

Abstract
Background
There is a lack of mechanism-driven, clinically relevant biomarkers in chronic obstructive pulmonary disease (COPD). Mitochondrial dysfunction, a proposed disease mechanism in COPD, is associated with the release of mitochondrial DNA (mtDNA), but plasma cell-free mtDNA has not been previously examined prospectively for associations with clinical COPD measures.
Methods
P-mtDNA, defined as copy number of mitochondrially-encoded NADH dehydrogenase-1 (MT-ND1) gene, was measured by real-time quantitative PCR in 700 plasma samples from participants enrolled in the Subpopulations and Intermediate Outcome Measures in COPD Study (SPIROMICS) cohort. Associations between p-mtDNA and clinical disease parameters were examined, adjusting for age, sex, smoking status, and for informative loss to follow-up.
Results
P-mtDNA levels were higher in participants with mild or moderate COPD, compared to smokers without airflow obstruction, and to participants with severe COPD. Baseline increased p-mtDNA levels were associated with better CAT scores in female smokers without airflow obstruction and female participants with mild or moderate COPD on 1-year follow-up, but worse 6MWD in females with severe COPD. Higher p-mtDNA levels were associated with better 6MWD in male participants with severe COPD. These associations were no longer significant after adjusting for informative loss to follow-up.
Conclusion
In this study, p-mtDNA levels associated with baseline COPD status but not future changes in clinical COPD measures after accounting for informative loss to follow-up. To better characterize mitochondrial dysfunction as a potential COPD endotype, these results should be confirmed and validated in future studies.
Trial Registration: ClinicalTrials.gov NCT01969344 (SPIROMICS)
Supplementary Information
The online version contains supplementary material available at 10.1186/s12931-021-01707-x.
Keywords: COPD, Mitochondrial dysfunction, mtDNA, SPIROMICS
Introduction
Chronic obstructive pulmonary disease (COPD) is a chronic lung disease that is defined by airflow limitation on spirometry and persistent respiratory symptoms and is a leading cause of morbidity and mortality worldwide [1, 2]. This devastating impact of COPD stems from both an underdiagnosis in the community [3] and to an underlying heterogeneity of the disease, with the latter both in terms of clinical phenotypes as well as disease progression [4]. Given the relative lack of biomarkers that could predict important clinical events or discriminate between patients with different disease trajectories [5, 6], there has been a more recent call for endotype-driven research, to focus on pathobiological mechanisms for patient monitoring and treatment [7].
Among the relevant proposed mechanisms is mitochondrial dysfunction, with supporting evidence from both COPD patients [8–14] and experimental COPD models [15–18]. This dysfunction, partly attributed to mitochondrial oxidative stress [12] and manifested differently in different cell types, is often accompanied by the release of mitochondrial DNA (mtDNA) into the cytosol and extracellular space [19]. Intracellular mtDNA acts as a critical second messenger that activates the NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome and the cyclic GMP-AMP synthase (cGAS)-Stimulator of Interferon Genes (STING) system, which triggers a profound type I interferon innate immune response [20, 21]. Although less is known about extracellular mtDNA, circulating extracellular plasma mtDNA (p-mtDNA) has been associated with mortality in studies of intensive care unit populations, and there is some evidence to suggest that extracellular mtDNA release may be critical for the pathogenesis of some inflammatory diseases [22, 23]. MtDNA has been previously examined in a number of extracellular fluid compartments in COPD, including blood, urine, and exhaled breath condensate; however, these studies have shown both increased and decreased mtDNA levels [24, 25], leaving the role of this biomarker in COPD unclear [26–31]. Additionally, to our knowledge, p-mtDNA has not been evaluated for associations with other clinical measures of COPD severity or has been utilized to monitor disease progression in a large prospective COPD cohort.
Based on the hypothesis that extracellular mtDNA is reflective of mitochondrial dysfunction in subjects with COPD, we sought to determine if circulating p-mtDNA levels associate with clinical measures of COPD severity, using the Subpopulations and Intermediate Outcome Measures in COPD Study (SPIROMICS) cohort. Furthermore, given that SPIROMICS is an ongoing, longitudinal cohort, we examined whether baseline p-mtDNA levels can forecast disease progression, as represented by lung function and exercise tolerance decline and worsening of respiratory symptoms. Here we show that p-mtDNA levels are lower in individuals with severe COPD when compared to those with mild/moderate disease and are associated with subsequent changes in respiratory symptom burden and exercise in female participants. These findings may inform us about the trajectory of COPD from mild to severe disease, especially in females, and provides further supportive evidence for a role for mitochondrial dysfunction in COPD pathobiology.
Methods
Study design and sample collection
SPIROMICS (ClinicalTrials.gov NCT01969344) is an on-going longitudinal, prospective, multicenter observational study that recruited never smokers (≤ 1 pack-year of tobacco-smoking history), current or former smokers (ever-smokers, ≥ 20 pack-years) without airflow obstruction, and ever-smokers with airflow obstruction [32]. The individual institutional review boards (IRBs) of all participating clinical centers approved all study protocols, and all participants provided written informed consent. Clinical data were collected at the baseline and at follow-up study visits, including demographics, comorbidities, questionnaires, cigarette smoke exposure, spirometry, and 6-min walk distance (6MWD) [32]. Symptom burden was quantified with the COPD Assessment Test (CAT) and health-related quality of life (HRQL) with the St. George’s Respiratory Questionnaire (SGRQ) total score [33, 34]. The extent of emphysema was characterized using CT scans of the lung using Imbio diagnostics software, with percent emphysema defined as low attenuation area (LAA) less than − 950 Hounsfield Units at total lung capacity [35].
Sample processing and quantification of P-mtDNA
Total DNA was isolated from plasma as described previously [20]. Briefly, 50 µL of plasma was diluted with 170 µL of PBS, and then centrifuged at 700×g at 4 °C for 5 min to remove cells and cellular debris. The obtained supernatant was further centrifuged at 18,000×g at 4 °C for 15 min, and the resulting supernatant (170 µl) was carefully saved. DNA was isolated from plasma using the DNeasy Blood and Tissue Kit (#69504; Qiagen). 200 µL of the provided elution buffer was used to collect the DNA.
Prior to quantification, the DNA solution was diluted 1:5 with nuclease-free deionized, distilled water. MtDNA levels were measured in triplicate by SYBR Green dye-based qPCR assay using a PRISM 7500 sequence detection system (Applied Biosystems), using the following primer sequences for human NADH dehydrogenase 1 gene: forward 5′-ATACCCATGGCCAACCTC-3′, reverse 5′-GGGCCTTTGCGTAGTTGTAT-3′ [20]. The thermal profile was as follows: 2 min at 50 °C, 10 min at 95 °C, 40 cycles for 15 s at 95 °C, and 1 min at 60 °C. For absolute quantitation of mtDNA, a standard curve was generated from DNA plasmid constructs in serial dilutions (ORIGENE #SC101172 and GenScript NM_173708).
