

Abstract
Effective control of COVID-19 requires antivirals directed against SARS-CoV-2. We assessed 10 hepatitis C virus (HCV) protease-inhibitor drugs as potential SARS-CoV-2 antivirals. There is a striking structural similarity of the substrate binding clefts of SARS-CoV-2 main protease (Mpro) and HCV NS3/4A protease. Virtual docking experiments show that these HCV drugs can potentially bind into the Mpro substrate-binding cleft. We show that seven HCV drugs inhibit both SARS-CoV-2 Mpro protease activity and SARS-CoV-2 virus replication in Vero and/or human cells. However, their Mpro inhibiting activities did not correlate with their antiviral activities. This conundrum is resolved by demonstrating that four HCV protease inhibitor drugs, simeprevir, vaniprevir, paritaprevir, and grazoprevir inhibit the SARS CoV-2 papain-like protease (PLpro). HCV drugs that inhibit PLpro synergize with the viral polymerase inhibitor remdesivir to inhibit virus replication, increasing remdesivir’s antiviral activity as much as 10-fold, while those that only inhibit Mpro do not synergize with remdesivir.
Keywords: COVID-19, SARS-CoV-2 3CL/Mpro protease, SARS-CoV-2 PL protease, HCV protease inhibitors, molecular docking, SARS-CoV-2 virus replication, synergism, remdesivir, antivirals
Graphical abstract
Bafna et al. report that several available hepatitis C virus drugs inhibit the SARS-CoV-2 Mpro and/or PLpro proteases and SARS-CoV-2 replication in cell culture. The four HCV drugs that inhibit PLpro enzyme activity also synergize with remdesivir to inhibit virus replication, increasing the antiviral activity of remdesivir and HCV drugs.
Introduction
Effective control of the SARS-CoV-2 coronavirus that causes COVID-19 requires antivirals. Considering the urgency to identify effective antiviral drugs, and the usually lengthy process involved in approving candidate drugs for human use, our goal is to identify existing drugs already approved for use in humans that can be repurposed as safe and effective therapeutics for treating COVID-19 infections, and which may also be useful as lead molecules for novel drug development.
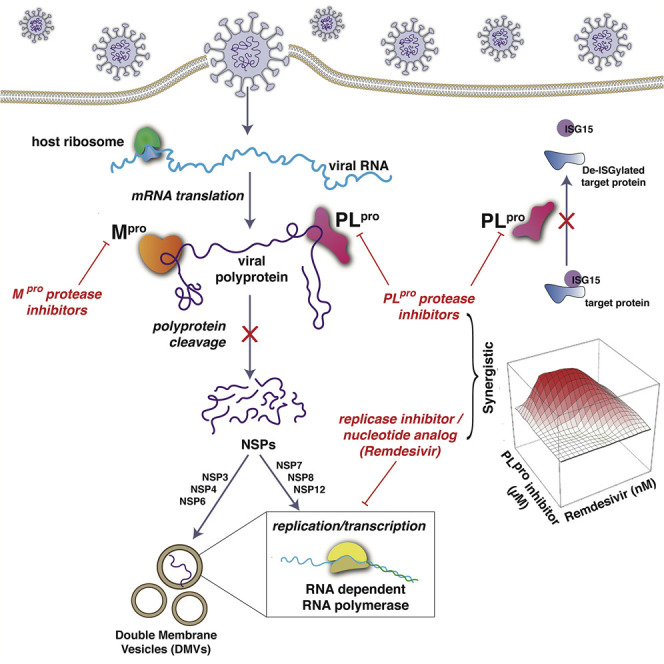
SARS-CoV-2 is an enveloped RNA virus, which causes COVID-19 (Wu et al., 2020). Its genome comprises a single, large positive-sense single-stranded RNA, which is directly translated by host cell ribosomes. The SARS-CoV-2 genome encodes 4 structural proteins, 16 non-structural proteins (NSPs), which carry out crucial intracellular functions, and 9 accessory proteins (Gordon et al., 2020; Wu et al., 2020). Many of these proteins, and their host binding partners, are potential targets for development of antivirals for SARS-CoV-2. For example, the repurposed drug remdesivir, which inhibits the viral RNA-dependent RNA polymerase, is the current FDA-approved antiviral standard of care for COVID-19 (Eastman et al., 2020; Pan et al., 2020).
Translation of the viral genomic RNA results in the biosynthesis of two polyproteins that are processed into the 16 separate NSPs by two virus-encoded cysteine proteases, the papain-like protease (PLpro) and a 3C-like protease (3CLpro). The latter is also referred to as the main protease (Mpro). Mpro and PLpro are essential for the virus life cycle and hence are attractive targets for antiviral development. These two viral proteases are required for the production of functional viral RNA polymerases. Mpro cleavages are predicted to generate several NSPs, including the three subunits nsp7, nsp8, and nsp12 that constitute the viral RNA polymerase complex (Peng et al., 2020), as well as integral membrane proteins nsp4 and nsp6. PLpro cleavages generate other NSPs, including nsp3 (Harcourt et al., 2004). The nsp3-nsp4-nsp6 complex is a key component of the replication organelles, also known as double-membrane vesicles (DMVs), that are required for the function of the viral polymerase in infected cells (Angelini et al., 2013; Gosert et al., 2002; Oudshoorn et al., 2017; Snijder et al., 2020; Wolff et al., 2020a, 2020b). Considering that both Mpro and PLpro generate either the RNA polymerase itself or the replication organelles required for polymerase function, we reasoned that inhibitors of one or both of these proteases might be synergistic with inhibitors of the viral polymerase, such as remdesivir.
We observed that the substrate binding cleft and active site of the SARS-CoV-2 Mpro have remarkable structural similarity with the active site of the hepatitis C virus (HCV) NS3/4A protease, suggesting that drugs that inhibit the HCV protease might also inhibit SARS-CoV-2 Mpro (Bafna et al., 2020). Consistent with this hypothesis, subsequent studies have reported that three of these HCV drugs, boceprevir, narlaprevir, and telaprevir, inhibit Mpro proteolytic activity and bind into its active site (Anson et al., 2020; Fu et al., 2020; Kneller et al., 2020a; Ma et al., 2020). Boceprevir has also been reported to inhibit SARS-CoV-2 replication in Vero cells (Anson et al., 2020; Fu et al., 2020; Ma et al., 2020). Other HCV protease inhibitors have also been reported to inhibit Mpro proteolytic activity (Anson et al., 2020; Fu et al., 2020; Lo et al., 2021; Ma et al., 2020) and/or viral replication (Lo et al., 2021) to various extents, while other studies report that some of these same HCV protease inhibitors did not significantly inhibit Mpro (Fu et al., 2020).
In this study, we assess the ability of 10 available HCV protease inhibitors to suppress SARS-CoV-2 replication. Virtual docking experiments predict that all 10 of these HCV drugs can bind snuggly into the Mpro binding cleft with docking scores comparable to a known Mpro inhibitor, suggesting that any of these 10 HCV drugs are potential inhibitors of Mpro. Seven of these HCV drugs inhibit both SARS-CoV-2 Mpro protease activity, and SARS-CoV-2 virus replication in Vero and/or human 293T cells expressing the SARS-CoV-2 ACE2 receptor. Surprisingly, we found that four HCV drugs also inhibit PLpro protease activity (including one that did not inhibit Mpro). Consequently, HCV drugs that inhibit Mpro and/or PLpro can suppress SARS-CoV-2 virus replication, viz, boceprevir (BOC), narlaprevir (NAR), vaniprevir (VAN), telaprevir (TEL), paritaprevir (PAR), simeprevir (SIM), grazoprevir (GRZ), and asunaprevir (ASU).
Further, we demonstrate that the four HCV drugs that inhibit the proteolytic activity of PLpro, SIM, GRZ, PAR, and VAN, also act synergistically with remdesivir to inhibit SARS-CoV-2 virus replication, thereby increasing remdesivir antiviral activity as much as 10-fold. In addition, the PLpro -specific inhibitor, GRL0617, also synergizes with remdesivir. In contrast, the HCV drugs BOC and NAR, which inhibit Mpro but not PLpro, as well as the Mpro-specific inhibitor GC-376, act additively rather than synergistically with remdesivir to inhibit virus replication. Our results suggest that the combination of a HCV protease inhibitor with a RNA polymerase inhibitor could potentially function as an antiviral against SARS-CoV-2. More generally, our results strongly motivate further studies of the potential use of PLpro protease inhibitors in combination with RNA polymerase inhibitors as antivirals against SARS-CoV-2.
Results
Similarity of the substrate-binding clefts and active sites of SARS-CoV-2 Mpro and HCV protease NS3/4A
The SARS-CoV-2 main protease (Mpro) is a 67.6 kDa homodimeric cysteine protease with three domains (Jin et al., 2020; Zhang et al., 2020b). Domains I and II adopt a double β-barrel fold, with the substrate binding site located in a shallow cleft between two antiparallel β-barrels of domains I and II. The fold architecture of domains I and II are similar to picornavirus cysteine proteases and chymotrypsin serine proteases (Anand et al., 2002; Gorbalenya et al., 1989). A three-dimensional structural similarity search of the Protein Data Bank using the DALI program (Holm and Sander, 1993, 1999), with domains I and II (excluding domain III) of the SARS-CoV-2 Mpro as the query, identified several proteases, including the HCV NS3/4A serine protease, as structurally similar. These HCV and SARS-CoV-2 enzymes have a structural similarity Z score (Holm and Sander, 1993, 1999) of +8.4 and overall backbone root-mean-square deviation for structurally similar regions of ∼3.1 Å. Superimposition of the analogous backbone structures of these two proteases results in superimposition of their substrate binding clefts and their active-site catalytic residues, His41/Cys145 of the SARS-CoV-2 Mpro cysteine protease and His57/Ser139 of the HCV NS3/4A serine protease (Figure 1 A). Because of these structural similarities, we proposed that some HCV protease inhibitors might bind well into the substrate-binding cleft of the SARS-CoV-2 Mpro and inhibit virus replication (Bafna et al., 2020).
Figure 1.

SARS-CoV-2 Mpro binds HCV NS3/4A protease inhibitors
(A) The active site cleft of the SARS-CoV-2 Mpro (green, PDB: 6Y2G) has remarkable structural similarity with the active site cleft of HCV protease NS3/4A (orange, PDB: 2P59). Both have a double β-barrel fold architecture, with a substrate binding site located in a shallow cleft between two the antiparallel β-barrels. The regions identified by DALI as structurally analogous are shown in color (green and orange), and the regions that are not structurally analogous are shown in gray. This superimposition of backbone atoms results in superimposition of the catalytic residues Cys145 and His41 of the SARS-CoV-2 Mpro with Ser139 and His57 of HCV protease. Asp81 of the HCV protease catalytic triad is also shown.
(B) Best AutoDock docking scores for 10 HCV protease inhibitors in the substrate binding cleft of SARS-CoV-2 Mpro. Each docking trajectory was run one-hundred times. The best docking score for Mpro inhibitor 13b is shown as a horizontal red dashed line.
(C) Comparison of the BOC binding pose in best-scoring AutoDock complex (magenta) with the X-ray crystal structure of the BOC-Mpro complex (Anson and Mesecar, 2020) (green, PDB: 6WNP).
