

Abstract
Angiosperms represent one of the most spectacular terrestrial radiations on the planet1, but their early diversification and phylogenetic relationships remain uncertain2–5. A key reason for this impasse is the paucity of complete genomes representing early-diverging angiosperms. Here, we present high-quality, chromosomal-level genome assemblies of two aquatic species—prickly waterlily (Euryale ferox; Nymphaeales) and the rigid hornwort (Ceratophyllum demersum; Ceratophyllales)—and expand the genomic representation for key sectors of the angiosperm tree of life. We identify multiple independent polyploidization events in each of the five major clades (that is, Nymphaeales, magnoliids, monocots, Ceratophyllales and eudicots). Furthermore, our phylogenomic analyses, which spanned multiple datasets and diverse methods, confirm that Amborella and Nymphaeales are successively sister to all other angiosperms. Furthermore, these genomes help to elucidate relationships among the major subclades within Mesangiospermae, which contain about 350,000 species. In particular, the species-poor lineage Ceratophyllales is supported as sister to eudicots, and monocots and magnoliids are placed as successively sister to Ceratophyllales and eudicots. Finally, our analyses indicate that incomplete lineage sorting may account for the incongruent phylogenetic placement of magnoliids between nuclear and plastid genomes.
Subject terms: Phylogenomics, Phylogenetics, Genome evolution
Genome assemblies of two aquatic species, prickly waterlily and rigid hornwort, clarify the early diversification and phylogeny of plants. Multiple independent polyploidization events are inferred in each of the five major angiosperm clades.
Main
The angiosperms, or flowering plants, represent one of the most diverse and species-rich clades on Earth. They provide the vast majority of food consumed by humans and contribute substantially to global photosynthesis and carbon sequestration1. The origin of angiosperms was famously coined ‘an abominable mystery’ owing to their sudden appearance and rapid diversification2–5. To date, angiosperms include more than 350,000 species6 and occupy nearly every habitat from forests and grasslands to sea margins and deserts; angiosperms encompass a considerable variety of life forms, including trees, herbs, submerged aquatics and epiphytes. Resolving early angiosperm phylogeny is therefore critical for our understanding of such diversifying processes1,7.
Decades of efforts have greatly resolved the angiosperm phylogeny, illuminating their evolutionary history and helping to delineate major groups2–5. It has been identified that the three early-diverging angiosperm orders Amborellales, Nymphaeales and Austrobaileyales, which constitute remarkable morphological disparity and low species diversity, represent the earliest diverged angiosperm lineages8 (that is, the so-called ANA grade). However, the vast majority of angiosperms belong to the Mesangiospermae clade, which includes approximately 99% of all extant angiosperms. Eudicots and monocots are the two largest Mesangiospermae subclades, including around 75% and 22% of all species, respectively9; magnoliids represent a third subclade with about 9,000 species10; and the remaining two subclades, Chloranthales and Ceratophyllales, are morphologically unusual with only 77 and 7 species, respectively10–12. Despite the elucidation and the strong support for each of the five subclades of Mesangiosperms4,13, phylogenetic relationships among these clades remain uncertain, and different topologies have been proposed on the basis of various morphological14 and/or molecular lines of evidence13,15–18 (Supplementary Fig. 1).
Genomic data provide a rich and convincing means to resolve such evolutionary uncertainties. Despite the availability of numerous sequenced genomes from eudicots and monocots, early-diverging angiosperms remain poorly sampled, therefore inhibiting insights into these fundamental questions. To date, no nuclear genome has been sequenced for the four key orders—Austrobaileyales, Ceratophyllales, Chloranthales and Nymphaeales—which exhibit diverse life histories, extreme morphological variation and great evolutionary divergence. This lack of critical taxon sampling probably exacerbates phylogenetic uncertainty when inferring early angiosperm relationships. For example, nuclear genomes of three magnoliids (that is, Cinnamomum kanehirae, Liriodendron chinense and Persea americana) have been subsequently published19–21; however, phylogenetic analyses in these two studies resulted in conflicting placement of magnoliids relative to monocots and eudicots—that is, either monocots as the sister to a clade of magnoliids and eudicots, or magnoliids as the sister to monocots and eudicots19–22. Moreover, cases of deep phylogenetic incongruence between nuclear and organellar genomes have been recently reported in angiosperms18–24, but their causation (such as hybridization and incomplete lineage sorting (ILS)) has not been fully evaluated.
Here we report the high-quality chromosomal-level genome assemblies of E. ferox Salisb. (prickly waterlily; estimated genome size of 768.2 Mb) and C. demersum L. (rigid hornwort; estimated genome size of 777.2 Mb), which are representatives of the two aquatic lineages Nymphaeales and Ceratophyllales, respectively (Supplementary Fig. 2, Supplementary Table 1). A total of 31.7 Gb of Oxford Nanopore Technologies (ONT) long reads and 47.4 Gb of Illumina short reads were generated for Euryale, and 80.5 Gb of ONT long reads and 46.4 Gb of short reads were generated for Ceratophyllum (Supplementary Fig. 3, Supplementary Table 2). ONT long reads were de novo assembled into contigs using the Canu assembler25, and two rounds of polishing were applied to the assembled contigs using Pilon26 with the Illumina short reads. The resulting genome assemblies of Euryale and Ceratophyllum were 725.2 Mb (N50 size of 4.75 Mb, where N50 corresponds to the minimum contig length needed to cover 50% of the genome) and 733.3 Mb (N50 size of 1.56 Mb), respectively (Supplementary Table 3). Moreover, a total of 84.4 Gb and 133.9 Gb of Hi-C data were generated using the Illumina platform for Euryale and Ceratophyllum, respectively. Assembled contigs were then clustered into 29 and 12 pseudochromosomes for Euryale and Ceratophyllum, respectively, using LACHESIS27 (Fig. 1a, Supplementary Tables 2 and 4). Both genome assemblies showed a high contiguity, completeness and accuracy (Fig. 1a, Supplementary Fig. 4, Supplementary Tables 5–7), and matched the chromosome counts obtained from cytological studies28. Using a combination of homology-based and transcriptome-based approaches, 40,297 and 30,138 protein-coding genes were predicted in the genomes of Euryale and Ceratophyllum, respectively (Supplementary Fig. 5, Supplementary Table 8). Moreover, 78.3% and 71.2% of all of the predicted protein-coding genes were clustered into gene families for Euryale and Ceratophyllum, respectively (Supplementary Note 1, Supplementary Fig. 6, Supplementary Table 9), and 85.6% and 89.8% of all of the predicted protein-coding genes were successfully annotated by at least one database (that is, SwissProt, TrEMBL, InterPro, GO or KEGG) for Euryale and Ceratophyllum, respectively (Supplementary Table 10). Furthermore, despite the similar genome size of these two species, the percentage of predicted repetitive elements was much higher in the genome of Ceratophyllum (that is, 38.35% versus 63.08% for Euryale and Ceratophyllum, respectively; Supplementary Table 11).
