

Abstract
The COVID-19 pandemic has been disastrous to society and effective drugs are urgently needed. The papain-like protease domain (PLpro) of SARS-CoV-2 (SCoV2) is indispensable for viral replication and represents a putative target for pharmacological intervention. In this work, we describe the development of a potent and selective SCoV2 PLpro inhibitor, 19. The inhibitor not only effectively blocks substrate cleavage and immunosuppressive function imparted by PLpro, but also markedly mitigates SCoV2 replication in human cells, with a submicromolar IC50. We further present a convenient and sensitive activity probe, 7, and complementary assays to readily evaluate SCoV2 PLpro inhibitors in vitro or in cells. In addition, we disclose the co-crystal structure of SCoV2 PLpro in complex with a prototype inhibitor, which illuminates their detailed binding mode. Overall, these findings provide promising leads and important tools for drug discovery aiming to target SCoV2 PLpro.
Keywords: COVID-19, papain-like protease, small-molecule inhibitor, assay development, high-throughput screening, structure-based design, SARS-CoV-2, lead optimization
Graphical abstract
PLpro represents a putative druggable target for SCoV2. Utilizing multiple approaches, including fluorogenic activity probes, HTS, co-crystallization, structure-based design, and other optimized cellular assays, Shan et al. successfully develop a potent and selective inhibitor of SCoV2 PLpro.
Introduction
The pandemic of coronavirus disease 2019 (COVID-19) has been ravaging for over a year, causing mounting infections and millions of deaths, as well as incalculable devastation of the global economy (Morens and Fauci, 2020). To overcome COVID-19, researchers worldwide are racing to discover effective drugs. Up to now, several vaccines have been approved, and the US FDA has granted Remdesivir, an antiviral agent targeting RNA polymerase (RdRP), approval for treatment of COVID-19. However, the agents currently applied in the clinic will most likely face anticipated drug resistance arising from the evolving virus (Thomson et al., 2021). Thus, developing more therapeutic strategies and drug candidates for COVID-19 treatment is an urgent demand.
The pathogen of COVID-19 is a novel coronavirus called severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2/SCoV2), a close relative of SARS-CoV (SCoV), which emerged in 2002 (Zhou et al., 2020). Based on their high genetic homology and precedent studies on SCoV, certain nonstructural but indispensable enzymatic domains of SCoV2, such as RdRP, the main protease (Mpro/3CLpro), and the papain-like protease (PLpro), are considered druggable targets (Li and De Clercq, 2020). Indeed, RdRP inhibitors, e.g., Remdesivir, have already been approved, while Mpro inhibitors have advanced into clinical trials (NCT04535167; NCT04627532). On the other hand, potent and selective inhibitors of SCoV2 PLpro remain to be discovered.
SCoV2 PLpro shares 83% sequence identity as well as proteolytic activity with SCoV PLpro. It can process nonstructural proteins, in particular, nsps 1–3, that are essential for virus replication, and can cleave K48-linked ubiquitin (Ub) chains or interferon-stimulated gene 15 (ISG15) modifications on host cell proteins to counteract the immune response during viral infection (Figure 1 A) (Li and De Clercq, 2020; Ratia et al., 2006; Shin et al., 2020). Previously, selective inhibitors had been developed for SCoV PLpro, which could restrain SCoV replication at high concentrations (Baez-Santos et al., 2014; Ratia et al., 2008). Some of these reported inhibitors were tested against SCoV2 and exhibited only weak to moderate antiviral effects in cells (Freitas et al., 2020; Klemm et al., 2020; Shin et al., 2020). Of note, given the functional similarity between PLpro and human deubiquitinating enzymes (DUBs), competitive inhibitors of PLpro might potentially hit off-targets in infected cells. Indeed, two peptidomimetic covalent PLpro inhibitors reported recently exhibited in vitro reactivity toward both SCoV2 and a DUB (Rut et al., 2020).
Figure 1.

Development of peptidomimetic activity probes for SCoV2 PLpro
(A) A cartoon summarizing the enzymatic functions of SCoV2 PLpro. As a part of nsp3, PLpro is responsible for the cleavage of viral nsp1–3 proteins, and can cleave K48-linked Ub chains or ISGylation in host cells.
(B) A diagram showing the cleavage-site residue sequences of PLpro substrates and the formulas of peptidomimetic fluorogenic probes.
(C) The chemical structure of 7 and a cartoon depicting its fluorogenic mechanism in the presence of PLpro.
(D) Bar graph showing relative sensitivity of each probe to SCoV2 PLpro. Data are presented as the mean ± SD (n = 3).
(E) Optimization of the enzymatic activity assays by titration of SCoV2 PLpro and probes. Data are presented as the mean ± SD (n = 3).
(F) Comparison of the enzymatic activities between SCoV2 PLproWT and PLproC112S (1 μM) with 5 or 7 (2.5 μM) as substrate. Data are presented as the mean ± SD (n = 3).
(G) Michaelis-Menten curves for 5 and 7 with 1 μM SCoV2 PLpro. Data are presented as the mean ± SD (n = 3).
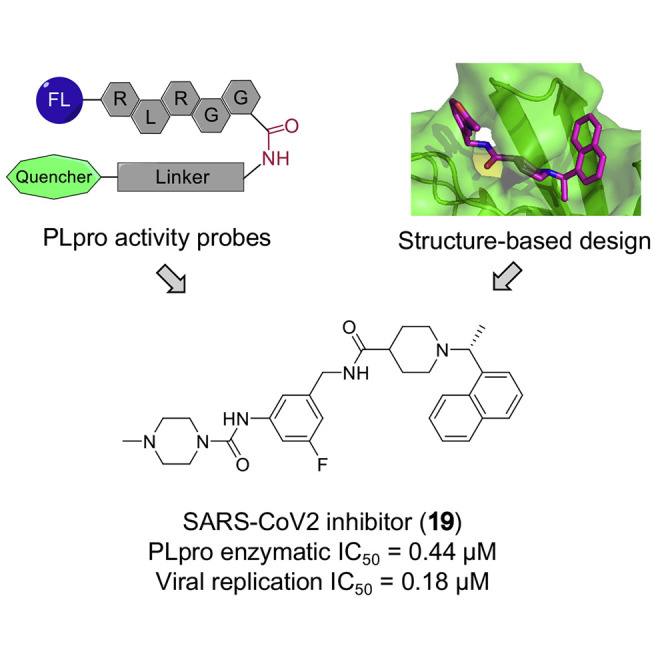
Herein, we present the discovery of a potent and selective SCoV2 PLpro inhibitor, i.e., 19, which efficiently suppressed viral replication with a submicromolar IC50. In addition, several optimized approaches are described to facilitate the identification and optimization of SCoV2 PLpro inhibitors, including drug design based on the co-crystal structure of SCoV2 PLpro in complex with an inhibitor; development of enzymatic activity assays that allow quick, large-scale, and affordable high-throughput screening (HTS); and implementation of a virus-free experimental system ready to assess cellular potency of lead compounds.
Results
Development of peptidomimetic activity probes for SCoV2 PLpro
As PLpro and DUBs share conserved target cleavage sites, we screened several reported DUB substrates to develop an enzymatic activity assay for SCoV2 PLpro (Figures 1B, 1C, and S1). The purified recombinant SCoV2 PLpro protein exhibited robust activity in cleaving K48-linked diUb, but weak or no activity against K63-linked or linear diUb, respectively (Figure S1). SCoV2 PLpro could readily cleave a Ub-rhodamine 110 conjugate (Ub-Rho/1) (Hassiepen et al., 2007) and release the fluorophore (Rho) from its C terminus. We demonstrated that it could likewise release the fluorogenic 7-amido-4-methylcoumarin (AMC) from the peptidomimetic probes 2 and 3, which harbored a conserved RLRGG sequence homologous to the C-terminal pentapeptide of Ub (Figures 1B and 1D) (Renatus et al., 2006).
To better mimic the isopeptide bond of SCoV2 PLpro substrates, we synthesized a hexapeptide probe (5) with a fluorogenic 7-hydroxy-3-carboxycoumarin (HCC) conjugated to its N terminus, and a 4-(dimethylaminoazo) benzene-4-carboxylic acid (DABCYL) to its C-terminal lysine (Figures 1B and S2). The maximal absorption of the HCC fluorophore in 5 is around 410 nm, and its emission peak is at 450 nm. In the absence of SCoV2 PLpro, the emission of HCC upon excitation will mostly be quenched by the proximate DABCYL subunit in 5 through a fluorescence resonance energy transfer (FRET) mechanism. In the presence of SCoV2 PLpro, 5 will be cleaved and the resultant fluorogenic pentapeptide (5′) will no longer be quenched (Figures 1C and S2). Compared with previous probes, 5 exhibited improved sensitivity to SCoV2 PLpro with lower background signal. We then replaced the N-terminal lysine of 5 with a methionine to mimic the linear diUb (6), or with an aliphatic chain (7), and observed that 6 was slightly less sensitive, whereas 7 was as sensitive as 5 (Figures 1B–1D).
To optimize the SCoV2 PLpro enzymatic activity assays with 5 and 7, we titrated each probe and confirmed that 5 or 7 at 2.5 μM was efficient enough (Figure 1E). Then we set probes at 2.5 μM and titrated SCoV2 PLpro, and confirmed 1 μM as an efficient concentration for the enzyme. Under the optimized conditions, both 5 and 7 exhibited excellent sensitivity to wild-type (WT) SCoV2 PLpro, but not to the catalytically inactive mutant, SCoV2 PLproC112S (Figure 1F). However, the Km value for 7 is 3-fold lower than that for 5 (Figure 1G), suggesting that 7 represents a better substrate for SCoV2 PLpro.
Screening for SCoV2 PLpro inhibitors
Having established the enzymatic activity assay with 7, we initially screened 25 reported inhibitors of DUB or DUB-like proteases for their inhibitory activity against SCoV2 PLpro at 10 μM (Figures 2A and 2B and Table S1). Most molecules were weak (≤30% inhibition) or inactive against SCoV2 PLpro, except for a UCHL3 inhibitor, TCID (Liu et al., 2003), which showed moderate inhibition (~50%). Among these compounds, the SCoV PLpro inhibitor GRL0617 (Ratia et al., 2008), recently reported to inhibit SCoV2 PLpro in vitro (Freitas et al., 2020; Shin et al., 2020), stood out, with over 80% inhibition of SCoV2 PLpro in our assays, as expected.
Figure 2.

Screening for SCoV2 PLpro inhibitors
(A) A bar graph showing inhibition of SCoV2 PLpro by reported DUB or DUB-like protease inhibitors (10 μM) in an enzymatic assay based on 7. Data are presented as the mean ± SD (n = 3).
(B) Chemical structures of exemplary compounds showing inhibition of SCoV2 PLpro in (A) or (C).
(C) HTS assay for SCoV2 PLpro inhibitor discovery based on probe 7. Screening data for a representative library of 9,071 small molecules are shown. Compounds with Z scores below −8 in both experiments were designated as hits for PLpro (red dots).
(D) A bar graph showing inhibition of SCoV2 PLpro or USP2 by selected hits (10 μM) in enzymatic assays based on 7. Data are presented as the mean ± SD (n = 3).
Using GRL0617 as a positive control, the SCoV2 PLpro enzymatic activity assay was further optimized to suit HTS, with a Z′-factor of 0.633 determined by 100 wells per control group (Zhang et al., 1999). Then we performed a primary screen on 35,360 diverse compounds, including lead-like fragments, FDA-approved drugs, and small molecules with reported biological activities. This HTS was repeated and resulted in a moderate hit rate of 0.037% with the Z-score threshold set at −8, representing a strong negative effect (Zhang, 2007). A total of 13 compounds were selected for further validation (Figures 2C and 2D and Table S1).
