MDSC, myeloid-derived suppressor cell; NK, natural killer.
CD73, which is thought to be largely responsible for adenosine accumulation, is highly expressed on a variety of tumor cells and stromal cells contributing to immune evasion, as well as directly regulating immune cell function through expression on immunosuppressive populations such as regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs) (Figure 2). In contrast, CD39 is widely expressed and is the predominant nucleotide-metabolizing enzyme expressed on immune cells in the TME due to the hypoxic environment, including Tregs, macrophages, dendritic cells (DCs), and neutrophils, as well as epithelial and endothelial cells (5–7). The expression of CD73 within the tumor is often associated with a poor clinical prognosis and has therefore gained significant attention as a potential metabolic checkpoint for immunotherapy (8, 9).
Figure 2.
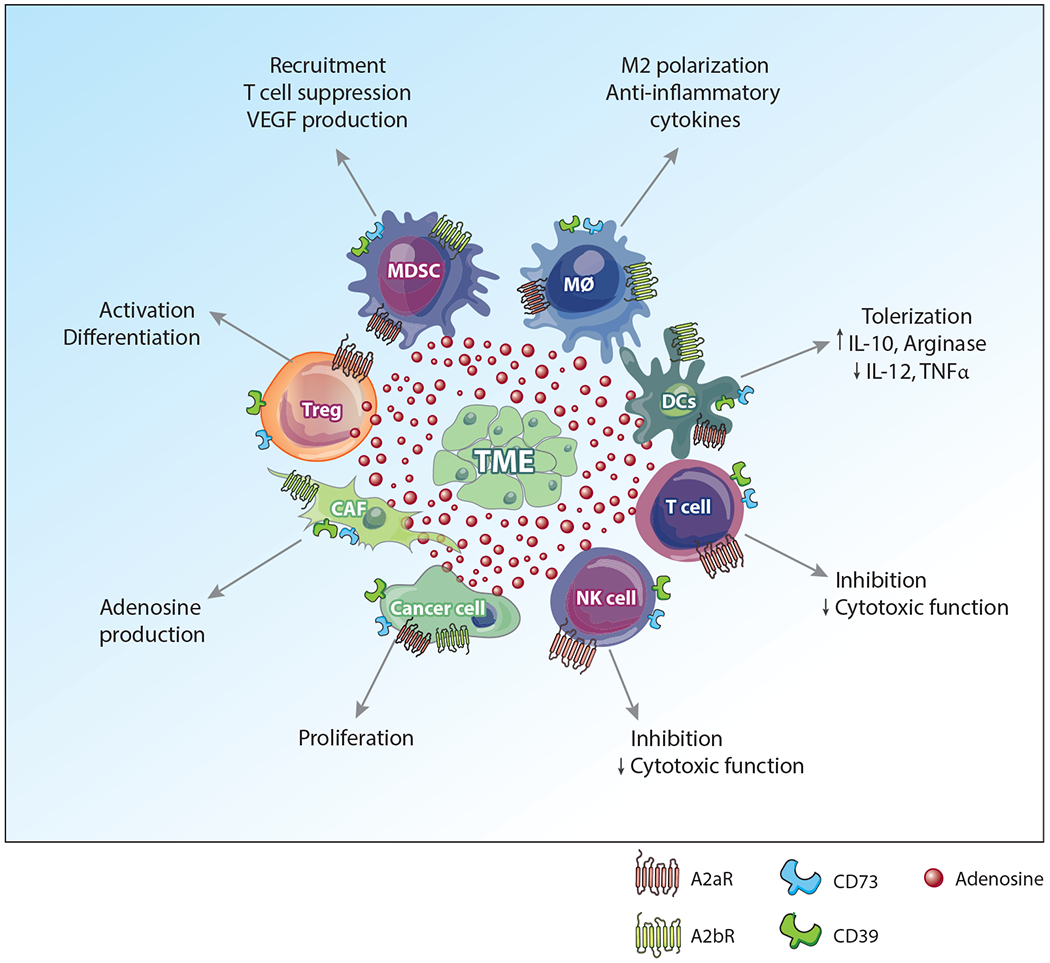
Adenosinergic targets within the TME. The TME is composed of a variety of cancer-associated and immune cells, each with differing expression of targetable molecules associated with the adenosine pathway; these include A2aR,A2bR,CD73, and CD39. While adenosine signaling induces a variety of immunosuppressive functions within these different cell types, targeting the various pathways may potently inhibit the immunosuppressive TME. Abbreviations: CAF, cancer-associated fibroblast; DC, dendritic cell; MDSC, myeloid-derived suppressor cell; MØ, macrophage; NK, natural killer; TME, tumor microenvironment; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor.
Although the CD39/CD73 axis is one of the most highly studied pathways and is thought to account for the bulk of adenosine production, alternative pathways are also present. CD38 and CD203a are able to sequentially convert NAD+ into AMP, which can again be converted into adenosine via CD73 (10). Further, adenosine can be generated through two additional pathways: (a) alkaline phosphatase (ALP), which can directly convert AMP, ATP, or ADP into adenosine, and (b) prostatic acid peptidase (PAP), which converts AMP into adenosine (11, 12). The multifaceted nature of adenosinergic signaling provides multiple potential targets that have been shown to alleviate the immunosuppressive TME in a variety of preclinical models (Figure 2). However, one consequence of this complex pathway that needs further investigation is the potential for compensatory mechanisms to mitigate blocked or altered signaling within another arm of the pathway. While this indicates that as a monotherapy adenosine blockade may not be sufficient, it provides rationale for targeting multiple arms of the adenosine pathway. In this article, we provide a brief overview of the current status of ongoing clinical trials involving adenosine blockade as cancer immunotherapy, as well as a rationale for targeting multiple arms of the adenosine pathway, highlighting several concepts that have yet to be fully incorporated into clinical trials.
CLINICAL TRIAL STATUS
There has been rapid development of clinical trials targeting multiple components of the adenosine pathway in recent years (Figure 3a). That said, most trials are still in early development, with the majority being phase I or combined phase I/phase II trials (Figure 3b, Table 1). Due to the early nature of these trials, efficacy data are limited; however, preliminary data presented at conferences indicate that adenosine targeting may be a viable treatment in a variety of cancer types (13–19).
Figure 3.
