

Abstract
Assessing the neurotoxicity of test chemicals has typically been performed using two-dimensionally (2D) cultured neuronal cell monolayers and animal models. The in vitro 2D cell models are simple and straightforward compared to animal models, which have the disadvantage of being relatively low throughput, expensive, and time-consuming. Despite their extensive use in this area of neurotoxicology research, both models often do not accurately recapitulate human outcomes. To bridge this gap and attempt to better replicate what happens in vivo, three-dimensionally (3D) cultured neural stem cells (NSCs) encapsulated in hydrogels have been developed on a 384-pillar plate via miniature 3D bioprinting. This technology allows users to print NSCs on a pillar plate for rapid 3D cell culture as well as high-throughput compound screening. For this, the bioprinted NSCs on the 384-pillar plate are sandwiched with standard 384-well plates with growth media for 3D culture, allowing researchers to expose the cells to test compounds and stain them with various fluorescent dyes, for a suite of high-content imaging (HCI) assays, including DNA damage, mitochondrial impairment, cell membrane integrity, intracellular glutathione level, and apoptosis. After acquiring cell images from automated fluorescence microscopes and extracting fluorescence intensities, researchers can obtain IC50 values of each compound to evaluate critical parameters in neurotoxicity. Here, we provide a detailed description of protocols for cell printing on the 384-pillar plate, 3D NSC culture, compound testing, 3D cell staining, and image acquisition and analysis, which altogether will allow researchers to investigate mechanisms of compound neurotoxicity with 3D-cultured NSCs in a high throughput manner.
Keywords: 384-Pillar plate, neural stem cell (NSC), neurotoxicity, high-throughput screening (HTS), high-content imaging (HCI)
INTRODUCTION
Neurodevelopmental disorders affect one in six children born in the U.S. each year, which could occur due to environmental risk factors such as the use of therapeutic and recreational drugs (e.g., opioids), alcohol, and tobacco during pregnancy, as well as prenatal or childhood exposure to environmental toxicants (Epa, 2015; Smirnova et al., 2014). The developing brain is known to be more vulnerable to chemical exposure compared to the adult brain due to the immature blood-brain barrier (BBB) and the complexity of brain development processes (Aschner et al., 2017; Mundy et al., 2015). Despite the potential vulnerability of the developing brain to environmental toxicants, only a few hundred compounds, among tens of thousands of commercially used chemicals, have been tested for their potential neurotoxicity (Llorens et al., 2012).
Traditionally, researchers have used animal models to evaluate mechanisms of compound neurotoxicity (Llorens et al., 2012; Taylor et al., 2020). This models, however, often fail to predict neurotoxicity potential in humans due to profound differences in genetic makeup and compound metabolism between animals and humans (Florio & Huttner, 2014; Fritsche et al., 2017; Olson et al., 2000). Therefore, alternative, in vitro cell-based models have become a complementary approach to evaluate the neurotoxicity potential of compounds, with an increasing need to be able to do this in a high-throughput and cost-effective manner (Crofton et al., 2012; Llorens et al., 2012). To address this issue, high-content imaging (HCI) assays have been developed on two-dimensional (2D) cell monolayers to analyze multiple cellular parameters and morphological changes (Grimm et al., 2015; Joshi & Lee, 2015; Van Vliet et al., 2014). However, 2D cell monolayers often lack cell-cell and cell-extracellular matrix (ECM) interactions, and cannot accurately recapitulate in vivo conditions, which could lead to poor predictability of chemical toxicity (Page et al., 2013; Joshi & Lee, 2015).
As compared to 2D-cultured cells, three-dimensional (3D) cell cultures can better mimic in vivo microenvironments, by supporting cell-cell and cell-ECM interactions (Astashkina & Grainger, 2014; Kriston-Vizi & Flotow, 2017). HCI assays have thus been coupled with 3D cell spheroids in ultralow attachment (ULA) 96-/384-well plates to investigate mechanisms of compound-induced toxicity (Justice et al., 2009). Nonetheless, ECM-free cell spheroids cannot truly mimic tissue structure in vivo, and it has been difficult to handle growth media and cell staining reagents in ULA well plates due to the small size of cell spheroids (Kriston-Vizi & Flotow, 2017).
To overcome these issues, human cell types have been cultured in biomimetic hydrogels on a 384-pillar plate and coupled with a 384-well plate for high-throughput screening (HTS) of compounds (Yu et al., 2018). In addition, the potential neurotoxicity of test compounds has recently been assessed using 3D-cultured ReNcell VM cells using this 384-pillar plate platform, which allows testing for potential developmental neurotoxicity (DNT) via HCI assays (Joshi et al., 2018; Bal-Price et al., 2018).
Here, we describe protocols for characterizing cellular responses to compounds through HCI assays, using miniature 3D neural stem cells (NSCs) on a 384-pillar plate for high-throughput assessment of neurotoxicity. Briefly, Basic Protocol 1 describes the methods of functionalizing the surface of the pillars for cell encapsulation and cell printing on the 384-pillar plate for 3D NSC culture. Basic Protocol 2 describes the methods of treating 3D-cultured NSCs with model compounds and staining 3D NSCs with various fluorescent dyes for HCI assays. Finally, Basic Protocol 3 describes the steps for cell image acquisition from the 384-pillar plate, image processing, data analysis (IC50 value, Z’ factor, and coefficient of variation (CV) value), and statistical analysis.
STRATEGIC PLANNING
Users need to be aware that the protocols described require access to a 3D bioprinter such as ASFA™ Spotter from Medical & Bio Decision (MBD) Korea (Fig. 1), as well as an automated fluorescence microscope such as BZ-X810 all-in-one fluorescence microscope from Keyence and S+ Scanner from MBD Korea. Since the 384-pilllar plate can only accommodate a small volume of cells in hydrogels on the pillars (typically 0.1 – 2 μL), ink-jet or microsolenoid-valve driven 3D printers will be better suited for rapid cell printing. Although cell images can be obtained from the 384-pillar plate using traditional fluorescence microscopes, it will be a daunting task to manually scan the entire 384 pillars. Thus, automated fluorescence microscopes are recommended for high-throughput image acquisition. In addition, we have selected ReNcell VM neural progenitor cell line as a cell model in the protocols described here due to its unique features, including high self-renewal in the presence of growth factors and differentiation into neurons, astrocytes, and oligodendrocytes (Choi et al., 2014; Donato et al., 2007; Joshi et al., 2018).
Figure 1.

The main components of the ASFA™ spotter for 3D bioprinting.
BASIC PROTOCOL 1: 3D NSC culture on a 384-pillar plate
This protocol explains how to functionalize the 384-pillar plate for cell spot attachment and hydrogel gelation, and rapidly print ReNcell VM cells in alginate on the 384-pillar plate using the ASFA™ Spotter for miniature 3D NSC culture. Users can generate 3D cell culture on the 384-pillar plate in a high-throughput manner without the excessive use of cells, hydrogels, and other reagents.
Materials
ReNcell VM human neural stem cell (EMD Millipore #SCC008, RRID:CVCL_E921)
Nunc EasYFlask 75 cm2 cell culture flask (Thermo Fisher Scientific, cat. no. 156499)
Complete NSC medium for culturing ReNcell VM (see Reagents and Solutions)
Laminin (Thermo Fisher Scientific, cat. no. 23017015)
DMEM/F-12 (Thermo Fisher Scientific, cat. no. 11320033)
Accutase™ (EMD Millipore, cat. no. A6964), Store at 4°C
Poly(maleic anhydride alt-1-octadecene) (PMA-OD; Sigma Aldrich, cat. no. 419117), dissolved in ethanol to prepare 1% (w/v) stock (see Reagents and Solutions)
Poly-L-lysine (PLL), 0.01% (w/v) stock (Sigma Aldrich, cat. no. P4707)
Calcium chloride dihydrate (CaCl2·2H2O) (Sigma Aldrich, cat. no. 223506), dissolved in sterile deionized water to prepare a 100 mM stock
3% (w/v) Alginate stock solution (see Reagents and Solutions)
Growth factor reduced (GFR) Geltrex® (ThermoFisher, cat. no. A1413302), 15 mg/mL, Store at −20°C until use
Cell culture incubator (5% CO2, 37°C)
Nunc™ 384-well, non-treated, flat-bottom microplate (Thermo Fisher Scientific, cat. no. 265202)
384-Pillar plate (MBD Korea, Suwon, Republic of Korea) (see Fig. 2)
Moxi Z mini automated cell counter (ORFLO Technologies, MXZ001)
Moxi Z cassette (ORFLO Technologies, cat. no. MXC002, type S)
Centrifuge (VWR, Eppendorf 5702 R)
ASFA™ Spotter (MBD Korea)
Class II, Type A2 Biosafety Cabinet (Nuaire, cat. no. NU-540–600)
Nunc™ Square BioAssay Dishes (ThermoFisher, cat. no. 166508)
Figure 2.
