

Abstract
Benign familial epilepsies that present themselves in the first year of life include benign familial neonatal epilepsy (BFNE), benign familial neonatal-infantile epilepsy (BFNIE) and benign familial infantile epilepsy (BFIE). We used Sanger sequencing and targeted next-generation sequencing to detect gene mutations in a Chinese cohort of patients with these three disorders. A total of 79 families were collected, including 4 BFNE, 7 BFNIE, and 68 BFIE. Genetic testing led to the identification of gene mutations in 60 families (60 out of 79, 75.9%). A total of 42 families had PRRT2 mutations, 9 had KCNQ2 mutations, 8 had SCN2A mutations, and 1 had a GABRA6 mutation. In total three of four BFNE families were detected with KCNQ2 mutations. Mutations were detected in all BFNIE families, including 3 KCNQ2 mutations, 3 SCN2A mutations, and 1 PRRT2 mutation. Gene mutations were identified in 50 out of 68 BFIE families (73.5%), including 41 PRRT2 mutations (41 out of 68, 60.3%), 5 SCN2A mutations, 3 KCNQ2 mutations, and 1 GABRA6 mutation. Our results confirmed that mutations in KCNQ2, SCN2A, and PRRT2 are major genetic causes of benign familial epilepsy in the first year of life in the Chinese population. KCNQ2 is the major gene related to BFNE. PRRT2 is the main gene responsible for BFIE.
Subject terms: Genetics research, Epilepsy
Introduction
Benign familial epilepsy with onset in the first year of life is an autosomal dominant form of genetic epilepsy characterized by unprovoked partial or secondary generalized seizures in neonates or infants with remission at a young age, and a generally benign outcome [1, 2]. All patients have a family history of early onset seizure. Three epileptic syndromes have been delineated so far, including benign familial neonatal epilepsy (BFNE: OMIM 121200), benign familial neonatal-infantile epilepsy (BFNIE: OMIM 607745) and benign familial infantile epilepsy (BFIE: OMIM 605751). These conditions are differentiated mainly for the age of onset. BFNE usually starts in neonatal period, typically before 5 days of life [3]. The seizure onset age is usually from 2 days to 3.5 months of age in BFNIE [4]. BFIE tend to occur between 3 and 20 months of onset age, mostly between 4 and 7 months [5]. During childhood or adolescence, family members in BFIE may develop paroxysmal kinesigenic dyskinesias (PKD). This clinical subtype of BFIE is known as infantile convulsions with paroxysmal choreoathetosis syndrome [6] (ICCA: OMIM 602066).
Defects in the genes encoding the voltage-gated potassium channel subunits KCNQ2 and KCNQ3 are responsible for BFNE [7, 8]. The voltage-gated sodium channel gene SCN2A was identified as the major causative gene for BFNIE [9, 10]. Recently studies also led to the identification of mutations in the proline-rich transmembrane protein 2 gene PRRT2 in BFIE [11, 12]. However, for some families, the causative genes have not been identified. Although these three disorders show a significant clinical overlap, a comprehensive mutational screening of the candidate genes on a large set of families has been not yet explored in the Chinese population. In this study, we performed genetic analysis of families diagnosed as BFNE, BFNIE, and BFIE in the Chinese Han population, and to explore genetic background behind them.
Materials and methods
Patients
This study was approved by the Ethics Committee of Peking University First Hospital. Written informed consent for the analysis and publication of clinical and genetic details was obtained from the patients or their parents. We recruited families with at least two family members affected by focal seizures starting within the first year of life at Peking University First Hospital from September 2006 to November 2016. Clinical information about age of seizure onset, seizure types, developmental milestones, and neurologic status of the patients and their relatives was collected using a pre-test questionnaire completed by the recruiting clinician by telephone or from medical records. Patients were followed up at a pediatric neurology clinic at our hospital or by telephone.
The diagnostic criterias for benign familial epilepsy were as follows [1, 2]: (1) unprovoked focal seizures with or without secondary generalization, usually manifesting motor arrest, deviation of the head and eyes to one side, generalized hypertonia, cyanosis, and limb jerks; (2) normal interictal electroencephalography; (3) normal brain imaging; (4) no underlying disorders or neurological disabilities; (5) normal psychomotor development before, during and after the onset of seizures; (6) family history of seizures (similar age at onset); (7) good response to treatment and, in many cases, cessation of seizures before the age of 2 years.
Seizures with onset in the first 28 days of life were defined as neonatal, and from the second month to 1 year, they were defined as infantile. Families were classified as BFNE, when all affected family members experienced neonatal seizures, BFNIE when the onset of seizures in family members was between 1 and 3 months of age or showed both neonatal and infantile seizures, and BFIE when all family members showed infantile seizures and at least one experienced seizure onset beyond 3 months of age. BFIE families with affected individuals showing PKD later in life were subclassified as ICCA.
Genetic analysis
DNA isolation
Blood samples were obtained from these probands and their family members when possible. Genomic DNA was extracted from peripheral blood by a standard method.
PRRT2 analysis
Mutation screening of PRRT2 was performed by using the polymerase chain reaction (PCR) and Sanger sequencing. The method has been described in our previous study [13]. Mutation found in a proband was examined for co-segregation in other family members.
Targeted next-generation sequencing
PRRT2 mutation negative probands were further screened for pathogenic mutations through a custom-designed gene panel in which a total of 149 candidate genes associated with epilepsy were selected as the genes of interest (Supplementary Appendix 1).