Fig. 1. Comparative genomics analyses.

a, The genomic features of E. ferox (pseudomolecules size: 721.2 Mb) and C. demersum (pseudomolecules size: 703.8 Mb). From inside to outside: GC content in 500-bp sliding windows (i; minimum–maximum, 0.2–0.8); repeat density in 10-kb sliding windows (ii; minimum–maximum: 0–1.0, coloured from white to dark green); gene density in 100-kb sliding windows (iii; minimum–maximum, 0–30, coloured from white to dark orange); and SNV density in 100-kb sliding windows (iv; minimum–maximum: 0–0.025). The links in the centre connect syntenic gene blocks that were detected using MCscan. Chr, chromosome. b, Distribution of average synonymous substitution levels (Ks) between syntenic blocks after evolutionary rate correction. c, Syntenic blocks (involving ≥10 colinear genes) between genomes. The corresponding median Ks value is shown for each block, and polyploidization events are represented by different colours.
By constructing the distribution of synonymous substitutions per synonymous site (Ks) using syntenic paralogues within each genome, we detected two and three polyploidization events in the genomes of Euryale and Ceratophyllum, respectively (Supplementary Fig. 7, Supplementary Table 12). After correction for evolutionary rate29, the two polyploidization events in the genome of Euryale were estimated to occur at approximately 16–18 million and 94–106 million years ago (Ma), respectively; the three polyploidization events in Ceratophyllum were estimated to occur approximately 13–15 Ma, 127–143 Ma and 157–177 Ma, respectively (Fig. 1b). Furthermore, we identified polyploidization events in the genomes of C. kanehirae, P. americana, L. chinense, Oryza sativa and Vitis vinifera. Interestingly, the Cinnamomum and Persea genomes share two recent polyploidization events, and multiple independent polyploidization events have occurred in each of five major clades (that is, Nymphaeales, magnoliids, monocots, Ceratophyllales and eudicots; Fig. 1b, Supplementary Fig. 7), paralleling recent studies demonstrating that whole-genome duplication (WGD) is a widespread and potentially important evolutionary feature in angiosperms30,31. To better elucidate the polyploidy of our newly assembled genomes, we conducted a more focused comparative genomic analysis using Amborella, Cinnamomum, Liriodendron and Vitis as placeholders. Syntenic depth ratios of 6:1, 6:4, 6:2 and 6:3 were inferred in the Euryale–Amborella, Euryale–Cinnamomum, Euryale–Liriodendron and Euryale–Vitis comparisons, respectively, and 8:1, 8:4, 8:2 and 8:3 in the Ceratophyllum–Amborella, Ceratophyllum–Cinnamomum, Ceratophyllum–Liriodendron and Ceratophyllum–Vitis comparisons, respectively (Fig. 1c, Supplementary Figs. 8 and 9). On the basis of the syntenic relationships between and within each species, our analyses collectively demonstrate that Euryale underwent an ancient WGD followed by one whole-genome triplication, and Ceratophyllum has undergone three WGDs.
For the first time, the genomic taxon sampling represents two of the three orders in the ANA grade and four of the five subclades of Mesangiospermae. To resolve early angiosperm phylogeny, a total of 1,374 single-copy nuclear genes (SSCGs) were first identified with SonicParanoid32 using whole-genome sequences from 14 seed plants—that is, four eudicots (Aquilegia coerulea, Arabidopsis thaliana, Prunus persica and Vitis), three monocots (Musa acuminate, Oryza and Phalaenopsis equestris), three magnoliids (Cinnamomum, Liriodendron and Persea), Ceratophyllum, two ANA-grade species (Amborella and Euryale) and one gymnosperm (Ginkgo biloba; Supplementary Table 13). Aligned protein-coding regions were concatenated and analysed using two methods—(1) including all three codon positions (SSCG-CDS) and (2) including only the first and second codon positions (SSCG-Codon12). Moreover, for coalescent-based analyses, gene trees were individually estimated from each of the two datasets (SSCG-CDS and SSCG-Codon12), which were then input into ASTRAL33 for species tree inference (Supplementary Fig. 10). Our estimated gene trees are generally well supported (Supplementary Fig. 11), and both concatenation and coalescent analyses produced an identical strongly supported topology (Fig. 2a,b, Supplementary Figs. 12–14). Here, Amborella and Euryale were placed as successively sister to all other angiosperms, and monocots and magnoliids were inferred as successively sister to Ceratophyllum and eudicots. Our phylogenetic placement of magnoliids differs from APG, but is consistent with other studies that used various molecular markers, such as the plastid inverted repeat region34 and transcriptome data18,35. To avoid potential errors in orthology inference, we also extracted single-copy genes using OrthoMCL36 from the 14 seed plants described above as well as another gymnosperm (Picea abies). Only those genes sampled from at least 11 species were selected for downstream analyses, and a total of 2,302 single-copy genes (OSCG) were retained with an average of 1,859 genes for each species. Concatenation and coalescent analyses were similarly conducted as got those described above, and corroborated our phylogenetic findings (Fig. 2a, Supplementary Fig. 15). Furthermore, we took advantage of the newly developed species-tree inference method STAG37, which was designed to leverage gene trees estimated from multi-copy gene families. Only those genes sampled from all 15 seed plants were included, and a total of 2,356 low-copy genes (LCG) were retained. Species trees inferred from datasets including all three codon positions (LCG-CDS) and the first and second codon positions only (LCG-Codon12) were topologically identical to the ones described above (Fig. 2a, Supplementary Fig. 16), suggesting that our findings are robust.