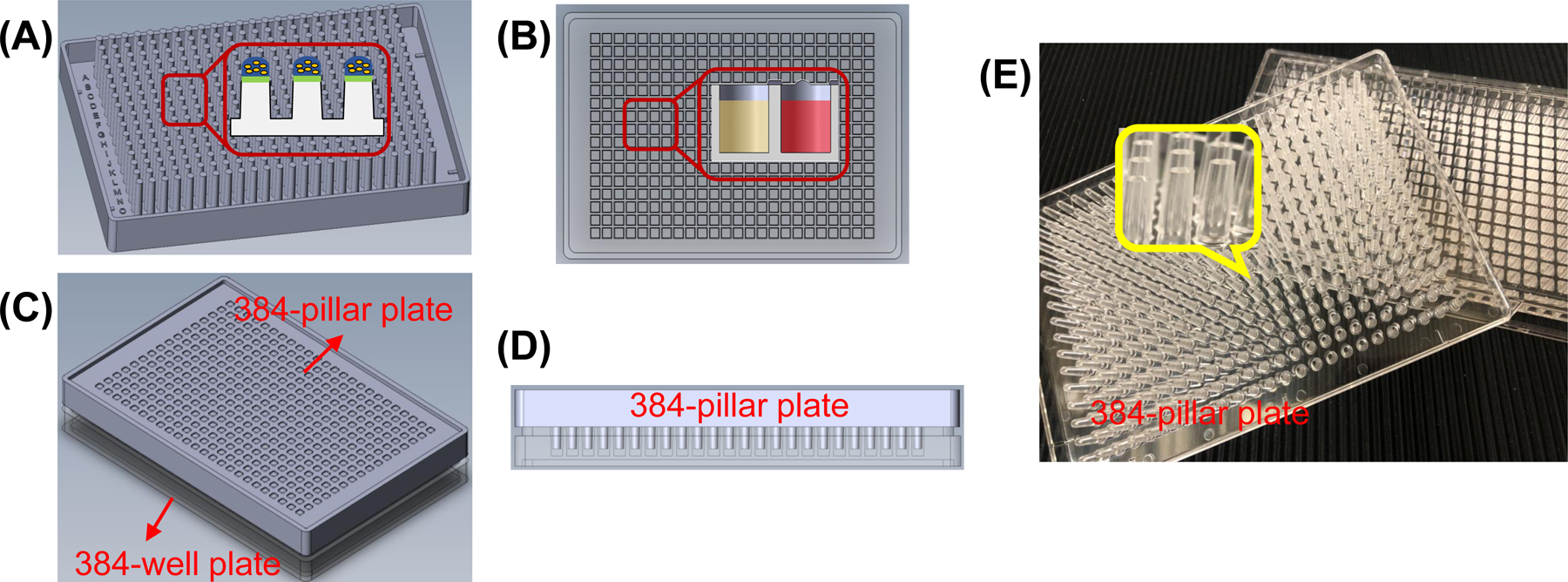
Schematics and pictures of the 384-pillar plate and a complementary 384-well plate: (A) Schematic of 384-pillar plate with printed cells in hydrogel. (B) A 384-well plate containing growth media. (C) The 384-pillar plate sandwiched onto the 384-well plate for 3D cell culture. (D) Cross sectional view of the sandwiched plates on C. (E) Pictures of an injection molded 384-pillar plate compared with a 384-well plate.
Protocol Steps
ReNcell VM culture and preparation of cell suspension (timing: 3 – 5 days)
-
1
Prepare a 20 μg/mL laminin solution in DMEM/F-12
-
2
Prepare a laminin-coated T75 flask by adding 5 mL of the laminin solution and incubating for 3 – 4 hours in a 5% CO2 incubator at 37°C.
-
3
Remove the laminin solution and seed 1.5 × 106 ReNcell VM cells in the laminin-coated T75 flask with 10 – 12 mL of complete NSC medium.
-
4
Place cells in the incubator, with medium change every other day until the cells reach 80 – 90% confluency (~ 3 – 4 days).
While the cells are growing, users can proceed to “Functionalization of the 384-pillar plate for 3D NSC culture”, so that the plate is ready when needed.
-
5
Remove the old medium from the flask and rinse the flask with 5 mL of 1× DPBS, taking care not to detach the cells.
-
6
Add 3 mL of Accutase™ to the flask and incubate for 3 – 5 minutes in the 5% CO2 incubator at 37°C to detach the cells from the surface of the flask.
-
7
Once the cells are completely detached, add 5 mL of complete NSC medium to stop the Accutase™ reaction and, using a 10 mL pipette tip, collect all of the cell suspension into a 15 mL conical tube.
-
8
Centrifuge the cell suspension for 4 minutes at 300 × g at room temperature and remove the supernatant without disturbing the cell pellet.
-
9
Add 1 mL of complete NSC medium and resuspend the cell pellet in the medium by gently pipetting up and down 3 – 5 times.
-
10
Take 4 μL of the cell suspension and mix with 196 μL of complete NSC medium to prepare a 200 μL cell suspension at 50-fold dilution for cell counting.
-
11
To count the cells, load 75 μL of the diluted cell suspension in a Moxi Z cassette inserted into Moxi Z mini automated cell counter.
Users can use a hemocytometer to count the cells if an automated cell counter is not available.
Note that in this protocol, the actual cell density is 50-fold higher than the density displayed on the cell counter because of the 50-fold dilution. Moxi Z cell counter gives cell density in the number of cells per milliliter. To obtain an accurate measurement, avoid using a too high-density cell suspension.
-
12
Prepare 3 mL of cell suspension at a final concentration of 3.5 × 106 cells/mL, by diluting the cell suspension with complete NSC medium.
Functionalization of the 384-pillar plate for 3D NSC culture (timing: 1 day)
Users can functionalize the plate while the cells are growing (Step 4).
-
13
Prepare 20 mL of 0.01% (w/v) PMA-OD and pour 10 mL in a lid of a 384-well plate.
20 mL of PMA-OD solution is enough for coating multiple 384-pillar plates.
-
14
Place a 384-pillar plate (with the pillars facing down) onto the lid of the 384-well plate containing the 10 mL of 0.01% (w/v) PMA-OD, to wet the pillars with PMA-OD.
Make sure that there are small droplets of PMA-OD on all of the pillars of the 384-pillar plate. Repeat this step to coat all 384-pillar plates needed with PMA-OD.
-
15
Dry the PMA-OD coated 384-pillar plate facing up for 4 – 6 hours at room temperature in a biosafety cabinet.
PMA-OD is necessary for covalent attachment of PLL on the 384-pillar plate.
-
16
Prepare a 3 mL mixture of 0.0033% (w/v) PLL and 25 mM CaCl2 in sterile deionized water by mixing 1000 μL of 0.001% (w/v) PLL, 750 μL of 100 mM CaCl2, and 1250 μL of sterile deionized water.
PLL is necessary for alginate attachment by ionic interactions, and CaCl2 is necessary for alginate gelation for cell encapsulation on the 384-pillar plate.
-
17
Place the PMA-OD coated 384-pillar plate on the chilling loading deck of the ASFA Spotter.
Maintain the temperature of the loading deck at 4 – 10°C to prevent water evaporation during sample printing.
-
18
Add the mixture of PLL and CaCl2 from Step 16 in a 96-well plate and print it onto the PMA-OD coated 384-pillar plate at a volume of 2 μL, by running an operating program.
The entire process of the mixture printing typically takes 5 minutes per 384-pillar plate. Users may be able to dispense a small volume of samples manually on the 384-pillar plate using a multi-channel pipette.
-
19
Dry the 384-pillar plate overnight at room temperature in a sterile Nunc™ square BioAssay dish.
PMA-OD coating as well as PLL and CaCl2 printing are necessary for cell encapsulation in alginate on the 384-pillar plate.
3D NSC culture on the 384-pillar plate (timing: 1 – 2 hours)
At this point, the cells (Step 12) are ready to be printed on the functionalized plate (Step 19).
-
20
Before printing the cells, prepare a 384-well plate containing 50 μL of complete NSC medium in each well and incubate in the 5% CO2 incubator at 37°C.
-
21
Prepare a 3 mL mixture of ReNcell VM in alginate and GFR Geltrex by mixing 1715 μL of the cell suspension from step 12 with 750 μL of 3% (w/v) alginate, 500 μL of 15 mg/mL GFR Geltrex, and 35 μL of complete NSC medium, to obtain a final concentration of 2 × 106 cells/mL in 0.75% (w/v) alginate and 2.5 mg/mL GFR Geltrex. Pipette 5 – 7 times to ensure that the cells are suspended uniformly in the hydrogels.
GFR Geltrex does not form a gel at this diluted concentration even at room temperature.
-
22
Place the dry PLL-CaCl2 treated 384-pillar plate (from Step 19) on the chilling loading deck of the ASFA Spotter.
Maintain the temperature of the loading deck at 4°C to prevent water evaporation during cell printing and hydrogel gelation for high cell viability.
-
23
Add 250 μL of cell suspension in alginate-Geltrex from step 21 in a 96-well plate and print it onto the PLL-CaCl2 treated 384-pillar plate at a volume of 2 μL (final 4,000 cells/pillar) by running an operating program in the ASFA Spotter.
-
24
Leave the 384-pillar plate with cells on the chilling deck inside the ASFA Spotter for 4 minutes for complete alginate gelation.
-
25
Insert the 384-pillar plate with cells from step 24 into the 384-well plate containing complete NSC medium from step 20 (Fig. 2) and incubate the sandwiched plates for 3 days in the 5% CO2 incubator at 37°C, with a medium change once after 2 days.
Check the sandwiched 384-pillar and well plates under a brightfield microscope to ensure uniform printing of the cells on all 384 pillars. Make sure the cells look healthy by observing their morphology and brightness. Viable and healthy cells exhibit bright round morphology.
BASIC PROTOCOL 2: Compound treatment and cell staining
This protocol describes how to use compounds against the 3D-cultured ReNcell VM cells on the 384-pillar plate generated in Basic Protocol 1, for evaluating compound toxicity mechanisms. The methods described in this protocol will enable users to prepare compound solutions in a 384-well plate, expose the 3D-cultured NSCs to the test compounds, and stain the cells with several fluorescent dyes to monitor the impact of the test compounds on various cellular processes. Model compounds, working concentrations, and mechanisms of neurotoxicity used in this example are shown in Table 1. The endpoint of fluorescent dyes and working concentrations are listed in Table 2.
Table 1.
List of model compounds used and their mechanism of toxicity.
| Model compounds | Concentration used (μM) | Compound type | Known mechanism of toxicity | References |
|---|---|---|---|---|
| Rotenone | 0.16 – 40 | Pesticide, insecticide, and piscicide | AP, MI, OS | (Jin et al., 2007; J. Li et al., 2005) |
| 4-Aminopyridine (4-AP) | 19.5 – 5000 | Vertebrate pesticide | MI, OS, PCB | (Glover, 1982; Jensen et al., 2014) |
| Digoxin | 0.04 – 10 | Inotropic agents | AP, OS | (Prassas et al., 2011) |
| Topotecan | 0.08 – 20 | Chemotherapeutic drug | AP, TI | (Staker et al., 2002; Sterzyńska et al., 2018) |
Apoptosis (AP), mitochondrial impairment (MI), oxidative stress (OS), potassium channel blocking (PCB), and topoisomerase inhibition (TI)
Table 2.