(D) Comparison of BOC binding poses in X-ray crystal structures of complexes with HCV NS3/4A protease (orange, PDB: 2OC8; (Prongay et al., 2007)) and SARS-CoV-2 Mpro protease (green, PDB: 6WNP).
(E) Dose-response curves based on FRET assay for inhibition of Mpro, by NAR, BOC, and TEL.
(F) 1D 1H-NMR assay for hydrolysis of the indicated peptide substrate. The amide proton doublets of Phe-10 prior to cleavage (F10_uncleaved) or of Gln-7 after cleavage (Q7_cleaved) provide well-resolved resonances for monitoring the proteolysis reaction. The amide proton doublet of Glu-14 (E14) is not perturbed by cleavage and provides an internal intensity calibration control. GRZ has resonances (labeled by ∗) that overlap with the upfield component of the E14 doublet.
(G) Percentage of cleavage of the indicated peptide substrate by Mpro after 30 min at 25°C. FRET data points are mean ± SD, n = 3; NMR resonance ratio uncertainties were based on signal-to-noise measurements.
See also Tables S1–S3 and Figures S1–S4 and S7.
HCV protease inhibitors are predicted to bind into the substrate binding cleft of Mpro
Based on these structural similarities, we carried out docking simulations of 10 HCV NS3/4A protease inhibitor drugs (Özen et al., 2019), using AutoDock (Forli et al., 2016; Morris et al., 2009). These 10 drugs have been approved for at least phase 1 clinical trials, and some are FDA-approved prescription drugs used to treat HCV-infected patients (Table 1 ). To test the validity of our docking protocol, we first carried out docking of a previously described inhibitor of the SARS-CoV-2 Mpro, compound 13b (Figure S1A), that also inhibits virus replication (Zhang et al., 2020a, 2020b). Details of these control docking studies are provided in the STAR Methods and Figures S1B–S1D. The resulting docking scores are summarized in Figure 1B and Table S1. These results demonstrate that all 10 of these HCV protease inhibitors have the potential to bind snuggly into the binding cleft of Mpro, with extensive hydrogen-bonded and hydrophobic contacts, and predicted AutoDock energies of −8.37 to −11.01 kcal/mol, comparable to those obtained for Mpro inhibitor 13b (∼ −9.0 kcal/mol).
Table 1.
HCV protease inhibitors
| Inhibitor (trade name) | Identifier of protease inhibitor | Trade name; manufacturer | Drug status |
|---|---|---|---|
| Vaniprevir | VAN | MK-7009; Merck | investigational drug |
| Simeprevir | SIM | Olysio/Medivir; Janssen | prescription drug |
| Paritaprevir | PAR | Veruprevir/ABT-450; Abbott Laboratories | prescription drug |
| Danoprevir | DAN | Ganovo; Array/Pfizer, Roche/Ascletis | investigational drug |
| Narlaprevir | NAR | Arlansa; Merck/R-Pharm | prescription drug |
| Grazoprevir | GRZ | Zepatier; Merck | prescription drug |
| Glecaprevir | GLE | Mavyreta Mavireta; AbbVie/Enanta | prescription drug |
| Boceprevir | BOC | Victrelis; Merck | prescription drug |
| Telaprevir | TEL | Incivek/Incivo; Vertex/J&J | prescription drug |
| Asunaprevir | ASU | Sunvepra; Bristol-Myers Squibb | investigational drug |
Mavyret (or Maviret) is a multidrug formulation including glecaprevir and pibrentasvir.
The 1.44-Å X-ray crystal structure of the complex of BOC bound to the SARS-CoV-2 Mpro was recently released in the Protein Data Bank [PDB: 6WNP (Anson et al., 2020; Anson and Mesecar, 2020)]. The BOC pose observed in this X-ray crystal structure is almost identical to the lowest energy pose (−9.13 kcal/mol) predicted by AutoDock (Figure 1C). In addition, both the docked and crystal structure binding poses for BOC near the active site of Mpro are very similar to its binding pose in the substrate binding cleft of HCV protease (Figure 1D), with an essentially identical hydrogen-bonding network between BOC and corresponding residues in each protease (shown in Figures S1E and S1F). Recently, Kneller et al. (2020a) also reported an X-ray crystal structure of the SARS-CoV2 Mpro-BOC complex, as well as the structures of Mpro complexed with HCV inhibitors NAR and TEL. The predicted binding modes of BOC and NAR are also an excellent match to these subsequently determined experimental structures (Figures S1G and S1H), while for TEL poses similar to the crystal structure are included among the best-scoring AutoDock poses (Figures S1I and S1J). BOC, NAR, and TEL are alpha-ketoamides which form covalent bonds with the active site residue Cys145 of Mpro. Although this docking protocol does not include energetics or restraints for covalent bond formation, in the low-energy poses of both BOC and NAR bound to Mpro, the alpha-keto amide carbon is positioned within 3.8 Å of the active site thiol sulfur atom. These blind tests support the predictive value of docking results for the other HCV protease inhibitors. From these virtual docking studies and comparison with subsequently determined X-ray crystal structures, we conclude that all 10 of these HCV protease inhibitors have the potential to bind into the substrate-binding cleft of Mpro, and to inhibit binding of its substrates.
Seven HCV drugs inhibit Mpro protease activity
Inhibition activity of HCV drugs against Mpro was initially assessed using a protease assay based on Föster resonance energy transfer (FRET) using the peptide substrate Dabsyl-KTSAVLQ/SGFRKME-(Edans), containing a canonical Mpro protease recognition site. Under the conditions of these FRET assays, there is little or no inner filter effect for most of the drug-peptide substrate assays (Table S3), and the half-life for the proteolytic reaction is about 30 min (Figures S2A and S2B). HCV protease inhibitors NAR, BOC, and TEL have significant enzyme inhibition activity, with IC50 values of 2.2 ± 0.4 μM, 2.9 ± 0.6 μM, and 18.7 ± 6.4 μM, respectively (Figure 1E). In contrast, little or no inhibition activity was detected in this FRET assay with the other seven HCV protease inhibitors (Figures S3A and S3B).
We also developed a 1D 1H-NMR assay, using the peptide substrate KTSAVLQ/SGFRKME that lacks the Dabsyl and Edans N-terminal and C-terminal tags (Figure S4). In this 1D 1H-NMR assay, NAR, BOC, and TEL have substantial enzyme inhibition activity, as was the case in the FRET assay. In addition, in the NMR assay substantial inhibitory activity was also observed for VAN, and moderate inhibitory activity was observed for GRZ, SIM, and ASU (Figures 1F and 1G). This significant inhibition activity of VAN, and moderate inhibition activities of GRZ, SIM, and ASU, was not detected in the FRET assay. DAN, PAR, and GLE had little or no detectable Mpro inhibitory activity. From these studies, we conclude that seven HCV drugs (viz BOC, NAR, TEL, VAN, GRZ, SIM, and ASU) inhibit SARS-CoV-2 Mpro strongly or moderately under the conditions tested.
Eight HCV drugs inhibit SARS-CoV-2 replication in Vero and/or human 293T cells
The motivation for the docking and biophysical studies described above was to identify HCV drugs with the potential to inhibit SARS-CoV-2 virus replication. For antiviral assays, Vero E6 cells or human 293T cells expressing the ACE2 receptor, were grown in 96-well plates and were incubated with various levels of a HCV protease inhibitor for 2 h. Cells were then infected with SARS-CoV-2 virus at the indicated multiplicity of infection (MOI, plaque-forming units [PFU)/cell) and incubated for the indicated times at 37°C in the presence of the inhibitor. Virus-infected cells were identified by immunofluorescence using an antibody specific for the viral nucleoprotein. Inhibition of viral replication was quantified by determining the percentage of positive infected cells at the end of the incubation period in the presence of the compound, as compared with the number of infected cells in its absence. To determine whether a HCV drug was cytotoxic, uninfected Vero E6 or human 293T cells were incubated with the same levels of the compounds for the same length of time, and cytotoxicity was measured using an MTT assay (Roche). In all of these replication assays, remdesivir was used as a positive control.
Viral replication inhibition data in Vero E6 cells are summarized in Figure 2 ; Table S2. Five of the HCV protease inhibitors tested, PAR, NAR, GRZ, ASU, and BOC, inhibited SARS-CoV-2 virus replication at concentrations significantly lower than the concentrations that cause cytotoxicity. Two other HCV protease inhibitors, SIM and VAN, inhibited virus replication with low IC50 values, but some cytotoxicity was also observed. These seven HCV drugs have IC50 values for inhibiting SARS-CoV-2 replication of 4.2 to 19.6 μM. The remaining three HCV drugs tested, TEL, GLE, and DAN, did not inhibit virus replication in Vero E6 cells, even at a drug concentration of 50 μM.
Figure 2.

Antiviral activity of HCV protease inhibitors in Vero E6 cells
(A–K) Inhibition of viral replication was determined in a concentration-dependent matter in Vero E6 cells. Replication assays were performed at MOI of 0.025 PFU/cell. In all panels, viral infectivity is shown as a solid line and cell toxicity as a dashed line. Data = mean ± SD; n = 3 independent samples. The estimated IC50 is labeled in the top-left corner of each plot. Remdesivir is included as a standard of care control. See also Table S2.
Next, we also determined whether the HCV drugs exhibited similar antiviral activities in human cells, specifically human 293T cells expressing the ACE2 receptor (Figure 3 ; Table S2). Again, PAR, SIM, and VAN were the most effective inhibitors of virus replication, with IC50 values of 0.55 to 3.0 μM, and with considerably reduced cytotoxicity as compared to assays in Vero cells. BOC and NAR, which are relatively strong Mpro inhibitors (Figures 1E–1G), were less effective inhibitors of virus replication, with IC50s of 5.4 and 15 μM. The other covalent Mpro inhibitor, TEL, which did not inhibit in Vero cells had a IC50 of 20.5 μM in human cells, while GRZ had less potent antiviral activity in human cells than in Vero cells (cf. Figures 2F, 2I, 3G, and 3H). Thus, seven HCV drugs inhibited SARS-CoV-2 replication in human 293T cells, with IC50 values ranging from 0.55 to 20.5 μM. GLE and DAN did not inhibit virus replication in human cells, as was also the case in Vero cells. ASU, a modest inhibitor in Vero cells, did not inhibit viral replication in human cells at concentrations lower than those exhibiting cytotoxicity.
Figure 3.

Antiviral activity of HCV protease inhibitors in HEK293T cells
(A–K) Inhibition of viral replication was determined in a concentration-dependent matter in human 293T cells. Replication assays were performed at MOI of 0.025 PFU/cell. In all panels, viral infectivity is shown as a solid line and cell toxicity as a dashed line. Calculated IC50 is indicated in the top-left corner of each plot. Remdesivir is included as a standard of care control.
(L) Time-of-addition assay in human 293T cells. Drugs, at the indicted non-cytotoxic concentrations, were added to cells at the indicated time points before (–2 h), at (0 h), or after (+2 or +4 h) viral infection, at MOI of 2 PFU/cell.
Data are mean ± SD; n = 3 independent samples.
See also Table S2.
