

SUMMARY
Discovering the interaction mechanism and location of RNA binding proteins (RBPs) on RNA is critical for understanding gene expression regulation. Here, we apply selective 2’-hydroxyl acylation analyzed by primer extension (SHAPE) on in vivo transcripts compared to protein-absent transcripts in four human cell lines to identify transcriptome-wide footprints (fSHAPE) on RNA. Structural analyses indicate that fSHAPE precisely detects nucleobases that hydrogen bond with protein. We demonstrate that fSHAPE patterns predict binding sites of known RBPs, such as iron response elements in both known loci and in previously unknown loci in CDC34, SLC2A4RG, COASY, and H19. Furthermore, by integrating SHAPE and fSHAPE with crosslinking and immunoprecipitation (eCLIP) of desired RBPs, we interrogate specific RNA-protein complexes, such as histone stem loop elements and their nucleotides that hydrogen bond with stem-loop binding protein. Together, these technologies greatly expand our ability to study and understand specific cellular RNA interactions in RNA-protein complexes.
Graphical Abstract
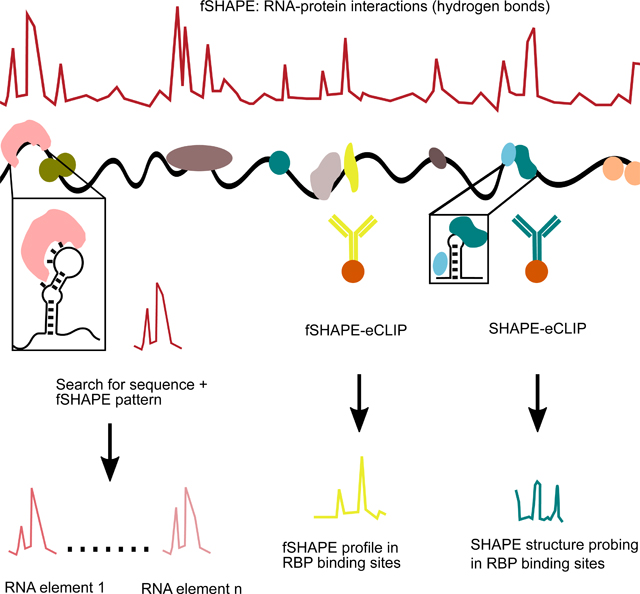
INTRODUCTION
RNA binding proteins (RBPs) modulate RNA transcripts through a myriad of interactions with binding sites on RNA. Detailed knowledge of RBP interactions with their binding sites greatly contributes to our understanding of gene expression regulation (Fiorini et al., 2015; Jackson et al., 2010; Loughlin et al., 2019; Tan et al., 2013; Tian et al., 2011; Walden et al., 2012). However, this knowledge does not extend to most transcripts in the cell. The innerworkings of RNA-protein interactions are routinely interrogated by a number of techniques. X-ray crystallography and nuclear magnetic resonance studies of RNA-protein complexes reveal key molecular interactions that comprise RBP binding (Cléry and Allain, 2011; Corley et al., 2020), but are limited to studying in vitro complexes comprised of fragments of RNA rather than full transcripts. Similarly, RNA Bind-N-Seq, which iteratively selects and identifies short RNA motifs bound by RBPs, is limited to in vitro interactions with small RNA oligonucleotides (Dominguez et al., 2018). Immunoprecipitation techniques have identified transcripts or portions of transcripts bound by hundreds of RBPs of interest, but lack the resolution to study the precise nucleotides that interact with proteins (Colombrita et al., 2012; Hafner et al., 2010; Licatalosi et al., 2008; Van Nostrand et al., 2016).
Here we describe an in-depth analysis of in vivo RNA-protein interactions transcriptome-wide and in a selectable manner by integrating footprinting, RNA structure probing, and immunoprecipitation technologies. The classic method of RNA footprinting uses RNA-reactive reagents in the presence and absence of protein to identify nucleotides that interact with protein residues, but has typically been limited to in vitro contexts (Tijerina et al., 2007). RNA structure probing techniques are similarly executed with RNA-reactive reagents, and several of these techniques have demonstrated footprinting of in vivo transcripts, including icSHAPE, SHAPE-MaP, and LASER (Feng et al., 2018; Lackey et al., 2018; McGinnis et al., 2009; Smola et al., 2015a; Smola et al., 2016; Spitale et al., 2015). Here we present a simplified strategy for in vivo footprinting with the SHAPE structure probing techniques (fSHAPE), and extract RNA-protein footprints transcriptome-wide in four cell lines. The size and scope of the dataset allows detailed structural analyses of the RNA-protein interactions revealed by fSHAPE. First, we demonstrate that fSHAPE detects nucleobases hydrogen bonded to proteins with high specificity and sensitivity. Hydrogen bonds comprise a significant portion of the molecular interactions that drive specific RNA-protein associations, determining transcript regulation by RBPs (Corley et al., 2020; Hu et al., 2018; Leulliot and Varani, 2001). Secondly, we show that the pattern of fSHAPE signal can serve as an identifier of specific RNA-protein complexes, detecting known RNA elements in FTL, TFRC, FTH1, and ALAS2 that bind iron response proteins (Stevens et al., 2011). fSHAPE also predicts previously unannotated iron response elements in CDC34, SLC2A4RG, COASY, and H19, which we independently validate, adding to the repertoire of transcripts whose regulation is linked to iron metabolism. To improve on the efficacy of interrogating RNA-protein complexes of interest, we combine enhanced crosslinking and immunoprecipitation (eCLIP)(Van Nostrand et al., 2016) to enrich for SHAPE and fSHAPE data in transcripts bound by specific RBPs. We validate SHAPE-eCLIP and fSHAPE-eCLIP by application to stem loop binding protein, which binds highly conserved stem loop elements at the 3’ ends of histone messenger RNAs (mRNAs). SHAPE- and fSHAPE-eCLIP successfully isolate and enrich for data among transcripts bound by stem loop binding protein, and correctly identify the stem loop elements and their nucleotides that hydrogen bond with protein. Altogether these analyses demonstrate the intricate details that SHAPE probing techniques reveal at RNA-protein interfaces. Integration of these techniques with eCLIP allows large-scale study of cellular RNA interactions with any protein of interest and will be instrumental for identifying the nucleotides and RNA elements that engage in protein interactions.
DESIGN
We initially approached studying protein binding sites using SHAPE probing techniques because of the overarching need to learn more details about the RNA elements that bind proteins. SHAPE probing techniques can be used for both RNA footprinting and structure probing and can be coupled to RNA-sequencing, making SHAPE our base-method of choice for technology development.
Footprinting with SHAPE (fSHAPE) transcriptome-wide in human cells
To identify nucleotides that interact with proteins, transcripts undergo structure probing either in vivo (“+protein”) or extracted and separated from proteins (“-protein”) (Figure 1a) (Feng et al., 2018; Flynn et al., 2016; Smola et al., 2015a). This is analogous to traditional RNA footprinting, in which RNA is treated with a structure probing reagent in the presence and absence of an RBP to identify portions of the molecule that are “protected” (Tijerina et al., 2007). In vivo click-SHAPE with the reagent NAI-N3 was our structure probing method of choice for its ability to penetrate cells and to enrich for transcripts modified by the probing reagent and thus enhance information over the entire transcriptome. Conditions are optimized to modify transcripts at a rate of once every ~100 nt for a given density of cells and can be robustly applied to multiple cell types (Flynn et al., 2016; Spitale et al., 2013). Typical to SHAPE probing techniques, modified transcripts in the “+protein” and “-protein” samples undergo reverse transcription to record truncation events at sites of NAI-N3-adduct formation, which are quantified by sequencing. fSHAPE takes a simplified approach compared to previous SHAPE-based footprinting methods as it does not require the un-probed sample typical to SHAPE probing experiments (Figure S1a), which controls for hotspots of sequence-based reverse transcriptase truncation events (Flynn et al., 2016; Smola et al., 2015a). We assume that “+protein” and “protein” samples will have equivalent sequence-based hotpots, and simply normalize “-protein” to “+protein” drop-off rates at each nucleotide to obtain “fSHAPE reactivity”. High fSHAPE reactivities indicate nucleotides that are more reactive with NAI-N3 in the absence of protein and thus likely interact with protein (Figure 1a). There is some chance that transcripts in the “-protein” sample undergo structural changes (relative to in vivo transcripts) and confound the fSHAPE signal that is due to eliminated protein interactions. We hope to minimize these occurrences by refolding these transcripts under cell-like conditions (see Methods) that are standard in the field when probing RNA in vitro (Busan et al., 2019; Flynn et al., 2016). fSHAPE also makes the assumption that proteins do not drastically alter RNA secondary structure when bound and thus that the changes we observe in protein-absent samples are due to lost interactions with protein. This is a necessary and reasonable assumption (Flores and Ataide, 2018; Hainzl et al., 2005; Leulliot and Varani, 2001; Yang et al., 2002), given that we currently have no method to simultaneously maintain protein and cell-induced RNA structures and probe RNA in the absence of said protein.
Figure 1.

SHAPE-based technologies for footprinting and structure probing of RNA-protein interfaces. (a) fSHAPE requires two RNA samples processed in parallel: a “+protein” samples in which cellular RNA is treated with the probing reagent (black star) and a “-protein” sample in which RNA is extracted from cells, stripped of protein, and treated with the probing reagent. Nucleotides that react with the reagent form adducts that result in drop-off events (colored square) during reverse transcription, such that the frequency of drop-off events at a given nucleotide is proportional to its reactivity rate with the probing reagent. “+protein” drop-off frequencies are subtracted from “-protein” drop-off frequencies and normalized to obtain an fSHAPE reactivity value at each nucleotide describing its degree of increased reactivity with the reagent in the absence of protein, akin to footprinting. (b) SHAPE-CLIP probes secondary structure in transcripts selected by CLIP. Cell samples are either treated with a structure probing reagent (black star) or an untreated negative control sample. Samples are UV crosslinked and extracted protein-bound transcripts are immunoprecipitated (IP) with an antibody to the desired protein. Nucleotides that react with the reagent form adducts that result in mutations (colored square) during a modified reverse transcription (Siegfried et al., 2014), such that the frequency of sequenced mutations at a given nucleotide is proportional to its reactivity rate with the probing reagent. “Treated” sample mutation rates are subtracted from “-control” mutation rates and normalized to obtain a SHAPE reactivity value at each nucleotide. Sequencing reads are also be used to determine protein binding sites (Van Nostrand et al., 2016). (c) fSHAPE-CLIP identifies nucleotides bound by protein in transcripts selected by CLIP. Cell samples are either initially treated with a structure probing reagent (“+protein”) or untreated (“-protein”). Samples are UV crosslinked and extracted protein-bound transcripts are immunoprecipitated (IP) with an antibody to the desired protein. RNA is protease-treated and refolded; the “-protein” samples is treated with the structure probing reagent. Nucleotides that react with the reagent form adducts that result in mutations (colored square) during a modified reverse transcription (Siegfried et al., 2014), such that the frequency of sequenced mutations at a given nucleotide is proportional to its reactivity rate with the probing reagent. “+protein” sample mutation rates are subtracted from “-protein” mutation rates and normalized to obtain an fSHAPE reactivity value at each nucleotide. Sequencing reads are also be used to determine protein binding sites (Van Nostrand et al., 2016). See also Table S1.