List of fluorescent dyes and their concentrations used in this protocol.
| Fluorescent dyes | Assays/endpoints | Stock conc. in DMSO (mM) | Working conc. in saline solution (μM) |
|---|---|---|---|
| Hoechst 33342 | DNA damage | 10 | 10 |
| Tetramethyl rhodamine methyl ester (TMRM) | Mitochondrial impairment | 50 | 0.5 |
| Monochlorobimane (mBCl) | Intracellular glutathione level/oxidative stress | 200 | 200 |
| Calcein AM | Cell membrane integrity/cell viability | 1 | 1 |
| YO-PRO-1 | Apoptosis | 1 | 5 |
Materials
- Test compounds (all from Sigma Aldrich) dissolved in DMSO:
- Rotenone (0.16 – 40 μM) (Sigma Aldrich, cat. no. A5000)
- 4-Aminopyridine (4-AP, 19.5 – 5,000 μM) (Sigma Aldrich, cat. no. 275875)
- Digoxin (0.04 – 10 μM) (Sigma Aldrich, cat. no. D6003)
-
Topotecan (0.08 – 20 μM) (Sigma Aldrich, cat. no. T2705)The concentration range of these compounds were selected based on the test concentrations used in the literature.
- Dimethyl sulfoxide (DMSO) as a negative control
- Fluorescent dyes:
- Hoechst 33342 (Thermo Fisher Scientific, cat. no. H1399), dissolved in DMSO, 10 mM stock (see Reagents and Solutions)
- Tetramethyl rhodamine methyl ester (TMRM, Thermo Fisher Scientific, cat. no. T-668), dissolved in DMSO, 50 mM stock (see Reagents and Solutions)
- Monochlorobimane (mBCl, Thermo Fisher Scientific, cat. no. M-1381MP), dissolved in DMSO, 200 mM stock (see Reagents and Solutions)
- Calcein AM (Thermo Fisher Scientific, cat. no. C1430), dissolved in DMSO, 1 mM stock (see Reagents and Solutions)
- YO-PRO1 (Thermo Fisher Scientific, cat. no. Y3603), dissolved in DMSO, 1 mM stock
Saline solution (see Reagents and Solutions)
Protocol Steps
Compound treatment of 3D-cultured NSCs (timing: 2 days)
-
1
Prepare a series of five 4-fold serial dilutions of test compounds and a separate DMSO control, at a volume of 1.5 mL per dilution in complete NSC medium.
Approximately 0.5 – 1.5 mL of compound dilution is required for one 384-well plate, depending on the number of replicates per concentration. Users are recommended to adjust the volume of compounds depending on the number of 384-pillar plates to be tested for HCI assays as well. In general, a series of five to nine 4-fold serial dilutions of a test compound and a DMSO control is used to generate a dose response curve. The final DMSO concentration should be below 0.5% (v/v) to avoid any basal toxicity issue of DMSO.
-
2
Dispense 50 μL/well of each compound dilution in a 384-well plate. Start with the DMSO control in column 1, moving to the highest concentration in column 6, for the first test compound. Repeat this process for all test compounds, moving to adjacent columns (e.g., compound 1 in columns 1 – 6, compound 2 in columns 7 – 12, and so on). This will result in 16 replicates per concentration, per compound (See Fig. 3).
-
3
Insert the 384-pillar plate with 3D-cultured NSCs (Basic Protocol 1, Step 25) onto the 384-well plate containing the serially diluted compound solutions, and incubate the sandwiched plates for 24 hours in the 5% CO2 incubator at 37°C to measure acute neurotoxicity of compounds.
Duration of test compound treatment depends on the drug and the mechanism of cell death that users are trying to investigate. Treatment time is typically between 12 and 72 hours.
Figure 3.

Map of a 384-well plate containing four compounds (each compound with five concentrations and a DMSO vehicle control, sixteen replicates per dosage).
Cell staining (timing: 2 – 3 hours)
-
4
Prepare stock solutions of fluorescent dyes in DMSO (except YO-PRO-1) at the following concentrations: 0.5 mM TMRM, 10 mM Hoechst 33342, 200 mM mBCl, and 1 mM calcein AM (see Reagents and Solutions).
-
5
Prepare five 384-well plates for cell rinsing, all plates containing 50 μL of the saline solution (see Reagents and Solutions) per well. This will be the “rinsing plate”.
-
6
After incubation with the compounds (Step 3), detach the 384-pillar plate and rinse twice for 10 minutes each, by sandwiching sequentially with two of the rinsing plates from Step 5.
-
7
Prepare working concentrations of the fluorescent dyes: 0.5 μM TMRM, 10 μM Hoechst 33342, 200 μM mBCl, 1 μM calcein AM, and 5 μM YO-PRO-1 in 20 mL of saline solution.
-
8
Prepare one 384-well plate for cell staining per dye by adding 50 μL of working concentrations of the fluorescent dyes in each well (“staining plate”) and keep it in the dark at room temperature.
Depending the number of replicates, users can adjust the number of staining plates. Staining plates with 2 dyes is usually recommended.
-
9
Separate the 384-pillar plate from the rinsing plate, sandwich the 384-pillar plate with the staining plate, and incubate the sandwiched plates for 1 hour at room temperature, protected from light.
-
10
Rinse stained cells on the 384-pillar plate to remove the dyes in excess on the pillars by sandwiching the 384-pillar plate sequentially with two rinsing plates (Step 5), for 10 minutes in each plate, in the dark.
-
11
Sandwich the 384-pillar plate with the remaining rinsing plate from Step 5 and proceed immediately to image acquisition.
BASIC PROTOCOL 3: Image acquisition, processing, and data analysis
This protocol provides detailed steps for high-content image acquisition with an automated fluorescence microscope (S+ scanner) and analysis of acquired images —using ImageJ and GraphPad Prism— for evaluating the neurotoxicity of the compounds tested in Basic Protocol 2.
Materials
Automated fluorescence microscope (S+ Scanner from MBD Korea)
The sandwiched plates from Basic Protocol 2, Step 11
ImageJ 1.8.0 (RRID:SCR_003070, NIH)
GraphPad Prism, version 4 or higher (RRID:SCR_002798, GraphPad Software, La Jolla, CA)
Protocol Steps
Acquisition of fluorescent cell images with S+ Scanner (Timing: 1 hour)
-
1
Load the sandwiched plates from Basic Protocol 2, Step 11 in an automated fluorescence microscope and open a plate layout file from the list.
The plate file will be selected based on the layout of the 384-pillar plate exposed to compounds and magnification of the objective lens. Typically, magnification of 4X is used to acquire entire cell images high throughput.
-
2
Choose the appropriate filter channels based on excitation and emission wavelengths of the fluorescent dyes used for cell staining.
-
3
Click the Auto button from the Brightness tab.
This built-in function in S+ Scanner finds the optimum brightness, but it can be adjusted This step is important to avoid potential photobleaching of the dyes.
-
4
Click the Batch Capture tab and acquire fluorescent images from each pillar with 3D-cultured ReNcell VM cells (Fig. 4).
Fluorescent images for one 384-pillar plate can be captured within 20 minutes.
-
5
Save the image files in the desired folder.
Figure 4.
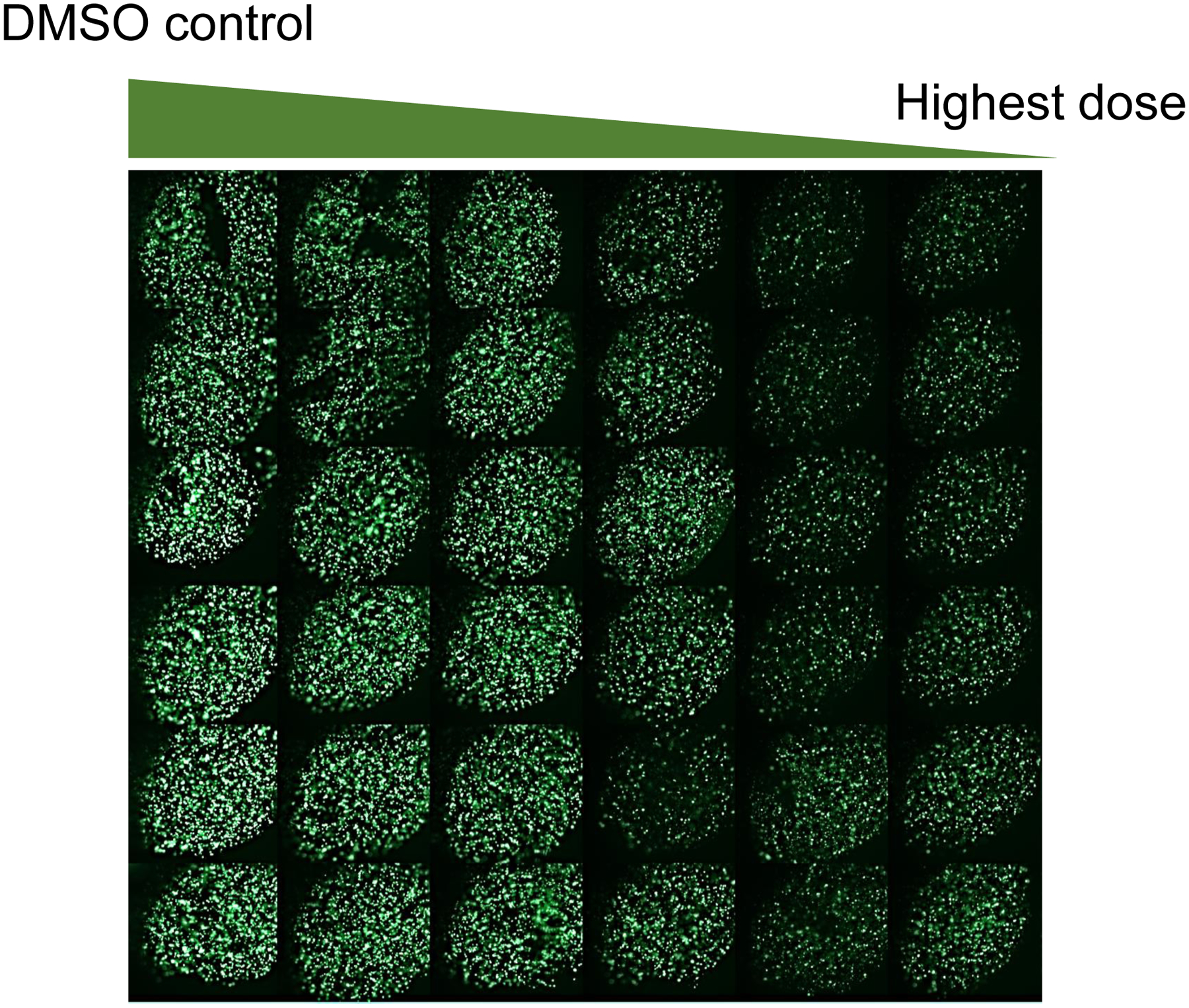
Fluorescent images of 3D-cultured ReNcell VM cells on the 384-pillar plate, obtained by exposing the cells to DMSO (vehicle control) and 0.08 – 20 μM of topotecan for 24 hours with 6 replicates in each row, staining with 1 μM of calcein AM for 1 hour, and scanning with a green fluorescence filter of S+ Scanner.