Fig. 2. Phylogenomic analyses of early-diverging angiosperms.

a, Chronogram of early-diverging angiosperms on the basis of the dataset SSCG-CDS inferred using MCMCTree. Bootstrap support percentages and posterior probabilities are indicated for each internal branch (from left to right, SSCG-CDS concatenation analysis using maximum likelihood (CA-ML), SSCG-CDS ASTRAL, LCG-CDS STAG, SSCG-Codon12 CA-ML, SSCG-Codon12 ASTRAL, LCG-Codon12 STAG), and an asterisk indicates 100 bootstrap support percentages and 1.0 posterior probabilities in all analyses. i, ii and iii indicate each internal branch. C, Carboniferous; J, Jurassic; K, Cretaceous; N, Neogene; P, Permian; 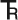 , Triassic; Q, Quaternary;
, Triassic; Q, Quaternary; 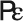 , Palaeogene. b, Species tree analysis using DiscoVista. Rows correspond to focal splits, and the spectrum indicates the support value for splits that are compatible with a species tree. Teal indicates themonophyly of a clade, and the different shades of teal indicate the level of its bootstrap support percentage (0 to 100%). Orange indicates rejection of a clade, and a 95% cut-off (instead of astandard 75%) was selected for strong rejection due to higher support values with genome-scale data. c, Superimposed ultrametric gene trees in a consensus DensiTree plot. The datasets SSCG-CDS and SSCG-Codon12 are shown in red and orange, respectively. d, The frequency of three topologies (q1–q3) around focal internal branches of ASTRAL species trees in the datasets SSCG-CDS and SSCG-Codon12. Each internal branch (labelled i, ii and iii) with four neighbouring branches can lead to three possible topologies (for example, q1, q2 and q3). e, Gene tree compatibility. The portion of gene trees for which focal splits are highly (or weakly) supported (or rejected). Weakly rejected splits are those that are not in the tree but are compatible if low support branches (below 75%) are contracted.
, Palaeogene. b, Species tree analysis using DiscoVista. Rows correspond to focal splits, and the spectrum indicates the support value for splits that are compatible with a species tree. Teal indicates themonophyly of a clade, and the different shades of teal indicate the level of its bootstrap support percentage (0 to 100%). Orange indicates rejection of a clade, and a 95% cut-off (instead of astandard 75%) was selected for strong rejection due to higher support values with genome-scale data. c, Superimposed ultrametric gene trees in a consensus DensiTree plot. The datasets SSCG-CDS and SSCG-Codon12 are shown in red and orange, respectively. d, The frequency of three topologies (q1–q3) around focal internal branches of ASTRAL species trees in the datasets SSCG-CDS and SSCG-Codon12. Each internal branch (labelled i, ii and iii) with four neighbouring branches can lead to three possible topologies (for example, q1, q2 and q3). e, Gene tree compatibility. The portion of gene trees for which focal splits are highly (or weakly) supported (or rejected). Weakly rejected splits are those that are not in the tree but are compatible if low support branches (below 75%) are contracted.
Despite the fact that the same set of phylogenetic relationships was consistently recovered when nuclear genes were analysed simultaneously, topological conflicts among gene trees were widely observed as visualized using DensiTree38 (Fig. 2c). A major discordance was identified in the datasets SSCG-CDS and SSCG-Codon12 involving the relationship between Amborella and Nymphaeales (Fig. 2c). For the datasets SSCG-CDS and SSCG-Codon12, 46.3% and 44.5% of all 1,374 gene trees supported Amborella and Euryale as successively sister to all other angiosperms, respectively; 27.2% and 26.0% supported Amborella as sister to Euryale, respectively; and the other 26.5% and 29.5% supported Euryale alone as the first lineage of angiosperms, respectively (Fig. 2d, Supplementary Figs. 17–19). We also summarized gene tree discordance using DiscoVista39, and similar results were observed for the datasets SSCG-CDS and SSCG-Codon12—that is, a substantial fraction of gene trees were incongruent with species trees regarding the placement of Amborella and Nymphaeales (Fig. 2e, Supplementary Figs. 17–19). Moreover, conflicting phylogenetic placements of Ceratophyllales were observed in the gene trees. For the datasets SSCG-CDS and SSCG-Codon12, 39.7% and 34.2% of 1,374 gene trees supported Ceratophyllum as sister to eudicots, respectively; 27.6% and 32.9% supported Ceratophyllum as sister to monocots, respectively; and the other 32.7% and 32.9% supported Ceratophyllum as sister to magnoliids, respectively (Fig. 2d, Supplementary Figs. 17–19). These analyses indicate that there is probably substantial ILS during early angiosperm evolution and greatly highlight the phylogenomic complexity of resolving early-diverging angiosperms.
