

Abstract
Identifying pathogenic mutations in BRCA1 and BRCA2 is a critical step for breast cancer prediction. Genome-wide association studies (GWAS) are the most commonly used method for inferring pathogenic mutations. However, identifying pathogenic mutations using GWAS can be difficult. The hypothesis of this study is that the pathogenic mutations in human BRCA1/BRCA2, which are present in many species, are more likely to be located in the evolutionarily conserved sites. This study defines the evolutionary conservativeness based on the previously developed Characteristic Attribute Organization System (CAOS) software. ClinVar is used to identify human pathogenic mutations in BRCA1 and BRCA2. Statistical tests suggest that compared to the non-pathogenic mutations, human pathogenic mutations were more likely to locate at the evolutionary conserved positions. The approach presented in this study shows promise in identifying pathogenic mutations in humans, suggesting that the methodology may be applied to other disease-related genes to identify putative pathogenic mutations.
Introduction
According to the Center for Disease Control and Prevention (CDC), breast cancer is one of the most common cancers in women in the United States.1 Mutations on genes such as breast cancer susceptibility gene 1 (BRCA1) and 2 (BRCA2) that connote an increased risk of breast cancer have been identified. According to a meta-analytic study, BRCA1 mutation carriers had 57% risk for developing breast cancer and 40% chance for ovarian cancer by the age of 70.2 The breast cancer risk for BRCA2 mutation carriers was 49% and ovarian cancer risk was 18% at the age of 70 years old. Identifying the pathogenic mutations in BRCA1 and BRCA2 is therefore critical for disease prediction and prevention. To date, the most widely used method to identify pathogenic mutations is through using high throughput sequencing and genome-wide association studies (GWAS).3,4 A limitation of GWAS studies is the requirement of sequencing data from an adequate number of cases. Distinguishing between pathogenic and benign mutations using GWAS can thus be difficult in rare diseases where the number of cases is less abundant.5
BRCA1 and BRCA2 are genes with long evolutionary history and present in many species.6 BRCA1 has been identified in plants and animals, while BRCA2 has also been found in fungi. Among the BRCA1 and BRCA2 genes, some positions are more conserved than others across evolutionary history.6 Research has shown that the pathogenic mutations in humans are more likely to exist in fixed sites of proteins.7 Several studies have shown the disease-associated missense mutations in BRCA1 are correlated with the conserved residues among different species.8,9,10 However, these studies have a limited number of species included and use nucleotide percentage similarity among sequences to determine conservativeness.
Different from the previous studies, this study proposes the use of a previously developed phylogenetic approach, called the Characteristic Attribute Organization System (CAOS), to determine the evolutionary conservativeness of different positions for a given gene.11 CAOS discovers rules associated with a given phylogenetic tree as shown in Figure 1a.11 A pure (Pu) rule or character attribute (CA) is a state that exists in all elements of a clade but not the alternate clade; a private (Pr) CA is present in some members of a clade but absent in the alternate clade. A variation number (VN) is defined as the number of occurrences of a position as a CA in all the tree clades. A flowchart of the VN calculation is shown in Figure 1d. For example, the VN of the first position in Figure 1a will be zero, the third position will be two, and the fifth position will be four. Evolutionary conservativeness in this study is defined as the positions with a relatively small VN. Thus, the first position in Figure 1a is more conserved than the third position. The clades in both Figure 1b and 1c have three sequences of cytosine and two sequences of adenine. The compositions are the same using conventional statistic method.9 By CAOS definition, however, the clade in Figure 1b has a VN of two while Figure 1c has a VN of five. The clade in Figure 1b is thus more conserved. The hypothesis of this study is that the positions of human pathogenic mutations in BRCA1 and BRCA2 genes are more likely to be evolutionarily conserved than the positions of the non-pathogenic mutations.
Figure 1.

Characteristic Attribute Organization System (CAOS)11. a. An example to define CAOS rules. b-c. Examples of CAOS differs from statistical methods. d. The flowchart for the VN calculation.