Image processing and data analysis (timing 3 – 4 hours)
Quantification of fluorescence intensity from the images is done with ImageJ. Standard dose-response curves are created by using GraphPad Prism or similar software. For processing a large number of images, an ImageJ macro can be created by recording the steps for a single sample instead of writing a Java script. The ImageJ macro can be saved either as a plugin, or used directly by copying the recorded steps and pasting them in a batch process window by selecting Batch → Macro under the Process tab. Users are recommended to try a desired option based on their individual needs.
-
6
Open one representative image in ImageJ software.
-
7
Eliminate background fluorescence (if any) from the image using the background subtraction function in ImageJ before extracting the fluorescence intensity from the image.
-
8
Go to Image → Type to select 8-bit to convert the RGB image into a gray scale.
-
9
Select a Moments threshold by going to Image → Adjust → Threshold and choosing Moments, to limit the intensity extraction from fluorescently labelled cellular area.
-
10
Select the necessary parameters such as integrated density and display label, under Analyze → Set Measurements.
These two parameters (display label and integrated density) should always be selected to measure the fluorescence intensity of the stained cellular region. Other parameters can be selected as per individual requirements.
-
11
Measure the fluorescence intensity from the images by using the Measure function under Analyze → Measure. A result window will show up with the quantified fluorescent intensities.
-
12
After recording the above steps for a single image, apply the batch processing macro to extract the fluorescent intensities from the entire batch of images (Fig. 5).
-
13
Save the result in an Excel file after batch processing all of the images.
-
14
Open the Excel sheet from Step 13 and normalize all fluorescent intensities obtained from the cells exposed to the compound with those from the cells exposed to DMSO alone (100% live cells), to obtain mean and SDs.
-
15
To plot dose-response curves and calculate IC50 values, copy normalized fluorescent intensities from Step 14 (with means and SDs) and paste them into GraphPad Prism, including the name of the compound and test concentrations of the compounds (dosage) (Fig. 6).
-
16
Go to Analysis→ Analyze and select “Nonlinear regression (curve fit)”.
-
17
Choose Log [inhibitor] vs. normalized response under “Dose-response - Inhibition” tab and press “OK” (Fig. 6).
-
18
A dose-response curve will appear in the Graph tab (Fig 7), and an IC50 value will be shown under the “Result” tab.
Dose-response curves and IC50 values will be obtained from the following equation:
X is the concentration of the compound in log scale, Y is the sigmoidal curve of the cellular response from the top plateau (Top) to the bottom plateau (Bottom), and H is the slop of the dose-response curve. The IC50 value is the concentration point in which 50% of the cellular response is inhibited.
Figure 5.
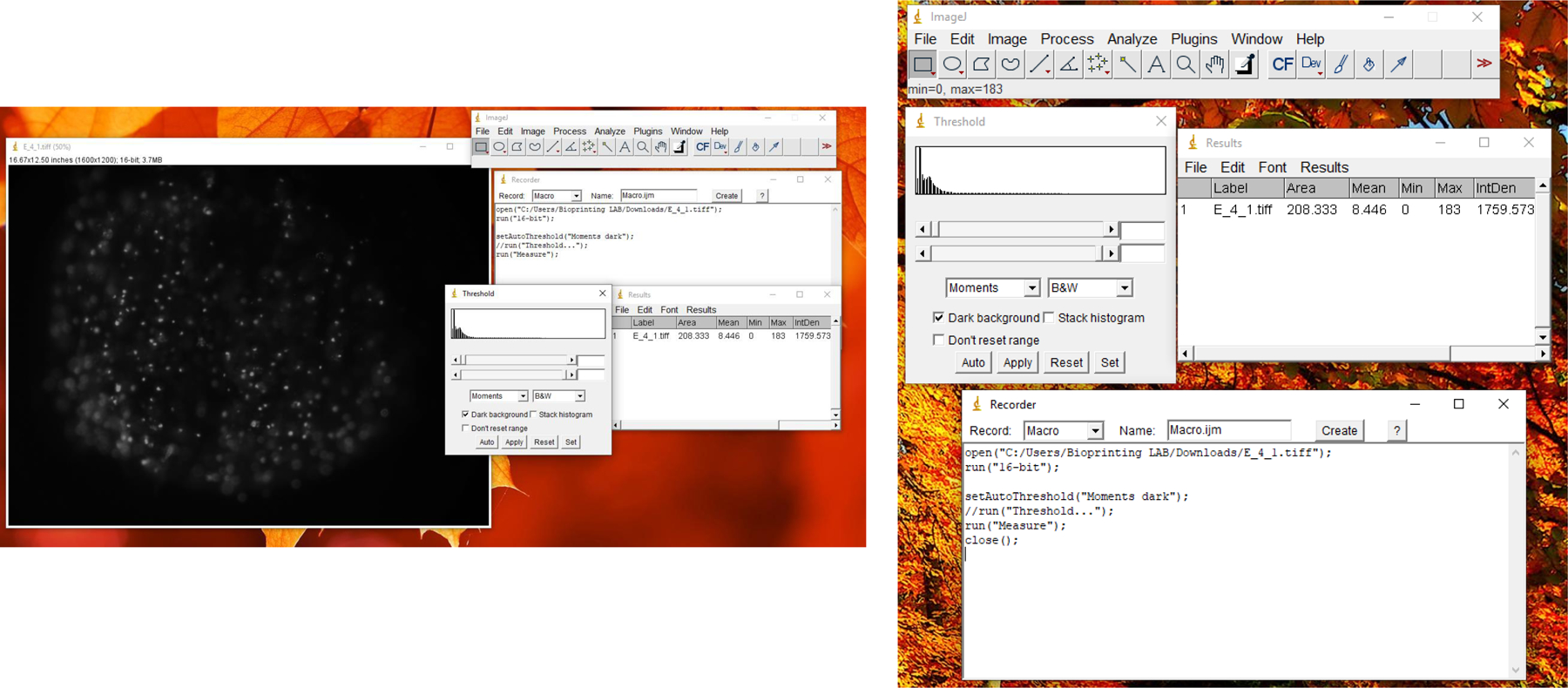
Screenshot of a single spot of ReNcell VM cells on the pillar and image processing in ImageJ software to acquire the fluorescence intensity from the fluorescent cell images.
Figure 6.
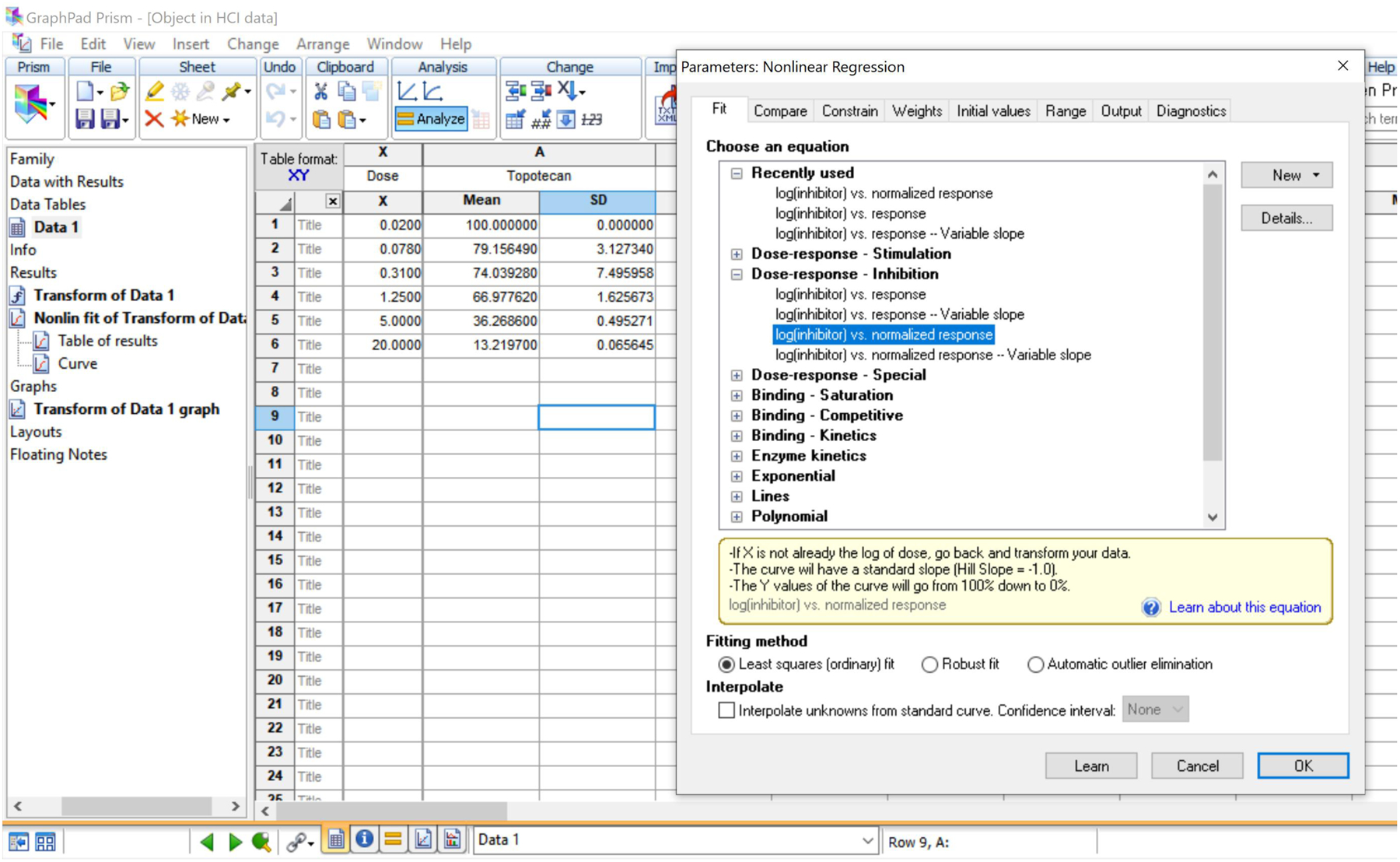
Screenshot of GraphPad Prism software, showing the options to plot dose-response curves and calculate IC50 values.
