

Abstract
Sporadic synchronous endometrial (ECs) and ovarian cancers (OCs), although clinically considered to be independent primaries, have been shown to be clonally related and likely constitute metastases from each other. We sought to define whether synchronous ECs/OCs in patients with DNA mismatch repair (MMR)-deficiency syndromes would be clonally-related. We subjected synchronous ECs/OCs from four patients (LS3-LS6) with clinically confirmed Lynch syndrome (LS) and one patient with constitutional mismatch repair deficiency syndrome (CMMRD) to massively parallel sequencing targeting 468 cancer-related genes. Somatic mutations, copy number alterations (CNAs), clonal relatedness and clonal decomposition analyses were performed using previously described bioinformatics methods. All synchronous ECs/OCs analyzed were considered independent primaries based on clinicopathologic criteria. Sequencing analysis revealed that the ECs/OCs of three cases (LS2-CMMRD, L3, L4) harbored similar repertoires of somatic mutations and CNAs and were clonally-related. In these three cases, a subset of subclonal mutations in the EC became clonal in the OC, suggesting that the EC was likely the substrate from which the OC developed. LS5’s EC/OC had distinct mutational profiles but shared specific CNAs. In contrast, LS6’s EC/OC harbored distinct somatic mutation and lacked CNAs, consistent with each tumor constituting an independent primary lesion. In LS5 and LS6, PTEN mutations and PTEN loss of protein expression were found to be restricted to the EC. Finally, re-analysis of sequencing data of sporadic synchronous ECs/OCs supported the observations made in the current study that the directionality of progression is likely from the endometrium to the ovary. In conclusion, contrary to sporadic synchronous ECs/OCs, which are almost invariably clonally-related, ECs/OCs simultaneously involving the uterus and ovary in LS patients may represent distinct primary tumors. A subset of MMR-deficiency syndrome-related synchronous ECs/OCs, however, may originate from a single primary tumor at variance with their clinical diagnosis, with the endometrium being the likeliest site of origin.
Keywords: synchronous endometrial and ovarian cancer, Lynch syndrome, sequencing, clonality
INTRODUCTION
Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer (HNPCC), is an autosomal dominant cancer predisposition syndrome caused by heterozygous germline mutations in the DNA mismatch repair (MMR) genes MLH1, MSH2, MSH6 or PMS2, or deletion of the 3’ end of EPCAM [1]. Lynch syndrome is associated with an increased risk of colorectal, endometrial and ovarian cancer, amongst others [1]. In contrast, biallelic germline mutations affecting the key MMR genes cause constitutional mismatch repair deficiency syndrome (CMMRD). CMMRD is a childhood cancer predisposition syndrome with a wide tumor spectrum including hematologic malignancies, brain tumors and Lynch syndrome-associated tumors, principally colorectal cancers, however ECs have also been described in this setting [2–4]. Patients with Lynch syndrome have a 10–60% cumulative lifetime risk of developing endometrial cancer (EC) and a 6–14% risk of developing ovarian cancer (OC) [1,5]. In the setting of Lynch syndrome, the vast majority of ECs occur at a younger age than sporadic cases, are of endometrioid histology and are characterized by a uniquely high prevalence of synchronous OCs [6]. Similarly, OCs in Lynch syndrome are most frequently of endometrioid/clear cell histology and are also diagnosed at an earlier age [7]. Synchronous EC and OC have been reported in the sporadic setting in approximately 10% of cases, but in up to 22% in the context of Lynch syndrome [6]. Conversely, approximately 7% of women with synchronous EC and OC meet clinical or molecular criteria for suspicion of Lynch syndrome [8]. In this clinical scenario, where carcinomas are simultaneously diagnosed in two separate organs, it is important to know the origin of these tumors and whether they are related or independent, given that this knowledge has an impact not only on the clinical diagnosis, but also on subsequent treatment and prognosis [9–11].
Given the implications for prognostication and patient management, we and others have performed massively parallel sequencing-based analyses of synchronous ECs/OCs and demonstrated that sporadic synchronous ECs and OCs are clonally related and likely represent dissemination from one site to the other [12–14]. We previously described, however, a synchronous EC/OC occurring in the setting of Lynch syndrome, which was the only case with independent, clonally unrelated synchronous EC and OC with distinct somatic mutation profiles and mutational signatures [12]. In contrast, Niskakoski et al recently reported on three synchronous ECs/OCs arising in women with Lynch syndrome subjected to targeted sequencing analysis, of which they found two cases with similar high-confidence somatic mutation profiles indicative of shared origins, and one case without [15]. Therefore, it remains to be determined whether synchronous ECs/OCs are clonally related or independent primary tumors in the context of Lynch syndrome. Furthermore, it is unknown whether synchronous ECs/OCs arising in the setting of CMMRD are clonally related.
In studies to date that have demonstrated shared origins between synchronous ECs/OCs, in the sporadic setting as well as within the context of Lynch syndrome, the directionality of spread could not be robustly inferred. Based on immunohistochemical characterization of synchronous ECs/OCs, it has been suggest that the synchronous lesions have greater similarity with ECs as compared to OCs, suggesting that the OCs likely constitute metastases from ECs (i.e. dissemination from the endometrium to the ovary) [16].
In this study, we sought to determine the repertoire of somatic mutations, mutational signatures, copy number alterations (CNAs) and clonality in synchronous ECs/OCs occurring in women with Lynch syndrome or CMMRD using targeted massively parallel sequencing. If a subset of synchronous ECs/ OCs were in fact unrelated, the finding of distinct immunohistochemical expression patterns or mutational profiles of these lesions during diagnostic work-up would support the need for assessment for DNA MMR-associated syndromes. Furthermore, we sought to infer the chronology of the development of the endometrial and ovarian tumors through (re-)analysis of clonally related sporadic and Lynch syndrome-related synchronous ECs/OCs.
MATERIALS AND METHODS
Cases
Cases with confirmed pathogenic germline DNA MMR mutations and synchronous EC/OC were identified at Memorial Sloan Kettering Cancer Center (MSK), New York, NY, USA (n=4), and IDIBELL-Hospital Universitari de Bellvitge, Barcelona, Spain (n=1). This study was approved by the Institutional Review Boards of the respective institutions, and patient consent was obtained, following the approved research protocols. Representative formalin-fixed paraffin-embedded (FFPE) tissue sections from these cases were reviewed by specialized gynecologic pathologists (KJP, APMS, AVB, LHE and/or XM-G). ECs and OCs were histologically subtyped according to the World Health Organization (WHO) classification [17], and staged and graded according to the International Federation of Gynecology and Obstetrics (FIGO) criteria [18]. The synchronous ECs/OCs were also classified into independent primary or metastatic disease based on the clinicopathologic features described by Scully et al [19,20]. The clinicopathologic criteria for synchronous independent primary tumors of the endometrium and ovary were as follows: 1) histologic dissimilarity of the tumors; 2) no or only superficial myometrial invasion of endometrial tumor; 3) no vascular space invasion of endometrial tumor; 4) atypical endometrial hyperplasia additionally present; 5) absence of other evidence of spread of endometrial tumor; 6) ovarian tumor unilateral (80–90% of cases); 7) ovarian tumor located in parenchyma; 8) no vascular space invasion, surface implants, or predominant hilar location in ovary; 9) absence of other evidence of spread of ovarian tumor; and 10) ovarian endometriosis present.