Of the 13 hits, 9 reproducibly inhibited SCoV2 PLpro, with at least 25% inhibitory activity, and 7 exhibited over 50% inhibition, including an essential nutrient, vitamin B12, and inhibitors reported for other targets, such as avasimibe and tiplaxtinin. An antibiotic commonly used in hair shampoo, zinc pyrithione, and an electrophilic α-chloroketone, compound 8, showed potency comparable to that of GRL0617. Given its high hydrolytic activity but low selectivity toward Ub chain linkages (Ritorto et al., 2014), the deubiquitinating protease USP2 was used in a counter-screen to assess the selectivity of all these hits, and the results indicated GRL0617 to be more selective than others (Figure 2D). GRL0617, which had already been optimized for SCoV PLpro, generally showed superior potency, selectivity, and molecular complexity than other hits. These findings are consistent with previously reported HTS results (Smith et al., 2020) and support that GRL0617 serves as a better lead for SCoV2 PLpro.
Determination of the co-crystal structure of the SCoV2 PLpro-12 complex
Encouraged by the potential of GRL0617, we synthesized and tested a series of reported SCoV PLpro inhibitors (11–13) that partially resemble GRL0617 with a shared naphthyl subunit (Baez-Santos et al., 2014). These inhibitors exhibited even better potency than GRL0617 in our assay based on 7, with IC50 values ranging between 2.6 and 4.3 μM (Figures 3A and S3). To study the pharmacophore of this series of inhibitors and to facilitate the following chemical optimization, we attempted to co-crystallize SCoV2 PLpro with these inhibitors (11–13). We first determined the high-resolution apo-form structure of the catalytically inactive mutant, SCoV2 PLproC112S (Figure S4A). More importantly, the co-crystal structure of the SCoV2 PLproC112S-12 complex was solved at 2.4 Å resolution as a dimer. Although the overall R free (0.326) and R work (0.272) values for the co-crystal are relatively high due to the anisotropy of diffraction data, particularly in the resolution range of 3.15–2.4 Å, the electron-density map of the binding site shows good quality, and 12 can be well traced in the map (Figures 3B and 3C and Table S2).
Figure 3.

Determination of the co-crystal structure of the SCoV2 PLpro-12 complex and optimization of inhibitors
(A) Structure-activity relationship of 11 and analogs. Each compound was titrated in enzymatic assays based on 7 or Ub-Rho. Data are presented as the mean ± SD (n = 3), assays were repeated at least three times with similar results.
(B) Surface structure of SCoV2 PLpro (chain A) in complex with 12 (purple sticks) (PDB: 7E35) showing that 12 resides in a channel in front of the catalytic site in the PLpro domain.
(C) Cartoon structures of the binding pockets of 12 (purple sticks) in two asymmetric SCoV2 PLpro units (green/azure ribbons for chains A/B). The key residues interacting with 12 are shown as sticks, and the 2mFo-DFc omit maps are shown as blue meshes.
(D) Superimposition and comparison of the binding sites of SCoV2 PLpro-12 (green/purple sticks, PDB: 7E35), SCoV2 PLpro-GRL0617 (gray/yellow sticks, PDB: 7JIR) (Osipiuk et al., 2021), and unbound SCoV2 PLpro (gray sticks, PDB: 7D6H).
Based on the co-crystal structure, 12 adopts a U-shape conformation to occupy a pocket between the S1 site and the catalytic site of SCoV2 PLpro, consequently blocking the N-terminal moiety of its substrates from accessing the S1 site (Figure 3B). Notably, the 2mFo-DFc electron-density maps indicated distinct binding modes for each unit of the asymmetrically dimerized complex (Figure 3C). Nevertheless, further analyses were based on the complex structure of chain A only, given its better quality of electron density compared with chain B. Upon closer examination, 12 interacts with the side chains of multiple surrounding residues, including Leu163, Pro248, Pro249, Tyr265, Tyr269, and Gln270, through van der Waals forces. In particular, the phenyl ring of Tyr269 simultaneously engages with all three hydrophobic rings of 12, acting as a latch of the binding pocket. In addition, the carbonyl of Tyr269 forms an H bond with the backbone amide of 12. Moreover, the electrostatic interaction between the negatively charged Asp165 and the positively charged tertiary amine of 12 further stabilizes the complex (Figures 3C and 3D).
Upon binding to SCoV2 PLpro, 12 pushes the side chain of Leu163 away from its binding pocket, but induces the side chain of Tyr269 to bend down steeply toward it (Figures 3D and S4A). Likewise, GRL0617 induces similar conformational changes in Leu163 and Tyr269, but to a lesser extent (PDB: 7IJR) (Osipiuk et al., 2021). In contrast, GRL0617 forms an H bond with the backbone nitrogen of Gln270, and induces significant conformational change in it (Figure 3D). Binding with Ub or ISG15 also induces the side chain of Tyr269 to bend down; however, it bends toward Pro248 rather than the binding pocket (Figure S4B) (Klemm et al., 2020; Shin et al., 2020). Collectively, 12 efficiently exploits the switchable Tyr269 to inhibit SCoV2 PLpro.
Optimization of SCoV2 PLpro inhibitors
Based on the co-crystal structure, we performed structure-activity relationship (SAR) studies to optimize the SCoV2 PLpro inhibitors. Given that derivatization of the naphthyl group is reportedly unfavorable (Baez-Santos et al., 2014), and as the piperidyl ring is tightly surrounded by Tyr269, Asp165, and other residues, we focused our efforts on the substitution of benzyl group (Figures 3 and S3). First, inspired by 11, we added a 5-fluorine to 12 to strengthen the interaction with Gln270, and the resultant 14 showed improved potency. Meanwhile, as the substituted benzyl group of 12 is exposed to the solution, we postulated that hydrophilic substituents might stabilize the complex. However, replacing the hydrophobic benzyl ring of 12 with a substituted piperidine ring (15) resulted in potency decrease. To introduce hydrophilic substitutions without disturbing the hydrophobic interaction between 12 and Gln270 and Leu163, we decided to modify the acetamide group, which seems to protrude toward the solvent in the co-crystal, rather than the whole benzyl ring. As expected, both 16 and 17, each introduced with a hydrophilic tertiary amine, showed improved potency compared with 12 and 15, while 17 showed a submicromolar IC50. Finally, we added 5-fluorine to 16 and 17 to generate 18 and 19 with further improved potency. All these analogs were also tested in a SCoV2 PLpro activity assay based on Ub-Rho, and their relative activities were overall consistent with those based on 7 (Figures 3A and S3).
Given the unsuccessful efforts to co-crystalize SCoV2 PLpro with 19 or other analogs, we performed molecular docking studies to elucidate the outstanding potency of 19 based on the co-crystal structure of SCoV2 PLpro-12 (Figures 4A–4C). The docking results predicted a binding mode for 19, which is partially similar to that of 12, but includes additional interactions for 19. Based on this mode, the substituted piperazine group and the 5-fluorine of 19 interact with the side chains of Tyr269 and Gln270, respectively, mainly through van der Waals forces. Compared with 12, which interacts with Tyr269 in three orthogonal directions, 19 almost encircles Tyr269 with interactions in four orthogonal directions, which provides a plausible explanation for its significantly improved potency (Figure 4C).
Figure 4.

Inhibitor 19 is a potent and selective SCoV2 PLpro inhibitor
(A) Chemical structures of 12 and 19.
(B and C) Inhibitor 19 was docked into the binding pocket of 12 in SCoV2 PLpro (chain A, PDB: 7E35). Superimposition of 19 (purple sticks) and 12 (azure sticks) indicated additional interactions between 19 and Tyr269-Gln 270 (green sticks in B) of SCoV2 PLpro (surface in C).
(D) SPR analysis of 19 binding to SCoV2 PLpro; GRL0617 was used as a control. Kd and SD were calculated based on three independent experiments.
(E) A bar graph showing the inhibition of 11 DUBs or DUB-like proteases by 19 (10 μM) (n = 3). Data are presented as the mean ± SD (n = 3).
To precisely characterize the affinity of 19 for SCoV2 PLpro, we performed surface plasma resonance (SPR) binding analysis of 19 in comparison with GRL0617. Based on the SPR results, 19 bound to SCoV2 PLpro with a K d of 2.6 μM, while that of GRL0617 was 10.8 μM (Figure 4D). To evaluate its selectivity, 19 was profiled against 10 DUBs or DUB-like proteases, including USP14 and OTUB1, which reportedly favor K48-linked Ub chains as substrates. Compared with SCoV2 PLpro, none of these 10 proteases were significantly inhibited by 19 at 10 μM (Figure 4E). Cumulatively, the results show that 19 is a potent and selective inhibitor of SCoV2 PLpro in vitro.
Inhibitor 19 and analogs effectively inhibited SCoV2 PLpro in live cells
To investigate if 19 and other analogs maintain their potency in live cells, we designed a cellular assay to assess the enzymatic activity of SCoV2 PLpro (Figure 5 A). We found that SCoV2 nsp1-nsp2 protein expressed in 293T cells is rather stable despite endogenous human DUBs, but can be readily cleaved into two parts by the co-expressed SCoV2 PLpro protein. However, such cleavage should be perturbed by SCoV2 PLpro inhibitors. And to our satisfaction, 17, 18, and 19 at 10 μM all significantly inhibited SCoV2 PLpro in 293T cells.
Figure 5.

Inhibitor 19 and analogs inhibited SCoV2 PLpro and viral replication in live cells
(A) Inhibitors 17, 18, and 19 inhibited the cleavage of recombinant nsp1-nsp2 by SCoV2 PLpro in a dose-dependent manner. 293T cells were pretreated with each inhibitor at the indicated concentrations and transfected with FLAG-tagged nsp1-nsp2 and SCoV2 PLpro for 24 h, followed by western blots.
(B) Inhibitors 17, 18, and 19 significantly recovered the activation level of NF-κB perturbed by SCoV2 PLpro. NF-κB-luc-reporting 293T cells were transfected with SCoV2 PLproWT or PLproC112S for 18 h in the presence of inhibitors (10 μM) and then stimulated by TNF-α for 6 h and measured for their NF-κB activity by the luciferase reporting system. Data are presented as the mean ± SD (n = 3, ∗∗∗p < 0.001).
(C) Inhibitors 17, 18, and 19 significantly recovered the level of ISGylation perturbed by SCoV2 PLpro. A549 cells stably expressing SCoV2 PLproWT or PLproC112S were preincubated with inhibitors for 6 h and then stimulated by IFN-α for 24 h and subjected to western blots.
(D) Inhibitors 15–17 at 5 μM abolished SCoV2 replication in hACE2-HeLa cells. hACE2-HeLa cells were infected with SCoV2 (0.3 MOI) for 1 h, then washed out with PBS, and cultured with normal medium and the indicated inhibitors (5 μM) for an additional 48 h, followed by western blots.
(E) Inhibitor 19 inhibited the replication of SCoV2 in human cells in a dose-dependent manner. hACE2-HeLa cells were cultured with SCoV2 for 1 h, then washed out with PBS, and cultured with normal medium and the indicated inhibitors for another 48 h, followed by immunostaining of spike protein (red) and nuclei (blue). Assays were repeated at least three times with similar results, and representative images are shown.
(F) Inhibition of SCoV2 replication curves based on the quantification of fluorescence, and the calculated IC50, CC50, and therapeutic indexes (CC50/IC50) for 17–19. Data are presented as the mean ± SD (n = 3).
As PLpro putatively impairs the immune response of host cells, to further evaluate our inhibitors for their cellular efficacy, we checked their modulatory effects on the immune signaling regulated by K48-linked Ub chains or ISGylation in live cells. Based on a luciferase-reporting assay, we confirmed that expression of SCoV2 PLproWT notably decreased NF-κB activation in 293T cells. In contrast, expression of catalytically inactive PLproC112S led to a much weaker effect, and 17, 18, or 19 at 10 μM could significantly recover the activation level of NF-κB (Figure 5B). We also confirmed that expression of SCoV2 PLpro significantly decreased the overall ISGylation induced by interferon α (IFN-α) in A549 cells, which could be efficiently restored by all three inhibitors (Figure 5C). These results further corroborated that 19 and analogs are effective SCoV2 PLpro inhibitors in human cells.