Furthermore, phylogenetic analyses of these 15 seed plants inferred from 72 concatenated plastid genes strongly support magnoliids as the first diverging lineage of Mesangiospermae (Supplementary Fig. 20, Supplementary Table 14). This placement of magnoliids is incongruent with our nuclear phylogeny, but consistent with a recent study that analysed 2,881 plastomes4. Thus, what might account for this deep phylogenetic incongruence between nuclear and plastid genomes? As multiple independent polyploidization events were identified in magnoliids, monocots, Ceratophyllales and eudicots (Fig. 1), allopolyploidization or hybridization is one probable source of genomic discordance. We first assessed putative hybridization events in our phylogeny using PhyloNetworks. Although three cases of hybridization were inferred, none involved the three species of magnoliids (that is, Cinnamomum, Liriodendron and Persea; Supplementary Fig. 21). Furthermore, very short internal branches among four subclades of Mesangiospermae were observed in all our analyses, corresponding to an estimated divergence time of around 20 Ma (Supplementary Figs. 12–16, 20 and 22). We therefore tested whether ILS might better explain this discordance. We simulated 20,000 gene trees under the multispecies coalescent model40 on the basis of the ASTRAL tree inferred from the dataset SSCG-CDS. We found considerable agreement between simulated and empirical gene trees (overall correlation coefficient, Spearman’s ρ = 0.97, P < 0.01; Supplementary Fig. 23), suggesting that the multispecies coalescent model is a good fit to our data. Here the relative frequencies of various topologies, including the topology inferred from plastomes, were consistent with frequencies of ILS as estimated from our coalescent analyses (Supplementary Fig. 23c,d). These results indicate that ILS may well account for the incongruent placement of magnoliids between nuclear and plastid genomes. Finally, as sparse taxon sampling could result in these discordant results41, we increased our taxon sampling in the nuclear phylogeny by adding taxa with published genomes. A total of 612 ‘mostly’ single-copy orthologous genes (SCOG) were extracted from 213 nuclear genomes, which included 211 angiosperms representing 33 orders and 67 families as well as two gymnosperms as outgroups (Supplementary Table 15), and the average number of genes per taxon was 545. Coalescent analyses of the datasets SCOG-CDS and SCOG-Codon12 recovered the same relationships among the four subclades of Mesangiospermae (Supplementary Figs. 24–26), suggesting that our results are robust to additional taxa sampling.
In summary, the high-quality genomes of prickly waterlily and rigid hornwort greatly help to clarify phylogenetic relationships of early-diverging angiosperms. Moreover, these genomic resources are essential for future comparative investigations of genic evolution that underpin the morphological, physiological and ecological diversification of early angiosperms (Supplementary Notes 1–3, Supplementary Figs. 27–28, Supplementary Tables 16–22).
Methods
Plant materials and DNA sequencing
Fresh leaves of E. ferox and whole plants of C. demersum were obtained for DNA extraction and sequencing. Total genomic DNA was extracted using the CTAB method42. The library for ONT sequencing was constructed using large (>20 kb) DNA fragments with the Ligation Sequencing Kit 1D (SQK-LSK108), and sequenced using the GridION X5 platform. Adapters and low-quality nucleotides (that is, mean quality score <7) were trimmed. Paired-end libraries with an insertion size of 350 bp were constructed according to the manufacturer’s protocols and sequenced using the Illumina HiSeq 2500 System. Illumina reads were filtered using following criteria: (1) containing more than 5% unidentified nucleotides, (2) more than 65% of bases with a Phred quality score <7 and (3) more than 10 bp adapter sequences (allowing 2 bp mismatches). For the high-throughput chromosome conformation capture (Hi-C) analysis, fresh leaves were fixed in formaldehyde solution (1%), and chromatin was cross-linked and digested using the restriction enzyme HindIII. The 5′ overhangs were filled-in with biotinylated nucleotides, and free blunt ends were ligated. After ligation, crosslinks were reversed and the DNA was purified to remove protein. Purified DNA was treated to remove biotin that was not internal to ligated fragments. DNA was sheared into fragments of ~350 bp, and sequenced using the Illumina platform.
Genome size estimation
Genome size was estimated using the k-mer analysis of Illumina 150-bp paired-end reads. The k-mer depth-frequency distribution was generated using SOAPec43 (v.2.0.1, https://sourceforge.net/projects/soapdenovo2/) with the following parameters: -k 17 -q 33 -t 10. The genome size was then calculated according to the following formula44: genome size = k-mer coverage/mean k-mer depth (Supplementary Table 1).
Genome assembly
ONT long reads were de novo assembled using the Canu assembler25 (v.1.7, https://github.com/marbl/canu/), and two rounds of polishing were applied to the assembled contigs using Pilon26 (v.1.22, https://github.com/broadinstitute/pilon/) with the Illumina short reads. HiC-Pro45 v.2.10.0 was used to evaluate the quality of Hi-C data. Valid interaction pairs were mapped to the contigs and anchored to the pseudochromosomes using LACHESIS27 (https://github.com/shendurelab/LACHESIS).
Transcriptome sequencing and assembly
For each species, total RNA was extracted from various plant organs (roots, leaves and stems), and residual DNA was removed using the DNA-free DNA Removal Kit. A total of 18.69 Gb and 5.85 Gb of reads were generated using the Illumina platform for E. ferox and C. demersum, respectively. Transcripts were assembled from filtered reads using Trinity46 v.2.8.4 with additional parameters including ‘--trimmomatic --normalize_reads’.
Annotation of repetitive elements
To annotate repetitive elements, we utilized a combination of evidence-based and de novo approaches. Genome assemblies were first searched using RepeatMasker47 (v.4.0.7, http://repeatmasker.org/) against the Repbase database (http://www.girinst.org/repbase). Next, a de novo repetitive-element library was constructed using RepeatModeler (v.1.0.11, http://repeatmasker.org/RepeatModeler.html). This de novo repetitive-element library was then utilized by RepeatMasker to annotate repetitive elements. Results from these two runs of RepeatMasker were merged.
Protein-coding gene prediction and functional annotation
The identification of protein-coding genes was based on transcriptome data and ab initio prediction. RNA transcripts were first mapped to the assembled genome using PASA48 (Program to Assemble Spliced Alignment v.2.3.3). Valid transcript alignments were clustered on the basis of mapping location and assembled into gene structures, and then the high-quality gene models were selected for training by AUGUSTUS49 v.3.2.3. Moreover, intron hints were generated using the script bam2hints provided by AUGUSTUS. Next, AUGUSTUS was utilized for ab initio gene prediction on the hard-masked genome assembly, and all of the predictions were integrated using EvidenceModeler49 (EVM, v.1.1.1) to generate consensus gene sets. For functional annotation, our predicted protein-coding genes were searched against the Swiss-Prot and TrEMBL databases, as well as the InterPro database using InterProScan50 release 5.33–72.0.