We also determined whether the inhibition of SARS-CoV-2 replication by representative HCV protease inhibitors occurs at steps after virus entry, as would be expected for inhibitors of viral proteases that are produced after infection. Accordingly, we performed time-of-addition assays in human cells, using BOC (50 μM), GRZ (25 μM), NAR (50 μM), VAN (5 μM), and SIM (5 μM), at concentrations that do not show detectable cytotoxicity as measured by MTT assay (Table S2) or by DAPI staining in infected cells. In a single cycle (MOI of 2 PFU/cell over a total infection time of 8 h) of infection, drugs were added 2 h prior to infection, at the time of infection, or at 2 or 4 h post-infection. Virus replication was inhibited by the addition of these drugs as late as 4 h post-infection (Figure L), indicating that these drugs can inhibit viral infection after the initial phase of virus entry. These results indicate that these HCV drugs inhibit virus-encoded proteases synthesized in infected cells. The results do not, however, rule out that these drugs may also inhibit other proteases or enzymes, including any involved in virus entry. In contrast, addition of the RNA polymerase inhibitor remdesivir 4 h after the initiation of infection decreased its ability to suppress virus infection, demonstrating the crucial role of viral RNA synthesis at early times of infection.
Simeprevir and grazoprevir synergize with remdesivir to increase inhibition of SARS-CoV-2 virus replication
Because Mpro and PLpro generate either the RNA polymerase itself or the proteins that constitute the replication organelles required for polymerase function, we predicted that HCV drugs that inhibit one or both of these viral proteases might be synergistic with inhibitors of the viral polymerase like remdesivir. To test this hypothesis, we carried out antiviral combination assays of SIM, GRZ, and BOC, respectively, with remdesivir in Vero cells. To assess synergy, two analyses are required. In one analysis the IC90 of the remdesivir was measured in the presence of increasing concentrations of each of these three HCV drugs (Figures 4 A–4C). These results demonstrate that SIM and GRZ increase the antiviral activity of remdesivir. For example, in the presence of 1.25 μM SIM, approximately 10-fold less remdesivir is required for the same antiviral effect achieved in the absence of SIM (Figure 4A). A similar 10-fold enhancement in antiviral activity of remdesivir is observed in the presence of GRZ, though at higher (6.25 μM) GRZ concentrations (Figure 4B). Surprisingly, although BOC is a much better inhibitor of Mpro than either SIM or GRZ, BOC did not significantly affect the antiviral activity of remdesivir (Figure 4C). In the second analysis, the IC90 concentration of each HCV drug was determined in the presence of increasing concentrations of remdesivir (Figures 4D–4F). Remdesivir increased the antiviral activity of SIM and GRZ. For example, addition of 1.25 μM remdesivir substantially reduces the concentration of SIM or GRZ needed to achieve IC90 conditions (Figures 4D and 4E). In contrast, remdesivir did not significantly affect the antiviral activity of BOC (Figure 4F).
Figure 4.

Simeprevir and grazoprevir are synergistic with remdesivir in Vero E6 cells
(A–C) SARS-CoV-2 inhibition by remdesivir in the presence of increasing concentrations of SIM, GRZ, or BOC.
(D–F) SARS-CoV-2 inhibition by SIM, GRZ, or BOC in the presence of increasing concentrations of remdesivir.
(G–I) Synergy landscapes and combination scores generated by the ZIP method using the program SynergyFinder (Ianevski et al., 2020).
These antiviral assays indicate that SIM and GRZ, but not BOC, act synergistically with remdesivir to inhibit virus replication. As confirmation, we subjected these results to analysis by the zero interaction potency (ZIP) model for synergy (Ianevski et al., 2020). In the landscapes generated by this model (Figures 4G–4I), red denotes a synergistic interaction, and green denotes an additive interaction. In this model a synergistic interaction between drugs has a score greater than +10, an additive interaction has a score between −10 to +10, and an antagonistic interaction has a score of less than −10. The landscapes for the interaction of remdesivir with both SIM and GRZ are red, with synergy scores of +30.2 and +25.0, respectively, denoting moderate synergism. In contrast, the landscape for the interaction of remdesivir with BOC does not indicate synergy (Figure 4I); the synergy score, −7.6, indicates an additive interaction. We also carried out combination antiviral assays in human 293T cells. The interaction between remdesivir and GRZ in inhibiting virus replication in the human cells was also synergistic, with a red landscape and a synergy score of +20.3 (Figure S5). Consequently, at least two HCV drugs, SIM and GRZ, act synergistically with remdesivir to inhibit SARS-CoV-2 virus replication in Vero and/or human 293T cells.
Four HCV protease inhibitors that are synergistic with remdesivir inhibit PLpro
The preceding results demonstrate that several HCV inhibitors inhibit viral replication, and that for some of these drugs inhibition is synergistic with the viral replication inhibition activity of remdesivir. Most of these drugs also inhibit SARS-CoV-2 Mpro. However, Mpro and SARS-CoV-2 inhibition by these drugs were not consistently correlated. For example, PAR does not inhibit Mpro in either the FRET or NMR assays but is an effective inhibitor of SARS-CoV-2 in both Vero (IC50 = 6 μM) and 293T (IC50 = 0.55 μM) assays. In addition, the HCV protease inhibitors SIM and GRZ, which are only moderate inhibitors of Mpro, synergize with remdesivir, while BOC, which is an excellent inhibitor of Mpro, acts additively rather than synergistically with remdesivir to inhibit virus replication. Although the lack of strong correlation between protease inhibition activity and viral inhibition activity could result from various effects, including the efficiency of transport of drugs into the cell and/or metabolism of the drugs in the cell-based assays, these results suggested that there might be a second viral target for some of these HCV drugs, for which inhibition may provide the basis for the observed synergy.
Aside from both being Cys proteases, the active site of PLpro does not share structural similarity with the HCV NS3/4A or Mpro proteases. However, it has recently been reported that SIM, GRZ, and ASU inhibit PLpro (Anson et al., 2020). Accordingly, we carried out virtual docking studies of these same 10 HCV drugs into the substrate-binding cleft of PLpro, using protocols similar to those developed in virtual docking studies with Mpro. The known PLpro inhibitor GRL0617 (Fu and Huang, 2020) was used to assess the docking protocol, providing a reference AutoDock score of −7.54 kcal/mol. The scores of docking poses for HCV drugs, summarized in Figure 5 A and Table S1, range from –5.56 kcal/mol for BOC and NAR, to much more favorable values of < –8 kcal/mol for others, including VAN, GRZ, SIM, and PAR. These proof-of-concept docking studies suggest that, surprisingly, some HCV protease inhibitors may bind in the substrate-binding clefts of both Mpro and PLpro.
Figure 5.

HCV protease inhibitors also inhibit SARS-CoV-2 PLpro
(A) Best AutoDock docking scores for 10 HCV protease inhibitors in the substrate binding cleft of SARS-CoV-2 PLpro. The docking score for PLpro inhibitor GRL0617 is also shown as a horizontal red dashed line. Each docking trajectory was run one-hundred times.
(B) Initial rates of proteolysis of a peptide substrate by PLpro in the presence of 20 μM inhibitor concentrations (vi) relative to initial rate in the absence of inhibitor (vi,o), at 25°C. GRZ inhibition of PLpro was even stronger (vo/vo,i = 32% ± 7%) at 100 μM drug concentration (Figure S6). Data are mean ± SD, n = 2 independent measurements.
(C and D) PLpro inhibitors PAR and VAN are synergistic with remdesivir.
(E) NAR does not inhibit PLpro and is also not synergistic with remdesivir.
See also Tables S2 and S3 and Figures S1 and S6.
Based on these docking predictions, we anticipated that several HCV protease inhibitors, not including BOC, NAR, or TEL, might effectively inhibit PLpro protease activity. To test this hypothesis, fluorescence assays of PLpro inhibition were carried out, using the substrate zRLRGG/AMC (z - carboxybenzyl; AMC - 7-Amino-4-methylcoumarin) (Anson et al., 2020) containing a natural canonical PLpro protease recognition site (XLXGG). Of the HCV drugs tested, four drugs predicted to bind into the active site of PLpro, VAN, SIM, PAR, and GRZ, do in fact inhibit PLpro protease activity (Figure 5B; Figure S6). Hence, VAN, SIM, and GRZ inhibit both Mpro and PLpro, while PAR inhibits PLpro, but not Mpro. Under these assay conditions, VAN and SIM are more effective PLpro inhibitors than GRZ or PAR.
These results strengthened our hypothesis that synergy between these HCV drugs and remdesivir arises primarily from their activities in inhibiting PLpro, rather than from inhibiting Mpro. To further test this, combination assays with remdesivir were carried out also for NAR, PAR, and VAN. As predicted, PAR which moderately inhibits PLpro but not Mpro, is synergistic with remdesivir (Figure 5C, synergy score +17.3). VAN, which inhibits both Mpro and PLpro, is also modestly synergistic, with synergy score + 10.9, while NAR, which inhibits Mpro but not PLpro, is additive with synergy score –3.6 (Figures 5D and 5E).
Data for HCV protease drugs on SARS-CoV-2 protease inhibition and synergy are summarized in Figure 6 A and in Table S2. Two drugs, BOC and NAR, which are relatively good inhibitors of Mpro but do not inhibit PLpro, have additive interactions with remdesivir. Three drugs, GRZ, SIM, and VAN, which inhibit both Mpro and PLpro, are synergistic, with synergy scores of +10.9 to +30.2. Interestingly, among these three, VAN, which is a relatively strong inhibitor of both Mpro and PLpro, has the weakest synergy. A fourth HCV drug, PAR, which inhibits PLpro but not Mpro, also has synergy with remdesivir, with synergy score +17.3. These data demonstrate a correlation between the PLpro inhibiting activities of these drugs and their ability to function synergistically with remdesivir to suppress viral replication.
Figure 6.

PLpro inhibition and synergy with remdesivir activities are correlated
(A) Summary of relative inhibition of SARS-COV-2 proteases by HCV protease inhibitors and their respective synergies with remdesivir. The inset key shows the color coding for strong (green, IC50 < 20 μM), moderate (yellow, 20 μM < IC50 < 50 μM), and no (red) enzyme inhibition activity.
(B) Initial rates of proteolysis of peptide substrates by selective Mpro (GC-376) and PLpro (compound 6) inhibitors at 20 μM concentrations (vi), relative to initial rate in the absence of inhibitor (vi,o), at 25°C. Data are mean ± SD, n = 2.
(C and D). Synergy landscapes and combination scores (Ianevski et al., 2020).
In order to further test this model of synergy, we also carried out biochemical and viral replicase assays with the molecule GC-376, an Mpro inhibitor (Fu et al., 2020; Ma et al., 2020), and an analog of GRL0617, referred to as compound 6, an established PLpro inhibitor (Fu and Huang, 2020; Osipiuk et al., 2021; Ratia et al., 2008). While GC-376 has been reported to be specific for Mpro relative to PLpro (Sacco et al., 2020), the relative specificity of GLR0617 (or compound 6) for PLpro relative to Mpro has not previously been reported. Using the same fluorescence assays outlined above, we validated the high specificity of GC-376 for Mpro inhibition compared to PLpro (Figure 6B, left) and documented high specificity of compound 6 for PLpro inhibition compared to Mpro (Figure 6B, right). As predicted, combination viral inhibition assay of GC-376 with remdesivir shows an additive interaction (Figure 6C; synergy score +5.9), while the GRL0617 analog (compound 6) has a synergistic interaction with remdesivir (Figure 6D; synergy score +18.6). We conclude that inhibitors of the SARS-CoV-2 PLpro protease function synergistically with remdesivir to inhibit viral replication, whereas specific inhibitors of the Mpro protease act additively with remdesivir.