Figure 7.
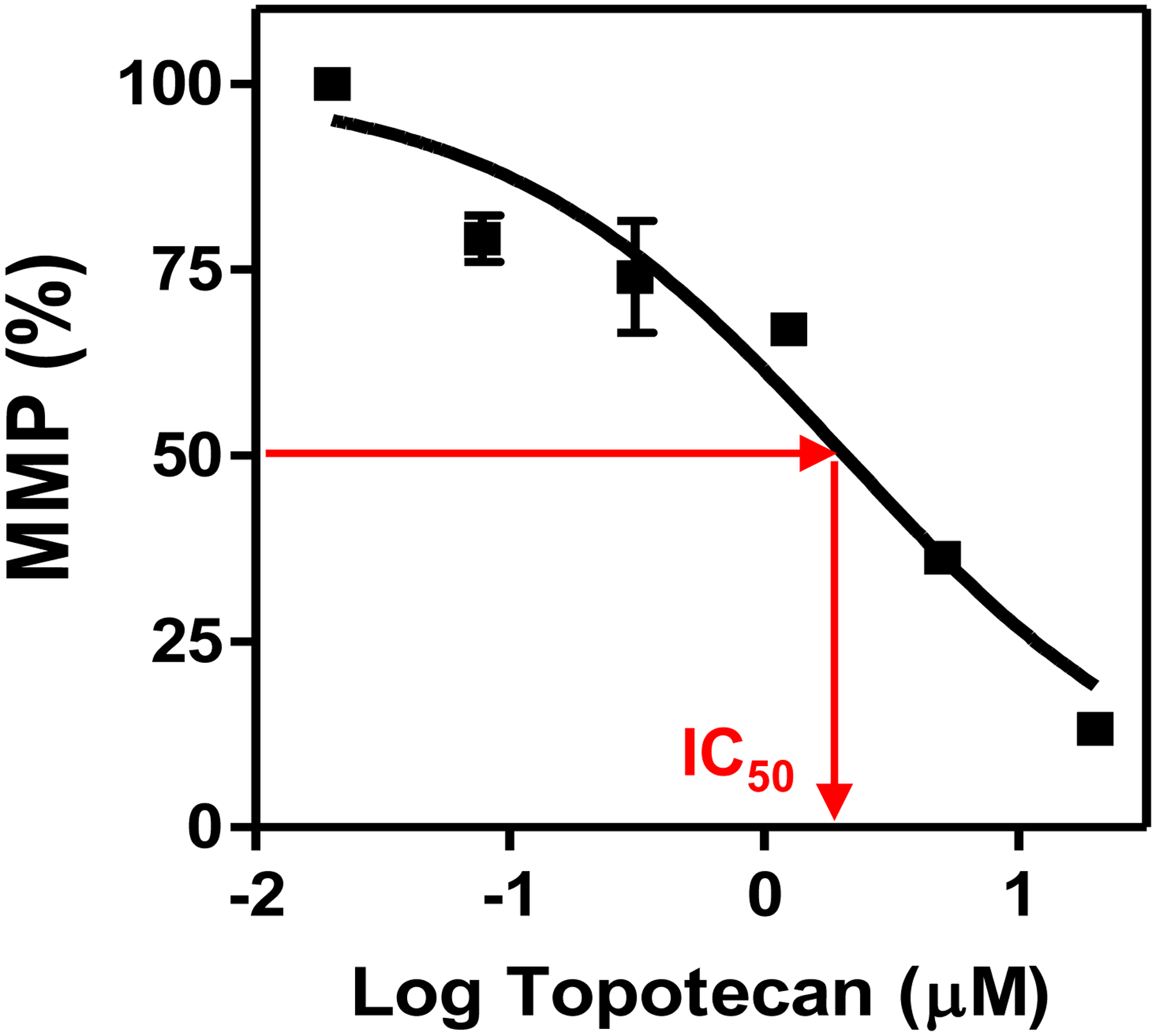
Dose-response curve of topotecan from the tetramethyl rhodamine methyl ester (TMRM) assay with ReNCell VM cells on the 384-pillar plate. The mitochondrial membrane potential (MMP) signals were obtained by calculating the mean and standard deviation (error bars) of six biological replicates, which were normalized with the signals from the DMSO alone control. The IC50 value is the compound concentration that affects MMP activity by 50%.
Assay validation (timing: 1 – 2 hours)
The Z’ factor and the coefficient of variation (CV) are critical to measure robustness and reproducibility of a new assay developed on the 384-pillar plate platform. A Z’ factor between 0.5 to 1 is accepted as “highly robust” for an assay (Sui & Wu, 2007; Yu, Kang, et al., 2018; Zhang et al., 1999). A CV value of less than 25% is acceptable for HTS assays (Sui & Wu, 2007; Yu, Kang, et al., 2018; Zhang et al., 1999). The day-to-day variation of the data obtained from the 384-pillar plate can be assessed by measuring CV values on different days.
-
19Evaluate the robustness of a new assay by measuring the Z’ factor using the following equation:
where AvgMax is the average fluorescence intensity from fully viable ReNcell VM cells on the 384-pillar plate (maximum fluorescence, DMSO control), AvgMin is the average fluorescence intensity from the cells treated by the highest dose of a compound (minimum fluorescence), SDMax is the standard deviation of maximum fluorescence intensity, and SDMin is a standard deviation of the minimum fluorescence intensity. -
20Assess the CV values on different day by calculating the ratio of the standard deviation (SD) to the average (Avg):
Statistical analysis (Timing: 1 – 2 hours)
One-way analysis of variance (ANOVA) can be performed to compare the IC50 values from the same compounds obtained from different fluorescent dyes (endpoints).
-
21
Open GraphPad Prism, go to New table & graph→ Grouped and select ‘Start with an empty data table’ under “Sample data” section.
-
22
Arrange at least three different sets of IC50 values from an individual endpoint in the data sheet. Row Y is converted into Log concentration.
-
23
Go to Analyze→Analyze these data and select “One-way ANOVA (and nonparametric)” under “Column analyses” (Fig 8).
-
24
Choose Test name as “One-way analysis of variance” under “Choose test”, select Test name “Bonferroni: Compare all pairs of columns” under “Post test”, select 0.05 (95% confidence intervals), select “4” under “Significant digits”, and press “OK”.
-
25
Go to “Data 1” under “Graphs” for checking the results. “Data 1” shows the table and displays an * symbol if any comparison is statistically significant (e.g., p < 0.05).
Figure 8.
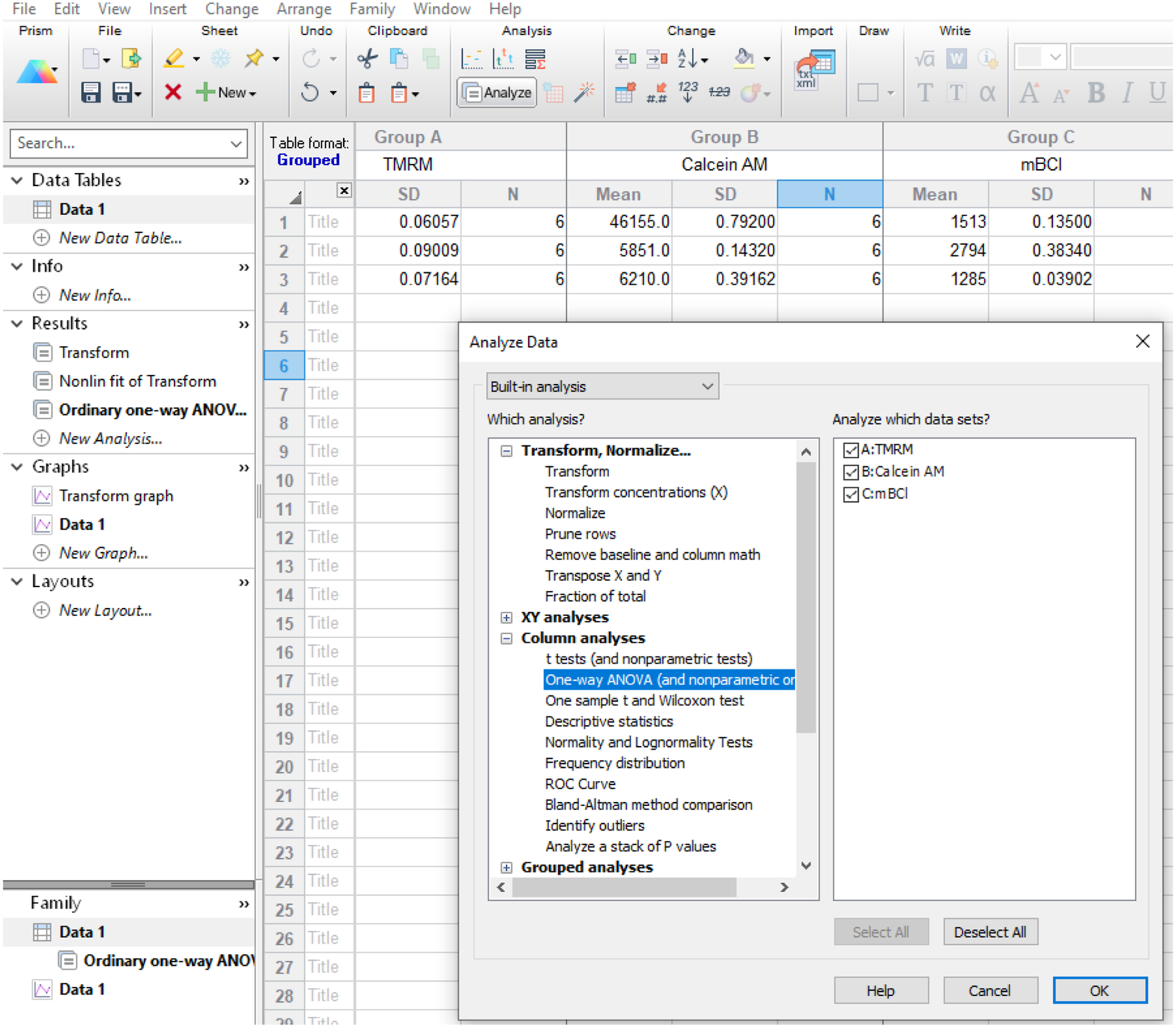
Screenshot of GraphPad Prism software, showing the options to calculate ANOVA using IC50 values from different end points (dyes).