Inhibitor 19 and analogs effectively mitigate the replication of SCoV2 in cells
As GRL0617 and other reported inhibitors exhibited sufficient antiviral effects only at rather high concentrations, we sought to test our inhibitors in hACE2-HeLa cells (Zhou et al., 2020) for their antiviral potency. As the nucleocapsid protein (NP) is a reliable biomarker for indicating the replication rate of SCoV2 in cells, we checked the NP level in infected hACE2-HeLa cells treated with each inhibitor. Based on western blot analysis, the reported inhibitors at 5 μM only moderately or weakly decreased the NP level, with 11 as the strongest. In contrast, our optimized analogs exhibited obviously improved potency, and 15, 16, or 17 almost completely diminished the expression of NP in infected cells (Figure 5D).
Immunostaining of the spike protein demonstrated that SCoV2 viruses replicated effectively in hACE2-HeLa cells, which consequently induced severe cell death (Figure 5E). Consistently, inhibitors 15–19 alleviated both replication of SCoV2 and the death of hACE2-HeLa cells in a dose-dependent manner, while 19 could significantly inhibit SCoV2 replication even at 400 nM (Figures 5E and S5B). On the other hand, at 10 μM, neither 17 nor 19 showed detectable cytotoxicity in hACE2-HeLa cells, whereas 18 did reduce the cell viability to nearly 60% compared with the control (Figure S5A). Based on the quantification of immunofluorescence, the IC50 values for 17–19 all fell within the nanomolar range, yet 19 remained the best, with a IC50 of 182 nM and a therapeutic index (CC50/IC50) over 55 (Figure 5F). Together, these results show that 19 can effectively and selectively inhibit the replication of SCoV2 in human cells.
Discussion
HTS could be an efficient approach to providing promising pharmacophores for the development of SCoV2 PLpro inhibitors. However, its success rate depends not only on the capacity and diversity of screened chemical libraries, but also on the robustness, convenience, and cost efficiency of the HTS assays. Among the reported fluorogenic activity probes for DUBs and SCoV PLpro, Ub-Rho is very sensitive, but too expensive for HTS. AMC-based peptidomimetic probes like 2 or 3 are moderately sensitive and much more cost effective. However, they often interfere with the screened fluorescent compounds with overlapping spectrums. In contrast, our FRET-based fluorogenic probe 7 exhibited excellent sensitivity with minimal background for SCoV2 PLpro and showed high reliability and low cost in HTS.
With probe 7 we screened reported inhibitors of DUBs or other proteases, performed a pilot HTS, and successfully identified several hits for SCoV2 PLpro. Some of these hits demonstrated encouraging chemical properties for further development. For example, avasimibe and tiplaxtinin could be repurposed and optimized for SCoV2 PLpro, whereas zinc pyrithione and 8 might potentially serve as covalent fragments in the design of covalent inhibitors of SCoV2 PLpro. Of particular importance, a series of SCoV PLpro inhibitors, including GRL0617 and 11–13, were identified as the best leads for SCoV2 PLpro in the current study. Thus, we determined the co-crystal structure of the 12-SCoV2 PLpro complex, which complemented well the recently reported structures of SCoV2 PLpro complexes (Fu et al., 2021; Gao et al., 2021; Osipiuk et al., 2021) and greatly facilitated our SAR studies. An optimized analog, 19, proved to be a potent and selective inhibitor of SCoV2 PLpro in vitro.
We have utilized several cellular assays in combination to evaluate our inhibitors in live cells. These assays are based on the cleavage of nsp proteins or IGSylation by SCoV2 PLpro, or based on its viral suppression of the host immune response. Indeed, 19 and other analogs exhibited robust inhibition of SCoV2 PLpro in these assays. Furthermore, we demonstrate that 19 is a remarkably more effective antiviral agent, compared with the reported analogs, and can mitigate the replication of SCoV2 in human cells with submicromolar IC50 values. Based on the preliminary evaluation, 19 exhibited moderate metabolic stability in mouse liver microsomes or plasma with half-lives of 28.0 or over 120 min, respectively (Figure S5C), and is warranted to be further assessed with animal models in the future.
In summary, we report the development of 19 as a potent and selective inhibitor of SCoV2 PLpro and demonstrate a repertoire of complementary approaches to facilitate drug discovery for SCoV2. Our sensitive and affordable activity probe, 7, could considerably expand the capacity of HTS and accelerate the identification of new hits; the virus-free cellular assays utilized in this work are feasible and can be very helpful in the early stage of development with limited resources, such as P3 laboratories; the detailed binding mode of 12-SCoV2 PLpro, revealed by a co-crystal structure, will certainly assist inhibitor design and optimization; and last, 19 has provided an optimized pharmacophore and a promising lead for further developments. Collectively, these findings represent a small but solid milestone in the arduous and prolonged battle against COVID-19.
Limitations of the study
Despite the favorable potency and selectivity of 19 in cultured cells, further evaluation of the efficacy and tolerability of 19 in vivo is necessary to establish its translational relevance to COVID-19. On the other hand, probe 7 may be incapable of screening for inhibitors that target other allosteric sites of SCoV2 PLpro. Thus, it should be considered as a complement to, rather than a substitute for, Ub-based probes. In addition, our efforts to co-crystallize SCoV2 PLpro bound to 19 were unsuccessful, and other advanced technologies, e.g., cryogenic electron microscopy, could help solve this obstacle and facilitate further optimization of 19.
Significance
COVID-19 has struck our society as a great calamity, and the need for effective drugs is more urgent than ever. PLpro is one of the most promising druggable targets of SCoV2; however, current small-molecule inhibitors of SCoV2 PLpro exhibit only weak to moderate inhibition of SCoV2 replication, precluding their further clinical translation. This study reports the development of potent and selective inhibitors of SCoV2 PLpro that effectively inhibit replication of SCoV2 and the death of infected cells, with a submicromolar IC 50 . We also describe a repertoire of complementary approaches to aid in drug development, including a fluorogenic probe with better sensitivity than reported peptidomimetic probes, which is more affordable than Ub-based probes and compatible with high-throughput screening to identify SCoV2 PLpro inhibitors. Several virus-free cellular assays are optimized to assess the enzymatic activity of SCoV2 PLpro, which can be very useful to evaluate the potency of candidate inhibitors in live cells outside of P3 labs. Structural studies reveal the detailed binding mode of the pharmacophore to be optimized, and our lead compounds effectively inhibit replication of SCoV2 and the death of infected cells with submicromolar IC 50 .
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal ANTI-FLAG® M2-Peroxidase (HRP) antibody produced in mouse | Sigma-Aldrich | Cat# A8592; RRID: AB_439702 |
| β-Tubulin Mouse MAb | Transgen | Cat# HC101 |
| ISG15 Recombinant Rabbit MAb | Invitrogen | Cat# 703131; RRID: AB_2784562 |
| SARS-CoV / SARS-CoV-2 Spike Rabbit PAb | SinoBiological | Cat# 40150-T62 |
| β-Actin Mouse MAb | Transgen | Cat# HC201 |
| SARS-CoV-2 nucleocapsid protein Rabbit MAb | SinoBiological | Cat# 40143-R019 |
| Bacterial and virus strains | ||
| E.coli BL21 (DE3) | Lifeng Pan’s laboratory, Shanghai Institute of Organic Chemistry | BL21 (DE3) |
| SARS-CoV2 (SZ02) | Shenzhen Third People’s Hospital | SZ02 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Human Ubiquitin Rhodamine 110 Protein, CF | BostonBiochem | Cat# U-555 |
| Probe 2-7 | Li Tan’s laboratory | N/A |
| SARS-CoV2-PLproWT | This paper | N/A |
| SARS-CoV2-PLproC112S | This paper | N/A |
| Recombinant Human FLAG-USP36 Protein, CF | BostonBiochem | Cat# E-628-025 |
| Recombinant Human His6 USP14 | Li Tan’s laboratory | N/A |
| Recombination Human His6 USP8 | BostonBiochem | Cat# E-520 |
| Recombination Human His6 USP7 | BostonBiochem | Cat# E-519 |
| Recombinant Human His6 USP2 | Li Tan’s laboratory | N/A |
| Recombination Human UCH-L1/PGP9.5 | BostonBiochem | Cat# E-340 |
| Recombination Human His6 SUMO-Specific Peptidase 1/SENP1 Catalytic Domain | BostonBiochem | Cat# E-700 |
| Recombinant Human His6 OTUB1 | Li Tan’s laboratory | N/A |
| Recombination Human Ataxin-3 Like | BostonBiochem | Cat# E-343 |
| Recombination Human AMSH/STAMBP protein | Novus Biologicals | Cat# NBP2-22825 |
| Recombinant Human TNFα | Novoprotein | Cat# C008 |
| Recombinant human Interferon alpha | Sigma-Aldrich | Cat# SRP4595 |
| K48-linked diUb | This paper | N/A |
| K63-linked diUb | This paper | N/A |
| M1-linked diUb | This paper | N/A |
| Aloxistatin | Selleck (Zang et al., 2020) | CAS 88321-09-9 |
| b-AP15 | Selleck (Ward et al., 2020) | CAS 1009817-63-3 |
| BAY11-7082 | MCE (Mori et al., 2002) | CAS 19542-67-7 |
| DUB-IN-1 | MCE (Colombo et al., 2010) | CAS 924296-18-4 |
| DUB-IN-2 | MCE (Colombo et al., 2010) | CAS 924296-19-5 |
| E-64 | Selleck (Wadhawan et al., 2014) | CAS 66701-25-5 |
| GRL0617 | MCE (Ratia et al., 2008) | CAS 1093070-16-6 |
| GW7647 | MCE (Luo et al., 2017) | CAS 265129-71-3 |
| HBX41108 | MCE | CAS 924296-39-9 |
| HY-13487 | MCE (Weinstock et al., 2012) | CAS 1247825-37-1 |
| HY-17541A | MCE | NA |
| IU1 | Selleck (Lee et al., 2010) | CAS 314245-33-5 |
| LDN57444 | MCE (Liu et al., 2003) | CAS 668467-91-2 |
| LY-3000328 | MCE (Jadhav et al., 2014) | CAS 1373215-15-6 |
| Mitoxantrone | Selleck (Takeuchi et al., 1992) | CAS 70476-82-3 |
| ML323 | MCE (Liang et al., 2014) | CAS 1572414-83-5 |
| NSC632839 | MCE (Nicholson et al., 2008) | CAS 157654-67-6 |
| Odanacatib | Selleck (Gauthier et al., 2008) | CAS 603139-19-1 |
| P22077 | MCE (Altun et al., 2011) | CAS 1247819-59-5 |
| Pimozide | MCE (Ybema et al., 1994) | CAS 2062-78-4 |
| PYR-41 | Selleck (Yang et al., 2007) | CAS 418805-02-4 |
| SJB3-019A | MCE (Mistry et al., 2013) | NA |
| Spautin-1 | Selleck (Shao et al., 2014) | CAS 1262888-28-7 |
| TCID | Selleck (Liu et al., 2003) | CAS 30675-13-9 |
| WP1130 | Selleck (Bartholomeusz et al., 2007) | CAS 856243-80-6 |
| 4E1Rcat | Selleck (Cencic et al., 2011) | CAS 328998-25-0 |
| Avasimibe | Selleck (Tardif et al., 2004) | CAS 166518-60-1 |
| CPI-169 | Selleck (Vidya Balasubramanian, 2014) | CAS 1450655-76-1 |
| GW4064 | Selleck (Akwabi-Ameyaw et al., 2008) | CAS 278779-30-9 |
| RG-7388 | MCE (Ding et al., 2013) | CAS 1229705-06-9 |
| Tiplaxtinin | Selleck (Hennan et al., 2008) | CAS 393105-53-8 |
| UMI-77 | Selleck (Abulwerdi et al., 2014) | CAS 518303-20-3 |
| Vitamin B12 | Selleck (Banerjee, 2003) | CAS 68-19-9 |
| Zafirlukast | Selleck (Finnerty et al., 1992) | CAS 107753-78-6 |
| Zinc Pyrithiene | Selleck (Ermolayeva adn Sanders, 1995) | CAS 13463-41-7 |
| 8 | ChemDiv | www.chemdiv.com |
| 9 | ChemDiv | www.chemdiv.com |
| 10 | ChemDiv | www.chemdiv.com |
| Critical commercial assays | ||
| SPR analysis | GE Healthcare | Cat# BR-1005-30 |
| NF-κB activity luciferase-reporting system. | Transgene | Cat# FR201-02 |
| CellTiter-Glo luminescent cell viability assays | Promega | Cat# G7570 |
| Microsomal stability assay | Corning® Gentest™ | Cat# 452701 |
| Deposited data | ||
| SCoV2 Plpro apo structure | This paper | PDB ID 7D6H |
| SCoV2 PLpro/12 structure | This paper | PDB ID 7E35 |
| SCoV2 PLpro/GRL0617 structure | Osipiuk et al., 2021 | PDB ID 7JIR |
| SCoV2 PLpro/Ub structure | Klemm et al., 2020 | PDB ID 6XAA |
| SCoV2 PLpro/ISG15 structure | Shin et al., 2020 | PDB ID 6YVA |
| Experimental models: cell lines | ||
| Human: 293T | Li Tan’s laboratory | N/A |
| Human: SCoV2 PLproWT A549 | This paper | N/A |
| Human: SCoV2 PLproC112S A549 | This paper | N/A |
| Human: hACE2-HeLa | Vitalstar | N/A |
| Recombinant DNA | ||
| pET32a-6xHis-3C-PLproWT | This paper | N/A |
| pET32a-6xHis-3C-PlproC112S | This paper | N/A |
| pET32a-6xHis-Ub2 | This paper | N/A |
| pCDNA3.1-NSP1-NSP2-FLAG | This paper | N/A |
| pCDNA3.1-3xFLAG- PLproWT | This paper | N/A |
| pCDNA3.1-3xFLAG- PlproC112S | This paper | N/A |
| Lenti-3xFLAG- PLproWT | This paper | N/A |
| Lenti-3xFLAG- PlproC112S | This paper | N/A |
| Software and algorithms | ||
| GraphPad Prism | Graphpad Software Inc | https://www.graphpad.com/scientific-software/prism/ |
| Schrödinger Glide software | Friesner et al., 2006 | https://www.schrodinger.com/products/glide |
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/ |
| XDS program | Kabsch, 2010 | https://xds.mr.mpg.de |
| autoPROC suite | Vonrhein et al., 2011 | https://www.globalphasing.com/autoproc |
| Phaser | McCoy et al., 2007 | https://www.phaser.cimr.cam.ac.uk/index.php/ Phaser_Crystallographic_Software |
| Coot | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Phenix | Liebschner et al., 2019 | https://www.phenix-online.org |
| MolProbity | Williams et al., 2018 | https//molprobity.manchester.ac.uk |
| PyMol | Schrödinger, Inc | http://www.pymol.org |
Resourse availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contact Li Tan (tanli@sioc.ac.cn )
Materiala availability
Plasmid, Probe 2-7 and all compounds can be requested through contacting the Lead Contact.