Statistical analysis
Clinical characteristics of SPIROMICS participants for whom p-mtDNA were measured were compared to those of all SPIROMICS participants summarized using medians and interquartile intervals or counts and percentages as appropriate. P-mtDNA was analyzed on the log2 scale to account for right-skewed distributions. Associations between p-mtDNA and baseline characteristics were first computed using unadjusted Pearson’s correlation coefficients for continuous baseline characteristics and ANOVA with Tukey’s post hoc adjustments for multiple comparisons for categorical baseline characteristics. We then examined whether p-mtDNA measured at baseline correlated with post-bronchodilator FEV1% predicted, 6MWD, SGRQ, CAT, and % emphysema measured at baseline using Pearson’s correlation coefficients and Pearson’s partial correlation coefficients holding age, sex, and current smoking status constant. Based on findings from our previous work which showed associations with urine mtDNA that differed among sex and COPD subgroups, we, where appropriate, stratified our p-mtDNA analyses by these same subgroups to determine if similar differential associations existed [30]. We next tested whether p-mtDNA measured at baseline correlated with changes between baseline and 1- and 3-year follow-up visit measurements of FEV1% predicted, 6MWD, SGRQ, and CAT, again using Pearson’s unadjusted and partial correlations, overall and within COPD and sex subgroups. These correlations with changes in COPD measures over time were further adjusted for informative loss to follow-up using Targeted Maximum Likelihood Estimation (TMLE) [36]. The mechanisms for loss to follow-up were explored by comparing baseline characteristics of patients who did and did not have 1- and 3-year follow up measurements using medians and interquartile intervals or counts and percentages as appropriate. The effect sizes of all estimates are reported as a correlation coefficient between -1 and 1 with a corresponding 95% confidence interval, representing the direction and strength of association between p-mtDNA and the clinical measures. Correlations reported as corrected for multiple comparisons were adjusted using the Benjamini and Hochberg False Discovery Rate (FDR) correction [37]. In a final analysis, we tested the association between baseline p-mtDNA and mortality using Cox Proportional Hazard models with robust standard errors, adjusting for age, sex, and smoking status. Analysis was conducted in R [38] and figures were produced using the package ggplot2 [39].
Results
Study population characteristics
A flow diagram for this study and the characteristics of the study participants for whom p-mtDNA levels were measured are shown in Fig. 1 and Table 1, respectively. Although samples were randomly selected, compared to the overall SPIROMICS population, the p-mtDNA study population was over-represented by non-smokers and participants with severe COPD, and under-represented in participants with mild or moderate COPD (p < 0.001, Additional file 1: Table 1); the study population was also less likely to be currently smoking compared to the overall SPIROMICS cohort (p = 0.002). Despite this difference, the sampled p-mtDNA population is otherwise representative of the overall SPIROMICS cohort in other clinical measures of COPD severity, including mean FEV1% predicted, 6MWD, SGRQ, CAT (Additional file 1: Table 1). As samples for p-mtDNA measurement were selected in 2018, these study participants have more complete long-term follow-up data compared to the overall SPIROMICS cohort (Additional file 1: Table 1).
Fig. 1.

Flow diagram for the participants in the SPIROMICS Plasma mtDNA study. COPD, chronic obstructive pulmonary disease
Table 1.
Baseline characteristics
| Parameter | Non-smokers | Smokers without airflow obstruction | Mild/Mod COPD | Severe COPD | P-value* |
|---|---|---|---|---|---|
| (n = 100) | (n = 199) | (n = 201) | (n = 200) | ||
| Age [IQI] | 56 [50, 65] | 61 [52, 67] | 66 [ 60, 71] | 65 [59, 71] | < 0.001 |
| Sex N (%) | |||||
| Male | 42 (42%) | 96 (48%) | 130 (65%) | 122 (61%) | < 0.001 |
| Race N (%) | < 0.001 | ||||
| Asian | 3 (3.1%) | 3 (1.5%) | 5 (2.5%) | 2 (1.0%) | |
| Black | 26 (27%) | 52 (27%) | 23 (12%) | 29 (15%) | |
| Other | 4 (4.1) | 8 (4.1%) | 4 (2.0%) | 4 (2.0%) | |
| White | 65 (66%) | 133 (68%) | 168 (84%) | 163 (82%) | |
| Current smoking N (%) | 0 (0%) | 98 (48%) | 77 (39%) | 40 (20%) | < 0.001 |
| FEV1% predicted [IQI] | 102 [95, 109] | 97 [86, 106] | 71 [61, 80] | 34 [27, 43] | < 0.001 |
| 6MWD [IQI] | 465 [392, 516] | 450 [390, 503] | 429 [366, 484] | 315 [249, 396] | < 0.001 |
| CAT [IQI] | 3 [1, 8] | 9 [5, 16] | 13 [8, 20] | 19 [13, 24] | < 0.001 |
| SGRQ [IQI] | 5 [4, 11] | 17 [8, 38] | 28 [16, 45] | 47 [39, 60] | < 0.001 |
IQI interquartile interval, FEV1 forced expiratory volume in 1 s, 6MWD six-minute walk distance, SGRQ St. George’s Respiratory Questionnaire, CAT COPD Assessment Test
*Kruskal–Wallis test, Chi-square test, or Fisher’s exact test comparing participants within each subgroup, as appropriate
P-mtDNA levels are lower in severe COPD compared to mild or moderate COPD
We first examined the relationship between baseline p-mtDNA and characterization of COPD severity, with mild/moderate COPD defined as FEV1% predicted greater than 50%, and severe COPD defined as less than 50% [32]. There were no significant differences in p-mtDNA levels between non-smokers and ever-smokers without COPD nor between non-smokers and participants with COPD (mild or severe) (Fig. 2). In unadjusted analyses, participants with mild or moderate COPD had higher p-mtDNA levels compared to ever-smokers without COPD (p = 0.029, Fig. 2), an association that was no longer significant after adjustment for age, sex, and current smoking status (Additional file 1: Table 2). Notably, participants with severe COPD had lower p-mtDNA levels compared with those with mild or moderate COPD (p < 0.001, Fig. 2), a finding which remained significant after adjustment and correction for multiple comparisons testing (Additional file 1: Table 2). These relationships were seen in both males-only and females-only subgroup analyses; p-mtDNA levels did not differ significantly between males and females overall (Additional file 1: Fig. 1A, B). P-mtDNA levels did not differ by smoking status (Additional file 1: Fig. 2). Associations between p-mtDNA levels and clinical COPD measures, such as FEV1% predicted, 6MWD, CAT and SGRQ scores, and extent of radiographic emphysema were examined, but did not reveal any statistically significant relationships (Additional file 1: Table 3). P-mtDNA levels were not associated with urine mtDNA levels in matched study participants (Additional file 1: Fig. 3).
Fig. 2.