Polyploidization analysis
Seven genomes were selected for our polyploidization analysis, that is, A. trichopoda (Amborellales; At), C. demersum (Ceratophyllales), C. kanehirae (magnoliids), E. ferox (Nymphaeales; Ef), L. chinense (magnoliids), O. sativa (monocots), P. Americana (magnoliids) and V. vinifera (eudicots; Vv). Colinear genes within each genome and between genomes were inferred using MCScan51 v.0.8 according to the combined information of gene similarity and gene order. Synonymous substitutions per synonymous site (Ks) between colinear genes were estimated using the Nei–Gojobori approach52 as implemented in the PAML53 package v.4.9 h. The median Ks values were selected to represent each syntenic block, and the probability density distribution curve of Ks was estimated using MATLAB with the kernel smoothing density function (ksdensity; bandwidth was typically set to 0.025). Multipeak fitting of the curve was performed using the Gaussian approximation function (cftool) in MATLAB, and the coefficient of determination (R2) was set as at least 0.95.
Furthermore, we performed a correction to the Ks values to distinguish the order of each polyploidization event using a similar method to a method used previously54. Here, supposing that the Ks values of colinear orthologues between two genomes i and j are , where N represents the normal distribution, μ represents the mean value and σ represents the standard deviation. We further supposing that the ratio of the evolutionary rate of species i to the assumed averaged evolutionary rate of angiosperms is ri, the correction coefficient λi is defined as and, accordingly, the correction coefficient factor of Xi − j is defined as
The mean of the corrected Xi − j-correction can be inferred to be
For E[tX] = tE[X] and D[tX] = t2D[X]
we can get
As Amborella and Euryale are basal angiosperms, the divergence between Amborella or Euryale and other plants occurred at the same time. Therefore, for genome i
The ai represents the mean ratio value among the observed Ks peak between Amboralla and Vitis, or Euryale and Vitis. After the divergence from the other studied plants, A. trichopoda has not been affected by polyploidization anymore; thus, we assumed that the evolutionary rate of Amborella genes is relatively stable and, therefore, set λAt = 1. The plant i with the slowest evolutionary rate is the most likely to have the same evolutionary rate as Amborella, that is, and . We determined the approximate value for V. vinifera (λVv) using the above estimator, and used it to assess the correction coefficient ratio for each species. The major-eudicot common hexaploidy 115–130 Ma (refs. 55,56), inferred by grape duplicated genes, was used as the reference to date the ages for the other polyploidization and speciation events (Supplementary Table 12).
Phylogenetic analyses
To infer the phylogenetic placements of E. ferox and C. demersum, SSCGs were first identified using SonicParanoid32 v.1.0 from 14 seed plants (SSCG; Supplementary Table 13). For each gene, amino acid sequences were aligned using MAFFT57 v.7.402, and then DNA sequences were aligned according to the corresponding amino acid alignments using PAL2NAL58 v.14. For datasets SSCG-CDS and SSCG-Codon12, the maximum likelihood (ML) trees were inferred from concatenated gene sequences using IQ-TREE59 v.1.6.9, which automatically selected the best-fit substitution model using ModelFinder60. Bootstrap support was estimated using 1,000 replicates of the ultrafast bootstrap approximation61 (-bb 1000 -m MFP). For coalescent-based analyses, gene trees were first estimated using IQ-TREE; the gene trees were then utilized by ASTRAL v.5.6.1 to infer species trees with quartet scores and posterior probabilities. Furthermore, SSCGs were identified using OrthoMCL36 v.2.0.9 (OSCG) with one more Gymnosperm (P. abies; Supplementary Table 13). Species trees were inferred from the datasets OSCG-CDS and OSCG-Codon12 using concatenation and coalescent methods as described above. Finally, we extracted low-copy genes from 15 seed plants (LCG). Here, each gene was required to include at least 1 sequence from each of the 15 species and less than 5 homologous sequences per species. For the datasets LCG-CDS and LCG-Codon12, gene trees were first estimated using IQ-TREE59; these gene trees were then utilized to construct species trees using STAG37 v.1.0.0.
For plastid genes, the 72 CDS of protein-coding genes were extracted from 15 seed plants (Supplementary Table 14), and aligned using MAFFT and PAL2NAL as described above. The ML trees were inferred from concatenated gene sequences using RAxML62 v.7.2.8 with 100 bootstraps.
For expanded taxon sampling, sequence similarity was first assessed for all of the amino acid sequences from 213 species (211 angiosperms and 2 gymnosperms; Supplementary Table 15) using MMseqs263 with an E-value threshold of 1 × 10−5, and then grouped using a Markov cluster algorithm64. Here, each gene was required to include sequences from more than 180 species. Next, ‘mostly’ single-copy orthologous genes (SCOG) were identified using a tree‐based method65,66. Each gene was aligned using MAFFT and PAL2NAL as described above, and species trees were inferred from datasets SCOG-CDS and SCOG-Codon12 using ASTRAL.
Visualizations of gene-tree discordance
Gene trees were first converted to ultrametric trees using the R package Phybase67, and then superimposed using DensiTree38 (Fig. 2c). Quartet frequencies of the internal branches in the species tree were calculated using ASTRAL33 with the parameter ‘-t=2’ (Supplementary Figs. 17 and 19). Furthermore, the analysis of gene-tree compatibility was conducted using DiscoVista39 v.1.0. Here, a total of 15 species groups were considered, and 7 of which are identified in our species tree, including: (1) 12 Mesagiospermae, (2) 4 eudicots, (3) 3 magnollids, (4) 3 monocots, (5) Euryale and all Mesangiospermaes, (6) Ceratophyllaceae and eudicots, and (7) Magnoliids and (Ceratophyllaceae and eudicots). The other 8 species groups were: (1) basal angiosperms (that is, Ambrorella and Euryale), (2) Ambrorella and Mesangiospermae, (3) Ceratophyllaceae and magnoliids, (4) Ceratophyllaceae and monocots, (5) magnoliids and eudicots, (6) magnoliids and monocots, (7) eudicots and monocots, and (8) Ceratophyllaceae, magnoliids and monocots (Fig. 2e, Supplementary Fig. 18). Bootstrap support values of at least 75% were interpreted as highly supported68 (Fig. 2e).