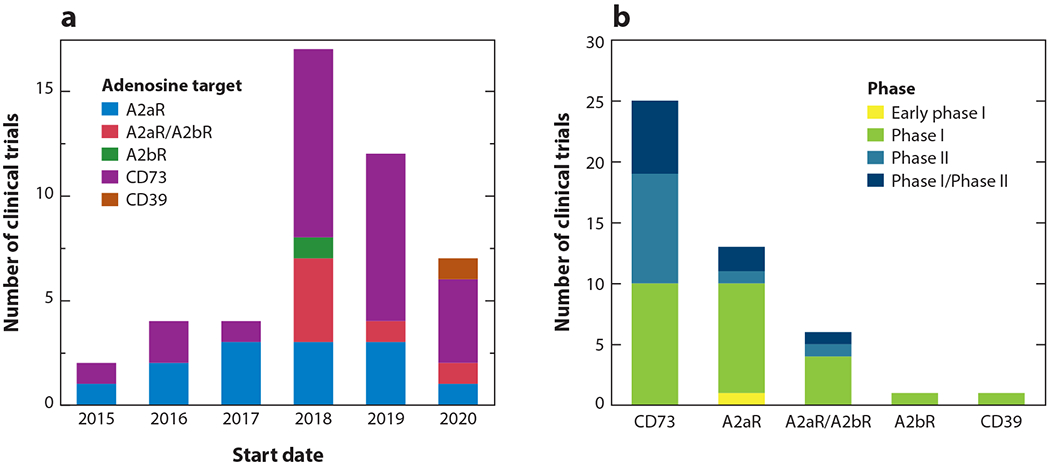
Clinical trials targeting the adenosine pathway. The figure displays trials according to (a) start date and (b) current phase as of May 2020.
Table 1.
Clinical trials targeting the adenosine pathway in cancer
| Target | NCT number | Start date | Conditions | Interventions | Phases | Company |
|---|---|---|---|---|---|---|
| A2aR | NCT02403193 | 2015 | NSCLC | PBF-509, PDR001 | Phase I, phase II | Palobiofarma SL |
| NCT03207867 | 2016 | NSCLC, RCC, pancreatic cancer, urothelial cancer, head and neck cancer, DLBCL, MSS, TNBC, melanoma | NIR178, PDR001 | Phase II | Novartis | |
| NCT02655822 | 2016 | RCC, mCRPC | Ciforadenant, ciforadenant + atezolizumab | Phase I | Corvus Pharmaceuticals, Inc. | |
| NCT03786484 | 2017 | Cancer | PBF-999 | Phase I | Palobiofarma SL | |
| NCT03237988 | 2017 | Healthy subjects | CPI-444 | Phase I | Corvus Pharmaceuticals, Inc. | |
| NCT03099161 | 2017 | Neoplasm | Preladenant, pembrolizumab | Phase I | Merck Sharp & Dohme Corp. | |
| NCT03710434 | 2018 | Healthy volunteers | AZD4635 | Phase I | AstraZeneca | |
| NCT03549000 | 2018 | NSCLC, TNBC, PDAC, MSS, ovarian cancer, RCC, mCRPC | NZV930, PDR001, NIR178 | Phase I | Novartis | |
| NCT03337698 | 2018 | Carcinoma, NSCLC | Atezolizumab, cobimetinib, docetaxel, CPI-444, pemetrexed, carboplatin, gemcitabine, linagliptin, tocilizumab, ipatasertib, idasanutlin | Phase I, phase II | Hoffmann–La Roche | |
| NCT03980821 | 2019 | Advanced solid malignancies | AZD4635 | Phase I | AstraZeneca | |
| NCT03873883 | 2019 | Solid tumor (adult) | EOS100850 | Phase I | iTeos Therapeutics | |
| NCT03742349 | 2019 | TNBC | Spartalizumab, LAG525, NIR178, capmatinib, MCS110, canakinumab | Phase I | Novartis | |
| NCT04237649 | 2020 | Solid tumor | KAZ954, PDR001, NIR178, NZV930 | Early phase I | Novartis | |
| A2aR and A2bR | NCT03720678 | 2018 | Gastroesophageal cancer, CRC | AB928, mFOLFOX | Phase I | Arcus Biosciences, Inc. |
| NCT03719326 | 2018 | TNBC, ovarian cancer | AB928, IPI-549, pegylated liposomal doxorubicin, nanoparticle albumin-bound paclitaxel | Phase I | Arcus Biosciences, Inc. | |
| NCT03629756 | 2018 | NSCLC, squamous cell carcinoma of head and neck, breast cancer, CRC, melanoma, bladder cancer, ovarian cancer, endometrial cancer, Merkel cell carcinoma, gastroesophageal cancer, RCC, CRPC | AB928, AB122 | Phase I | Arcus Biosciences, Inc. | |
| NCT03555149 | 2018 | CRC | Regorafenib, atezolizumab, imprime PGG, bevacizumab, isatuximab, selicrelumab, idasanutlin, AB928 | Phase I, phase II | Hoffmann–La Roche | |
| NCT03846310 | 2019 | Metastatic NSCLC, NSCLC, nonsquamous non–small cell neoplasm of lung, sensitizing EGFR gene mutation | AB928, AB122, carboplatin, pemetrexed, pembrolizumab | Phase I | Arcus Biosciences, Inc. | |
| NCT04262856 | 2020 | NSCLC, nonsquamous NSCLC, squamous NSCLC, lung cancer | Zimberelimab, AB154, AB928 | Phase II | Arcus Biosciences, Inc. | |
| A2bR | NCT03274479 | 2018 | Locally advanced or metastatic NSCLC | PBF-1129 | Phase I | Palobiofarma SL |
| CD73 | NCT02503774 | 2015 | Solid tumor | MEDI9447, MEDI9447, MEDI4736 | Phase I | MedImmune LLC |
| NCT02754141 | 2016 | Malignant solid tumor | BMS-986179, nivolumab, rHuPH20 | Phase I, phase II | Bristol-Myers Squibb | |
| NCT02740985 | 2016 | Advanced solid malignancies, NSCLC, mCRPC, CRC | AZD4635, durvalumab, abiraterone acetate, enzalutamide, oleclumab, docetaxel | Phase I | AstraZeneca | |
| NCT03334617 | 2017 | NSCLC | Durvalumab, AZD9150, AZD6738, vistusertib, olaparib, oleclumab, trastuzumab deruxtecan, cediranib | Phase II | AstraZeneca | |
| NCT03822351 | 2018 | Stage III NSCLC (unresectable) | Durvalumab + oleclumab, durvalumab, durvalumab + monalizumab | Phase II | MedImmune LLC | |
| NCT03819465 | 2018 | Metastatic NSCLC | Durvalumab, danvatirsen, oleclumab, pemetrexed, carboplatin, gemcitabine, cisplatin, nab-paclitaxel | Phase I | AstraZeneca | |
| NCT03742102 | 2018 | Triple-negative breast neoplasms | Durvalumab, capivasertib, danvatirsen, oleclumab, paclitaxel | Phase I, phase II | AstraZeneca | |
| NCT03736473 | 2018 | Advanced solid malignancies | MEDI9447 (oleclumab) | Phase I | AstraZeneca | |
| NCT03677973 | 2018 | Healthy volunteers | AB680 | Phase I | Arcus Biosciences, Inc. | |
| NCT03616886 | 2018 | TNBC | Paclitaxel, carboplatin, MEDI4736, MEDI9447 | Phase I, phase II | AstraZeneca | |
| NCT03611556 | 2018 | Carcinoma, metastatic pancreatic adenocarcinoma | Oleclumab, durvalumab, gemcitabine, nab-paclitaxel, oxaliplatin, leucovorin, 5-FU | Phase I, phase II | MedImmune LLC | |
| NCT03454451 | 2018 | NSCLC, RCC, CRC, TNBC, cervical cancer, ovarian cancer, pancreatic cancer, endometrial cancer, sarcoma, squamous cell carcinoma of head and neck, bladder cancer, mCRPC, non-Hodgkin lymphoma | CPI-006, CPI-006 + ciforadenant, CPI-006 + pembrolizumab | Phase I | Corvus Pharmaceuticals, Inc. | |
| NCT03381274 | 2018 | Carcinoma, NSCLC | MEDI9447, osimertinib, AZD4635 | Phase I, phase II | MedImmune LLC | |
| NCT04148937 | 2019 | Advanced cancer | LY3475070, pembrolizumab | Phase I | Eli Lilly and Company | |