REAGENTS AND SOLUTIONS
Complete NSC medium
Supplement ReNcell NSC maintenance medium (EMD Millipore, cat. no. SCM005) with 20 ng/mL of epidermal growth factor (EGF, EMD Millipore, cat. no. GF144), 20 ng/mL of basic fibroblast growth factor (bFGF, EMD Millipore, cat. no. GF003-AF), and 1% (v/v) of penicillin/streptomycin (Thermo Fisher, cat. no. 15140122). Prepare aliquots and store at −20°C up to the expiration date provided by the manufacturer.
PMA-OD solution
Dissolve 100 mg of PMA-OD (Sigma Aldrich, cat. no. 419117) in 10 mL of 100% ethanol to achieve 1% (w/v) PMA-OD stock solution. To prepare a working concentration of 0.01% (w/v) PMA-OD solution, dilute 1% (w/v) PMA-OD stock solution 100-fold with 100% ethanol.
3% Alginate stock solution
Dissolve 0.3 g of alginic acid sodium salt (Sigma Aldrich, cat. No. A1112) in 10 mL of sterile deionized water in a sterile glass vial and stir for 2 days on a magnetic stirring plate to achieve 3% (w/v) alginate stock solution. Store the alginate stock solution at 4°C for up to 12 months.
Compound stock solutions
Test compound stock solutions should be prepared at 200-fold higher concentrations than the desired final concentrations. The concentrations of compound stock solutions will vary depending on neurotoxicity of the compounds.
-
To prepare the test compounds solutions used in this protocol (rotenone, 4-aminopyridine (4-AP), digoxin, and topotecan), dissolve them in dimethyl sulfoxide (DMSO, Fisher scientific, cat. no. D128–500). If necessary, sonicate the compounds in DMSO to dissolve completely. Take aliquots of the compound stock solutions and store them at −20°C for up to 12 months.
Do not vortex the powder form of compounds in DMSO, to avoid undissolved compounds to attach to the side of the vial. Check solubility and use the DMSO as solvent for compounds. Alternative solvents such as ethanol, acetonitrile, or others can be used if a compound is insoluble in DMSO. Follow the manufacturer’s instructions.
Saline solution
-
Dissolve 8.1 g of NaCl and 2.9 g of CaCl2·H2O in 1 L of sterile deionized water to make a saline solution containing 140 mM NaCl and 20 mM CaCl2.
CaCl2 is supplemented to prevent alginate degradation during cell staining and rinsing.
Hoechst 33342 solution
Prepare a 10 mM stock solution of Hoechst 33342 (Thermo Fisher Scientific, cat. no. H1399 by dissolving 10 mg of Hoechst 33342 in 1.62 mL DMSO. Prepare aliquots of 20 μL and store at −20°C protected from light.
To prepare a final concentration of 10 μM Hoechst 33342 for cell staining, dilute 10 μL of the Hoechst stock solution in 10 mL of the saline solution.
Tetramethyl rhodamine methyl ester (TMRM) solution
Prepare a 50 mM stock solution of TMRM (TMRM, Thermo Fisher Scientific, cat. no. T-668) by dissolving 50 mg TMRM in 2 mL DMSO. Vortex for 1 minute if needed. Prepare 40 μL aliquots and store them at −20°C protected from light.
Dilute the stock solution of TMRM 100-fold in DMSO to get a 0.5 mM working stock solution. Further dilute the working stock of 0.5 mM TMRM to a final concentration of 0.5 μM by diluting 10 μL of 0.5 mM TMRM solution in 10 mL of the saline solution.
Calcein AM solution
Dissolve 1 mg of calcein AM (Thermo Fisher Scientific, cat. no. C1430) in 1 mL DMSO to prepare a 1 mM stock solution of calcein AM. Prepare aliquots of 20 μL and store them at −20°C protected from light.
Add 10 μL of the calcein AM stock solution in 10 mL of the saline solution to get a final concentration of 1 μM calcein AM.
Monochlorobimane (mBCl) solution
Prepare a 200 mM stock solution of mBCl (Thermo Fisher Scientific, cat. no. M-1381MP) by dissolving 25 mg of mBCl in 550 μL DMSO. Make 20 μL aliquots of and store them at −20°C protected from light.
Dilute 10 μL of the mBCl stock solution in 10 mL saline solution to achieve a final concentration of 200 μM mBCl.
YO-PRO-1 solution
YO-PRO-1 dye comes in a stock concentration of 1 mM in 1 mL DMSO (ThermoFisher, cat. no. Y3603). Add 50 μL of the YO-PRO-1 stock solution to 10 mL of the saline solution to achieve a final concentration of 5 μM.
COMMENTARY
Background Information
Conventionally, HCI assays have been performed on 2D cell monolayers, which may have limited predictability of in vivo toxicity due to limited cell-to-cell and cell-to-ECM interactions (Page et al., 2013). Although 3D cell models can mimic complex biological systems and provide more reliable information on functional tissues, existing 3D culture models such as 3D-printed tissue constructs have low throughput (requiring relatively large volume of samples) and poor imaging capability for HTS of compounds (Kriston-Vizi & Flotow, 2017). Here, we describe a protocol for an assay that uses a 384-pillar plate that can be coupled with standard 384-well plates for HCI assays (Yu, Kang, et al., 2018; Joshi et al., 2020). Although we have described here the use of this assay for the assessment of neurotoxicity using 3D-cultured NSCs, the cell printing and encapsulation technique can be applicable to any mammalian cell type. Cells can simply be mixed with alginate and Geltrex (or other hydrogels) and printed on the 384-pillar plate for cell encapsulation and 3D culture, which can be followed by compound treatment and cell staining, all in situ. In this protocol, Geltrex supports cell growth and spheroid formation by providing necessary ECM components, whereas alginate provides structural rigidity for cell encapsulation, because it is biologically inert (Datar et al., 2015). We have previously reported cell printing and encapsulation in several hydrogels including alginate, methacrylated alginate, Matrigel, and PuraMatrix on microarray chip platforms for miniature 3D cell culture (Kwon et al., 2014; Lee, 2016; Lee et al., 2008; Roth et al., 2018; Yu et al., 2018). In addition, we have also shown that 3D-cultured NSCs on the 384-pillar plate can be used to facilitate high-throughput spheroid culture in hydrogels while offering HCI capability (Joshi et al., 2020).
The 384-pillar plate offers several advantages over more conventional 3D cell culture platforms, including ultra-low attachment (ULA) well plates, Transwell® inserts, and hanging droplet plates. The 384-pillar plate requires fewer cells and less reagents, including hydrogels, ECMs, growth factors, growth media, compounds, and reagents for creating and evaluating 3D spheroids. Cell printing and encapsulation protocols developed are flexible and allow for culturing any mammalian cell type in biomimetic hydrogels on the pillar plate. The 384-pillar plate built on the footprint of standard 384-well plates is compatible with existing HTS equipment such as fully automated fluorescence microscopes and microtiter well plate readers, which is an important feature for compound screening. Unlike other traditional 3D cell culture platforms, the 384-pillar plate with 3D-cultured cells can easily be detached from a 384-well plate and sandwiched onto another 384-well plate containing growth media or cell-staining reagents, without disturbing or damaging the cells, for high-throughput, 3D cell-based HCI assays. It can be adopted for highly reproducible, high-throughput precision printing to test a variety of 3D cell culture conditions and individual compounds/mixtures of compounds in combination, which makes it well suited for early-stage HTS of compound libraries. Cell image acquisition from 3D-cultured cells is easy and straightforward because the whole sample depth fits within the focus depth of a normal objective (4x and 10x). The entire 384-pillar plate can be scanned with an automated florescence microscope, and 384 images can be obtained within 20 – 40 minutes, depending on exposure time. Thus, our miniature 3D bioprinting approach on the 384-pillar plate could be used for predictive screening of compounds, which can ultimately lead to safer chemical product development and use.
Critical Parameters
For reproducible and uniform cell printing on the 384-pillar plate, it is critical to maintain good suspension of the cells in hydrogels during printing, which can be achieved by selecting appropriate viscosity of the hydrogel (typically 50 – 100 cP) and quick pipetting up and down prior to aspiration of the cell suspension for 3D bioprinting. Unlike extrusion-based 3D bioprinters, ink-jet and micro-solenoid valve-driven 3D bioprinters can often have difficulty handling high concentrations of cells in hydrogels (typically greater than 15 million cells/mL) due to clogging of ink-jet nozzles and micro-solenoid valves.