Targeted capture massively parallel sequencing
Representative 8μm-thick sections of EC and OC FFPE blocks were microdissected to enrich for tumor cells, and genomic DNA was extracted from micro-dissected tumor and normal tissue/ blood samples using the DNeasy Blood and Tissue Kit (Qiagen), as previously described [21]. Tumor and normal DNAs were subjected to massively parallel sequencing at MSK’s Integrated Genomics Operation (IGO) using the Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) platform, targeting all exons and selected introns of 468 key cancer genes, as previously described [22,23]. The median depth of coverage was 737x (range 292–1296x) for tumor and 393x (range 179–700x) for matched normal samples. Sequencing data were analyzed as previously described [23,24]. In brief, somatic single nucleotide variants (SNVs) were identified using MuTect (v1.17)[25], small insertions and deletions (indels) using Strelka (v1.0.15), VarScan 2 (v2.3.7), Lancet (v1.0.0) and Scalpel (v0.5.3)[26–29] and further curated by manual inspection. In addition, mutations detected in the EC or OC from a given patient were subsequently interrogated in the matched respective EC or OC by manual inspection of BAM files using mpileup files generated from SAMtools (version 1.2 htslib 1.2.1)[30]. Copy number alterations (CNAs) and loss of heterozygosity (LOH) were defined using FACETS [31], as previously described [23,24]. The cancer cell fractions (CCFs) of somatic mutations were computed using ABSOLUTE (v1.0.6)[32], and a mutation was classified as clonal if its probability of being clonal was >50% or if the lower bound of the 95% confidence interval of its CCF was >90%, as previously described [23,24]. Mutational hotspots were annotated according to Chang et al [33]. For the construction of phylogenetic trees based on CNAs, major and minor copy numbers computed by FACETS [31] were modeled using transducer-based pairwise comparison functions using MEDICC [34] assuming a diploid state with no CNAs to root the phylogenies. In addition to the analysis of the synchronous ECs/OCs arising in MMR-deficiency associated syndrome patients, we retrieved and re-analyzed sequencing data from 22 sporadic synchronous ECs/OCs previously published by our group elsewhere [12]. The sporadic synchronous EC/OC patients were confirmed not to harbor germline MMR gene mutations. Of these 22 cases, 17 were subjected to MSK-IMPACT and the remaining 5 to whole-exome sequencing (WES)[12]. Sequencing data were re-analyzed utilizing the analytical pipeline described above.
Clonality analysis
Clonal relatedness between the synchronous EC and OC of a given case was calculated using “clonality index” (CI), based on a previously validated method [12,35], as the probability of the mutations shared between the two lesions not to have co-occurred by chance. Adopting a previously established approach [12,35], we defined . Given the repertoire of mutations of the two samples, the probability of observing a given mutation in both samples is defined by the binomial probability , n=2, k=2, where p is the frequency of a given mutation and n is the number of shared mutations between a pair of components or the average number of mutations found in the two samples in the target regions divided by the size of the target regions. Thus, the probability of observing a given set of M identical mutations in the two samples is given by .
MSIsensor score and mutational signatures
MSIsensor was used to assess microsatellite instability (MSI), as described by Niu et al. [36]. Samples with an MSIsensor score of <3 were considered microsatellite stable, 3–10 indeterminate and ≥10 MSI-high, as previously described [37,38]. Mutational signatures were inferred from mutations present in each EC and OC using Signature Multivariate Analysis (SigMA) at default parameters [39], as previously described [38].
Immunohistochemical analysis
Immunohistochemical analysis for the DNA MMR proteins MLH1, MSH2, MSH6 and PMS2 was performed in the clinical setting, as previously described [40]. Loss of DNA MMR protein expression was defined as the complete absence from all tumor cell nuclei in the presence of a positive internal control, including blood vessels, stromal cells and/or lymphocytes. Cases LS5 and LS6 were also subjected to PTEN immunohistochemistry (IHC) using the monoclonal 138G6 antibody (#9559; Cell Signaling), as previously described [41]. All assays were performed on a Leica Bond 3 (Leica) automated stainer platform.
RESULTS
Clinicopathologic features of synchronous ECs/ OCs in germline DNA MMR gene mutation carriers
Five ECs and OCs synchronously diagnosed in patients with Lynch syndrome (germline MSH2, n=2; MSH6, n=1; and PMS2, n=1) and in one patient with CMMRD (homozygous PMS2 germline mutation) were included in this study (Table 1). Age at diagnosis ranged from 39 to 56 years. All patients were considered to have clinically independent primary tumors (synchronous primaries) based on published criteria [17,19,42], and were diagnosed with early stage disease (Table 1). The ECs and OCs of all cases were of endometrioid histology. The endometrioid EC of LS3 displayed focal papillary features, which were more prominent in the corresponding OC (Table 1). The patterns of loss of DNA MMR protein expression by IHC were in concordance with the respective underlying germline DNA MMR gene alteration (see below; Supplementary Figure S1). As expected in synchronous ECs/OCs arising in Lynch syndrome patients (n=4), targeted massively parallel sequencing analysis revealed a relatively high somatic mutation burden in the ECs/OCs analyzed (median 71.5, range 27 – 390; Supplementary Table S1), few if any CNAs with the exception of case LS5 (see below, Supplementary Figure S2), and high MSIsensor scores (median 31.5, range 15 – 55, Table 1). In contrast, the synchronously diagnosed EC/OC of LS2-CMMRD had fewer somatic mutations (EC, n=13; OC, n=18; Supplementary Table S1) and had an indeterminate microsatellite status based on MSIsensor (EC=5; OC=8; Table 1).
Table 1.