Data and code availability
The accession numbers for the crystal structures reported in this paper are PDB: 7D6H and PDB: 7E35.
Experimental model and subject details
Cell lines and cell culture
HEK293T / SCoV2 PLproWT A549 / SCoV2 PLproC112S A549
HEK293T (human embryonic kidney cells; hypotriploid; female ; fetal) were obtained from Junhao Hu’s laboratory and were not further authenticated. A549 expressing WT SCoV2 PLpro or C112S mutant SCoV2 PLpro were constructed in this paper. hACE2-Hela cells were purchased from Vitalstar. Cells were grown in Dulbecco’s Modified Eagle’s medium (DMEM, Gibco) supplemented with 10% (v/v) fetal bovine serum (FBS, Gibco), 50 units/mL penicillin, 50 units/mL streptomycin. Cells were incubated in 5% CO2 at 37 oC. Cell lines were passaged every 72 hours with trypsin and not allowed to reach confluency.
293T cells / A549 lung adenocarcinoma cells
293T cells (female) and A549 lung adenocarcinoma cells (male) were obtained from ATCC. hACE2-HeLa cells (female) were obtained from Vitalstar. All the cells were cultured in Dulbecco Modified Eagle medium (DMEM) with 10% FBS and 100 U/mL penicillin-streptomycin at 37°C/5% CO2 in a humidified environment.
Method details
Protein expression and purification
DNA fragment that encodes full-length PLpro of SCoV2 was synthesized and cloned into a modified pET32a vector which fuses a 6✕His tag and an HRV 3C protease cutting sequence to the N-terminal of PLpro. C112S point mutation was introduced by the standard PCR-based mutagenesis method and verified by Sanger sequencing.
PLpro wild-type and C112S mutant proteins were expressed in E. coli BL21(DE3). Specifically, when the OD600 of bacteria culture reached about 0.8, zinc chloride was supplemented to the culture with a final concentration of 0.1 mM and protein expression was induced with 0.1 mM IPTG at 16°C for 18 hours. The bacterial cells were pelleted by centrifugation and resuspended in binding buffer (50 mM Tris-HCl at pH 7.9, 500 mM NaCl and 5 mM imidazole) and disrupted with a high-pressure homogenizer FB-110S (Shanghai Litu Machinery Equipment Engineering Co., Ltd.). Supernatant of the lysate after centrifugation at 16,000g for 30 min was subjected to His-tag affinity purification with Ni2+-NTA agarose (GE Healthcare). The N-terminal 6✕His tag was cut by homemade HRV 3C protease and removed by size-exclusion chromatography on an HiLoad Superdex 26/60 column (GE healthcare) equilibrated in a buffer containing 20 mM Tris-HCl at pH 7.5, 100 mM NaCl and 1 mM DTT.
The wild-type ubiquitin and its mutants (K48R, K63R or Ub-D77 which contains a glutamate following the last G76 residue of Ub) were expressed as 6✕His-tagged recombinant proteins in E. coli BL21(DE3) and purified in a similar manner as SCoV2 PLpro.
Preparation of Di-Ubiquitin proteins
K48- and K63-linked di-ubiquitin were synthesized according to published protocols (Hofmann and Pickart, 2001; Piotrowski et al., 1997). For M1-linked di-ubiquitin preparation, two tandem repeats of coding sequence for human ubiquitin were cloned into a modified pET32a vector which fuses a 6✕His tag and an HRV 3C protease cutting sequence to the N-terminal of insert. The M1-linked di-ubiquitin was expressed and purified in a similar way as mono-ubiquitin.
Protein crystallization and structural determination
Crystallization of apo-form SCov-2 PLproC112S was set up in hanging-drop manner at 16 oC by mixing fresh protein (10 or 15 mg/mL in buffer containing 20 mM Tris-HCl at pH 7.5, 100 mM NaCl and 1 mM DTT) with equal volume of reservoir solution (0.1 M acetate buffer pH 4.5, 0.8 M NaH2PO4/1.2M K2HPO4) and incubated at 16°C.
For the crystal growth of PLproC112S/compound complex, PLproC112S protein (10 mg/mL) was incubated with 3 mM compound 12 at 4°C overnight and then used to set up crystallization in sitting-drops by mixing protein/compound with equal volume of reservoir solution which contains 0.1 M sodium citrate tribasic dihydrate at pH 5.5 and 16% (v/v) PEG 8000.
After about 10 days, both apo-form and complex crystals appeared and were harvested in reservoir solution supplemented with 25% (v/v) glycerol as cryo-protectant and flash frozen in liquid nitrogen. Diffraction datasets were collected at the beamline BL17U1 of the Shanghai Synchrotron Radiation Facility (SSRF). The diffraction data sets were processed with XDS program and the autoPROC suite (Kabsch, 2010; Tickle et al., 2018; Vonrhein et al., 2011).
The initial phase for the structure of both apo-form and complex form were solved by molecular replacement using the PDB entry 6W9C as the search model with program Phaser (McCoy et al., 2007). The structural model was further manually re-built in Coot and refined with Phenix (Emsley et al., 2010; Liebschner et al., 2019). The qualities of final models were validated by MolProbity and listed in Table S2 (Williams et al., 2018). All the structural figures were prepared in program PyMol.
Molecular docking study
The docking study of 19 was performed based on coordinates of SCoV2 PLpro-12 (Chain A, PDB 7E35) and Schrödinger Glide software (Friesner et al., 2006). Meanwhile, 12 as a control was docked back with same parameters. GlideScores simulate binding free energies, more negative values represent tighter binders, and scores of -10 or lower usually represent very good binding. In this study, the GlideScore for the top-ranking predicted binding mode of 19 was -11.814, in comparison, that for for 12 was -10.851.
Absorption and emission spectra
Absorption and emission spectra were recorded by a Enspire Multimode plate reader (Perkin Elmer). Solutions of probes (20 μM, 20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 5 mM 2-Hydroxy-1-ethanethiol) were added to the wells. The absorption was recorded between 300-600 nm, and then the Emission was recorded between 420-650 nm at an excitation of 405 nm.
Kinetic assays
Kinetic assay was performed in assay buffer (20 mM Tris-HCl, 100 mM NaCl, 5 mM 2-hydroxy-1-ethanethiol, pH 7.8) with 5 mM 2-hydroxy-1-ethanethiol as the reducing agent to eliminate cysteine-reactive compounds. 15 min was selected as the initial condition to compare these probes with different λex and λem (probe 1: λex = 485 nm; λem = 535nm; probe 2: λex =345 nm; λem = 445nm; probe 3: λex =330 nm; λem = 500nm; probe 5/6/7: λex = 405 nm; λem = 450nm). PLproC112S was used to determine whether signal generation was dependent on enzyme activity. Enzyme concentration (PLpro 1 μM) and incubation time with substrate were optimized to yield a linear response in a 60-mintime frame (probe 5, 2.5μM), and a 30-mintime frame (probe 7, 2.5 μM) for HTS Screening. The enzyme activity was monitored using the Enspire Multimode Plate Reader (Perkin Elmer).
KM value determination
The assay was performed in a 40 μL reaction volume using a black flat-bottom 384-well plates (Greiner Bio-One, #781076). We used the substrate after cleaving the quencher to make the standard curve of substrate concentration and signal value. Initial velocity of enzymatic reaction was plotted over time within the linear range in various concentrations of enzyme and substrates. According to the Michaelis equation, the initial velocity and substrate concentration were plotted to get the K M value:
Where is initial velocity of enzymatic reaction, is the reaction rate when the enzyme is saturated with the substrate, is substrate concentration, and is Michaelis constant.
HTS screening
A compound library consisting of 35,360 structurally diverse small molecules was purchased from Chemdiv and maintained as 5 mg/ml stock solutions dissolved in DMSO and stored at -20°C. The automated liquid dispensing of primary screen was performed on Echo 550 (Labcyte) and Viafill (Integra). Fluorescence values were measured on Enspire Multimode Plate Reader (PerkinElmer). All assays were performed in duplicate at room temperature, using black flat-bottom 384-well plates (Greiner Bio-One, #784076) containing a final reaction volume of 20 μL. The assays were assembled as follows: 20 nL compounds (final concentration 5 μg/ml) was dispensed into wells and then incubated with 10 μL of 2 μM PLpro (final concentration 1 μM) in Buffer A (20 mM Tris-HCl, 100 mM NaCl, 5 mM 2-hydroxy-1-ethanethiol, pH 7.8) for 30 min. Reactions were initiated with 10 μL of 5 μM probe 5 (final concentration 2.5 μM) in Buffer A, shaken vigorously for 10 sec, and then incubated for 60 min, before measuring fluorescence emission intensity (λex = 405 nm; λem = 450 nm). Each 384-well plate contained 11 positive control wells (20 nL of DMSO replacing 20 nL of inhibitor in DMSO), 12 negative control wells (assay components lacking PLpro) and 1 positive inhibitor control well (20 nL of 10 mM GRL0617). Compounds that showed Z-scores of less than -8 were cherry picked for further analysis.
IC50 value determination
IC50 values for all inhibitors were determined in 384-well format. The final enzyme concentrations were 1 μM and 0.5 μM for probe 7 and Ub-Rho, respectively, and the final substrate concentrations were 2.5 μM and 0.5 μM in a 30 min time frame and a 5 min time frame. The assays were performed at room temperature, and the enzyme activity was monitored as described above.
