

Abstract
Cephalopods are pivotal components of marine food webs, but biodiversity studies are hampered by challenges to sample these agile marine molluscs. Metabarcoding of environmental DNA (eDNA) is a potentially powerful technique to study oceanic cephalopod biodiversity and distribution but has not been applied thus far. We present a novel universal primer pair for metabarcoding cephalopods from eDNA, Ceph18S (Forward: 5′-CGC GGC GCT ACA TAT TAG AC-3′, Reverse: 5′-GCA CTT AAC CGA CCG TCG AC-3′). The primer pair targets the hypervariable region V2 of the nuclear 18S rRNA gene and amplifies a relatively short target sequence of approximately 200 bp in order to allow the amplification of degraded DNA. In silico tests on a reference database and empirical tests on DNA extracts from cephalopod tissue estimate that 44–66% of cephalopod species, corresponding to about 310–460 species, can be amplified and identified with this primer pair. A multi-marker approach with the novel Ceph18S and two previously published cephalopod mitochondrial 16S rRNA primer sets targeting the same region (Jarman et al. 2006 Mol. Ecol. Notes. 6, 268–271; Peters et al. 2015 Mar. Ecol. 36, 1428–1439) is estimated to amplify and identify 89% of all cephalopod species, of which an estimated 19% can only be identified by Ceph18S. All sequences obtained with Ceph18S were submitted to GenBank, resulting in new 18S rRNA sequences for 13 cephalopod taxa.
Keywords: metabarcoding, environmental DNA, Cephalopoda, universal primer
1. Introduction
Cephalopods, the molluscan class to which squids, octopods, cuttlefish and vampire squids belong, occur in all the world's oceans from the intertidal zone to the deep sea [1–3]. Their high protein content and large populations make them important in commercial fisheries and food–web interactions as both predator and prey [4–8]. Cephalopods are among the giants of the ocean (giant squid Architeuthis spp., colossal squid Mesonychoteuthis hamiltoni), and the highest diversity at the family level is found in the deep sea [9].
Deep-sea cephalopods have evolved specialized traits to cope with life in continuous darkness, but basic biological data are still lacking for the majority of species. One of the reasons for this paucity of information is that cephalopods are difficult to study with traditional sampling methods like net catches or video surveys [2]. The size of trawled cephalopods is biased by the used mesh and net size and specimens often get entangled and damaged in the mesh which may make it difficult to identify them morphologically [10]. Additional difficulties in sampling cephalopods result from their possibly patchy distribution and their agility which allows them to avoid or escape sampling gear. Video surveys with submersibles and towed cameras require lights that are easily detectable by the well-developed cephalopod eyes, which may result in avoidance behaviour [3]. The study of cephalopod remains in the stomachs of predators provides indirect evidence of their presence [11–13], but the digested state of cephalopod remains may hamper species identification, and the selectivity of the predators and limited knowledge of their foraging area introduces bias. The challenges associated with cephalopod sampling, particularly in remote areas such as deep pelagic environments, raise an urgent need for novel monitoring methods.
Environmental DNA (eDNA) analysis constitutes a promising tool to study the distribution and diversity of cephalopods. This technique is based on the idea that organisms leave DNA in the environment, and that this DNA can be extracted and sequenced to identify the species from which it originates [14]. PCR amplification of eDNA in a sample (e.g. filtered from water) can detect the presence of a species or of multiple taxa by targeting a variable gene with either a species-specific or universal primer set, respectively. Biodiversity assessments of an eDNA sample using a universal primer set fall under the broader term metabarcoding [15,16], i.e. parallel identification of multiple taxa from one complex DNA sample. Metabarcoding has its origins in microbiology, palaeoecology and diet analysis [17–21]. Sampling eDNA is relatively easy in the marine environment (from water or sediment) and developments in the field of next-generation sequencing have greatly reduced sequencing costs and dramatically increased sequencing output. Therefore, eDNA analysis represents a non-invasive and cost-effective method for biodiversity assessments, especially for rare or elusive species and in remote areas [14,22–25].
Metabarcoding of eDNA from seawater has mostly been used to identify fishes [26–28] and assess overall (metazoan) eukaryotic diversity [29–32], but to our knowledge has not been used to focus on specific taxonomic groups like cephalopods. Metabarcoding has only been applied to cephalopod eDNA in studies focused on larger taxonomic groups from coastal areas, e.g. eukaryotes [29], metazoans [31] and invertebrates [33]. Even though these studies did not specifically focus on cephalopod distribution, they were able to provide valuable insights on cephalopod distribution. For example, the distribution of Sepiola tridens' eDNA suggested that the species’ distribution might extend further into the coastal zones of Northern Europe than previously thought [31]. In recent years, there has been a sharp increase in the number of studies that employed eDNA analysis in the deep sea [22,34,35], but none to specifically investigate cephalopod diversity. Recently, a species-specific primer has been developed and used to detect the giant squid, Architeuthis dux, in the photic zone of the Sea of Japan [25].
Universal metabarcoding primers, i.e. a single primer set that targets multiple taxa, should have a common annealing site in all taxa and amplify a sequence with enough variation to distinguish between groups at the desired taxonomic resolution, which often is the species level [36]. Ideally, a pair of universal primers will target the largest possible taxonomic group of interest, unambiguously identify all species, while not amplifying non-target taxa. However, due to the often degraded state of eDNA, a target sequence size of less than 300 bp, sometimes referred to as a mini-barcode [37], is preferable [36]. Therefore, the primer pair that amplifies approximately 650 bp of the cytochrome-oxidase-1 (COI) [38,39], which is most commonly used for species identification, is unsuitable for eDNA metabarcoding. In practice, there is a trade-off between the taxonomic range of amplifiable species (i.e. universality), indicated by the coverage index (Bc = n. of amplified taxa/n. of target taxa), and the resolution for identification at species level, indicated by the specificity index (Bs = n. of identified taxa/n. of amplified taxa) [40]. The analysis of eDNA using multiple markers, preferably from different genes, can increase the sensitivity of the technique and overcome the specificity issues of a single pair of universal primers [29]. So far, two sets of universal primers specifically targeting cephalopods have been published, one targeting the mitochondrial 16S rDNA [41,42] and one targeting the cytochrome b region [43]. However, the latter is not suitable for eDNA metabarcoding due to the long target amplicon size. This study describes the development of a novel set of universal primers for cephalopods targeting the nuclear 18S rRNA region and describes its complementarity to the mitochondrial 16S rDNA primer set [41,42] in a multi-marker approach to study the diversity and distribution of cephalopods through metabarcoding of eDNA.
2. Material and methods
The process for the development of a metabarcoding primer pair encompassed four steps: (i) assembling a reference database, (ii) identifying potential primer sets, (iii) in silico testing, and (iv) empirical testing.
2.1. Reference database
Two cephalopod 18S rRNA databases were generated from GenBank and SILVA. The results for a general GenBank query ‘18S Cephalopoda’ included environmental samples and partial sequences from other 18S subregions. To ensure exclusion of environmental samples and inclusion of sequences from the same subregion, the first sequence of the largest subset of partial sequences (the longfin inshore squid Doryteuthis pealeii, MH586846, 760 bp) was aligned against the full GenBank database (discontinuous megablast, 9 October 2018) [44] and all matches with full query cover were downloaded. This approach resulted in 31 partial cephalopod 18S rRNA sequences from 24 species. For the SILVA database, all cephalopod 18S rRNA reference sequences were downloaded (11 October 2018) and included 146 sequences from 88 species ranging from 423 to 2610 bp. The NCBI taxonomy dump file was downloaded (18 September 2018) and used for sequence annotation of both the GenBank and SILVA database with unique taxonomic identifications using Python v. 2.7.15 and OBITools v. 1.2.10 [45]. The annotated extended Fasta files were converted to the reference ecoPCR database v. 0.2 format [40].
2.2. Identification of potential primer sets
The ecoPrimer v. 0.3 algorithm [16] was used to identify potential primer pairs of 20 bp each that amplify a target sequence of 50–200 bp [46] with ample variation for taxonomic resolution to species level. The settings used on the GenBank reference database did not return any potential primer sets when applied to the SILVA reference database; hence for the SILVA database, the settings were somewhat relaxed (algorithm parameters in table 1). The potential primers were filtered for lowest melting temperature (Tm) between 59 and 69°C, maximum difference between lowest melting temperatures less than 3°C, GC count of 50–60%, a GC clamp with less than four G and/or C, less than four nucleotide repeats and less than four dinucleotide repeats [46]. The relaxed settings for the SILVA database may have resulted in suboptimal primers. Therefore, only primer sets with a Bc equal to or higher than the GenBank derived primer sets when tested in silico against the SILVA database were considered and filtered for lowest melting temperature (Tm) (between 45 and 70°C) and a GC count between 45 and 65%.