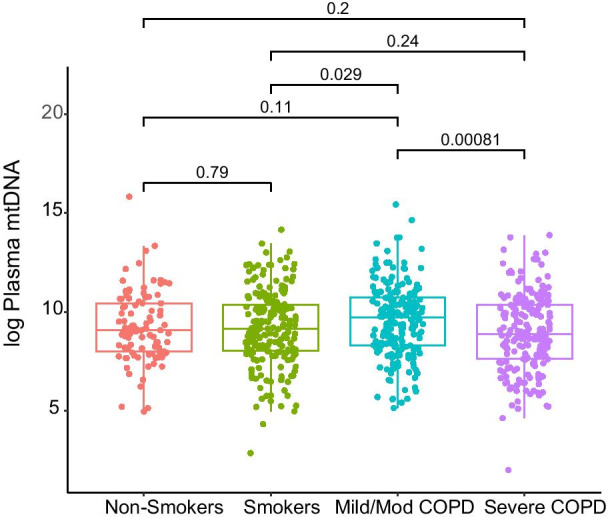
Plasma mtDNA levels were higher in subjects with mild/moderate COPD but falls in severe disease. P-mtDNA was measured in SPIROMICS participants, including never smokers (n = 100; red), smokers without airflow obstruction (n = 199; green), participants with mild/moderate COPD (n = 201; blue), and severe COPD (n = 200; purple). Data are presented as median with box indicating upper and lower quartiles, whiskers indicating extrema, and with p-values calculated by Tukey’s range test
Higher baseline p-mtDNA levels are associated with slower symptom burden progression in female smokers and female participants with COPD
SPIROMICS is an ongoing, prospective COPD cohort, and has extensive longitudinal follow-up data of its study participants. We utilized these follow-up time points to assess if baseline p-mtDNA levels associated with changes in clinical COPD measures between the baseline and one year after baseline visit (700/700 eligible for follow-up). Overall, we did not find any significant correlations between p-mtDNA with changes in clinical COPD measures at one year (Table 2). In subgroup analyses, we found that higher baseline p-mtDNA associated with improved (lower) CAT scores in females (r = − 0.15, CI [− 0.28, − 0.02], p = 0.02), especially in female smokers without airflow obstruction (r = − 0.28, CI [− 0.50, − 0.06], p = 0.01) and females with mild or moderate COPD (r = − 0.29, CI [− 0.54, − 0.03], p = 0.01); these associations remained significant after adjustment for age and smoking status (Table 2). However, higher baseline p-mtDNA was also associated with a decline in 6MWD in female participants with severe COPD (r = − 0.33, CI [− 0.61, − 0.05], p = 0.02) in unadjusted and adjusted analyses, a relationship that was not observed in female smokers or females with mild/moderate disease (Table 2). In males, higher baseline p-mtDNA did not associate with CAT but associated with better (longer) 6MWD in male participants with severe COPD (r = 0.27, CI [0.06, 0.48], p = 0.02), an association that remained significant after adjustment for age and smoking status (Table 2).
Table 2.
P-mtDNA and disease progression at 1-year follow-up
| Unadjusted | Adjusted for age & smoking status | |
|---|---|---|
| (CI)* | (CI)* | |
| FEV1% predicted | ||
| Alla | 0 (− 0.08, 0.07) | − 0.01 (− 0.09, 0.06) |
| Males | − 0.01 (− 0.1, 0.09) | − 0.01 (− 0.12, 0.09) |
| Male smokers | − 0.08 (− 0.25, 0.1) | − 0.08 (− 0.27, 0.1) |
| Male mild/mod | − 0.03 (− 0.23, 0.17) | − 0.05 (− 0.26, 0.15) |
| Male severe | 0.15 (− 0.07, 0.36) | 0.14 (− 0.08, 0.35) |
| Females | 0 (− 0.11, 0.11) | − 0.01 (− 0.12, 0.1) |
| Female smokers | 0.04 (− 0.16, 0.23) | 0.04 (− 0.16, 0.23) |
| Female mild/mod | − 0.09 (− 0.32, 0.15) | − 0.11 (− 0.35, 0.13) |
| Female severe | 0.10 (− 0.16, 0.36) | 0.08 (− 0.18, 0.34) |
| 6MWD (meters) | ||
| Alla | − 0.02 (− 0.1, 0.06) | − 0.01 (− 0.09, 0.07) |
| Males | 0.03 (− 0.07, 0.14) | 0.04 (− 0.07, 0.15) |
| Male smokers | − 0.01 (− 0.19, 0.16) | − 0.01 (− 0.18, 0.16) |
| Male mild/mod | − 0.06 (− 0.28, 0.16) | − 0.05 (− 0.27, 0.16) |
| Male severe | 0.27 (0.06, 0.48)b | 0.27 (0.05, 0.48) |
| Females | − 0.09 (− 0.21, 0.03) | − 0.09 (− 0.21, 0.04) |
| Female smokers | 0.03 (− 0.17, 0.22) | 0.03 (− 0.17, 0.22) |
| Female mild/mod | 0.08 (− 0.16, 0.32) | 0.08 (− 0.16, 0.32) |
| Female severe | − 0.33 (− 0.61, − 0.05)c | − 0.30 (− 0.59, − 0.02) |
| SGRQ | ||
| Alla | − 0.02 (− 0.1, 0.07) | − 0.03 (− 0.12, 0.06) |
| Males | − 0.02 (− 0.13, 0.09) | − 0.04 (− 0.16, 0.08) |
| Male smokers | − 0.07 (− 0.27, 0.13) | − 0.1 (− 0.3, 0.1) |
| Male mild/mod | 0 (− 0.2, 0.2) | 0 (− 0.2, 0.2) |
| Male severe | − 0.14 (− 0.39, 0.1) | − 0.16 (− 0.41, 0.08) |
| Females | − 0.02 (− 0.14, 0.11) | − 0.01 (− 0.14, 0.11) |
| Female smokers | − 0.15 (− 0.38, 0.08) | − 0.15 (− 0.38, 0.08) |
| Female mild/mod | 0.08 (− 0.2, 0.35) | 0.10 (− 0.18, 0.37) |
| Female severe | − 0.02 (− 0.29, 0.25) | − 0.10 (− 0.39, 0.18) |
| CAT | ||
| Alla | − 0.07 (− 0.15, 0.01) | − 0.07 (− 0.15, 0.02) |
| Males | − 0.01 (− 0.11, 0.09) | − 0.01 (− 0.11, 0.1) |
| Male smokers | 0.01 (− 0.16, 0.19) | 0.04 (− 0.14, 0.21) |
| Male mild/mod | − 0.08 (− 0.29, 0.13) | − 0.08 (− 0.29, 0.12) |
| Male severe | − 0.05 (− 0.26, 0.17) | − 0.05 (− 0.27, 0.17) |
| Females | − 0.15 (− 0.28, − 0.02) | − 0.14 (− 0.27, − 0.01) |
| Female smokers | − 0.28 (− 0.5, − 0.06) | − 0.30 (− 0.52, − 0.08) |
| Female mild/mod | − 0.29 (− 0.54, − 0.03) | − 0.27 (− 0.52, − 0.01) |
| Female severe | 0.05 (− 0.21, 0.31) | 0.03 (− 0.22, 0.29) |
r correlation coefficient, CI 95% confidence interval, FEV1 forced expiratory volume in 1 s, 6MWD six-minute walk distance, SGRQ St. George’s Respiratory Questionnaire, CAT COPD Assessment Test
*Pearson’s correlation coefficients with log2 p-mtDNA as among all participants and within each group
aAdjusting for age, sex, and smoking status
bItalic—statistically significant with positive correlation coefficient
cBold—statistically significant with negative correlation coefficient
At 3-year follow-up (566/700 eligible for follow-up), there was significant loss of study participants, and many of the associations observed at 1-year were no longer present. There were no statistically significant associations between p-mtDNA and the examined COPD outcomes, overall or in subgroups stratified by sex (Table 3). Additionally, we examined for associations between p-mtDNA and COPD exacerbation risk and did not find any significant relationships for either moderate (defined as exacerbations requiring treatment with systemic corticosteroids, antibiotics, or both) or severe (defined as exacerbations requiring emergency room visit or hospitalization) COPD exacerbations (Additional file 1: Fig. 4A, B). P-mtDNA levels did not differ in study participants who died during this follow-up period compared to those who did not, in unadjusted analysis or in the Cox proportional hazards model, adjusting for age, sex, and COPD status (Additional file 1: Fig. 5, Table 4).
