

Abstract
Objective
To describe the phenotypic spectrum in patients with MBD5-associated neurodevelopmental disorder (MAND) and seizures; features of MAND include intellectual disability, epilepsy, psychiatric features of aggression and hyperactivity, and dysmorphic features including short stature and microcephaly, sleep disturbance, and ataxia.
Methods
We performed phenotyping on patients with MBD5 deletions, duplications, or point mutations and a history of seizures.
Results
Twenty-three patients with MAND and seizures were included. Median seizure onset age was 2.9 years (range 3 days–13 years). The most common seizure type was generalized tonic-clonic; focal, atypical absence, tonic, drop attacks, and myoclonic seizures occurred frequently. Seven children had convulsive status epilepticus and 3 nonconvulsive status epilepticus. Fever, viral illnesses, and hot weather provoked seizures. EEG studies in 17/21 patients were abnormal, typically showing slow generalized spike-wave and background slowing. Nine had drug-resistant epilepsy, although 3 eventually became seizure-free. All but one had moderate-to-severe developmental impairment. Epilepsy syndromes included Lennox-Gastaut syndrome, myoclonic-atonic epilepsy, and infantile spasms syndrome. Behavioral problems in 20/23 included aggression, self-injurious behavior, and sleep disturbance.
Conclusions
MBD5 disruption may be associated with severe early childhood-onset developmental and epileptic encephalopathy. Because neuropsychiatric dysfunction is common and severe, it should be an important focus of clinical management.
MBD5 (methyl-CpG-binding domain protein 5; OMIM #611472), located on chromosome 2q23.1, belongs to a family of genes involved in DNA methylation and chromatin remodeling.1 Disruption of this gene through heterozygous deletion or point mutation leads to MBD5-associated neurodevelopmental disorder (MAND), with some patients described as having a Kleefstra syndrome phenotypic spectrum.2 Intellectual disability occurs in all patients, with seizures, dysmorphic features including short stature and microcephaly, sleep disturbance, ataxia, aggressive behavior, and hyperactivity frequently observed.3–5 MBD5 deletions are not especially rare, found in 0.05% (1 in 2,000) of 17,477 samples that underwent clinical microarray testing.3 Deletions or mutations are almost always de novo although inheritance from mildly affected or mosaic parents has been reported.6
Seizures occur in over 80% of patients with MAND; however, the epileptology has not yet been delineated.3–5,7 Here, we analyzed the phenotypic spectrum in 23 patients with heterozygous deletion, duplication, or point mutation of MBD5 and a history of seizures.
Methods
We searched our epilepsy genetics research databases for patients with pathogenic variants involving MBD5 and identified 9 individuals. Two patients were identified through the Epi4K research testing program.8 Fourteen additional families volunteered to participate after social media patient groups brought attention to our research. Patients were ascertained from Australia, Italy, New Zealand, Finland, Canada, Germany, the United Kingdom, and the United States. We conducted personal interviews with all patients' families and reviewed medical records, EEG, neuroimaging, and genetic testing results. For medication response, we classified drugs as effective if caregivers reported a clear reduction in seizure frequency, even if seizures were not completely controlled. Wherever possible, epilepsy syndromes were classified according to the International League Against Epilepsy classification.9,10 Genetic variants were classified per American College of Medical Genetics and Genomics guidelines.11,12
Standard Protocol Approvals, Registrations, and Patient Consents
Written informed consent was provided for all patients by a parent or legal guardian. This study was approved by the Human Research Ethics Committee, Austin Health, or the local ethics committee.
Data Availability
Anonymized data will be shared by request from any qualified investigator.
Results
Twenty-three patients from 22 families were identified with a history of seizures in the context of a MBD5 molecular lesion.
Genetic Findings
Nineteen of the 23 patients had heterozygous 2q23.1 deletions: 15 resulted in partial deletion of MBD5 (11 proximal, 3 distal, and 1 data not available) and 4 had complete loss of MBD5 (figure 1, table 1). Two patients had 2q23 microduplications with partial MBD5 duplication (1 distal and 1 intragenic). The remaining 2 patients had point mutations resulting in truncation: p.Arg200* and p.Thr157Glnfs*4 (previously published13).
Figure 1. Copy Number Variants of Patients With MBD5-Associated Neurodevelopmental Disorder.

Green denotes deletion; yellow denotes duplication. This figure includes a screenshot from UCSC genome browser (genome.ucsc.edu).
Table 1.