The target regions included all exons and the adjacent +50 bp and −50 bp segments of the introns of the selected genes. The DNA of peripheral blood was fragmented, and libraries were prepared following recommended protocols of Illumina; The library DNA was captured by using a GenCap exome capture kit (MyGenostics). The capture experiment was conducted according to the manufacturer’s protocol. Enriched capture libraries were sequenced on a HiSeq X-ten or HiSeq 2500 platform (Illumina) for 150 bp pair-end sequencing. The average sequencing depth on the target regions was ×200, and the coverage of the target regions (introns and exons) was 99.8%.
Raw sequencing data were saved in FASTQ format, then analyzed using the following bioinformatics workflow: First, Illumina sequencing adapters and low quality reads (<80 bp) were filtered by using fastq_mcf. After quality control, the clean reads were mapped to the UCSC hg19 human reference genome using BWA. Duplicated reads were removed using Picard Tools, and only uniquely mapping reads were used for variation detection. Second, SNP and indel variants were detected by using the GATK HaplotypeCaller. Then, the GATK VariantFiltration tool was used to filter the variants. The filtered standards were described as follows: (a) variants with mapping qualities <30; (b) the Total Mapping Quality Zero Reads < 4; (c) approximate read depth <5; (d) QUAL <50.0; (e) phred-scaled p-value using Fisher’s exact test to detect strand bias >10.0. After these two steps, the data were transformed to VCF format, and variants were further annotated by ANNOVAR and associated with multiple databases, such as the 1000 genomes project, ESP6500, dbSNP, EXAC, Inhouse (MyGenostics), and HGMD. Variant functions were predicted by SIFT, PolyPhen-2, MutationTaster, and GERP++. The related software and database are available in Supplementary Appendix 2.
Five steps were used to select potential pathogenic mutations in the downstream analysis: (a) Mutation reads should be >5, and the fraction of mutant alleles should be no <30%; (b) Common variants with alternative allele frequencies greater than 5% in the 1000 Genomes, ESP6500, or Inhouse database were removed; (c) Variants present in the InNormal database (MyGenostics) were removed; (d) Synonymous variants were labeled. (e) After (a), (b), and (c), only synonymous variants reported in HGMD were kept. After the above filters, the remaining variants were considered to be potential pathogenic mutations.
Copy number variants (CNVs) were also detected by using the next-generation sequencing capture panel. Copy numbers were first determined from the clean bam files. Then, the copy numbers were corrected for GC content and normalized according to population information. CNV candidates satisfying the combined statistical tests were annotated by using information from OMIM, GeneReviewer, Decipher, and ClinVar. CNV candidates were also filtered by using the DGV and MyGenostics control databases. CNV candidates reported in Decipher and those overlapping with the Decipher database were determined to be pathogenic CNVs. CNV candidates annotated as pathogenic or likely pathogenic in dbVAR, and those covering functionally related genes, were determined to be likely pathogenic. Other CNVs were uncertain. The related software and database are also available in Supplementary Appendix 2.
The potential pathogenic variations and CNVs suggested by the targeted next-generation sequencing were validated using Sanger sequencing, multiplex ligation-dependent probe amplification (MLPA), or Real-time Quantitative PCR (qPCR). The DNA of other family members was only analyzed for potentially pathogenic variants and CNVs. Next, we performed segregation analysis in these families. For novel variants, we analyzed the variant in 104 healthy Chinese controls. Variations and CNVs that showed evidence of cosegregation with the families’ phenotypes or fulfilled genetic models were considered very likely to be causal.
Results
Clinical findings
A total of 79 unrelated families with benign familial epilepsies in the first year of life were collected. Of the 79 families, 255 family members were affected, with a range of 2–19 affected individuals per family (average 3.2). Seizure onset beyond 1 year of age in some affected members was found in 3 families (family 37, 47, and 58). The latest onset age of seizure in these 3 families were 13 months, 3 years and 14 months, respectively. Of the 79 families, 4 were classified as BFNE, 7 as BFNIE, and 68 as BFIE. A total of 15 out of 68 BFIE families were subclassified as ICCA. Family pedigrees of all 79 families are shown in Supplementary Appendix 3.
Genetic analysis
Gene mutations were found in 60 of 79 families (75.9%) . Of the mutations found, 42 affected PRRT2, 9 affected KCNQ2, 8 affected SCN2A, and one affected GABRA6. In total 14 mutations were novel (14/60, 23.3%). Sequence chromatograms of novel mutations are shown in Fig. 1a.
Fig. 1.

Sequence chromatograms. a Sequence chromatograms showing the 14 novel gene mutations detected in families with benign familial epilepsies in the first year of life, compared with wild-type traces. b Sequence chromatograms of family 50. The proband was found with a compound heterozygous mutation of PRRT2 which consists of c.649dupC inherited from his father and c.593_594delCT from his mother. The arrow shows the position of the mutation
In the 4 BFNE families, causative mutations were only found in KCNQ2, which was identified as the affected gene in 3 of the families. All 7 BFNIE families had identifiable gene mutations, PRRT2 was found in one family, KCNQ2 in 3 families, and SCN2A in 3 families. In the 68 BFIE families, gene mutations were identified in 50 families (50 out of 68, 73.5%), with PRRT2 mutations found in 41 families (41 out of 68, 60.3%), SCN2A mutations found in 5 families, KCNQ2 mutations found in 3 families, and a GABRA6 mutation found in one family. In total 14 of 15 ICCA families were found to have PRRT2 mutations (14 out of 15, 93.3%). The remaining ICCA family was not detected with any pathogenic mutation. The detection rates of PRRT2, KCNQ2, SCN2A, and GABRA6 mutations in families with BFNE, BFNIE, and BFIE are shown in Fig. 2. The clinical features and genetic testing results of all 79 families are summarized in Table 1.