Table 1.
Parameters settings used for the ecoPrimer v. 0.3 analysis [16]. GB, GenBank reference database (the analysis was run twice with different mismatch and 3′ match settings). SV, SILVA reference database (quorum parameters were relaxed relative to the GenBank settings).
| parameter | value | description |
|---|---|---|
| primer length (bp) | 20 | required length of forward and reverse primer |
| target amplicon length (bp) | 50–200 | required length range of the amplified sequence |
| strict matching quorum | GB: 0.7 SV: 0.5 |
minimum fraction of sequence records in the reference database with an exact match between the primer and target sequence |
| sensitivity quorum | GB: 0.9 SV: 0.7 |
minimum fraction of sequence records in the reference database that exactly match the specified parameters |
| no. mismatches | GB: 0 and 3 SV: 3 |
number of allowed mismatches between primer and target sequence |
| no. 3′ matches | GB: NA and 2 SV: 2 |
number of strict matches required at the 3′ end |
2.3. In silico testing
From the filtered list, eight primer sets with the highest coverage index (Bc) and specificity index (Bs), as estimated by ecoPrimer [16], were chosen for further tests in silico, i.e. to predict their effectiveness in PCR amplification. The primers were analysed for secondary structures (hairpins and primer-dimers) using the online Oligonucleotide Properties Calculator v. 3.27 [47], and for self-complementarity and in silico amplification using online Primer-BLAST [48]. The Bc and Bs of the primer set was calculated with an ecoPCR v. 0.2 [40] in silico test against the SILVA database (no mismatches allowed). Three primer sets with optimal characteristics, i.e. no or limited secondary structures and highest Bc and Bs indices, were ordered for empirical testing.
The Bc and Bs of the two mitochondrial 16S rRNA primer sets, CephMLS (CephMLSf1: 5′-TGC GGT ATT WTA ACT GTA CT-3′, CephMLSr1: 5′-TTA TTC CTT RAT CAC CC-3′) [41] and S_Cephalopoda (S_Cephelapoda-F: 5′- GCT RGA ATG AAT GGT TTG AC-3′; S_Cephalopoda-R: 5′-TCA WTA GGG TCT TCT CGT CC-3′) [42] were estimated to determine the complementarity of the newly developed and existing primer sets. The target sequence size of S_Cephalopoda is 70–73 bp [42] and falls right within the targeted region of CephMLS, which has a target sequence size of 212–244 bp [41]. A 16S reference database was obtained by using Primer-BLAST [48] with the respective primer sets, and subsequently blasting the first match (blastn protocol, limit to cephalopod taxon in nucleotide database, exclude uncultured/environmental samples, query cover greater than 50%) [44] to obtain sequences that were known to contain the targeted region. An ecoPCR v. 0.2 [40] in silico test (no mismatches allowed) was used to determine Bc and Bs indices for S_Cephalopoda and CephMLS using this 16S GenBank reference database.
The 18S primer development process was based on two reference databases: one from SILVA with 146 sequences, and one from GenBank with 31 sequences. The latter has significantly less sequences than the SILVA database, caused by our specific filtering choices to avoid non-overlapping sequences which would have obstructed the development process. We calculated the Bc and Bs indices for our new primer set from the SILVA database during the development process. However, we felt that a comparison between these SILVA-derived 18S indices and the Primer-BLAST-derived 16S indices would be biased. SILVA is specific about which GenBank sequences are admitted into the alignment, and some GenBank sequences might have been left out. To ensure an unbiased comparison between 16S and 18S coverage and specificity indices, we obtained a third 18S database by using the newly developed 18S primer sequence in a GenBank Primer-BLAST [48]. This third GenBank database could not have been obtained at the start when we had not yet developed the primer set.
2.4. Empirical testing
Cephalopod specimens were collected in the eastern tropical Atlantic in waters near the Republic of Cabo Verde by the R.v. Walter Herwig III in March and April 2015 (cruise ID WH383, permissions obtained from Ministério da Agricultura e Ambiente and Agência Marítima e Portuária of Cape Verde). The sampling net was a pelagic trawl (Engel Netze, Bremerhaven, Germany, length 18 m, 16 × 30 m mouth opening, cod end 20 mm stretched mesh-opening, 1.8 mm inlet sewn into last 1 m of cod end) with a multi-sampler allowing depth-stratified sampling (electronic supplementary material, table S1). The specimens were morphologically identified by H.-J.T.H., and the full specimen (for small individuals) or a part of an arm (for larger individuals) was stored in a 2 ml tube with ethanol. DNA was extracted from the identified cephalopod tissue samples with the DNeasy Blood and Tissue Kit (Qiagen) following the manufacturer's protocol.
DNA purity and concentration were measured with NanoDrop (Thermo Fisher Scientific) and Qubit (dsDNA broad range assay kit, Thermo Fisher Scientific), respectively. DNA extracts diluted to approximately 10 ng µl−1 of three species from different families (Bathyteuthis abyssicola, Heteroteuthis dispar and Liocranchia reinhardtii), a mixture of these three extracts, negative extraction controls (no tissue) and negative PCR controls, i.e. with PCR grade water instead of DNA template, were amplified with a temperature gradient for all selected potential new primer pairs. A PCR mixture of 40 µl reaction volume of which 10 µl DNA template was prepared with the KAPA Hifi kit (Kapa Biosystems, Roche Inc.) (1× Fidelity buffer [which corresponds to 0.4 mM MgCl2], 0.3 mM KAPA dNTPs, 5% DMSO, 0.02 U µl−1 KAPA Hotstart Polymerase, 0.5 µM of each forward and reverse primer). An Applied Biosystems Veriti Thermal Cycler (Thermo Fisher Scientific) was used for the PCR reaction. The PCR programme consisted of a 5 min initial denaturation step at 95°C followed by 35 cycles of denaturing at 98°C for 20 s, annealing temperature gradient for 15 s and extension at 72°C for 1 min, followed by a final elongation step at 72°C for 10 min and 4°C on hold. The temperature gradient started at 3°C below the lowest Tm of the primer set and increased with five steps of 3°C each. This temperature gradient was chosen to account for the expected increase in optimal annealing temperature due to the KAPA Hifi kit, and an expected decrease in optimal annealing temperature due to the DMSO in the PCR mix, which together could cause deviation from the theoretical optimal annealing temperature by several degrees Celsius. The PCR products were visualized under UV light on a 1.2% agarose gel using loading dye, GelRed (Biotium) and a 100 bp ladder (Thermo Fisher Scientific). Once the optimal annealing temperature was determined, the same PCR procedure was conducted on more cephalopod tissue DNA extracts (30 species, figure 4; electronic supplementary material, table S1), DNA extract mixture of the newly tested species, and negative extraction and PCR controls.
Figure 4.

BLAST results of sequences of 75 tissue DNA extracts obtained with the Ceph18S primer. Morphological ID names in bold indicate that there was a reference sequence to the same taxonomic resolution (genus or species) in GenBank. Grey specimens could not be amplified. Name colours indicate whether the match between morphological and BLAST ID was to species level (dark blue), genus level (light blue), family level (orange) or other taxonomic level (red). A ∼ (tilde) indicates there was another BLAST match with the exact same score, i.e. that the taxon could not be unambiguously identified.
PCR products from the primer set producing the most promising results, i.e. clear bands and limited smearing and non-specific bands on the agarose gel, were used for Sanger sequencing. Sanger sequencing was performed in GEOMAR and at the Institute of Clinical Molecular Biology (IKMB) in Kiel using different protocols. In GEOMAR, the PCR products were prepared for sequencing using the Sanger Sequencing Kit (Applied Biosystems). A reaction volume of 12 µl (10 µl PCR product, 0.03 U µl−1 FastAP, 0.33 U µl−1 ExoI) was used to remove unincorporated primers with the following PCR conditions: 37°C for 20 min, 80°C for 15 min and 4°C on hold. The sequencing reaction was conducted in a volume of 10 µl (0.5 µl cleaned PCR product, 0.5× Sequencing buffer, 2.5% BigDye Terminator Mix 3.1, 0.25 µM forward or reverse primer) with the following PCR conditions: 1 min at 96°C, 28 cycles at 96°C for 10 s, primer annealing temperature for 5 s, 60°C for 4 min and hold at 8°C. Finally, the sequencing reaction was purified with a bead-based reagent (62% Sam solution and 13% BigDye XTerminator) by shaking the mixture for 30 min at full speed and centrifuging for 2 min at 1000 r.p.m. At the IKMB, the PCR products were purified in a reaction volume of 10 µl (8 µl PCR product, 0.06 U µl−1 FastAP, 0.30 U µl−1 ExoI) with the following PCR conditions: 37°C for 10 min, 75°C for 15 min and 10°C on hold. The sequencing reaction was conducted in a volume of 10 µl (2 µl purified PCR product, 0.75× Sequencing buffer, 7% BigDye Terminator Mix 3.1, 0.32 µM forward or reverse primer) with the following PCR conditions: 1 min at 96°C, 25 cycles at 96°C for 10 s, 50°C for 5 s, 60°C for 4 min and hold at 10°C. The sequencing products were purified through Sephadex G-50 fine gel filtration (GE Healthcare Buchler).