Table 3.
P-mtDNA and disease progression at 3-year follow-up
| Unadjusted | Adjusted for age & smoking status | |
|---|---|---|
| (CI)* | (CI)* | |
| FEV1% predicted | ||
| Alla | 0.04 (− 0.05, 0.14) | 0.04 (− 0.06, 0.14) |
| Males | 0.08 (− 0.05, 0.22) | 0.09 (− 0.05, 0.23) |
| Females | 0 (− 0.14, 0.13) | − 0.01 (− 0.15, 0.13) |
| 6MWD (meters) | ||
| All | − 0.04 (− 0.14, 0.07) | − 0.02 (− 0.12, 0.08) |
| Males | 0 (− 0.13, 0.13) | 0.03 (− 0.11, 0.17) |
| Females | − 0.08 (− 0.23, 0.07) | − 0.07 (− 0.23, 0.08) |
| SGRQ | ||
| All | − 0.05 (− 0.16, 0.06) | − 0.06 (− 0.18, 0.05) |
| Males | − 0.05 (− 0.19, 0.10) | − 0.07 (− 0.22, 0.08) |
| Females | − 0.06 (− 0.22, 0.10) | − 0.06 (− 0.23, 0.10) |
| CAT | ||
| All | 0.01 (− 0.09, 0.11) | 0 (− 0.09, 0.10) |
| Males | 0.01 (− 0.12, 0.13) | − 0.01 (− 0.15, 0.12) |
| Females | 0.02 (− 0.13, 0.16) | 0.02 (− 0.12, 0.17) |
aAdjusting for age, sex, and smoking status
r correlation coefficient, CI 95% confidence interval, FEV1 forced expiratory volume in 1 s, 6MWD six-minute walk distance, SGRQ St. George’s Respiratory Questionnaire, CAT COPD Assessment Test
*Pearson’s correlation coefficients with log2 p-mtDNA as among all participants and within each group
P-mtDNA is not associated with future changes in clinical COPD measures after adjusting for informative loss to follow-up
All longitudinal prospective cohort studies are prone to subject drop-out and non-response, and while the reason for dropping out can be either practical or biological, if missing data occur in a selective, non-random manner, the “missingness” can be informative and significantly impact data analysis and interpretation. When comparing the characteristics of participants with complete follow-up data with those missing data, we discovered that participants with missing follow-up FEV1% predicted at 1-year follow-up have more severe disease, by baseline FEV1% predicted (79% vs. 45%, p < 0.001, Additional file 1: Table 5) or by a number of other clinical measures, including 6MWD, CAT, and SGRQ (p < 0.001 in all comparisons, Additional file 1: Table 5). Similar differences were observed comparing participants with complete follow-up with those with missing FEV1% predicted at 3-year follow-up (Additional file 1: Table 6). This was consistently seen if we evaluated missingness by other variables, such as absent 6MWD, CAT, or SGRQ values at 1 or 3 years (not shown). A fraction of the loss to follow-up can be attributed to deaths, but death as an outcome represents only a minority of the those with missing follow-up data (Additional file 1: Tables 5–6). When we adjusted our analyses to account for missingness, the statistically significant associations observed at 1-year follow-up were no longer statistically significant (Additional file 1: Tables 7, 8). A simulated example showing the true data and the data observed (a subset dependent on severity of COPD) was generated for illustration (Fig. 3). The relevance of adjusting for missing data is apparent: if p-mtDNA is associated with COPD severity, and participants with more severe disease are more likely to be lost to follow-up, then analyzing only complete cases without adjusting for loss to follow-up would result in a biased conclusion.
Fig. 3.

Illustrative example of informative missingness. A simulated correlation between p-mtDNA levels and FEV1% predicted in the true population (a) in the hypothetical, ideal situation where all participants had complete follow-up (b) in the hypothetical, more likely situation where patients with worse FEV1% were also less likely to have complete follow-up. Black dots represent complete cases, red dots indicate cases lost to follow-up
Discussion
In this large study of 700 participants from the SPIROMICS cohort, we tested the hypothesis that p-mtDNA, a surrogate for mitochondrial dysfunction, would associate with COPD severity and progression. To the best of our knowledge, this is the largest COPD population for which p-mtDNA has been evaluated. Here, we found that p-mtDNA levels were higher in participants with mild or moderate COPD compared to smokers without airflow obstruction, and to those with severe COPD. These findings are interesting and potentially supportive of a role for mitochondrial dysfunction in the transition from mild to severe disease in COPD.
The observation that p-mtDNA levels were lower in those with severe COPD when compared to individuals with mild/moderate disease has yet to be demonstrated in previous studies but is consistent with findings which show decreased mtDNA levels in the tissues of patients with severe COPD [24, 25]. P-mtDNA levels also trended higher in mild/moderate participants when compared to healthy smokers, suggesting a possible biphasic release of mtDNA that peaks in individuals with early disease but declines upon progression to severe disease. MtDNA is well recognized as a danger-associated molecular pattern (DAMP) that is an important component of the innate immune response [40], and is released by lung epithelial cells in response to cigarette smoke extract, likely as a result of mitochondrial stress [28, 29]. In vivo, short term smoke exposure increases bronchoalveolar lavage fluid mtDNA levels, and the number of abnormal mitochondria inside the lung epithelium increases with the length of smoke exposure [15, 28, 29]. The effect of prolonged cigarette smoke exposure on mtDNA levels has not been experimentally modeled, but perhaps with continued mitochondrial stress, and consequently, mtDNA release, mitochondrial exhaustion may occur. This phenomenon has been suggested in a number of diseases of dysregulated immunity, such as sepsis [41], as well as in diseases of accelerated aging, of which COPD is considered to be a prime example [42, 43]. Despite the lack of differences in p-mtDNA levels between healthy controls and individuals with mild/moderate or severe COPD, our findings may inform us as to the dynamic nature of p-mtDNA release in COPD disease trajectory whereby active p-mtDNA release plays a role in the course of the disease from healthy smokers to individuals with mild COPD, followed by a decline in p-mtDNA release that can be attributed to mitochondrial exhaustion in severe COPD.
Despite the above observations, we otherwise found no significant associations between p-mtDNA levels and baseline clinical measures of COPD and only minor associations between p-mtDNA and clinical correlates (CAT, 6MWD) in subgroup analyses at the one-year follow-up visit. We have previously demonstrated in a subset of this same cohort that higher urine mtDNA was associated with respiratory symptom burden and worse exercise tolerance, particularly in smokers without COPD and in women [30]. In this study, some of the statistically significant associations between p-mtDNA and 6MWD were similar to what we observed in our study of urine mtDNA, such as the association between higher baseline p-mtDNA levels and a lower 6MWD in women at 1 year [30]. Clinically, women with COPD report more dyspnea than men, and develop earlier COPD onset with more rapid lung function loss despite overall lower smoke exposure burden [44, 45]. In light of these observations, it is perhaps not coincidental that mtDNA is maternally inherited, which imparts a genetic risk and potentially provides some biological rationale for the sex-dependent associations we have found between mtDNA, CAT and 6MWD in this study.