Fig. 2.
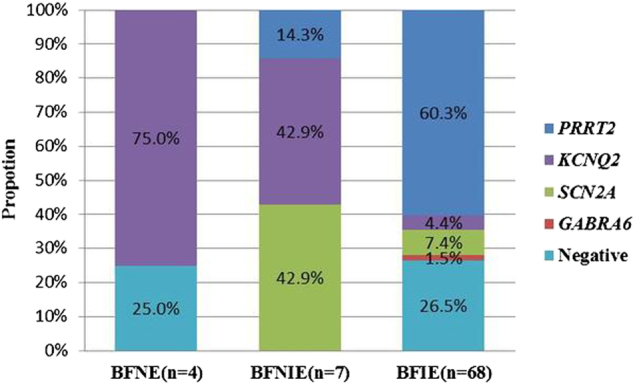
Detection rates of PRRT2, KCNQ2, SCN2A, and GABRA6 mutations in families with BFNE, BFNIE, and BFIE
Table 1.
The clinical features and genetic testing results of 79 families with benign familial epilepsies with onset in the first year of life
| Family ID | Affected members | Age range at seizure oneset | Diagnosis | Gene | Transcript | Mutation | Reported/Novel |
|---|---|---|---|---|---|---|---|
| 1 | 2 | 1–4 d | BFNE | KCNQ2 | NM_172107.2 | c.1048A>C[p.Asn350His] | Novel |
| 2 | 3 | 2–3 d | BFNE | KCNQ2 | NM_172107.2 | c.242T>C[p.Leu81Pro] | Novel |
| 3 | 3 | 3 d | BFNE | KCNQ2 | NM_172107.2 | c.2506G>T[p.Glu836*] | Novel |
| 4 | 2 | 1–2 d | BFNE | ND | — | — | — |
| 5 | 3 | 25 d–2.5 m | BFNIE | KCNQ2 | NM_172107.2 | c.958G>A[p.Val320Ile] | Novel |
| 6 | 8 | 2 d–6 m | BFNIE | KCNQ2 | NM_172107.2 | c.998G>A[p.Arg333Gln] | Reported |
| 7 | 2 | 5 d–3.5 m | BFNIE | KCNQ2 and CHRNA4 | NM_172107.2 | Deletion | Reported |
| NM_000744.6 | |||||||
| 8 | 2 | 6 d–2 m | BFNIE | SCN2A | NM_001040142.1 | c.2674G>A[p.Val892Ile] | Reported |
| 9 | 6 | 3 d–3 m | BFNIE | SCN2A | NM_001040142.1 | c.2872A>G[p.Met958Val] | Novel |
| 10 | 6 | 2.5–3 m | BFNIE | SCN2A | NM_001040142.1 | c.2627A>G[p.Asn876Ser] | Reported |
| 11 | 2 | 2–2.5 m | BFNIE | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 12 | 2 | 1–3.5 m | BFIE | PRRT2 | NM_145239.2 | c.649delC[p.Arg217Glufs*12] | Reported |
| 13 | 4 | 3–6 m | BFIE | KCNQ2 | NM_172107.2 | c.775G>A[p.Asp259Asn] | Reported |
| 14 | 19 | 4.5–12 m | BFIE | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 15 | 3 | 4–5 m | BFIE | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 16 | 3 | 2–6 m | BFIE | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 17 | 3 | 3–6 m | BFIE | PRRT2 | NM_145239.2 | c.904dupG[p.Asp302Glyfs*39] | Reported |
| 18 | 5 | 4.5–6 m | BFIE | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 19 | 3 | 3–12 m | BFIE | PRRT2 | NM_145239.2 | c.649delC[p.Arg217Glufs*12] | Reported |
| 20 | 3 | 3–12 m | BFIE | PRRT2 | NM_145239.2 | c.514_517delTCTG[p.Ser172Argfs*3] | Reported |
| 21 | 2 | 4.5–6 m | BFIE | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 22 | 2 | 6–7 m | BFIE | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 23 | 10 | 4–6 m | BFIE | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 24 | 2 | 3.5 m | BFIE | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 25 | 2 | 4–4.5 m | BFIE | PRRT2 | NM_145239.2 | c.649delC[p.Arg217Glufs*12] | Reported |
| 26 | 8 | 2–12 m | BFIE | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 27 | 3 | 4–4.5 m | BFIE | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 28 | 3 | 5–8 m | BFIE | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 29 | 2 | 3.5–4.5 m | BFIE | PRRT2 | NM_145239.2 | c.323_324delCA[p.Thr108Serfs*25] | Reported |
| 30 | 3 | 6–6.5 m | BFIE | PRRT2 | NM_145239.2 | Deletion | Reported |
| 31 | 2 | 4 m | BFIE | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 32 | 2 | 3.5 m | BFIE | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 33 | 2 | 5 m | BFIE | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 34 | 2 | 4–6 m | BFIE | PRRT2 | NM_145239.2 | c.629dupC[p.Ala211Serfs*14] | Reported |
| 35 | 2 | 5–6 m | BFIE | PRRT2 | NM_145239.2 | C.679C>T[p.Arg227*] | Novel |
| 36 | 2 | 5 m | BFIE | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 37 | 2 | 11–13 m | BFIE | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 38 | 2 | 7–8 m | BFIE | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 39 | 4 | 6–8 m | BFIE | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 40 | 2 | 4–4.5 m | BFIE/ICCA | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 41 | 3 | 4–5.5 m | BFIE/ICCA | PRRT2 | NM_145239.2 | c.649delC[p.Arg217Glufs*12] | Reported |
| 42 | 2 | 7–12 m | BFIE/ICCA | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 43 | 2 | 4 m | BFIE/ICCA | PRRT2 | NM_145239.2 | c.649delC[p.Arg217Glufs*12] | Reported |