Clinicopathologic characteristics of synchronous endometrial and ovarian cancer cases arising in patients with DNA mismatch repair-deficiency associated syndromes
| Case ID | Age at diagnosis (years) | Germline mutation | Site | Bilateral ovarian disease | Histology | Tumor grade | FIGO Stage | Myometrial invasion | Cervical invasion | LVI | Endometriosis | Clinical diagnosis | Somatic MMR LOH | MSIsensor score | Status | Follow up (years) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LS2- CMMRD | 39 | PMS2 p.S8fs* homozygous | Uterus | Endometrioid | G1 | IIIC1 | <50% | Yes | Yes | Synchronous primaries | No | 5 | DOC | 7 | ||
| Ovary | Yes | Endometrioid | G1 | IB | No | 8 | ||||||||||
| LS3 | 41 | MSH2 p.Q324fs* heterozygous | Uterus | Endometrioid; clear cell features | G2 | IB | >50% | No | Yes | Synchronous primaries | No | 55 | NED | 13 | ||
| Ovary | No | Clear cell | IA | No | 32 | |||||||||||
| LS4 | 49 | PMS2 p.Q30* heterozygous | Uterus | Endometrioid | G1 | IA | None | No | No | Synchronous primaries | Yes | 35 | AWD | 9 | ||
| Ovary | No | Endometrioid | G2 | IC | Yes | 31 | ||||||||||
| LS5 | 49 | MSH2 p.Y521* heterozygous | Uterus | Endometrioid | G1 | II | Superficial | Yes | No | Synchronous primaries | No | 15 | NED | 2 | ||
| Ovary | No | Endometrioid | G1 | IA | Yes | 26 | ||||||||||
| LS6 | 56 | MSH6 p.R1334P heterozygous | Uterus | Endometrioid | G1 | IA | None | No | No | Synchronous primaries | No | ND | NED | 1 | ||
| Ovary | No | Endometrioid | G1 | IA | No | ND |
AWD, alive with disease. CMMRD, constitutional mismatch repair deficiency; DOC, dead of other cause; LOH, loss of heterozygosity; LS, Lynch syndrome; LVI, lymphovascular space invasion; MMR, mismatch repair; NED, no evidence of disease; ND, not defined.
Clonally unrelated synchronously diagnosed EC and OC in Lynch syndrome patients
LS6 harbored a pathogenic germline MSH6 p.R1334P mutation (Table 1, Figure 1). The well-differentiated endometrioid carcinoma in the uterus lacked myoinvasion and was associated with extensive atypical hyperplasia, whereas the well-differentiated endometrioid carcinoma in the ovary displayed focal squamous differentiation. No endometriosis was observed. Targeted massively parallel sequencing of the two FIGO grade 1 endometrioid tumors, revealed a total of 63 somatic mutations in the EC and 27 somatic mutations in the OC, none of which were shared (Figure 1A, Supplementary Table S1). A formal clonality analysis, together with the fact that no shared mutations were identified and that both lesions lacked CNAs, provide evidence to suggest that the endometrioid EC and OC arising in this Lynch syndrome patient LS6 were independent, clonally unrelated tumors (Figure 1A, Supplementary Figure S2). We further noted that despite the presence of a germline MSH6 mutation and a high mutation burden, the EC and OC both displayed a dominant (77% EC, 80% OC) aging-related mutational signature 1 rather than a DNA MMR-related signature. Also, of all cases included, LS6 was the oldest at the age of diagnosis of this synchronous EC/OC (Table 1).
Figure 1: Somatic mutations, mutational signatures and histologic features of the synchronous endometrial and ovarian cancers from Lynch syndrome case LS6.
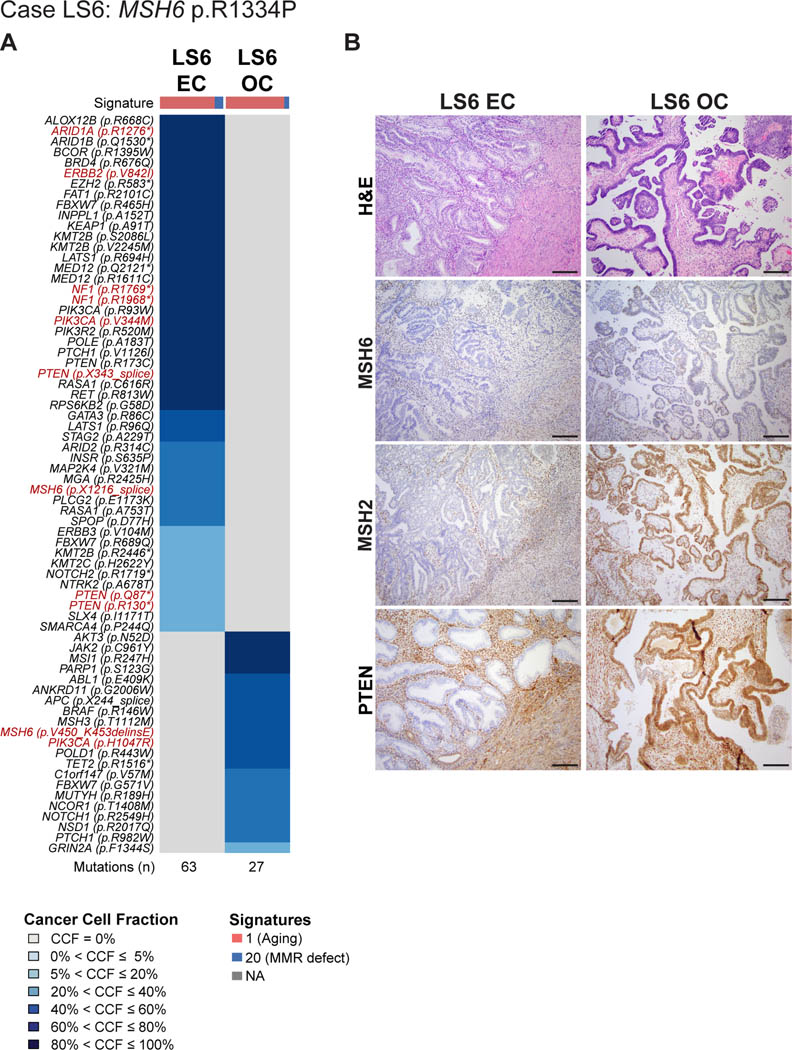
(A) Cancer cell fractions of somatic mutations identified in the endometrial cancer (EC) and synchronous ovarian cancer (OC) of case LS6 harboring a germline MSH6 p.R1334P mutation, color coded according to the legend. The EC and OC both display a dominant mutational signature related to aging (Signature 1). Selected pathogenic mutations are highlighted in red font. The total number of somatic mutations identified in the EC and OC are shown below the heatmap. (B) Micrographs of representative hematoxylin and eosin (H&E), MSH6, MSH2 and PTEN stained sections of the endometrioid EC and OC (scale bar, 100 μm). Loss of MSH6 and MSH2 protein expression was observed in the EC, whereas the OC displayed heterogeneous/ partial loss of MSH6 expression with retention of MSH2 expression. PTEN expression was found to be lost only in the EC, harboring multiple PTEN somatic mutations. CCF, cancer cell fraction; EC, endometrial carcinoma; OC, ovarian carcinoma.