Divergence time estimation
Divergence time was estimated for the dataset SSCG-CDS using the program MCMCTree in the PAML53 package v.4.9 h. After a burn-in of 5,000,000 iterations, the MCMC process was performed 20,000 times with sample frequency of 5,000. Convergence was assessed using two independent runs. We used the following age constraints in our estimation procedure: the divergence between angiosperms and gymnosperms (330–289 Ma; http://www.timetree.org/), the crown group of angiosperms (267–132.9 Ma)4, the crown group of monocots (184–113 Ma)4 and the crown group of eudicots (161–125 Ma)4.
Hybridization inference and ILS simulation
Hybridization was detected for the dataset OSCG-CDS using the maximum pseudolikelihood estimation of phylogenetic networks, as implemented in PhyloNetworks69 v.0.9.0. The maximum allowed number of hybridizations was set from hmax=0 to hmax=10, each with 100 runs. The ILS simulation was performed as previously described70. We simulated 200,000 gene trees under the multispecies coalescent model using the R function sim.coaltree.sp as implemented in the package Phybase67 v.1.5. The internal branch lengths of the ASTRAL tree were used for the simulation, and all terminal branches were set to 1 (as 1 allele was generated for each species). It should be noted that internal branch lengths (in coalescent units) in our simulation might have been overestimated, as the cause of gene tree heterogeneity was assumed to result from only ILS. Gene-tree quartet frequencies were calculated for simulated and empirical datasets, and the correlation test was performed using the cor.test function in R.
Demographic inference
The pairwise sequentially Markovian coalescent (PSMC) model71 v.0.6.4-r49 was used to infer the demographic history of seven species, that is, A. trichopoda (Amborellales), E. ferox (Nymphaeales), C. demersum (Ceratophyllales), C. kanehirae (magnoliids), L. chinense (magnoliids), P. equestris (monocots) and V. vinifera (eudicots). The genome of E. ferox showed very low heterozygosity (about 0.02%; Supplementary Table 22) and, therefore, two individuals were included in the PSMC analysis72. For each species, whole-genome resequencing data (at least 30-fold coverage) were obtained from NCBI (Supplementary Table 22). Reads were mapped to the assembled genome, and the consensus sequences were extracted. The analysis was performed using the following parameters: -N25 -t15 -r5 -p ‘4+25×2+4+6’. Here, for A. trichopoda, C kanehirae, L. chinense and V. vinifera, the generation time and mutation rate were obtained from previous studies7,19,20,73. For other three species (that is, E. ferox, C. demersum and P. equestris), the mutation rate was first estimated using r8s74. Furthermore, as E. ferox is an annual species, the generation time was set to 1. For perennial species, as the generation time is difficult to determine precisely75, we tested the generation time for both 3 and 5 years, and similar results were obtained.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Supplementary notes, figures, tables and references.
Acknowledgements
This work was supported equally by the Strategic Priority Research Program of Chinese Academy of Sciences (XDB31010300) and the National Key Research and Development Program of China (2017YFC0505203), and further by the National Natural Science Foundation of China (grant numbers 31590821, 91731301 and 31561123001), the Fundamental Research Funds for the Central Universities 2018CDDY-S02-SCU and SCU2019D013, and National High-Level Talents Special Support Plans.
Author contributions
J.L. was the leader of this study. J.L., Z.X., X.W. and C.C.D. designed the experiments and coordinated the project. L.Z., D.W. and Q.W. performed field work and collected samples. Y.Y., L.L., D.W. and D.R. performed the assembly of the two genomes. Y.Y., L.L., T.M. and D.R. carried out the repeat and gene annotations. X.W., P.S., F.M., B.J., L.S., M.L. and Z.Q. performed the polyploidization analysis. Y.Y., L.L., D.R., Y.L. and X.S. carried out the gene family analysis and the phylogenomic analysis. Y.Y. and Y.L. performed the PSMC analysis. Y.Y., J.L., Z.X., C.C.D. and X.W. wrote and edited most of the manuscript. All of the authors read and approved the final manuscript.
Data availability
All of the raw sequence reads used in this study have been deposited at NCBI under the BioProject accession numbers PRJNA552436 (E. ferox) and PRJNA552433 (C. demersum). The assemblies and annotations are available from the CoGe comparative genomics platform at https://genomevolution.org/CoGe/GenomeInfo.pl?gid=56574 (E. ferox chromosome assembly), https://genomevolution.org/CoGe/GenomeInfo.pl?gid=56571 (E. ferox contig assembly), https://genomevolution.org/CoGe/GenomeInfo.pl?gid=56572 (C. demersum chromosome assembly) and https://genomevolution.org/CoGe/GenomeInfo.pl?gid=56569 (C. demersum contig assembly).
Code availability
The custom scripts have deposited in GitHub (https://github.com/yongzhiyang2012/Euryale_ferox_and_Ceratophyllum_demersum_genome_analysis).
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Nature Plants thanks Victor Albert and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Yongzhi Yang, Pengchuan Sun.
Contributor Information
Zhenxiang Xi, Email: zxi@scu.edu.cn.
Xiyin Wang, Email: wangxiyin@vip.sina.com.
Charles C. Davis, Email: cdavis@oeb.harvard.edu
Jianquan Liu, Email: liujq@nwipb.ac.cn.
Supplementary information
Supplementary information is available for this paper at 10.1038/s41477-020-0594-6.
References
- 1.Judd, W. S., Campbell, C. S., Kellogg, E. A., Stevens, P. F. & Donoghue, M. J. Plant Systematics (Sinauer Sunderland, 2002).
- 2.Friedman WE. The meaning of Darwin’s ‘abominable mystery’. Am. J. Bot. 2009;96:5–21. doi: 10.3732/ajb.0800150. [DOI] [PubMed] [Google Scholar]
- 3.Buggs RJA. The deepening of Darwin’s abominable mystery. Nat. Ecol. Evol. 2017;1:0169. doi: 10.1038/s41559-017-0169. [DOI] [PubMed] [Google Scholar]
- 4.Li HT, et al. Origin of angiosperms and the puzzle of the Jurassic gap. Nat. Plants. 2019;5:461–470. doi: 10.1038/s41477-019-0421-0. [DOI] [PubMed] [Google Scholar]
- 5.Steemans P, et al. Origin and radiation of the earliest vascular land plants. Science. 2009;324:353. doi: 10.1126/science.1169659. [DOI] [PubMed] [Google Scholar]
- 6.The Plant List. The Plant List – A working list of all plant species. Royal Botanic Gardens, Kew and Missouri Botanical Garden (2019). Available online at http://www.theplantlist.org/ (last retrieved 20 Aug 2019).