Low-quality ends and primers were trimmed manually from the Sanger sequences, which were then manually checked and edited using 4Peaks v. 1.8 [49], and subsequently assembled using AliView v. 1.24 [50]. The assembled sequences (or single forward or reverse sequences in cases of failed sequencing) were checked against the online GenBank reference database with BLAST (megablast algorithm, nucleotide collection nr/nt, 29 June 2020) [44].
3. Results
3.1. Potential primer pairs
All three empirically tested primer pairs amplified cephalopod DNA of the expected length. However, two primer pairs produced electrophoresis gels with side products, i.e. smearing and non-specific bands, potentially due to false priming or greater propensity to form secondary structures. The primer pair with the cleanest amplification was named ‘Ceph18S’ (table 2) and selected for sequencing. The other two alternatives were not tested beyond this point (alternative 1: F-5′-GCA CTT AAC CGA CCG TCG AC-3′ and R-5′-GTC GCG GCG CTA CAT ATT AG-3′; alternative 2: ATT AGA CTG AGA CCG ATG CG-3′ and R-5′-GAC CGT CGA CAG TTG ATA GG-3′).
Table 2.
Properties of the developed universal cephalopod primer pair targeting the 18S rRNA region to be used in eDNA metabarcoding.
| name | Ceph18S |
|---|---|
| forward | 5′-CGCGGCGCTACATATTAGAC-3′ |
| reverse | 5′-GCACTTAACCGACCGTCGAC-3′ |
| forward Tma | 59.3°C |
| reverse Tma | 61.7°C |
| target length | ∼150–190 bp |
| Bcb | 0.85 |
| Bsb | 0.78 |
aThe Tm is estimated using the SantaLucia method [51] for a salt concentration of 0.05.
bBc is in silico coverage index (amplified taxa/target taxa) and Bs is in silico specificity index (identified taxa/amplified taxa) based on the SILVA database with 146 sequences of 97 unique taxa.
The Ceph18S primer pair targets the V2 variable region of the small-subunit 18S rRNA gene [52] (figure 1). Alignment against Loligo formosana (accession code AY557478) shows that the target sequence is located at positions 258–442 (figure 1), but the exact location will depend on the species due to the variation in this region. The in silico PCR with Ceph18S on the SILVA database shows a target sequence length ranging from 131 bp for Watasenia scintillans to 196 bp for Sepia elegans (table 2). The alignments of these ecoPCR target sequences and sequences obtained in the laboratory clearly show conserved flanking regions and a highly variable internal region (figure 2). The optimal annealing temperature for Ceph18S in the PCR master mix used in this study was found to be 62°C. This differs from the calculated Tm (table 2) as both the KAPA reagents and DMSO in our PCR master mix alter the annealing temperature.
Figure 1.

Position of the forward and reverse Ceph18S primers on the cephalopod 18S rRNA gene. Nucleotide diversity (π) was calculated with a sliding-window analysis (window size = 99 bp, step size = 10 bp) over the SILVA alignment (after removal of large indels 0–572, 2004–2411, 6255–6529) and shown based on the reference sequence of L. formosana (accession code: AY557478).
Figure 2.

Target sequences alignment. Alignment of (a) target sequences obtained through in silico PCR and (b) sequences of extracted cephalopod DNA, both amplified with Ceph18S. Alignment using MUSCLE and image creation done in AliView [50].
3.2. Ceph18S resolution
The power of the Ceph18S primer pair to identify cephalopod species was examined with an in silico PCR on 88 species plus nine unique genera (i.e. genera without species identification which complemented the genera of the 88 identified species) in the SILVA database. From the 97 unique taxa in the SILVA database, 82 could be amplified in silico (Bc = 0.85). Of these 82 amplified taxa, 64 could be identified unambiguously (Bs = 0.78, figure 3). Therefore, 18 taxa could not be identified unambiguously, i.e. their target sequence was equal to the target sequence of another species: (i) Leachia atlantica and Leachia lemur, (ii) Octopoteuthis megaptera and Taningia danae, (iii) Chiroteuthis veranyi and Chiroteuthis calyx, (iv) Discoteuthis laciniosa and Discoteuthis discus, (v) Onykia carriboea and Ancistroteuthis lichtensteinii, (vi) Selenoteuthis scintillans and Lycoteuthis lorigera, (vii) Histioteuthis miranda and Histioteuthis bonellii, (viii) Gonatus antarcticus and Gonatopsis sp., and (ix) Gonatopsis octopedatus, and Okutania anonycha. Taxa for which some reference sequences were amplified but not all, were Chtenopteryx sicula, Loligo forbesi, Sepia elegans, Sepiella inermis and Todaropsis eblanae. Further inspection revealed that some reference sequences of these species were incomplete, i.e. omitting at least the V2 region around which the Ceph18S primer set anneals. Taxa that were not amplified at all due to a lacking reference V2 region were Eledone cirrhosa, Euprymna scolopes, Hapalochlaena maculosa, Loligo vulgaris, Octopus vulgaris, Opisthoteuthis sp. and Rossia macrosoma. Taxa that were not amplified even though a reference V2 region was available were Alloteuthis sp., Bathypolypus sp., Cirrothauma murrayi, Pyroteuthis margaritifera, Sepia pharaonis, Sepioloidea lineolata, Spirula spirula and Vampyroteuthis infernalis.
Figure 3.
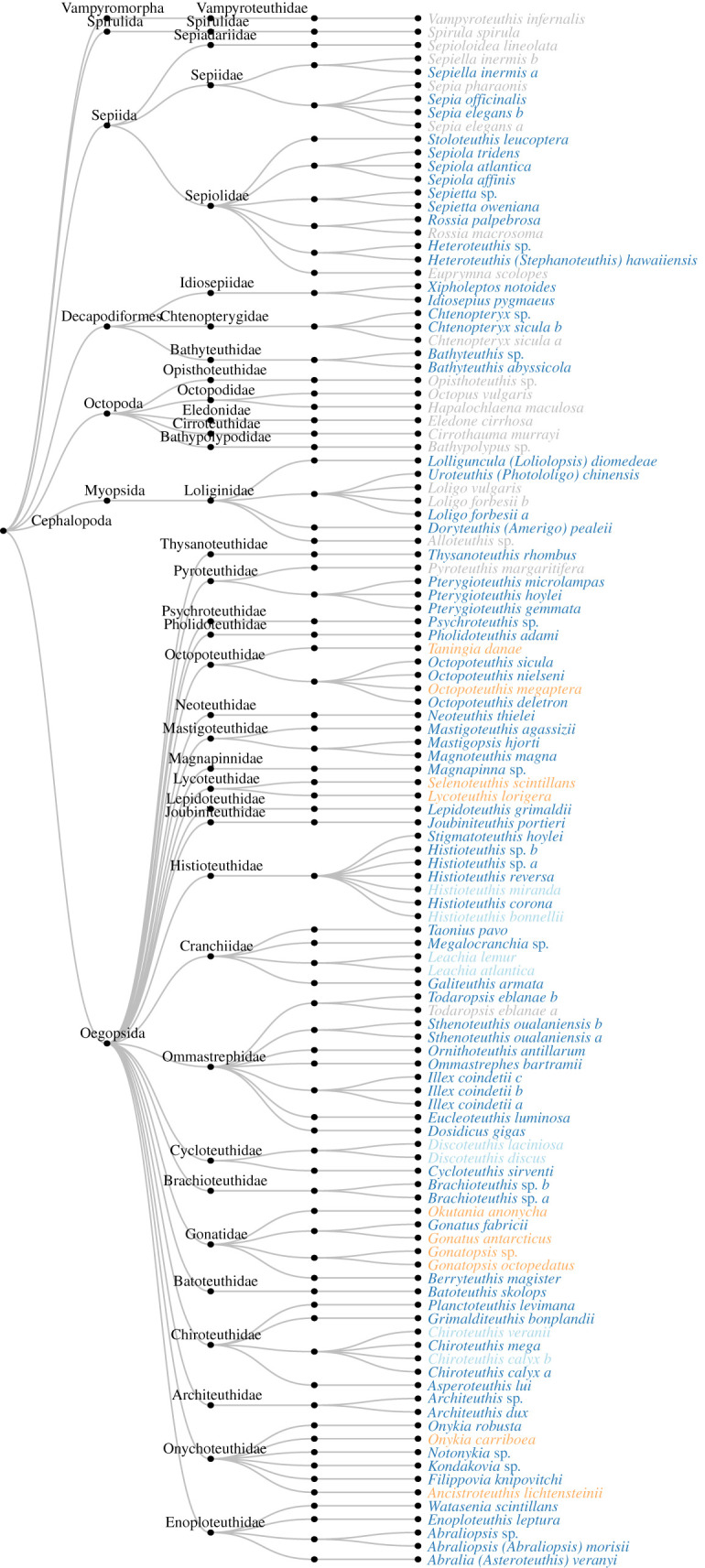
Results of in silico amplification of the SILVA database (146 sequences, 97 taxa) with Ceph18S. Grey taxa were not amplified in silico, whereas dark blue taxa were amplified and unambiguously identified. When taxa had equal target sequences and hence could not be unambiguously identified, colour indicates whether the shared taxonomic level was genus (light blue) or family (orange).