IHC analysis revealed that the EC lacked both MSH6 and MSH2 protein expression, whereas the OC displayed heterogeneous/ partial loss of MSH6 expression but MSH2 expression was retained (Figure 1B, Supplementary Figure S1). Although we observed these distinct MSH6 protein expression patterns, both, the EC and the OC harbored MSH6 somatic mutations, however these were distinct (EC: MSH6 p.X1216 splice site; OC: MSH6 p.V450_K453delinsE). Overall, the somatic mutations present in the EC were characteristic of those described in pure non-synchronous endometrioid cancers, including ARID1A, PIK3CA and PTEN mutations in the EC [43], whereas the OC only displayed a PIK3CA mutation characteristic of endometroid histology. Notably, the PIK3CA mutations were distinct; the EC was found to harbor two PIK3CA mutations (p.R93W and p.V344M hotspot), whereas the OC displayed a PIK3CA p.H1047R hotspot mutation. Additionally, PTEN mutations were present in the EC (p.R130*, p.Q87*, p.R173C, and p.X343_splice) but not detected in the OC. Consistent with this genetic finding, immunohistochemical analysis revealed loss of PTEN expression in the EC but not in the OC (Figure 1B).
Mutationally un-related/ copy number-related synchronously diagnosed EC and OC in a Lynch syndrome patient
LS5 harbored a germline nonsense MSH2 (p.Y521*) mutation (Table 1, Figure 2). Both synchronously diagnosed endometrioid tumors were well-differentiated. The ovarian tumor arose in an adenofibroma with areas of borderline endometrioid tumor, with foci of endometrial-like stroma within the adenofibroma. Sequencing analysis revealed at total of 35 somatic mutations in the EC and 42 in the OC, none of which were shared. Furthermore, whilst the EC component harbored a somatic MSH2 frameshift mutation (p.A771Lfs*38), no second somatic alteration of MSH2 in the form of a mutation or LOH was identified in the OC of LS5 (Figure 2A). Despite the absence of somatic inactivation of the MSH2 wild-type allele in the OC, both the EC and OC displayed a dominant mutational signature 6 associated with defective DNA MMR (89% EC, 95% OC), had high MSI-sensor scores (15 EC, 26 OC; Table 1, Figure 2A) and IHC revealed MSH2 and MSH6 loss of protein expression in the EC and OC (Supplementary Figure S1). PI3K pathway mutations were present in both, the EC and OC; whilst the OC harbored a PIK3CA p.E545D hotspot mutation, the EC harbored a PIK3R2 p.A444T mutation. The EC also displayed a somatic PTEN p.T319* truncating mutation, which was associated with distinct PTEN protein expression pattern between the two lesions (Figure 2B).
Figure 2: Somatic mutations, mutational signatures, copy number alterations and histologic features of the synchronous endometrial and ovarian cancers from Lynch syndrome case LS5.

(A) Cancer cell fractions of somatic mutations identified in the endometrial cancer (EC) and synchronous ovarian cancer (OC) of case LS5 harboring a germline MSH2 p.Y521* mutation, color coded according to the legend. The EC and OC both display a dominant mutational signature related to DNA mismatch repair defects (Signature 6). Selected pathogenic mutations are highlighted in red font. The total number of somatic mutations identified in the EC and OC are shown below the heatmap. (B) Micrographs of representative hematoxylin and eosin (H&E) and PTEN stained sections of the endometrioid EC and OC (scale bar, 100 μm). Loss of PTEN expression was found only in the EC harboring a PTEN somatic mutation. (C) Chromosome plots of the EC and OC, with the Log2-ratios plotted on the y-axis and the genomic positions on the x-axis (top). Phylogenetic tree based on the copy number alterations depicting the evolution of the EC and OC (bottom). CCF, cancer cell fraction; EC, endometrial carcinoma; OC, ovarian carcinoma.
Analysis of the copy number profiles revealed, however, that although the mutational profiles were distinct, the repertoire of CNAs of the EC/OC of LS5 were similar and shared the same gains on chromosomes 9q, 16q and 19 (Figure 2C). These findings are consistent with the notion that the CNAs in the EC and OC of LS5 preceded the acquisition of somatic mutations. These data further suggest that unlike LS6, the EC and OC of LS5 are likely clonally related given their similar patterns of CNAs, irrespective of their distinct mutational profiles, derived from the analysis of 468 genes.
Clonally related synchronously diagnosed ECs and OCs in patients with DNA mismatch repair-deficiency associated syndromes
Unlike the Lynch syndrome case previously reported by our team [12] and LS6 from this study, three of the five synchronous ECs/OCs were clonally related, lacking CNAs and harboring similar mutation profiles (i.e. LS2-CMMRD, LS3, and LS4; Figure 3, Supplementary Figure 2).
Figure 3: Somatic mutations, mutational signatures, directionality of progression and clonality of synchronous endometrial and ovarian cancers from DNA mismatch repair germline mutation carriers LS2-CMMRD, LS3 and LS4.

Cancer cell fractions of somatic mutations identified in the endometrial cancer (EC) and synchronous ovarian cancer (OC) of cases LS2-CMMRD (A), LS3 (B) and LS4 (C), color coded according to the legend. Detailed view of subclonal mutations in the EC that became clonal in the OC on the right. Total number of somatic mutations identified in a given EC and OC are shown below the heatmaps. Micrographs of representative hematoxylin and eosin (H&E) stained sections of the EC and OC (scale bars, 100 μm; top right). Directionality of progression from the EC to the OC based on the clonality of mutations (capital letters, clonal; lower case letters, subclonal) are shown in the bottom right. (A) Case LS2-CMMRD harboring a homozygous germline PMS2 p S8fs* mutation (constitutional mismatch repair deficiency syndrome). The endometrioid EC and endometrioid OC both display a dominant mutational signature related to DNA mismatch repair defects (Signature 26), and harbor a clonal FBXW7 mutation. (B) Lynch syndrome case LS3 harboring a MSH2 p.Q324fs* mutation. The endometrioid EC with clear cell features and primarily clear cell OC both display a dominant mutational signature related to DNA mismatch repair defects (Signature 20), and harbor a clonal PIK3CA mutation. (C) Lynch syndrome case LS4 harboring a homozygous germline PMS2 p.Q30* mutation. The endometrioid EC and endometrioid OC both display a dominant mutational signature related to DNA mismatch repair defects (Signature 15), and harbor a clonal APC mutation. CCF, cancer cell fraction; EC, endometrial carcinoma; OC, ovarian carcinoma.