Integration of SHAPE and fSHAPE with eCLIP
Despite saturating modification of all cellular transcripts, many sequencing-based methods, including fSHAPE and SHAPE, only produce high quality data across the most highly abundant transcripts in the cell. This poses a challenge for RNA-protein studies for which the RNA is not highly expressed. Orthogonally, eCLIP can efficiently isolate RNA bound by RBPs through immunoprecipitation with specific antibodies, less sensitive to their cellular abundance (Van Nostrand et al., 2016). Thus, we explored integrating eCLIP with SHAPE methods for the purpose of enhancing their return on desired RNA-protein interfaces. Notably, SHAPE- and fSHAPE-eCLIP are not more time or labor intensive than eCLIP alone.
We first combined SHAPE with eCLIP (SHAPE-eCLIP) in order to specifically probe the RNA secondary structures that contextualize RBP binding sites. SHAPE-eCLIP also served as a stepping stone for producing a combined fSHAPE with eCLIP (fSHAPE-eCLIP) protocol, which requires more extensive modifications to eCLIP. As in SHAPE structure probing (Ding et al., 2013; Flynn et al., 2016; Smola and Weeks, 2018), SHAPE-eCLIP requires a sample treated with a structure probing reagent and a mock-treated control sample (Figure 1b). Cells are immediately crosslinked with UV light and protein-RNA complexes are extracted, immunoprecipitated, and further treated as in the described eCLIP protocol (Van Nostrand et al., 2016). Reverse transcription is modified according to the mutational profiling method (Siegfried et al., 2014), in which reagent-formed adducts on nucleotides are recorded as mutations in the cDNA rather than truncation events, as in icSHAPE (Flynn et al., 2016). (Note that icSHAPE and fSHAPE could also be implemented with mutational profiling, which would allow higher transcript probing rates without sacrificing read lengths of the resulting sequencing libraries.) Since mutational profiling enables longer read lengths, we reasoned it would avoid exacerbating the short read lengths produced by eCLIP. The resulting library is both a structure probing and eCLIP library, containing mutation rates from SHAPE structure probing at each nucleotide in RBP binding sites enriched by immunoprecipitation. Normalizing mutation rates at each nucleotide in the treated sample to the untreated control yields a SHAPE reactivity profile that describes the secondary structure landscape in RBP binding sites (Figure 1b).
Upon confirming that SHAPE and eCLIP were successfully combined, we imposed additional modifications to perform fSHAPE-eCLIP (Figure 1c). As in fSHAPE, a “+protein” cell sample is treated with the structure probing reagent, while a “-protein” cell sample is mock-treated. Cells are UV crosslinked and protein-RNA complexes are extracted and immunoprecipitated with an antibody for the desired RBP. The isolated RBP-RNA interactions are then treated with protease to remove bound RBPs, followed by treatment of the “-protein” sample with the probing reagent. As in SHAPE-eCLIP, reverse transcription with the mutational profiling method encodes adduct formation events across transcripts and sequencing delivers the rate of these events at each nucleotide in RBP binding sites. Normalizing “-protein” to “+protein” mutation rates reveals fSHAPE reactivities across RBP binding sites, where high fSHAPE reactivities indicate precise nucleotides that hydrogen bond with protein (Figure 1c). There was some concern that RNA lesions induced by the crosslinking step in eCLIP would produce interfering signal during reverse transcription. However, since the sample pairs compared to each other in both SHAPE- and fSHAPE-eCLIP are UV-crosslinked in the same manner, any replicable lesions that interfere with reverse transcription are controlled for. We will note that due to the similarity in execution between SHAPE-eCLIP and fSHAPE-eCLIP, it is possible to combine the two methods by simply adding an mock-treated control in parallel with fSHAPE-eCLIP, thus returning RBP binding sites, secondary structure information, and nucleobases that contact protein in a single experiment. Refer to Table S1 for a summary of the purpose of each technique discussed in this manuscript.
RESULTS
Footprinting with SHAPE (fSHAPE) transcriptome-wide in human cells
fSHAPE reactivities were measured transcriptome-wide in duplicate on K562, HepG2, 293T (Lu et al., 2016), and HeLa cell lines (Table S2), with good correlations between replicates (Figure S1b). The fSHAPE strategy successfully produces protein footprints on RNA by comparing in vivo (+protein) probed transcripts to protein-removed (-protein) probed transcripts (Figure 1a). fSHAPE dispenses the need for the additional untreated sample used in other SHAPE footprinting experiments (Flynn et al., 2016; Smola et al., 2015a), as correlations between fSHAPE reactivities produced with the untreated sample versus without are very high (Figure S2). We first studied fSHAPE reactivities in relation to known examples of RNA-protein interaction sites, such as the iron response element (IRE) in ferritin light chain (FTL) which binds iron response proteins (Fillebeen et al., 2014; Walden et al., 2012). fSHAPE reactivity profiles in the FTL IRE display consistently prominent spikes in the RNA element’s highly conserved apical loop and bulge, which have been structurally characterized contacting iron response protein (Walden et al., 2006) (Figure 2).
Figure 2.

fSHAPE identifies sites bound by proteins. (a) Example replicate averaged fSHAPE reactivity profiles from four cell types across the iron response element in the Ferritin light chain (FTL) transcript. (b) The predicted secondary structure of the FTL iron response element (left), bases colored according to fSHAPE reactivity and numbered by position in FTL transcript NM_000146. Asterisks indicate bases known to hydrogen bond with iron response element binding protein (IRP1), based on the crystal structure (right; PDBID: 3SNP)(Walden et al., 2006) of IRP1 (green) bound to the FTL iron response element (black). See also Tables S2–3.
Interpreting fSHAPE reactivities
Existing structures of human RBP-RNA complexes allow us to evaluate how accurately fSHAPE reactivities detect RBP interactions with RNA nucleotides. We curated 10 RNA-protein x-ray crystallography structures corresponding to 12 transcript regions that also have fSHAPE data (Table S3). We determined the hydrogen bonds and their bond lengths formed with the base, 2’-OH, or backbone atoms (Fig. 3a) of each nucleotide in the structures in order to quantify the “ground-truth” set of RNA-protein interactions. To determine which types of interactions (protein-RNA base, protein-RNA backbone, and/or protein-RNA sugar) fSHAPE values most correlate with, we compared fSHAPE reactivities against several models of hydrogen bond interactions in the ground-truth structures. Optimal hydrogen bond lengths for each model were fit to fSHAPE reactivities by maximizing receiver operator characteristic (ROC) curve performance (Fig 3b. The best performing yet most parsimonious model achieves an area under the curve (AUC) of 0.82 and indicates that high fSHAPE values are highly correlated to RNA nucleotides that do not pair with other RNA nucleotides (within 3.0 Å) and whose base moieties hydrogen bond with protein within 3.0 Å (Fig 3c and Table S4). This model sheds light on how the probing reagent used to generate fSHAPE data, NAI-N3, reacts with RNA in the presence of protein. Consistent with its use in structure probing, the reagent does not appear to react with nucleotides whose bases hydrogen bond with other RNA bases in a base-pair, but also does not react with RNA whose bases hydrogen bond with protein. Thus, protein residues that interact with RNA “protect” RNA bases from the reagent by pairing with them, rather than stearic hindrance alone. The reagent does not differentially detect bases that pair with both protein and RNA, or backbone and 2’-OH moieties that pair with protein. Thus fSHAPE does not detect binding sites that typically use these modes of interaction, such as sites bound by double-stranded RNA binding proteins (Corley et al., 2020; Sugimoto et al., 2015). It is somewhat surprising that fSHAPE does not appear to detect sugar moieties that hydrogen bond with proteins, given that the probing reagent directly reacts with the 2’-OH. However, this is consistent with the reagent’s use in structure probing, in which reactivity with the 2’-OH is highly correlated with the flexibility of the adjacent base and is used to indicate the paired state of the base (Wilkinson et al., 2006).
Figure 3.

Fitting fSHAPE values to hydrogen bond information from RNA-protein x-ray crystallography structures. (a) Nucleotides known to interact with protein (green) tend to correlate with high fSHAPE reactivities, likely determined by the combination of protein hydrogen bonds (dashed lines) with the backbone, base, and 2’-OH RNA moieties as well as pairing between RNA bases. This set of hydrogen bonds can be quantified in RNA-protein crystal structures as hydrogen bond lengths “BackP”, “BaseP”, “SugarP”, and “BaseR”, respectively. (b) Models made up of combinations of hydrogen bonds lengths were constructed to describe each nucleotide in human RNA-protein crystal structures and bond length threshold (θ) was adjusted to maximize models’ fit to corresponding fSHAPE reactivities. Models were fit to fSHAPE with receiver operator characteristic (ROC) curves; maximum area under the curve (AUC) and corresponding θ in angstroms (Å) shown for each model. The best model indicates excellent agreement between high fSHAPE reactivities and nucleotides whose base moieties form hydrogen bonds under 3.0 Å in length with protein AND do not form hydrogen bonds with other RNA moieties within 3.0 Å. (c) The ROC curve (black) of the model with best agreement between fSHAPE reactivities and crystal structure hydrogen bonds and bounding ROC curves from cross-validation (grey). AUC of each curve indicated.
fSHAPE reactivities in the context of RNA structure
Consistent with its ability to detect protein-bound bases that are otherwise unpaired (Figure 3), we asked if high fSHAPE values correspond to unpaired bases transcriptome-wide (Figure 4). For example, the structural model of the human MYC (c-myc) internal ribosome entry site (IRES) shows high fSHAPE reactivities almost exclusively in bulges and loops (Figure 4a), particularly in the apical loops with demonstrated contributions to IRES-mediated translational control (Le Quesne et al., 2001). Selecting for 200 nucleotides regions around bases with the strongest fSHAPE signals, base pairing probabilities were calculated for these regions via computational structure prediction supported by structure probing data in the same cell lines (see Methods) (Deigan et al., 2009; Lorenz et al., 2011). This yielded ~10,000 non-overlapping regions with K562 fSHAPE data and 3,000–5,000 regions in the other cell lines. Separating bases in these regions into low, middle, and high fSHAPE reactivities, we observe that these bases occupy dramatically different sets of corresponding base pairing probabilities (Figure 4b and Figure S2). As expected, bases with the highest fSHAPE reactivities are predominantly predicted to have a low probability of base pairing, i.e. are unpaired (Figure 4b). Bases with intermediate fSHAPE reactivities, that is, no difference in reactivity between in vivo and protein-removed conditions of structure probing, predominantly correspond to high base pairing probabilities (Figure 4b). Bases with very low fSHAPE reactivities are more difficult to interpret. They may represent structurally dynamic nucleotides that become single-stranded upon RBP binding, and thus these types of nucleotides occupy both paired and unpaired states with equal frequencies (Figure 4b). Additionally, previous structure probing experiments have observed that A and U nucleotides tend to be less frequently base-paired than G and C, and this result is reproduced among fSHAPE reactivities (Figure S3) (Spitale et al., 2015). However, higher fSHAPE reactivities among A and U nucleotides may also suggest that these bases hydrogen bond with protein more frequently, as observed among large numbers of RNA-protein crystal structures and in binding motifs determined by RNA Bind-N-Seq experiments (Corley et al., 2020; Dominguez et al., 2018). fSHAPE reactivities are thus in line with the known chemistry of SHAPE structure probing, while also revealing trends in the chemistry of RBP binding.
Figure 4.

fSHAPE reactivities in the context of RNA structure. (a) Example functional RNA structure, the internal ribosome entry site (IRES) of human MYC (c-myc) (Le Quesne et al., 2001), overlaid with corresponding fSHAPE reactivities in K562 cells. Nucleotides numbered by position in MYC transcript NM_002467. (b) Predicted base pairing probability densities for nucleotides grouped by low, medium, and high fSHAPE reactivities. Median and interquartile range displayed in white. Average base pairing probability indicated above each group. (c) Shannon entropy values predicted for 50-nucleotide regions containing high fSHAPE reactivities compared to 50-nucleotide flanking regions show a downward shift in Shannon entropy (p < 0.01). Median and interquartile range displayed in black. Average Shannon entropy indicated above each type of region. See also Figure S2.