Heterozygous Pathogenic Variants and Affecting MBD5
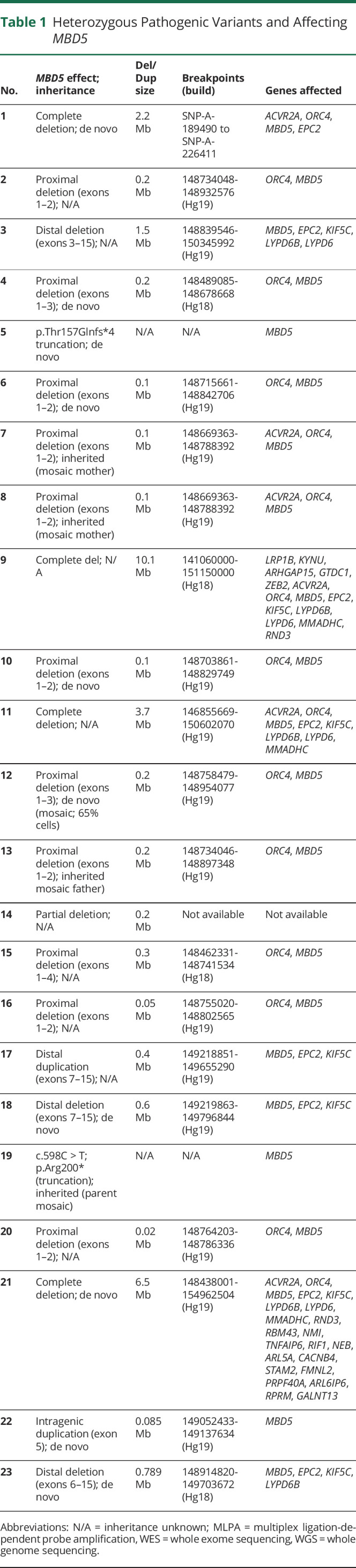
Pathogenic variants were de novo in 9 patients, with results from both parents not available in 10. Mutations were inherited from a mosaic parent in 4 patients from 3 families (figure 2). Patients 7 and 8 were brothers whose mosaic mother (20%–30% mosaicism in blood) had mild-moderate intellectual disability, but no history of seizures. Patient 13 had a mosaic carrier father (36% mosaicism in blood) of normal intellect and no history of seizures. Her older brother also inherited the deletion and had autism spectrum disorder and intellectual disability, without seizures. Patient 19 had parents who both tested negative for his mutation on blood-derived DNA; however, chorionic villus sampling of their next pregnancy showed that the fetus carried the mutation and the pregnancy was terminated. This suggested that 1 parent must be mosaic, with the mutation possibly limited to gonadal tissue. Patient 12 was mosaic for his deletion, affecting ∼65% of cells in saliva. The results of other genetic testing performed are given in table e-1, links.lww.com/NXG/A402.
Figure 2. Pedigrees of Families With Inheritance From Mosaic Parent.

The parents of patient 19 (right) both tested negative for the mutation on blood sequencing; however, chorionic villus sampling of their second pregnancy showed that the fetus carried the same mutation, so one of the parents is assumed to have low-level (likely gonadal) mosaicism. “m” = deletion/truncation mutation affecting MBD5; “m0.x” = mosaic with x% of cells having deletion; “+” = wild type. Central black circle indicates mosaic carrier; central question mark indicates possible mosaic carrier.
Seizures
Seizures began at median age 2.9 years (range 3 days to 13 years; table 2). Bilateral tonic-clonic seizures occurred in 19/23 (83%) patients, with focal impaired awareness seizures (FIAS; 9/23; 39%), tonic (8/23; 35%), unclassified drop attacks (7/23; 30%), myoclonic (7/23; 30%), atypical absences (7/23; 30%), myoclonic-atonic (1/23; 4%), atonic (1/23; 4%), hemiclonic (1/23; 4%), unclassified staring spells (absences vs FIAS; 1/23; 4%), and epileptic spasms (1/23; 4%) also observed. Convulsive status epilepticus occurred in 7/23 (30%) patients and nonconvulsive status epilepticus in 3/23 (13%). Fever and viral illnesses provoked seizures in 11 patients; 1 patient's seizures were triggered by painful stimuli (e.g., mild accidental falls to the ground).
Table 2.
Epilepsy Features

The most common interictal EEG findings were diffuse background slowing (11/23; 48%) and generalized spike-wave or polyspike-wave activity (10/23; 43%). Focal slowing and/or multifocal epileptiform discharges occurred in 9/23 (39%) patients. Epilepsy syndromes were defined in 7 patients: 3 had Lennox-Gastaut syndrome (LGS), 2 had myoclonic-atonic epilepsy (MAE), 1 had infantile spasms syndrome, and 1 had febrile seizures plus (FS+).