Subsequently, Ceph18S was tested empirically on 75 tissue DNA extracts (figure 4) from 68 specimens (electronic supplementary material, table S1) representing 30 cephalopod species plus two unique genera from specimens that could not be identified to species level (sequences are deposited in GenBank, accession numbers MT680727–MT680790 and MT680792–MT680800). Of the 32 taxa, 14 taxa did not have corresponding 18S rRNA reference sequences in GenBank (figure 4). All tested taxa, except for Discoteuthis discus and Vitreledonella richardi, could be amplified with Ceph18S, although sometimes with suboptimal sequence quality, so empirical Bc = 0.94. The Ceph18S barcodes submitted to GenBank add previously non-existent 18S rRNA reference sequences for 13 taxa: Abraliopsis atlantica, Abralia redfieldi, Bathothauma lyromna, Chiroteuthis cf. joubini, Egea inermis, Helicocranchia pfefferi, Heteroteuthis dispar (the existing partial 18S sequence accession code AF034565 is from a different 18S subregion), Histioteuthis meleagroteuthis, Liocranchia reinhardtii, Ornithoteuthis volatilis, Sthenoteuthis pteropus, Stigmatoteuthis arcturi and Teuthowenia megalops.
Of the 15 amplified species with a reference sequence in GenBank (i.e. excluding the two genus-only taxa and D. discus), seven could be unambiguously matched to species level (Bs = 0.47): Bathyteuthis abyssicola, Cranchia scabra, Enoploteuthis leptura, Histioteuthis reversa, Mastigopsis hjorti, Ornithoteuthis antillarum and Vampyroteuthis infernalis. Some species had an ambiguous BLAST match, i.e. the BLAST match returned multiple species with the same top alignment score including the expected morphologically identified species which, therefore, could not be genetically distinguished from other species. Leachia atlantica could not be distinguished from Leachia lemur, Octopoteuthis danae could not be distinguished from Octopoteuthis megaptera and Selenoteuthis scintillans could not be distinguished from Lycoteuthis lorigera. Species with a GenBank reference sequence that could not be identified were Ancistrocheirus lesueurii (best match with Identity = 96, E-val = 2.00 × 10−67 to Mastigoteuthis hjorti which belongs to a different family, as identified in GenBank accession code EU735291 by Lindgren [53], but accepted in WoRMS as Mastigopsis hjorti [54]), Histioteuthis corona (best match with Identity = 100, E-val = 1.00 × 10−63 to Histioteuthis hoylei as identified in GenBank accession code AY557500 by Lindgren et al. [55], but accepted in WoRMS as Stigmatoteuthis hoylei [56]), Octopoteuthis sicula (best match with Identity = 100, E-val = 1.00 × 10−83 to Octopoteuthis danae and Octopoteuthis megaptera), Pterygioteuthis gemmata (best match with Identity = 99, E-val = 3.00 × 10−47 to Pterygioteuthis microlampas) and Taningia danae (best match with Identity = 93, E-val = 2.00 × 10−67 to Lepidoteuthis grimaldii, both belonging to octopoteuthid families).
Of the 20 amplified genera with a reference sequence at genus level in GenBank, 15 could be identified unambiguously (Bs = 0.75). Of the six species that did not have a reference sequence in GenBank at either species or genus level, four could be identified to family level: in the Cranchiidae family (i) Helicocranchia pfefferi matched to Taonius pavo and Megalocranchia sp., (ii) Liocranchia reinhardtii matched to Cranchia scabra, and (iii) Teuthowenia megalops matched to Taonius pavo, whereas in the Histioteuthidae family (iv) Stigmatoteuthis arcturi matched to Histioteuthis hoylei (as identified in GenBank accession code AY557500 by Lindgren et al. [55], but accepted in WoRMS as Stigmatoteuthis hoylei [56]). Additionally, Taningia danae was identified as Lepidoteuthis grimaldii, which belong to closely related but different families [57].
Taking into account the availability of reference sequences, a total of 18 of the 30 amplified taxa had the best possible expected outcome (60%). An additional seven taxa could be matched to the next best taxonomic resolution (23%), and three taxa could not be identified to species, genus or family level despite reference sequences being available (10%). A final two taxa could not be identified to species, genus or family level, but also no reference sequences were available, so it is unknown if the lack of references or poor resolution of the primer is responsible (7%).
3.3. Comparison to 16S rRNA primer sets
The GenBank nuclear 18S rRNA reference database obtained through Primer-BLAST [48] with Ceph18S contained 107 taxa, and was, therefore, similar in size to the SILVA 18S rRNA reference database with 97 taxa. The coverage index and specificity index for Ceph18S was similar for this GenBank (Bc = 0.80, Bs = 0.80) and SILVA database (Bc = 0.85, Bs = 0.78).
The GenBank mitochondrial 16S rRNA database for both CephMLS [41] and S_Cephalopoda [42] contained 367 taxa. The coverage index for CephMLS and S_Cephalopoda was Bc = 0.82 and Bc = 0.72, respectively, i.e. of similar size and lower than for Ceph18S. The specificity index for CephMLS and S_Cephalopoda was Bs = 0.69 and Bs = 0.46, i.e. 11–34% lower than for Ceph18S. A Venn diagram analysis shows that 95% and 94% of cephalopod taxa can be amplified (figure 5a) and identified (figure 5b), respectively, by a multi-marker approach with Ceph18S, CephMLS and S_Cephalopoda for eDNA surveys. There is 1% of taxa that can only be amplified by Ceph18S and not by CephMLS or S_Cephalopoda, and 19% of cephalopod taxa can be identified unambiguously by Ceph18S but not by CephMLS or S_Cephalopoda. For comparison, 7% and 0% of taxa can be unambiguously identified only by CephMLS or S_Cephalopoda, respectively, and 9% can be identified by both 16S primer sets but not by Ceph18S. In other words, while the primer sets complement each other only moderately in terms of amplification success, the Ceph18S target sequences have a greater taxonomic resolution so that 19% additional taxa can be identified.
Figure 5.

Venn diagram of complementarity in (a) amplification and (b) identification between Ceph18S, CephMLS [41] and S_Cephalopoda [42]. N represents (a) the number of taxa that were present in both the 16S and 18S GenBank reference databases, and (b) the number of taxa amplified by all three primer sets (i.e. the middle plane of (a) is the subset used for (b)). Grey percentage in the box is the number of taxa that could not be (a) amplified and (b) identified by either primer set. Sum of percentages may deviate ±1% due to rounding.
4. Discussion
A novel universal primer pair named Ceph18S targeting the cephalopod nuclear 18S rRNA region was characterized. Due to the relatively small target sequence length of approximately 200 bp, the primer pair is suitable for metabarcoding of cephalopod eDNA and can be applied in field studies. We will discuss the Ceph18S primer in the context of metabarcoding, where reliable primers are important to avoid false positives (i.e. wrongly identified taxa) and false negatives (i.e. undetected species) [58].
4.1. Application of Ceph18S in field studies
The Ceph18S primer pair targets the V2 variable region of the small-subunit 18S rRNA [52] flanked by relatively conserved regions. This region is suitable for taxonomic assignment [59], with enough variation to allow the identification of a variety of taxa. However, clustering algorithms in metabarcoding pipelines can have difficulties with ribosomal target sequences due to the inherent length variation of rRNA variable regions [60]. As this length variation is present in Ceph18S target sequences (131–196 bp), it is recommended to omit clustering and use sequence variants directly, for example, with DADA2 [61], so no clustering threshold has to be chosen and a higher taxonomic resolution can be obtained [62].