We further assessed the Shannon entropy of nucleotides with high fSHAPE reactivities in order to understand the larger structural context of regions that bind RBPs. Shannon entropy describes the density of the ensemble of secondary structures that an RNA region forms, where low Shannon entropy values indicate stable structural regions and high Shannon entropies indicate more dynamic regions of RNA. We calculated Shannon entropies in the same regions used for base pairing probability regions and averaged the Shannon entropies in a 50 nucleotide window around bases with high fSHAPE values, as well as in their 50 nucleotide flanking regions for comparison. We find that Shannon entropies are significantly lower in transcript regions with high fSHAPE values as compared to flanking regions (Figure 4c and Figure S2). This suggests that the RNA-protein interactions detected by fSHAPE tend to occur in the overarching context of stable structural elements. Indeed, stable RNA stems presenting RBP binding sites in unstructured loops is a common mode of interaction with proteins (Diribarne and Bensaude, 2009; Leppek et al., 2018; Loughlin et al., 2019; Mukhopadhyay et al., 2009; Tan et al., 2013; Walden et al., 2006).
fSHAPE reactivity patterns predict RBP interaction sites
We used patterns in fSHAPE reactivity profiles to predict interaction sites with iron response proteins 1 and 2 (IRP1 and IRP2) transcriptome-wide. IRP1 and IRP2 binding to the iron response element (IRE) in FTL is well-characterized, the former of which is measured binding the IRE with picomolar affinity (Fillebeen et al., 2014; Walden et al., 2012). The IRE consists of a bulge-stem-loop structure with conserved bases in the bulge and apical loop that hydrogen bond with iron response protein, whose binding to an IRE in the 5’UTR regulates translation and binding in the 3’UTR regulates degradation of the transcript (Walden et al., 2012; Walden et al., 2006). Additional IREs have been discovered in the untranslated regions of multiple genes, implying that many more IREs may await detection (Stevens et al., 2011). We reasoned that the clear pattern of fSHAPE reactivities in the IRE of FTL (Figure 2) and its highly conserved sequence would enable a simple search for IREs transcriptome-wide. This strategy first searches the transcriptome for the conserved [GC]NNNNNCAG[AU]G sequence, then compares the pattern of fSHAPE reactivities for each match to the pattern of fSHAPE reactivities in the FTL IRE. Matches whose correlation with the FTL IRE exceeds 0.8 and whose bulge and apical loop bases have sufficiently high fSHAPE reactivities were selected as candidate IREs (Figure 5a). IREs identified by this algorithm include known IREs in FTH1, ALAS2, and multiple IREs in TFRC (5 out of 7 known human IREs with available fSHAPE data) in addition to novel putative IREs (Figure 5b and Figure S4). Putative IREs from CDC34, H19, SLC2A4RG, and COASY were selected and tested for IRP binding via electrophoretic mobility shift assay (EMSA), all demonstrating binding to IRP1 and/or IRP2 by comparison to FTL, which reliably binds either IRP1 or IRP2 depending on cellular conditions (Figure 5c)(Fillebeen et al., 2014; Stys et al., 2011). The IREs in CDC34 and SLC2A4RG are surprisingly found in the coding regions, while the IRE is in the 3’UTR of COASY and near the 3’ end of H19, which is noncoding. All previously known IREs have been found in UTRs, although IREs have been predicted in the coding sequence of a few transcripts (Stevens et al., 2011). To further test how these IRE candidates respond at the transcriptional level to cellular iron levels, we supplemented K562 cells with either an iron source (ferric ammonium citrate; FAC) or an iron chelator (deferoxamine mesylate; DFOM) for 24 hours and measured CDC34, COASY, and SLC2A4RG transcript abundance via quantitative RT-PCR (H19 is not expressed in K562 cells). TFRC, which is known to be negatively regulated by high cellular iron at the transcriptional level, was also measured as a positive control. TFRC transcript abundance relative to housekeeping gene RPL4 strongly increased in response to DFOM and decreased in response to FAC, as expected (Figure S4) (Popovic and Templeton, 2004). CDC34 responded in a similar manner as TFRC, indicating that this transcript is protected by IRPs under low iron conditions. CDC34 is an E2 ubiquitin conjugating enzyme (Williams et al., 2019) without reported links to iron metabolism, except that IRP2 itself is degraded by the ubiquitin pathway in the presence of high cellular iron (Stys et al., 2011). We find that CDC34 levels increase in the presence of DFOM (Figure S4). CDC34 binding by IRP2 suggests a feedback loop whereby CDC34 transcripts are protected from decay by IRP2 (Figure 5c) and also indirectly check IRP2 protein levels, and CDC34 transcript levels increase following extended low iron conditions that increase IRP2 availability. SLC2A4RG and COASY also demonstrate significant changes under low iron conditions, albeit in the opposite direction compared to TFRC and CDC34 (Figure S4). Decreased transcript abundance under low iron conditions—when IRPs are available for binding—suggests a mechanism by which these transcripts are degraded upon IRP recruitment by their IREs, which is contrary to known IRP mechanisms (Popovic and Templeton, 2004; Stys et al., 2011; Walden et al., 2012). SLC2A4RG produces a transcription factor that regulates SLC2A4 expression, reflecting several members of the solute carrier (SLC) gene family known to harbor IREs (Stevens et al., 2011). COASY produces an enzyme whose loss is linked to brain iron accumulation through an unknown mechanism (Levi and Tiranti, 2019). H19 as a noncoding RNA is an unconventional IRE candidate, although the known IRE-containing gene SLC11A2 also produces a non-coding transcript variant (NCBI ID: NR_033421) that contains the same IRE sequence as its coding variant siblings. Recent evidence linking H19 to an inverse relationship with known iron regulator FTH1 argues for a functional role for its IRE, which likely recruits IRP1 to its 3’ end to regulate H19 abundance (Di Sanzo et al., 2018).
Figure 5.

fSHAPE predicts uncharacterized iron response elements (IREs) bound by iron response protein 1/2 (IRP1 1/2). (a) A simple workflow for discovering IREs. Transcript sequences that match the conserved IRE sequence and have fSHAPE data are compared to the FTL IRE’s fSHAPE profile via correlation coefficient (R). R above 0.8 and fSHAPE reactivities at key positions above threshold t (dashed line) are selected as candidate IREs. (b) Selected fSHAPE profiles of IREs predicted by the workflow. Profiles are replicate averages. Pearson correlation compared to FTL is indicated in top left corner, gene name and sequence indicated above each plot. IREs in FTH1, TFRC, and ALAS2 (top row) have been previously verified (Stevens et al., 2011); predicted IREs in CDC34, COASY, H19, and SLC2A4RG (bottom row) are novel. Threshold (t) indicated with dashed line. (c) Electromobility shift assays testing predicted IREs for binding to IRP1 1/2. Biotin-labeled RNA is shown alone, incubated with liver cytosolic extract, or with antibodies (red) to IRP1, IRP2, or Immunoglobulin G (IgG; negative control). FTL IRE, which tightly binds IRP proteins, is shown as a positive control (Fillebeen et al., 2014); h3 stem loop of RN7SK shown as a negative control. Shifted bands in the presence of liver cytosol indicate RNA binding to protein. The release of RNA in the presence of antibodies indicates disruption of RNA-protein binding. See also Figure S4.
SHAPE-eCLIP and fSHAPE-eCLIP application
We next explored the use of enhanced crosslinking and immunoprecipitation (eCLIP) to selectively probe transcript regions bound by an RBP of interest with SHAPE- and fSHAPE-eCLIP (Figure 1b–c). We developed fSHAPE-eCLIP to identify protein-interacting nucleotides in transcripts specifically bound by an RBP of interest, and SHAPE-eCLIP to more effectively interrogate the structural motifs that recruit RBPs, since RNA sequence motifs alone do not account for many protein binding events (Dominguez et al., 2018; Kazan et al., 2010; Maticzka et al., 2014; Pan et al., 2018). (For a summary of the purpose of each technique presented in this manuscript see Table S1.) To validate SHAPE- and fSHAPE-eCLIP approaches we applied them to stem loop binding protein (SLBP) (Figure S5). SLBP has structurally well-characterized binding stem loop elements at the 3’ ends of histone mRNAs (Tan et al., 2013), and published eCLIP binding sites for SLBP reiterates this association (Van Nostrand et al., 2020). Binding sites identified for both SHAPE-eCLIP and fSHAPE-eCLIP closely match known eCLIP binding sites for SLBP, demonstrating that modifications to eCLIP implemented for SHAPE and fSHAPE techniques did not interfere with immunoprecipitation of SLBP-bound transcripts (Figure 6a–b). Two structure probing reagents, dimethyl sulfate (DMS) and 2-methylnicotinic acid imidazolide (NAI), were tested with SHAPE-CLIP to gauge the compatibility of various reagents with eCLIP. DMS yields structure information on adenine and cytosine nucleotides; NAI yields on all four nucleotides (Spitale et al., 2013; Zubradt et al., 2017). Both reagents were successfully implemented in SHAPE-eCLIP (Figure 6), but because NAI returns information on all nucleotides we chose this reagent for implementation in fSHAPE-eCLIP. SHAPE-eCLIP accurately and consistently returns low SHAPE reactivities in the stems of histone mRNA stem loop elements (Figure 6c and Figure S8c), as expected. However, SHAPE-eCLIP reactivities are also consistently low in the apical loop of histone stem loop elements which are unpaired and thus expected to display high SHAPE reactivities (Siegfried et al., 2014). The loop reactivities are much lower than expected because the bases hydrogen bond with SLBP and 3’hExonuclease (Figure 6d), dampening their reactivity with the probing reagent in the same manner as a base-pair. High fSHAPE-eCLIP reactivities in these loops confirm this interpretation (Figure 6d). fSHAPE-eCLIP reactivities across multiple histone mRNA stem loops reveal higher reactivities in the apical loop and the single-stranded region 5’ to the loop, whose bases hydrogen bond with SLBP and its binding partner 3’hExonuclease (Figure 6d–e, lower) (Martin et al., 2012; Tan et al., 2013). Additionally, fSHAPE reactivities in the apical loop are their maximal at the uracil nucleotides previously identified to be most sequence-conserved in the context of SLBP binding (Martin et al., 2012). By comparison, CLIP-seq methods use nucleotide crosslinking rates in RBP binding sites as a proxy indicator of the nucleotide-specific protein interaction site (Lee and Ule, 2018). However, the occurrence of crosslinking is restricted to aromatic amino acids and predominantly uracil and cytosine nucleotides (Hockensmith et al., 1986; Poria and Ray, 2017). Thus crosslinking sites do not necessarily coincide with the select nucleotides that form molecular bonds with protein. For example, nucleotides that display the highest crosslinking rate in previously published eCLIP SLBP binding sites are upstream of the stem loop elements that actually binds SLBP (Figure 6e, upper), contrasting with fSHAPE-eCLIP reactivities that peak in the stem loops of histone transcripts (Figure 6e, lower). In summary, SHAPE-eCLIP and fSHAPE-eCLIP successfully select for and probe transcript regions bound SLBP, corroborating details of the regions’ structure and protein interactions, which are intimately linked.
Figure 6.