Ten patients had drug-resistant epilepsy. Although no drug was clearly superior, valproate showed the most consistent beneficial effect (12/14 cases), while carbamazepine exacerbated seizures in patient 2. Patient 2 became seizure-free during periods of illness and had dramatic reduction in seizure frequency on the ketogenic diet. Ketogenic diet therapy was also trialled in patient 5 with no benefit.
Twenty-one patients had available brain MRI results, with normal findings in 17/21 (85%). Of the patients with abnormal MRI, none had epileptogenic lesions (table 3). Patient 12 had a normal MRI, but fluorodeoxyglucose PET showed severe hypometabolism in the temporoparietal and occipital regions bilaterally.
Table 3.
Developmental Impairment, Behavioral Issues, and Other Clinical Features

Development and Behavior
Developmental impairment was present in all patients: severe in 14, moderate in 8, and mild in 1 (table 3). Regression with seizures occurred in 5 patients. The most dramatic regression occurred in patient 2 whose early developmental milestones were normal to mildly delayed (sat at 8 months, walked at 17 months, and first word at 12 months). When seizures began at age 2 years, there was marked developmental regression, particularly involving language. By age 4 years, he had only single words that were mostly unintelligible.
Marked behavioral difficulties were reported in 16 patients, with hyperactivity and aggression most common. Self-injurious behaviors occurred frequently, including finger and nail biting, scratching and picking lips until they bled. Behavioral difficulties were not controlled by stimulants, antipsychotics, and sedatives although methylphenidate elicited some benefit. Sleep disturbance during childhood was reported in 17 patients and involved frequent nocturnal awakenings. Nine patients had microcephaly.
Three patients had signs of metabolic dysfunction, which were considered coincidental. Patient 2 had decreased biotinidase activity suggesting a partial biotinidase deficiency; his parents reported some improvement in seizure control with biotin therapy. Patient 5 had borderline hypoglycorrhachia on lumbar punctures at 8 and 10 years (CSF glucose 2.9 and 2.2 mM with CSF:serum ratios of 0.41 and 0.43, respectively). Patient 20 was diagnosed with galactosemia on newborn screening.
Although our cohort is relatively small, we looked for genotype-phenotype correlations. Surprisingly, phenotypic severity was not more severe in patients with deletions affecting multiple genes. For example, patient 5 with a simple MBD5 truncation point mutation had one of the most severe phenotypes with a refractory developmental and epileptic encephalopathy and severe behavioral disturbance. The lack of strong genotype-phenotype correlation was further emphasized by the 2 families in which sibling pairs inherited the same deletion from a mosaic parent but had marked phenotypic differences, with 1 not having epilepsy at all. We did, however, observe greater phenotypic severity in patients with complete deletion of MBD5 compared with those with only partial gene deletion. Of those with partial deletions, the mildest phenotypes (patients 10, 16, and 20) were seen in individuals with loss of the proximal end of MBD5.
Discussion
With increasing access to molecular testing including high-resolution chromosomal microarray, more patients with disruption of MBD5 resulting in MAND will be identified. Understanding the epileptology of this genetic disease is critical for prompt diagnosis and optimal management. We analyzed the phenotypes of a global cohort of patients with MBD5 deletion, duplication, or point mutation and a history of seizures. We found a spectrum of phenotypes associated with MBD5 pathogenic variants, which often resulted in a severe early childhood-onset developmental and epileptic encephalopathy.
A broad spectrum of phenotypes was observed, and no genotype-phenotype correlation could be identified. The size of the cohort limits this analysis, and as the epileptology of further cases is described, correlations may become apparent. Phenotypes were more severe in patients with complete MBD5 gene deletion and milder in those with deletion of only the proximal end of the gene. This phenomenon could be explained by the deletion of fewer and different contiguous genes in the patients with proximal deletions. However, substantial phenotypic differences were noted in siblings carrying the same deletion, suggesting that variable expressivity in MAND reflects the involvement of modifier genes, epigenetic, or environmental influences.
Our molecular data highlight the importance of mosaicism, both in patient 12 and, even more critically, in 3/22 (14%) families who had 2 affected pregnancies. In 1 family, antenatal testing identified a second affected child despite negative testing of parental blood, strongly implicating gonadal mosaicism or low-level mosaicism in 1 parent that was missed on conventional sequencing. This suggests that inheritance from parents with low-level mosaicism may be more frequent than previously thought with key implications for reproductive counseling.14
Multiple seizure types were usual, including both generalized and focal, with tonic-clonic, focal, absence, atonic, myoclonic, and tonic seizures. EEG studies showed generalized and multifocal epileptiform activity. Seizures were often initially medically refractory but sometimes spontaneously resolved in childhood (age 4–7 years). Neuropsychiatric and developmental features were prominent including moderate-to-severe developmental impairment, language deficits, sleep disturbance, hyperactivity, and aggression.