Methods
The pipeline for analysis developed for this study is shown in Figure 2. The overall process includes sequence retrieval, multiple sequence alignment, phylogenetic tree construction, and analysis of the conservativeness of gene positions. The results were assessed relative to previously annotated clinical pathogenicity of positions in the genes of interest. Homo sapiens DNA repair associated breast cancer 1 (BRCA1) and breast cancer 2 (BRCA2) reference transcripts were retrieved from National Center for Biotechnology Information (NCBI) nucleotide database (Accession numbers NM_007294 and NM_000059, respectively).12
Figure 2.

Flowchart of the pipeline.
Sequence retrieval and alignments
The Basic Local Alignment Search Tool (BLAST) was used to retrieve sequences that were similar to Homo sapiens BRCA1 and BRCA2.13 Both BRCA1 and BRCA2 were searched over the nucleotide collection database for all animal organisms. The max target sequences were set to 20,000 with expected threshold of one. The result sequences were filtered such that, if there were multiple sequences for one organism, the entire sequence with the best hit and with the highest max score in the BLAST results was kept. Synthetic sequences were removed.
In addition to using BLAST for finding similar sequences, BRCA1 and BRCA2 orthologs from multiple vertebrates were retrieved from the NCBI Orthologs database. Multiple sequences may be available for a given species, but only one sequence with the longest length for each species was retrieved. Sequences from BLAST and NCBI Orthologs were used separately for multiple sequence alignment. Multiple sequence alignments were performed using multiple sequence comparison log-expectation (MUSCLE) software with the default settings.14
Tree building
Maximum parsimony trees were generated using the phylogenetic analysis using parsimony (PAUP*) software and the aligned sequence sets.15 A heuristic search method with two-hundred replicates and tree bisection reconnection (TBR) branch swapping was applied and the best tree was retained. A total of four trees were generated, two each for BRCA1 and BRCA2. The “BLAST tree” refers to the maximum parsimony tree using the sequences from the BLAST search; the “orthologs tree” refers to the maximum parsimony tree using the sequences retrieved from NCBI orthologs. The trees were then used for CAOS analysis.
Clinical variation retrieval
Clinical variations for BRCA1 and BRCA2 were downloaded from ClinVar.16 Only the single nucleotide variations were included. For BRCA1, the variations in the coding region of accession NM_007294.3 were kept. The coding region variations of NM_000059.3 were kept for BRCA2. Clinical significance was used to categorize whether a variation is considered pathogenic or non-pathogenic. This study only considered the variation position (i.e., not the specific variation). If the clinical significance was noted as “Pathogenic” then the variation position would be categorized as pathogenic. Otherwise, it was considered non-pathogenic. If there were multiple variations at the same position and at least one variation at that position is pathogenic, then the position was categorized as pathogenic.
Characteristic attribute organization system (CAOS) analysis
The Characteristic Attribute Organization System (CAOS) system was used to identify positions of interest (“rules”) for each tree node.11 Each position of the aligned sequences was assigned a VN, which was calculated based on the breadth first search and was described in Figure 1d. A Student’s T-test was performed using the VNs of pathogenic positions and non-pathogenic positions. Two control groups were generated by randomly picking positions and their corresponding VNs from the aligned sequences. The size of the control groups matched the number of pathogenic and non-pathogenic positions, correspondingly. The randomly chosen control groups were then used to perform a T-test. This process was repeated randomly five times. These T-tests served as negative controls to examine the effect of size differences on the T-test results.
The conventional statistical method, which calculates the nucleotide percentage similarity for a given position, was also used to find the conservativeness of the pathogenic and non-pathogenic positions. For each position, the percentage similarity was calculated by dividing the number of occurrences of the most abundant nucleotide by the total number of sequences. A Student’s T-test using the percentage similarity was conducted as a comparison to the T-test using the VNs. In addition to the T-test, the sequences at pathogenic positions and non-pathogenic positions were extracted to build phylogenetic trees separately. The number of clades was counted in each tree for comparison. Because there were more pathogenic than non-pathogenic positions, N non-pathogenic positions were randomly selected to generate 20 random trees as controls for sizes, where N was the total number of pathogenic positions.