Discussion
To provide antiviral drugs that can be rapidly deployed to combat the COVID-19 pandemic, we carried out the present study to identify currently available drugs that could potentially be repurposed as inhibitors of the SARS-CoV-2 virus that causes the COVID-19 disease. A total of eight HCV drugs were identified that inhibit virus replication in Vero and/or human 293T cells expressing the ACE2 receptor.
We initiated our search based on the striking similarity of the substrate binding clefts of the SARS-CoV-2 Mpro and HCV NS3/4A proteases (Bafna et al., 2020). The substrate-binding clefts and active sites of Mpro and HCV proteases superimpose remarkably well (Figure 1A), despite having very low sequence similarity (Figure S7) and significantly different structural topologies (Bafna et al., 2020). Our virtual docking experiments showed that 10 HCV protease inhibitors can be docked snuggly into the substrate binding cleft of Mpro and hence have the potential to inhibit binding of the Mpro substrate, thereby inhibiting proteolytic cleavage of the viral polyprotein to form NSPs. For BOC and NAR, these predicted docking poses (Bafna et al., 2020) are consistent with the subsequently determined X-ray crystal structures (Anson et al., 2020; Anson and Mesecar, 2020; Kneller et al., 2020a); for TEL, some predicted binding poses are also similar to the corresponding crystal structure (Kneller et al., 2020a). Four of these HCV drugs, BOC, NAR, TEL, and VAN, are relatively strong inhibitors of SARS-CoV-2 Mpro protease activity (IC50 of 2 to ∼20 μM), and three other HCV drugs, GRZ, SIM, and ASU, moderately inhibit Mpro activity. BOC, NAR, and TEL are α-keto amides, which can form a covalent bond with the active site Cys thiol of Mpro . The other four HCV drugs are non-covalent inhibitors of the Mpro protease and cannot form a covalent bond with the active site Cys thiol.
Other groups have also recently reported that some of these same HCV protease inhibitors can inhibit Mpro protease activities (Anson et al., 2020; Fu et al., 2020; Kneller et al., 2020a; Lo et al., 2021; Ma et al., 2020). While all of these studies report BOC as a moderately potent inhibitor of Mpro, there are inconsistent reports of the effectiveness of some of the other HCV protease inhibitors reported here as inhibitors of Mpro and/or PLpro. These inconsistencies likely arise from details of the different assays, including the specific protein constructs and polypeptide substrates.
The significant intrinsic fluorescence of these non-covalent inhibitor drugs complicates the Mpro FRET assay (see STAR Methods), particularly for VAN, SIM, and GRZ (see Figure S3A). For this reason, we also used 1D 1H-NMR assay for Mpro inhibition, which confirmed that BOC, NAR, and TEL inhibit Mpro. In the NMR assay VAN also has inhibitory activity comparable to TEL, while GRZ, SIM, and ASU are moderate inhibitors of Mpro. The ability of the NMR assay to detect inhibitory activity that was not detected by the FRET assay is attributable to several factors, including differences in substrate and enzyme concentrations used in these assays, and differences in the substrates themselves. The ability of HCV drugs to inhibit Mpro also depends on other details of the assay conditions, most notably the enzyme, substrate, and drug concentrations and details of the Mpro construct (Grum-Tokars et al., 2008).
Although the active site of PLpro does not share structural similarity with the HCV NS3/4A protease, we observe that four HCV drugs, SIM, GRZ, VAN, and PAR, inhibit PLpro protease activity in vitro. None of these four inhibitors can form covalent bonds with the active-site Cys residue of PLpro. VAN is a good inhibitor of both Mpro and PLpro, presumably accounting for its strong inhibition of virus replication. All four of these HCV drugs function synergistically with remdesivir to inhibit SARS-CoV-2 virus replication in Vero and/or human cells.
Particularly interesting in this set is PAR, which has the strongest potency in the human cell assay (IC50 = 0.55 μM), strong synergy with remdesivir (synergy score + 17.3), and low cytotoxicity (CC50 > 100 μM) in both the Vero and human cell assays (Table S2). However, PAR only moderately inhibits PLpro and does not inhibit Mpro . One possibility is that inhibition of virus replication by PAR could result, at least in part, from its inhibition of a third target. Inhibition of such a putative third target might also play some role in the inhibition of virus replication by the other HCV drugs.
In addition to its function in cleavage of viral polyproteins to generate crucial viral non-structural proteins, PLpro also removes the ubiquitin-like ISG15 protein from viral proteins (Daczkowski et al., 2017; Lindner et al., 2007). ISG15 conjugation in infected cells results in a dominant-negative effect on the functions of viral proteins (Zhao et al., 2016); i.e., ISGlyation disrupts a wide range of viral functions. In addition, the resulting free ISG15 is secreted from infected cells and binds to the LFA-1 receptor on immune cells, causing the release of interferon-γ and inflammatory cytokines (Swaim et al., 2017, 2020). The release of these cytokines could contribute to the strong inflammatory response, the so-called cytokine storm, that has been implicated in the severity of COVID-19 disease (McGonagle et al., 2020). Inhibition of PLpro by a HCV drug should also inhibit the release of interferon-γ and inflammatory cytokines, potentially mitigating the cytokine storm.
Viral replication assays using combinations of drugs allowed us to assess whether the interactions between HCV drugs and remdesivir are additive or synergistic. We found that these inhibitory effects are additive or synergistic depending on which HCV drug is used to inhibit virus replication. In particular, HCV drugs that inhibit PLpro synergize with remdesivir to inhibit SARS-CoV-2 replication in Vero and 293T cells. These HCV drugs include SIM, VAN, GRZ, and PAR. The conclusion that inhibition of PLpro alone is sufficient for synergy with remdesivir was confirmed by a combination assay with compound 6 (a GRL0617 analog) that specifically inhibits PLpro but not Mpro. On the other hand, we show that the HCV drugs BOC and NAR that inhibit only Mpro have an additive rather than a synergistic interaction with remdesivir in inhibiting SARS-CoV-2 replication. The conclusion that selective inhibition of Mpro has an additive interaction with remdesivir was confirmed by a synergy assay with compound GC-376, that specifically inhibits Mpro but not PLpro. Another Mpro inhibitor (PF-00835231) has been reported to act in combination with remdesivir, but it was not clear whether this interaction was additive or synergistic (Boras et al., 2020). It was also recently reported that SIM acts synergistically with remdesivir but that this synergy results from inhibition of Mpro and/or other targets (Lo et al., 2021).
The mechanism through which PLpro inhibition, but not Mpro inhibition, results in synergy with remdesivir is not yet known. One rational mechanism involves the critical role of PLpro in the formation of replication organelles (DMVs). Studies of DMV formation by SARS-CoV nsp3, nsp4, and nsp6 proteins demonstrate a requirement for all three proteins, and for a catalytically active PLpro nsp3 construct (Angelini et al., 2013). HCV drugs that inhibit PLpro in infected cells should therefore inhibit formation of DMVs that are required for polymerase function, reducing the amount of functional viral RNA polymerases, and hence reducing the amount of remdesivir needed for inhibition of virus replication. This hypothetical mechanism could explain why drugs that inhibit PLpro (e.g., SIM, VAN, PAR, and GRZ) act synergistically with remdesivir. In contrast, the drugs that inhibit Mpro, but not PLpro (e.g., BOC and NAR), are not synergistic with remdesivir. Mpro inhibitors are expected to reduce the amount of the three subunits, nsp7, nsp8, and nsp12, of the viral polymerase in infected cells. However, the reduction in the amounts of these polymerase subunits might not reduce the level of the viral polymerase sufficiently to exhibit synergy with remdesivir if there is relatively large pool of these subunits in infected cells. While Mpro also generates the nsp4 and nsp6 proteins that contribute to DMV formation (Angelini et al., 2013), it is not known whether this function of Mpro is required for DMV formation.
Synergy between PLpro and viral polymerase inhibitors could also involve other viral or host targets of these protease inhibitors. Removal of ISG15 from viral or host proteins by PLpro could potentially restore their functions, and inhibition of the de-ISGlyaton function of PLpro could provide another mechanism for synergy between inhibitors of PLpro and inhibitors of other viral or host protein functions, including remdesivir.