SHAPE-eCLIP and fSHAPE-eCLIP applied to stem loop binding protein (SLBP). (a) Read densities mapped to histone mRNA HIST1H1C for published eCLIP datasets (ENCODE)(Van Nostrand et al., 2020). SHAPE-eCLIP under two probing reagent conditions (DMS and NAI), and fSHAPE-eCLIP. Each eCLIP dataset includes an immunoprecipitated sample (IP) and a non-immunoprecipitated negative control (Input). Binding sites inferred in each dataset are indicated as rectangles under read densities. (b) Percent of binding sites inferred in each eCLIP dataset that occur in histone transcripts and compared to previously published (ENCODE) SLBP binding sites. The percent overlap with ENCODE of an equivalent number of randomized binding sites is also shown for each dataset as a negative control. (c) Predicted structure and overlaid SHAPE reactivities from two SHAPE-eCLIP datasets for the stem loop motif of HIST1H1C. Nucleotides numbered by position in transcript. (d) Predicted structure and overlaid fSHAPE reactivities from fSHAPE-eCLIP dataset for the stem loop motif of HIST1H1C. Nucleotides numbered by position in transcript. Higher reactivities indicate bases that hydrogen bond with protein. The crystal structure of SLBP and 3’hExonuclease with canonical stem loop motif (PDBID: 4L8R)(Tan et al., 2013). Bases known to hydrogen bond with either SLBP or 3’hExonuclease are circled in red. (e) Upper: Average crosslinking rates (percent*10 for scale), inferred from truncation events in published SLBP eCLIP datasets, in multiple histone transcripts. Centered by stem loop motif. Lower: Replicate averaged fSHAPE reactivity profiles from fSHAPE-eCLIP in multiple histone transcripts centered by stem loop motif. Average of all profiles shown as dashed line. Stars indicate bases that are known to hydrogen bond with protein. See also Figure S5.
DISCUSSION
We demonstrate here that RNA footprinting with SHAPE structure probing directly detects hydrogen bonds at sites of RNA-protein interfaces across the transcriptome. Many protein interaction sites on RNA occur within structured RNA elements, arguing for the existence of numerous such functional RNA structures. Thus clusters or patterns of hydrogen bonds with protein detected by fSHAPE additionally signals for functional RNA elements and reveals the crucial nucleobases that coordinate regulation by RBPs. We illustrate this concept with a simple fSHAPE pattern and sequence motif search that successfully identifies previously uncharacterized RNA element binding by iron response proteins. Additional such elements may be discovered by a combination of sequence, structure, and fSHAPE pattern searches for bipartite protein binding motifs, which are common platforms for RBP binding (Loughlin et al., 2019; Tan et al., 2013; Walden et al., 2006). More sophisticated strategies for pattern discovery (Saria et al., 2011) within fSHAPE reactivities may accelerate these discoveries, aided by abundant fSHAPE-eCLIP data for numerous RBPs. fSHAPE-eCLIP further enhances fSHAPE study on RBP binding sites of interest, where amassing such datasets for numerous RBPs will provide an atlas of the RNA elements most salient to transcripts’ regulation by proteins. SHAPE probing of RNA secondary structures is similarly enhanced by integration with eCLIP. SHAPE-eCLIP could aid discovery of the structural motifs preferred by any RBP of interest (Dominguez et al., 2018; Kazan et al., 2010; Maticzka et al., 2014; Pan et al., 2018), given that RNA structure predictions are vastly improved by incorporating structure probing data (Deigan et al., 2009; Lotfi et al., 2015; Low and Weeks, 2010; Ramachandran et al., 2013). However, we note that in vivo structure probing signal is convoluted by bases that form hydrogen bonds with protein and thus appear to be base-paired (with RNA) to the naïve observer. Thus, it may be the case that probing secondary structures in vitro delivers information that is more reminiscent of the inherent structure of a given transcript. Deconvolving structural and protein-interaction signals will be enabled by novel strategies for probing RNA (Feng et al., 2018), which themselves could further benefit by coupling with eCLIP. The collection of analyses presented here complement current techniques for the study of RNA-protein complexes, while offering enhanced resolution on the transcripts bound by desired RBPs. In the future these techniques will be instrumental for studying cellular RNA-protein interactions and functional RNA structures at single nucleotide resolution, for any RBP of interest.
Limitations
fSHAPE, like many transcriptome-wide techniques, yields limited data on transcripts that are not highly abundant. By coupling to immunoprecipitation, SHAPE-eCLIP and fSHAPE-eCLIP improve data yield on less abundant transcripts, but are limited by the availability of an antibody for the RNA binding protein of interest. All SHAPE structure probing techniques are usually limited to returning structural information averaged over the ensemble of conformations that an RNA may adopt (Mustoe et al., 2019), thus structures that are more stable yield more clear results with SHAPE-eCLIP. By extension, this also means that the most stable RNA-protein complexes will likely be the easiest to detect with fSHAPE and fSHAPE-eCLIP. It should be noted that the protein-interacting nucleotides detected by fSHAPE and fSHAPE-eCLIP do not reveal the identity of the bound protein, although with fSHAPE-eCLIP it is most likely the protein that we immunoprecipitated for or a partner protein. fSHAPE and fSHAPE-eCLIP do not detect protein interactions with nucleotides that are typically double-stranded. Additionally, fSHAPE and fSHAPE-eCLIP assume, based on evidence, that the structural context of RNA-protein interaction sites is not substantially altered upon protein removal (see Design). However, in cases in which a region’s structure does change in the absence of protein, the fSHAPE signal will be confounded by these structural changes.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Gene W. Yeo, Ph.D. (geneyeo@ucsd.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
All fSHAPE, fSHAPE-eCLIP, and SHAPE-eCLIP raw sequencing reads and processed data are available for download in the Gene Expression Omnibus (GEO), accession number GSE149767. All custom software used to process this data is available at github.com/meracorley and github.com/YeoLab/clipper, and upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
Human K562, Hepg2, and HeLa cells were acquired from ATCC. K562 cells were cultured in RPMI 1640 medium (Gibco) with 10% FBS (Corning) and 1% penicillin/streptomycin (Gibco). HepG2 and HeLa cells were cultured in DMEM media (Gibco) with 10% FBS 1% penicillin/streptomycin. All cells were grown at 37 °C in 5% CO2 and routinely tested with MycoAlert PLUS (Lonza) for mycoplasma contamination.
METHOD DETAILS
fSHAPE
in vivo click selective 2-hydroxyl acylation and profiling experiment (icSHAPE) was performed on cells from K562, HepG2, and HeLa cell lines (293T previously published (Lu et al., 2016)) under both +protein (“in vivo”) and -protein (“in vitro”) conditions, as previously described in detail (Flynn et al., 2016). Briefly, in the +protein condition, 20 million cells were treated with 100mM NAI-N3 at 37 °C for 5 minutes. Cells were centrifuged and supernatant removed to stop the reaction. RNA was extracted from cells with a standard Trizol extraction. Ethanol was added to the aqueous phase, which was isolated and column-purified (Zymo). In the -protein condition, RNA was Trizol extracted from cells as above and column-purified, leaving RNA purified from protein. Purified RNA was heated in water to 95 °C for 2 minutes, then flash-cooled on ice. Denatured RNA was added to SHAPE folding buffer (333 mM HEPES, pH 8.0, 20 mM MgCl2, 333 mM NaCl) and RNA allowed to re-fold at 37 °C for 10 min. Re-folded -protein RNA was probed with 100 mM NAI-N3 for 10 minutes, Reaction was stopped with the addition of buffer RLT (Qiagen) and ethanol, followed by column purification. Treated RNA from +protein and -protein conditions were poly(A)+ selected twice, then biotin-labeled on NAI-N3 adducts via click reaction with DIBO-biotin (Molecular Probes). RNA was fragmented and end repaired, followed by 3’ end ligation with RNA linker (/5rApp/AGAUCGGAAGAGCGGUUCAG/3Biotin/) and size selection. RNA underwent reverse transcription (RT primer: /5phos/DDDNNXXXXNNNNAGATCGGAAGAGCGTCGTGGA/iSp18/GGATCC/iSp18/TACTGAACCGC; XXXX=sample-specific barcode) and magnetic streptavidin bead selection for biotin-labeled RNA:cDNA hybrids. cDNA was circularized, amplified, and size-selected. Libraries were sequenced to a depth of approximately 200 million reads. Untreated samples were also prepared for each cell line as above (although this is not necessary for fSHAPE), in which cells were treated with DMSO rather than NAI-N3 and RNA ligated to a biotin-conjugated RNA linker (/5rApp/AGAUCGGAAGAGCGGUUCAG/3Biotin/) to facilitate RNA pull-down in the absence the click reaction with DBO-biotin. “in vitro”/”in vivo” icSHAPE data from 293T cells (Lu et al., 2016) was re-analyzed here in parallel with K562, HepG2, and HeLa to produce fSHAPE reactivities. Raw sequencing reads as well as processed reactivities are available on the Gene Expression Omnibus (GEO) as GSE149767.
fSHAPE data analysis
All code for the processing of sequencing data is made available at github.com/meracorley/fSHAPE. Reads were barcode trimmed with cutadapt 1.14 (parameters: -a AGATCGGAAGAGCGGTTCAGCAGGAATGCCGAGACCGATCTCGTATGCCGTCTTCTGCTTG --minimum-length=23 --overlap=5), mapped to GRCh38 with Star aligner version 2.4.0i (parameters: --outSAMstrandField intronMotif --outFilterIntronMotifs RemoveNoncanonical), and de-duplicated with UMItools 0.5.0 (parameters: --bc-pattern=NNNNCCCCNNNNN (for HeLa and 293T samples) --bc-pattern=NNNCCCCNNNNN (for K562 and HepG2 samples); –spliced-is-unique -S). Aligned, unique reads were separated based on chromosome and strand (genome build GrCh38). To calculate the frequency of reverse transcription-induced truncation events, 5’ end read coverage (truncation events) and total read coverage at each position across the genome was counted via bedtools 2.25.0 (parameters: genomcov −5 -strand -dz and genomcov -split -strand -dz, respectively) (Quinlan and Hall, 2010). Script bedReactivities.py (github.com/meracorley/fSHAPE) was used to calculate normalized drop-off frequencies (fSHAPE reactivities) at each nucleotide in every transcript (NCBI RefSeq Hg38). Special normalization procedures were implemented to handle the artifact of drop-off events dropping to 0 immediately 5’ to a gap in total read coverage (Figure S6), which erroneously reports these regions’ drop-off frequencies as 0 rather than “no data.” To address the “5’ drop-off” artifact, a Hidden Markov Model was trained on K562 “-protein” SHAPE total coverage data from several transcripts with high coverage interrupted by gaps. Each nucleotide was categorized as state ‘0’ if its coverage was below 200, and state ‘1’ otherwise, 200 being the previously determined cutoff of coverage that produces acceptable reproducibility between replicates. States were used to train a two-state multinomial Hidden Markov Model with hmmlearn 0.2.1 in scikit-learn, which labels regions as “1” (covered) or “0” (no coverage). Total read coverage tends to be monotonic, such that when total read coverage drops to 0 it tends to do so gradually. We found that the 5’ drop-off artifacts tended to occur where the total coverage peaked before a gap in total coverage. Thus, once total coverage regions are categorized via the Hidden Markov Model, the local maxima of total coverage occurring before any ‘no coverage’ regions is set as the “true” starting point of the coverage gap and the drop-off rate is assessed as “no data.” fSHAPE reactivities are calculated as the drop-off frequency in the +protein sample minus drop-off rate in the -protein sample for each nucleotide, divided by a normalization factor based on all the raw reactivities in the given region as in the ShapeMapper 2.0 pipeline (Smola et al., 2015b). Briefly, the normalization factor for each transcript is calculated as the average of the top 10% of values below a cutoff--either the highest 10% of values or 1.5*(the value at the top of the third quartile minus the value at the top of the first quartile), whichever includes fewer values. Final fSHAPE reactivities are output in the form of .map and .rx files for each transcript in the human reference transcriptome (NCBI RefSeq Hg38), GEO access GSE149767. Files denote the fSHAPE reactivity at each nucleotide for each replicate (.rx format) or the nucleotide number, average reactivity, variance, and base identity (.map format). Nucleotides without reactivity data are denoted as “−999.”