One major strength of this current study is the assessment between p-mtDNA and longitudinal changes in COPD taking into consideration informative missingness in our analysis. This is important because significant changes in longitudinal measures may be missed or incorrect if only study participants with complete follow-up data are examined. We verified in our cohort that severity of illness was related to the likelihood of observing follow-up measures in our p-mtDNA cohort, as participants with worse baseline measures were significantly less likely to have subsequent 1- or 3-year COPD measurements recorded. We accounted for this in our correlations with TMLE by modeling the probability of observing the components of our longitudinal correlations (p-mtDNA measurements and the difference in COPD outcomes from baseline to follow-up) given the other baseline measures of severity of illness, before computing the correlation coefficient. Although some of our findings were no longer statistically significant after this process, our statistical adjustment for an informative missingness mechanism ensures that we are not overlooking important correlations or drawing biased conclusions in the relationship between p-mtDNA and COPD progression. In addition to modeling informative missingness, we chose our analytical method carefully, opting to examine differences between COPD measures at baseline and a single subsequent time point rather than a pooled model with more time points. Our modeling strategy was in response to our initial questions of interest, of whether p-mtDNA might associate with a change in COPD symptoms over two potential time frames: one relatively immediate (1-year follow-up) and one longer (3-year follow-up). Although there were data available to potentially examine the relationship between baseline p-mtDNA and COPD progression trajectories in more detail, given the several near-zero correlations in our current progression analyses, it is unlikely that a model pooling time points would add relevant new information.
There are still many other important unknowns regarding mtDNA and COPD, including the source of extracellular p-mtDNA. Although cigarette smoke is introduced in the airway, the multimorbidity of COPD suggests that the effect of cigarette smoke is systemic, and thus mtDNA could be released as a result of mitochondrial stress in epithelial cells [13, 29], circulating immune cells [8, 10], or endothelial cells [46] in the lung or elsewhere, or possibly a combination of these sources; the fact that mtDNA was detected in the urine of smokers and COPD patients is provocative supportive evidence for an extrapulmonary source of mtDNA [30]. Notably, urine mtDNA and p-mtDNA available on the same patients did not linearly associate in this study, indicating that the presence of mtDNA in urine is not simply a result of circulating p-mtDNA filtered by the kidney. We also did not find similar sex differences in p-mtDNA levels which we observed with females exhibiting higher urine mtDNA across the subgroups in the SPIROMICS cohort [30]. Thus, p-mtDNA and urine mtDNA may come from entirely different sources and represent distinct biological phenomena driving the results of these two studies.
Our study has several limitations. The smoking status was determined by self-report at baseline, corresponding to the baseline blood sample from which p-mtDNA levels were measured, and it is possible that a study participant could have stopped smoking at a future study visit, when additional clinical measurements were taken. In addition, we did not examine clinical parameters that may be more reflective of cachexia or sarcopenia (e.g. grip strength), phenotypes which are commonly observed in patients with severe COPD and which may be attributable to mitochondrial dysfunction [47, 48]. We performed many statistical tests in this study and recognize the potential for type I errors; we adjusted for multiple comparisons and the great majority of associations we observed were no longer statistically significant. We are also limited by the lack of a validation cohort, despite measuring p-mtDNA in a large number of SPIROMICS study participants; our findings should be examined and confirmed in future studies and in other COPD cohorts.
Over forty years after the seminal work of Fletcher and Peto, it is clear now that beyond accelerated lung function loss in smokers, there are multiple lung function trajectories that give rise to COPD, and that the natural history of COPD is not uniform but individualized in different patients [49, 50]. One of the aims of COPD research should be the development of tools and biomarkers that reflect the underlying pathobiology but could also track individualized disease progression; this is particularly important early at disease onset [7, 51]. While some studies have examined biomarkers in association with COPD exacerbations [5], fewer have evaluated biomarkers in relation to disease progression, given the difficulty in maintaining a large longitudinal cohort [52]. This study evaluated p-mtDNA as a conceptual marker of COPD progression, and observed possible interesting differences between p-mtDNA levels in individuals with mild COPD when compared to those with severe COPD. In subgroup analyses stratifying by sex, a number of associations between p-mtDNA and symptom burden as measured by 6MWD and CAT were observed, particularly in females; however, these observations must be considered as preliminary and require further testing. Nevertheless, the findings here add to the ever-expanding literature that suggests a role for mitochondrial dysfunction in severe COPD and advocates for future investigations measuring p-mtDNA in longitudinal studies.
Supplementary Information
Additional file 1: Figure 1. P-mtDNA levels do not differ between males and females. Figure 2. P-mtDNA is not associated with smoking status. Figure 3. P-mtDNA is not associated with u-mtDNA. Figure 4. Baseline p-mtDNA levels do not differ between study participants with and without exacerbations. Figure 5. Baseline p-mtDNA levels do not differ by study participant mortality. Table 1. P-mtDNA cohort compared to overall SPIROMICS cohort. Table 2. P-mtDNA between subgroups. Table 3. P-mtDNA and clinical COPD measures. Table 4. P-mtDNA and all-cause mortality. Table 5. Baseline characteristics of participant with and without 1-year follow-up Spirometry Data. Table 6. Baseline characteristics of participant with and without 3-year follow-up Spirometry Data. Table 7. P-mtDNA and disease progression at 1-year follow-up, adjusted for missingness. Table 8. P-mtDNA and disease progression at 3-year follow-up, adjusted for missingness.
Acknowledgements
The authors thank the SPIROMICS participants and participating physicians, investigators and staff for making this research possible. More information about the study and how to access SPIROMICS data is available at www.spiromics.org. The authors would like to acknowledge the University of North Carolina at Chapel Hill BioSpecimen Processing Facility for sample processing, storage, and sample disbursements (http://bsp.web.unc.edu/). We would like to acknowledge the following current and former investigators of the SPIROMICS sites and reading centers: Neil E. Alexis, MD; Wayne H. Anderson, PhD; Mehrdad Arjomandi, MD; Igor Barjaktarevic, MD, PhD; R. Graham Barr, MD, DrPH; Patricia Basta, PhD; Lori A. Bateman, MSc; Surya P. Bhatt, MD; Eugene R. Bleecker, MD; Richard C. Boucher, MD; Russell P. Bowler, MD, PhD; Stephanie A. Christenson, MD; Alejandro P. Comellas, MD; Christopher B. Cooper, MD, PhD; David J. Couper, PhD; Gerard J. Criner, MD; Ronald G. Crystal, MD; Jeffrey L. Curtis, MD; Claire M. Doerschuk, MD; Mark T. Dransfield, MD; Brad Drummond, MD; Christine M. Freeman, PhD; Craig Galban, PhD; MeiLan K. Han, MD, MS; Nadia N. Hansel, MD, MPH; Annette T. Hastie, PhD; Eric A. Hoffman, PhD; Yvonne Huang, MD; Robert J. Kaner, MD; Richard E. Kanner, MD; Eric C. Kleerup, MD; Jerry A. Krishnan, MD, PhD; Lisa M. LaVange, PhD; Stephen C. Lazarus, MD; Fernando J. Martinez, MD, MS; Deborah A. Meyers, PhD; Wendy C. Moore, MD; John D. Newell Jr, MD; Robert Paine III, MD; Laura Paulin, MD, MHS; Stephen P. Peters, MD, PhD; Cheryl Pirozzi, MD; Nirupama Putcha, MD, MHS; Elizabeth C. Oelsner, MD, MPH; Wanda K. O’Neal, PhD; Victor E. Ortega, MD, PhD; Sanjeev Raman, MBBS, MD; Stephen I. Rennard, MD; Donald P. Tashkin, MD; J. Michael Wells, MD; Robert A. Wise, MD; and Prescott G. Woodruff, MD, MPH. The project officers from the Lung Division of the National Heart, Lung, and Blood Institute were Lisa Postow, PhD, and Lisa Viviano, BSN.