HCV drugs that are strongly synergistic with remdesivir are most pertinent for the goal of the present study. Repurposed drugs may not have sufficient inhibitory activity on their own to achieve clinical efficacy. Synergy with remdesivir increases the potency of both the proposed repurposed HCV drugs and remdesivir. We identified four HCV drugs, SIM, VAN, PAR, and GRZ, that act synergistically with remdesivir to inhibit SARS-CoV-2 virus replication. Of these four, SIM, PAR, and VAN are particularly interesting as repurposed drugs because they effectively inhibit SARS-CoV-2 virus replication in human cells at lower concentrations than GRZ. Consequently, the combination of an FDA-approved PLpro inhibitor, such as SIM or PAR, and remdesivir, could potentially function as an antiviral against SARS-CoV-2, while more specific and potent SARS-CoV-2 antivirals are being developed. SIM, VAN, PAR, and GRZ are orally administered drugs that might also be combined with an oral polymerase inhibitor rather than with remdesivir, which has to be administered intravenously. One such oral polymerase inhibitor, molnupiravir (MK-4482) (Sheahan et al., 2020), which is currently in late-stage clinical trials, could potentially be combined with one of these four HCV protease inhibitors for clinical applications. For example, a combination of SIM and molnupiravir could be assessed for outpatient use. Beyond the proposed repurposing of these FDA-approved HCV inhibitors as antivirals for COVID-19, our results indicate that the SARS-CoV-2 PLpro is an important target for future antiviral drug development that when used in conjunction with polymerase inhibitors could provide potent efficacy and protection from SARS-CoV-2, especially for virus variants that are resistant to vaccine-generated antibodies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-N protein | Dr. Thomas Moran (MSSM) | mAb 1C7 |
| Bacterial and virus strains | ||
| USA-WA1/2020 | BEI Resources | NR-52281 |
| Biological samples | ||
| Not applicable | ||
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant SARS-CoV-2 PLpro protein; residues 1564 to 1881 of the SARS-CoV-2 replicase polyprotein 1a (Uniprot id P0DTD1 (R1A_SARS2)), with a C-terminal purification tag LEHHHHHH | Laboratory of Prof. J. Hunt, Columbia University | N/A |
| KTSAVLQ/SGFRKME NMR assay substrate | GenScript Inc; custom synthesis this paper | N/A |
| Dabcyl-KTSAVLQ/SGFRKME-Edans FRET assay substrate | GenScript Inc; custom synthesis this paper | N/A |
| zRLRGG/AMC Fluorescence assay substate | Bachem America Inc. | Cat# 4027158.0025 |
| Ampicillin | Research Products International | Cat# A40040 |
| IPTG | Research Products International | Cat# 156000 |
| asunaprevir HCV protease inhibitor | Selleckchem Inc. | Cat# S4935 |
| boceprevir HCV protease inhibitor | Selleckchem Inc. | Cat# S3733 |
| danoprevir HCV protease inhibitor | Selleckchem Inc. | Cat# S1183 |
| glecaprevir HCV protease inhibitor | Selleckchem Inc. | Cat# S5720 |
| grazoprevir HCV protease inhibitor | Selleckchem Inc. | Cat# S3728 |
| paritaprevir HCV protease inhibitor | Selleckchem Inc. | Cat# S5404 |
| simeprevir HCV protease inhibitor | Selleckchem Inc. | Cat# S5015 |
| telaprevir HCV protease inhibitor | Selleckchem Inc. | Cat# S1538 |
| narlaprevir HCV protease inhibitor | MedChemExpress | Cat# HY-10300 |
| vaniprevir HCV protease inhibitor | Addoq | Cat# A11600-5 |
| GC-376 | Selleckchem Inc. | Cat# S0457 |
| compound 6 (GRL0617 analog) | MedChemExpress | Cat# HY-17542 |
| remdesivir | Medkoo Bioscience Inc. | Cat# 329511 |
| Critical commercial assays | ||
| Not applicable | ||
| Deposited data | ||
| 1H and 15N assignments for 14-residues peptide that is cleaved by SARS-CoV-2 Mpro. | This paper. | BioMagResDB accession number: 50568 |
| 1H and 15N assignments for 14-residue peptide after cleavage by SARS-CoV-2 Mpro | This paper | BioMagResDB accession number: 50569 |
| Experimental models: Cell lines | ||
| E. coli BL21(DE3) | New England Biolabs Inc | Cat# C2527 |
| Vero E6 cells | ATCC | Cat# CRL-1586 |
| HEK293T cells | ATCC | Cat# CRL-3216 |
| HEK293T cells transduced with a lentiviral vector expressing human ACE2. Cells were puromycin selected and then single-cell-cloned and screened for their ability to support SARS-CoV-2 replication. | This paper | |
| Experimental models: Organisms/strains | ||
| Not applicable | ||
| Oligonucleotides | ||
| Not applicable | ||
| Recombinant DNA | ||
| Mpro expression vector GTM_COV2_NSP5_001, corresponding to residues 3264 to 3567 of the SARS-CoV-2 replicase polyprotein 1a (Uniprot id P0DTD1 (R1AB_SARS2) cloned into the pGEX-6P-1 vector with a C-terminal His6 tag, | GenScript Inc; custom synthesis this paper | N/A |
| Software and algorithms | ||
| Topspin 3.2.6 | Bruker Biospin, Inc. | https://www.bruker.com/en.html |
| AutoDock 4.2 | Morris et al., 2009 | http://autodock.scripps.edu/ |
| AutoDockTools 1.5.6 | Sanner, 1999 | http://autodock.scripps.edu/resources/adt |
| Synergy Finder | Ianevski et al., 2020 | https://synergyfinder.fimm.fi/ |
| DALI structure alignment server | Holm and Sander, 1993, 1999 | http://ekhidna2.biocenter.helsinki.fi/dali/ |
| Prism 8 graphing software | GraphPad, Inc. | https://www.graphpad.com/ |
| Other | ||
| Bruker Avance II 600 MHz NMR System | Bruker Biospin, Inc. | https://www.bruker.com/en.html |
| Infinite M1000 TECAN plate reader fluorimeter | ThermoFisher Scientific. Inc. | https://www.tecan.com/hubfs/Tecan_Journal/200801/Tecan_Journal_01_08_page16-17.pdf |
Resource Availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the designated contact, Gaetano T. Montelione (monteg3@rpi.edu).
Materials availability
All materials generated in this study are available upon request.
Data and code availability
The following datasets generated during this study are available at the BioMagResDB:
BioMagResDB accession number: 50568. Title: “1H and 15N assignments for 14-residues peptide that is cleaved by SARS-CoV-2 Mpro.” DOI: 10.13018/BMR50568
BioMagResDB accession number: 50569. Title: “1H and 15N assignments for 14-residue peptide after cleavage by SARS-CoV-2 Mpro.” DOI: 10.13018/BMR50569
Experimental models and subject details
Vero E6 (ATCC, CRL-1586) and 293T cells (ATCC, CRL-3216; kind gift of Dr. Viviana Simon), were maintained in DMEM (Corning) supplemented with 10% FB (Peak Serum) and penicillin/streptomycin (Corning) at 37°C and 5% CO2. hACE2-293T cells were generated for this study. Briefly, 293T cells were transduced with a lentiviral vector expressing human ACE2. Puromycin resistant cells with hACE2 surface expression were sorted after staining with AlexaFluor 647-conjugated goat anti-hACE2 antibodies. Cells were then single-cell-cloned and screened for their ability to support SARS-CoV-2 replication. All cell lines used in this study were regularly screened for mycoplasma contamination using the Universal Detection Kit (ATCC, 30-1012K).
Method details
Molecular docking
We used the free open source AutoDock suite (Morris et al., 2009). AutoDockTools (Sanner, 1999) was used for coordinate preparation, docking, and analysis of results. The computational docking program AutoDock v4.2.6. is based on an empirical free-energy force field and uses a search method based on Lamarckian genetic algorithm (Morris et al., 1998). Target protein coordinates were obtained from SARS-CoV-2 Mpro X-ray crystal structure (PDB id 6Y2G) (Zhang et al., 2020b), and structural water was removed. Three-dimensional coordinates for ligand molecules were obtained from PDB (http://www.rcsb.org/) or from chemical structure databases, ChemSpider (http://www.chemspider.com/) and DrugBank (https://go.drugbank.com/). Protein and ligand coordinates were prepared using AutoDockTools; polar hydrogens were added to protein structures, and Gasteiger-Marsili empirical atomic partial charges were added to ligands. Torsional degrees of freedom (dihedral angles) were identified for each ligand. These data and parameters for each protein and ligand were saved as individual PDBQT files. In these studies, ligand dihedral angles were allowed to vary (except where stated otherwise), and all protein dihedral angles were kept rigid. The program Autogrid (Sanner, 1999) was used to prepare affinity maps for all atom types in the receptor and ligands. A grid of 56, 40 and 48 points in x, y, and z direction, with a grid spacing of 0.375 Å was used to compute electrostatic maps. The grid center was placed on the center of the inhibitor 13b molecule in its complex with Mpro (PDB id 6Y2G) (Zhang et al., 2020b). The Lamarckian genetic algorithm (LGA) method was used for sampling ligand binding conformation (Morris et al., 1998), with the following LGA parameters: 150 individuals in population; 2,500,000 energy evaluations; 27,000 maximum number of generations; and with mutation and crossover rates of 0.02 and 0.08, respectively. A maximum of 300 iterations per local search was used. The calculations were repeated for 100 docking simulations for each HCV drug. All docking simulations were analyzed using the AutoDockTools. Atomic coordinates for best scoring conformation obtained in each docking simulation, for each drug-protein complex, were saved in PDB format for analysis. In addition, because the experimentally-determined pose is often not the one with the lowest docking energy, but rather is found among other highly-ranked poses (Kolb et al., 2009; Kolb and Irwin, 2009), we also examined other low-energy poses. These protein-ligand complexes were analyzed in detailed using open source PyMol molecular visualization tool (DeLano, 2009) and fully automated Protein-Ligand Interaction Profiler (Salentin et al., 2015) (https://plip-tool.biotec.tu-dresden.de/plip-web/plip/index).
For a comparative analysis, docking simulations of the α-ketoamide inhibitor 13b (Zhang et al., 2020b) were also performed using the same protocol used for docking HCV protease inhibitor drugs. In the 1.75-Å X-ray crystal structure of the 13b-Mpro complex (Zhang et al., 2020b), 13b makes hydrogen-bonded interactions with backbone amides of key residues, Gly143, Ser144 and Cys145, in the canonical oxyanion hole of the active site. To estimate the docking score for the pose observed in this X-ray crystal structure, we first carried out docking using a modified protocol in which the dihedral angles of 13b were fixed to the values observed in the crystal structure. The lowest energy pose obtained with this protocol matches the crystal structure almost exactly (Figure S1B), with AutoDock binding energy of −7.19 kcal/mol. Next, we assessed the docking protocol using flexible ligand dihedral angles, as was used for all the HCV protease inhibitors. The best-scoring docked conformation (Figure S1C), has an AutoDock binding energy of −9.17 kcal/mol. A pose with slightly higher binding energy of −9.03 kcal/mol, is almost identical to the pose observed in the crystal structure (Figure S1D). Hence, binding poses very similar to that observed in the crystal structure are indeed included among the low energy poses generated by the AutoDock protocol.
For PLpro, inhibitor complexes in the PDB revealed that the BL2 loop present at the entrance of active site adopts significantly different conformations depending on the size of the inhibitor bound to the PLpro . This plasticity in the BL2 loop suggests an induced fit mechanism of ligand binding to PLpro active site. To avoid a closed conformation of the BL2 loop found in protein ligand complex we chose a SARS-CoV-2 PLpro X-ray crystal structure in its apo form (PDB id 6W9C) as the target to dock HCV protease inhibitors. The docking protocol was same as above, except a larger grid of size 56, 56, and 58 points in the x, y, and z direction respectively was used to compute electrostatic maps for PLpro target. For a comparative analysis, docking simulations of PLpro inhibitor GRL0617 (PDB id 7CJM) were also performed using the same protocol.
Mpro expression and purification
The full-length SARS-CoV-2 Mpro gene, corresponding to residues 3264 to 3567 of the SARS-CoV-2 replicase polyprotein 1a [Uniprot id P0DTD1 (R1AB_SARS2)], was obtained from GenScript USA, Inc. and cloned into the pGEX-6P-1 vector with a C-terminal His6 tag, as previously described (Zhang et al., 2020b). This expression vector is designated GTM_COV2_NSP5_001. This plasmid, which expresses SARS-CoV-2 Mpro as a self-cleaving (using its native cleavage site) GST-fusion, was transformed into competent E. coli BL21(DE3) cells. A single colony was picked and inoculated in 2 mL LB supplemented with 0.1 mg/ml ampicillin at 37°C and 225 rpm. The 2 mL inoculum was added to 1L LB broth with 0.1 mg/mL ampicillin. The cells were allowed to grow to an optical density of 0.6 at 600 nm at 37°C and 225 rpm, and were induced with 1 mM IPTG. The induced cells were incubated overnight at 18°C and 225 rpm. The cells were harvested and resuspended in lysis buffer (20 mM Tris pH 8.0, 300 mM NaCl) and then lysed by sonication. The cell debris was removed by centrifugation at 14,000 rpm for 40 mins. The supernatant was added to a Ni-NTA column pre-equilibrated with loading buffer (20 mM Tris pH 8.0, 300 mM NaCl, 10 mM imidazole), and bound proteins were eluted with 20 mM Tris pH 8.0, containing 100 mM NaCl and100 mM imidazole. The elution fractions that contained Mpro were buffer exchanged (20 mM Tris pH 8.0, 100 mM NaCl, 1 mM EDTA, 1 mM DTT) and concentrated using a swinging bucket centrifuge at 4000 rpm. The final Mpro concentration was 23.3 μM, determined by absorbance at 280 nm using a calculated extinction coefficient of 33,640 M-1 cm-1. Homogeneity was validated by SDS-PAGE (> 95% homogeneous), and the construct was validated by MALDI-TOF mass spectrometry. The enzymatic activity of freshly purified Mpro was measured using Michealis Menten equation best fit values of K M and V max; these were 55.5 μM and 0.018 μM/sec, respectively. The calculated k cat was 1.80 s-1, and k cat /K M was 32,400 s-1 M-1.