Correlations between replicates
Correlation coefficients between fSHAPE replicates were calculated for transcripts within each cell sample as follows. Transcript per million (TPM) expression was calculated for each transcript in each cell line (K562, HepG2, HeLa, 293T) with Sailfish 0.10.0. For each gene, a single transcript with highest TPM was selected. If TPM>150 and the transcript contained data across >30% of its length, the correlation coefficient between the cell sample’s replicates was taken in rolling 50 nucleotide windows across the transcript and averaged. Similarly for SHAPE-eCLIP and fSHAPE-eCLIP replicates, correlation coefficients were calculated in rolling 50 nucleotide windows between all transcripts with data covering >30% of their length.
Hydrogen bond analyses
10 human RNA-protein x-ray crystallography structures were selected for their representations of portions of human transcripts (rather than random RNA fragments) that also have fSHAPE reactivity data in at least one cell line. Structures were downloaded from the protein databank (PDB) and matched to 12 regions in transcripts that are represented by the RNA fragments in the structures (Table S2). fSHAPE reactivities from all four cell lines were extracted (where available) for the matching transcript regions and re-normalized by region and outliers removed. Hydrogen bonds in PDB files were assessed by HBPLUS (McDonald and Thornton, 1994), which outputs all detected hydrogen bonds between any two moieties, including water molecules (parameters: -d 3.35 -h 2.7). RNA-protein and RNA-RNA hydrogen bonds occurring with each moiety (backbone, 2’-OH, or base) of each nucleotide in RNA-protein structures were compiled and quantified by bond length (θ) (script: process_hb2.py from github.com/meracorley/hbplus_tools, parameters: -R). For a given model of hydrogen bonds (Figure 2b), nucleotides in each structure were labeled as “cases” if their set of hydrogen bonds fulfilled the model and “controls” if not. fSHAPE reactivities corresponding to cases and controls were assessed with receiver operator characteristic (ROC) curves (R package pROC 1.14.0; expect controls<cases), modulating θ to maximize the area under each curve.
Base-pairing probabilities and Shannon entropy
High quality transcripts were selected as above (correlations between replicates). Nucleotides with fSHAPE reactivities above 4.0 (top 1%) were selected and 200 base regions around them were defined. Sequences in the 200 base regions were extracted and their base pairing probability matrix predicted with RNAfold 2.4.14 (parameters: -p), supported by icSHAPE reactivities calculated with “-protein” samples normalized to untreated samples in each cell line (available in GEO, GSE149767). Base pairing probabilities and Shannon entropies were calculated for each nucleotide (script: shannonEntropy_rnafold.py from github.com/meracorley/RNAstructure_tools) in these transcript regions using the predicted base pairing probability matrices. The central 50 bases around the high fSHAPE-valued base(s) were taken as the “high fSHAPE region” while the flanking 50 base regions were taken as “flanking regions,” and Shannon entropies were averaged in each of these sub-regions for each transcript and plotted. Similarly, The base pairing probability matrices for the above 200 base transcript regions were used to calculate the sum of base pairing probabilities for each nucleotide (script:getBPprobs_rnafold.pyfromgithub.com/meracorley/RNAstructure_tools). Bases were grouped by high (fSHAPE > 2.5), medium (−2.0 < fSHAPE < 2.0), or low fSHAPE (fSHAPE < −2.5) reactivity and plotted according to base pairing probability.
Predicting Putative Iron Response Elements (IREs)
Sequence matches to the IRE motif ([CG]NNNNNCAG[AU]G) were searched transcriptome-wide. For multiple matching transcript isoforms per gene, the transcript with the highest expression was selected. Sequence matches with fSHAPE data were compared to the FTL IRE fSHAPE profile in the form of a Pearson correlation coefficient. Matches that exceeded a correlation coefficient of 0.8 and whose fSHAPE values at positions 1 and 7, 8, or 9 of the sequences motif were greater than 1.0 were selected as candidate IREs. Minimum free energy structures for each candidate IRE were predicted with RNAfold 2.4.14 using default settings to further select for candidates for electromobility shift assays.
Electromobility Shift Assays
To test for putative iron response element (IRE) binding to IRP1, RNA oligonucleotides were obtained for four predicted IREs. CDC34: rGrUrGrGrArCrGrArCrCrCrCrCrArGrArGrCrGrGrGrGrArGrCrUrGrC COASY: rCrUrGrGrUrUrCrArGrGrCrCrCrArGrArGrGrUrCrCrArArGrCrUrArUrA SLC2A4RG: rGrArUrGrGrGrArCrCrUrGrGrCrCrArGrUrGrArCrCrArGrUrCrCrUrCrUrC, H19: rGrCrArCrUrGrGrCrCrUrCrCrArGrArGrCrCrCrGrUrGrGrCrCrArArG. RNA oligonucleotides were 3’ biotinylated (Fisher Scientific cat# 20160MI) at 16 °C for 2 hours and purified (Zymo Research cat# R1080). The labeled RNA control from biotin labeling kit (Fisher Scientific cat# 20160MI), which is the FTL IRE, was used as the positive control RNA: rUrCrCrUrGrCrUrUrCrArArCrArGrUrGrCrUrUrGrGrArCrGrGrArArC. The “Hairpin 3” from RN7SK (Diribarne and Bensaude, 2009) served as a negative control: DNA oligonucleotide TAATACGACTCACTATAGGGTATACGAGTAGCTGCGCTCCCCTGCTAGAACCTCCAAACAA GCTCTCAAGGTCCATTTGTAGGAGAACGTAGGGTAGTCAAGCTTCCAACAACAACA was in vitro transcribed (NEB T7 cat# E2040S), purified and size selected with 6% urea-PAGE, then biotin labeled and purified as above. IRP1 protein was supplied in the form of human liver cytosolic extract (Life Technologies cat# HMCYPL). Conditions for all 20 uL EMSA binding reactions, 2 uL 10X “RNA EMSA” buffer (100 mM HEPES, pH=7.3, 200 mM KCl, 10mM MgCl2, 10mM DTT), 2uL 50% glycerol. Each RNA was tested under two conditions: biotin-labeled RNA alone, and biotin-labeled RNA plus cytosolic liver extract. The FTL positive control reactions contained 125 fmol biotin-labeled FTL IRE RNA and 2 ug cytosolic liver extract, RN7SK negative control contained 100 fmol and 2 ug liver cytosol. 650 fmol CDC34 and COASY with 2 ug liver cytosol, 650 fmol SLC2A4RG and H19 biotin-labeled RNA with 40 ug liver cytosol. Supershift assays were performed on FTL and CDC34 IREs, where 125 fmol and 650 fmol of biotin-labeled RNA was incubated alone or with 0.5 ug and 20 ug liver cytosol, respectively. Reactions were assembled and incubated at 25 °C for 30 minutes. 2ug of either IRP1 (SCBT E-12 lot #H0117), IRP2 (SCBT 4G11 lot # F317), or Immunoglobulin G antibodies (mouse) were added to FTL and CDC34 samples and incubated for 10 minutes further. All assays were then loaded onto a 6% native TBE gel with TBE loading buffer (Life Technologies cat# LC6678) and run at 100V for 40 minutes in 0.5X TBE buffer. Gel was subsequently transferred to nylon membrane (Amersham Hybond -XL GE Healthcare) via standard transfer setup in cold 0.5X TBE buffer at 35V for 30 minutes. RNA was crosslinked to membrane with UV light at 120 mJ/cm2 for 1 minute. Membrane was processed with chemiluminescent nucleic acid detection module (Thermo Fisher cat# 89880) followed by exposure to film.
Quantification of candidate IRE-containing transcripts in response to iron
K562 cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum. At a density of 5 × 105 per mL, 1.25M cells were treated for 24 hr, in biological triplicates, with 20 mg/ml ammonium iron(III) citrate (FAC, Acros Organics) or 0.1 mM deferoxamine mesylate (DFOM, Sigma-Aldrich). K562 cells were collected, centrifuged at 300 x g for 3 min, washed with DPBS, and centrifuged again. Cell pellets were resuspended in TRIzol Reagent (Invitrogen) and RNA was extracted using the Direct-zol RNA Miniprep Kit (Zymo Research). Concentrations of purified RNA were determined using a Nanodrop spectrophotometer. Equal amounts of cDNA were synthesized using the SuperScript III First-Strand Synthesis System (Invitrogen) and 25 pmol oligo-dT and 25 ng random hexamer primers. qPCR was performed, in technical triplicates, using a cDNA equivalent of approximately 25 ng of total RNA, 10 uM each of gene-specific forward and reverse primers (see Table S4), and Power SYBR Green Master Mix (Applied Biosystems). Quantitative PCR was performed at 95°C for 10 min and 40 cycles of 95°C for 15 s and 60°C for 1 min. Treatment-dependent target gene fold expression change was calculated using the ΔΔCt method by first normalizing technical triplicates to a housekeeping gene, RPL4, then normalizing treated to untreated technical triplicates. The resulting ΔΔCt values were averaged and used to calculate fold change in expression (2^(-ΔΔCt)) for each set of technical triplicates. These were subsequently averaged to calculate fold expression change for each gene target and in each treatment condition. Significance was calculated using a paired t-test.