LS2-CMMRD harbored a homozygous germline PMS2 (p.S8fs*) mutation, a case of CMMRD (Table 1). The histologic features of the synchronous EC/OC were identical; both lesions were low-grade endometrioid carcinomas displaying squamous differentiation (Figure 3). No endometriosis was observed. The mutational burden of LS2-CMMRD’s EC/OC (EC, n=13; OC, n=18) were lower than those found in the Lynch syndrome cases in this study (median 71.5, range 27–390) and not MSI-high, but rather MSI-indeterminate. Of the total 13 somatic mutations in the EC and 18 in the OC, 8 were shared (Figure 3A), one of which was a clonal FBXW7 p.R505C mutation. Among the remaining shared mutations, a subset was subclonal in the EC, including AR p.Q803Pfs*27, SPEN p.D1364G and CTNNB1 p.S33P mutations, while fully clonal in the OC (Figure 3A, Supplementary Table S1); suggesting that a minor subclone of the EC likely constituted the substrate from which the OC originated. As for the mutations restricted to each component, the OC was found to harbor pathogenic mutations affecting ARID1A (p.M1634Hfs*14) and PIK3R1 (p.X582_splice, Supplementary Table S1), while the EC had a private pathogenic mutation in NOTCH1 (p.L573P). Although LS2-CMMRD harbored a lower number of somatic mutations, also in this case a dominant mutational signature 26, associated with defective DNA mismatch repair (MSI), was present in both the EC (46%) and OC (44%; Figure 3A).
LS3 harbored a germline frameshift MSH2 (p.Q324fs*) mutation and was considered to be MSI-high (Table 1). The endometrial tumor exhibited a glandular and papillary architecture with scattered foci of marked cytologic atypia (grade 2), and both myometrial and lymphovascular invasion were present. The ovarian tumor was embedded in fibrous stroma and exhibited a tubulocystic architecture with moderate to focal marked nuclear atypia and clear cytoplasm, consistent with a clear cell carcinoma arising in the background of an adenofibroma. The EC/OC both displayed loss of MSH6 and MSH2 protein expression (Supplementary Figure S1). Similar to LS2-CMMRD, clonality analysis of this case showed that both the EC and OC components were clonally related (Figures 3B–3C). Of the 335 total somatic mutations in the EC and 80 in the OC, 51 were found to be shared, whereas 284 and 28 were found to be restricted to the EC and OC, respectively (Supplementary Table S1). Amongst others, a clonal hotspot mutation in PIK3CA (p.Q546R) was shared between the EC/OC as was a somatic MSH2 frameshift (p.F887Sfs*5) mutation. A dominant mutational signature 20 associated with defective DNA mismatch repair (MSI) was present in the EC (61%) and OC (52%; Figure 3B). Akin to LS2-CMMRD, clonality analysis provided evidence to suggest that the OC likely originated from a minor subclone of the EC as supported by a subset of 8 subclonal mutations in the EC becoming clonal in the OC, including a pathogenic KIT p.K710N mutation.
LS4 harbored a germline nonsense PMS2 pQ30* mutation associated with LOH of the wild-type allele and was considered to be MSI-high (Table 1). The EC demonstrated cribriform growth characteristics of a grade 1 endometrioid carcinoma, and the ovarian tumor was composed of back-to-back glands with an endometrioid appearance with focal areas of solid growth (grade 2), and a small cyst with features of endometriosis. Among all cases included in this study, LS4 encompassed the largest total number of mutations. A total of 390 somatic mutations were identified in the EC and 324 in the OC, of which 286 were shared, whereas 104 and 38 where private to the EC and OC, respectively (Figure 3C, Supplementary Table S1). A clonal hotspot mutation in SOS1 (p.N233Y) as well as a pathogenic APC (p.R2439C) missense mutation were shared between the EC/OC. LS4, akin to LS2 and LS3, not only demonstrated a clonal origin between the two synchronous lesions (Figure 3D), but also provided evidence to suggest that a minor subclone of the EC was likely the origin of the OC. Specifically, 48 subclonal mutations in EC were found to be clonal in the OC, including ARID1A (p.M156Hfs*8), PIK3CA (p.V344A), PIK3R1 (p.I82Nfs*24) and ATM (p.R2832C). Both the EC and OC had a dominant mutational signature 15 associated with defective DNA mismatch repair (MSI; Figure 3C, Supplementary Table S1).
All women with Lynch syndrome-associated synchronous EC/OC had no evidence of disease (n=3) or were alive with disease (n=1) at last follow-up (range 1–13 years; Table 1), irrespective of the clonal relatedness of the synchronously diagnosed tumors in the uterus and ovary. The CMMRD patient developed other malignancies and died of other cause 7 years following the synchronous EC/OC diagnosis (Table 1).
Assessment of the directionality of progression in sporadic synchronous ECs and OCs
Given that we observed a likely progression from the EC to the OC in the clonally related DNA MMR-deficiency associated cases, we sought to define whether the same chronology of events would be applicable to sporadic synchronous ECs/OCs. Sequencing data reported in Schultheis et al [12], including 5 sporadic ECs/OCs subjected to WES and 17 sporadic ECs/OCs subjected to targeted massively parallel sequencing, were re-analyzed and utilized to infer the clonal evolutionary pattern of these 22 sporadic synchronous ECs/OCs. The clonality analysis provided objective information on the directionality of progression in 2/5 cases subjected to WES (i.e. SYN2, SYN5) and 6/17 cases subjected to MSK-IMPACT sequencing (i.e. SP1, SP2, SP3, SP9, SV5, and SP5). Uniformly, in these 8 cases, the most parsimonious explanation for their evolution was that the OC likely stemmed from a minor subclone of the EC (Figure 4). The directionality in these cases was inferred on the basis of the presence of multiple subclonal mutations present in the EC that became clonal in the respective OC. This includes KMT2A and PIK3CA mutations that were subclonal in the EC that became clonal in both the left and right OCs in SP1. In SP9, KRAS hotspot and ARID1A mutations were subclonal in the EC and clonal in the OC. In SP3, POLE, ARID1A, PIK3CA and two PTEN mutations were all subclonal in the EC and clonal in the OC. In SP5, in addition to a clonal PTEN (p.R130Q) hotspot mutation present in both components, a PTEN (p.L42R) mutation as well as two APC mutations were subclonal in the EC and clonal in the OC. Furthermore, subclonal BCL6 (SV5), CREBBP (SP2), BAI3 (SYN2) and PIK3R1 (SYN5) mutations in ECs became clonal their respective OC (Figure 4). The remainder of the cases re-analyzed also demonstrated clonal relatedness [12], however the chronology of the development of the two synchronous lesions could not be deduced based on the targeted sequencing results (Supplementary Figure S3).
Figure 4: Clonal composition analysis and directionality of progression of sporadic synchronous endometrioid endometrial and endometrioid ovarian cancers.