| 44 | 4 | 5 m | BFIE/ICCA | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 45 | 4 | 4.5–6 m | BFIE/ICCA | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 46 | 3 | 3–4 m | BFIE/ICCA | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 47 | 2 | 8 m–3 y | BFIE/ICCA | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 48 | 3 | 6–12 m | BFIE/ICCA | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 49 | 2 | 5–6 m | BFIE/ICCA | PRRT2 | NM_145239.2 | c.560dupT[p.Gln188Alafs*4] | Novel |
| 50 | 8 | 1–3.5 m | BFIE/ICCA | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 51 | 2 | 2.5–6 m | BFIE/ICCA | PRRT2 | NM_145239.2 | c.649dupC[p.Arg217Profs*8] | Reported |
| 52 | 2 | 5–8 m | BFIE/ICCA | PRRT2 | NM_145239.2 | c.649delC[p.Arg217Glufs*12] | Reported |
| 53 | 4 | 3–4 m | BFIE/ICCA | PRRT2 | NM_145239.2 | c.1023A>T[p.*341Cys] | Reported |
| 54 | 3 | 3.5–4 m | BFIE | SCN2A | NM_001040142.1 | c.668G>A[p.Arg223Gln] | Reported |
| 55 | 11 | 3–5.5 m | BFIE | SCN2A | NM_001040142.1 | c.752T>C[p.Val251Ala] | Novel |
| 56 | 2 | 3–3.5 m | BFIE | SCN2A | NM_001040142.1 | c.1307T>C[p.Leu436Ser] | Novel |
| 57 | 2 | 3 –3.5 m | BFIE | SCN2A | NM_001040142.1 | c.4835C>G[p.Ala1612Gly] | Novel |
| 58 | 3 | 14 m | BFIE | SCN2A | NM_001040142.1 | c.1737C>G[p.Ser579Arg] | Novel |
| 59 | 2 | 7–12 m | BFIE | KCNQ2 | NM_172107.2 | c.237T>G[p.Asn79Lys] | Novel |
| 60 | 2 | 9 m | BFIE | KCNQ2 | NM_172107.2 | c.1510C>T[p.Arg504Trp] | Novel |
| 61 | 2 | 8.5–9 m | BFIE | GABRA6 | NM_000811.2 | c.523G>T[p.Gly175Trp] | Novel |
| 62 | 3 | 7 m | BFIE | ND | — | — | — |
| 63 | 2 | 6 m | BFIE | ND | — | — | — |
| 64 | 4 | 11–12 m | BFIE | ND | — | — | — |
| 65 | 2 | 10 m | BFIE | ND | — | — | — |
| 66 | 2 | 4 m | BFIE | ND | — | — | — |
| 67 | 2 | 3–11 m | BFIE | ND | — | — | — |
| 68 | 2 | 7 –8 m | BFIE | ND | — | — | — |
| 69 | 2 | 8–12 m | BFIE | ND | — | — | — |
| 70 | 2 | 6–8 m | BFIE | ND | — | — | — |
| 71 | 3 | 3–12 m | BFIE | ND | — | — | — |
| 72 | 2 | 9–12 m | BFIE | ND | — | — | — |
| 73 | 2 | 6–7 m | BFIE | ND | — | — | — |
| 74 | 5 | 8 m | BFIE | ND | — | — | — |
| 75 | 2 | 9.5–12 m | BFIE | ND | — | — | — |
| 76 | 3 | 7–8 m | BFIE | ND | — | — | — |
| 77 | 2 | 6–7 m | BFIE | ND | — | — | — |
| 78 | 2 | 4 m | BFIE | ND | — | — | — |
| 79 | 2 | 7.5 m | BFIE/ICCA | ND | — | — | — |
d day, m month, y year, ND Not detected
Of the 42 families with PRRT2 mutations, 39 had frameshift mutations, one had a nonsense mutation, one showed a loss of a stop codon, and one was a microdeletion of the gene (family 30). The size of the deletion detected by the targeted next-generation sequencing was about 1583 bp (chr16: 29824376-29825959). The deletion was verified by qPCR (the qPCR results of the family members are shown in Supplementary Appendix 4). The mutation c.649dupC[p.Arg217Profs*8] and c.649delC[p.Arg217Glufs*12] were hot spot mutations of PRRT2. The mutation c.649dupC was found in 28 families (28 out of 42, 66.7%). The mutation c.649delC was found in 6 families. In total 19 of the 42 families with PRRT2 mutation were described previously [13]. The mutations c.560dupT[p.Gln188Alafs*4] and c.679C>T[p.Arg227*] are novel. We found a proband from an ICCA family (family 50) with a compound heterozygous mutation of PRRT2 consisting of a mutation c.649dupC[p.Arg217Profs*8] inherited from his father, who had PKD; this proband also inherited a novel mutation c.593_594delCT [p.Pro198Argfs*26] from his asymptomatic mother, who did not have a family history of seizures or PKD (see family pedigrees in Fig. 3a1 and a2). The sequence chromatograms of the proband and her parents are shown in Fig. 1b. The proband is a girl. She had seizure onset in 2.5 months. Her seizures were well-controlled with valproate until she was 2 years old, when the valproate therapy stopped. The seizures relapsed 2 days after valproate withdrawal. The duration of seizures were about 2–3 min. The seizures were so frequent that recurred once every hour. Then carbamazepine was used for this patient and her seizures were controlled. She was 28 months old at last follow-up and had a light developmental delay in speech.