SHAPE-eCLIP
40 million K562 cells per sample were resuspended in 4mL RPMI media (Gibco) in 10 cm plates. Cells were injected with 100 uL pure DMS (treated samples) or left untreated, mixed, and incubated at 37 C for 3 minutes. Cells were placed on chilled metal plate and crosslinked (lids removed) with UV-C light at 4000 U for 2 minutes. All samples were treated with 2 mL 40% 2-mercaptoethanol to quench excess DMS. (SHAPE-eCLIP samples treated with NAI rather than DMS used the same starting material and volumes, but were treated with 200 uL 2M NAI in DMSO (Neta biosciences) or 200 uL DMSO, mixed, and incubated at 37 °C for 10 minutes, then crosslinked as above.) Crosslinked cells were spun down, supernatant removed, and resuspended in cold phosphate buffered saline (PBS). PBS wash was repeated twice; cell pellets were flash frozen on dry ice and stored at −80 C. Cell pellets from treated and untreated samples were used as the starting point for single-end enhanced crosslinking and immunoprecipitation on SLBP, as previously described (Van Nostrand et al., 2016), with modifications. Briefly, cells lysates were sonicated and briefly RNase treated to select for RBP protected RNA fragments, then immunoprecipitated overnight with SLBP antibody (MBLI) and anti-rabbit secondary antibody-conjugated magnetic beads. 2% of each immunoprecipitated (IP) sample was saved as Input control. (Input controls are not needed for both reagent treated and untreated IP samples; one set is sufficient). IP samples were washed on magnet and underwent alkaline phosphatase and polynuceotide kinase treatment followed by RNA 3’ linker ligation (InvRiL19: /5Phos/rArGrArUrCrGrGrArArGrArGrCrArCrArCrGrUrC/3SpC3/). IP samples were decoupled from beads both IP and Input samples run on a 4–12% Bis-Tris gel. Samples were transferred from gel to nitrocellulose membrane at 4 °C. Bands at the appropriate SLBP protein size plus 75 kDa above were cut from the nitrocellulose membrane. RNA was eluted and protein removed with proteinase K treatment and RNA spin column clean-up (Zymo). Input samples then underwent alkaline phosphatase and polynuceotide kinase treatment followed by RNA 3’ linker ligation. Both IP and Input samples then underwent cDNA synthesis. Importantly, RNA reverse transcription was modified to perform mutational profiling of the DMS-probed transcripts, as described previously (Siegfried et al., 2014). Specifically, 9 uL of each RNA sample was added to 1 uL of 5 uM reverse primer (InvAR17: CAGACGTGTGCTCTTCCGA) and 1 uL of 10mM dNTPs, heated to 65 °C for 2 minutes, then placed on ice. 5.56 uL water, 2 uL 10X SHAPE buffer (500 mM Tris-HCl, pH 8.0, 750 mM KCl), 1 uL 0.1 M DTT, 0.2 uL RNase inhibitor, 1 uL Superscript II, and 0.24 uL 500 mM manganese chloride (to a concentration of 6 mM) was added to each sample and incubated at a temperature of 45 °C for 3 hours. cDNA was cleaned with Silane beads, ligated to a 5’ Illumina compatible linker (InvRand3Tr3: /5Phos/NNNNNNNNNNAGATCGGAAGAGCGTCGTGT/3SpC3/), and quantified via qPCR. Libraries were PCR amplified with barcoded Illumina compatible primers (Forward: AATGATACGGCGACCACCGAGATCTACACNNNNNNNNACACTCTTTCCCTACACGACGCTCTTCCGATCT, Reverse: CAAGCAGAAGACGGCATACGAGATNNNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC barcode positions underlined) based on individual qPCR quantification, cleaned with Ampure xP beads, and size selected to a final size of 180–350 nucleotides with a 3% low melting temperature agarose gel (NuSieve GTG, cat# 50080). Each sample library was sequenced to a depth of approximately 40 million reads.
fSHAPE-eCLIP
40 million K562 cells per sample were resuspended in 4mL RPMI media (Gibco) in 10 cm plates. Cells were injected with 200 uL 2M NAI in DMSO (Neta biosciences) for the “+protein” sample or 200 uL DMSO for the “-protein” sample, mixed, and incubated at 37 °C for 10 minutes. Cells were placed on chilled metal plate and crosslinked (lids removed) with UV light at 4000 U for 2 minutes. Crosslinked cells were spun down, supernatant removed, and resuspended in cold PBS. PBS wash was repeated twice; cell pellets were flash frozen on dry ice and stored at −80 °C. Cell pellets from NAI-treated and untreated samples were used as the starting point for single-end enhanced crosslinking and immunoprecipitation on SLBP and ACO1 combined with structure probing. Cells lysates were sonicated and briefly RNase treated to select for RBP protected RNA fragments, then immunoprecipitated overnight with SLBP antibody (MBLI) and anti-rabbit secondary antibody-conjugated magnetic beads. 4% of each immunoprecipitated (IP) sample was saved as Input control. (Technically Input controls are not needed for both +protein and -protein IP samples; one set is sufficient). IP samples were washed on magnet. 20% of each IP sample and 50% of each Input control was saved for test western blot confirming successful pull-down of protein. The remainder of each sample was treated with proteinase K to remove protein, and the resulting RNA was column purified (Zymo). +protein samples treated with NAI at the cell stage were set aside. Samples that were not treated with NAI are the “-protein” samples, and were refolded and probed with NAI. In detail, 11.4 uL “in vitro” samples were heated to 95 °C for 2 minutes, placed on ice to cool, then added to 6.6 uL 3.3X SHAPE folding buffer (333 mM HEPES, pH 8.0, 20 mM MgCl2, 333 mM NaCl), with 1 uL RNase inhibitor and folded at 37 °C for 5 minutes. 1 uL 2M NAI was added, and samples were further incubated at 37 °C for 10 minutes. Samples were cleaned with a Zymo column to remove excess NAI. As in eCLIP, both “+protein” and “-protein” RNA was then FastAP and PNK treated, followed by Zymo column purification. Samples underwent 3’ RNA linker ligation (InvRiL19: /5Phos/rArGrArUrCrGrGrArArGrArGrCrArCrArCrGrUrC/3SpC3/), followed by denaturation at 65 C for 3 minutes in RNA running buffer and purification on a 6% TBE Urea gel (180V for 40 minutes). Each sample was spaced with a low-range RNA ladder (NEB). Gel was stained for 5 minutes in a SYBR Gold solution. Samples were cut from the gel in the range of 50–200 bases, and RNA was isolated using a Zymo small RNA PAGE recovery kit. RNA was reverse transcribed according to the mutational profiling method. Specifically, 9 uL of each RNA sample was added to 1 uL of 5 uM reverse primer (InvAR17: CAGACGTGTGCTCTTCCGA) and 1 uL of 10mM dNTPs, heated to 65 C for 2 minutes, then placed on ice. 5.56 uL water, 2 uL 10X SHAPE buffer [500 mM Tris-HCl, pH 8.0, 750 mM KCl], 1 uL 0.1 M DTT, 0.2 uL RNase inhibitor, 1 uL Superscript II, and (importantly) 0.24 uL 500 mM manganese chloride (to a concentration of 6 mM) was added to each sample and incubated at a temperature of 45 °C for 3 hours. cDNA was cleaned with Silane beads, ligated to a 5’ Illumina compatible linker (InvRand3Tr3: /5Phos/NNNNNNNNNNAGATCGGAAGAGCGTCGTGT/3SpC3/), and quantified via qPCR. Libraries were PCR amplified with barcoded Illumina compatible primers (Forward: AATGATACGGCGACCACCGAGATCTACACNNNNNNNNACACTCTTTCCCTACACGACGCTCTTCCGATCT, Reverse: CAAGCAGAAGACGGCATACGAGATNNNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC barcode positions underlined) based on individual qPCR quantification, cleaned with Ampure xP beads, and size selected to a final size of 180–350 nucleotides with a 3% low melting temperature agarose gel (NuSieve GTG, cat# 50080). Each sample library was sequenced to a depth of approximately 40 million reads. Raw sequencing reads are available on GEO GSE149767.
SHAPE-eCLIP and fSHAPE-eCLIP data analysis
All code for the processing of sequencing data is made available at github.com/meracorley/f-SHAPE-eCLIP. IP and Input sample reads from SHAPE-eCLIP and fSHAPE-eCLIP were trimmed, mapped to the human genome (GrCh37, converted to GrCh38), and de-duplicated with the eCLIP pipeline (available at github.com/YeoLab/eclip) (Van Nostrand et al., 2016), which also calls RBP binding peaks in IP samples given the background of Input samples. De-duplicated reads mapped by the eCLIP pipeline from IP samples in SHAPE-eCLIP or fSHAPE-eCLIP were the starting point for SHAPE or fSHAPE data analysis. Total read coverage and mutation events were counted across the genome using sorted, uniquely mapped reads (script countMutationsBam.py), and stored by chromosome. Mutation events indicate nucleotides that formed an adduct with the probing reagent (DMS or NAI). Mutation frequencies at each nucleotide across transcripts were calculated as in fSHAPE analysis, except the hmmlearn adjustment was not necessary and G and U bases are ignored for DMS-treated samples (script bedReactivities.py). Among SHAPE-eCLIP experiments, the untreated sample mutation rates are subtracted from treated mutation rates and normalized to produce SHAPE reactivities. For the fSHAPE-eCLIP experiment, the +protein sample mutation rates are subtracted from -protein mutations rates and normalized to produce fSHAPE reactivities at each nucleotide. Final reactivities are output in the form of .map and .rx files for each transcript in the human reference transcriptome (NCBI RefSeq Hg38), GEO access GSE149767. Files denote the (f)SHAPE reactivity at each nucleotide for each replicate (.rx format) or the nucleotide number, average reactivity, variance, and base identity (.map format). Nucleotides without reactivity data are denoted as “−999.”
Crosslinking rates in SLBP binding sites
5’ end coverage and total read coverage at each nucleotide across histone transcripts was calculated (bedtools, parameters: genomcov −5 -strand -dz and genomcov -split -strand -dz, respectively) from mapped SLBP eCLIP reads from the ENCODE project (Van Nostrand et al., 2016). 5’ ends represent RT drop-off (truncation) events and occur more frequently at nucleotides crosslinked to protein (analogous to SHAPE probing). Crosslinking rate at each nucleotide was calculated as the 5’ end coverage divided by total coverage and averaged across eCLIP replicates.
QUANTIFICATION AND STATISTICAL ANALYSIS
Profiles for fSHAPE, fSHAPE-eCLIP, and SHAPE-eCLIP were determined by averaging reactivities across duplicates, using the software pipeline appropriate to each method (see Method Details). For fSHAPE and fSHAPE-eCLIP, we generally consider values above ~2.0 to strongly indicate nucleotides that hydrogen bond with protein, where these values represent the top 2.5% of fSHAPE reactivities. Receiver operator characteristic and area under the curve analyses (Figure 3) were calculated with the pROC package in R (see Method Details). The statistical significance of regions’ average Shannon entropies compared to flanking regions (Figure 4) was calculated by shuffling each region and its flanking regions 100 times and counting the number of instances in which the shuffled differential was equal to or more than actual differential. Where <1/100 instances yields a p-value of < 0.01. The correlation between candidate iron response elements’ fSHAPE reactivities and the FTL iron response element (Figure 5) was calculated with a simple Pearson correlation coefficient (numPy) between fSHAPE profiles. Quantitative PCR results for candidate iron response elements (Figure S4) were determined by averaging technical triplicates followed by further averaging biological triplicates (see Method Details). Standard deviation of biological replicates and t-tests were used to identify significant differences between conditions.