- 7.Amborella Genome Project. The Amborella genome and the evolution of flowering plants. Science342, 1241089 (2013). [DOI] [PubMed]
- 8.Qiu Y-L, et al. The earliest angiosperms: evidence from mitochondrial, plastid and nuclear genomes. Nature. 1999;402:404–407. doi: 10.1038/46536. [DOI] [PubMed] [Google Scholar]
- 9.Drinnan, A. N., Crane, P. R. & Hoot, S. B. in Early Evolution of FlowersSupplement 8, Vol. 8 (eds Endress, P. K. & Friis, E. M.) 93–122 (Springer, 1994).
- 10.Cronquist, A. & Takhtadzhian, A. L. An Integrated System of Classification of Flowering Plants (Columbia Univ. Press, 1981).
- 11.Friis EM, Pedersen KR, Crane PR. Diversity in obscurity: fossil flowers and the early history of angiosperms. Proc. R. Soc. B. 2010;365:369–382. doi: 10.1098/rstb.2009.0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dilcher DL, Wang H. An early cretaceous fruit with affinities to Ceratophyllaceae. Am. J. Bot. 2009;96:2256–2269. doi: 10.3732/ajb.0900049. [DOI] [PubMed] [Google Scholar]
- 13.Chase MW, et al. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016;181:1–20. [Google Scholar]
- 14.Endress PK, Doyle JA. Reconstructing the ancestral angiosperm flower and its initial specializations. Am. J. Bot. 2009;96:22–66. doi: 10.3732/ajb.0800047. [DOI] [PubMed] [Google Scholar]
- 15.Moore MJ, Bell CD, Soltis PS, Soltis DE. Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl Acad. Sci. USA. 2007;104:19363–19368. doi: 10.1073/pnas.0708072104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin-Long Q, et al. Angiosperm phylogeny inferred from sequences of four mitochondrial genes. J. Sys. Evol. 2010;48:391–425. [Google Scholar]
- 17.Zhang N, Zeng L, Shan H, Ma H. Highly conserved low‐copy nuclear genes as effective markers for phylogenetic analyses in angiosperms. N. Phytol. 2012;195:923–937. doi: 10.1111/j.1469-8137.2012.04212.x. [DOI] [PubMed] [Google Scholar]
- 18.Zeng L, et al. Resolution of deep angiosperm phylogeny using conserved nuclear genes and estimates of early divergence times. Nat. Commun. 2014;5:4956. doi: 10.1038/ncomms5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chaw S-M, et al. Stout camphor tree genome fills gaps in understanding of flowering plant genome evolution. Nat. Plants. 2019;5:63–73. doi: 10.1038/s41477-018-0337-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen J, et al. Liriodendron genome sheds light on angiosperm phylogeny and species–pair differentiation. Nat. Plants. 2019;5:18–25. doi: 10.1038/s41477-018-0323-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rendon-Anaya M, et al. The avocado genome informs deep angiosperm phylogeny, highlights introgressive hybridization, and reveals pathogen-influenced gene space adaptation. Proc. Natl Acad. Sci. USA. 2019;116:17081–17089. doi: 10.1073/pnas.1822129116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soltis DE, Soltis PS. Nuclear genomes of two magnoliids. Nat. Plants. 2019;5:6–7. doi: 10.1038/s41477-018-0344-1. [DOI] [PubMed] [Google Scholar]
- 23.Wickett NJ, et al. Phylotranscriptomic analysis of the origin and early diversification of land plants. Proc. Natl Acad. Sci. USA. 2014;111:E4859–E4868. doi: 10.1073/pnas.1323926111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun M, et al. Deep phylogenetic incongruence in the angiosperm clade Rosidae. Mol. Phylogenet. Evol. 2015;83:156–166. doi: 10.1016/j.ympev.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 25.Koren S, et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walker BJ, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE. 2014;9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burton JN, et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 2013;31:1119–1125. doi: 10.1038/nbt.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rice A, et al. The Chromosome Counts Database (CCDB)—a community resource of plant chromosome numbers. N. Phytol. 2015;206:19–26. doi: 10.1111/nph.13191. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, et al. Statistical inference of chromosomal homology based on gene colinearity and applications to Arabidopsis and rice. BMC Bioinform. 2006;7:447. doi: 10.1186/1471-2105-7-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Estep MC, et al. Allopolyploidy, diversification, and the Miocene grassland expansion. Proc. Natl Acad. Sci. USA. 2014;111:15149–15154. doi: 10.1073/pnas.1404177111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cai L, et al. Widespread ancient whole-genome duplications in Malpighiales coincide with Eocene global climatic upheaval. N. Phytol. 2019;221:565–576. doi: 10.1111/nph.15357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cosentino S, Iwasaki W. SonicParanoid: fast, accurate and easy orthology inference. Bioinformatics. 2019;35:149–151. doi: 10.1093/bioinformatics/bty631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang C, Rabiee M, Sayyari E, Mirarab S. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 2018;19:153. doi: 10.1186/s12859-018-2129-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore MJ, et al. Phylogenetic analysis of the plastid inverted repeat for 244 species: insights into deeper-level angiosperm relationships from a long, slowly evolving sequence region. Int. J. Plant Sci. 2011;172:541–558. [Google Scholar]
- 35.One Thousand Plant Transcriptomes Initiative One thousand plant transcriptomes and the phylogenomics of green plants. Nature. 2019;574:679–685. doi: 10.1038/s41586-019-1693-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Emms, D. & Kelly, S. STAG: species tree inference from all genes. Preprint at 10.1101/267914 (2018).