Fig. 3.

Family pedigrees. a1 Paternal pedigree of family 50; a2 Maternal pedigree of family 50; The proband had a compound heterozygous mutation of PRRT2 which consists of a frameshift mutation c.649dupC from her father who had shown a phenotype of PKD and another frameshift mutation c.593_594delCT from his asymptomatic mother who didn’t have any clinical history of seizure and PKD. b Pedigree of family 6 which had an affected family member (IV7) suffered from intractable epilepsy from the second day of life and evolved to epileptic encephalopathy. c Pedigree of family 61 which was detected with a GABRA6 mutation c.523G>T
Of the 9 families with KCNQ2 mutations, one had a nonsense mutation, 7 had missense mutations, and one had a gene deletions of KCNQ2, that also extended to the adjacent gene, CHRNA4 (family 7). The deletions included whole exons of KCNQ2 and CHRNA4 (MLPA results are shown in Supplementary Appendix 5). Six KCNQ2 mutations in our study were novel mutations, including c.1048A>C[p.Asn350His], c.242T>C[p.Leu81Pro], c.2506G>T[p.Glu836*], c.958G>A[p.Val320Ile], c.237T>G[p.Asn79Lys], and c.1510C>T[p.Arg504Trp]. Family 6, with the reported KCNQ2 mutation c.998G>A[p.Arg333Gln], was diagnosed as BFNIE, with 7 affected members who experienced seizures within the first 2 days to 6 months of life and had seizure remission before 1 year of life (see family pedigree in Fig. 3b). However, one affected individual (IV7) in this family has suffered intractable epilepsy from the second day after birth. The seizures were resistant to multiple antiepileptic drugs and led to severe developmental delay. He is 8 years old now and cannot walk alone. He also carries the KCNQ2 mutation Arg333Gln, which was inherited from his father, who had benign neonatal epilepsy.
All 8 SCN2A mutations found in our study are missense mutations. Five SCN2A mutations in our study were novel mutations, including c.2872A>G[p.Met958Val], c.752T>C[p.Val251Ala], c.1307T>C[p.Leu436Ser], c.4835C>G[p.Ala1612Gly], and c.1737C>G[p.Ser579Arg].
We identified a BFIE family (family 61) with a novel GABRA6 mutation c.523G>T[p. Gly175Trp]. The proband and her mother showed seizure onset at 8.5–9 months of life and showed remission before 1 year. The proband was 5 years old at last follow up. Both the proband and her mother had no seizure relapsed and showed normal intelligence. The clinical history of infantile seizure could not be obtained for the grandmother of the proband, who also carried the same mutation. The family pedigree is shown in Fig. 3c. The position of p.G175 codes for a highly conserved amino acid. The in silico analysis of the Gly175Trp mutation based on SIFT (score 0.000), Polyphen-2 (score 0.99), and Mutation Taster (score 0.99) predicted that this substitution at position 175 from Gly to Trp could be pathogenic. The change is not reported in Single Nucleotide Polymorphism database (dbSNP), and is neither found in ExAC nor 1000 G. This mutation is also absent in the 104 healthy Chinese controls.
Discussion
We aimed to establish the genetic spectrum in Chinese families with benign familial epilepsies of the first year of life. Our results showed that in 79 families, the detection rate for gene mutations was 75.9%, which was lower than in a study by Zara et al. [14], who had a rate of 89%. It might be because the proportion of BFNE, BFNIE, and BFIE families in our study were different from the study by Zara et al.
KCNQ2 is the only gene related to BFNE in our study. This indicates that KCNQ2 is a major causative gene of BFNE. Although the sample size of our BFNE families was small, this result was consistent with a larger study by Grinton et al. [15], consisting of ~80% BFNE families with KCNQ2 mutations. Grinton et al. also identified one BFNE family with a KCNQ3 mutation and 2 families with SCN2A mutations. However, we did not identify any mutations in KCNQ3 or SCN2A in our BFNE families. This suggests that BFNE families with causative mutations in KCNQ3 and SCN2A are rare.
In our study, all 7 BFNIE families were detected with gene mutations. The genes involved were KCNQ2, SCN2A, and PRRT2. SCN2A had been proved to be a main causative gene of BFNIE [9, 10]. KCNQ2 mutations were previously identified in several families fulfill the diagnosis of BFNIE [14, 16]. We identified 3 families with SCN2A mutations and 3 families with KCNQ2 mutations in our BFNIE families. This indicates that both SCN2A and KCNQ2 are important genes in BFNIE families. KCNQ2 and CHRNA4 deletions have been reported in typical BFNE families, as well as patients with neonatal seizures and developmental delay in previous studies [17, 18]. We found one BFNIE family with KCNQ2 and CHRNA4 deletions. Both of the two affected members in this family showed typical benign neonatal or infantile epilepsy. We found one BFNIE family with a PRRT2 hotspot mutation, c.649dupC. The onset of seizures in affected members was between 2 and 2.5 months of age in this family. The earliest age of onset of seizures in individuals with PRRT2 mutations in a previous study was during the first few days of life [19, 20].