ADDITIONAL RESOURCES
Detailed Protocol
Methods S1 in the Supplement provides bench protocol details for the methods fSHAPE, fSHAPE-eCLIP, and SHAPE-eCLIP.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Stem loop binding protein | MBLI | RN045P Lot# 001 |
| Iron response protein 1 | SCBT | E-12 lot #H0117 |
| Iron response protein 2 | SCBT | 4G11 lot #F317 |
| Immunoglobulin G (mouse) | Thermo Fisher | Catalog # 02–6502 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| TRIzol Reagent | Invitrogen | Cat# 15596026 |
| NAI-N3 | Synthesized in house | N/A |
| Buffer RLT | Qiagen | cat # 79216 |
| DIBO-biotin | Molecular Probes | Cat# 10130670 |
| D-Biotin | Molecular Probes | Cat# B20656 |
| CircLigase II ssDNA ligase | Epicentre | Cat# CL9025K |
| DMSO | Sigma-Aldrich | Cat# D2650 |
| Dynabeads MyOne streptavidin C1 | Life Technologies | Cat# 65002 |
| Dynabeads M-280 sheep anti-rabbit 10 mg/ml | LifeTech | Cat# 11204D |
| Dynabeads MyOne Silane 40 mg/ml | LifeTech | Cat# 37002D |
| Agencourt AMPure XP beads | Beckman Coulter | Cat# A63881 |
| RNA fragmentation reagents | Ambion | Cat# AM8740 |
| T4 polynucleotide kinase | New England BioLabs | Cat# M0201L |
| FastAP thermosensitive alkaline phosphatase | Thermo Scientific | Ct# EF0651 |
| Proteinase K | New England BioLabs | Cat# P8107S |
| SuperScript III Reverse Transcriptase | LifeTech | Cat# 18080044 |
| Turbo DNase | LifeTech | Cat# AM2239 |
| T4 RNA ligase 1, high concentration | New England BioLabs | Cat# M0437M |
| Phusion high-fidelity PCR master mix | New England BioLabs | Cat# M0531L |
| RiboLock RNase inhibitor | Thermo Scientific | Cat# EO0384 |
| RNase H | Enzymatics | Cat# Y9220L |
| RNase cocktail enzyme mix | Ambion | Cat# AM2286 |
| Q5 PCR Master Mix | New England BioLabs | Cat# M0492L |
| Protease Inhibitor Cocktail III | EMD Millipore | Cat# 539134–1SET |
| Exo-SAP-IT | Affymetrix | Cat# 78201 |
| UltraPure 1 M Tris-HCI buffer, pH 7.5 | Invitrogen | Cat# 15567–027 |
| Tween-20 | Sigma Aldrich | Cat# P1379–500ML |
| Chloroform | Ricca Chemical | Cat# RSOC0020–500C |
| HEPES, 1 M | Life Technologies | Cat# 15630–080 |
| Magnesium chloride, 1 M | Ambion | Cat# AM9530G |
| Sodium chloride, 5 M | Ambion | Cat# AM9759 |
| Buffer RWT | Qiagen, | Cat# 1067933 |
| Nonidet P40 (NP-40) | Roche | Cat# 11332473001 |
| N-Lauroylsarcosine sodium salt solution (20%, for molecular biology) | Sigma Aldrich | Cat# L7414–10ML |
| Deoxycholic acid sodium salt | Fisher Scientific | Cat# BP349–100 |
| eCLIP Lysis Buffer | 50 mM Tris-HCl pH 7.4, 100mM NaCl, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate | N/A |
| eCLIP High Salt Wash Buffer | 50 mM Tris-HCl pH 7.4, 1 M NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate |
N/A |
| eCLIP Wash Buffer | 20 mM Tris-HCl pH 7.4, 10 mM MgCl2, 0.2% Tween-20, 5 mM NaCl |
N/A |
| eCLIP RLTW Buffer | RLT Buffer, 0.025% Tween-20 | N/A |
| eCLIP PKS Buffer | 100mM Tris-HCl pH 7.4, 50mM NaCl, 10mM EDTA, 0.2% SDS |
N/A |
| 3.3X SHAPE Folding Buffer | 333mM HEPES pH 8.0, 20mM MgCl2, 333mM NaCl | N/A |
| 10X SHAPE FS Buffer | 500mM TrisHCl pH 8.0, 750 mM KCl | N/A |
| Cytosolic liver extract | Life Technologies | Cat# HMCYPL |
| Ammonium iron(III) citrate | Acros Organics | Cat# AC612215000 |
| Deferoxamine mesylate | Sigma Aldrich | Cat# D9533 |
| Dimethyl sulfate | EMD Millipore | Cat# D186309 |
| 2-methylnicotinic acid imidazolide (NAI) | EMD Millipore | Cat# 03–310 |
| Manganese (II) chloride solution 1M | Sigma Aldrich | Cat# M1787 |
| 10X RNA EMSA Binding Buffer | 100mM HEPES (7.3), 200mM KCl, 10mM MgCl2, 10mM DTT |
N/A |
| Glycerol | Thermo Scientific | Cat# 15514011 |
| Power SYBR Green PCR Master Mix | Applied Biosystems | Cat# 4368577 |
| Critical Commercial Assays | ||
| RNA Clean and Concetrator Kit | Zymo Research | Cat# R1019 |
| Small-RNA PAGE Recovery Kit | Zymo Research | Cat# R1070 |
| Direct-zol RNA Miniprep Kit | Zymo Research | Cat# R2050R2051 |
| Poly(A)Purist MAG kit | Ambion | Cat# AM1922 |
| RNA 3’ End Biotinylation Kit | Pierce | Cat# 20160 |
| Chemiluminescent Nucleic Acid Detection Module | Thermo Scientific | Cat# 89880 |
| Deposited Data | ||
| Sequencing data and processed fSHAPE and SHAPE reactivities | This paper | Gene expression omnibus (GEO), GSE149767 |
| Experimental Models: Cell Lines | ||
| K562 | ATCC | CCL-243 |
| HepG2 | ATCC | HB-8065 |
| HeLa | ATCC | CCL-2 |
| Oligonucleotides | ||
| /5rApp/ rArGrArUrCrGrGrArArGrArGrCrGrGrUrUrCrArG /3ddC/ | IDT | N/A |
| /5rApp/rArGrArUrCrGrGrArArGrArGrCrGrGrUrUrCrArG/3Biotin/ | IDT | N/A |
| 5′-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGA CGCTCTTCCGATCT-3′ | IDT | N/A |
| 5′-CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTG CTGAACCGCTCTTCCGATCT-3’ | IDT | N/A |
| /5phos/DDDNNXXXXNNNNAGATCGGAAGAGCGTCGTGGA/iSp18/GGATCC/iSp18/TACTGAACCGC; XXXX=sample barcode | IDT | N/A |
| 5’-rGrUrGrGrArCrGrArCrCrCrCrCrArGrArGrCrGrGrGrGrArGrCrUrGrC-3’ | IDT | N/A |
| 5’- rGrArUrGrGrGrArCrCrUrGrGrCrCrArGrUrGrArCrCrArGrUrCrCrUrCrUrC-3’ | IDT | N/A |
| 5’- rGrCrArCrUrGrGrCrCrUrCrCrArGrArGrCrCrCrGrUrGrGrCrCrArArG-3’ | IDT | N/A |
| 5’- rCrUrGrGrUrUrCrArGrGrCrCrCrArGrArGrGrUrCrCrArArGrCrUrArUrA-3’ | IDT | N/A |
| 5’-rUrCrCrUrGrCrUrUrCrArArCrArGrUrGrCrUrUrGrGrArCrGrGrArArC-3’ | IDT | N/A |
| 5’-AATACGACTCACTATAGGGTATACGAGTAGCTGCGCTCCCCTGCTAGAACCTCCAAACAAGCTCTCAAGGTCCATTTGTAGGAGAACGTAGGGTAGTCAAGCTTCCAACAACAACA-3’ | IDT | N/A |
| Primers for qPCR, see Table S4 | ||
| /5Phos/rArGrArUrCrGrGrArArGrArGrCrArCrArCrGrUrC/3SpC3/ | IDT | N/A |
| 5’-CAGACGTGTGCTCTTCCGA-3’ | N/A | |
| /5Phos/NNNNNNNNNNAGATCGGAAGAGCGTCGTGT/3SpC3/ | IDT | N/A |
| 5’-AATGATACGGCGACCACCGAGATCTACACNNNNNNNNACACTCTTTCCCTACACGACGCTCTTCCGATCT-3’ | IDT | N/A |
| 5’-CAAGCAGAAGACGGCATACGAGATNNNNNNNNGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC-3’ | IDT | N/A |
| Software and Algorithms | ||
| Cutadapt 1.14 | (Martin, 2011) | https://cutadapt.readthedocs.io/en/stable/ |
| UMItools 0.5.0 | (Smith et al., 2017) | https://github.com/CGATOxford/UMI-πtools |
| STAR 2.4.0i | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| Bedtools 2.25.0 | (Quinlan and Hall, 2010) | https://bedtools.readthedocs.io/en/latest/ |
| Samtools 1.9 | (Li et al., 2009) | https://samtools.github.io |
| RNAfold 2.4.14 | (Lorenz et al., 2011) | https://www.tbi.univie.ac.at/RNA |
| Clipper pipeline | (Van Nostrand et al., 2016) | https://github.com/YeoLab/clipper |
| fSHAPE reactivity software | This paper | github.com/meracorley/fSHAPE |
| fSHAPE- and SHAPE-eCLIP reactivity pipelines | This paper | github.com/meracorley/f-SHAPE-eCLIP |
| Hmmlearn 0.2.1 | Scikit-learn | https://hmmlearn.readthedocs.io/en/latest/ |
| HBPLUS | (McDonald and Thornton, 1994) | https://www.ebi.ac.uk/thornton-srv/software/HBPLUS/ |
| Hbplus_tools | (Corley et al., 2020) | github.com/meracorley/hbplus_tools |
| RNAstructure_tools | This paper | github.com/meracorley/RNAstructure_tools |
| Other | ||
| Detailed protocols | This paper | Methods S1 |
HIGHLIGHTS.
fSHAPE compares protein-absent and -present conditions to probe RNA-protein interfaces.
fSHAPE identifies nucleobases that hydrogen bond with protein.
Patterns in fSHAPE signal detect specific protein-binding RNA elements.
SHAPE and fSHAPE with eCLIP selectively probes RNA bound by proteins of interest.
RNA is universally regulated by RNA binding proteins (RBPs), which interact with specific sequence and structural RNA elements. Corley et al develop several technologies to probe specific RNA-protein complexes, revealing the nucleotides that hydrogen bond with RBPs and the structural context of RBP binding.
ACKNOWLEGMENTS
We would like to thank Dr. Stefan Aigner in the Yeo lab for his expert advice on experiments and critical reading of this manuscript. This work was partially supported by NIH grants R01-HG004659, U41-HG009889, R01-HL137219, R01-HL137223 and R01-HD085902 to G.W.Y., and R01-HG004361 R35-CA209919 to H.Y.C. M.C. was supported by the ALS Association Milton Safenowitz Post-doctoral Fellowship.