Fever provoked seizures in 10 patients in our cohort, a pattern reported in 4 published cases.4,5,15,16 An additional study described hemiclonic seizures with alternating sides beginning at 10 months of age, a feature classically associated with Dravet syndrome (DS), a well-recognized developmental and epileptic encephalopathy associated with SCN1A mutations.17,18 Our cases, together with those reported, suggest that epilepsy in MAND sometimes has phenotypes on the genetic epilepsy with febrile seizures plus spectrum, including DS, MAE, FS+, and febrile seizures.19
MAND is typically associated with normal neuroimaging or thin corpus callosum with mild hypomyelination in rare cases. There are rare reports of focal cerebral malformations with MBD5 pathogenic variants, but these were associated with relatively large heterozygous deletions involving loss of many genes other than MBD5.4,20,21
The neuropsychiatric and behavioral abnormalities commonly observed in MBD5 pathogenic variants included sleep disturbance, developmental disability, language impairment, aggressive, and hyperactive behavior.4 When these occur, families should be counseled that these features are likely intrinsic to the genetic syndrome rather than secondary to medications or uncontrolled seizures. This is an important observation because some patients may undergo unnecessary investigations or medication changes, potentially jeopardizing seizure control, when the etiology of these behaviors is poorly understood.
Our findings should, however, be considered with some caution, given that this study had several limitations. Given the small size of the cohort, it was not possible to conduct statistical analyses or to make meaningful comments regarding genotype-phenotype correlation. As a retrospective study in which many patients self-referred, there are potential biases, most notably selection and recall biases. There was a preponderance of patients with copy number variants in our cohort because only 2 individuals had truncating MBD5 variants. This may relate to the accessibility of different investigations because CGH microarrays are far more readily available than gene panels around the world so that point mutations may be being missed in patients who lack access to next-generation sequencing.
In summary, patients with MBD5 pathogenic variants and seizures may have a range of phenotypes, including early childhood-onset developmental and epileptic encephalopathy. Epilepsy syndromes include infantile spasms syndrome, LGS, and MAE. Convulsive status epilepticus and nonconvulsive status epilepticus are fairly frequent, and seizures are often provoked by fever or environmental hyperthermia. Parental genetic testing should be offered because inheritance from mosaic parents may be more common than currently appreciated with important implications for genetic counseling.
Disclosure
K.A. Myers has received a travel grant from Zynerba and receives/has received research support from Fonds de Recherches du Québec—Santé, Citizens United for Research in Epilepsy (CURE), Research Institute of the McGill University Health Centre, the Savoy Foundation, Koolen-de Vries Foundation, and Dravet Canada. I.E. Scheffer serves on the editorial boards of Neurology® and Epileptic Disorders; may accrue future revenue on a pending patent re: Therapeutic compound; has received speaker honoraria from Athena Diagnostics, UCB, GSK, Eisai, and Transgenomics; has received scientific advisory board honoraria from Nutricia and GSK; has received funding for travel from Athena Diagnostics, UCB, and GSK; and receives/has received research support from the NHMRC, ARC, NIH, Health Research Council of New Zealand, March of Dimes, the Weizmann Institute, CURE, US Department of Defense, and the Perpetual Charitable Trustees. The other authors have indicated they have no relevant competing interests to disclose. Go to Neurology.org/NG for full disclosures.
Acknowledgment
The authors thank the patients and their families for their participation in this research. Amy Schneider assisted with data collection. This study was supported by funding from the National Health and Medical Research Council and the Research Institute of the McGill University Health Centre.
Glossary
- DS
Dravet syndrome
- FS+
febrile seizures plus
- LGS
Lennox-Gastaut syndrome
- MAE
myoclonic-atonic epilepsy
- MAND
MBD5-associated neurodevelopmental disorder
Appendix. Authors
Contributor Information
Carla Marini, Email: carla.marini@ospedaliriuniti.marche.it.
Gemma L. Carvill, Email: gemma.carvill@northwestern.edu.
Amy McTague, Email: a.mctague@ucl.ac.uk.
Julie Panetta, Email: julpul99@gmail.com.
Chloe Stutterd, Email: chloe.stutterd@vcgs.org.au.
Thorsten Stanley, Email: thorsten.stanley@otago.ac.nz.