Re-analysis of sporadic synchronous endometrial (ECs) and ovarian cancers (OCs) [12] subjected to whole-exome sequencing (SYN2, SYN5) or targeted MSK-IMPACT sequencing (SP1, SP2, SP3, SP9, SV5, SP5). Cases for which chronology of development could be inferred based on clonal composition analysis are shown. For each case, cancer cell fractions of somatic mutations identified in the EC and synchronous OC are depicted, color coded according to the legend, with detailed view of subclonal mutations in the EC that became clonal in the OC on the right. Total number of somatic mutations identified in a given EC and OC are below the heatmaps. Directionality of progression from the EC to the OC based on the clonality of mutations (capital letters, clonal; lower case letters, subclonal) are shown in the bottom right. CCF, cancer cell fraction; EC, endometrial carcinoma; OC, ovarian carcinoma.
DISCUSSION
Here we demonstrate that within the context of Lynch syndrome but not CMMRD there is a subset of synchronously diagnosed ECs/OCs that are clonally unrelated and represent two separate independent primaries. Combining our previously published synchronous EC/OC Lynch syndrome case, SV2 [12], with the analyses performed here, we report a 40% (2/5) rate of independent primary tumors versus metastatic lesions in patients with Lynch syndrome. This is in contrast to sporadic synchronous clinically independent ECs/OCs, which have been consistently shown to be clonally related and metastases from each other, as demonstrated by both our group and others [13–16,41,44,45].
Using single cell sequencing of breast cancers, Navin and colleagues have shown that the majority of CNAs are acquired at the earliest stages of tumor evolution, in short punctuated bursts, followed by stable clonal expansions, whilst point mutations evolved gradually over long periods of time [46,47]. MSI-high ECs are generally characterized by a high mutational burden and the lack of CNAs [43]. The synchronous EC/OC of LS5 analyzed in this study, from a MSH2 germline mutation carrier, was found to harbor CNAs (fraction of genome altered: EC=8%, OC=12%), many of which were shared between the EC and OC, whereas none of the somatic mutations were shared. One may hypothesize that these ECs/OCs have a common origin, with CNAs acquired as early events and the acquisition of mutations at later stages of tumor evolution. Alternatively, given that samples from this patient were analyzed with MSK-IMPACT, we cannot rule out that somatic mutations present in both the EC and OC were present but not assessed in the genomic regions covered by this targeted capture sequencing assay.
The somatic genetics of ECs arising in the context of CMMRD have yet to be characterized. One of the cases included in this study, LS2-CMMRD, harbored a homozygous PMS2 germline mutation. We noted that the synchronous EC/OC of LS2-CMMRD harbored a lower mutational burden than the Lynch syndrome cases studied here, and although tumors from this patient were not deemed MSI-high by MSI-sensor, they did harbor dominant mutational signatures associated with DNA MMR defects. Mutations that were shared between the synchronously diagnosed EC/OC in LS2-CMMRD included ARID5B, AR and PTEN frameshift mutations as well as CTNNB1 hotspot, FBXW7 and SPEN mutations.
In case LS5, which harbored a germline nonsense MSH2 p.Y521* mutation, the EC had a somatic MSH2 mutation, however in the OC no second somatic alteration of MSH2 in the form of a mutation or LOH was identified. Notably, both EC and OC of LS5 harbored dominant mutational signatures associated with defective DNA MMR, had high MSI-sensor scores and showed MSH2 and MSH6 loss of protein expression. These findings are consistent with the notion that yet another mechanism not detectable by targeted massively parallel sequencing (e.g. MSH2 promoter hypermethylation as described in Lynch syndrome colorectal cancers [48]) may serve as the ‘second hit’.
Using clonal composition analyses, we assessed the directionality of the evolutionary pattern in the clonally-related DNA MMR-deficiency syndrome cases. In these three cases, we found evidence to suggest that a minor subclone of the EC gave rise to the OC. This was further validated by a re-analysis of sporadic synchronous EC/OCs from Schultheis et al [12]. In the clonally related Lynch syndrome, CMMRD, and sporadic synchronous ECs/OCs analyzed here, and for which we could bioinformatically infer the directionality from primary EC to ovarian metastasis, 4/12 were associated with endometriosis (2/3 Lynch; 0/1 CMMRD; 2/8 sporadic). Endometriosis has been suggested to play a role in the development of synchronous ECs/OCs and the absence of ovarian endometriosis is one of the pathologic criteria used to support primary EC with ovarian metastasis [19]. These data suggest that the presence of endometriosis may not be necessarily required for the development of an endometrioid OC synchronously diagnosed with EC in the context of clonally-related lesions, as the OC likely constitutes dissemination of the EC. It should be noted, however, that although this is the most parsimonious explanation for our findings where directionality of the progression could be inferred from EC to OC, we cannot rule out other biological processes. Clonally related ECs/OCs could also stem from endometrium already harboring early somatic genetic alterations giving rise to the endometriosis. In this scenario both the eutopic endometrium and endometriosis would harbor genetic alterations in common, and the EC and OC developing from these different areas would be de facto clonally related, however the EC and OC would harbor a substantial number of somatic genetic alterations restricted to each lesion, given the divergent evolution after the implantation of the ectopic endometrium. Further studies are required, to assess whether the endometriosis harbors mutations found in the synchronous EC/OC.
Based on these findings one could hypothesize a model in the setting of Lynch syndrome, where the likelihood of transformation happening in the endometrium, either eutopic or ectopic, is high. Hence, in this context, synchronous ECs/OCs can either constitute i) clonally-related lesions in the form of a primary EC giving rise to the OC, or ii) independent lesions, whereby the endometrium and the ectopic endometrial cells would transform independently and have no somatic genetic alterations in common (Figure 5). In the context of sporadic cancers, the likelihood of transformation of the eutopic or ectopic endometrium is low; therefore, the majority of synchronous ECs/OCs are clonally related and likely derived from a primary EC “metastasizing” to the ovary. It should be noted, however, that in the context of clonally related synchronous ECs/OCs, both in the sporadic and Lynch/CMMRD settings, another possibility that could be entertained includes one where the endometrium that gave rise to the endometriosis, albeit not entirely transformed, already harbored somatic genetic alterations acquired early in the development of ECs [49,50]. In this case, there would be a subset of somatic genetic alterations shared between the EC and the OC, hence the lesions would be clonal, however a high number of genetic alterations that diverge between the two lesions would be expected (Figure 5).
Figure 5. Model of synchronous endometrial and ovarian cancer development in the Lynch syndrome and sporadic settings.