| NCT04104672 | 2019 | Advanced pancreatic cancer | AB680, AB122 | Phase I | Arcus Biosciences, Inc. | |
| NCT04089553 | 2019 | Prostate cancer, mCRPC | AZD4635, oleclumab, durvalumab | Phase II | AstraZeneca | |
| NCT04068610 | 2019 | Metastatic MSS | FOLFOX + bevacizumab, FOLFOX + bevacizumab + durvalumab + oleclumab | Phase I, phase II | MedImmune LLC | |
| NCT03875573 | 2019 | Luminal B | Durvalumab, stereotactic body radiotherapy, oleclumab | Phase II | AstraZeneca | |
| NCT03833440 | 2019 | NSCLC | Durvalumab (MEDI4736), monalizumab (IPH2201), oleclumab (MEDI9447), AZD6738, docetaxel | Phase II | ||
| NCT03794544 | 2019 | Resectable early-stage NSCLC | Durvalumab, durvalumab + oleclumab, durvalumab + monalizumab, durvalumab + danvatirsen | Phase II | MedImmune LLC | |
| NCT03773666 | 2019 | Muscle-invasive bladder cancer | Durvalumab, oleclumab | Phase I | AstraZeneca | |
| NCT04262388 | 2020 | PDAC, NSCLC, squamous cell carcinoma of head and neck | Durvalumab, oleclumab | Phase II | AstraZeneca | |
| NCT04262375 | 2020 | NSCLC, RCC | Durvalumab, oleclumab | Phase II | AstraZeneca | |
| NCT04145193 | 2020 | MSS | mFOLFOX6, mFOLFOX + durvalumab, mFOLFOX6 + durvalumab + oleclumab, mFOLFOX6 + durvalumab + monalizumab | Phase II | MedImmune LLC | |
| NCT03884556 | 2019 | Solid tumor, lymphoma | TTX-030, TTX-030 + pembrolizumab, TTX-030 + docetaxel, TTX-030 + gemcitabine + nab-paclitaxel | Phase I | Tizona Therapeutics Inc. | |
| CD39 | NCT04306900 | 2020 | Solid tumor (adult) | TTX-030 + budigalimab + mFOLFOX6, TTX-030 + budigalimab + docetaxel, TTX-030 + mFOLFOX6, TTX-030 + budigalimab | Phase I | Tizona Therapeutics Inc. |
| NCT04261075 | 2020 | Advanced solid tumor | IPH5201, durvalumab, oleclumab | Phase I | MedImmune LLC |
Abbreviations: CRC, colorectal carcinoma; CRPC, castration-resistant prostate cancer; DLBCL, diffuse large B cell lymphoma; mCRPC, metastatic castration-resistant prostate cancer; MSS, microsatellite-stable colon cancer; NCT, National Clinical Trial; NSCLC, non–small cell lung cancer; PDAC, pancreatic ductal adenocarcinoma; RCC, renal cell cancer; TNBC, triple-negative breast cancer.
Anti-CD73
The most commonly targeted molecule in clinical trials is CD73, with four distinct monoclonal antibodies currently being tested. The first antibody to be tested in clinical trials was oleclumab (MEDI9447), developed by MedImmune/AstraZeneca, which initiated its first clinical trial in 2015 (NCT02503774). This antibody is a human IgG1λ that noncompetitively binds to and inactivates the ectonuclease activity of CD73 on the surface of cells and can be internalized (20, 21). As this antibody is cross reactive with murine CD73, it has been shown in a preclinical mouse model of cancer to reduce tumor burden; however, clinical results have not yet been published (21). Preliminary results, however, indicate that it reduced CD73 expression in tumor cells and increased CD8 T cell infiltration; partial responses occurred in three patients receiving combined oleclumab with durvalumab (anti-PD-L1) (22). Oleclumab is now included in 18 additional clinical trials and is currently the most widely tested adenosine blockade therapy. BMS-986179 is a hybrid IgG1-IgG2 antibody that binds to surface CD73, inhibiting its activity, and also induces internalization to downregulate tumor CD73 expression. An ongoing clinical trial, initiated in 2016, is evaluating BMS-986179 with or without anti-PD-1 (nivolumab) in patients with advanced solid tumors (NCT02754141). Preliminary results indicate a safety profile similar to that of nivolumab monotherapy and antitumor activity in a subset of patients with a variety of solid tumors including head and neck, pancreatic, prostate, anal, and renal cancers (23). Corvus Pharmaceuticals initiated clinical trials in 2018 evaluating the anti-CD73 antibody CPI-006 (NCT03454454), which is a humanized IgG1 FcγR-binding-deficient antibody that competes with the active binding site of AMP. Preliminary data indicate that this antibody is overall well tolerated and shows signs of rapid lymphocyte distribution following injection and prolonged receptor occupancy (24). Finally, Surface Oncology in collaboration with Novartis began investigation in 2018 into NZV930, a fully human monoclonal antibody targeting CD73 (NCT03549000). To date, there is limited information on the mechanism of NZV930 or clinical progress. In addition to the more traditional monoclonal antibodies targeting CD73, small-molecule CD73 inhibitors are also being developed. Both AB680 and LY3475070, developed by Arcus Biosciences and Eli Lilly, respectively, are currently being tested in clinical trials (NCT04104672, NCT04148973, NCT04381832). In summary, there is a growing portfolio of anti-CD73 antibodies that are being tested as monotherapy or in combination with other immunotherapies and standard of care. While data are still limited, this class of drugs appears to be overall well tolerated and shows moderate efficacy in a subset of patients. Results are encouraging but indicate possible avenues for improved responses.