SPR analysis
SPR experiment was performed by Biacore T200 (GE Healthcare) at 20°C. PBS-P buffer was filtered through a 0.22 μm membrane filter and degassed prior to use. First, PLpro protein was immobilized on the CM5 chip surface. PLpro protein was diluted to 50 μg/mL using sodium acetate solution at pH 5.5. The diluted protein flowed at 10 μL/min and was soon covalently immobilized to a flow cell of CM5 sensor chip via primary amine group, using standard Amine Coupling Kit. The final level of immobilized protein was 13537 RU. A flow cell without coupling proteins was used as a blank reference. Small molecules were diluted in PBS-P buffer in a series of concentrations (1.25-125 μM for GRL0617, 0.24-125 μM for 19) and flowed over the immobilized PLpro or blank cell at 30 μL/min with 90 s contact time and 120 s dissociation time. The equilibrium dissociation constant (K d) was used to evaluate the binding activity of each compound. Considering the influence of non-specific binding, we used non-specific binding function to fit the curve in GraphPad Prism. Then subtract the non-specific contribution from the original data, and we got the actual specific binding response. K d was then calculated this response (R) and the corresponding concentration ([S]) with the formula below.
Inhibitor specificity assay
To verify the specificity of inhibitor, Ub-Rho was used as substrate of PLpro and the release of Rhodamine 110 was measured by increase of fluorescence intensity (Ex./Em. 485/535 nm) on 384-well microplate reader (Enspire, PerkinElmer). Compound 19 (10 μM) was incubated with 10 μL different DUBs (suitable concentration) for 1 h at RT respectively and reaction was initiated by addition of 10 μL Ub-Rho (1 μM) to the well. The reaction was maintained at 37°C until the appropriate fluorescence intensity value was reached. The experiment was repeated three times.
Nsp1-Nsp2 cleavage assay
4 x 105 HEK293T cells per well were seeded onto 12-well plates a day before pretreated with compounds for 1 h, and then transiently transfected with 500 ng nsp1-nsp2-flag and 250 ng w/o Flag-SCoV2-PLpro plasmid. 24h after transfection, cell lysates were collected and analyzed for nsp1-nsp2-flag cleavage by immunoblot analysis.
NF-κB activity luciferase-reporting assay
1x105 HEK293T cells per well were seeded onto 96-well plates and transiently transfected with 0.02 μg p65 reporter construct bearing firefly luciferase, 0.01 μg construct bearing renilla luciferase downstream of CMV promoter and w/o Flag-SCoV2 PLpro plasmid. 18 h after seeding, HEK293T cells were stimulated with TNF-α (200 ng/mL) for 6 h and then firefly and renilla luciferase activity was determined with TransDetect double-luciferase reporter assay kit (Transgene, FR201-02) according to the manufacturers’ instruction.
DeISGylation assay
A549 cells stably expressing of PLproWT or PLproC112S mutant were pretreated with compounds (10 μM) for 6 h, and treated with 1.25 nM IFN-α for 24 h to induce ISGylation. Cells were lysed with lysis buffer (100 mM Tris-HCl (pH 8.0), 1% (v/v) SDS), boiled at 95°C for 10 min and protein concentration was measured with BCA assay (Thermo Fischer). 10 μg cell lysates were loaded onto 13.5% Tris-Glycin PAGE gel, transferred to nitrocellulose using a semi-dry transfer apparatus (0.2 A for 90 mins), and blocked for 30 mins at RT. The membrane was incubated with anti-ISG15 (1:1000 in 3% BSA, Invitrogen #703131) at 4°C overnight and incubated with secondary antibody (1:5000) for 1 h at RT.
SARS-CoV2 infection and evaluation of antiviral activities
A clinically isolated SARS-CoV2 (SZ02) was propagated in Vero-E6 cells, viral titer was determined by 50% tissue culture infective dose (TCID50). All the infection experiments were performed in a biosafety level-3 (BLS-3) laboratory. To evaluate the antiviral efficacy of these inhibitors, hACE2-Hela cells were cultured in 24-well plate (1 x 105 cells/well) or in 96-well plate (1 x 104 cells/well) overnight, and then infected with 0.3 MOI virus for 2 h. The supernatant was removed and the cells were washed 3 times with PBS, then cultured with fresh medium plus inhibitors at indicated doses. At 48 h post infection, cells were collected with 2X SDS sample buffer for immunoblotting with antibody against SCoV2 NP (Sino Biological 40143-R019), or fixed with 4% PFA and immunostained with anti-Spike antibody (Sinobiologica, 40150-T62). The nuclei were stained with DAPI (Beyotime, China). The images were recorded with fluorescence microscopy.
To evaluate the cytotoxicity of the inhibitors, 2 x 103 cells/well hACE2-HeLa cells cultured in 384-well plate were treated with each inhibitor at indicated concentrations for 72 h, then subjected to viability assays (CellTiter-Glo, Promega) followed with measuring and quantification.
Metabolic stability studies
Inhibitor 19 or Verapamil (control) and mouse liver microsomes (MLM) were primarily incubated in a thermostat water bath maintained at 37°C for 5 min (250 μL, containing 1.5 μM compound and 750 ng/mL MLM in PBS). The primary incubations were divided into 7 tubes with 30 uL which was added into 15 μL NADPH (3 mM) or 15 μL PBS, and incubated in a thermostat water bath maintained at 37°C. At various time points, e.g., 0, 5, 10, 20, 30, 60 min (all above with 15 μL NADPH), 60 min (with 15 μL PBS), incubations were quenched with 90 μL ice-cold acetonitrile containing 100 ng/mL IS. These mixtures were centrifuged at 10,000 rpm for 10 min, the liquid supernatants were transferred to clean tubes. After diluted with water (1:1), the incubations were analyzed by LC-MS.
Inhibitor 19 (0.25 mM in PBS) and mouse plasma were incubated in a thermostat water bath maintained at 37°C (200 μL, containing 8.35 μM compound). At various time points, e.g., 0, 15, 30, 60, 30, 120 min, 30 μL incubations were quenched with 120 μL ice-cold acetonitrile containing 100 ng/mL IS. These mixtures were centrifuged at 10,000 rpm for 10 min, the liquid supernatants were transferred to clean tubes. After diluted with water (1:1), the incubations were analyzed by LC-MS.
Chemical synthesis
General synthetic procedures
Unless otherwise noted, reagents and solvents were obtained from commercial suppliers and were used without further purification. Reactions were run in roundbottom flasks or glass vials and stirred with Teflon-coated magnetic stir bars. Solvent evaporation was performed on rotary evaporator (Büchi) under reduced pressure. Reactions were monitored by thin layer chromatography or LC-MS (Waters). Acquity H-Class UPLC/MS with QDa mass spectrometer using water + 0.05% formic acid (solvent A) and acetonitrile + 0.05% formic acid (solvent B). Preparative HPLC was performed on a Waters Sunfire C18 column (19 x 100 mm, 5μM) using a gradient of 10-95% acetonitrile in water containing 0.05% trifluoroacetic acid (TFA) over 10 min (15 min run time) at a flow rate of 15 mL/min. 1H/13C NMR spectra were recorded at ambient temperature on Bruker DMX 400 (400 MHz for 1H NMR and 101 MHz for 13C NMR) instruments in the specified deuterated solvents. Observed proton absorptions were reported as δ units of parts per million (ppm) relative to tetramethylsilane (δ 0.0). Multiplicities were reported: s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets) and m (multiplet). Coupling constants were reported as a J value in Hertz (Hz).
Abbreviations used in this section: THF = tetrahydrofuran; DCM = dichloromethane; DMF = N,N-dimethylformamide; DMSO = dimethyl sulfoxide; DCE = 1,2-dichloroethane; DIPEA = diisopropylethylamine; EA = ethyl acetate; TFA = trifluoroacetic acid; HCl = hydrochloric acid; RT = room temperature.
Reaction schemes

Reagents and conditions: (a) Ms2O, DIEA, DCM, 40 min, 0°C; (b) ethyl isonipecotate, DCM, RT, 16 h; (c) 1 M NaOH, MeOH, RT, 16 h; (d) EDCI, HOBt, DIEA, DCM, RT, 16 h.

Reagents and conditions: (a) triphosgene, DCM, RT; (b) methylpiperazine, RT, 16 h; (c) dimethylglycine, HATU, DIEA, DCM, RT, 10 h; (d) Acetic anhydride, THF, RT, 2 h; (f) H2, Raney Ni, MeOH (NH3), RT, 16 h.
Ethyl (R)-1-(1-(naphthalen-1-yl) ethyl) piperidine-4-carboxylate (21)
(S)-1-(naphthalen-1-yl) ethan-1-ol (500 mg, 2.9 mmol) was dissolved in dry DCM (14.5 mL) at 0°C. With continuous stirring at 0°C, DIEA (2 mL, 11.6 mmol) was added, and Ms2O (758 mg, 4.35 mmol) was added dropwise. The solution was stirred for 40 min at 0°C, at which point ethyl piperidine-4-carboxylate (0.32 mL, 2.03 mmol) was added. The mixture was then stirred for 16 h at room temperature. The reaction mixture was quenched with water and extracted with CH2Cl2, washed with 10% aqueous Na2CO3. The organic layer was then dried (Na2SO4), filtered, and removed in vacuo. The residue was purified by silica gel column chromatography to furnish compound 21 in 54% yield (slightly yellow oil). 1H NMR (400 MHz, DMSO-d6) δ 8.46 – 8.40 (m, 1H), 7.93 – 7.88 (m, 1H), 7.79 (dd, J = 8.1, 1.3 Hz, 1H), 7.54 – 7.44 (m, 4H), 4.15 (q, J = 6.7 Hz, 1H), 4.06 – 4.01 (m, 2H), 2.96 (d, J = 11.3 Hz, 1H), 2.76 – 2.68 (m, 1H), 2.25 (ddt, J = 11.2, 8.0, 4.0 Hz, 1H), 2.08 (tdd, J = 11.2, 5.5, 2.8 Hz, 2H), 1.84 – 1.77 (m, 1H), 1.74 – 1.67 (m, 1H), 1.55 – 1.45 (m, 2H), 1.39 (d, J = 6.7 Hz, 3H), 1.15 (t, J = 7.1 Hz, 3H).
(R)-1-(1-(naphthalen-1-yl) ethyl) piperidine-4-carboxylic acid (22)
21 (300 mg, 0.96 mmol) was dissolved in methanol (15 mL), followed by the addition of 1 N sodium hydroxide (2 mL, 2 mmol) in one portion. The resulting mixture was stirred for 16 h at room temperature, then concentrated in vacuo, and dissolved in small amount of water. The solution was treated with AcOH dropwise until the solution was acidic by pH paper. The residue was purified by reversed phase column chromatography to furnish compound 22 in 98% yield (white solid). 1H NMR (400 MHz, DMSO-d6) δ 8.38 (d, J = 8.5 Hz, 1H), 8.07 – 8.03 (m, 2H), 7.88 (dt, J = 7.4, 2.1 Hz, 1H), 7.73 – 7.57 (m, 4H), 5.46 – 5.38 (m, 1H), 3.90 (d, J = 12.2 Hz, 2H), 3.10 (s, 1H), 2.90 (q, J = 11.2, 10.7 Hz, 1H), 2.44 (s, 1H), 2.13 (d, J = 14.2 Hz, 1H), 1.94 (d, J = 14.1 Hz, 1H), 1.83 (d, J = 13.0 Hz, 1H), 1.75 (d, J = 6.7 Hz, 3H), 1.64 (d, J = 11.7 Hz, 1H).
N-(3-(aminomethyl)-5-fluorophenyl) acetamide (14c)
3-amino-5-fluorobenzonitrile (500 mg, 4 mmol) was dissolved in THF (3 mL), followed by the addition of acetic anhydride (0.58 mL, 6 mmol) in one portion. The resulting mixture was stirred for 2 h at room temperature, then concentrated in vacuo. The residue was purified by silica gel column chromatography to furnish compound 14a in 92% yield (white solid). 1H NMR (400 MHz, DMSO-d6) δ 10.46 (s, 1H), 7.80 – 7.76 (m, 2H), 7.50 (ddd, J = 8.4, 2.4, 1.4 Hz, 1H), 2.09 (s, 3H).