One could hypothesize a model in the setting of Lynch syndrome, where the likelihood of transformation happening in the endometrium, either eutopic or ectopic, is high (⚡). Synchronous endometrial carcinomas (ECs) and ovarian carcinomas (OCs) can either constitute independent lesions, whereby the endometrium and the ectopic endometrial cells would transform independently and have no somatic genetic alterations in common (left) or be clonally related lesions in the form of a primary EC giving rise to the OC (middle). In the context of sporadic cancers, the likelihood of transformation of the eutopic or ectopic endometrium is lower; therefore, the majority of synchronous ECs/OCs are clonally related and likely derived from a primary EC “metastasizing” to the ovary (middle). It should be noted, however, that in the context of clonally related synchronous ECs/OCs, both in the sporadic and Lynch/CMMRD settings, another possibility that could be entertained includes one where the endometrium that gave rise to the endometriosis, albeit not entirely transformed, already harbored somatic genetic alterations acquired early in the development of ECs. In this case, there would be a subset of somatic genetic alterations shared between the EC and the OC, hence the lesions would be clonal, however a high number of genetic alterations that diverge between the two lesions would be expected. CMMRD, constitutional mismatch repair deficiency.
Our study is limited by small sample size, however we are providing important additional data in this space, given the rarity of synchronous ECs/OCs occurring in the setting of Lynch syndrome/ CMMRD. We suggest there is a subset of synchronous ECs/OCs in women with Lynch syndrome that are not clonally related and constitute independent primary tumors. Targeted massively parallel sequencing with MSK-IMPACT was the basis for the genomic profiling of these samples, which targets a limited number of genes (n=468). Whole-exome or whole-genome sequencing analyses in a larger series of cases are warranted to provide more definitive results regarding the clonality of these cases. Despite these limitations, we further show that synchronous ECs/OCs most often originate from the EC and give rise to the OC in the Lynch syndrome, CCMRD and sporadic settings.
Supplementary Material
ACKNOWLEDGEMENTS
JSR-F was funded in part by a Breast Cancer Research Foundation grant, BW in part by Breast Cancer Research Foundation, Cycle for Survival and Stand Up To Cancer grants, F.P. partially by a National Institutes of Health/National Cancer Institute K12 CA184746 grant, XM-G by AECC (grupos estables) and GC and MP by the Spanish Ministry of Economy and Competitiveness and co-funded by FEDER funds -a way to build Europe- (grant SAF2015-68016-R), CIBERONC and the Government of Catalonia (grant 2017SGR1282). XM-G, GC and MP thank CERCA Programme for institutional support. Research reported in this publication was supported in part by a Cancer Center Support Grant of the NIH/NCI (Grant No. P30CA008748).
Footnotes
CONFLICTS OF INTEREST
N.R.A-R reports institutional grants from Stryker/Novadaq, Olympus and GRAIL, outside the submitted work. JSR-F reports receiving personal/consultancy fees from Goldman Sachs and REPARE Therapeutics, membership of the scientific advisory boards of VolitionRx and Paige.AI, and ad hoc membership of the scientific advisory boards of Roche Tissue Diagnostics, Ventana Medical Systems, Novartis, Genentech and InVicro, outside the scope of this study. ZKS’s immediate family member holds consulting/advisory roles in Ophthalmology with Allergan, Adverum Biotechnologies, Alimera Sciences, Biomarin, Fortress Biotech, Genentech/Roche, Novartis, Optos, Regeneron, Regenxbio, Spark Therapeutics, outside of this work. The remaining authors have no conflicts of interest to declare.
REFERENCES
- 1.Aarnio M, Sankila R, Pukkala E, Salovaara R, Aaltonen LA, de la Chapelle A, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer. 1999;81:214–8. [DOI] [PubMed] [Google Scholar]
- 2.Wimmer K, Kratz CP, Vasen HF, Caron O, Colas C, Entz-Werle N, et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium ‘care for CMMRD’ (C4CMMRD). J Med Genet. 2014;51:355–65. [DOI] [PubMed] [Google Scholar]
- 3.Lavoine N, Colas C, Muleris M, Bodo S, Duval A, Entz-Werle N, et al. Constitutional mismatch repair deficiency syndrome: clinical description in a French cohort. J Med Genet. 2015;52:770–8. [DOI] [PubMed] [Google Scholar]
- 4.Bakry D, Aronson M, Durno C, Rimawi H, Farah R, Alharbi QK, et al. Genetic and clinical determinants of constitutional mismatch repair deficiency syndrome: report from the constitutional mismatch repair deficiency consortium. Eur J Cancer. 2014;50:987–96. [DOI] [PubMed] [Google Scholar]
- 5.Dominguez-Valentin M, Sampson JR, Seppala TT, Ten Broeke SW, Plazzer JP, Nakken S, et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genet Med. 2020;22:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rossi L, Le Frere-Belda MA, Laurent-Puig P, Buecher B, De Pauw A, Stoppa-Lyonnet D, et al. Clinicopathologic Characteristics of Endometrial Cancer in Lynch Syndrome: A French Multicenter Study. Int J Gynecol Cancer. 2017;27:953–60. [DOI] [PubMed] [Google Scholar]
- 7.Helder-Woolderink JM, Blok EA, Vasen HF, Hollema H, Mourits MJ, De Bock GH. Ovarian cancer in Lynch syndrome; a systematic review. Eur J Cancer. 2016;55:65–73. [DOI] [PubMed] [Google Scholar]
- 8.Soliman PT, Broaddus RR, Schmeler KM, Daniels MS, Gonzalez D, Slomovitz BM, et al. Women with synchronous primary cancers of the endometrium and ovary: do they have Lynch syndrome? J Clin Oncol. 2005;23:9344–50. [DOI] [PubMed] [Google Scholar]
- 9.Lim YK, Padma R, Foo L, Chia YN, Yam P, Chia J, et al. Survival outcome of women with synchronous cancers of endometrium and ovary: a 10 year retrospective cohort study. J Gynecol Oncol. 2011;22:239–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams MG, Bandera EV, Demissie K, Rodriguez-Rodriguez L. Synchronous primary ovarian and endometrial cancers: a population-based assessment of survival. Obstet Gynecol. 2009;113:783–9. [DOI] [PubMed] [Google Scholar]
- 11.Ramus SJ, Elmasry K, Luo Z, Gammerman A, Lu K, Ayhan A, et al. Predicting clinical outcome in patients diagnosed with synchronous ovarian and endometrial cancer. Clin Cancer Res. 2008;14:5840–8. [DOI] [PubMed] [Google Scholar]
- 12.Schultheis AM, Ng CK, De Filippo MR, Piscuoglio S, Macedo GS, Gatius S, et al. Massively Parallel Sequencing-Based Clonality Analysis of Synchronous Endometrioid Endometrial and Ovarian Carcinomas. J Natl Cancer Inst. 2016;108:djv427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anglesio MS, Wang YK, Maassen M, Horlings HM, Bashashati A, Senz J, et al. Synchronous Endometrial and Ovarian Carcinomas: Evidence of Clonality. J Natl Cancer Inst. 2016;108:djv428. [DOI] [PubMed] [Google Scholar]