Abbreviations
- 6MWD
Six-minute walk distance
- CAT
COPD assessment test
- COPD
Chronic obstructive pulmonary disease
- FEV1
Forced expiratory volume in one second
- p-mtDNA
Plasma mitochondrial DNA
- SGRQ
St. George’s Respiratory Questionnaire
- TMLE
Targeted maximum likelihood estimation
Authors' contributions
SMC, AMKC, KN, and MEC contributed to the conception and design of the study. IB, SPP, NP, RPP, JMW, JLC, MKH, RPIII, PGW, GJC, NHH. FJM contributed to the acquisition of the plasma samples and clinical data. KTS performed the measurements. KLH, CO, ID, and KVB performed the statistical analysis. WZZ, KLH, and SMC drafted the initial manuscript. WZZ, KLH, SPP, DJC, WWL, MKH, RPIII, PGW, NHH, ID, KN, MEC, FJM, AMKC, and SMC contributed to revisions of the manuscript for critically important intellectual content. All of the authors approved this version of the manuscript to be published. WZZ and SMC are guarantors of the manuscript and take responsibility for the integrity of the data and the accuracy of the data analysis. All authors read and approved the final manuscript.
Funding
This work was supported by National Institutes of Health grants P01 HL114501 (A.M.K.C.), R01 HL132198 (A.M.K.C. and M.E.C.), R00 HL125899 (S.M.C), KL2-TR-002385 (K.N.), U01 HL128964, U01 HL137880, P01 HL114501, R01 HL122438, R01 HL136682 (F.J.M.) and T32-HL134629 and a research grant from the Stony Wold-Herbert Fund (W.Z.Z). SPIROMICS was supported by contracts from the NIH/NHLBI (HHSN268200900013C, HHSN268200900014C, HHSN268200900015C, HHSN268200900016C, HHSN268200900017C, HHSN268200900018C, HHSN268200900019C, HHSN268200900020C), grants from the NIH/NHLBI (U01 HL137880 and U24 HL141762), and supplemented by contributions made through the Foundation for the NIH and the COPD Foundation from AstraZeneca/MedImmune; Bayer; Bellerophon Therapeutics; Boehringer-Ingelheim Pharmaceuticals, Inc.; Chiesi Farmaceutici S.p.A.; Forest Research Institute, Inc.; GlaxoSmithKline; Grifols Therapeutics, Inc.; Ikaria, Inc.; Novartis Pharmaceuticals Corporation; Nycomed GmbH; ProterixBio; Regeneron Pharmaceuticals, Inc.; Sanofi; Sunovion; Takeda Pharmaceutical Company; and Theravance Biopharma and Mylan.
Availability of data and materials
Interested investigators may request access to available clinical data through processes outlined on the SPIROMICS website (https://www.spiromics.org/spiromics); additional data are available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
The individual institutional review boards (IRBs) of all participating clinical centers approved all study protocols, and all participants provided written informed consent.
Consent for publication
Not applicable.
Competing interests
IB reports grants from NHLBI, during the conduct of the study; grants and personal fees from Theravance and Mylan, grants from Amgen, personal fees from Astra Zeneca, personal fees from GSK, personal fees from Boehringer Ingelheim, personal fees from Verona Pharma, personal fees from Grifols, outside the submitted work. AMKC is a cofounder, stockholder, and serves on the Scientific Advisory Board for Proterris, which develops therapeutic uses for carbon monoxide. A.M.K.C. also has a use patent on CO. A.M.K.C. served as a consultant for Teva Pharmaceuticals in July 2018. MEC reports grants from NIH, during the conduct of the study. DJC reports grants from NHLBI, grants from COPD Foundation, during the conduct of the study. GJC reports grants and personal fees from Galaxo Smith Kline, grants and personal fees from Boehringer Ingelheim, grants and personal fees from Chiesi, grants and personal fees from Mereo, personal fees from Verona, grants and personal fees from Astra Zeneca, grants and personal fees from Pulmonx, grants and personal fees from Pneumrx, personal fees from BTG, grants and personal fees from Olympus, grants and personal fees from Broncus, personal fees from EOLO, personal fees from NGM, grants and personal fees from Lungpacer, grants from Alung, grants and personal fees from Nuvaira, grants and personal fees from ResMed, grants and personal fees from Respironics, grants from Fisher Paykel, grants and personal fees from Patara, grants from Galapgos, outside the submitted work. JLC reports a grant from NIH/NHLBI during the conduct of the study; grants from NIH/NHLBI, NIH/NIAID, the Department of Veterans Affairs, and the Department of Defense, outside the submitted work; and personal fees from AstraZeneca, Ltd. and Novartis AG, outside the submitted work. MKH reports grants from NHLBI, during the conduct of the study; personal fees from GlaxoSmithKline, personal fees from Boehringer Ingelheim, personal fees from AstraZeneca, personal fees from Merck, personal fees from Mylan, other from Sunovion, other from Novartis, outside the submitted work. NHH reports grants and personal fees from AstraZeneca, grants from Boehringer Ingelheim, grants from NIH, grants from COPD Foundation, personal fees from Mylan, outside the submitted work. WWL reports grants from National Institutes of Health, non-financial support from Pulmonx, personal fees from Konica Minolta, outside the submitted work. FJM reports personal fees and non-financial support from American College of Chest Physicians, personal fees and non-financial support from AstraZeneca, personal fees and non-financial support from Boehringer Ingelheim, non-financial support from ProterrixBio, personal fees from Columbia University, personal fees and non-financial support from ConCert, personal fees and non-financial support from Genentech, personal fees and non-financial support from GlaxoSmithKline, personal fees and non-financial support from Inova Fairfax Health System, personal fees from Integritas, personal fees from MD Magazine, personal fees from Methodist Hospital Brooklyn, personal fees and non-financial support from Miller Communicatinos, personal fees and non-financial support from National Association for Continuing Education, personal fees and non-financial support from Novartis, personal fees from New York University, personal fees and non-financial support from Pearl Pharmaceuticals, personal fees and non-financial support from PeerView Communications, personal fees and non-financial support from Prime Communications, personal fees and non-financial support from Puerto Rican Respiratory Society, personal fees and non-financial support from Chiesi, personal fees and non-financial support from Sunovion, personal fees and non-financial support from Theravance, personal fees from UpToDate, personal fees from WebMD/MedScape, personal fees from Western Connecticut Health Network, other from Afferent/Merck, non-financial support from Gilead, non-financial support from Nitto, personal fees from Patara/Respivant, personal fees from PlatformIQ, personal fees and non-financial support from Potomac, other from Biogen, personal fees and non-financial support from University of Alabama Birmingham, other from Veracyte, non-financial support from Zambon, personal fees from American Thoracic Society, grants from NIH, personal fees and non-financial support from Physicians Education Resource, personal fees from Rockpointe, other from Prometic, personal fees from Rare Disease Healthcare Communications, other from Bayer, other from Bridge