For high cell viability after printing on the 384-pillar plate, it is critically important to avoid water evaporation during cell printing (which typically takes 5 – 10 minutes depending on the number of 384-pillar plates printed) and hydrogel gelation (5 – 20 minutes). Thus, always maintain the plate loading deck at a low temperature (typically 4 – 10°C depending on relative humidity in the room) in 3D bioprinters, so that slight water condensation on the pillar surface can be achieved. Maintaining too low a temperature of the plate loading deck, however, at high relative humidity in the summer, will lead to excessive water condensation on the 384-pillar plate, which is also not desired. Ink-jet and micro-solenoid valve-driven 3D bioprinting does not induce high cell death because of low shear stress and pressure applied in the system. With non-cytotoxic hydrogels selected, users can obtain cell viability greater than 95%.
Proper cell seeding density on the 384-pillar plate (typically 1,000 – 5,000 cells/pillar) is a critical parameter, which should be optimized for cell growth, compound treatment, and image acquisition. Too high cell seeding on the pillars may cause overgrowth of the cells, leading to frequent growth medium change, rapid ECM remodeling, and difficulty in cell staining and imaging. Overgrowth of cells may lead to hydrogel degradation and detachment from the 384-pillar plate due to excessive secretion of matrix metalloproteinases (Ha et al., 2004; Mason & Joyce, 2011; Weng et al., 2012).
The selection of biomimetic hydrogels for cell encapsulation and 3D culture depends on several critical factors, including gelation mechanism, compatibility with the functionalized surface of the 384-pillar plate for robust cell spot attachment, appropriate viscosity for 3D bioprinting, and basal cytotoxicity. The gelation mechanism is one of the most critical parameters in selecting hydrogels for robust cell encapsulation and long-term culture. For example, temperature-sensitive hydrogels such as Matrigel® and Geltrex are difficult to handle at room temperature due to spontaneous gelation during printing, thus requiring the printing head and tubing in 3D bioprinters to be chilled at less than 7°C. Other hydrogels, which form a gel by ionic crosslinking, photo-polymerization, covalent bonding, or pH-induced phase transition may be a better alternative to temperature-sensitive hydrogels if users do not want to deal with chilling the equipment and pipette tips for cell dispensing and encapsulation. In addition, certain gelation mechanisms are more toxic to cells due to additives, UV light, and chemical reactions. For example, photo-polymerization is cytotoxic to NSCs due to UV light and the oxidative radicals generated.
Pre-incubation of the cells on the 384-pillar plate prior to compound exposure is another important parameter that can affect cellular responses to compounds. Users are recommended to pre-incubate the bioprinted cells for 3 – 7 days before exposure to test compounds, so that the cells can recover from trypsinization and form spheroids in hydrogels.
The duration of test compound treatment is critical to obtain desired cellular responses from compounds in cell-based assays, since some mechanisms of cell death such as apoptosis are time-dependent and cannot be detected after certain time points. In general, it is required to incubate the cells with compounds for 4 – 24 hours for the measurement of apoptosis, whereas the measurement of cell growth inhibition by compounds requires at least two doubling times of cell incubation with compounds (48 – 72 hours).
To obtain high quality images from cell spheroids on the 384-pillar plate, it is necessary to optimize the concentration of fluorescent dyes and the incubation time with fluorescent dyes. Users are suggested to use the manufacturer’s recommended concentration first and then optimize the cell staining conditions depending on the signal-to-noise ratio obtained from fluorescent cell images
Troubleshooting
Please refer to Table 3 for a troubleshooting guide for robust 3D cell culture on the 384-pillar plate. The most common problem users may encounter is the detachment of cell spots from the 384-pillar plate. Spot detachment occurs mainly due to degradation of hydrogels over time and inappropriate functionalization of the pillar surface. Hydrogels such as alginate and Matrigel are degraded mainly because of excessive chelating agents (e.g., phosphate ions and EDTA) in culture media and/or the secretion of matrix metalloproteinases. Therefore, it is important to select a strong ionic crosslinker (e.g., barium chloride over calcium chloride for alginate crosslinking) and optimize cell seeding density to suppress excessive secretion of matrix metalloproteinases. To avoid spot detachment due to incompatibility between the pillar surface and the hydrogel, the selection of surface chemistry is highly important. The surface of the 384-pillar plate should be functionalized with amine-reactive amphiphilic polymers such PMA-OD, so that the positively charged PLL covalently attached on the surface can interact with the negatively charged alginate for robust spot attachment.
Table 3.
Troubleshooting guide for robust 3D cell culture on the 384-pillar plate
| Problem | Possible cause | Solution |
|---|---|---|
| Cell spot detachment from the pillars | Degradation of hydrogels over time by chelating agents | Select a strong ionic crosslinker |
| Cell spot detachment from the pillars | Degradation of hydrogels over time by matrix metalloproteinases | Lower cell seeding density or increase hydrogel concentration |
| Cell spot detachment from the pillars | Inappropriate functionalization of the pillar surface | Use fresh reagents for surface functionalization (e.g., PMA-OD and PLL) |
| Nonuniform cell printing | Precipitation of cells in hydrogels in a 96-well plate | Resuspend cells in hydrogels prior to aspiration by a 3D bioprinter or adjust viscosity of hydrogels |
| Cell death after printing | Evaporation of water during cell printing and hydrogel gelation | Maintain low temperature of the plate loading deck in a 3D bioprinter |
Understanding Results
The dose-response curve generated from this protocol provides information on the change or effect in cells caused by a wide concentration range of a test compound in a certain period of time. The IC50 value (half-maximal inhibitory concentration) of a compound obtained from the dose-response curve determines the strength of the compound in affecting various morphological and functional features of interest in cells (Fig. 7). By comparing IC50 values from different end points, users could understand the main mechanisms of compound neurotoxicity (Joshi et al., 2020). In addition, IC50 values obtained from a set of 50 – 200 model compounds can be used to determine predictivity of new cell models by calculating sensitivity and specificity (Yu, Nadanaciva, et al., 2018).
Time Considerations
- BASIC PROTOCOL 1: 3D NSC culture on a 384-pillar plate
- ReNcell VM culture and preparation of cell suspension (timing: 3 – 5 days)
- Functionalization of the 384-pillar plate for cell encapsulation in alginate (timing: 1 day)
- Cell printing on the 384-pillar plate (timing: 1 – 2 hours)
- BASIC PROTOCOL 2: Compound treatment and cell staining
- Compound treatment against 3D-cultured NSCs (timing: 2 days)
- Cell staining (timing: 2 – 3 hours)
- BASIC PROTOCOL 3: Image acquisition, processing, and data analysis
- Acquisition of fluorescent cell images with S+ Scanner (Timing: 1 hour)
- Image processing and data analysis (timing 3 – 4 hours)
In the example outlined here, the entire process takes about 1 week due to the rapid doubling time of ReNcell VM cells. Cell printing on the 384-pillar plate takes less than 5 minutes per plate, and alginate gelation for cell encapsulation on the pillars takes about 5 minutes. In addition, cell staining and image acquisition take 2 hours and 0.5 hours, respectively. Excluding time-consuming pre-incubation of cells after printing and compound treatment, it takes 2 hours to obtain hundreds of fluorescent cell images. Thus, these 3D cell-based assays on the 384-pillar plate are well suited for HTS of compounds.
ACKNOWLEDGEMENTS
This study was supported by the National Institutes of Health (NIEHS R01ES025779) and institutional grants from Cleveland State University (Faculty Research Development and Faculty Innovation Fund).
Footnotes
CONFLICT OF INTEREST STATEMENT:
The authors declare no conflict of interest.