Results
Sequence retrieval and alignments
The length of the Homo sapiens BRCA1 transcript is 7088 base pairs (bp). The coding region is 5592 bp long, spanning from position 114 to 5705. Homo sapiens BRCA2 transcript is 11,386 bp long, with the coding region spanning from position 228 to 10,484. Sequences like human BRCA1 and BRCA2 were found using BLAST and filtered such that only one sequence was kept for each organism. After filtering, 808 organisms were found to have sequences similar to Homo sapiens BRCA1 and 382 organisms had sequences similar to Homo sapiens BRCA2 as shown in Table 1. In the NCBI orthologs database, there are 273 BRCA1 orthologs from jawed vertebrates other than Homo sapiens and 266 BRCA2 orthologs. Among the 809 BRCA1-like sequences found using BLAST, 62 are present in the BRCA1 orthologs database. The overlap between BRCA2 BLAST and orthologs sequences are 93.
Table 1.
Number of species included for BRCA1 and BRCA2 multiple sequence alignment using either BLAST13 or NCBI Orthologs, and the number of overlapped sequences between BLAST and orthologs sequences.
| BLAST | Orthologs | Overlapped | |
| Total BRCA1 sequences Total BRCA2 sequences |
809 383 |
274 267 |
62 93 |
The BLAST sequences of both BRCA1 and BRCA2 were mainly from the class Mammalia. The orthologs sequences were from jawed vertebrates consisted of several classes as shown in Table 2. All BRCA1 BLAST sequences were from the class Mammalia consisted of 21 different orders. Fifty-one sequences were from the order Primates. Among the 383 BRCA2 BLAST sequences, 373 were from the class Mammalia, six from Archelosauria, two from Testudines and Archosauria group, and one each from Actinopterygii and Lepidosauria. The BRCA2 BLAST sequences were from 27 different orders with 138 Primates sequences. BRCA1 orthologs sequences were from nine different classes with 125 mammal sequences and BRCA2 orthologs sequences were from the same nine classes with 124 mammal sequences. MUSCLE was used for multiple sequence alignment. After alignment, the length of BRCA1 BLAST, BRCA1 orthologs, BRCA2 BLAST, BRCA2 orthologs sequences were 13,945 bp, 19,727 bp, 18,170 bp, and 29,216 bp, respectively.
Table 2.
The total number of species from each order or class and the maximum number of organisms from the same order of the BLAST sequences or the same class of the orthologs sequences grouped together in the corresponding phylogenetic tree.
a. BRCA1 sequences obtained using BLAST.
| Order | Total* | Max* | Order | Total | Max | Order | Total | Max |
| Carnivora | 134 | 104 | Eulipotyphla | 1 | 1 | Pilosa | 11 | 11 |
| Cetartiodactyla | 46 | 45 | Glires | 316 | 258 | Primates | 51 | 44 |
| Chiroptera | 128 | 77 | Hyracoidea | 3 | 3 | Proboscidea | 2 | 2 |
| Chrysochloridae | 3 | 3 | Insectivora | 58 | 39 | Scandentia | 6 | 6 |
| Cingulata | 14 | 11 | Macroscelidea | 4 | 3 | Sirenia | 4 | 4 |
| Dermoptera | 2 | 2 | Perissodactyla | 16 | 8 | Tenrecidae | 3 | 2 |
| Didelphidae | 1 | 1 | Pholidota | 4 | 3 | Tubulidentata | 2 | 1 |
Total is the total number of species from each order or class.