To a solution of 14a (300 mg, 1.68 mmol) in saturated ammonia in MeOH, Raney-Ni (30 mg, 10%) was added, and the mixture was stirred under H2 atmosphere at room temperature for 16 h. The reaction was filtered through a celite pad and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography to furnish compound 14c in 98% yield (slightly yellow oil).
N-(3-(aminomethyl) phenyl)-2-(dimethylamino) acetamide (16c)
To a solution of dimethylglycine (600 mg, 5.82 mmol), EDCI (1.12 g, 5.82 mmol), and HOBt (0.79 g, 5.82 mmol) in dry DCM (20 mL) were added a solution of DIEA (4.2 mL, 24.2 mmol) and 3-aminobenzonitrile (573 mg, .4.85 mmol) in dry CH2Cl2 (20 ml) at 0°C under argon atmosphere, and it was allowed to stir for 16 h at room temperature. The reaction mixture was quenched with water and extracted with CH2Cl2. The organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to furnish compound 16a in 36% yield (slightly yellow oil). 1H NMR (400 MHz, Chloroform-d) δ 9.31 (s, 1H), 8.03 – 7.97 (m, 1H), 7.81 (ddd, J = 8.0, 2.2, 1.4 Hz, 1H), 7.48 – 7.41 (m, 1H), 7.38 (dt, J = 7.7, 1.4 Hz, 1H), 3.10 (s, 2H), 2.39 (s, 6H).
To a solution of 16a (200 mg, 0.98 mmol) in saturated ammonia in MeOH, Raney-Ni (20 mg, 10%) was added, and the mixture was stirred under H2 atmosphere at room temperature for 16 h. The reaction was filtered through a celite pad and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography to furnish compound 16c in 98% yield (slightly yellow oil).
N-(3-(aminomethyl) phenyl)-4-methylpiperazine-1-carboxamide (17c)
A solution of 3-isocyanatobenzonitrile (100 mg, 0.69 mmol) and 1-methylpiperazine (76 mg, 0.76 mmol) in diethylether (1.7 ml) prepared at room temperature, and was stirred at the same temperature. The reaction was concentrated under reduced pressure. The residue was purified by silica gel column chromatography to furnish compound 17b in 83% yield (colorless oil). 1H NMR (400 MHz, DMSO-d6) δ 8.84 (s, 1H), 7.93 (t, J = 1.9 Hz, 1H), 7.74 (ddd, J = 8.4, 2.3, 1.2 Hz, 1H), 7.44 (t, J = 7.9 Hz, 1H), 7.37 (dt, J = 7.6, 1.4 Hz, 1H), 3.45 (t, J = 5.0 Hz, 4H), 2.32 (t, J = 5.0 Hz, 4H), 2.20 (s, 3H).
To a solution of 17b (140 mg, 0.57 mmol) in saturated ammonia in MeOH, Raney-Ni (14 mg, 10%) was added, and the mixture was stirred under H2 atmosphere at room temperature for 16 h. The reaction was filtered through a celite pad and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography to furnish compound 17c in 98% yield (slightly yellow oil).
N-(3-(aminomethyl)-5-fluorophenyl)-2-(dimethylamino) acetamide (18c)
To a solution of dimethylglycine (500 mg, 3.67 mmol), and HATU (1.67 g, 4.4 mmol) in dry DCM (20 mL) were added a solution of DIEA (3.2 mL, 18.3 mmol) and 3-amino-5-fluorobenzonitrile (454 mg, 4.4 mmol) in dry CH2Cl2 (17 ml) at 0°C under argon atmosphere, and it was allowed to stir for 16 h at room temperature. The reaction mixture was quenched with water and extracted with CH2Cl2. The organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to furnish compound 18a in 37% yield (white solid).
To a solution of 18a (110 mg, 0.5 mmol) in saturated ammonia in MeOH, Raney-Ni (11 mg, 10%) was added, and the mixture was stirred under H2 atmosphere at room temperature for 16 h. The reaction was filtered through a celite pad and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography to furnish compound 18c in 98% yield (slightly yellow oil).
N-(3-(aminomethyl)-5-fluorophenyl)-4-methylpiperazine-1-carboxamide (19c)
3-amino-5-fluorobenzonitrile (500 mg, 3.67 mmol) was dissolved in CH2Cl2 (10 ml), and added dropwise in portions to a solution of triphosgene (546 mg, 1.84 mmol) in CH2Cl2 (20 ml). The reaction was stirred at room temperature for 6 h, and was concentrated under reduced pressure to get the compound 19a.
A solution of 19a (3.67 mmol) and 1-methylpiperazine (407 mg, 3.67 mmol) in CH2Cl2 (44 ml) prepared at room temperature, and was stirred at the same temperature for 16 h. The reaction was concentrated under reduced pressure. The residue was purified by silica gel column chromatography to furnish compound 19c in 52% yield (slightly yellow solid).
To a solution of 19c (200 mg, 0.76 mmol) in saturated ammonia in MeOH, Raney-Ni (20 mg, 10%) was added, and the mixture was stirred under H2 atmosphere at room temperature for 16 h. The reaction was filtered through a celite pad and the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography to furnish compound 19c in 98% yield (slightly yellow oil).
General Amide Coupling Method. To a solution of (R)-1-(1(naphthalen-1-yl)ethyl) piperidine-4-carboxylic acid (0.05 g, 0.18 mmol), EDCI (0.04 g, 0.21 mmol), and HOBt (0.03 g, 0.21 mmol) in dry DCM (1 mL) were added a solution of DIEA (0.154 mL, 0.88 mmol) and amine (0.21 mmol) in dry CH2Cl2 at 0°C under argon atmosphere, and it was allowed to stir for 16 h at room temperature. The reaction mixture was quenched with water and extracted with CH2Cl2. The organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to furnish compound.
(R)-N-(3-fluorobenzyl)-1-(1-(naphthalen-1-yl)ethyl)piperidine-4-carboxamide (11)
The title compound was obtained as described in the general procedure in 53% yield (white solid). 1H NMR (400 MHz, DMSO-d6) δ 8.46 (d, J = 8.2 Hz, 1H), 8.30 (t, J = 6.0 Hz, 1H), 7.91 (dd, J = 7.2, 2.4 Hz, 1H), 7.80 (d, J = 8.1 Hz, 1H), 7.55 – 7.43 (m, 4H), 7.33 (td, J = 7.9, 6.1 Hz, 1H), 7.07 – 6.96 (m, 3H), 4.24 (d, J = 6.0 Hz, 2H), 4.15 (q, J = 6.5 Hz, 1H), 3.07 (d, J = 11.2 Hz, 1H), 2.79 (d, J = 11.4 Hz, 1H), 2.18 – 2.12 (m, 1H), 2.05 – 1.97 (m, 2H), 1.72 (d, J = 12.7 Hz, 1H), 1.63 – 1.52 (m, 3H), 1.40 (d, J = 6.6 Hz, 3H).
(R)-N-(3-acetamidobenzyl)-1-(1-(naphthalen-1-yl) ethyl) piperidine-4-carboxamide (12)
The title compound was obtained as described in the general procedure in 52% yield (white solid). 1H NMR (400 MHz, DMSO-d6) δ 9.87 (s, 1H), 8.46 (d, J = 8.1 Hz, 1H), 8.24 (t, J = 5.9 Hz, 1H), 7.94 – 7.88 (m, 1H), 7.80 (d, J = 8.1 Hz, 1H), 7.56 – 7.40 (m, 6H), 7.19 (t, J = 7.8 Hz, 1H), 6.86 (d, J = 7.6 Hz, 1H), 4.19 (d, J = 5.9 Hz, 2H), 4.14 (d, J = 6.7 Hz, 1H), 3.07 (d, J = 10.9 Hz, 1H), 2.79 (d, J = 11.3 Hz, 1H), 2.13 (d, J = 6.6 Hz, 1H), 2.01 (s, 5H), 1.73 (d, J = 12.7 Hz, 1H), 1.57 (d, J = 12.2 Hz, 3H), 1.40 (d, J = 6.6 Hz, 3H).
(R)-N-((2-methoxypyridin-4-yl) methyl)-1-(1-(naphthalen-1-yl) ethyl) piperidine-4-carboxamide (13)
The title compound was obtained as described in the general procedure in 66% yield (white solid). 1H NMR (400 MHz, DMSO-d6) δ 8.46 (d, J = 8.1 Hz, 1H), 8.32 (t, J = 6.0 Hz, 1H), 8.05 (d, J = 5.2 Hz, 1H), 7.91 (dd, J = 7.9, 1.8 Hz, 1H), 7.80 (d, J = 8.1 Hz, 1H), 7.56 – 7.44 (m, 4H), 6.80 (dd, J = 5.3, 1.4 Hz, 1H), 6.59 – 6.56 (m, 1H), 4.20 (d, J = 6.0 Hz, 2H), 4.15 (q, J = 6.6 Hz, 1H), 3.81 (s, 3H), 3.07 (d, J = 11.2 Hz, 1H), 2.79 (d, J = 11.4 Hz, 1H), 2.17 (ddd, J = 11.6, 7.5, 4.1 Hz, 1H), 2.02 (tdd, J = 8.5, 5.6, 2.9 Hz, 2H), 1.73 (d, J = 12.9 Hz, 1H), 1.63 – 1.50 (m, 3H), 1.40 (d, J = 6.6 Hz, 3H).
(R)-N-(3-acetamido-5-fluorobenzyl)-1-(1-(naphthalen-1-yl) ethyl) piperidine-4-carboxamide (14)
The title compound was obtained as described in the general procedure in 41% yield (white solid). 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H), 8.46 (d, J = 8.1 Hz, 1H), 8.30 (d, J = 6.0 Hz, 1H), 7.91 (d, J = 7.2 Hz, 1H), 7.80 (d, J = 8.1 Hz, 1H), 7.50 (m, 5H), 7.11 (s, 1H), 6.68 (d, J = 9.4 Hz, 1H), 4.19 (d, J = 5.9 Hz, 2H), 4.15 (d, J = 6.5 Hz, 1H), 3.08 (d, J = 10.9 Hz, 1H), 2.80 (d, J = 11.2 Hz, 1H), 2.13 (d, J = 9.2 Hz, 1H), 2.02 (d, J = 1.1 Hz, 5H), 1.74 (d, J = 13.0 Hz, 1H), 1.58 (m, 3H), 1.40 (d, J = 6.5 Hz, 3H).
(R)-N-((1-methylpiperidin-4-yl) methyl)-1-(1-(naphthalen-1-yl) ethyl) piperidine-4-carboxamide (15)
The title compound was obtained as described in the general procedure in 58% yield (white solid). 1H NMR (400 MHz, DMSO-d6) δ 8.45 (d, J = 8.0 Hz, 1H), 7.91 (dd, J = 7.2, 2.3 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.68 (t, J = 5.7 Hz, 1H), 7.55 – 7.43 (m, 4H), 4.13 (q, J = 6.7 Hz, 1H), 3.05 (d, J = 11.1 Hz, 1H), 2.89 (t, J = 6.3 Hz, 2H), 2.73 (d, J = 12.5 Hz, 3H), 2.14 (s, 2H), 2.09 (s, 3H), 2.02 – 1.94 (m, 2H), 1.81 (t, J = 11.6 Hz, 2H), 1.65 (d, J = 12.4 Hz, 1H), 1.55 (s, 1H), 1.54 – 1.50 (m, 3H), 1.39 (d, J = 6.6 Hz, 3H), 1.33 – 1.26 (m, 1H), 1.13 – 1.03 (m, 2H).
(R)-N-(3-(2-(dimethylamino) acetamido) benzyl)-1-(1-(naphthalen-1-yl) ethyl) piperidine-4-carboxamide (16)
The title compound was obtained as described in the general procedure in 54% yield (white solid). 1H NMR (400 MHz, DMSO-d6) δ 9.70 (s, 1H), 8.46 (d, J = 8.1 Hz, 1H), 8.25 (t, J = 6.0 Hz, 1H), 7.94 – 7.89 (m, 1H), 7.80 (d, J = 8.1 Hz, 1H), 7.56 – 7.45 (m, 6H), 7.21 (t, J = 7.8 Hz, 1H), 6.89 (dt, J = 7.7, 1.4 Hz, 1H), 4.19 (d, J = 5.9 Hz, 2H), 4.15 (s, 1H), 3.09 (s, 3H), 2.79 (d, J = 11.1 Hz, 1H), 2.28 (s, 6H), 2.15 (s, 1H), 2.01 (s, 2H), 1.73 (d, J = 12.5 Hz, 1H), 1.58 (d, J = 13.0 Hz, 3H), 1.41 (d, J = 6.6 Hz, 3H).