DATA AVAILABILITY STATEMENT:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Literature Cited
- Aschner M, Ceccatelli S, Daneshian M, Fritsche E, Hasiwa N, Hartung T, Hogberg HT, Leist M, Li A, Mundy WR, Padilla S, Piersma AH, Bal-Price A, Seiler A, Westerink RH, Zimmer B, & Lein PJ (2017). Reference compounds for alternative test methods to indicate developmental neurotoxicity (DNT) potential of chemicals: Example lists & criteria for their selection & use. Altex, 34(1), 49–74. 10.14573/altex.1604201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astashkina A, & Grainger DW (2014). Critical analysis of 3-D organoid in vitro cell culture models for high-throughput drug candidate toxicity assessments. Advanced Drug Delivery Reviews, 69–70, 1–18. 10.1016/j.addr.2014.02.008 [DOI] [PubMed] [Google Scholar]
- Bal-Price A, Pistollato F, Sachana M, Bopp SK, Munn S, & Worth A (2018). Strategies to improve the regulatory assessment of developmental neurotoxicity (DNT) using in vitro methods. Toxicology and Applied Pharmacology, 354, 7–18. 10.1016/j.taap.2018.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Kim YH, Hebisch M, Sliwinski C, Lee S, D’Avanzo C, Chen H, Hooli B, Asselin C, Muffat J, Klee JB, Zhang C, Wainger BJ, Peitz M, Kovacs DM, Woolf CJ, Wagner SL, Tanzi RE, & Kim DY (2014). A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature, 515(7526), 274–278. 10.1038/nature13800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crofton KM, Mundy WR, & Shafer TJ (2012). Developmental neurotoxicity testing: A path forward. Congenital Anomalies, 52(3), 140–146. 10.1111/j.1741-4520.2012.00377.x [DOI] [PubMed] [Google Scholar]
- Datar A, Joshi P, & Lee MY (2015). Biocompatible hydrogels for microarray cell printing and encapsulation. Biosensors, 5(4), 647–663. 10.3390/bios5040647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato R, Miljan EA, Hines SJ, Aouabdi S, Pollock K, Patel S, Edwards FA, & Sinden JD (2007). Differential development of neuronal physiological responsiveness in two human neural stem cell lines. BMC Neuroscience, 8(1), 36. 10.1186/1471-2202-8-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epa, U. S. (2015). America’s Children and the Environment: Neurodevelopmental Disorders. October,1–32.https://www.epa.gov/sites/production/files/2015-10/documents/ace3_neurodevelopmental.pdf
- Florio M, & Huttner WB (2014). Neural progenitors, neurogenesis and the evolution of the neocortex. Development (Cambridge), 141(11), 2182–2194. 10.1242/dev.090571 [DOI] [PubMed] [Google Scholar]
- Fritsche E, Alm H, Baumann J, Geerts L, Håkansson H, Masjosthusmann S, & Witters H (2017). Literature review on in vitro and alternative Developmental Neurotoxicity (DNT) testing methods.EFSASupportingPublications, 12(4), 1–186. 10.2903/sp.efsa.2015.en-778 [DOI] [Google Scholar]
- Glover WE (1982). 2 pyridine ‘ ~ NH2 ~NH. 13(c). [Google Scholar]
- Grimm FA, Iwata Y, Sirenko O, Bittner M, & Rusyn I (2015). High-content assay multiplexing for toxicity screening in induced pluripotent stem cell-derived cardiomyocytes and hepatocytes. Assay and Drug Development Technologies, 13(9), 529–546. 10.1089/adt.2015.659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha K-T, Kim J-K, Lee Y-C, & Kim C-H (2004). Inhibitory effect of Daesungki-Tang on the invasiveness potential of hepatocellular carcinoma through inhibition of matrix metalloproteinase-2 and −9 activities. Toxicology and Applied Pharmacology, 200(1), 1–6. 10.1016/j.taap.2004.03.012 [DOI] [PubMed] [Google Scholar]
- Jensen HB, Ravnborg M, Dalgas U, & Stenager E (2014). 4-Aminopyridine for symptomatic treatment of multiple sclerosis: A systematic review. Therapeutic Advances in Neurological Disorders, 7(2), 97–113. 10.1177/1756285613512712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Davis J, Zhu D, Kashima DT, Leroueil M, Pan C, Montine KS, & Zhang J (2007). Identification of novel proteins affected by rotenone in mitochondria of dopaminergic cells. BMC Neuroscience, 8(1), 67. 10.1186/1471-2202-8-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi P, Kang S-Y, Yu K-N, Kothapalli C, & Lee M-Y (2020). High-content imaging of 3D-cultured neural stem cells on a 384-pillar plate for the assessment of cytotoxicity. Toxicology in Vitro, 65, 104765. 10.1016/j.tiv.2020.104765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi P, & Lee MY (2015). High content imaging (HCI) on miniaturized three-dimensional (3D) cell cultures. In Biosensors (Vol. 5, Issue 4, pp. 768–790). 10.3390/bios5040768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi P, Yu K-N, Kang S-Y, Kwon SJ, Kwon PS, Dordick JS, Kothapalli CR, & Lee M-Y (2018). 3D-cultured neural stem cell microarrays on a micropillar chip for high-throughput developmental neurotoxicology. Experimental Cell Research, 370(2), 680–691. 10.1016/j.yexcr.2018.07.034 [DOI] [PubMed] [Google Scholar]
- Justice BA, Badr NA, & Felder RA (2009). 3D cell culture opens new dimensions in cell-based assays. Drug Discovery Today, 14(1–2), 102–107. 10.1016/j.drudis.2008.11.006 [DOI] [PubMed] [Google Scholar]
- Kriston-Vizi J, & Flotow H (2017). Getting the whole picture: High content screening using three-dimensional cellular model systems and whole animal assays. Cytometry Part A, 91(2), 152–159. 10.1002/cyto.a.22907 [DOI] [PubMed] [Google Scholar]
- Kwon SJ, Lee DW, Shah DA, Ku B, Jeon SY, Solanki K, Ryan JD, Clark DS, Dordick JS, & Lee MY (2014). High-throughput and combinatorial gene expression on a chip for metabolism-induced toxicology screening. Nature Communications, 5(May), 1–12. 10.1038/ncomms4739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M-Y, Kumar RA, Sukumaran SM, Hogg MG, Clark DS, & Dordick JS (2008). Three-dimensional cellular microarray for high-throughput toxicology assays. Proceedings of the National Academy of Sciences, 105(1), 59–63. 10.1073/pnas.0708756105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M (2016). Microarray Bioprinting Technology. In Microarray Bioprinting Technology. 10.1007/978-3-319-46805-1 [DOI] [Google Scholar]
- Li J, Spletter ML, Johnson DA, Wright LS, Svendsen CN, & Johnson JA (2005). Rotenone-induced caspase 9/3-independent and -dependent cell death in undifferentiated and differentiated human neural stem cells. Journal of Neurochemistry, 92(3), 462–476. 10.1111/j.1471-4159.2004.02872.x [DOI] [PubMed] [Google Scholar]
- Llorens J, Li AA, Ceccatelli S, & Suñol C (2012). Strategies and tools for preventing neurotoxicity: To test, to predict and how to do it. NeuroToxicology, 33(4), 796–804. 10.1016/j.neuro.2012.01.019 [DOI] [PubMed] [Google Scholar]
- Mason SD, & Joyce JA (2011). Proteolytic networks in cancer. Trends in Cell Biology, 21(4), 228–237. 10.1016/j.tcb.2010.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundy WR, Padilla S, Breier JM, Crofton KM, Gilbert ME, Herr DW, Jensen KF, Radio NM, Raffaele KC, Schumacher K, Shafer TJ, & Cowden J (2015). Expanding the test set: Chemicals with potential to disrupt mammalian brain development. Neurotoxicology and Teratology, 52, 25–35. 10.1016/j.ntt.2015.10.001 [DOI] [PubMed] [Google Scholar]
- Olson H, Betton G, Robinson D, Thomas K, Monro A, Kolaja G, Lilly P, Sanders J, Sipes G, Bracken W, Dorato M, Van Deun K, Smith P, Berger B, & Heller A (2000). Concordance of the Toxicity of Pharmaceuticals in Humans and in Animals. Regulatory Toxicology and Pharmacology, 32(1), 56–67. 10.1006/rtph.2000.1399 [DOI] [PubMed] [Google Scholar]
- Page H, Flood P, & Reynaud EG (2013). Three-dimensional tissue cultures: current trends and beyond. Cell and Tissue Research, 352(1), 123–131. 10.1007/s00441-012-1441-5 [DOI] [PubMed] [Google Scholar]
- Prassas I, Karagiannis GS, Batruch I, Dimitromanolakis A, Datti A, & Diamandis EP (2011). Digitoxin-induced cytotoxicity in cancer cells is mediated through distinct kinase and interferon signaling networks. Molecular Cancer Therapeutics, 10(11), 2083–2093. 10.1158/1535-7163.MCT-11-0421 [DOI] [PubMed] [Google Scholar]
- Roth AD, Lama P, Dunn S, Hong S, & Lee M-Y (2018). Polymer coating on a micropillar chip for robust attachment of PuraMatrix peptide hydrogel for 3D hepatic cell culture. Materials Science & Engineering. C, Materials for Biological Applications, 90, 634–644. 10.1016/j.msec.2018.04.092 [DOI] [PubMed] [Google Scholar]
- Smirnova L, Hogberg HT, Leist M, & Hartung T (2014). Food for thought…: Developmental neurotoxicity - Challenges in the 21st century and in vitro opportunities. Altex, 31(2), 129–156. 10.14573/altex.1403271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, & Stewart L (2002). The mechanism of topoisomerase I poisoning by a camptothecin analog. Proceedings of the National Academy of Sciences, 99(24), 15387 LP – 15392. 10.1073/pnas.242259599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterzyńska K, Klejewski A, Wojtowicz K, Świerczewska M, Andrzejewska M, Rusek D, Sobkowski M, Kędzia W, Brazert J, Nowicki M, & Januchowski R (2018). The role of matrix gla protein (MGP) expression in paclitaxel and topotecan resistant ovarian cancer cell lines. International Journal of Molecular Sciences, 19(10). 10.3390/ijms19102901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Y, & Wu Z (2007). Alternative statistical parameter for high-throughput screening assay quality assessment. Journal of Biomolecular Screening, 12(2), 229–234. 10.1177/1087057106296498 [DOI] [PubMed] [Google Scholar]
- Taylor CA, Tuschl K, Nicolai MM, Bornhorst J, Gubert P, Varão AM, Aschner M, Smith DR, & Mukhopadhyay S (2020). Maintaining Translational Relevance in Animal Models of Manganese Neurotoxicity. The Journal of Nutrition, 150(6), 1360–1369. 10.1093/jn/nxaa066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vliet E, Daneshian M, Beilmann M, Davies A, Fava E, Fleck R, Julé Y, Kansy M, Kustermann S, Macko P, Mundy WR, Roth A, Shah I, Uteng M, Van De Water B, Hartung T, & Leist M (2014). Current approaches and future role of high content imaging in safety sciences and drug discovery. Altex, 31(4), 479–493. 10.14573/altex.1405271 [DOI] [PubMed] [Google Scholar]
- Weng CJ, Chou CP, Ho CT, & Yen GC (2012). Molecular mechanism inhibiting human hepatocarcinoma cell invasion by 6-shogaol and 6-gingerol. Molecular Nutrition and Food Research, 56(8), 1304–1314. 10.1002/mnfr.201200173 [DOI] [PubMed] [Google Scholar]
- Yu KN, Kang SY, Hong S, & Lee MY (2018). High-throughput metabolism-induced toxicity assays demonstrated on a 384-pillar plate. Archives of Toxicology, 92(8), 2501–2516. 10.1007/s00204-018-2249-1 [DOI] [PubMed] [Google Scholar]
- Yu KN, Nadanaciva S, Rana P, Lee DW, Ku B, Roth AD, Dordick JS, Will Y, & Lee MY (2018). Prediction of metabolism-induced hepatotoxicity on three-dimensional hepatic cell culture and enzyme microarrays. Archives of Toxicology, 92(3), 1295–1310. 10.1007/s00204-017-2126-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, & Oldenburg KR (1999). A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. Journal of Biomolecular Screening, 4(2), 67–73. 10.1177/108705719900400206 [DOI] [PubMed] [Google Scholar]