Max is the maximum number of organisms from the same order of the BLAST sequences or the same class of the ortholog sequences grouped together.
b. BRCA1 sequences obtained using NCBI orthologs database.
| Class | Total | Max | Class | Total | Max | Class | Total | Max |
| Alligators | 4 | 4 | Bony fishes | 58 | 40 | Lizards | 9 | 9 |
| Amphibians | 3 | 2 | Cartilaginous fishes | 1 | 1 | Mammals | 125 | 124 |
| Birds | 69 | 69 | Coelacanths | 1 | 1 | Turtles | 4 | 4 |
c. BRCA2 sequences obtained using BLAST.
| Order | Total | Max | Order | Total | Max | Order | Total | Max |
| Alligatoridae | 2 | 2 | Glires | 54 | 19 | Primates | 138 | 136 |
| Carnivora | 33 | 18 | Hyracoidea | 1 | 1 | Proboscidea | 2 | 2 |
| Cetartiodactyla | 41 | 41 | Insectivora | 33 | 25 | Saurischia | 2 | 1 |
| Chiroptera | 28 | 28 | Longirostris | 2 | 2 | Scandentia | 3 | 3 |
| Chrysochloridae | 1 | 1 | Macroscelidea | 1 | 1 | Sirenia | 3 | 2 |
| Cingulata | 7 | 7 | Ostariophysi | 1 | 1 | Tenrecidae | 3 | 3 |
| Dermoptera | 2 | 2 | Perissodactyla | 6 | 6 | Testudinoidea | 1 | 1 |
| Durocryptodira | 2 | 1 | Pholidota | 3 | 3 | Tubulidentata | 1 | 1 |
| Eulipotyphla | 3 | 1 | Pilosa | 11 | 11 | Unidentata | 2 | 2 |
d. BRCA2 sequences obtained using NCBI orthologs database.
| Class | Total | Max | Class | Total | Max | Class | Total | Max |
| Alligators | 4 | 4 | Bony fishes | 54 | 53 | Lizards | 9 | 9 |
| Amphibians | 3 | 3 | Cartilaginous fishes | 2 | 2 | Mammals | 124 | 119 |
| Birds | 67 | 67 | Coelacanths | 1 | 1 | Turtles | 3 | 3 |
Tree building
Four maximum parsimony trees were generated using the four sets of sequences and were shown in Figure 3 using the interactive tree of life (iTOL) software.17 Each tree leaf was a species. The tree leaves were colored according to the taxonomical order or class they belong to and the legend was shown on the left of each graph. The trees were unrooted. The total number of species from each order of BLAST sequences or each class of orthologs sequences were listed in Table 2 column Total. The maximum number of organisms, which were from the same order of the BLAST sequences or the same class of the orthologs sequences, grouped in the same clade was shown in Table 2 column Max. Such clades were the largest clades in the tree that only contained taxa from the same order for BLAST sequences or the same class for the orthologs sequences. Most organisms from the same order or class were clustered into a single clade as suggested by Table 2 and shown in Figure 3, suggesting the trees were taxonomically valid. These results also indicated the evolutionary importance of BRCA1 and BRCA2 in different species, as the phylogenetic tree generated using simply the transcripts can cluster most species from the same order or class into the same group.
Figure 3.

Brief tree visualization using iTOL17.
Clinical variation retrieval
The number of pathogenic and non-pathogenic positions for BRCA1 and BRCA2 were listed in Table 3. There was a total of 4834 single nucleotide variations identified for BRCA1 transcript variant 1, accession NM_007294.3 in ClinVar (accessed November 19th, 2019).16 Among the 4834 variations, 586 were deemed pathogenic and 4248 were deemed non-pathogenic variations. Four hundred and eight-two of the 586 pathogenic variations were found in the coding region, while 3102 of the 4248 non-pathogenic variations were identified in the coding region. For the variation positions in the coding region, 441 positions were categorized as pathogenic and 2402 were non-pathogenic.
Table 3.