Biotherapeutics, personal fees and non-financial support from Canadian Respiratory Network, other from ProMedior, personal fees and non-financial support from Teva, personal fees from France Foundation, personal fees and non-financial support from Dartmouth, outside the submitted work. RP reports grants from NHLBI, grants from COPD Foundation, during the conduct of the study; grants from US Department of Veterans Affairs, outside the submitted work. SPP reports personal fees from AstraZeneca, personal fees from Novartis, personal fees from TEVA, personal fees from Wolters Kluwer—UpToDate, personal fees from Palladian Partners, NIAID, personal fees from Syneos Health, personal fees from Syneos Health, personal fees from Genentech, personal fees from GSK, personal fees from Integrity CE, personal fees from Respiratory Medicine—Elsevier, outside the submitted work. NP reports grants from NIH, during the conduct of the study. JMW reports grants from NIH/NHLBI, during the conduct of the study; grants from NIH/NCATS, grants from NIH/NHLBI, grants from Vertex Pharmaceuticals, Inc, grants from Bayer AG, grants from ARCUS-MED, LLC, grants from Mereo BioPharma, other from AstraZeneca, other from GlaxoSmithKline, other from Boehringer Ingelheim, other from Takeda, outside the submitted work. PGW reports grants from NIH, during the conduct of the study; personal fees from Sanofi, personal fees from Regeneron, personal fees from Glenmark, personal fees from Theravance, personal fees from GSK, personal fees from NGM Pharma, personal fees from Genentech, outside the submitted work. All other authors report no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.GBD 2015 Chronic Respiratory Disease Collaborators. Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Respir Med. 2017;5(9):691–706. [DOI] [PMC free article] [PubMed]
- 2.Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. GOLD executive summary. Am J Respir Crit Care Med. 2017;195(5):557–82. [DOI] [PubMed]
- 3.Ford ES, Mannino DM, Wheaton AG, Giles WH, Presley-Cantrell L, Croft JB. Trends in the prevalence of obstructive and restrictive lung function among adults in the United States: findings from the National Health and Nutrition Examination surveys from 1988–1994 to 2007–2010. Chest. 2013;143(5):1395–1406. doi: 10.1378/chest.12-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agusti A, Hogg JC. Update on the pathogenesis of chronic obstructive pulmonary disease. N Engl J Med. 2019;381(13):1248–1256. doi: 10.1056/NEJMra1900475. [DOI] [PubMed] [Google Scholar]
- 5.Keene JD, Jacobson S, Kechris K, et al. Biomarkers predictive of exacerbations in the SPIROMICS and COPDGene Cohorts. Am J Respir Crit Care Med. 2017;195(4):473–481. doi: 10.1164/rccm.201607-1330OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stockley RA, Halpin DMG, Celli BR, Singh D. Chronic obstructive pulmonary disease biomarkers and their interpretation. Am J Respir Crit Care Med. 2019;199(10):1195–1204. doi: 10.1164/rccm.201810-1860SO. [DOI] [PubMed] [Google Scholar]
- 7.Agusti A, Celli B, Faner R. What does endotyping mean for treatment in chronic obstructive pulmonary disease? Lancet. 2017;390(10098):980–987. doi: 10.1016/S0140-6736(17)32136-0. [DOI] [PubMed] [Google Scholar]
- 8.Bialas AJ, Liberski PP, Zielinska A, et al. Morphometric analysis of mitochondria in lymphocytes of patients with exacerbations of chronic obstructive pulmonary disease—pilot study. Int J Chron Obstruct Pulmon Dis. 2018;13:2313–2318. doi: 10.2147/COPD.S163249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bewley MA, Preston JA, Mohasin M, et al. Impaired mitochondrial microbicidal responses in chronic obstructive pulmonary disease macrophages. Am J Respir Crit Care Med. 2017;196(7):845–855. doi: 10.1164/rccm.201608-1714OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belchamber KBR, Singh R, Batista CM, et al. Defective bacterial phagocytosis is associated with dysfunctional mitochondria in COPD macrophages. Eur Respir J. 2019;54(4):1802244. doi: 10.1183/13993003.02244-2018. [DOI] [PubMed] [Google Scholar]
- 11.Even B, Fayad-Kobeissi S, Gagliolo JM, et al. Heme oxygenase-1 induction attenuates senescence in chronic obstructive pulmonary disease lung fibroblasts by protecting against mitochondria dysfunction. Aging Cell. 2018;17(6):e12837. doi: 10.1111/acel.12837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wiegman CH, Michaeloudes C, Haji G, et al. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2015;136(3):769–780. doi: 10.1016/j.jaci.2015.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffmann RF, Zarrintan S, Brandenburg SM, et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir Res. 2013;14:97. doi: 10.1186/1465-9921-14-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puente-Maestu L, Perez-Parra J, Godoy R, et al. Abnormal mitochondrial function in locomotor and respiratory muscles of COPD patients. Eur Respir J. 2009;33(5):1045–1052. doi: 10.1183/09031936.00112408. [DOI] [PubMed] [Google Scholar]
- 15.Mizumura K, Cloonan SM, Nakahira K, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. 2014;124(9):3987–4003. doi: 10.1172/JCI74985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cloonan SM, Glass K, Laucho-Contreras ME, et al. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat Med. 2016;22:163–174. doi: 10.1038/nm.4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aravamudan B, Kiel A, Freeman M, et al. Cigarette smoke-induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2014;306(9):L840–854. doi: 10.1152/ajplung.00155.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aravamudan B, Thompson M, Sieck GC, Vassallo R, Pabelick CM, Prakash YS. Functional effects of cigarette smoke-induced changes in airway smooth muscle mitochondrial morphology. J Cell Physiol. 2017;232(5):1053–1068. doi: 10.1002/jcp.25508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lieber T, Jeedigunta SP, Palozzi JM, Lehmann R, Hurd TR. Mitochondrial fragmentation drives selective removal of deleterious mtDNA in the germline. Nature. 2019;570(7761):380–384. doi: 10.1038/s41586-019-1213-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12(3):222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.West AP, Khoury-Hanold W, Staron M, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520(7548):553–557. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakahira K, Kyung SY, Rogers AJ, et al. Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS Med. 2013;10(12):e1001577; discussion e1001577. doi: 10.1371/journal.pmed.1001577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J, Gupta R, Blanco LP, et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science. 2019;366(6472):1531–1536. doi: 10.1126/science.aav4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leermakers PA, Schols A, Kneppers AEM, et al. Molecular signalling towards mitochondrial breakdown is enhanced in skeletal muscle of patients with chronic obstructive pulmonary disease (COPD) Sci Rep. 2018;8(1):15007. doi: 10.1038/s41598-018-33471-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kosmider B, Lin CR, Karim L, et al. Mitochondrial dysfunction in human primary alveolar type II cells in emphysema. EBioMedicine. 2019;46:305–316. doi: 10.1016/j.ebiom.2019.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J, Wang J, Wang X, Liu Z, Ren J, Sun T. Early surgery increases mitochondrial DNA release and lung injury in a model of elderly hip fracture and chronic obstructive pulmonary disease. Exp Ther Med. 2017;14(5):4541–4546. doi: 10.3892/etm.2017.5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heijink IH, Pouwels SD, Leijendekker C, et al. Cigarette smoke-induced damage-associated molecular pattern release from necrotic neutrophils triggers proinflammatory mediator release. Am J Respir Cell Mol Biol. 2015;52(5):554–562. doi: 10.1165/rcmb.2013-0505OC. [DOI] [PubMed] [Google Scholar]