A2aR Antagonist
The second most widely tested modality for adenosine blockade is A2aR antagonists. These small-molecule drugs were initially developed for neurological disorders, with some having been tested in phase III clinical trials for Parkinson’s disease, allowing them to be more readily integrated into clinical trials for cancer therapy. There are currently five different agents in this class being tested in clinical trials (Table 1). Again, results are limited, but preclinical evaluations have found that pharmacologic or genetic inhibition of A2aR can reduce tumor growth and metastasis with a corresponding increase in survival (25–29). This is thought to occur through immune activation, as tumors showed increased infiltration of activated CD8 T cells and natural killer (NK) cells (27–29). Recently, Fong and colleagues published clinical results of the Corvus Pharmaceuticals A2aR antagonist ciforadenant (CPI-444) (30, 31). In total, 68 patients with refractory renal cell cancer were treated with ciforadenant as a monotherapy or in combination with anti-PD-L1 (atezolizumab). The trial was able to establish safety and feasibility and showed signs of efficacy in certain patients. Importantly, 72% of the patients in this trial had previously failed anti-PD1 or anti-PDL1 therapy as a monotherapy. As assessed by RECIST criteria, a partial response occurred in 1 of 35 patients receiving monotherapy and 4 of 35 patients in the combination group. While these numbers are relatively small, 39% of patients showed disease control for at least 6 months, and it is important to remember that this cohort of patients had already failed several other therapies, indicating that adenosine targeting has the ability to overcome ICB resistance. Similar to preclinical results, responses were associated with CD8 T cell infiltration and T cell receptor repertoire diversification. Although clinical results have not been published, other trials examining A2aR antagonists have preliminarily presented similar data (13, 16–19). As with the anti-CD73 trials, results indicate that A2aR blockade can induce immunological responses; however, whether biomarker-driven patient selection or combinatorial approaches will be necessary to increase response rates remains to be seen.
Dual A2aR/A2bR Inhibitors and Anti-CD39
While the vast majority of clinical trials are evaluating anti-CD73 and A2aR antagonists, there has recently been an expansion in alternative agents entering clinical trials. A novel compound developed by Arcus Biosciences that is a dual-specific inhibitory molecule of both A2aR and A2bR (AB928) has already been incorporated into several clinical studies. The first trials were initiated in 2018 and there are currently no published data on efficacy, making it hard to assess the potency of the drug. However, the rationale behind dual A2aR/A2bR blockade is logical, and it will be highly interesting to see how this class of compounds will perform in the clinic. Phase I trials in healthy volunteers and patients with solid tumors did not establish any safety concerns and showed significant adenosine receptor inhibition (15, 32). One of the newest additions to the adenosine portfolio is an antibody targeting CD39. TTX-030 is a human monoclonal antibody developed by Tizona Therapeutics. A clinical trial has recently started, posted in March 2019 (NCT03884556), in which anti-CD39 is delivered alone or in combination with pembrolizumab or chemotherapy. Similarly, Innate Pharma has a CD39 blocking antibody, IPH5201, which has entered clinical study alone, combined with durvalumab (anti-PD-L1), and, in an effort to more fully silence adenosine production, combined with durvalumab and oleclumab (anti-CD73) (NCT04261075). Data from this trial have not yet been reported, but preclinical data suggested substantial synergy with PD-L1 blockade.
Anti-CD38
CD38 is responsible for the conversion of NAD+ into AMP, which can feed into the ultimate production of adenosine. Although several clinical trials are evaluating anti-CD38 antibodies such as daratumumab and isatuximab, they are predominantly focused on CD38+ multiple myeloma and used to mediate killing of tumor cells. This antibody functions via Fc-dependent ADCC (antibody-dependent cellular cytotoxicity) to eliminate CD38+ tumor cells and is therefore largely outside of the scope of this review, but it has been reviewed elsewhere (33). However, evidence suggests that this antibody can also eliminate CD38+ immunosuppressive cells such as Tregs and MDSCs (34). Although not designed as such, these trials may also give insight into the function of CD38 and the ability to target this component of the adenosine pathway. The availability of a clinically tested anti-CD38 antibody also offers potential combinatorial approaches with other adenosinergic targeting agents.
COMBINATORIAL APPROACHES
Anti-CD73: A Need to Supplement?
As described above, anti-CD73 antibodies are to date the most commonly tested modality for adenosine blockade in clinical trials. However, blocking CD73 as a monotherapy may not be sufficient to achieve full adenosine blockade. Although CD73 is an important mechanism for adenosine production, it is not the sole pathway. Even simultaneous blockade of CD39 and CD73 is not able to improve antitumor responses in some settings, indicating possible other sources of adenosine (35, 36). Alternatively, ALP is able to convert AMP, ATP, and ADP into adenosine (Figure 1). Similar to CD73, ALP is expressed on cancer cells, and it has been shown that both cellular and serum ALP levels correlate with disease stage (37–39). These findings indicate that CD73 is not the sole contributor to adenosine production.
This may become particularly relevant within the context of CD73 blockade. PAP is another molecule that can produce adenosine. PAP is predominantly found in the prostate, although it can be elevated in other cancerous tissues (40). Within the context of prostate cancer, PAP may play a more dominant role, and it has been shown that serum levels of PAP increase during cancer progression (11, 40–42). There is rather limited information on the relative contribution of alternative pathways, particularly within the context of CD73 blockade. Several questions remain to be answered, including the half-life of AMP in the context of anti-CD73, and whether ALP or other alternative pathways play a compensatory role in continuing to convert ATP/AMP into adenosine to compensate for loss of CD73 function. The proliferation of anti-CD73 trials enables assessment of these questions in a clinical setting, allowing for more informed trial design in the future. The answers to these questions may indicate the need to block both CD73 and ALP or PAP to achieve a complete shutdown of extracellular adenosine production.
An alternative, and to date more clinically relevant, way to combat incomplete adenosine blockade is by simultaneously blocking adenosine production and receptor binding. This approach is currently being tested in several clinical trials combining anti-CD73 antibodies with A2aR antagonists. This approach has the added advantage of being able to accommodate intracellular adenosine, which could be released in the same manner as ATP under cellular stress or death. The nucleotides ATP and ADP are in continual metabolic flux within a cell to accommodate the cell’s energy needs. As such, there is an intracellular pool of adenosine; however, under steady-state conditions, concentrations remain relatively low, largely through the activity of adenosine kinase (AK), which converts adenosine back into AMP (Figure 1). However, under conditions of metabolic stress or high cellular activity, there can be elevated levels of intracellular adenosine (43). Intracellular adenosine can also be produced by the metabolism of the amino acid l-homocysteine by the enzyme S-adenosyl-homocysteine (SAH) hydrolase. Finally, CD73 can also be present and active intracellularly; however, not all anti-CD73 antibodies have the ability to be internalized. If CD73 is not efficiently blocked at all locations, there can be continual adenosine production. Targeting adenosine receptors alone may prove difficult without reducing adenosine levels, but a combined approach may be a potent way to suppress downstream immunosuppressive signaling, regardless of the source of adenosine production. Although clinical data on responses are limited, there is preclinical evidence demonstrating the benefit of this approach (44). Genetic deletion of both CD73 and A2aR effects greater tumor reduction than deletion of either one alone, indicating they possess distinct immunosuppressive activities. These findings could be replicated pharmacologically using an anti-CD73 antibody combined with a small-molecule A2aR inhibitor (44). Interestingly, the same study also observed increased expression of CD73 in A2aR-deficient mice, again highlighting potential compensatory mechanisms when only one component is blocked, as may be the case in single-agent A2aR trials.