- 38.Bouckaert RR. DensiTree: making sense of sets of phylogenetic trees. Bioinformatics. 2010;26:1372–1373. doi: 10.1093/bioinformatics/btq110. [DOI] [PubMed] [Google Scholar]
- 39.Sayyari E, Whitfield JB, Mirarab S. DiscoVista: interpretable visualizations of gene tree discordance. Mol. Phylogenet. Evol. 2018;122:110–115. doi: 10.1016/j.ympev.2018.01.019. [DOI] [PubMed] [Google Scholar]
- 40.Rannala B, Yang ZH. Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics. 2003;164:1645–1656. doi: 10.1093/genetics/164.4.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hillis DM, Pollock DD, McGuire JA, Zwickl DJ. Is sparse taxon sampling a problem for phylogenetic inference? Syst. Biol. 2003;52:124–126. doi: 10.1080/10635150390132911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tel-Zur N, Abbo S, Myslabodski D, Mizrahi Y. Modified CTAB procedure for DNA isolation from epiphytic cacti of the genera Hylocereus and Selenicereus (Cactaceae) Plant Mol. Biol. Rep. 1999;17:249–254. [Google Scholar]
- 43.Luo R, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1:18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi J, et al. Chromosome conformation capture resolved near complete genome assembly of broomcorn millet. Nat. Commun. 2019;10:464. doi: 10.1038/s41467-018-07876-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Servant N, et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015;16:259. doi: 10.1186/s13059-015-0831-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009;25:4.10.1–4.10.14. doi: 10.1002/0471250953.bi0410s25. [DOI] [PubMed] [Google Scholar]
- 48.Haas BJ, et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003;31:5654–5666. doi: 10.1093/nar/gkg770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stanke M, Diekhans M, Baertsch R, Haussler D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 2008;24:637–644. doi: 10.1093/bioinformatics/btn013. [DOI] [PubMed] [Google Scholar]
- 50.Jones P, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tang HB, et al. Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps. Genome Res. 2008;18:1944–1954. doi: 10.1101/gr.080978.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 53.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 54.Wang X, et al. Genome alignment spanning major Poaceae lineages reveals heterogeneous evolutionary rates and alters inferred dates for key evolutionary events. Mol. Plant. 2015;8:885–898. doi: 10.1016/j.molp.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 55.Vekemans D, et al. Gamma paleohexaploidy in the stem lineage of core eudicots: significance for MADS-box gene and species diversification. Mol. Biol. Evol. 2012;29:3793–3806. doi: 10.1093/molbev/mss183. [DOI] [PubMed] [Google Scholar]
- 56.Jiao YN, et al. Ancestral polyploidy in seed plants and angiosperms. Nature. 2011;473:97–100. doi: 10.1038/nature09916. [DOI] [PubMed] [Google Scholar]
- 57.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34:W609–W612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods. 2017;14:587–589. doi: 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018;35:518–522. doi: 10.1093/molbev/msx281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Steinegger M, Soding J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017;35:1026–1028. doi: 10.1038/nbt.3988. [DOI] [PubMed] [Google Scholar]
- 64.Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang Y, Smith SA. Orthology inference in nonmodel organisms using transcriptomes and low-coverage genomes: improving accuracy and matrix occupancy for phylogenomics. Mol. Biol. Evol. 2014;31:3081–3092. doi: 10.1093/molbev/msu245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.De Smet R, et al. Convergent gene loss following gene and genome duplications creates single-copy families in flowering plants. Proc. Natl Acad. Sci. USA. 2013;110:2898–2903. doi: 10.1073/pnas.1300127110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu L, Yu L. Phybase: an R package for species tree analysis. Bioinformatics. 2010;26:962–963. doi: 10.1093/bioinformatics/btq062. [DOI] [PubMed] [Google Scholar]
- 68.Jarvis ED, et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science. 2014;346:1320–1331. doi: 10.1126/science.1253451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Solis-Lemus C, Bastide P, Ane C. PhyloNetworks: a package for phylogenetic networks. Mol. Biol. Evol. 2017;34:3292–3298. doi: 10.1093/molbev/msx235. [DOI] [PubMed] [Google Scholar]
- 70.Wang K, et al. Incomplete lineage sorting rather than hybridization explains the inconsistent phylogeny of the wisent. Commun. Biol. 2018;1:169. doi: 10.1038/s42003-018-0176-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475:493–496. doi: 10.1038/nature10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thomas CG, et al. Full-genome evolutionary histories of selfing, splitting, and selection in Caenorhabditis. Genome Res. 2015;25:667–678. doi: 10.1101/gr.187237.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou Y, Massonnet M, Sanjak JS, Cantu D, Gaut BS. Evolutionary genomics of grape (Vitis vinifera ssp. vinifera) domestication. Proc. Natl Acad. Sci. USA. 2017;114:11715–11720. doi: 10.1073/pnas.1709257114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sanderson MJ. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics. 2003;19:301–302. doi: 10.1093/bioinformatics/19.2.301. [DOI] [PubMed] [Google Scholar]
- 75.Petit RJ, Hampe A. Some evolutionary consequences of being a tree. Annu. Rev. Ecol. Evol. Syst. 2006;37:187–214. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary notes, figures, tables and references.
Data Availability Statement
All of the raw sequence reads used in this study have been deposited at NCBI under the BioProject accession numbers PRJNA552436 (E. ferox) and PRJNA552433 (C. demersum). The assemblies and annotations are available from the CoGe comparative genomics platform at https://genomevolution.org/CoGe/GenomeInfo.pl?gid=56574 (E. ferox chromosome assembly), https://genomevolution.org/CoGe/GenomeInfo.pl?gid=56571 (E. ferox contig assembly), https://genomevolution.org/CoGe/GenomeInfo.pl?gid=56572 (C. demersum chromosome assembly) and https://genomevolution.org/CoGe/GenomeInfo.pl?gid=56569 (C. demersum contig assembly).
The custom scripts have deposited in GitHub (https://github.com/yongzhiyang2012/Euryale_ferox_and_Ceratophyllum_demersum_genome_analysis).