Mpro was observed to be unusually sensitive to active site Cys oxidation (Kneller et al., 2020b), requiring special care in preparing samples, maintaining them under reducing conditions, and checking samples for time-dependent loss of activity over the course of enzyme activity and drug inhibition measurements. Purified samples of Mpro were prepared in 20 mM TRIS buffer, pH 8.0, containing 100 mM NaCl, 1 mM EDTA, and 1 mM DTT, flash frozen in 50 μL aliquots, and stored at −80°For enzyme assays, freshly thawed enzyme aliquots were prepared in buffers containing 3 mM TCEP, and assayed for specific activity at the beginning and end of the data collection session, or back-to-back with each measurement, in order to avoid spurious results due to enzyme inactivation during a measurement session.
Mpro proteolysis inhibition assays
The proteolysis of substrate KTSAVLQ/SGFRKME was studied using Fluorescence Resonance Energy Transfer (FRET) and nuclear magnetic resonance (NMR) assays. Fluorescence studies were carried out using an Infinite M1000 TECAN plate reader, with 3 mm path lengths, and Magellan™ software. NMR assays were carried out using a Bruker Avance II 600 MHz NMR spectrometer system. For the FRET assay the substrate was Dabsyl-KTSAVLQ/SGFRKME-Edans, labeled with a Dabcyl and Edans FRET pair on the N and C-termini of the peptide, respectively, as described elsewhere (Ma et al., 2020). The NMR assay used the same peptide substrate without the fluorescence dyes attached. Both labeled and unlabeled peptide substrates were obtained from GenScript USA, Inc.
As part of our initial development of the FRET assay for Mpro activity, we assessed catalysis rates over the range of 1 nM to 200 nM enzyme concentration, with the fluorophore-labeled peptide substrate. We observed significant proteolytic activity over this whole range (Figure S2). As rates of hydrolysis at enzyme concentrations > ∼50 nM are quite fast, the concentration of 10 nM was selected in order to provide the most accurate kinetic data. Other FRET-based assays of Mpro activity have been reported using enzyme concentrations ranging from 20 nM to 500 nM (Fu et al., 2020; Grum-Tokars et al., 2008; Hung et al., 2020; Lo et al., 2021; Ma et al., 2020). However, it is well established that the kinetic properties of SARS CoV Mpro are significantly influenced by the construct and assay conditions utilized (Grum-Tokars et al., 2008). For SARS CoV Mpro, the monomeric form is inactive and dimerization is required for its enzymatic activity (Grum-Tokars et al., 2008). Reports of the dimer dissociation constant Kd for SARS-CoV Mpro range from 1 nM to 200 μM; these dimer dissociation constants are also very sensitive to the presence of non-native N- or C-terminal residues (Grum-Tokars et al., 2008). The dimer dissociation constant Kd for a similar construct of SARS-CoV-2 Mpro has been reported to be ∼2.5 μM (Zhang et al., 2020b). Hence, the fraction of active enzyme present at 10 nM concentration is expected to be quite low. However, other groups have also reported SARS-CoV-2 Mpro enzyme activity assays at 20 nM enzyme concentration (Hung et al., 2020). Assuming the substrate binds more tightly to the dimer than to the monomer, the thermodynamic equilibrium between monomer and dimer would be expected to be shifted by the substrate, resulting in the observed protease activity even at enzyme concentrations well below the homodimer Kd. In this case, in the right concentration range there would be a non-linear dependence of activity versus enzyme concentration. This was not observed over the range of 0 to 20 nM enzyme concentration (Figure S2). Alternatively, the monomeric form of the SARS-CoV-2 Mpro may in fact have significant protease activity.
Mpro FRET Assay
All Mpro protease assays were carried out in Reaction Buffer containing 20 mM HEPES pH 6.5, 120 mM NaCl, 0.4 mM EDTA and 3 mM TCEP. Additionally, 1 mg/mL BSA was added to the buffer for FRET assays. For the FRET assay, 10 nM Mpro was incubated with 20 μM of HCV drugs. The reaction was initiated by addition of 20 μM FRET substrate and monitored for 2 hours on an Infinite M1000 TECAN plate reader, exciting at 360 nm and detecting donor emission at 460 nm. The initial velocity of the reaction was calculated as the slope obtained from linear fits of emission intensity versus time plots for the first 15 minutes of the reaction. All FRET data were analyzed and plotted for initial velocity on Microsoft Excel.
In the FRET assays, the percent proteolytic activity in the presence of each drug was calculated as a ratio of initial velocity in presence of inhibitor (v i) to initial velocity in absence of inhibitor (v i0) i.e., v i / v i0. A histogram plot of v i / v i0 for each inhibitor was used to compare relative inhibition activities. All FRET proteolysis reaction curves were measured twice, and the uncertainty in v i / v i0, estimated from the standard deviation of 2 independent measurements is shown as error bars. Short (< 3 min) time points exhibiting equilibration artifacts (Figure S2A) were excluded from this analysis.
For IC50 measurements of HCV inhibitors BOC, NAR and TEL, 10 nM of Mpro was incubated with a range of inhibitor concentrations in the same Reaction Buffer described above. The inhibitor concentration ranges were 0.1 – 200 μM for BOC, 0.1 to 100 μM for NAR, and 0.2 to 100 μM for TEL. The reaction was initiated by adding 20 μM FRET substrate and monitored for 1 hr. Each measurement was repeated three times. The percent inhibition at each inhibitor concentration was calculated as:
where vi = initial velocity at a given inhibitor concentration
vi0 = initial velocity in absence of inhibitor
vimax = initial velocity at maximum inhibition
The percent inhibition was plotted as a function of inhibitor concentration to obtain a dose-response curve using Prism 8 (GraphPad Software) software. IC50 was calculated from fitting to the equation:
For several of the HCV drugs, intrinsic fluorescence of the drugs appears to compromise the accuracy of the FRET kinetic curves. Even though little or no inner filter effects were observed (Kasparek and Smyk, 2018) in these measurements, simple subtraction of the drug fluorescence from the final values of kinetic curves results in curves which do not match the final values for curves obtained in the absence of drugs, suggesting possible energy transfer between the drug and the FRET fluorophores on the peptide substrate.
The inner filter effect results from absorbance of the sample at the fluorescence excitation wavelength, attenuating fluorescence excitation (Kasparek and Smyk, 2018). Boceprevir, narleprevir, and telaprevir, which do not have significant intrinsic fluorescence but have similar extinction coefficients to the other drugs at 360 nm, did not present a problem for the assay, indicating that inner filter effects are not significant. The inner filter effect (Kasparek and Smyk, 2018) was experimentally assessed by measuring fluorescence and emission for the fluorescent dye diethylamino naphthalene sulfonate (DENS) over the range 0.01 to 1.0 O.D. units, at the excitation wavelength (360 nm), using the Infinite M1000 TECAN plate reader fluorimeter. These data show that in this system the inner filter effects are negligible for samples with total OD360 < 0.025, similar to results reported elsewhere (Kasparek and Smyk, 2018). As summarized in Table S3, with the exception of the vaniprevir study, these reaction mixtures had OD360 < 0.025. Hence, under the conditions of these assays, most of the assay samples have no significant inner filter effect; only the vaniprevir assay had a small effect, which was appropriately corrected for.
Mpro NMR Assay
For the Mpro 1H NMR proteolysis assay the reaction was performed at 100 nM Mpro in the same assay buffer described above, along with 5% D2O and 50 μM HCV inhibitors dissolved in d6-DMSO. For the control experiments where no inhibitor is added, the same quantity of d6-DMSO was added. 50 μM of unlabeled peptide substrate was added and immediately transferred to a 5-mm NMR tube. The final volume of each reaction mixture was 600 μL. The NMR tube was quickly placed in a 600 MHz Bruker Avance II spectrometer equipped with a 5-mm TCI cryoprobe, equilibrated at 298 K. The homogeneity of the magnetic field was adjusted by gradient shimming on the z axis, and in each case an array of 24 1H experiments was acquired with 1D 1H NMR using excitation sculpting for water suppression. The probe had previously been tuned and matched with a sample of similar composition. The delay between initiation of the reaction and starting acquisition was ∼5 mins for most of the reaction conditions. The duration of each NMR experiment was also taken into account to obtain accurate time values. All 1H spectra were acquired, processed, and analyzed in Bruker TopSpin 3.6.2 software. The regions of interest were integrated, and the values obtained were used for further analysis and plotting.
These 1H NMR spectra were used to monitor the evolution of substrate and product as a function of time. Resonance assignments (discussed below) of the cleaved and uncleaved KTSAVLQ/SGFRKME peptide identified the amide HN resonances that were monitored during the reaction. The HN resonances for amino-acid residues Phe-10 (uncleaved) and Gln-7 (cleaved) were used to quantify substrate utilization and product formation, respectively, during the reaction. The HN peak intensity of residue Glu-14, which did not shift upon cleavage, was monitored as an internal control. The percent substrate cleavage in the presence of inhibitors at 30 min was calculated as a ratio of the HN resonance integrals of Gln-7 in presence of inhibitor to the corresponding resonance integral with no inhibitor.
Amide 1H and 15N chemical shift assignments for Mpro peptide substrate
Chemical shift assignments of backbone amide 1H and 15N resonances in the 14-residue peptide KTSAVLQSGFRKME in 20 mM HEPES pH 6.5, 100 mM NaCl, 0.4 mM EDTA, 3 mM TCEP and 5% 2H2O were determined at 298 K using 2D COSY, TOCSY, and 1H-15N HSQC, along with 1D 1H NMR experiments, and referenced to internal DSS. Backbone amide 1H and 15N resonances were assigned for 12/14 and 10/14 residues in the uncleaved and cleaved peptide respectively. These assignments have been deposited in the BioMagResDB as BMRB entries 50568 and 50569, respectively.
PLpro proteolysis inhibition assay
SARS-CoV-2 PLpro enzyme was provided as a generous gift by Prof. John Hunt (Columbia University). The construct, residues 1564 to 1881 of the SARS-CoV-2 replicase polyprotein 1a (Uniprot id P0DTD1 (R1A_SARS2)), was produced in expression vector pET21_NESG with a C-terminal purification tag LEHHHHHH. Homogeneity (> 95%) was validated by SDS-PAGE. The fluorogenic substrate zRLRGG/AMC was obtained from Bachem. All PLpro proteolysis assays were carried out in buffer containing 50 mM HEPES, pH 7.5, 5 mM DTT, 1 mg/ml BSA. For the fluorescence assay, 20 nM of PLpro was incubated with 20 μM of HCV drugs. 20 μM substrate was added and the reaction was monitored for 2 hr using the Infinite M1000 TECAN plate reader with Magellan ™ software, with filters for excitation at 360 nm and emission at 460 nm. No anomalous fluorescence interactions were observed in this assay for any of the drugs. We also assessed the PLpro fluorescence assay for inner filter effects. As shown in Table S3, inner filter effects were also not significant in this assay, except for the vaniprevir study for which appropriate corrections were made.