A2bR: An Overlooked Receptor?
Of the two primary adenosine receptors responsible for immunosuppressive activity, A2bR has received substantially less attention than the high-affinity A2aR. A2bR is a low-affinity receptor expressed primarily on myeloid cells including DCs, macrophages, and MDSCs, as well as cancer-associated fibroblasts (CAFs). In contrast, A2aR is predominantly expressed on T cells and NK cells. Much attention has focused on adenosine-mediated inhibition of T cells, and therefore clinical efforts have primarily focused on A2aR. The mechanisms behind A2aR signaling in T cell suppression have been reviewed extensively elsewhere (2, 45–47). Although A2bR is a low-affinity receptor, it has been shown to be significantly engaged in adenosine-rich environments like the TME. Therefore, this receptor may play a larger role than previously appreciated, with each receptor playing a nonredundant role in immunosuppression. Most clinical trials take a targeted approach to block A2aR exclusively. While this may successfully block adenosine signaling within T cells, the TME is often a complex milieu consisting of a variety of immunosuppressive cells, including populations of myeloid cells and even CAFs (Figure 2). Therefore, blocking both the A2aR and the A2bR could provide a more comprehensive target, for reasons described below.
Adenosine signaling within the myeloid compartment has also been shown in preclinical models to contribute to immune suppression. In order to generate robust and protective T cell responses, it is critical to have efficient and stimulatory antigen presentation by DCs. Binding of A2bR on DCs can convert them to a tolerogenic phenotype and lead to so-called alternative priming of T cells. In mice, A2b signaling can decrease DCs’ production of inflammatory cytokines, including tumor necrosis factor alpha (TNFα) and interleukin (IL)-12, critical for effective CD8 T cell generation (48, 49). These DCs become more tolerogenic, with a concomitant increase in immune-suppressive cytokines and molecules, including IL-10 and arginase, and limited up-regulation of costimulatory molecules (48, 49). Blockade or deletion of A2bR leads to indirect suppression of CD8 T cells (50). These findings have been repeated using in vitro models with human monocytes and DCs, indicating a translatability to human clinical trials (51, 52). A2bR signaling in macrophages can also skew toward an anti-inflammatory M2 phenotype through a post-transcriptional mechanism relieving the translational repressive effect of the IL-10 3′UTR (53).
Another cell type that has recently gained increased attention is MDSCs. Although described previously, these cells were termed MDSCs only in 2007 and represent a heterogenous population of myeloid cells that exert immunosuppression through a variety of mechanisms, including PD-L1 expression, inducible nitric oxide synthase (iNOS) production, and elaboration of arginase (54, 55). While less is known about adenosine signaling within these populations, it has been shown that they may rely predominantly on A2bR signaling. Signaling through A2bR, but not the other adenosine receptors, led to an increase in MDSCs, predominantly with a granulocytic phenotype (56). It has also been shown that polymorphonuclear MDSCs express high levels of CD73 and become increasingly suppressive in vitro with elevated AMP levels (56). Adenosine signaling can also increase production of vascular endothelial growth factor (VEGF) by MDSCs while increasing angiogenesis and MDSC recruitment (57). Therefore, focusing only on A2aR signaling on T cells neglects a large component of the immune landscape that may have substantial impact on immunotherapy response rates.
Finally, nonhematopoietic cells also play a key role in dictating the immune landscape of a tumor. Non-hematopoietic cells provide scaffolding and can either aid or impede immune cell invasion into the tumor. Mesenchymal stromal cells and CAFs produce extracellular matrix and growth factors, cytokines, and chemokines that contribute to the TME. The critical role of these cells in maintaining an immunosuppressive environment has often been overlooked. CAFs also express CD73 and contribute to the generation of adenosine in the TME. A feedforward loop involving A2bR signaling on CAFs induces upregulation of CD73, further contributing to adenosine production (58). Importantly, the upregulation of CD73 was seen in the absence of A2aR signaling, signifying a clear function for A2bR signaling. Increased extracellular adenosine strongly activated the A2b pathway on CAFs and increased CD73 expression. It is important to note that adenosine levels can rapidly increase in response to either pathological or therapeutically induced tissue damage or cell death. Therefore, this feedforward mechanism involving CAFs not only may play a role in the context of adenosine blockade but should also be considered in combination with other immunotherapies, such as ICB.
Adenosine Blockade in Combination with Immune Checkpoint Blockade
Due to the success of ICB, almost all ongoing clinical trials blocking adenosine contain an arm in combination with standard ICB, chemotherapy, or radiation. Despite the remarkable ICB responses seen in a subset of patients, we know that using current strategies, in lung cancer for example, only approximately 30% of patients will respond to ICB as a monotherapy (59). It is also becoming more evident that even in patients whose cancers progress on immunotherapy, some have initiated a substantial antitumor T cell response (60). This indicates that there may be a subset of patients who are capable of mounting an antitumor response yet fail to overcome the immunosuppressive environment. Combining ICB with adenosine targeting may be able to bridge this gap and bring successful immunotherapy to a wider cohort of patients. To date, there is substantial preclinical evidence that combining ICB with adenosine blockade can improve efficacy. The effects of anti-CD73 can be improved by combining it with either anti-PD-1 or anti-CTLA-4 (21, 61). This has also been found true for A2aR antagonists (26, 62, 63). As tumor cells die in response to immunotherapy, they are able to release extracellular AMP/adenosine and could promote a second wave of immunosuppression. To this end, anti-PD-1 efficacy has been shown to be limited by tumor CD73 expression and also to cause an increase in A2aR expression on tumor-infiltrating CD8 T cells (62). By targeting both pathways in clinical trials, we may be able to combat some of the inhibitory feedback induced by a successful immune response. Adenosine targeting may also prove useful for enhancing adoptive cellular therapy (ACT), although this has yet to be explored in the clinic. ACT utilizes either tumor-infiltrating T cells expanded ex vivo and reinfused into the patient or T cells engineered with either a chimeric antigen receptor or exogenous T cell receptor specific for the tumor. Without addressing the immunosuppressive TME, infused T cells may find their way to the tumor but lose the ability to become activated or kill target cells. Several lines of preclinical evidence indicate that concomitant delivery of ACT with anti-CD73 or A2aR antagonists can improve antitumor responses and survival (26, 64–66). This improvement is based on increased T cell infiltration into the tumor and superior T cell activation. Alternatively, transferred T cells can be engineered to be deficient in A2aR expression, rendering them resistant to the immunosuppressive signaling mediated by high adenosine concentrations in the TME (67, 68). Together these data indicate a promising future for combined adenosine targeting and ICB or ACT.