Samantha Marin, Email: smarin2@exchange.hsc.mb.ca.
John Nguyen, Email: janguyen735@gmail.com.
Carmen Barba, Email: carmen.barba@meyer.it.
Anna Rosati, Email: anna.rosati@meyer.it.
Richard H. Scott, Email: richard.scott@genomicsengland.co.uk.
Heather C. Mefford, Email: hmefford@uw.edu.
Renzo Guerrini, Email: r.guerrini@meyer.it.
Ingrid E. Scheffer, Email: i.scheffer@unimelb.edu.au.
Study Funding
NHMRC (1091593), Research Institute of the McGill University Health Centre.
References
- 1.Talkowski ME, Mullegama SV, Rosenfeld JA, et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am J Hum Genet 2011;89:551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kleefstra T, Kramer JM, Neveling K, et al. Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am J Hum Genet 2012;91:73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hodge JC, Mitchell E, Pillalamarri V, et al. Disruption of MBD5 contributes to a spectrum of psychopathology and neurodevelopmental abnormalities. Mol Psychiatry 2014;19:368–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Bon BW, Koolen DA, Brueton L, et al. The 2q23.1 microdeletion syndrome: clinical and behavioural phenotype. Eur J Hum Genet 2010;18:163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wagenstaller J, Spranger S, Lorenz-Depiereux B, et al. Copy-number variations measured by single-nucleotide-polymorphism oligonucleotide arrays in patients with mental retardation. Am J Hum Genet 2007;81:768–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tadros S, Wang R, Waters JJ, et al. Inherited 2q23.1 microdeletions involving the MBD5 locus. Mol Genet Genomic Med 2017;5:608–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mullegama SV, Mendoza-Londono R, Elsea SH. MBD5 haploinsufficiency. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews®, Seattle: University of Washington, 1993. Available at https://www.ncbi.nlm.nih.gov/books/NBK1116/. [PubMed] [Google Scholar]
- 8.Consortium EK. Epi4K: gene discovery in 4,000 genomes. Epilepsia 2012;53:1457–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia 2017;58:512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fisher RS, Cross JH, French JA, et al. Operational classification of seizure types by the international League against epilepsy: position paper of the ILAE commission for classification and terminology. Epilepsia 2017;58:522–530. [DOI] [PubMed] [Google Scholar]
- 11.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and Genomics and the association for molecular pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riggs ER, Andersen EF, Cherry AM, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med 2020;22:245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carvill GL, Heavin SB, Yendle SC, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet 2013;45:825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Myers CT, Hollingsworth G, Muir AM, et al. Parental mosaicism in "de novo" epileptic encephalopathies. N Engl J Med 2018;378:1646–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bravo-Oro A, Lurie IW, Elizondo-Cardenas G, et al. A novel interstitial deletion of 2q22.3 q23.3 in a patient with dysmorphic features, epilepsy, aganglionosis, pure red cell aplasia, and skeletal malformations. Am J Med Genet A 2015;167A:1865–1871. [DOI] [PubMed] [Google Scholar]
- 16.Shichiji M, Ito Y, Shimojima K, et al. A cryptic microdeletion including MBD5 occurring within the breakpoint of a reciprocal translocation between chromosomes 2 and 5 in a patient with developmental delay and obesity. Am J Med Genet A 2013;161A:850–855. [DOI] [PubMed] [Google Scholar]
- 17.Dravet C. The core Dravet syndrome phenotype. Epilepsia 2011;52(Suppl 2):3–9. [DOI] [PubMed] [Google Scholar]
- 18.Motobayashi M, Nishimura-Tadaki A, Inaba Y, et al. Neurodevelopmental features in 2q23.1 microdeletion syndrome: report of a new patient with intractable seizures and review of literature. Am J Med Genet A 2012;158A:861–868. [DOI] [PubMed] [Google Scholar]
- 19.Scheffer IE, Berkovic SF. Generalized epilepsy with febrile seizures plus. A genetic disorder with heterogeneous clinical phenotypes. Brain 1997;120:479–490. [DOI] [PubMed] [Google Scholar]
- 20.Du X, An Y, Yu L, et al. A genomic copy number variant analysis implicates the MBD5 and HNRNPU genes in Chinese children with infantile spasms and expands the clinical spectrum of 2q23.1 deletion. BMC Med Genet 2014;15:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaillard S, Dubourg C, Gerard-Blanluet M, et al. 2q23.1 microdeletion identified by array comparative genomic hybridisation: an emerging phenotype with Angelman-like features?. J Med Genet 2009;46:847–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data will be shared by request from any qualified investigator.