The number of pathogenic and non-pathogenic positions for BRCA1 and BRCA2
| Pathogenic | Non-Pathogenic | |
| BRCA1 (NM_007294.3) | 441 | 2402 |
| BRCA2 (NM_000059.3) | 596 | 4668 |
For BRCA2 transcript with accession NM_000059.3, 7723 single nucleotide variations were in ClinVar as of November 19th, 2019.16 Seven hundred and sixty-one variations were found to be pathogenic with 670 in the coding region. There were 6962 non-pathogenic variations identified and 5875 in the coding region. In the coding region, 596 positions were found to be pathogenic and 4668 positions were deemed non-pathogenic. The analysis revealed that there were five times more non-pathogenic positions than pathogenic positions in BRCA1 and seven times more in BRCA2.
Characteristic attribute organization system (CAOS) analysis
For a given tree, the CAs for each clade of the tree found by CAOS were collected to find the time of appearance or the VN of each sequence position. The VNs at the pathogenic positions and non-pathogenic positions were extracted to perform a Student’s T-test. The histogram and the normalized histogram of the VNs of pathogenic and non-pathogenic positions from BRCA1 sequences obtained using BLAST were shown in Figure 4. The x-axis was the VN and the y-axis was the number or percentage of positions with that VN. The pathogenic positions were marked in pink and the non-pathogenic positions were marked in blue. The pathogenic positions had a mean VN of 189.23 while the non-pathogenic positions had a mean VN of 210.82. The smaller mean VN suggested that the pathogenic positions were more conserved evolutionarily compared to the non-pathogenic positions. The t-value was 8.57 and the p-value was 8.37E-17. These results suggested a significant difference between the VNs of pathogenic and non-pathogenic positions. The T-tests for the BRCA1 orthologs sequences and BRCA2 BLAST and orthologs sequences showed similar results, as listed in Table 4.
Figure 4.

Histogram and normalized histogram of the VNs of CAOS rules at pathogenic and non-pathogenic positions in BRCA1 BLAST sequences. The x-axis represents the variation number. The y-axis represents the number of gene positions (top) and the percentage of gene positions (bottom) with a specific variation number.
Table 4.
The T-test results for VNs’ comparison between pathogenic and non-pathogenic positions in each set of sequences
| Pathogenic | Non-Pathogenic | T-test | ||||||
| Mean | Median | STD | Mean | Median | STD | t-Value | p-Value | |
| BRCA1 BLAST | 189.23 | 181.00 | 48.39 | 210.82 | 205.00 | 49.76 | -8.57 | 8.37e-17 |
| BRCA1 Orthologs | 104.86 | 112.00 | 48.13 | 133.05 | 140.00 | 47.40 | -11.32 | 4.31e-27 |
| BRCA2 BLAST | 154.48 | 157.00 | 34.98 | 169.81 | 172.00 | 36.51 | -10.02 | 2.66e-22 |
| BRCA2 Orthologs | 97.03 | 104.00 | 46.37 | 117.66 | 125.00 | 44.87 | -10.25 | 3.60e-23 |
Two sets of randomly picked positions, with each set matched the size of corresponding pathogenic and non-pathogenic positions, and their VNs were used to perform T-tests as controls to examine the effect of size difference. This process was repeated five times. As a result, there were no significant differences from the controls, as shown in Figure 5. The control groups demonstrated that the significant T-test results shown in Figure 4 were not caused by the size differences between the pathogenic and non-pathogenic positions.
Figure 5.

Control normalized histograms of the VNs of CAOS rules at pathogenic and non-pathogenic positions in BRCA1 BLAST sequences.
As a comparison to VNs’ results, T-test was also performed using the percentage similarities at the pathogenic and non-pathogenic positions. The percentage similarity was defined as the frequency of the nucleotide with the most occurrence in a given position. A larger percentage similarity indicated that a given position is more evolutionarily conserved. The T-test results of BRCA1 BLAST sequences using percentage similarity were shown in Figure 6. There was no significant difference between the pathogenic and non-pathogenic positions when using percentage similarity. Similarly, the T-test results for the BRCA1 orthologs, BRCA2 BLAST, and BRCA2 orthologs sequences using percentage similarity were less significant than using the VNs. These results were not shown here. Such differences in significance showed the advantage of CAOS over the conventional statistical method.