Footnotes
DECLARATION OF INTERESTS
G.W.Y. is co-founder, member of the Board of Directors, equity holder, and paid consultant for Locana and Eclipse BioInnovations. G.W.Y is a Distinguished Visiting Professor at the National University of Singapore. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. MC and GWY are inventors on a patent disclosure to UCSD on technologies described in this manuscript. H.Y.C. is an inventor on a patent owned by Stanford University on in vivo SHAPE. H.Y.C. is a co-founder of Accent Therapeutics, Boundless Bio, and an advisor for 10x Genomics, Arsenal Biosciences, and Spring Discovery. H.Y.C. is an Investigator of the Howard Hughes Medical Institute.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Busan S, Weidmann CA, Sengupta A, and Weeks KM (2019). Guidelines for SHAPE Reagent Choice and Detection Strategy for RNA Structure Probing Studies. Biochemistry 58, 2655–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cléry A, and Allain FHT (2011). From structure to function of RNA binding domains. In RNA Binding Proteins, Zdravko L, ed. (Landes Bioscience Springer Science+Business Media; ). [Google Scholar]
- Colombrita C, Onesto E, Megiorni F, Pizzuti A, Baralle FE, Buratti E, Silani V, and Ratti A. (2012). TDP-43 and FUS RNA-binding proteins bind distinct sets of cytoplasmic messenger RNAs and differently regulate their post-transcriptional fate in motoneuron-like cells. J Biol Chem 287, 15635–15647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corley M, Burns MC, and Yeo GW (2020). How RNA-Binding Proteins Interact with RNA: Molecules and Mechanisms. Mol Cell 78, 9–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deigan KE, Li TW, Mathews DH, and Weeks KM (2009). Accurate SHAPE-directed RNA structure determination. Proc Natl Acad Sci U S A 106, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Sanzo M, Chirillo R, Aversa I, Biamonte F, Santamaria G, Giovannone ED, Faniello MC, Cuda G, and Costanzo F. (2018). shRNA targeting of ferritin heavy chain activates H19/miR-675 axis in K562 cells. Gene 657, 92–99. [DOI] [PubMed] [Google Scholar]
- Ding Y, Tang Y, Kwok CK, Zhang Y, Bevilacqua PC, and Assmann SM (2013). In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 505, 696–700. [DOI] [PubMed] [Google Scholar]
- Diribarne G, and Bensaude O. (2009). 7SK RNA, a non-coding RNA regulating P-TEFb, a general transcription factor. RNA Biol 6, 122–128. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez D, Freese P, Alexis MS, Su A, Hochman M, Palden T, Bazile C, Lambert NJ, Van Nostrand EL, Pratt GA, et al. (2018). Sequence, Structure, and Context Preferences of Human RNA Binding Proteins. Mol Cell 70, 854–867 e859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng C, Chan D, Joseph J, Muuronen M, Coldren WH, Dai N, Correa IR Jr., Furche F, Hadad CM, and Spitale RC (2018). Light-activated chemical probing of nucleobase solvent accessibility inside cells. Nat Chem Biol 14, 325. [DOI] [PubMed] [Google Scholar]
- Fillebeen C, Wilkinson N, and Pantopoulos K. (2014). Electrophoretic mobility shift assay (EMSA) for the study of RNA-protein interactions: the IRE/IRP example. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorini F, Bagchi D, Le Hir H, and Croquette V. (2015). Human Upf1 is a highly processive RNA helicase and translocase with RNP remodelling activities. Nature communications 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores JK, and Ataide SF (2018). Structural Changes of RNA in Complex with Proteins in the SRP. Front Mol Biosci 5, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RA, Zhang QC, Spitale RC, Lee B, Mumbach MR, and Chang HY (2016). Transcriptome-wide interrogation of RNA secondary structure in living cells with icSHAPE. Nat Protoc 11, 273–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr., Jungkamp AC, Munschauer M, et al. (2010). Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141, 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hainzl T, Huang S, and Sauer-Eriksson AE (2005). Structural insights into SRP RNA: an induced fit mechanism for SRP assembly. RNA 11, 1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockensmith JW, Kubasek WL, Vorachek WR, and Vonhippel PH (1986). Laser Cross-Linking of Nucleic-Acids to Proteins - Methodology and 1st Applications to the Phage-T4 DNA-Replication System. Journal of Biological Chemistry 261, 3512–3518. [PubMed] [Google Scholar]
- Hu W, Qin L, Li ML, Pu XM, and Guo YZ (2018). A structural dissection of protein-RNA interactions based on different RNA base areas of interfaces. Rsc Adv 8, 10582–10592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CUT, and Pestova TV (2010). The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Bio 11, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazan H, Ray D, Chan ET, Hughes TR, and Morris Q. (2010). RNAcontext: a new method for learning the sequence and structure binding preferences of RNA-binding proteins. PLoS Comput Biol 6, e1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackey L, Coria A, Woods C, McArthur E, and Laederach A. (2018). Allele-specific SHAPE-MaP assessment of the effects of somatic variation and protein binding on mRNA structure. RNA 24, 513–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Quesne JP, Stoneley M, Fraser GA, and Willis AE (2001). Derivation of a structural model for the c-myc IRES. J Mol Biol 310, 111–126. [DOI] [PubMed] [Google Scholar]
- Lee FCY, and Ule J. (2018). Advances in CLIP Technologies for Studies of Protein-RNA Interactions. Mol Cell 69, 354–369. [DOI] [PubMed] [Google Scholar]
- Leppek K, Das R, and Barna M. (2018). Functional 5’ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat Rev Mol Cell Biol 19, 158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leulliot N, and Varani G. (2001). Current topics in RNA-protein recognition: control of specificity and biological function through induced fit and conformational capture. Biochemistry 40, 7947–7956. [DOI] [PubMed] [Google Scholar]
- Levi S, and Tiranti V. (2019). Neurodegeneration with Brain Iron Accumulation Disorders: Valuable Models Aimed at Understanding the Pathogenesis of Iron Deposition. Pharmaceuticals (Basel) 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang X, et al. (2008). HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456, 464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, and Hofacker IL (2011). ViennaRNA Package 2.0. Algorithms Mol Biol 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotfi M, Zare-Mirakabad F, and Montaseri S. (2015). RNA secondary structure prediction based on SHAPE data in helix regions. J Theor Biol 380, 178–182. [DOI] [PubMed] [Google Scholar]
- Loughlin FE, Lukavsky PJ, Kazeeva T, Reber S, Hock EM, Colombo M, Von Schroetter C, Pauli P, Clery A, Muhlemann O, et al. (2019). The Solution Structure of FUS Bound to RNA Reveals a Bipartite Mode of RNA Recognition with Both Sequence and Shape Specificity. Molecular Cell 73, 490-+. [DOI] [PubMed] [Google Scholar]
- Low JT, and Weeks KM (2010). SHAPE-directed RNA secondary structure prediction. Methods 52, 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Zhang QC, Lee B, Flynn RA, Smith MA, Robinson JT, Davidovich C, Gooding AR, Goodrich KJ, Mattick JS, et al. (2016). RNA Duplex Map in Living Cells Reveals Higher-Order Transcriptome Structure. Cell 165, 1267–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L, Meier M, Lyons SM, Sit RV, Marzluff WF, Quake SR, and Chang HY (2012). Systematic reconstruction of RNA functional motifs with high-throughput microfluidics. Nat Methods 9, 1192–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17. [Google Scholar]
- Maticzka D, Lange SJ, Costa F, and Backofen R. (2014). GraphProt: modeling binding preferences of RNA-binding proteins. Genome Biol 15, R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald IK, and Thornton JM (1994). Satisfying hydrogen bonding potential in proteins. J Mol Biol 238, 777–793. [DOI] [PubMed] [Google Scholar]
- McGinnis JL, Duncan CD, and Weeks KM (2009). High-throughput SHAPE and hydroxyl radical analysis of RNA structure and ribonucleoprotein assembly. Methods Enzymol 468, 67–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay R, Jia J, Arif A, Ray PS, and Fox PL (2009). The GAIT system: a gatekeeper of inflammatory gene expression. Trends Biochem Sci 34, 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustoe AM, Lama NN, Irving PS, Olson SW, and Weeks KM (2019). RNA base-pairing complexity in living cells visualized by correlated chemical probing. Proc Natl Acad Sci U S A 116, 24574–24582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan XY, Rijnbeek P, Yan JC, and Shen HB (2018). Prediction of RNA-protein sequence and structure binding preferences using deep convolutional and recurrent neural networks. Bmc Genomics 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic Z, and Templeton DM (2004). Iron accumulation and iron-regulatory protein activity in human hepatoma (HepG2) cells. Mol Cell Biochem 265, 37–45. [DOI] [PubMed] [Google Scholar]
- Poria DK, and Ray PS (2017). RNA-protein UV-crosslinking Assay. Bio-Protocol 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran S, Ding F, Weeks KM, and Dokholyan NV (2013). Statistical analysis of SHAPE-directed RNA secondary structure modeling. Biochemistry 52, 596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saria S, Duchi A, and Koller D. (2011). Discovering Deformable Motifs in Continuous Time Series data. Proceedings of the Twenty-Second international joint conference on Artificial Intelligence 2, 1465–1471. [Google Scholar]
- Siegfried NA, Busan S, Rice GM, Nelson JA, and Weeks KM (2014). RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nat Methods 11, 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T, Heger A, and Sudbery I. (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res 27, 491–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smola MJ, Calabrese JM, and Weeks KM (2015a). Detection of RNA-Protein Interactions in Living Cells with SHAPE. Biochemistry 54, 6867–6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smola MJ, Christy TW, Inoue K, Nicholson CO, Friedersdorf M, Keene JD, Lee DM, Calabrese JM, and Weeks KM (2016). SHAPE reveals transcript-wide interactions, complex structural domains, and protein interactions across the Xist lncRNA in living cells. Proc Natl Acad Sci U S A 113, 10322–10327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smola MJ, Rice GM, Busan S, Siegfried NA, and Weeks KM (2015b). Selective 2’-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nat Protoc 10, 1643–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smola MJ, and Weeks KM (2018). In-cell RNA structure probing with SHAPE-MaP. Nat Protoc 13, 1181–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitale RC, Crisalli P, Flynn RA, Torre EA, Kool ET, and Chang HY (2013). RNA SHAPE analysis in living cells. Nat Chem Biol 9, 18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitale RC, Flynn RA, Zhang QC, Crisalli P, Lee B, Jung JW, Kuchelmeister HY, Batista PJ, Torre EA, Kool ET, et al. (2015). Structural imprints in vivo decode RNA regulatory mechanisms. Nature 519, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens SG, Gardner PP, and Brown C. (2011). Two covariance models for iron-responsive elements. RNA Biol 8, 792–801. [DOI] [PubMed] [Google Scholar]
- Stys A, Galy B, Starzynski RR, Smuda E, Drapier JC, Lipinski P, and Bouton C. (2011). Iron regulatory protein 1 outcompetes iron regulatory protein 2 in regulating cellular iron homeostasis in response to nitric oxide. J Biol Chem 286, 22846–22854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto Y, Vigilante A, Darbo E, Zirra A, Militti C, D’Ambrogio A, Luscombe NM, and Ule J. (2015). hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature 519, 491–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan D, Marzluff WF, Dominski Z, and Tong L. (2013). Structure of histone mRNA stem-loop, human stem-loop binding protein, and 3’hExo ternary complex. Science 339, 318–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Simanshu DK, Ma JB, and Patel DJ (2011). Structural basis for piRNA 2’-O-methylated 3’-end recognition by Piwi PAZ (Piwi/Argonaute/Zwille) domains. Proc Natl Acad Sci U S A 108, 903–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tijerina P, Mohr S, and Russell R. (2007). DMS footprinting of structured RNAs and RNA-protein complexes. Nat Protoc 2, 2608–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, et al. (2016). Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat Methods 13, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Pratt GA, Yee BA, Wheeler EC, Blue SM, Mueller J, Park SS, Garcia KE, Gelboin-Burkhart C, Nguyen TB, et al. (2020). Principles of RNA processing from analysis of enhanced CLIP maps for 150 RNA binding proteins. Genome Biol 21, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walden WE, Selezneva A, and Volz K. (2012). Accommodating variety in iron-responsive elements: Crystal structure of transferrin receptor 1 B IRE bound to iron regulatory protein 1. FEBS Lett 586, 32–35. [DOI] [PubMed] [Google Scholar]
- Walden WE, Selezneva AI, Dupuy J, Volbeda A, Fontecilla-Camps JC, Theil EC, and Volz K. (2006). Structure of dual function iron regulatory protein 1 complexed with ferritin IRE-RNA. Science 314, 1903–1908. [DOI] [PubMed] [Google Scholar]
- Wilkinson KA, Merino EJ, and Weeks KM (2006). Selective 2’-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat Protoc 1, 1610–1616. [DOI] [PubMed] [Google Scholar]
- Williams KM, Qie S, Atkison JH, Salazar-Arango S, Alan Diehl J, and Olsen SK (2019). Structural insights into E1 recognition and the ubiquitin-conjugating activity of the E2 enzyme Cdc34. Nature communications 10, 3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YS, Declerck N, Manival X, Aymerich S, and Kochoyan M. (2002). Solution structure of the LicT-RNA antitermination complex: CAT clamping RAT. Embo Journal 21, 1987–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubradt M, Gupta P, Persad S, Lambowitz AM, Weissman JS, and Rouskin S. (2017). DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nat Methods 14, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All fSHAPE, fSHAPE-eCLIP, and SHAPE-eCLIP raw sequencing reads and processed data are available for download in the Gene Expression Omnibus (GEO), accession number GSE149767. All custom software used to process this data is available at github.com/meracorley and github.com/YeoLab/clipper, and upon request.