According to the coverage index estimated in silico, Ceph18S should be able to amplify approximately 80–85% of cephalopod species. This coverage index might be slightly underestimated due to missing V2 regions in SILVA reference sequences for some species. Coverage of these species by Ceph18S could be checked using tissue DNA extracts if specimens are available. The variation in the annealing sites of the primers indicates that there will be mismatches between the primer and some target species. A small number of mismatches are not always problematic, as amplification might still occur although perhaps suboptimally [63]. This is confirmed by the fact that all but two tested cephalopod DNA extracts (94%) could be amplified with Ceph18S empirically. Case in point is the observation that Vampyroteuthis infernalis was not amplified in silico due to a mismatch between the primer and annealing site but was amplified and correctly identified to species level empirically. The distinctive phylogenetic position of v. infernalis in the cephalopod phylogenetic tree [53], and thus its distinctive target sequence, allowed the alignment algorithm to distinguish between v. infernalis and target sequences from other taxonomic groups even though the annealing mismatch might potentially have resulted in suboptimal amplification. A possibility to increase the universality of the primer would be to create a degenerate version of Ceph18S, i.e. a mixture of very similar primers where one or more nucleotide bases are varied in order to limit mismatches with target species [64].
Overall, the tests indicate that an estimated 44–66% (Bc × Bs) of all cephalopod species can be amplified and correctly identified with the Ceph18S primer. In comparison, S_Cephalopoda [42] and CephMLS [41] are estimated to amplify and identify 33% and 56% of all cephalopod species, respectively. Both the in silico and empirical tests indicate that the Ceph18S primer pair is not suitable for the detection of octopods, and can give ambiguous results for sepiids, myopsids, octopotheuthids and gonatids. Based on the empirical results, it might appear that the cranchiids are also problematic, but this may also be caused by a severe under-representation of this group in GenBank. If only specificity to genus level is required, Ceph18S performs well with an empirically obtained genus-level specificity of 75%. Furthermore, filtering procedures in the metabarcoding pipeline can increase the number of identified taxa. Any assumptions in these filtering procedures should, however, be considered when interpreting the results. For example, it might be possible to filter based on known biogeographic area, i.e. there are multiple possible species matches to a global reference database, but only one species is known to occur in the studied region and this would omit the detection of, for example, any invasive species.
4.2. Reference sequences
The specificity index of Ceph18S was 0.78–0.80 as estimated in silico, and 0.47 as estimated empirically. However, this estimated empirical specificity index is based on a limited number of identified species (n = 15) for which reference sequences were available in GenBank. Of the approximately 700 known cephalopod species, only 88 species are present in the SILVA database and fewer than 300 species have a complete or partial 18S rRNA sequence in GenBank. The reference sequences for cephalopods in GenBank that are currently available are mostly mitochondrial sequences, whereas the 18S rRNA gene is nuclear. For almost half of the taxa that were empirically tested with the Ceph18S primer, no 18S rRNA reference sequences were available in GenBank. Our barcoding efforts with Ceph18S add new 18S rRNA reference sequences for 13 taxa to GenBank. A comprehensive high-quality reference database is important for metabarcoding primer development and testing because it provides a good overview of nucleotide diversity over the gene and can reduce the occurrence of misidentifications and false negatives [58]. An effort to sequence additional species, as we present here, is, therefore, recommended to aid further primer development and the taxonomic resolution of eDNA metabarcoding surveys [65]. If a proper reference library is not available, alternative biodiversity estimates based on sequence diversity (i.e. omitting direct taxonomic assignment) could be used [66].
There are five species for which the Ceph18S target sequences did not match to the expected species in GenBank, even though a representative reference sequence was available.
A first explanation could be a wrong morphological identification assigned to the DNA sequence. For example, the taxa in the Histioteuthidae family are relatively difficult to distinguish, which may have caused a misidentification of our Histioteuthis corona or its matching GenBank sequence Histioteuthis hoylei. However, we deem this explanation unlikely, as all morphological identifications in both this paper and for the GenBank reference sequences were done by cephalopod experts (H.-J.T.H. and Annie Lindgren, respectively).
A second explanation could be the existence of cryptic species, where species are morphologically similar but genetically different. Although widespread existence of cryptic oceanic species has been suggested [67] and has been shown for some cephalopod taxa [68,69], no cryptic species complexes have been reported for the species with GenBank mismatches. Additionally, the taxonomy of the Octopoteuthidae is problematic with evidence of genetic similarity between Octopoteuthis sicula, O. danae and O. megaptera, which does not support the distinction of multiple species [70] and explains our 100% match of O. sicula to O. danae and O. megaptera with our relatively short Ceph18S target sequence.
A third explanation for the mismatches is that the relatively short target sequence length of Ceph18S in some cases cannot provide enough resolution to account for natural variability for a reliable identification, especially if the species is under-represented in GenBank. Three of the five mismatched species did match to the correct genus. Target sequences within a taxon can be expected to be relatively similar, so that a couple of different nucleotide bases, either due to natural variability or erroneous base calls in the sequencing process, can induce mismatches especially in short target sequences. The remaining two mismatched species with hits outside the expected genus had low identities to their best match (93%, 96%) and only one representative reference sequence available in GenBank. The quality of all our barcoded sequences was reviewed and approved, and repeated sequencing of the same individuals gave consistent results. For example, the same specimen of Taningia danae, which was reliably identified morphologically, was sequenced twice with consistent target sequences and closest match of 93% to Lepidoteuthis grimaldii. Therefore, it is likely this sequence of T. danae reflects natural variability in this partial 18S rRNA region for the species.
4.3. Biodiversity surveys with a multi-marker approach
Extrapolating the Ceph18S Bc × Bs value to the known approximately 700 cephalopod species, it is estimated about 310–460 species can be amplified and identified with Ceph18S. Additionally, 19% of taxa that are amplified by all three primers can only be unambiguously identified by Ceph18S and 16% can only be unambiguously identified by the 16S primer sets. Therefore, a multi-marker approach, where multiple universal cephalopod primers are combined, can increase overall coverage and specificity when applying eDNA analysis for cephalopod biodiversity studies [29]. Note that the 16S rRNA primer set S-Cephalopoda from [42] targets a subregion of the target sequence amplified by the 16S primer set CephMLS from [41], which are, therefore, similar in their resolution.
Employing a multi-marker approach has several advantages. Firstly, the highest confidence for species detection is obtained when a species is found in multiple samples and by multiple markers [71]. However, solely applying such a stringent definition would omit species that can be detected by one marker only. In the complementary case of 18S and 16S cephalopod primer sets, 35% of cephalopod species would not be detected with confidence under this stringent definition as they are only identified by either one, and not by both. Therefore, we recommend appreciating the complementary nature of primers sets and assigning a confidence value to a species detection rather than fully discarding single marker detections. Secondly, employing multiple markers is useful with limited availability of reference sequences [72]. By searching for multiple markers, the reference database can include species that might have a reference sequence for one region, but not the other. To successfully employ the multi-marker approach, it is important to understand the limitations of each marker. For example, markers may have different affinities to different taxonomic groups [32] and there might be differences in detectability between nuclear and mitochondrial eDNA [73–75]. Studies into marker specificity and limitations, like the current study, can, therefore, help interpret eDNA metabarcoding results.
If the specificity of certain cephalopod groups is still low even with the combined marker approach, additional universal primer pairs can be developed (e.g. based on the 28S region, [55]), potentially targeting only a subgroup within the cephalopods. Additionally, the usage of slightly varying primer annealing temperatures, like in a touch-down PCR [76], can reveal more taxa than using a single annealing temperature. High temperatures favour rare but perfectly matching sequences, but lower temperatures generally recover broader diversity as it allows annealing mismatches, and taxa found at different temperatures are not strictly subsets of each other and thus add to overall richness [77].
Even though primer sets for specific cephalopod taxa allow identification of specific elusive species [25] and might complement a universal cephalopod primer for problematic taxa, it would be difficult to do cephalopod biodiversity assays using only species-specific primers. For example, regional reported species diversity is 32 in the Arctic [78], 54 in the Antarctic [78], 68 at the Southwest Indian Ocean Ridge [79], 70 around the Kermadec Islands [80], 77 near Bear Seamount [81], up to 85 around the Canary Islands [82]. Additionally, such an approach would require pre-existing knowledge of local cephalopod diversity, which is often lacking. Therefore, an eDNA metabarcoding approach with multiple markers of which the strengths and limitations are tested is a suitable complementary method for further studies into local cephalopod biodiversity patterns.