FUTURE PERSPECTIVES
The exciting expansion of adenosine blockade in the clinic leads us to several questions that we are now poised to answer. As alluded to already, a main concern moving forward will be the feedback mechanisms and compensatory responses that occur within the adenosinergic pathway in response to blockade of single nodes. Many of these questions can readily be answered in preclinical models; however, the availability of biospecimens from ongoing clinical trials offers a unique opportunity to answer these questions in a highly translatable manner. Recent papers have touched upon the idea of increased CD73 expression in response to A2bR signaling and also in A2aR knockout mice (44, 58), but this concept has not yet been fully explored. Based on a better understanding of these intertwined signaling pathways, future trials exploring simultaneous blockade of multiple points within the adenosine pathway may be warranted.
It also remains to be seen if current drugs represent the optimal formulation to induce potent responses. Many of the A2aR antagonists were initially developed for neurological disorders, which has facilitated their rapid deployment in the clinic. However, it is possible that these molecules may need to be reformulated for increased activity within the TME. For example, alternative delivery methods or routes of administration may be required to optimize bioavailability within the tumor itself. Since the site of action is within the tumor and may involve adenosine production by both tumor cells and stromal support cells, it is critical that the drug is able to enter and distribute within the TME and not be sequestered in the periphery. Perhaps nanoparticle delivery would be able to induce a more localized and sustained delivery, as has been shown previously for other small molecules such as Toll-like receptor agonists (69–71). The combined A2aR and A2bR antagonist developed by Arcus Biosciences has significant promise; however, it remains to be seen if this is the optimal drug and formulation for bispecific targeting. Antibody engineering could also improve the activity of monoclonal anti-CD73 antibodies. Because many tumors also express high levels of CD73, these antibodies could be used to directly target tumor cells in addition to the adenosine pathway. Young et al. (44) demonstrated that optimal tumor rejection required the Fc portion of the antibody, indicating antibodies optimized for ADCC or other measures may perform better in the clinic (72). The importance of specific Fc receptors has been demonstrated in several other monoclonal antibodies, whether or not ADCC is a requirement for efficacy (73, 74). Altered antibody isotypes or glycosylation patterns should therefore be explored in order to optimize delivery in CD73-expressing tumors.
Finally, a critical issue for developing these therapies will be identifying patients who will respond to adenosine-targeting therapy. Ideally, extracellular adenosine could be measured to identify tumors highly enriched for adenosine signaling. However, extracellular adenosine has an exceedingly short half-life of approximately 10 s, and therefore detecting it does not represent a viable clinical procedure for identifying patients (75). An alternative approach, being pursued by both Arcus Biosciences and Corvus Pharmaceuticals, is to identify an adenosine gene signature, which will reflect increased levels of adenosine. Fong and colleagues (30) recently published preliminary findings on an adenosine gene signature that was developed using peripheral blood mononuclear cells stimulated in vitro with adenosine. Of note, when evaluated at the protein level, most of these changes appeared in the monocyte/myeloid compartment as opposed to CD8 T cells. The identified gene set (AdenoSig) was applied to tumor biopsy pretreatment and used to predict response rates with the A2aR antagonist ciforadenant. Patients with a high AdenoSig pretreatment showed significantly higher tumor regression and longer progression-free survival, indicating that this could function as a biomarker in renal cell cancer as tested. Arcus Biosciences is also aiming to develop a so-called adenosine fingerprint that combines mRNA transcript levels, CD73 levels, and AMP-ase enzymatic activity (76). These methods can now be tested retrospectively in a variety of clinical cohorts to determine if rates of response to adenosine blockade could be predicted prior to initiation of therapy.
CONCLUSIONS
To date, the dramatic preclinical data observed in mouse models have translated into only modest results in early clinical trials. Nonetheless, the ability of small-molecule inhibitors of A2aR and CD73 to demonstrate some efficacy in previously heavily treated patients suggests that the adenosinergic axis is involved in promoting tumor immune evasion. Critical issues remain concerning compensatory mechanisms and the potency of the current agents, which in the case of A2aR were originally developed for neurological disorders. Finally, combination therapy that seeks to block adenosine production, as well as receptor blockade, along with ICB, holds high theoretical promise. At this point, it remains to be seen if more efficient targeting of this pathway can lead to a therapeutic “home run” or whether the efficacy of the current agents represents a therapeutic plateau for adenosine inhibition.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (R01CA226765 to J.D.P., R01CA229451 to J.D.P., and 1P41EB028239-01 to J.D.P.) and The Bloomberg–Kimmel Institute for Cancer Immunotherapy.
Footnotes
DISCLOSURE STATEMENT
J.D.P. is a scientific founder and paid consultant of Dracen Pharmaceuticals. He has been a paid consultant of and received researching funding from Corvus Pharmaceuticals. He holds equity in Dracen Pharmaceuticals and Corvus Pharmaceuticals.