Figure 6.
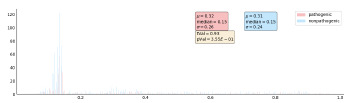
Percentage similarity histogram of pathogenic and non-pathogenic positions in BRCA1 BLAST sequences.
The pathogenic positions and non-pathogenic positions characterized for BRCA1 and BRCA2 transcripts were extracted to build phylogenetic trees separately. The number of clades for BRCA1 BLAST tree using pathogenic and non-pathogenic positions is shown in Table 5. Because there are more non-pathogenic positions than pathogenic positions, 20 trees were built using randomly selected non-pathogenic positions matching the number of pathogenic positions, and the average number of branches was calculated to serve as a control. These trees served as controls to test the effect of size on trees. The pathogenic tree resulted in fewer tree clades than the non-pathogenic tree. The average number of branches in the control group trees is less than in the non-pathogenic tree, suggesting the number of positions included can affect the tree topology. However, with the same number of positions included, there were still more clades in the control group trees than the pathogenic tree, suggesting the pathogenic positions contain less information to build complex phylogenetic trees. The number of clades for other sequence sets showed similar results and were not shown here.
Table 5.
Number of clades in pathogenic, non-pathogenic, and the average of twenty non-pathogenic trees where the number of positions selected equaled to the number of positions in the BRCA1 BLAST pathogenic tree.
| Pathogenic | Non-pathogenic | Controls | |
| Number of branches | 571 | 759 | 645.9 |
Discussion
Mutations in BRCA1 or BRCA2 greatly increase the risk of human breast cancer and identifying pathogenic mutations in humans can help to detect breast cancer in the early stages, thus increasing the patient survival rate. GWAS is widely used to infer pathogenic mutations. However, many mutations can be family specific and hard to detect. This study proposed a method to study the correlation between the evolutionarily conservativeness of mutations and their pathogenicity in human breast cancer. The method proposed in this study may be applied to other complex diseases with a genetic component, and it can be beneficial especially to rare diseases where limited patient data are available.
Transcript variants of BRCA1 and BRCA2 were used instead of whole genome or protein sequences for several reasons. Transcript variants were the intermediate between DNA and mRNA with introns removed. The short length of the transcript variants compared to the whole gene sequence makes it computationally efficient while retain the most important information in the genes. They contain information needed to produce the respective proteins, but are more specific than protein sequences. NM_007294.3 for BRCA1 and NM_000059.3 for BRCA2 were the reference variants commonly seen in ClinVar. Using transcript variants made it convenient to compare the data in ClinVar. The current version of BRCA1 transcript variant on NCBI is NM_007294.4 instead of NM_007294.3, but the coding region of NM_007294.4 and NM_007294.3 are the same. It will also be of future interest to further separate the pathogenic and non-pathogenic variants into synonymous and non-synonymous groups.
Instead of using pure statistical methods, an evolutionary approach using CAOS was taken in this study. This approach was chosen to preserve potentially more biologically meaningful relationships, such as illustrated in Figures 1b and 1c. Both Figure 1b and 1c have three sequences of cytosine and two sequences of adenine for a given position. Intuitively, one might consider that the position in Figure 1c is less informative as it can change freely among different species. The clade in Figure 1b, on the other hand, shows the conservativeness of the position because one subgroup would have the same sequence while the other subgroup has another sequence. By CAOS definition, the clade in Figure 1b has a VN of two while Figure 1c has a VN of five, meaning the clade in Figure 1b is more conserved than the clade in Figure 1c. However, such a conclusion cannot be made when using the conventional statistical method as the percentage similarity in both trees are the same.