5. Conclusion
Despite the pivotal role of cephalopods in marine food webs, knowledge on their diversity and distribution still has major gaps, especially in oceanic regions. Metabarcoding of eDNA is a proven powerful technique for biodiversity surveys, but it has not yet been applied to the study of these elusive organisms. This study characterized a new universal metabarcoding primer pair for the analysis of cephalopod eDNA targeting the nuclear 18S rRNA gene in order to complement published mitochondrial 16S rRNA primer sets [41,42]. The developed Ceph18S primer pair amplifies an approximately 200 bp target sequence estimated to be able to identify about 310–460 cephalopod species. Ceph18S is estimated to amplify and identify 8–31% more cephalopod species than the 16S rRNA primer sets. Furthermore, 19% of taxa amplified by both the 16S and 18S rRNA primer sets can only be identified with Ceph18S, thereby increasing overall taxonomic resolution in a multi-marker metabarcoding approach. Ceph18S is not suitable for the detection of octopods, and should be used with caution on sepiids, myopsids, octopotheuthids and gonatids. The submitted barcodes to GenBank add new 18S rRNA partial sequences for 13 cephalopod taxa previously absent in the GenBank database.
The Ceph18S primer pair is currently being applied in metabarcoding studies of cephalopod eDNA from epi-, meso- and bathypelagic depths [83]. The preliminary results of this study show that Ceph18S can successfully be applied in a metabarcoding workflow on a large dataset and can successfully amplify and identify cephalopod eDNA. Therefore, the Ceph18S primer pair, potentially in degenerate form or combined with other universal cephalopod primers, is a useful new molecular tool that can be used alongside other sampling methods for studying cephalopod diversity and distribution from shallow, coastal waters to the pelagic and deep-sea environments.
Supplementary Material
Acknowledgements
We thank the Institute of Clinical Molecular Biology in Kiel for providing Sanger sequencing as supported in part by the DFG Clusters of Excellence ‘Precision Medicine in Chronic Inflammation’ and ‘ROOTS’. We thank T. Naujoks, Dr D. Langfeldt and Dr B. Löscher for technical support. We thank Dr Heino Fock (chief scientist of WH383) and Dr Stephanie Czudaj for the collaboration and opportunity to collect cephalopods in the eastern Atlantic which were used in this study.
Ethics
Cabo Verde has not ratified the Nagoya protocol. To fulfil the national ABS regulations of Cabo Verde, we obtained the required permit for the publication of results based on samples collected in Cabo Verde waters from the Direccao Nacional do Ambiente (National Directorate for the Environment of Cabo Verdes). Permission for fieldwork and publication of results was granted by Ministério da Agricultura e Ambiente of Cape Verde and Agência Marítima e Portuária of Cape Verde.
Data accessibility
All barcoded sequences are accessible via GenBank accession numbers MT680727–MT680790 and MT680792–MT680800. The electronic supplementary material includes the Supplementary Data and Code zip-file, which contains the code and data used for data analysis and the creation of figures 1, 3 and 4, and Supplementary Table S1, which contains sample station information.
Authors' contributions
D.S.W.d.J. carried out the molecular laboratory work, performed the bioinformatics and data analysis and drafted the manuscript; V.M. participated in the design of the study, collected field data, helped with the laboratory work and participated in data analysis. T.B. helped with the laboratory work and data analysis. O.P. participated in conceiving and designing the study. T.B.H.R. participated in conceiving and designing the study. H.-J.T.H. conceived and designed the study, collected field data, performed morphologies species identifications and coordinated the study. All authors critically revised the manuscript, gave final approval for publication and agree to be held accountable for the work performed therein.
Competing interests
We declare we have no competing interests.
Funding
This research is funded by the Deutsche Forschungsgemeinschaft (DFG) under grant HO 5569/2-1 (Emmy Noether Junior Research Group) awarded to H.-J.T.H., and by GEOMAR's Programme Oriented Funding III OCEANS programme.
References
- 1.Hoving H-JT, Robison BH. 2017. The pace of life in deep-dwelling squids. Deep. Res. Part I Oceanogr. Res. Pap. 126, 40-49. ( 10.1016/j.dsr.2017.05.005) [DOI] [Google Scholar]
- 2.Hoving H-JT, et al. 2014. Chapter three: the study of deep-sea cephalopods. Adv. Mar. Biol. 67, 235-359. ( 10.1016/B978-0-12-800287-2.00003-2) [DOI] [PubMed] [Google Scholar]
- 3.Villanueva R, Perricone V, Fiorito G. 2017. Cephalopods as predators: a short journey among behavioral flexibilities, adaptions, and feeding habits. Front. Physiol. 8, 598. ( 10.3389/fphys.2017.00598) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clarke MR. 1996. Cephalopods as prey. III Cetaceans. Phil. Trans. R. Soc. B 351, 1053-1065. [Google Scholar]
- 5.de la Chesnais T, Fulton EA, Tracey SR, Pecl GT.. 2019. The ecological role of cephalopods and their representation in ecosystem models. Rev. Fish Biol. Fish. 29, 313-334. ( 10.1007/s11160-019-09554-2) [DOI] [Google Scholar]
- 6.Gasalla MA, Rodrigues AR, Postuma FA. 2010. The trophic role of the squid Loligo plei as a keystone species in the South Brazil Bight ecosystem. ICES J. Mar. Sci. 67, 1413-1424. ( 10.1093/icesjms/fsq106) [DOI] [Google Scholar]
- 7.Hoving H-JT, Bush SL, Haddock SHD, Robison BH. 2017. Bathyal feasting: post-spawning squid as a source of carbon for deep-sea benthic communities. Proc. R. Soc. B 284, 20172096. ( 10.1098/rspb.2017.2096) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smale MJ. 1996. Cephalopods as prey. Iv. Fishes. Phil. Trans. R. Soc. B 351, 1067-1081. ( 10.1098/rstb.1996.0094) [DOI] [Google Scholar]
- 9.Young RE, Vecchione M, Donovan DT. 1998. The evolution of coleoid cephalopods and their present biodiversity and ecology. South Afr. J. Mar. Sci. 20, 393-420. ( 10.2989/025776198784126287) [DOI] [Google Scholar]
- 10.Vecchione M, Young RE, Piatkowski U. 2010. Cephalopods of the northern Mid-Atlantic Ridge. Mar. Biol. Res. 6, 25-52. ( 10.1080/17451000902810751) [DOI] [Google Scholar]
- 11.Clarke MR. 2006. Oceanic cephalopod distribution and species diversity in the eastern north Atlantic. Arquipélago 23A, 27-246. [Google Scholar]
- 12.Clarke MR, Martins HR, Pascoe P. 1993. The diet of sperm whales (Physeter macrocephalus Linnaeus 1758) off the Azores. Phil. Trans. R. Soc. Lond. 339, 67-82. ( 10.1098/rstb.1993.0005) [DOI] [PubMed] [Google Scholar]
- 13.Pereira JM, Paiva VH, Xavier JC. 2017. Using seabirds to map the distribution of elusive pelagic cephalopod species. Mar. Ecol. Prog. Ser. 567, 257-262. ( 10.3354/meps12020) [DOI] [Google Scholar]
- 14.Ficetola GF, Miaud C, Pompanon F, Taberlet P. 2008. Species detection using environmental DNA from water samples. Biol. Lett. 4, 423-425. ( 10.1098/rsbl.2008.0118) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pompanon F, Coissac E, Taberlet P. 2011. Metabarcoding a new way to analyze biodiversity. Biofutur 319, 30-32. [Google Scholar]
- 16.Riaz T, Shehzad W, Viari A, Pompanon F, Taberlet P, Coissac E. 2011. EcoPrimers: inference of new DNA barcode markers from whole genome sequence analysis. Nucleic Acids Res. 39, 1-11. ( 10.1093/nar/gkr732) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giovannoni SJ, Britschgi TB, Moyer CL, Field KG. 1990. Genetic diversity in Sargasso Sea bacterioplankton. Nature 345, 60-63. ( 10.1038/345060a0) [DOI] [PubMed] [Google Scholar]
- 18.Ogram A, Sayler GS, Barkay T. 1987. The extraction and purification of microbial DNA from sediments. J. Microbiol. Methods. 7, 57-66. ( 10.1016/0167-7012(87)90025-X) [DOI] [Google Scholar]
- 19.Valentini A, et al. 2009. New perspectives in diet analysis based on DNA barcoding and parallel pyrosequencing: the trnL approach. Mol. Ecol. Resour. 9, 51-60. ( 10.1111/j.1755-0998.2008.02352.x) [DOI] [PubMed] [Google Scholar]
- 20.Willerslev E, et al. 2003. Diverse plant and animal genetic records from holocene and pleistocene sediments. Science 300, 791-795. ( 10.1126/science.1084114) [DOI] [PubMed] [Google Scholar]
- 21.Venter JC, et al. 2004. Environmental genome shotgun sequencing of the Sargasso Sea. Science 304, 66-74. ( 10.1126/science.1093857) [DOI] [PubMed] [Google Scholar]