The Annual Review of Medicine is online at med.annualreviews.org
LITERATURE CITED
- 1.Di Virgilio F, Sarti AC, Falzoni S, et al. 2018. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat. Rev. Cancer 18:601–18 [DOI] [PubMed] [Google Scholar]
- 2.Vigano S, Alatzoglou D, Irving M, et al. 2019. Targeting adenosine in cancer immunotherapy to enhance T-cell function. Front. Immunol 10:925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jacobson KA, Gao ZG. 2006. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov 5:247–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohta A, Sitkovsky M. 2001. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 414:916–20 [DOI] [PubMed] [Google Scholar]
- 5.Borsellino G, Kleinewietfeld M, Di Mitri D, et al. 2007. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood 110:1225–32 [DOI] [PubMed] [Google Scholar]
- 6.Mizumoto N, Kumamoto T, Robson SC, et al. 2002. CD39 is the dominant Langerhans cell-associated ecto-NTPDase: modulatory roles in inflammation and immune responsiveness. Nat. Med 8:358–65 [DOI] [PubMed] [Google Scholar]
- 7.Koziak K, Sevigny J, Robson SC, et al. 1999. Analysis of CD39/ATP diphosphohydrolase (ATPDase) expression in endothelial cells, platelets and leukocytes. Thromb. Haemost 82:1538–44 [PubMed] [Google Scholar]
- 8.Jiang T, Xu X, Qiao M, et al. 2018. Comprehensive evaluation of NT5E/CD73 expression and its prognostic significance in distinct types of cancers. BMC Cancer 18:267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leone RD, Emens LA. 2018. Targeting adenosine for cancer immunotherapy. J. Immunother. Cancer 6:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horenstein AL, Chillemi A, Zaccarello G, et al.2013.A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology 2:e26246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zimmermann H 2009. Prostatic acid phosphatase, a neglected ectonucleotidase. Purinerg. Signal 5:27375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zimmermann H, Zebisch M, Strater N. 2012. Cellular function and molecular structure of ectonucleotidases. Purinerg. Signal 8:437–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiappori A, Williams CC, Creelan BC, et al. 2018. Phase I/II study of the A2AR antagonist NIR178 (PBF-509), an oral immunotherapy, in patients (pts) with advanced NSCLC. J. Clin. Oncol 36:9089 [Google Scholar]
- 14.Bendell J, Bauer T, Patel M, et al. 2019. Abstract CT026: Evidence of immune activation in the first-inhuman Phase Ia dose escalation study of the adenosine 2a receptor antagonist, AZD4635, in patients with advanced solid tumors. Cancer Res. 79:CT026 [Google Scholar]
- 15.Powderly JD, de Souza PL, Gutierrez R, et al. 2019. AB928, a novel dual adenosine receptor antagonist, combined with chemotherapy or AB122 (anti-PD-1) in patients (pts) with advanced tumors: preliminary results from ongoing phase I studies. J. Clin. Oncol 37:2604 [Google Scholar]
- 16.Fong L, Powderly J II, Luke J, et al. 2018. Refractory renal cell cancer (RCC) exhibits high adenosine A2A receptor (A2AR) expression and prolonged survival following treatment with A2AR antagonist CPI-444. Paper presented at Society for Immunotherapy of Cancer (SITC) 33rd Annual Meeting, Nov. 7–11, Washington, DC [Google Scholar]
- 17.Willingham SB. 2017. Inhibition of A2AR induces anti-tumor immunity alone and in combination with anti-PD-L1 in preclinical and clinical studies. Paper presented at American Association for Cancer Research (AACR) 2017 Annual Meeting, Dec. 5–9, San Antonio, TX [Google Scholar]
- 18.Emens L, Powderly J, Fong L, et al. 2017. Abstract CT119: CPI-444, an oral adenosine A2a receptor (A2AR) antagonist, demonstrates clinical activity in patients with advanced solid tumors. Cancer Res. 77:CT119 [Google Scholar]
- 19.Fong LCM, George S, Hughes B, et al. 2020. Adenosine receptor blockade with ciforadenant + atezolizumab in advanced metastatic castration resistant prostate cancer (mCRPC). J. Clin. Oncol 38:129 [Google Scholar]
- 20.Geoghegan JC, Diedrich G, Lu X, et al. 2016. Inhibition of CD73 AMP hydrolysis by a therapeutic antibody with a dual, non-competitive mechanism of action. MAbs 8:454–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hay CM, Sult E, Huang Q,et al.2016. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology 5:e1208875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Overman MJ, LoRusso P, Strickler JH, et al. 2018. Safety, efficacy and pharmacodynamics (PD) of MEDI9447 (oleclumab) alone or in combination with durvalumab in advanced colorectal cancer (CRC) or pancreatic cancer (panc). J. Clin. Oncol 36:4123 [Google Scholar]
- 23.Siu LL, Burris H, Le DT, et al. 2018. Abstract CT180: Preliminary phase 1 profile of BMS-986179, an anti-CD73 antibody, in combination with nivolumab in patients with advanced solid tumors. Cancer Res. 78:CT180 [Google Scholar]
- 24.Luke JJ, Powderly JD, Merchan JR, et al. 2019. Immunobiology, preliminary safety, and efficacy of CPI-006, an anti-CD73 antibody with immune modulating activity, in a phase 1 trial in advanced cancers. J. Clin. Oncol 37:2505 [Google Scholar]
- 25.Ohta A, Gorelik E, Prasad SJ, et al. 2006. A2A adenosine receptor protects tumors from antitumor T cells. PNAS 103:13132–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leone RD, Sun IM, Oh MH, et al. 2018. Inhibition of the adenosine A2a receptor modulates expression of T cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and ACT in murine cancer models. Cancer Immunol. Immunother 67:1271–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mittal D, Young A, Stannard K, et al. 2014. Antimetastatic effects of blocking PD-1 and the adenosine A2A receptor. Cancer Res. 74:3652–58 [DOI] [PubMed] [Google Scholar]
- 28.Beavis PA, Divisekera U, Paget C, et al. 2013. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. PNAS 110:14711–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waickman AT, Alme A, Senaldi L, et al. 2012. Enhancement of tumor immunotherapy by deletion of the A2A adenosine receptor. Cancer Immunol. Immunother 61:917–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fong L, Hotson A, Powderly JD, et al. 2019. Adenosine 2A receptor blockade as an immunotherapy for treatment-refractory renal cell cancer. Cancer Discov. 10:40–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sitkovsky MV 2020. Lessons from the A2A adenosine receptor antagonist-enabled tumor regression and survival in patients with treatment-refractory renal cell cancer. Cancer Discov. 10:16–19 [DOI] [PubMed] [Google Scholar]
- 32.Seitz L,Jin L, Leleti M, et al. 2018. Safety, tolerability, and pharmacology of AB928, a novel dual adenosine receptor antagonist, in a randomized, phase 1 study in healthy volunteers. Investig. New Drug. 37:711–21 [DOI] [PubMed] [Google Scholar]
- 33.Morandi F, Horenstein AL, Costa F, et al. 2018. CD38: a target for immunotherapeutic approaches in multiple myeloma. Front. Immunol 9:2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feng X, Zhang L, Acharya C, et al. 2017. Targeting CD38 suppresses induction and function of T regulatory cells to mitigate immunosuppression in multiple myeloma. Clin. Cancer Res 23:4290–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hausler SF, Del Barrio IM, Diessner J, et al. 2014. Anti-CD39 and anti-CD73 antibodies A1 and 7G2 improve targeted therapy in ovarian cancer by blocking adenosine-dependent immune evasion. Am. J. Transl. Res 6:129–39 [PMC free article] [PubMed] [Google Scholar]
- 36.Hausler SF, Montalban del Barrio I, Strohschein J, et al. 2011. Ectonucleotidases CD39 and CD73 on OvCA cells are potent adenosine-generating enzymes responsible for adenosine receptor 2A-dependent suppression of T cell function and NK cell cytotoxicity. Cancer Immunol. Immunother 60:1405–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saif MW, Alexander D, Wicox CM. 2005. Serum alkaline phosphatase level as a prognostic tool in colorectal cancer: a study of 105 patients. J. Appl. Res 5:88–95 [PMC free article] [PubMed] [Google Scholar]