As with any evolutionary analysis framework, there are several methodological choices that one can make to identify positions of interest in a given gene. This study chose to use maximum parsimony as the optimality criterion for inferring the phylogenetic trees. The choice for maximum parsimony was largely for convenience, computational efficiency, and previous experience with the phylogenetic inferencing technique in the context of CAOS. However, CAOS could be adapted to any character-based phylogenetic inferring technique, including those using a maximum likelihood or Bayesian optimality criterion. The methodology for VN determination would need to be adjusted to accommodate these additional optimality criteria (the VN in this study only accommodated for counts of differences between groups, which is in alignment with how maximum parsimony determines similarity). Similarly, the choice of BLAST and MUSCLE as the sequence retrieval and alignment tools could also be substituted with other tools. BLAST was chosen because it can identify more sequences like BRCA1/BRCA2 than using the NCBI orthologs alone. However, the sequences identified using might not be BRCA1/BRCA2 related sequences, thus the BRCA1/BRCA2 homologous sequences from the NCBI orthologs database were also used. MUSCLE was used for multiple sequence alignment in this study. However, a more sophisticated alignment method such as codon-alignment will be considered for future studies. T-tests were used in this study to compare the pathogenic and non-pathogenic positions. However, additional statistical analysis may be used to further validate the findings (e.g., using non-parametric tests, such as the Mann-Whitney U test) to avoid the assumption that the variation numbers of the pathogenic and non-pathogenic positions are normally distributed. The results of this study suggest that the general framework for gathering and organizing complex disease genes and subjecting them to an evolutionary analysis is a promising approach to identify putatively pathogenic mutations.
The method proposed in this study might be used to prioritize potential pathogenic mutations in human breast cancer solely based on sequencing data from different species. Although there are pathogenic mutations that seemed not to be conserved throughout evolution, as indicated by large VNs, the mutations in conserved positions do have a larger chance of being pathogenic. One limitation of the VN is that for a given set of sequences, a relatively smaller VN is defined as more conserved than a larger VN. But since the VNs depend on a specific set of sequences, no numerical cutoff for a VN can be given. The approach taken in this study alone may not be powerful enough to identify human pathogenic variants, however, it can be used as an important feature for a more sophisticated method. Our next goal is to develop a model/tool for calculating the probability of a variant being pathogenic or not, and the variant number described in the manuscript will be an important component for our future study. For the future study, we will also take into consideration other measurements such as the protein folding and intermolecular interactions. We will then compare the performance between our method to other existing methods. The method proposed can also be applied to rare diseases, in which GWAS study can be difficult because of the limited number of cases.
Human pathogenic alleles can be wild type in other species18. It is interesting to study why some positions are pathogenic in human while wild type in other species in BRCA1 and BRCA2 genes. It may be explained by compensated pathogenic deviations (CPD) and their compensated differences18. Studying CPDs are clinically important as it may help to explain why people carry BRCA1/BRCA2 mutations do not develop breast cancer. If people carrying BRCA1/BRCA2 pathogenic mutations as well as mutations in the compensated sites never developed breast cancer, then these sites can serve as markers and therapeutic targets for breast cancer. The specific CPDs and the compensatory differences will be the focus of future studies. One plausible way to find the CPDs is by finding the positions in the genes where the conservativeness is seen in some subgroups of the tree as shown in Figure 1b using CAOS. Such CPDs may also explain why some species were not clustered together with the species in the same order or class.
Conclusion
This study intended to apply an evolutionary framework to study BRCA1/BRCA2 human pathogenicity in relation to evolution. The requirement of sequences from different species instead of different patients made this study unique compared to other GWAS studies. Statistical analysis showed the advantage of using CAOS to define conservativeness than using nucleotide percentage similarity. Future works based on this study may be clinically meaningful.
Acknowledgements
The authors thank Dr. Daniel Weinreich, Dr. Dilber Ece Uzun, and Dr. Wafik El-Diery, all from Brown University, for discussion of the general concepts that underpin the methodology described here.