The data points for the first 10 mins of the proteolysis reaction progression curves were used to calculate the initial velocity (v i) in the presence and absence of the inhibitor. The percent proteolytic activity in presence of each drug is calculate as a ratio of initial velocity in presence of inhibitor (v i) to initial velocity in absence of inhibitor (v i0), i.e., v i / v i0. A histogram plot of v i / v i0 for each inhibitor was used to compare relative inhibition activities. All proteolysis reaction curves were measured twice, and the standard deviation (s.d.) in v i / v i0 is shown as error bars.
Cells and viruses
Vero E6 (ATCC, CRL-1586) and 293T cells (ATCC, CRL-3216; kind gift of Dr. Viviana Simon), were maintained in DMEM (Corning) supplemented with 10% FB (Peak Serum) and penicillin/streptomycin (Corning) at 37°C and 5% CO2. hACE2-293T cells were generated for this study. Briefly, 293T cells were transduced with a lentiviral vector expressing human ACE2. Puromycin resistant cells with hACE2 surface expression were sorted after staining with AlexaFluor 647-conjugated goat anti-hACE2 antibodies. Cells were then single-cell-cloned and screened for their ability to support SARS-CoV-2 replication. All cell lines used in this study were regularly screened for mycoplasma contamination using the Universal Detection Kit (ATCC, 30-1012K). Cells were infected with SARS-CoV-2, isolate USA-WA1/2020 (BEI Resources NR-52281) under biosafety level 3 (BSL3) containment in accordance to the biosafety protocols developed by the Icahn School of Medicine at Mount Sinai. Viral stocks were grown in Vero E6 cells as previously described (Amanat et al., 2020), and were validated by genome sequencing.
Viral growth and cytotoxicity assays in the presence of inhibitors
2,000 Vero E6 or hACE2-293T cells were seeded into 96-well plates in DMEM (10% FBS) and incubated for 24 h at 37C, 5% CO2. Two hours before infection, the medium was replaced with 100 μL of DMEM (2% FBS) containing the compound of interest at concentrations 50% greater than those indicated, including a DMSO control. Plates were then transferred into the BSL3 facility and 100 PFU (MOI = 0.025) was added in 50 μL of DMEM (2% FBS), bringing the final compound concentration to those indicated. Plates were then incubated for 48 h at 37°C. After infection, supernatants were removed and cells were fixed with 4% formaldehyde for 24 hours prior to being removed from the BSL3 facility. The cells were then immunostained for the viral NP protein [an in-house mAb 1C7, provided by Dr. Thomas Moran (MSSM)] with a DAPI counterstain. Infected cells (488 nM) and total cells (DAPI) were quantified using the Celigo (Nexcelcom) imaging cytometer. Infectivity was measured by the accumulation of viral NP protein in the nucleus of the Vero E6 cells (fluorescence accumulation). Percent infection was quantified as
and the DMSO control was then set to 100% infection for analysis. The IC50 and IC90 for each experiment were determined using the Prism (GraphPad Software) software. Cytotoxicity was also performed using the MTT assay (Roche), according to the manufacturer’s instructions. Cytotoxicity was performed in uninfected VeroE6 cells with same compound dilutions and concurrent with viral replication assay. All assays were performed in biologically independent triplicates. Remdesivir was purchased from Medkoo Bioscience inc. Time of addition experiments were performed using the same immunofluorescence-based assay with the following alterations: Vero E6 cells were infected with 8000 PFU (moi of 2) of SARS-CoV-2, the drug was added at different times relative to infection as indicated, and the infection was ended by fixation with 4% formaldehyde after 8 hours of infection (single cycle assay).
Antiviral combination assay
Like the previous antiviral assay, 2,000 Vero E6 cells were seeded into 96-well plates in DMEM (10% FBS) and incubated for 24 h at 37°C, 5% CO2. Two hours before infection, the medium was replaced with 100 μL of DMEM (2% FBS) containing the combination of HCV protease inhibitors and remdesivir following a dilution combination matrix. A 6 by 6 matrix of drug combinations was prepared in triplicate by making serial two-fold dilutions of the drugs on each axis, including a DMSO control column and row. The resulting matrix had no drug in the right upper well, a single drug in rising 2-fold concentrations in the vertical and horizontal axes starting from that well, and the remaining wells with rising concentrations of drug mixtures reaching maximum concentrations of the drugs at the lower left well. Plates were then transferred into the BSL3 facility and SARS-CoV-2 (MOI 0.025) was added in 50 μL of DMEM (2% FBS), bringing the final compound concentration to those indicated in the figures. Plates were then incubated for 48 h at 37°C. After infection, cells were fixed with final concentration of 5% formaldehyde for 24 hours prior to being removed from the BSL3 facility. The cells were then immunostained for the viral NP protein using the in-house mAb 1C7 provided by Dr. Thomas Moran, MSSM) with a DAPI counterstain. Infected cells (AlexaFluor 488) and total cells (DAPI) were quantified using the Celigo (Nexcelcom) imaging cytometer. Infectivity was measured by the accumulation of viral NP protein in the nucleus of the Vero E6 cells (fluorescence accumulation). Percent infection was quantified as [(Infected cells/Total cells) – Background] ∗100, and the DMSO control was then set to 100% infection for analysis. The combination antiviral assay was performed in biologically independent triplicates.
The apparent IC90 for each combination in the matrix was determined using the Prism (GraphPad Software) software. The IC90 for HCV drugs and remdesivir were calculated for each drug treatment alone and in combination. This combination data were analyzed using SynergyFinder by the ZIP method (Ianevski et al., 2020), and combination indices were calculated as previously described (Amanat et al., 2020).
Quantification and statistical analysis
Viral replication measurements were each done in triplicate (n = 3), and reported as mean ± s.d. Fluorescence measurements were done in triplicate (n = 3) or duplicate (n = 2) and reported as mean ± s.d. Estimates of uncertainties in NMR intensity measurements were determined from the spectral noise, and propagated to uncertainties in peak ratios ΔR as: (ΔR/R)2 = (ΔA/A)2 + (ΔB/B)2, where A, B, and R are the intensities of peaks A and B and their ratio, respectively, and ΔA and ΔB are the noise associated with each intensity measurement (https://nmr.chem.ucsb.edu/protocols/SNR.html).
Acknowledgments
We thank Drs. Y.P. Huang, G. Liu, L. Ma, S. Shukla, G.V.T. Swapna, and R. Xiao for helpful discussions and comments on the manuscript. We also thank J. Hunt and S. Krishna for a generous gift of SARS-CoV-2 PLpro enzyme and R. Albrecht for support with the BSL3 facility at the Icahn School of Medicine at Mount Sinai. This research was supported by grants from the National Institutes of Health (R01-GM120574 to G.T.M.) and RPI Center for Computational Innovations (to K.B. and G.T.M.). This research was also partly funded by CRIP (Center for Research for Influenza Pathogenesis), a NIAID-supported Center of Excellence for Influenza Research and Surveillance (HHSN272201400008C), DARPA grant HR0011-19-2-0020, NIAID grants U19AI142733 and U19AI135972, DOD grant W81XWH-20-1-0270, and by the generous support of the JPB Foundation, the Open Philanthropy Project (2020-215611 [5384)), and anonymous donors to A.G.-S.
Author contributions
Conceptualization, K.B., K.W., B.H., C.A.R., A.G.-S., R.M.K., and G.T.M.; methodology, K.B., K.W., B.H., T.B.A., T.R., R.R., C.A.R., A.G.-S., R.M.K., and G.T.M.; validation, K.B., K.W., B.H., C.A.R., A.G.-S., R.M.K., and G.T.M.; formal analysis, K.B., K.W., B.H., T.R., C.A.R., A.G.-S., R.M.K., and G.T.M.; investigation, K.B., K.W., B.H., R.R., T.R., E.M., T.K., and L.M.; resources, C.A.R., A.G.-S., and G.T.M.; writing – original draft, K.B., K.W., A.G.-S., R.M.K., and G.T.M.; writing, review, and editing, K.B., K.W., B.H., C.A.R., A.G.-S., R.M.K., and G.T.M.; visualization, K.B., K.W., B.H., C.A.R., A.G.-S., R.M.K., and G.T.M.; supervision, K.W., C.A.R., A.G.-S., and G.T.M.; funding acquisition, C.A.R., A.G.-S., and G.T.M.
Declaration of interests
A provisional patent application related to these studies has been filed. G.T.M. is a founder of Nexomics Biosciences, Inc. The A.G.-S. laboratory has received research support from Pfizer, Senhwa Biosciences, 7Hills Pharma, Avimex, Blade Therapeutics, Dynavax, ImmunityBio, Nanocomposix, Pharmamar, and Kenall Manufacturing, and A.G.-S. has consulting agreements for the following companies involving cash and/or stock: Vivaldi Biosciences, Pagoda, Contrafect, 7Hills Pharma, Avimex, Vaxalto, Accurius, Pfizer, and Esperovax. These relationships have no conflict of interest with respect to this study.
Published: May 18, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109133.