- 22.Laroche O, Kersten O, Smith CR, Goetze E. 2020. From sea surface to seafloor: a benthic allochthonous eDNA survey for the abyssal ocean. Front. Mar. Sci. 7, 1-16. ( 10.3389/fmars.2020.00682)32802822 [DOI] [Google Scholar]
- 23.Harper KJ, Goodwin KD, Harper LR, LaCasella EL, Frey A, Dutton PH. 2020. Finding crush: environmental DNA analysis as a tool for tracking the green sea turtle Chelonia mydas in a marine estuary. Front. Mar. Sci. 6, 810. ( 10.3389/fmars.2019.00810) [DOI] [Google Scholar]
- 24.Nguyen BN, et al. 2020. Environmental DNA survey captures patterns of fish and invertebrate diversity across a tropical seascape. Sci. Rep. 10, 6729. ( 10.1038/s41598-020-63565-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wada T, et al. 2020. Exploring a legendary giant squid: an environmental DNA approach. Mar. Biol. 167, 3-8. ( 10.1007/s00227-020-03773-z) [DOI] [Google Scholar]
- 26.Andruszkiewicz EA, Starks HA, Chavez FP, Sassoubre LM, Block BA, Boehm AB. 2017. Biomonitoring of marine vertebrates in Monterey Bay using eDNA metabarcoding. PLoS ONE 12, 1-20. ( 10.1371/journal.pone.0176343) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sigsgaard EE, Nielsen IB, Carl H, Krag MA, Knudsen SW, Xing Y, Holm-Hansen TH, Møller PR, Thomsen PF. 2017. Seawater environmental DNA reflects seasonality of a coastal fish community. Mar. Biol. 164, 128. ( 10.1007/s00227-017-3147-4) [DOI] [Google Scholar]
- 28.Thomsen PF, Kielgast J, Iversen LL, Møller PR, Rasmussen M, Willerslev E. 2012. Detection of a diverse marine fish fauna using environmental DNA from seawater samples. PLoS ONE 7, 1-10 ( 10.1371/journal.pone.0041732) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stat M, Huggett MJ, Bernasconi R, Dibattista JD, Berry TE, Newman SJ, Harvey ES, Bunce M. 2017. Ecosystem biomonitoring with eDNA: metabarcoding across the tree of life in a tropical marine environment. Sci. Rep. 7, 1-11. ( 10.1038/s41598-017-12501-5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Djurhuus A, et al. 2018. Evaluation of marine zooplankton community structure through environmental DNA metabarcoding. Limnol. Oceanogr. Methods 16, 209-221. ( 10.1002/lom3.10237) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Günther B, Knebelsberger T, Neumann H, Laakmann S, Martínez Arbizu P. 2018. Metabarcoding of marine environmental DNA based on mitochondrial and nuclear genes. Sci. Rep. 8, 14822. ( 10.1038/s41598-018-32917-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stefanni S, Stanković D, Borme D, de Olazabal A, Juretić T, Pallavicini A, Tirelli V. 2018. Multi-marker metabarcoding approach to study mesozooplankton at basin scale. Sci. Rep. 8, 12085. ( 10.1038/s41598-018-30157-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Borrell YJ, Miralles L, Do Huu H, Mohammed-Geba K, Garcia-Vazquez E. 2017. DNA in a bottle—rapid metabarcoding survey for early alerts of invasive species in ports. PLoS ONE 12, e0183347. ( 10.1371/journal.pone.0183347) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cowart DA, Matabos M, Brandt MI, Marticorena J, Sarrazin J. 2020. Exploring environmental DNA (eDNA) to assess biodiversity of hard substratum faunal communities on the Lucky Strike Vent Field (Mid-Atlantic Ridge) and investigate recolonization dynamics after an induced disturbance. Front. Mar. Sci. 6, 783. ( 10.3389/fmars.2019.00783) [DOI] [Google Scholar]
- 35.Kitahashi T, Sugime S, Inomata K, Nishijima M, Kato S, Yamamoto H. 2020. Meiofaunal diversity at a seamount in the pacific ocean: a comprehensive study using environmental DNA and RNA. Deep. Res. Part I Oceanogr. Res. Pap. 160, 103253. ( 10.1016/j.dsr.2020.103253) [DOI] [Google Scholar]
- 36.Taberlet P, Bonin A, Zinger L, Coissac E. 2018. Environmental DNA: for biodiversity research and monitoring, 1st edn. New York, NY: Oxford University Press. [Google Scholar]
- 37.Meusnier I, Singer GAC, Landry JF, Hickey DA, Hebert PDN, Hajibabaei M. 2008. A universal DNA mini-barcode for biodiversity analysis. BMC Genomics. 9, 1-4. ( 10.1186/1471-2164-9-214) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R. 1994. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3, 294-299. [PubMed] [Google Scholar]
- 39.Geller J, Meyer C, Parker M, Hawk H. 2013. Redesign of PCR primers for mitochondrial cytochrome c oxidase subunit I for marine invertebrates and application in all-taxa biotic surveys. Mol. Ecol. Resour. 13, 851-861. ( 10.1111/1755-0998.12138) [DOI] [PubMed] [Google Scholar]
- 40.Ficetola GF, Coissac E, Zundel S, Riaz T, Shehzad W, Bessière J, Taberlet P, Pompanon F. 2010. An in silico approach for the evaluation of DNA barcodes. BMC Genomics. 11, 434. ( 10.1186/1471-2164-11-434) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jarman SN, Redd KS, Gales NJ. 2006. Group-specific primers for amplifying DNA sequences that identify Amphipoda, Cephalopoda, Echinodermata, Gastropoda, Isopoda, Ostracoda and Thoracica. Mol. Ecol. Notes 6, 268-271. ( 10.1111/j.1471-8286.2005.01172.x) [DOI] [Google Scholar]
- 42.Peters KJ, Ophelkeller K, Bott NJ, Goldsworthy SD. 2015. PCR-based techniques to determine diet of the Australian sea lion (Neophoca cinerea): a comparison with morphological analysis. Mar. Ecol. 36, 1428-1439. ( 10.1111/maec.12242) [DOI] [Google Scholar]
- 43.Kim E-B, Lee SR, Il Lee C, Park H, Kim H-W. 2019. Development of the cephalopod-specific universal primer set and its application for the metabarcoding analysis of planktonic cephalopods in Korean waters. PeerJ 7, e7140. ( 10.7717/peerj.7140) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Z, Schwartz S, Wagner L, Miller W. 2000. A greedy algorithm for aligning DNA sequences. Comput. Biol. 7, 203-214. ( 10.1089/10665270050081478) [DOI] [PubMed] [Google Scholar]
- 45.Boyer F, Mercier C, Bonin A, Taberlet P, Coissac E. 2014. OBITools: a Unix-inspired software package for DNA metabarcoding. Mol. Ecol. Resour. 16, 176-182. ( 10.1111/1755-0998.12428) [DOI] [PubMed] [Google Scholar]
- 46.Bradburn SL. 2017. How to create real-time PCR primers using Primer-BLAST. Top Tip Bio. See https://toptipbio.com/real-time-pcr-primer-blast/.
- 47.Kibbe W. 2007. OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids Resour. 35, W43-46. ( 10.1093/nar/gkm234) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jian Y, George C, Irena Z, Ioana C, Steve R, Madden Thomas L. 2012. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinf. 13, 134. ( 10.1186/1471-2105-13-134) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Griekspoor A, Groothuis T. 2004. 4Peaks. Nucleobytes. See https://nucleobytes.com/4peaks.
- 50.Larsson A. 2014. AliView: a fast and lightweight alignment viewer and editor for large data sets. Bioinformatics 30, 3276-3278. ( 10.1093/bioinformatics/btu531) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.SantaLucia J. 1998. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl Acad. Sci. USA 95, 1460-1465. ( 10.1073/pnas.95.4.1460) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hadziavdic K, Lekang K, Lanzen A, Jonassen I, Thompson EM, Troedsson C. 2014. Characterization of the 18s rRNA gene for designing universal eukaryote specific primers. PLoS ONE 9, e0087624. ( 10.1371/journal.pone.0087624) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lindgren AR. 2010. Molecular inference of phylogenetic relationships among Decapodiformes (Mollusca: Cephalopoda) with special focus on the squid Order Oegopsida. Mol. Phylogenet. Evol. 56, 77-90. ( 10.1016/j.ympev.2010.03.025) [DOI] [PubMed] [Google Scholar]
- 54.MolluscaBase (eds). 2020. Mastigopsis hjorti (Chun, 1913) [Internet]. MolluscaBase. World Register of Marine Species. See http://www.marinespecies.org/aphia.php?p=taxdetails&id=759125 (accessed 29 June 2020) [Google Scholar]
- 55.Lindgren AR, Giribet G, Nishiguchi MK. 2004. A combined approach to the phylogeny of Cephalopoda (Mollusca). Cladistics 20, 454-486. ( 10.1111/j.1096-0031.2004.00032.x) [DOI] [PubMed] [Google Scholar]
- 56.MolluscaBase (eds). 2020. Stigmatoteuthis hoylei (Goodrich, 1896). 2020 [Internet]. MolluscaBase. World Register of Marine Species; 2020. See http://www.marinespecies.org/aphia.php?p=taxdetails&id=410403 (accessed 23 June 2020). [Google Scholar]
- 57.Vecchione M, Young RE. 2019. Lepidoteuthid families. Version 26 (under construction). See http://tolweb.org/Lepidoteuthid_families/19416/2019.03.26 in The Tree of Life Web Project, http://tolweb.org/.