BFIE families were the most genetically heterogeneous. PRRT2, KCNQ2, SCN2A, and GABRA6 were found to be involved. The rate of identifiable gene mutations in the BFIE families was 73.5% in our study. PRRT2 mutations were clustered in families with BFIE (41 out of 68, 60.3%). It may be due to regional and racial differences, however, our rate was lower than those rates found by other research groups [11, 12, 14, 21]. In our study, most PRRT2 mutations were frameshift mutations. And we did not find any missense mutation in PRRT2. Both c.649dupC[p.Arg217Profs*8] and c.649delC[p.Arg217Glufs*12] were hot spot mutations of PRRT2. The mutation c.649dupC was most common in PRRT2 mutations, accounting for 66.7% (28 out of 42) families with PRRT2 mutations in our study. The ICCA families exhibited only PRRT2 mutations. The positive rate of PRRT2 mutations in our ICCA families was 93.3%. This result was consistent with the rate that had been reported by other research groups [19, 21–24]. The identification of SCN2A mutations in 5 BFIE families in our cohort further proved that this gene is also involved in families with a delayed age of onset [25]. KCNQ2 was the only gene that could be related to these three benign familial epilepsies in our study. However, KCNQ2 mutations showed a progressively decreasing rate as the age of seizure onset increased. We found 3 families with KCNQ2 mutations in our 68 BFIE families (4.4%, 3 out of 68). Zara et al. [14] found one family with KCNQ2 mutation in 29 BFIE families (3.4%, 1 out of 29). These two rates are similar. In our study, 3 BFIE families have affected individuals with seizure onset beyond 1 year of age. All three families were found to have a mutation in PRRT2 or SCN2A.
Labate et al. [26] have reported homozygous mutation in PRRT2 in a family with intellectual disability and seizures with onset in infancy. Delcourt et al. [27] also reported that patients with biallelic mutations in PRRT2 gene had diverse forms of paroxysmal neurological disorders, including status epilepticus, paroxysmal non-kinesigenic dyskinesia, learning abilities, behavioural problems, episodes of ataxia, and cerebellar atrophy. In our study, an affected member in an ICCA family (family 50) was identified with a compound heterozygous PRRT2 mutation. This patient was more dependent on anti-epileptic drugs compared to the other affected members with a heterozygous c.649dupC mutation in her family and showed a slight language development delay. It may due to our short follow-up time (28 months at last follow up), however, this girl had not yet showed a distinct phenotype of infantile epileptic encephalopathy or other severe paroxysmal neurological disorders.
Both benign epilepsy and epileptic encephalopathy have been reported to be associated with KCNQ2 mutations [28]. Families with KCNQ2 mutations and variable phenotypes have been described previously [29, 30]. We identified a BFNIE family (family 6) with 7 affected members who showed benign epilepsy as neonates or infants, and one affected boy presented epileptic encephalopathy. This boy was found to have a familial Arg333Gln mutation in KCNQ2, which had been reported in BFNE [31]. This further demonstrates that the correlation between genotype and phenotype is inconsistent, even within a family. Just as most other epilepsy associated with ion channelopathies, the genotype-phenotype relation for KCNQ2 can be substantially modified by other genes and environmental factors.
We detected a novel c.523 G > T[p. Gly175Trp] mutation in the GABRA6 gene in a BFIE family. GABRA6 is a member of the GABA-A receptor gene family of heteromeric pentameric ligand-gated ion channels through which GABA, the major inhibitory neurotransmitter in the mammalian brain, acts. GABRA6 mutations have been shown to be related to the childhood absence epilepsy in a previous study and polymorphisms in this gene were important risk factors for the development of the idiopathic generalized epilepsy [32, 33]. As far as we know, we have identified the first GABRA6 mutation in a BFIE family. Further functional research is necessary to strongly associate the mutation with the BFIE phenotype. However, the pathogenic bioinformatics prediction and the cosegregation of mutation allow us to speculate on a deleterious effect of this variation in our family. The absence of the mutation in SNP/variant databases, and in our control population also supports this hypothesis.
Previous studies showed that SCN8A and CHRNA2 mutations was identified in sporadic families with BFIE [34, 35]. These two genes are also included in our gene panel. However, we did not reveal any causative mutations in these two genes in our BFNE, BFNIE, and BFIE families. This suggests that mutations of these genes are rare as a cause of benign familial epilepsies.
In summary, mutations in KCNQ2, SCN2A, and PRRT2 are major genetic causes of benign familial epilepsies of the first year of life in Chinese population. KCNQ2 is the major gene related to BFNE. PRRT2 is the main gene responsible for BFIE. GABRA6 mutations might be involved in families with BFIE.
Electronic supplementary material
Acknowledgements
This study was supported by grants from the omics-based precision medicine of epilepsy being entrusted by Key Research Project of the Ministry of Science and Technology of China (2016YFC0904400 and 2016YFC0904401) and the Beijing Key Laboratory of Molecular Diagnosis and Study on Pediatric Genetic Diseases (Z141107004414036). We thank the patients and their family members for taking part in this study.