Supplemental information
References
- Amanat F., White K.M., Miorin L., Strohmeier S., McMahon M., Meade P., Liu W.C., Albrecht R.A., Simon V., Martinez-Sobrido L., et al. An In Vitro Microneutralization Assay for SARS-CoV-2 Serology and Drug Screening. Curr. Protoc. Microbiol. 2020;58:e108. doi: 10.1002/cpmc.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K., Palm G.J., Mesters J.R., Siddell S.G., Ziebuhr J., Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelini M.M., Akhlaghpour M., Neuman B.W., Buchmeier M.J. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. MBio. 2013;4:e00524-13. doi: 10.1128/mBio.00524-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anson B., Mesecar A. 2020. X-ray structure of SARS-CoV-2 main protease bound to boceprevir at 1.45 A. PDB ID 6WNP. Published online April 23, 2020. [DOI] [Google Scholar]
- Anson B., Chapman M., Lendy E., Pshenychnyi S., Richard T., Satchell K., Mesecar D.A. 2020. Broad-spectrum inhibition of coronavirus main and papain-like proteases by HCV drugs. Published online May 1, 2020. [DOI] [Google Scholar]
- Bafna K., Krug R.M., Montelione G.T. 2020. Structural similarity of SARS-CoV2 M(pro) and HCV NS3/4A proteases suggests new approaches for identifying existing drugs useful as COVID-19 therapeutics. Published online April 21, 2020. [DOI] [Google Scholar]
- Boras B., Jones R.M., Anson B.J., Arenson D., Aschenbrenner L., Bakowski M.A., Beutler N., Binder J., Chen E., Eng H., et al. [DOI]
- Daczkowski C.M., Dzimianski J.V., Clasman J.R., Goodwin O., Mesecar A.D., Pegan S.D. Structural Insights into the Interaction of Coronavirus Papain-Like Proteases and Interferon-Stimulated Gene Product 15 from Different Species. J. Mol. Biol. 2017;429:1661–1683. doi: 10.1016/j.jmb.2017.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W.L. Schrödinger, LLC; 2009. The PyMOL Molecular Graphics System, Version 1.2r3pre. [Google Scholar]
- Eastman R.T., Roth J.S., Brimacombe K.R., Simeonov A., Shen M., Patnaik S., Hall M.D. Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS Cent. Sci. 2020;6:672–683. doi: 10.1021/acscentsci.0c00489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forli S., Huey R., Pique M.E., Sanner M.F., Goodsell D.S., Olson A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016;11:905–919. doi: 10.1038/nprot.2016.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z., Huang H. 2020. SARS CoV-2 PLpro in complex with GRL0617. PDB: 7CJM. [DOI] [Google Scholar]
- Fu L., Ye F., Feng Y., Yu F., Wang Q., Wu Y., Zhao C., Sun H., Huang B., Niu P., et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020;11:4417. doi: 10.1038/s41467-020-18233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbalenya A.E., Koonin E.V., Donchenko A.P., Blinov V.M. Coronavirus genome: prediction of putative functional domains in the non-structural polyprotein by comparative amino acid sequence analysis. Nucleic Acids Res. 1989;17:4847–4861. doi: 10.1093/nar/17.12.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O’Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L., et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosert R., Kanjanahaluethai A., Egger D., Bienz K., Baker S.C. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 2002;76:3697–3708. doi: 10.1128/JVI.76.8.3697-3708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grum-Tokars V., Ratia K., Begaye A., Baker S.C., Mesecar A.D. Evaluating the 3C-like protease activity of SARS-Coronavirus: recommendations for standardized assays for drug discovery. Virus Res. 2008;133:63–73. doi: 10.1016/j.virusres.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harcourt B.H., Jukneliene D., Kanjanahaluethai A., Bechill J., Severson K.M., Smith C.M., Rota P.A., Baker S.C. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J. Virol. 2004;78:13600–13612. doi: 10.1128/JVI.78.24.13600-13612.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L., Sander C. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- Holm L., Sander C. Protein folds and families: sequence and structure alignments. Nucleic Acids Res. 1999;27:244–247. doi: 10.1093/nar/27.1.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung H.C., Ke Y.Y., Huang S.Y., Huang P.N., Kung Y.A., Chang T.Y., Yen K.J., Peng T.T., Chang S.E., Huang C.T., et al. Discovery of M Protease Inhibitors Encoded by SARS-CoV-2. Antimicrob. Agents Chemother. 2020;64:e00872-20. doi: 10.1128/AAC.00872-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianevski A., Giri A.K., Aittokallio T. SynergyFinder 2.0: visual analytics of multi-drug combination synergies. Nucleic Acids Res. 2020;48(W1):W488–W493. doi: 10.1093/nar/gkaa216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Kasparek A., Smyk B. A new approach to the old problem: Inner filter effect type I and II in fluorescence. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2018;198:297–303. doi: 10.1016/j.saa.2018.03.027. [DOI] [PubMed] [Google Scholar]
- Kneller D.W., Galanie S., Phillips G., O’Neill H.M., Coates L., Kovalevsky A. Malleability of the SARS-CoV-2 3CL Mpro Active-Site Cavity Facilitates Binding of Clinical Antivirals. Structure. 2020;28:1313–1320. doi: 10.1016/j.str.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneller D.W., Phillips G., O’Neill H.M., Tan K., Joachimiak A., Coates L., Kovalevsky A. Room-temperature X-ray crystallography reveals the oxidation and reactivity of cysteine residues in SARS-CoV-2 3CL Mpro: insights into enzyme mechanism and drug design. IUCrJ. 2020;7:1028–1035. doi: 10.1107/S2052252520012634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb P., Irwin J.J. Docking screens: right for the right reasons? Curr. Top. Med. Chem. 2009;9:755–770. doi: 10.2174/156802609789207091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb P., Ferreira R.S., Irwin J.J., Shoichet B.K. Docking and chemoinformatic screens for new ligands and targets. Curr. Opin. Biotechnol. 2009;20:429–436. doi: 10.1016/j.copbio.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner H.A., Lytvyn V., Qi H., Lachance P., Ziomek E., Ménard R. Selectivity in ISG15 and ubiquitin recognition by the SARS coronavirus papain-like protease. Arch. Biochem. Biophys. 2007;466:8–14. doi: 10.1016/j.abb.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo H.S., Hui K.P.Y., Lai H.-M., Khan K.S., Kaur S., Huang J., Li Z., Chan A.K.N., Cheung H.H.-Y., Ng K.-C., et al. Simeprevir potently suppresses SARS-CoV-2 replication and synergizes with remdesivir. ACS Cent. Sci. 2021 doi: 10.1021/acscentsci.0c01186. Published online April 15, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C., Sacco M.D., Hurst B., Townsend J.A., Hu Y., Szeto T., Zhang X., Tarbet B., Marty M.T., Chen Y., Wang J. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30:678–692. doi: 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGonagle D., Sharif K., O’Regan A., Bridgewood C. The Role of Cytokines including Interleukin-6 in COVID-19 induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun. Rev. 2020;19:102537. doi: 10.1016/j.autrev.2020.102537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G.M., Goodsell D.S., Halliday R.S., Huey R., Hart W.E., Belew R.K., Olson A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998;19:1639–1662. [Google Scholar]
- Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipiuk J., Azizi S.A., Dvorkin S., Endres M., Jedrzejczak R., Jones K.A., Kang S., Kathayat R.S., Kim Y., Lisnyak V.G., et al. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021;12:743. doi: 10.1038/s41467-021-21060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oudshoorn D., Rijs K., Limpens R.W.A.L., Groen K., Koster A.J., Snijder E.J., Kikkert M., Bárcena M. Expression and Cleavage of Middle East Respiratory Syndrome Coronavirus nsp3-4 Polyprotein Induce the Formation of Double-Membrane Vesicles That Mimic Those Associated with Coronaviral RNA Replication. MBio. 2017;8:e01658-17. doi: 10.1128/mBio.01658-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özen A., Prachanronarong K., Matthew A.N., Soumana D.I., Schiffer C.A. Resistance outside the substrate envelope: hepatitis C NS3/4A protease inhibitors. Crit. Rev. Biochem. Mol. Biol. 2019;54:11–26. doi: 10.1080/10409238.2019.1568962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H., Peto R., Abdool Karim Q., Alejandria M., Henao-Restrepo A.M., Hernández García C., Kieny M.-P., Malekzadeh R., Murthy S., Preziosi M.-P., et al. Repurposed antiviral drugs for COVID-19 – interim WHO SOLIDARITY trial results. MedRciv. 2020 doi: 10.1101/2020.10.15.20209817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Q., Peng R., Yuan B., Zhao J., Wang M., Wang X., Wang Q., Sun Y., Fan Z., Qi J., et al. Structural and Biochemical Characterization of the nsp12-nsp7-nsp8 Core Polymerase Complex from SARS-CoV-2. Cell Rep. 2020;31:107774. doi: 10.1016/j.celrep.2020.107774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prongay A.J., Guo Z., Yao N., Pichardo J., Fischmann T., Strickland C., Myers J., Jr., Weber P.C., Beyer B.M., Ingram R., et al. Discovery of the HCV NS3/4A protease inhibitor (1R,5S)-N-[3-amino-1-(cyclobutylmethyl)-2,3-dioxopropyl]-3- [2(S)-[[[(1,1-dimethylethyl)amino]carbonyl]amino]-3,3-dimethyl-1-oxobutyl]- 6,6-dimethyl-3-azabicyclo[3.1.0]hexan-2(S)-carboxamide (Sch 503034) II. Key steps in structure-based optimization. J. Med. Chem. 2007;50:2310–2318. doi: 10.1021/jm060173k. [DOI] [PubMed] [Google Scholar]
- Ratia K., Pegan S., Takayama J., Sleeman K., Coughlin M., Baliji S., Chaudhuri R., Fu W., Prabhakar B.S., Johnson M.E., et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. USA. 2008;105:16119–16124. doi: 10.1073/pnas.0805240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco M.D., Ma C., Lagarias P., Gao A., Townsend J.A., Meng X., Dube P., Zhang X., Hu Y., Kitamura N., et al. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against Mpro and cathepsin L. Sci. Adv. 2020;6:eabe0751. doi: 10.1126/sciadv.abe0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salentin S., Schreiber S., Haupt V.J., Adasme M.F., Schroeder M. PLIP: fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015;43(W1):W443–W447. doi: 10.1093/nar/gkv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanner M.F. Python: a programming language for software integration and development. J. Mol. Graph. Model. 1999;17:57–61. [PubMed] [Google Scholar]
- Sheahan T.P., Sims A.C., Zhou S., Graham R.L., Pruijssers A.J., Agostini M.L., Leist S.R., Schäfer A., Dinnon K.H., 3rd, Stevens L.J., et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Transl. Med. 2020;12:eabb5883. doi: 10.1126/scitranslmed.abb5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Limpens R.W.A.L., de Wilde A.H., de Jong A.W.M., Zevenhoven-Dobbe J.C., Maier H.J., Faas F.F.G.A., Koster A.J., Bárcena M. A unifying structural and functional model of the coronavirus replication organelle: Tracking down RNA synthesis. PLoS Biol. 2020;18:e3000715. doi: 10.1371/journal.pbio.3000715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaim C.D., Scott A.F., Canadeo L.A., Huibregtse J.M. Extracellular ISG15 Signals Cytokine Secretion through the LFA-1 Integrin Receptor. Mol. Cell. 2017;68:581–590. doi: 10.1016/j.molcel.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaim C.D., Canadeo L.A., Monte K.J., Khanna S., Lenschow D.J., Huibregtse J.M. Modulation of Extracellular ISG15 Signaling by Pathogens and Viral Effector Proteins. Cell Rep. 2020;31:107772. doi: 10.1016/j.celrep.2020.107772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff G., Limpens R.W.A.L., Zevenhoven-Dobbe J.C., Laugks U., Zheng S., de Jong A.W.M., Koning R.I., Agard D.A., Grünewald K., Koster A.J., et al. A molecular pore spans the double membrane of the coronavirus replication organelle. Science. 2020;369:1395–1398. doi: 10.1126/science.abd3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff G., Melia C.E., Snijder E.J., Bárcena M. Double-Membrane Vesicles as Platforms for Viral Replication. Trends Microbiol. 2020;28:1022–1033. doi: 10.1016/j.tim.2020.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Lin D., Kusov Y., Nian Y., Ma Q., Wang J., von Brunn A., Leyssen P., Lanko K., Neyts J., et al. α-Ketoamides as Broad-Spectrum Inhibitors of Coronavirus and Enterovirus Replication: Structure-Based Design, Synthesis, and Activity Assessment. J. Med. Chem. 2020;63:4562–4578. doi: 10.1021/acs.jmedchem.9b01828. [DOI] [PubMed] [Google Scholar]
- Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C., Sridharan H., Chen R., Baker D.P., Wang S., Krug R.M. Influenza B virus non-structural protein 1 counteracts ISG15 antiviral activity by sequestering ISGylated viral proteins. Nat. Commun. 2016;7:12754. doi: 10.1038/ncomms12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The following datasets generated during this study are available at the BioMagResDB:
BioMagResDB accession number: 50568. Title: “1H and 15N assignments for 14-residues peptide that is cleaved by SARS-CoV-2 Mpro.” DOI: 10.13018/BMR50568
BioMagResDB accession number: 50569. Title: “1H and 15N assignments for 14-residue peptide after cleavage by SARS-CoV-2 Mpro.” DOI: 10.13018/BMR50569