- 58.Cristescu ME, Hebert PDN. 2018. Uses and misuses of environmental DNA in biodiversity science and conservation. Annu. Rev. Ecol. Evol. Syst. 49, 209-230. ( 10.1146/annurev-ecolsys-110617-062306) [DOI] [Google Scholar]
- 59.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261-5267. ( 10.1128/AEM.00062-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Flynn JM, Brown EA, Chain FJJ, MacIsaac HJ, Cristescu ME. 2015. Toward accurate molecular identification of species in complex environmental samples: testing the performance of sequence filtering and clustering methods. Ecol. Evol. 5, 2252-2266. ( 10.1002/ece3.1497) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. 2016. DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581-583. ( 10.1038/nmeth.3869) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Callahan BJ, McMurdie PJ, Holmes SP. 2017. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 11, 2639-2643. ( 10.1038/ismej.2017.119) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sipos R, Szekely AJ, Palatinszky M, Revesz S, Marialigeti K, Nikolausz M. 2007. Effect of primer mismatch, annealing temperature and PCR cycle number on 16S rRNA gene-targetting bacterial community analysis. FEMS Microbiol. Ecol. 60, 341-350. ( 10.1111/j.1574-6941.2007.00283.x) [DOI] [PubMed] [Google Scholar]
- 64.Lear G, et al. 2018. Methods for the extraction, storage, amplification and sequencing of DNA from environmental samples. N Z J. Ecol. 42, 10. ( 10.20417/nzjecol.42.9) [DOI] [Google Scholar]
- 65.Stat M, John J, DiBattista JD, Newman SJ, Bunce M, Harvey ES. 2018. Combined use of eDNA metabarcoding and video surveillance for the assessment of fish biodiversity. Conserv. Biol. 33, 196-205. ( 10.1111/cobi.13183) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cordier T, Forster D, Dufresne Y, Martins CIM, Stoeck T, Pawlowski J. 2018. Supervised machine learning outperforms taxonomy-based environmental DNA metabarcoding applied to biomonitoring. Mol. Ecol. Resour. 18, 1381-1391. ( 10.1111/1755-0998.12926) [DOI] [PubMed] [Google Scholar]
- 67.Miya M, Nishida M. 1997. Speciation in the open ocean. Nature 389, 803-804. ( 10.1038/39774) [DOI] [Google Scholar]
- 68.Xu L, Liu P, Wang X, Van Damme K, Du F.. 2020. Phylogenetic relationships and cryptic species in the genus Sthenoteuthis (Cephalopoda: Ommastrephidae) in the South China Sea. Mol. Phylogenet. Evol. 149, 106846. ( 10.1016/j.ympev.2020.106846) [DOI] [PubMed] [Google Scholar]
- 69.Allcock AL, et al. 2011. Cryptic speciation and the circumpolarity debate: a case study on endemic Southern Ocean octopuses using the COI barcode of life. Deep Sea Res. Part II Top. Stud. Oceanogr. 58, 242-249. ( 10.1016/j.dsr2.2010.05.016) [DOI] [Google Scholar]
- 70.Jereb P, et al. 2016. The deep-water squid Octopoteuthis sicula Rüppell, 1844 (Cephalopoda: Octopoteuthidae) as the single species of the genus occurring in the Mediterranean Sea. Mar. Biol. 163, 192. ( 10.1007/s00227-016-2965-0) [DOI] [Google Scholar]
- 71.Evans NT, et al. 2017. Fish community assessment with eDNA metabarcoding: effects of sampling design and bioinformatic filtering. Can. J. Fish. Aquat. Sci. 74, 1362-1374. ( 10.1139/cjfas-2016-0306) [DOI] [Google Scholar]
- 72.Topstad L, Guidetti R, Majaneva M, Ekrem T. 2020. Multi-marker DNA metabarcoding reflects tardigrade diversity in different habitats. Genome. 0, 1-15. ( 10.1139/gen-2019-0218) [DOI] [PubMed] [Google Scholar]
- 73.Bylemans J, Furlan EM, Gleeson DM, Hardy CM, Duncan RP. 2018. Does size matter? An experimental evaluation of the relative abundance and decay rates of aquatic environmental DNA. Environ. Sci. Technol. 52, 6408-6416. ( 10.1021/acs.est.8b01071) [DOI] [PubMed] [Google Scholar]
- 74.Moushomi R, Wilgar G, Carvalho G, Creer S, Seymour M. 2019. Environmental DNA size sorting and degradation experiment indicates the state of Daphnia magna mitochondrial and nuclear eDNA is subcellular. Sci. Rep. 9, 12500. ( 10.1038/s41598-019-48984-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jo T, Arimoto M, Murakami H, Masuda R, Minamoto T. 2020. Estimating shedding and decay rates of environmental nuclear DNA with relation to water temperature and biomass. Environ. DNA 2, 140-151. ( 10.1002/edn3.51) [DOI] [Google Scholar]
- 76.Hecker KH, Roux KH. 1996. High and low annealing temperatures increase both specificity and yield in touchdown and stepdown PCR. Biotechniques 20, 478-485. ( 10.2144/19962003478) [DOI] [PubMed] [Google Scholar]
- 77.Schmidt PA, Bálint M, Greshake B, Bandow C, Römbke J, Schmitt I. 2013. Illumina metabarcoding of a soil fungal community. Soil Biol. Biochem. 65, 128-132. ( 10.1016/j.soilbio.2013.05.014) [DOI] [Google Scholar]
- 78.Xavier JC, Cherel Y, Allcock L, Rosa R, Sabirov RM, Blicher ME, Golikov AV. 2018. A review on the biodiversity, distribution and trophic role of cephalopods in the Arctic and Antarctic marine ecosystems under a changing ocean. Mar. Biol. 165, 93. ( 10.1007/s00227-018-3352-9) [DOI] [Google Scholar]
- 79.Laptikhovsky V, Boersch-Supan P, Bolstad K, Kemp K, Letessier T, Rogers AD. 2017. Cephalopods of the Southwest Indian OceanRidge: a hotspot of biological diversity and absence of endemism. Deep Res. Part II Top. Stud. Oceanogr. 136, 98-107. ( 10.1016/j.dsr2.2015.07.002) [DOI] [Google Scholar]
- 80.Braid HE, Bolstad KSR. 2019. Cephalopod biodiversity of the Kermadec Islands: implications for conservation and some future taxonomic priorities. Invertebr. Syst. 33, 402-425. ( 10.1071/IS18041) [DOI] [Google Scholar]
- 81.Shea EK, Judkins H, Staudinger MD, Dimkovikj VH, Lindgren A, Vecchione M. 2017. Cephalopod biodiversity in the vicinity of Bear Seamount, western North Atlantic based on exploratory trawling from 2000 to 2014. Mar. Biodivers. 47, 699-722. ( 10.1007/s12526-017-0633-3) [DOI] [Google Scholar]
- 82.Escánez A, Guerra Á, Riera R, Rocha FJ.. 2020. Revised species records reveal the Canary Islands as a cephalopod biodiversity hotspot. Reg. Stud. Mar. Sci. 41, 101541. ( 10.1016/j.rsma.2020.101541) [DOI] [Google Scholar]
- 83.Merten V, Bayer T, de Jonge D, Puebla O, Reusch TBH, Hoving H-JT. 2020. Use of environmental DNA metabarcoding to trace deep-sea cephalopod distribution and biodiversity in the Eastern Tropical Atlantic. In Ocean Sciences Meeting 2020. San Diego, CA, 19 February. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All barcoded sequences are accessible via GenBank accession numbers MT680727–MT680790 and MT680792–MT680800. The electronic supplementary material includes the Supplementary Data and Code zip-file, which contains the code and data used for data analysis and the creation of figures 1, 3 and 4, and Supplementary Table S1, which contains sample station information.