- 28.Pouwels SD, Zijlstra GJ, van der Toorn M, et al. Cigarette smoke-induced necroptosis and DAMP release trigger neutrophilic airway inflammation in mice. Am J Physiol Lung Cell Mol Physiol. 2016;310(4):L377–386. doi: 10.1152/ajplung.00174.2015. [DOI] [PubMed] [Google Scholar]
- 29.Pouwels SD, Hesse L, Faiz A, et al. Susceptibility for cigarette smoke-induced DAMP release and DAMP-induced inflammation in COPD. Am J Physiol Lung Cell Mol Physiol. 2016;311(5):L881–l892. doi: 10.1152/ajplung.00135.2016. [DOI] [PubMed] [Google Scholar]
- 30.Zhang WZ, Rice MC, Hoffman KL, et al. Association of urine mitochondrial DNA with clinical measures of COPD in the SPIROMICS cohort. JCI Insight. 2020;5(3):e133984. doi: 10.1172/jci.insight.133984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carpagnano GE, Lacedonia D, Carone M, et al. Study of mitochondrial DNA alteration in the exhaled breath condensate of patients affected by obstructive lung diseases. J Breath Res. 2016;10(2):026005. doi: 10.1088/1752-7155/10/2/026005. [DOI] [PubMed] [Google Scholar]
- 32.Couper D, LaVange LM, Han M, et al. Design of the subpopulations and intermediate outcomes in COPD Study (SPIROMICS) Thorax. 2014;69(5):491–494. doi: 10.1136/thoraxjnl-2013-203897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones PW, Quirk FH, Baveystock CM. The St George's Respiratory Questionnaire. Respir Med. 1991;85(Suppl B):25–31; discussion 33–27. doi: 10.1016/S0954-6111(06)80166-6. [DOI] [PubMed] [Google Scholar]
- 34.Jones PW, Harding G, Berry P, Wiklund I, Chen WH, Kline LN. Development and first validation of the COPD Assessment Test. Eur Respir J. 2009;34(3):648–654. doi: 10.1183/09031936.00102509. [DOI] [PubMed] [Google Scholar]
- 35.Galban CJ, Han MK, Boes JL, et al. Computed tomography-based biomarker provides unique signature for diagnosis of COPD phenotypes and disease progression. Nat Med. 2012;18(11):1711–1715. doi: 10.1038/nm.2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van der Laan MJ, Rose S. Targeted learning: causal inference for observational and experimental data. New York: Springer; 2011. [Google Scholar]
- 37.Haynes W. Benjamini–Hochberg Method. In: Encyclopedia of systems biology. New York: Springer; 2013. p. 78.
- 38.R Core Team. R: a language and environment for statistical computing. 2018; https://www.R-project.org/.
- 39.Wickham H. ggplot2: elegant graphics for data analysis. New York, NY: Springer-Verlag; 2016. [Google Scholar]
- 40.West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol. 2017;17(6):363–375. doi: 10.1038/nri.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ho J, Yu J, Wong SH, et al. Autophagy in sepsis: degradation into exhaustion? Autophagy. 2016;12(7):1073–1082. doi: 10.1080/15548627.2016.1179410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barnes PJ. Senescence in COPD and its comorbidities. Annu Rev Physiol. 2017;79:517–539. doi: 10.1146/annurev-physiol-022516-034314. [DOI] [PubMed] [Google Scholar]
- 43.Mengel-From J, Thinggaard M, Dalgard C, Kyvik KO, Christensen K, Christiansen L. Mitochondrial DNA copy number in peripheral blood cells declines with age and is associated with general health among elderly. Hum Genet. 2014;133(9):1149–1159. doi: 10.1007/s00439-014-1458-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Han MK, Postma D, Mannino DM, et al. Gender and chronic obstructive pulmonary disease: why it matters. Am J Respir Crit Care Med. 2007;176(12):1179–1184. doi: 10.1164/rccm.200704-553CC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sorheim IC, Johannessen A, Gulsvik A, Bakke PS, Silverman EK, DeMeo DL. Gender differences in COPD: are women more susceptible to smoking effects than men? Thorax. 2010;65(6):480–485. doi: 10.1136/thx.2009.122002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Polverino F, Laucho-Contreras ME, Petersen H, et al. A pilot study linking endothelial injury in lungs and kidneys in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2017;195(11):1464–1476. doi: 10.1164/rccm.201609-1765OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boengler K, Kosiol M, Mayr M, Schulz R, Rohrbach S. Mitochondria and ageing: role in heart, skeletal muscle and adipose tissue. J Cachexia Sarcopenia Muscle. 2017;8(3):349–369. doi: 10.1002/jcsm.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Migliavacca E, Tay SKH, Patel HP, et al. Mitochondrial oxidative capacity and NAD(+) biosynthesis are reduced in human sarcopenia across ethnicities. Nat Commun. 2019;10(1):5808. doi: 10.1038/s41467-019-13694-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fletcher C, Peto R. The natural history of chronic airflow obstruction. Br Med J. 1977;1(6077):1645–1648. doi: 10.1136/bmj.1.6077.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lange P, Celli B, Agusti A, et al. Lung-function trajectories leading to chronic obstructive pulmonary disease. N Engl J Med. 2015;373(2):111–122. doi: 10.1056/NEJMoa1411532. [DOI] [PubMed] [Google Scholar]
- 51.Martinez FJ, Han MK, Allinson JP, et al. At the root: defining and halting progression of early chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2018;197(12):1540–1551. doi: 10.1164/rccm.201710-2028PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vestbo J, Edwards LD, Scanlon PD, et al. Changes in forced expiratory volume in 1 second over time in COPD. N Engl J Med. 2011;365(13):1184–1192. doi: 10.1056/NEJMoa1105482. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure 1. P-mtDNA levels do not differ between males and females. Figure 2. P-mtDNA is not associated with smoking status. Figure 3. P-mtDNA is not associated with u-mtDNA. Figure 4. Baseline p-mtDNA levels do not differ between study participants with and without exacerbations. Figure 5. Baseline p-mtDNA levels do not differ by study participant mortality. Table 1. P-mtDNA cohort compared to overall SPIROMICS cohort. Table 2. P-mtDNA between subgroups. Table 3. P-mtDNA and clinical COPD measures. Table 4. P-mtDNA and all-cause mortality. Table 5. Baseline characteristics of participant with and without 1-year follow-up Spirometry Data. Table 6. Baseline characteristics of participant with and without 3-year follow-up Spirometry Data. Table 7. P-mtDNA and disease progression at 1-year follow-up, adjusted for missingness. Table 8. P-mtDNA and disease progression at 3-year follow-up, adjusted for missingness.
Data Availability Statement
Interested investigators may request access to available clinical data through processes outlined on the SPIROMICS website (https://www.spiromics.org/spiromics); additional data are available from the corresponding author on reasonable request.