- 38.Ren HY, Sun LL, Li HY, Ye ZM. 2015. Prognostic significance of serum alkaline phosphatase level in osteosarcoma: a meta-analysis of published data. Biomed. Res. Int 2015:160835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rao SR, Snaith AE, Marino D, et al.2017. Tumour-derived alkaline phosphatase regulates tumour growth, epithelial plasticity and disease-free survival in metastatic prostate cancer. Br. J. Cancer 116:227–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Graddis TJ,McMahan CJ, Tamman J, et al. 2011. Prostatic acid phosphatase expression in human tissues. Int. J. Clin. Exp. Pathol 4:295–306 [PMC free article] [PubMed] [Google Scholar]
- 41.Torres Á, Erices JI, Sanchez F, et al. 2019. Extracellular adenosine promotes cell migration/invasion of glioblastoma stem-like cells through A3 adenosine receptor activation under hypoxia. Cancer Lett. 446:112–22 [DOI] [PubMed] [Google Scholar]
- 42.Liu T-Z, Wang X, Bai Y-F, et al. 2014. The HIF-2alpha dependent induction of PAP and adenosine synthesis regulates glioblastoma stem cell function through the A2B adenosine receptor. Int. J. Biochem. Cell Biol 49:8–16 [DOI] [PubMed] [Google Scholar]
- 43.Spychala J 2000. Tumor-promoting functions of adenosine. Pharmacol. Ther 87:161–73 [DOI] [PubMed] [Google Scholar]
- 44.Young A, Ngiow SF, Barkauskas DS, et al. 2016. Co-inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell 30:391–403 [DOI] [PubMed] [Google Scholar]
- 45.Sitkovsky M, Lukashev D, Deaglio S, et al. 2008. Adenosine A2A receptor antagonists: blockade of adenosinergic effects and T regulatory cells. Br. J. Pharmacol 153(Suppl. 1):S457–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Allard D, Turcotte M, Stagg J. 2017. Targeting A2 adenosine receptors in cancer. Immunol. Cell Biol 95:333–39 [DOI] [PubMed] [Google Scholar]
- 47.Leone RD, Lo YC, Powell JD. 2015. A2aR antagonists: next generation checkpoint blockade for cancer immunotherapy. Comput. Struct. Biotechnol. J 13:265–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Addi AB, Lefort A, Hua X, et al. 2008. Modulation of murine dendritic cell function by adenine nucleotides and adenosine: involvement of the A2B receptor. Eur. J. Immunol 38:1610–20 [DOI] [PubMed] [Google Scholar]
- 49.Wilson JM, Ross WG, Agbai ON, et al. 2009. The A2B adenosine receptor impairs the maturation and immunogenicity of dendritic cells. J. Immunol 182:4616–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen S, Akdemir I, Fan J, et al. 2020. The expression of adenosine A2B receptor on antigen-presenting cells suppresses CD8+ T-cell responses and promotes tumor growth. Cancer Immunol. Res 8(8):1064–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sciaraffia E, Riccomi A, Lindstedt R, et al. 2014. Human monocytes respond to extracellular cAMP through A2A and A2B adenosine receptors. J. Leukoc. Biol 96:113–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Novitskiy SV, Ryzhov S, Zaynagetdinov R, et al. 2008. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood 112:1822–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nemeth ZH, Lutz CS, Csoka B, et al. 2005. Adenosine augments IL-10 production by macrophages through an A2B receptor-mediated posttranscriptional mechanism. J. Immunol 175:8260–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gabrilovich DI, Bronte V, Chen SH, et al. 2007. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 67:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Talmadge JE, Gabrilovich DI. 2013.History of myeloid-derived suppressor cells. Nat. Rev. Cancer 13:739–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ryzhov S, Novitskiy SV, Goldstein AE, et al. 2011. Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b + Gr1 + cells. J. Immunol 187:6120–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sorrentino C,Miele L, Porta A, et al. 2015.Myeloid-derived suppressor cells contribute to A2B adenosine receptor-induced VEGF production and angiogenesis in a mouse melanoma model. Oncotarget 6:27478–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu M, Guo G, Huang L, et al. 2020. CD73 on cancer-associated fibroblasts enhanced by the A2B-mediated feedforward circuit enforces an immune checkpoint. Nat. Commun 11:515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Topalian SL, Drake CG, Pardoll DM. 2015. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27:450–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zappasodi R, Merghoub T, Wolchok JD. 2018. Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell 33:581–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Allard B, Pommey S, Smyth MJ, Stagg J. 2013. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin. Cancer Res 19:5626–35 [DOI] [PubMed] [Google Scholar]
- 62.Beavis PA, Milenkovski N, Henderson MA, et al. 2015. Adenosine receptor 2A blockade increases the efficacy of anti-PD-1 through enhanced antitumor T-cell responses. Cancer Immunol. Res 3:506–17 [DOI] [PubMed] [Google Scholar]
- 63.Mittal D, Sinha D, Barkauskas D, et al. 2016. Adenosine 2B receptor expression on cancer cells promotes metastasis. Cancer Res. 76:4372–82 [DOI] [PubMed] [Google Scholar]
- 64.Beavis PA, Henderson MA, Giuffrida L, et al. 2017. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J. Clin. Investig 127:929–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jin D, Fan J, Wang L, et al. 2010. CD73 on tumor cells impairs antitumor T-cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res. 70:2245–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang L, Fan J, Thompson LF, et al. 2011. CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J. Clin. Investig 121:2371–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cekic C, Linden J. 2014. Adenosine A2A receptors intrinsically regulate CD8+ T cells in the tumor microenvironment. Cancer Res. 74:7239–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kjaergaard J, Hatfield S, Jones G, et al. 2018. A2A adenosine receptor gene deletion or synthetic A2A antagonist liberate tumor-reactive CD8+ T cells from tumor-induced immunosuppression. J. Immunol 201:782–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thompson EA, Ols S,Miura K, et al. 2018.TLR-adjuvanted nanoparticle vaccines differentially influence the quality and longevity of responses to malaria antigen Pfs25. JCI Insight 3:e120692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lynn GM, Sedlik C, Baharom F, et al. 2020. Peptide-TLR-7/8a conjugate vaccines chemically programmed for nanoparticle self-assembly enhance CD8 T-cell immunity to tumor antigens. Nat. Biotechnol 38:320–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moyer TJ, Zmolek AC, Irvine DJ. 2016. Beyond antigens and adjuvants: formulating future vaccines. J. Clin. Investig 126:799–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dahan R, Ravetch JV 2016. Co-targeting of adenosine signaling pathways for immunotherapy: potentiation by Fc receptor engagement. Cancer Cell 30:369–71 [DOI] [PubMed] [Google Scholar]
- 73.Dahan R, Barnhart BC, Li F, et al. 2016. Therapeutic activity of agonistic, human anti-CD40 monoclonal antibodies requires selective FcγR engagement. Cancer Cell 29:820–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Graziano RF, Engelhardt JJ. 2019. Role of FcγRs in antibody-based cancer therapy. Curr. Top. Microbiol. Immunol 423:13–34 [DOI] [PubMed] [Google Scholar]
- 75.Moser GH, Schrader J, Deussen A. 1989. Turnover of adenosine in plasma of human and dog blood. Am. J. Physiol 256:C799–806 [DOI] [PubMed] [Google Scholar]
- 76.DiRenzo D, Ashok D, Anderson AE, et al. 2019. Abstract 3168: Methods for assessment of the “adenosine fingerprint” in clinical trials of AB928. Cancer Res. 79:3168 [Google Scholar]