Compliance with Ethical Standards
Conflict of Interest
All authors declare that there is no conflict of interest.
Electronic supplementary material
The online version of this article (10.1038/s10038-017-0359-x) contains supplementary material, which is available to authorized users.
References
- 1.Vigevano F, Fusco L, Di Capua M, Ricci S, Sebastianelli R, Lucchini P. Benign infantile familial convulsions. Eur J Pediatr. 1992;151:608–12. doi: 10.1007/BF01957732. [DOI] [PubMed] [Google Scholar]
- 2.Okumura A, Hayakawa F, Kato T, Kuno K, Negoro T, Watanabe K. Early recognition of benign partial epilepsy in infancy. Epilepsia. 2000;41:714–7. doi: 10.1111/j.1528-1157.2000.tb00233.x. [DOI] [PubMed] [Google Scholar]
- 3.Tomlinson SE, Bostock H, Grinton B, Hanna MG, Kullmann DM, Kiernan MC, et al. In vivo loss of slow potassium channel activity in individuals with benign familial neonatal epilepsy in remission. Brain. 2012;135:3144–52. doi: 10.1093/brain/aws241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaplan RE, Lacey DJ. Benign familial neonatal-infantile seizures. Am J Med Genet. 1983;16:595–9. doi: 10.1002/ajmg.1320160417. [DOI] [PubMed] [Google Scholar]
- 5.Vigevano F. Benign familial infantile seizures. Brain Dev. 2005;27:172–7. doi: 10.1016/j.braindev.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 6.Szepetowski P, Rochette J, Berquin P, Piussan C, Lathrop GM, Monaco AP. Familial infantile convulsions and paroxysmal choreoathetosis: a new neurological syndrome linked to the pericentromeric region of human chromosome 16. Am J Hum Genet. 1997;61:889–98. doi: 10.1086/514877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh NA, Charlier C, Stauffer D, DuPont BR, Leach RJ, Melis R, et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18:25–29. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- 8.Charlier C, Singh NA, Ryan SG, Lewis TB, Reus BE, Leach RJ, et al. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat Genet. 1998;18:53–55. doi: 10.1038/ng0198-53. [DOI] [PubMed] [Google Scholar]
- 9.Heron SE, Crossland KM, Andermann E, Phillips HA, Hall AJ, Bleasel A, et al. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet. 2002;360:851–2. doi: 10.1016/S0140-6736(02)09968-3. [DOI] [PubMed] [Google Scholar]
- 10.Berkovic SF, Heron SE, Giordano L, Marini C, Guerrini R, Kaplan RE, et al. Benign familial neonatal-infantile seizures: characterization of a new sodium channelopathy. Ann Neurol. 2004;55:550–7. doi: 10.1002/ana.20029. [DOI] [PubMed] [Google Scholar]
- 11.Heron SE, Grinton BE, Kivity S, Afawi Z, Zuberi SM, Hughes JN, et al. PRRT2 mutations cause benign familial infantile epilepsy and infantile convulsions with choreoathetosis syndrome. Am J Hum Genet. 2012;90:152–60. doi: 10.1016/j.ajhg.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schubert J, Paravidino R, Becker F, Berger A, Bebek N, Bianchi A, et al. PRRT2 mutations are the major cause of benign familial infantile seizures. Hum Mutat. 2012;33:1439–43. doi: 10.1002/humu.22126. [DOI] [PubMed] [Google Scholar]
- 13.Yang X, Zhang Y, Xu X, Wang S, Yang Z, Wu Y, et al. Phenotypes and PRRT2 mutations in Chinese families with benign familial infantile epilepsy and infantile convulsions with paroxysmal choreoathetosis. BMC Neurol. 2013;13:209. doi: 10.1186/1471-2377-13-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zara F, Specchio N, Striano P, Robbiano A, Gennaro E, Paravidino R, et al. Genetic testing in benign familial epilepsies of the first year of life: clinical and diagnostic significance. Epilepsia. 2013;54:425–36. doi: 10.1111/epi.12089. [DOI] [PubMed] [Google Scholar]
- 15.Grinton BE, Heron SE, Pelekanos JT, Zuberi SM, Kivity S, Afawi Z, et al. Familial neonatal seizures in 36 families: clinical and genetic features correlate with outcome. Epilepsia. 2015;56:1071–80. doi: 10.1111/epi.13020. [DOI] [PubMed] [Google Scholar]
- 16.Zhou X, Ma A, Liu X, Huang C, Zhang Y, Shi R, et al. Infantile seizures and other epileptic phenotypes in a Chinese family with a missense mutation of KCNQ2. Eur J Pediatr. 2006;165:691–5. doi: 10.1007/s00431-006-0157-5. [DOI] [PubMed] [Google Scholar]
- 17.Kurahashi H, Wang JW, Ishii A, Kojima T, Wakai S, Kizawa T, et al. Deletions involving both KCNQ2 and CHRNA4 present with benign familial neonatal seizures. Neurology. 2009;73:1214–7. doi: 10.1212/WNL.0b013e3181bc0158. [DOI] [PubMed] [Google Scholar]