- 14.Chao A, Wu RC, Jung SM, Lee YS, Chen SJ, Lu YL, et al. Implication of genomic characterization in synchronous endometrial and ovarian cancers of endometrioid histology. Gynecol Oncol. 2016;143:60–7. [DOI] [PubMed] [Google Scholar]
- 15.Niskakoski A, Pasanen A, Porkka N, Eldfors S, Lassus H, Renkonen-Sinisalo L, et al. Converging endometrial and ovarian tumorigenesis in Lynch syndrome: Shared origin of synchronous carcinomas. Gynecol Oncol. 2018;150:92–8. [DOI] [PubMed] [Google Scholar]
- 16.Kelemen LE, Rambau PF, Koziak JM, Steed H, Kobel M. Synchronous endometrial and ovarian carcinomas: predictors of risk and associations with survival and tumor expression profiles. Cancer Causes Control. 2017;28:447–57. [DOI] [PubMed] [Google Scholar]
- 17.Kurman RJ, Carcangiu ML, Herrington CS, et al. WHO Classification of Tumours of Female Reproductive Organs. IARC: Lyon, 2014. [Google Scholar]
- 18.FIGO staging for carcinoma of the vulva, cervix, and corpus uteri. International Journal of Gynecology & Obstetrics. 2014;125:97–8. [DOI] [PubMed] [Google Scholar]
- 19.Scully RE YR, Clement PB. Tumors of the ovary, maldeveloped gonads, fallopian tube, and broad ligament. Armed Forces Institute of Pathology: Washington DC, 1998. [Google Scholar]
- 20.Soliman PT, Slomovitz BM, Broaddus RR, Sun CC, Oh JC, Eifel PJ, et al. Synchronous primary cancers of the endometrium and ovary: a single institution review of 84 cases. Gynecol Oncol. 2004;94:456–62. [DOI] [PubMed] [Google Scholar]
- 21.Martelotto LG, De Filippo MR, Ng CK, Natrajan R, Fuhrmann L, Cyrta J, et al. Genomic landscape of adenoid cystic carcinoma of the breast. J Pathol. 2015;237:179–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn. 2015;17:251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weigelt B, Bi R, Kumar R, Blecua P, Mandelker DL, Geyer FC, et al. The Landscape of Somatic Genetic Alterations in Breast Cancers From ATM Germline Mutation Carriers. J Natl Cancer Inst. 2018;110:1030–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Da Cruz Paula A, da Silva EM, Segura SE, Pareja F, Bi R, Selenica P, et al. Genomic profiling of primary and recurrent adult granulosa cell tumors of the ovary. Mod Pathol. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–7. [DOI] [PubMed] [Google Scholar]
- 27.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Narzisi G, Corvelo A, Arora K, Bergmann EA, Shah M, Musunuri R, et al. Genome-wide somatic variant calling using localized colored de Bruijn graphs. Commun Biol. 2018;1:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Narzisi G, O’Rawe JA, Iossifov I, Fang H, Lee YH, Wang Z, et al. Accurate de novo and transmitted indel detection in exome-capture data using microassembly. Nat Methods. 2014;11:1033–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012;30:413–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang MT, Bhattarai TS, Schram AM, Bielski CM, Donoghue MTA, Jonsson P, et al. Accelerating Discovery of Functional Mutant Alleles in Cancer. Cancer Discov. 2018;8:174–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwarz RF, Trinh A, Sipos B, Brenton JD, Goldman N, Markowetz F. Phylogenetic quantification of intra-tumour heterogeneity. PLoS Comput Biol. 2014;10:e1003535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JY, Schizas M, Geyer FC, Selenica P, Piscuoglio S, Sakr RA, et al. Lobular Carcinomas In Situ Display Intralesion Genetic Heterogeneity and Clonal Evolution in the Progression to Invasive Lobular Carcinoma. Clin Cancer Res. 2019;25:674–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics. 2014;30:1015–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Middha S, Yaeger R, Shia J, Stadler ZK, King S, Guercio S, et al. Majority of B2M-Mutant and -Deficient Colorectal Carcinomas Achieve Clinical Benefit From Immune Checkpoint Inhibitor Therapy and Are Microsatellite Instability-High. JCO Precis Oncol. 2019;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith ES, Paula ADC, Cadoo KA, Abu-Rustum NR, Pei X, Brown DN, et al. Endometrial Cancers in BRCA1 or BRCA2 Germline Mutation Carriers: Assessment of Homologous Recombination DNA Repair Defects. JCO Precision Oncology. 2019:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gulhan DC, Lee JJ, Melloni GEM, Cortes-Ciriano I, Park PJ. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat Genet. 2019;51:912–9. [DOI] [PubMed] [Google Scholar]
- 40.Modica I, Soslow RA, Black D, Tornos C, Kauff N, Shia J. Utility of immunohistochemistry in predicting microsatellite instability in endometrial carcinoma. Am J Surg Pathol. 2007;31:744–51. [DOI] [PubMed] [Google Scholar]
- 41.Geyer FC, Pareja F, Burke KA, Schultheis AM, Hussein YR, Ye J, et al. Genetic analysis of a morphologically heterogeneous ovarian endometrioid carcinoma. Histopathology. 2017;71:480–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ulbright TM, Roth LM. Metastatic and independent cancers of the endometrium and ovary: a clinicopathologic study of 34 cases. Hum Pathol. 1985;16:28–34. [DOI] [PubMed] [Google Scholar]
- 43.Cancer Genome Atlas Research Network, Kandoth CN, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hajkova N, Ticha I, Hojny J, Nemejcova K, Bartu M, Michalkova R, et al. Synchronous endometrioid endometrial and ovarian carcinomas are biologically related: A clinico-pathological and molecular (next generation sequencing) study of 22 cases. Oncol Lett. 2019;17:2207–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guerra F, Girolimetti G, Perrone AM, Procaccini M, Kurelac I, Ceccarelli C, et al. Mitochondrial DNA genotyping efficiently reveals clonality of synchronous endometrial and ovarian cancers. Mod Pathol. 2014;27:1412–20. [DOI] [PubMed] [Google Scholar]
- 46.Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol. 2016;34:155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi X, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512:155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nagasaka T, Rhees J, Kloor M, Gebert J, Naomoto Y, Boland CR, et al. Somatic hypermethylation of MSH2 is a frequent event in Lynch Syndrome colorectal cancers. Cancer Res. 2010;70:3098–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lac V, Nazeran TM, Tessier-Cloutier B, Aguirre-Hernandez R, Albert A, Lum A, et al. Oncogenic mutations in histologically normal endometrium: the new normal? J Pathol. 2019;249:173–81. [DOI] [PubMed] [Google Scholar]
- 50.Moore L, Leongamornlert D, Coorens THH, Sanders MA, Ellis P, Dentro SC, et al. The mutational landscape of normal human endometrial epithelium. Nature. 2020;580:640–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.