(R)-4-methyl-N-(3-((1-(1-(naphthalen-1-yl) ethyl) piperidine-4-carboxamido) methyl) phenyl) piperazine-1-carboxamide (17)
The title compound was obtained as described in the general procedure in 43% yield (white solid). 1H NMR (400 MHz, DMSO-d6) δ 8.46 (d, J = 6.0 Hz, 2H), 8.20 (t, J = 6.0 Hz, 1H), 7.91 (dd, J = 7.9, 1.7 Hz, 1H), 7.80 (d, J = 8.1 Hz, 1H), 7.56 – 7.44 (m, 4H), 7.29 (dt, J = 7.1, 1.8 Hz, 2H), 7.16 – 7.10 (m, 1H), 6.77 (dt, J = 7.5, 1.4 Hz, 1H), 4.17 (d, J = 5.9 Hz, 2H), 4.14 (d, J = 6.5 Hz, 1H), 3.40 (d, J = 10.1 Hz, 3H), 3.07 (d, J = 10.9 Hz, 1H), 2.78 (d, J = 11.2 Hz, 1H), 2.28 (t, J = 5.0 Hz, 4H), 2.18 (s, 3H), 2.09 (s, 3H), 1.99 (dd, J = 10.6, 6.7 Hz, 2H), 1.72 (d, J = 12.8 Hz, 1H), 1.57 (d, J = 11.3 Hz, 2H), 1.40 (d, J = 6.6 Hz, 3H).
(R)-N-(3-(2-(dimethylamino) acetamido)-5-fluorobenzyl)-1-(1-(naphthalen-1-yl) ethyl) piperidine-4-carboxamide (18)
The title compound was obtained as described in the general procedure in 55% yield (white solid). 1H NMR (400 MHz, DMSO-d6) δ 9.88 (s, 1H), 8.46 (d, J = 8.0 Hz, 1H), 8.28 (t, J = 6.0 Hz, 1H), 7.91 (dd, J = 7.9, 1.7 Hz, 1H), 7.80 (d, J = 8.1 Hz, 1H), 7.56 – 7.44 (m, 5H), 7.30 (d, J = 1.7 Hz, 1H), 6.69 (dd, J = 9.5, 2.2 Hz, 1H), 4.19 (d, J = 6.0 Hz, 2H), 4.15 (d, J = 6.7 Hz, 1H), 3.05 (s, 3H), 2.79 (d, J = 11.3 Hz, 1H), 2.24 (s, 6H), 2.18 – 2.12 (m, 1H), 2.06 – 1.98 (m, 2H), 1.73 (d, J = 12.6 Hz, 1H), 1.63 – 1.52 (m, 3H), 1.40 (d, J = 6.6 Hz, 3H).
(R)-N-(3-fluoro-5-((1-(1-(naphthalen-1-yl) ethyl) piperidine-4-carboxamido) methyl) phenyl)-4-methylpiperazine-1-carboxamide (19)
The title compound was obtained as described in the general procedure in 32% yield (white solid). 1H NMR (400 MHz, DMSO-d6) δ 8.68 (s, 1H), 8.48 – 8.44 (m, 1H), 8.25 (t, J = 6.0 Hz, 1H), 7.93 – 7.89 (m, 1H), 7.80 (d, J = 8.1 Hz, 1H), 7.55 – 7.45 (m, 4H), 7.28 (dt, J = 11.9, 2.3 Hz, 1H), 7.11 (t, J = 1.7 Hz, 1H), 6.56 (ddd, J = 9.5, 2.5, 1.4 Hz, 1H), 4.16 (d, J = 6.0 Hz, 2H), 4.14 (s, 1H), 3.41 (t, J = 5.0 Hz, 4H), 3.07 (d, J = 11.2 Hz, 1H), 2.79 (d, J = 11.1 Hz, 1H), 2.29 (t, J = 5.0 Hz, 4H), 2.19 (s, 3H), 2.15 (d, J = 5.9 Hz, 1H), 2.01 (s, 2H), 1.73 (d, J = 12.9 Hz, 1H), 1.55 (m, 3H), 1.40 (d, J = 6.6 Hz, 3H).
Quantification and statistical analysis
All statistical analyses and curves were produced using GraphPad Prism 8. All the enzymatic activity assay were performed in triplicate and quantified data were presented as mean ± SD.
For microscopy experiments, fluorescence intensities were quantified with ImageJ software. Data was presented as mean ± SD of three experimental replicates.
IC50 values were calculated by means of non-linear regression using Graphpad Prism 8 and were shown as data points representing as mean ± SD.
Acknowledgments
We thank Dr. Sara J. Buhrlage (Harvard) for her generous help with the reported DUB and DUB-like protease inhibitors, Dr. Sheng-Ce Tao (Shanghai Jiao Tong University) for the cDNA of SCoV2 PLpro, Dr. Zairong Zhang (IRCBC, CAS) for DUB plasmids, Dr. Deju Ye (Nanjing University) for help with the development of fluorogenic probes, and Dr. Zhaoyin Wang (IRCBC, CAS) for help with the metabolic stability studies. This work was supported by grants from the National Natural Science Foundation of China (21778066 to L.T., 31801163 to Y.L., 21822705 to J.L. and L.P., 32071297 to J.L., and 21621002 to L.P.), Shanghai Municipal Science and Technology Commission (18ZR1448400 to Y.L., 20XD1425200 to L.P., 18JC1420500 and Major Project 2019SHZDZX02 to L.T.), and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB39030500 to L.T. and XDB20000000 to L.P.).
Author contributions
Conceptualization, L.T., L.P., and Y.L.; methodology, S.H., J.L., J.S., G.X., and Y.L.; software, L.P. and Y.L.; formal analysis, H.S., J.L., J.S., Y.T., H.X., Y.L., and L.T.; investigation, H.S., J.L., J.S., J.D., G.X., K.L., C.H., Y.W., X.X., Y.T., and Z.A.; resources, L.P. and Z.Z.; writing – original draft, L.T., Y.L., and G.Z.; supervision, L.T., L.P., Y.L., Z.Z., and J.H.; funding acquisition, L.T., L.P., and Y.L.
Declaration of interests
The authors declare no competing interest.
Published: May 12, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.chembiol.2021.04.020.
Supplemental information
References
- Abulwerdi F., Liao C., Liu M., Azmi A.S., Aboukameel A., Mady A.S., Gulappa T., Cierpicki T., Owens S., Zhang T., et al. A novel small-molecule inhibitor of mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer Ther. 2014;13:565–575. doi: 10.1158/1535-7163.MCT-12-0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akwabi-Ameyaw A., Baas J.Y., Caldwell R.D., Caravella J.A., Chen L., Creech K.L., Deaton D.N., Jones S.A., Kaldor I., Liu Y., Madauss K.P., et al. Conformationally constrained farnesoid X receptor (FXR) agonists: Naphthoic acid-based analogs of GW 4064. Bioorg. Med. Chem. Lett. 2008;18:4339–4343. doi: 10.1016/j.bmcl.2008.06.073. [DOI] [PubMed] [Google Scholar]
- Altun M., Kramer H.B., Willems L.I., McDermott J.L., Leach C.A., Goldenberg S.J., Kumar K.G., Konietzny R., Fischer R., Kogan E., et al. Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chem. Biol. 2011;18:1401–1412. doi: 10.1016/j.chembiol.2011.08.018. [DOI] [PubMed] [Google Scholar]
- Baez-Santos Y.M., Barraza S.J., Wilson M.W., Agius M.P., Mielech A.M., Davis N.M., Baker S.C., Larsen S.D., Mesecar A.D. X-ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain-like proteases. J. Med. Chem. 2014;57:2393–2412. doi: 10.1021/jm401712t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R., Ragsdale S.W. The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Annu. Rev. Biochem. 2003;72:209–247. doi: 10.1146/annurev.biochem.72.121801.161828. [DOI] [PubMed] [Google Scholar]
- Bartholomeusz G.A., Talpaz M., Kapuria V., Kong L.Y., Wang S., Estrov Z., Priebe W., Wu J., Donato N.J. Activation of a novel Bcr/Abl destruction pathway by WP1130 induces apoptosis of chronic myelogenous leukemia cells. Blood. 2007;109:3470–3478. doi: 10.1182/blood-2006-02-005579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cencic R., Hall D.R., Robert F., Du Y., Min J., Li L., Qui M., Lewis I., Kurtkaya S., Dingledine R., et al. Reversing chemoresistance by small molecule inhibition of the translation initiation complex eIF4F. Proc. Natl. Acad. Sci. U S A. 2011;108:1046–1051. doi: 10.1073/pnas.1011477108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo M., Vallese S., Peretto I., Jacq X., Rain J.C., Colland F., Guedat P. Synthesis and biological evaluation of 9-oxo-9H-indeno[1,2-b]pyrazine-2,3-dicarbonitrile analogues as potential inhibitors of deubiquitinating enzymes. ChemMedChem. 2010;5:522–528. doi: 10.1002/cmdc.200900409. [DOI] [PubMed] [Google Scholar]
- Ding Q., Zhang Z., Liu J.J., Jiang N., Zhang J., Ross T.M., Chu X.J., Bartkovitz D., Podlaski F., Janson C., et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. J. Med. Chem. 2013;56:5979–5983. doi: 10.1021/jm400487c. [DOI] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermolayeva E., Sanders D. Mechanism of pyrithione-induced membrane depolarization in Neurospora crassa. Appl. Environ. Microbiol. 1995;61:3385–3390. doi: 10.1128/aem.61.9.3385-3390.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnerty J.P., Wood-Baker R., Thomson H., Holgate S.T. Role of leukotrienes in exercise-induced asthma. Ihibitory effect of ICI 204219, a potent leukotriene D4 receptor antagonist. Am. Rev. Respir. Dis. 1992;145:746–749. doi: 10.1164/ajrccm/145.4_Pt_1.746. [DOI] [PubMed] [Google Scholar]
- Freitas B.T., Durie I.A., Murray J., Longo J.E., Miller H.C., Crich D., Hogan R.J., Tripp R.A., Pegan S.D. Characterization and noncovalent inhibition of the deubiquitinase and deISGylase activity of SARS-CoV-2 papain-like protease. ACS Infect Dis. 2020;6:2099–2109. doi: 10.1021/acsinfecdis.0c00168. [DOI] [PubMed] [Google Scholar]
- Friesner R.A., Murphy R.B., Repasky M.P., Frye L.L., Greenwood J.R., Halgren T.A., Sanschagrin P.C., Mainz D.T. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- Fu Z., Huang B., Tang J., Liu S., Liu M., Ye Y., Liu Z., Xiong Y., Zhu W., Cao D., et al. The complex structure of GRL0617 and SARS-CoV-2 PLpro reveals a hot spot for antiviral drug discovery. Nat. Commun. 2021;12:488. doi: 10.1038/s41467-020-20718-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X., Qin B., Chen P., Zhu K., Hou P., Wojdyla J.A., Wang M., Cui S. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B. 2021;11:237–245. doi: 10.1016/j.apsb.2020.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier J.Y., Chauret N., Cromlish W., Desmarais S., Duong L.T., Falgueyret J.P., Kimmel D.B., Lamontagne S., Léger S., LeRiche T., et al. The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg. Med. Chem. Lett. 2008;18:923–928. doi: 10.1016/j.bmcl.2007.12.047. [DOI] [PubMed] [Google Scholar]
- Hassiepen U., Eidhoff U., Meder G., Bulber J.F., Hein A., Bodendorf U., Lorthiois E., Martoglio B. A sensitive fluorescence intensity assay for deubiquitinating proteases using ubiquitin-rhodamine110-glycine as substrate. Anal Biochem. 2007;371:201–207. doi: 10.1016/j.ab.2007.07.034. [DOI] [PubMed] [Google Scholar]
- Hennan J.K., Morgan G.A., Swillo R.E., Antrilli T.M., Mugford C., Vlasuk G.P., Gardell S.J., Crandall D.L. Effect of tiplaxtinin (PAI-039), an orally bioavailable PAI-1 antagonist, in a rat model of thrombosis. J. Thromb. Haemost. 2008;6:1558–1564. doi: 10.1111/j.1538-7836.2008.03063.x. [DOI] [PubMed] [Google Scholar]
- Hofmann R.M., Pickart C.M. In vitro assembly and recognition of Lys-63 polyubiquitin chains. J. Biol. Chem. 2001;276:27936–27943. doi: 10.1074/jbc.M103378200. [DOI] [PubMed] [Google Scholar]