- 18.Pascual FT, Wierenga KJ, Ng YT. Contiguous deletion of KCNQ2 and CHRNA4 may cause a different disorder from benign familial neonatal seizures. Epilepsy Behav Case Rep. 2013;1:35–38. doi: 10.1016/j.ebcr.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Becker F, Schubert J, Striano P, Anttonen AK, Liukkonen E, Gaily E, et al. PRRT2-related disorders: further PKD and ICCA cases and review of the literature. J Neurol. 2013;260:1234–44. doi: 10.1007/s00415-012-6777-y. [DOI] [PubMed] [Google Scholar]
- 20.Guerrero-Lopez R, Ortega-Moreno L, Giraldez BG, Alarcon-Morcillo C, Sanchez-Martin G, Nieto-Barrera M, et al. Atypical course in individuals from Spanish families with benign familial infantile seizures and mutations in the PRRT2 gene. Epilepsy Res. 2014;108:1274–8. doi: 10.1016/j.eplepsyres.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 21.Ishii A, Yasumoto S, Ihara Y, Inoue T, Fujita T, Nakamura N, et al. Genetic analysis of PRRT2 for benign infantile epilepsy, infantile convulsions with choreoathetosis syndrome, and benign convulsions with mild gastroenteritis. Brain Dev. 2013;35:524–30. doi: 10.1016/j.braindev.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 22.Scheffer IE, Grinton BE, Heron SE, Kivity S, Afawi Z, Iona X, et al. PRRT2 phenotypic spectrum includes sporadic and fever-related infantile seizures. Neurology. 2012;79:2104–8. doi: 10.1212/WNL.0b013e3182752c6c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cloarec R, Bruneau N, Rudolf G, Massacrier A, Salmi M, Bataillard M, et al. PRRT2 links infantile convulsions and paroxysmal dyskinesia with migraine. Neurology. 2012;79:2097–103. doi: 10.1212/WNL.0b013e3182752c46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee HY, Huang Y, Bruneau N, Roll P, Roberson ED, Hermann M, et al. Mutations in the gene PRRT2 cause paroxysmal kinesigenic dyskinesia with infantile convulsions. Cell Rep. 2012;1:2–12. doi: 10.1016/j.celrep.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wolff M, Johannesen KM, Hedrich UB, Masnada S, Rubboli G, Gardella E, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A-related disorders. Brain. 2017;140:1316–1336. doi: 10.1093/brain/awx054. [DOI] [PubMed] [Google Scholar]
- 26.Labate A, Tarantino P, Viri M, Mumoli L, Gagliardi M, Romeo A, et al. Homozygous c.649dupC mutation in PRRT2 worsens the BFIS/PKD phenotype with mental retardation, episodic ataxia, and absences. Epilepsia. 2012;53:e196–199. doi: 10.1111/epi.12009. [DOI] [PubMed] [Google Scholar]
- 27.Delcourt M, Riant F, Mancini J, Milh M, Navarro V, Roze E, et al. Severe phenotypic spectrum of biallelic mutations in PRRT2 gene. J Neurol Neurosurg Psychiatry. 2015;86:782–5. doi: 10.1136/jnnp-2014-309025. [DOI] [PubMed] [Google Scholar]
- 28.Hortiguela M, Fernandez-Marmiesse A, Cantarin V, Gouveia S, Garcia-Penas JJ, Fons C, et al. Clinical and genetic features of 13 Spanish patients with KCNQ2 mutations. J Hum Genet. 2017;62:185–9. doi: 10.1038/jhg.2016.104. [DOI] [PubMed] [Google Scholar]
- 29.Steinlein OK, Conrad C, Weidner B. Benign familial neonatal convulsions: always benign? Epilepsy Res. 2007;73:245–9. doi: 10.1016/j.eplepsyres.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 30.Al Yazidi G, Shevell MI, Srour M. Two novel KCNQ2 mutations in 2 families with benign familial neonatal convulsions. Child Neurol Open. 2017;4:2329048X17691396. doi: 10.1177/2329048X17691396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh NA, Westenskow P, Charlier C, Pappas C, Leslie J, Dillon J, et al. KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain. 2003;126:2726–37. doi: 10.1093/brain/awg286. [DOI] [PubMed] [Google Scholar]
- 32.Hernandez CC, Gurba KN, Hu N, Macdonald RL. The GABRA6 mutation, R46W, associated with childhood absence epilepsy, alters 6beta22 and 6beta2 GABA(A) receptor channel gating and expression. J Physiol. 2011;589:5857–78. doi: 10.1113/jphysiol.2011.218883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prasad DK, Shaheen U, Satyanarayana U, Prabha TS, Jyothy A, Munshi A. Association of GABRA6 1519 T>C (rs3219151) and Synapsin II (rs37733634) gene polymorphisms with the development of idiopathic generalized epilepsy. Epilepsy Res. 2014;108:1267–73. doi: 10.1016/j.eplepsyres.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 34.Anand G, Collett-White F, Orsini A, Thomas S, Jayapal S, Trump N, et al. Autosomal dominant SCN8A mutation with an unusually mild phenotype. Eur J Paediatr Neurol. 2016;20:761–5. doi: 10.1016/j.ejpn.2016.04.015. [DOI] [PubMed] [Google Scholar]
- 35.Trivisano M, Terracciano A, Milano T, Cappelletti S, Pietrafusa N, Bertini ES, et al. Mutation of CHRNA2 in a family with benign familial infantile seizures: potential role of nicotinic acetylcholine receptor in various phenotypes of epilepsy. Epilepsia. 2015;56:e53–57. doi: 10.1111/epi.12967. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.