Funding
This work was possible with resources partially funded by U54GM115677.
Conflict of Interest: none declared.
Figures & Table
References
- 1.Center for Disease Control and Prevention 2019. Breast Cancer. Retrieved from https://www.cdc.gov/cancer/breast/statistics/index.htm .
- 2.Chen S, Parmigiani G. Meta-Analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol. 2007;25:1329–1333. doi: 10.1200/JCO.2006.09.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Turnbull C, Ahmed S, Morrison J, et al. Genome-Wide association study identifies five new breast cancer susceptibility loci. Nature Genetics. 2010;42:504–507. doi: 10.1038/ng.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghoussaini M, Fletcher O, Michailidou K, et al. Genome-Wide association analysis identifies three new breast cancer susceptibility loci. Nat Genet. 2012;44:312–318. doi: 10.1038/ng.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salgado D, Bellgard MI, Desvignes JP, Beroud C. How to identify pathogenic mutations among all those variations: variant annotation and filtration in the genome sequencing era. Human Mutation. 2016;37:1272–1282. doi: 10.1002/humu.23110. [DOI] [PubMed] [Google Scholar]
- 6.Pfeffer CM, Ho BN, Singh AT. The evolution, functions and applications of the breast cancer genes BRCA1 and BRCA2. Cancer Genomics & Proteomics. 2017;14:293–298. doi: 10.21873/cgp.20040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kondrashov AS, Sunyaev S, Kondrashov FA. Dobzhansky-Muller incompatibilities in protein evolution. PNAS. 2002;99:14878–14883. doi: 10.1073/pnas.232565499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pavlicek A, Noskov VN, Kouprina N, Barrett JC, Jurka J, Larionov V. Evolution of the tumor suppressor BRCA1 locus in primates: implications for cancer predisposition. Hum Mol Genet. 2004;15:2737–51. doi: 10.1093/hmg/ddh301. [DOI] [PubMed] [Google Scholar]
- 9.Szabo CI, Wagner LA, Francisco LV, Roach JC, Argonza R, King MC, Ostrander EA. Human, canine and murine BRCA1 genes: sequence comparison among species. Hum Mol Genet. 1996;5:1289–98. doi: 10.1093/hmg/5.9.1289. [DOI] [PubMed] [Google Scholar]
- 10.Fleming MA, Potter JD, Ramirez CJ, Ostrander GK, Ostrander EA. Understanding missense mutations in the BRCA1 gene: An evolutionary approach. PNAS. 2003;100:1151–1156. doi: 10.1073/pnas.0237285100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarkar I., Planet PJ, DeSalle R. CAOS software for use in character-based DNA barcoding. Molecular Ecology Resources. 2008;8:1256–1259. doi: 10.1111/j.1755-0998.2008.02235.x. [DOI] [PubMed] [Google Scholar]
- 12.Nucleotide [Internet]. Bethesda (MD): National Library of Medicine (US). National Center for Biotechnology Information. 2004. [cited 2019 Nov 19]. Available from: https://www.ncbi.nlm.nih.gov/nuccore .
- 13.BLAST [software]. Bethesda (MD). National Library of Medicine (US), National Center for Biotechnology Information. 2004. Available from: https://blast.ncbi.nlm.nih.gov/Blast.cgi .
- 14.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004;32:1792–97. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swofford DL. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods) (Version 4) [computer software] 2003. Available from https://paup.phylosolutions.com .
- 16.Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018. PubMed PMID: 29165669. [DOI] [PMC free article] [PubMed]
- 17.Letunic I, Bork P. Interactive tree of life(iTOL) v4: recent updates and new developments. Nucleic Acids Research. 2019;47:256–259. doi: 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kondrashov AS, Sunyaev S. Kondrashov FA. Dobzhansky-Muller incompatibilities in protein evolution. PNAS. 2002;99:14878–14883. doi: 10.1073/pnas.232565499. [DOI] [PMC free article] [PubMed] [Google Scholar]