- Jadhav P.K., S M, Gavardinas K., Kim E.J., Matthews D.P., Staszak M.A., Coffey D.S., Shaw B.W., Cassidy K.C., Brier R.A., et al. Discovery of cathepsin S inhibitor LY3000328 for the treatment of abdominal aortic aneurysm. ACS Med. Chem. Lett. 2014;5:1138–1142. doi: 10.1021/ml500283g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm T., Ebert G., Calleja D.J., Allison C.C., Richardson L.W., Bernardini J.P., Lu B.G., Kuchel N.W., Grohmann C., Shibata Y., et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020;39:e106275. doi: 10.15252/embj.2020106275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B.H., Lee M.J., Park S., Oh D.C., Elsasser S., Chen P.C., Gartner C., Dimova N., Hanna J., Gygi S.P., et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467:179–184. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., De Clercq E. Therapeutic options for the 2019 novel coronavirus (2019-nCoV) Nat. Rev. Drug Discov. 2020;19:149–150. doi: 10.1038/d41573-020-00016-0. [DOI] [PubMed] [Google Scholar]
- Liang Q., Dexheimer T.S., Zhang P., Rosenthal A.S., Villamil M.A., You C., Zhang Q., Chen J., Ott C.A., Sun H., et al. A selective USP1-UAF1 inhibitor links deubiquitination to DNA damage responses. Nat. Chem. Biol. 2014;10:298–304. doi: 10.1038/nchembio.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D., Afonine P.V., Baker M.L., Bunkoczi G., Chen V.B., Croll T.I., Hintze B., Hung L.W., Jain S., McCoy A.J., et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 2019;75:861–877. doi: 10.1107/S2059798319011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Lashuel L., Choi S., Xing X., Case A., Ni J., Yeh L.A., Cuny G.D., Stein R.L., Lansbury P.T., Jr. Discovery of inhibitors that elucidate the role of UCH-L1 activity in the H1299 lung cancer cell line. Chem. Biol. 2003;10:837–846. doi: 10.1016/j.chembiol.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Luo M., Y S, Liu Y., Chodisetti G., Riederer B., Menon M.B., Tachibana K., Doi T., Seidler U.E. IL-1β-Induced downregulation of the multifunctional PDZ adaptor PDZK1 is attenuated by ERK inhibition, RXRα, or PPARα stimulation in enterocytes. Front Physiol. 2017;8:61. doi: 10.3389/fphys.2017.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry H., Hsieh G., Buhrlage S.J., Huang M., Park E., Cuny G.D., Galinsky I., Stone R.M., Gray N.S., D'Andrea A.D., Parmar K. Small-molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol. Cancer Ther. 2013;12:2651–2662. doi: 10.1158/1535-7163.MCT-13-0103-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morens D.M., Fauci A.S. Emerging pandemic diseases: how we got to COVID-19. Cell. 2020;183:837. doi: 10.1016/j.cell.2020.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori N., Yamada Y., Ikeda S., Yamasaki Y., Tsukasaki K., Tanaka Y., Tomonaga M., Yamamoto N., Fujii M. Bay 11-7082 inhibits transcription factor NF-kappaB and induces apoptosis of HTLV-I-infected T-cell lines andprimary adult T-cell leukemia cells. Blood. 2002;100:1828–1834. doi: 10.1182/blood-2002-01-0151. [DOI] [PubMed] [Google Scholar]
- Nicholson B., Leach C.A., Goldenberg S.J., Francis D.M., Kodrasov M.P., Tian X., Shanks J., Sterner D.E., Bernal A., Mattern M.R., et al. Characterization of ubiquitin and ubiquitin-like-protein isopeptidase activities. Protein Sci. 2008;17:1035–1043. doi: 10.1110/ps.083450408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipiuk J., Azizi S.A., Dvorkin S., Endres M., Jedrzejczak R., Jones K.A., Kang S., Kathayat R.S., Kim Y., Lisnyak V.G., et al. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021;12:743. doi: 10.1038/s41467-021-21060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotrowski J., Beal R., Hoffman L., Wilkinson K.D., Cohen R.E., Pickart C.M. Inhibition of the 26 S proteasome by polyubiquitin chains synthesized to have defined lengths. J. Biol. Chem. 1997;272:23712–23721. doi: 10.1074/jbc.272.38.23712. [DOI] [PubMed] [Google Scholar]
- Ratia K., Pegan S., Takayama J., Sleeman K., Coughlin M., Baliji S., Chaudhuri R., Fu W., Prabhakar B.S., Johnson M.E., et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. U S A. 2008;105:16119–16124. doi: 10.1073/pnas.0805240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratia K., Saikatendu K.S., Santarsiero B.D., Barretto N., Baker S.C., Stevens R.C., Mesecar A.D. Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. U S A. 2006;103:5717–5722. doi: 10.1073/pnas.0510851103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renatus M., Parrado S.G., D'Arcy A., Eidhoff U., Gerhartz B., Hassiepen U., Pierrat B., Riedl R., Vinzenz D., Worpenberg S., et al. Structural basis of ubiquitin recognition by the deubiquitinating protease USP2. Structure. 2006;14:1293–1302. doi: 10.1016/j.str.2006.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritorto M.S., Ewan R., Perez-Oliva A.B., Knebel A., Buhrlage S.J., Wightman M., Kelly S.M., Wood N.T., Virdee S., Gray N.S., et al. Screening of DUB activity and specificity by MALDI-TOF mass spectrometry. Nat. Commun. 2014;5:4763. doi: 10.1038/ncomms5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rut W., Lv Z., Zmudzinski M., Patchett S., Nayak D., Snipas S.J., El Oualid F., Huang T.T., Bekes M., Drag M., et al. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: a framework for anti-COVID-19 drug design. Sci. Adv. 2020;6:eabd4596. doi: 10.1126/sciadv.abd4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S., Li S., Qin Y., Wang X., Yang Y., Bai H., Zhou L., Zhao C., Wang C. Spautin-1, a novel autophagy inhibitor, enhances imatinib-induced apoptosis in chronic myeloid leukemia. Int. J. Oncol. 2014;44:1661–1668. doi: 10.3892/ijo.2014.2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D., Mukherjee R., Grewe D., Bojkova D., Baek K., Bhattacharya A., Schulz L., Widera M., Mehdipour A.R., Tascher G., et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587:657–662. doi: 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith E., Davis-Gardner M.E., Garcia-Ordonez R.D., Nguyen T.T., Hull M., Chen E., Baillargeon P., Scampavia L., Strutzenberg T., Griffin P.R., et al. High-throughput screening for drugs that inhibit papain-like protease in SARS-CoV-2. SLAS Discov. 2020;25:1152–1161. doi: 10.1177/2472555220963667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi N., Nakamura T., Takeuchi F., Hashimoto E., Yamamura H. Inhibitory effect of mitoxantrone on activity of protein kinase C and growth of HL60 cells. J. Biochem. J. Biochem. 1992;112:762–767. doi: 10.1093/oxfordjournals.jbchem.a123972. [DOI] [PubMed] [Google Scholar]
- Tardif J.C., Grégoire J., L'Allier P.L., Anderson T.J., Bertrand O., Reeves F., Title L.M., Alfonso F., Schampaert E., Hassan A., et al. Effects of the acyl coenzyme A:cholesterol acyltransferase inhibitor avasimibe on human atherosclerotic lesions. Circulation. 2004;110:3372–3377. doi: 10.1161/01.CIR.0000147777.12010.EF. [DOI] [PubMed] [Google Scholar]
- Thomson E.C., Rosen L.E., Shepherd J.G., Spreafico R., da Silva Filipe A., Wojcechowskyj J.A., Davis C., Piccoli L., Pascall D.J., Dillen J., et al. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell. 2021;184:1171–1187.e20. doi: 10.1016/j.cell.2021.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tickle I.J., Flensburg C., Keller P., Paciorek W., Sharff A., Vonrhein C., Bricogne G. Global Phasing Ltd; 2018. STARANISO. [Google Scholar]
- Vidya Balasubramanian E.A. CPI-169, a novel and potent EZH2 inhibitor, synergizes with CHOP in vivoand achieves complete regression in lymphoma xenograft models. Cancer Res. 2014;74:1697. [Google Scholar]
- Vonrhein C., Flensburg C., Keller P., Sharff A., Smart O., Paciorek W., Womack T., Bricogne G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 2011;67:293–302. doi: 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadhawan M., Singh N., Rathaur S. Inhibition of cathepsin B by E-64 induces oxidative stress and apoptosis in filarial parasite. PLoS One. 2014;9:e93161. doi: 10.1371/journal.pone.0093161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward J.A.-O., Pinto-Fernandez A., Cornelissen L., Bonham S., Díaz-Sáez L., Riant O.A.-O., Huber K.A.-O., Kessler B.M., Feron O., Tate E.A.-O. Re-evaluating the mechanism of action of α,β-unsaturated carbonyl DUB inhibitors b-AP15 and VLX1570: a paradigmatic example of unspecific protein cross-linking with Michael acceptor motif-containing drugs. J. Med. Chem. 2020;63:3756–3762. doi: 10.1021/acs.jmedchem.0c00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstock J., Wu J., Cao P., Kingsbury W.D., McDermott J.L., Kodrasov M.P., McKelvey D.M., Suresh Kumar K.G., Goldenberg S.J., Mattern M.R., Nicholson B. Selective dual inhibitors of the cancer-related deubiquitylating proteases USP7 and USP47. ACS Med. Chem. Lett. 2012;3:789–792. doi: 10.1021/ml200276j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams C.J., Headd J.J., Moriarty N.W., Prisant M.G., Videau L.L., Deis L.N., Verma V., Keedy D.A., Hintze B.J., Chen V.B., et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 2018;27:293–315. doi: 10.1002/pro.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Kitagaki J., Dai R.M., Tsai Y.C., Lorick K.L., Ludwig R.L., Pierre S.A., Jensen J.P., Davydov I.V., Oberoi P., et al. Inhibitors of ubiquitin-activating enzyme (E1), a new class of potential cancer therapeutics. Cancer Res. 2007;67:9472–9481. doi: 10.1158/0008-5472.CAN-07-0568. [DOI] [PubMed] [Google Scholar]
- Ybema C.E., Olivier B., Mos J., Tulp M.T., Slangen J.L. Adrenoceptors and dopamine receptors are not involved in the discriminative stimulus effect of the 5-HT1A receptor agonist flesinoxan. Eur. J. Pharmacol. 1994;256:610–624. doi: 10.1016/0014-2999(94)90238-0. [DOI] [PubMed] [Google Scholar]
- Zang R., Gomez Castro M.F., McCune B.T., Zeng Q., Rothlauf P.W., Sonnek N.M., Liu Z., Brulois K.F., Wang X., Greenberg H.B., et al. TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci. Immunol. 2020;5:eabc3582. doi: 10.1126/sciimmunol.abc3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.H., Chung T.D., Oldenburg K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Zhang X.D. A new method with flexible and balanced control of false negatives and false positives for hit selection in RNA interference high-throughput screening assays. J. Biomol. Screen. 2007;12:645–655. doi: 10.1177/1087057107300645. [DOI] [PubMed] [Google Scholar]
- Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the crystal structures reported in this paper are PDB: 7D6H and PDB: 7E35.