

Abstract
Brain derived neurotrophic factor (BDNF), a member of the neurotrophin family, has an extensively studied classical role in neuronal growth, differentiation, survival, and plasticity. Neurotrophic, from the Greek neuro and trophos, roughly translates as “vital nutrition for the brain.” During development, BDNF and its associated receptor tyrosine receptor kinase B (TrkB) are tightly regulated as they influence the formation and maturation of neuronal synapses. Preclinical research investigating the role of BDNF in neurological disorders has focused on the effects of decreased BDNF expression on the development and maintenance of neuronal synapses. In contrast, heightened BDNF-TrkB activity has received less scrutiny for its role in neurological disorders. Recent studies suggest that excessive BDNF-TrkB signaling in the developing brain may promote the hyperexcitability that underlies refractory neonatal seizures. This review will critically examine BDNF-TrkB signaling in the immature brain, its role in the emergence of refractory neonatal seizures, and the potential of targeting BDNF-TrkB signaling as a novel anti-seizure strategy.
Keywords: Seizures, BDNF, TrkB, KCC2, Epilepsy, Development
The Bdnf gene
Bdnf has a complex transcriptome consisting of nine functional promoters that permit at least twenty different transcripts. The Bdnf coding sequence resides in exon IX with eight other upstream exons encoding promoter elements. Each Bdnf transcript consists of a 5’ untranslated region exon that is alternatively spliced with the conserved common coding region within exon IX at the 3’ end, which results in each transcript encoding for an identical pre-proBDNF protein [1–3]. The nine promoters of Bdnf are suggested to permit its spatial, temporal, and stimulus specific production [1]. Future research will establish the structural differences of the human BDNF gene as well as the presence of antisense RNA.
BDNF transcripts are widely distributed in the rodent brain with notably high levels in the hippocampus and entorhinal cortex [2]. The majority of BDNF is produced from Bdnf promoters I, II, IV, or VI. Promoters IV and VI are the greatest contributors to total BDNF production in contrast to promoters I and II [1]. Promoter IV is associated with activity-dependent expression after physiological stimuli and structural long-term potentiation of glutamatergic synapses. The prevention of promoter I- or II-specific BDNF results in elevated aggressive behavior during young adulthood, suggesting that individual Bdnf transcripts are functionally relevant for the regulation of specific behaviors [1]. The increase of (pan) Bdnf expression and BDNF protein occurs after seizures [2] however the promoter, cell type, and age-dependent differences in neonatal post-seizure Bdnf expression are currently unknown (Figure 1).
Figure 1.
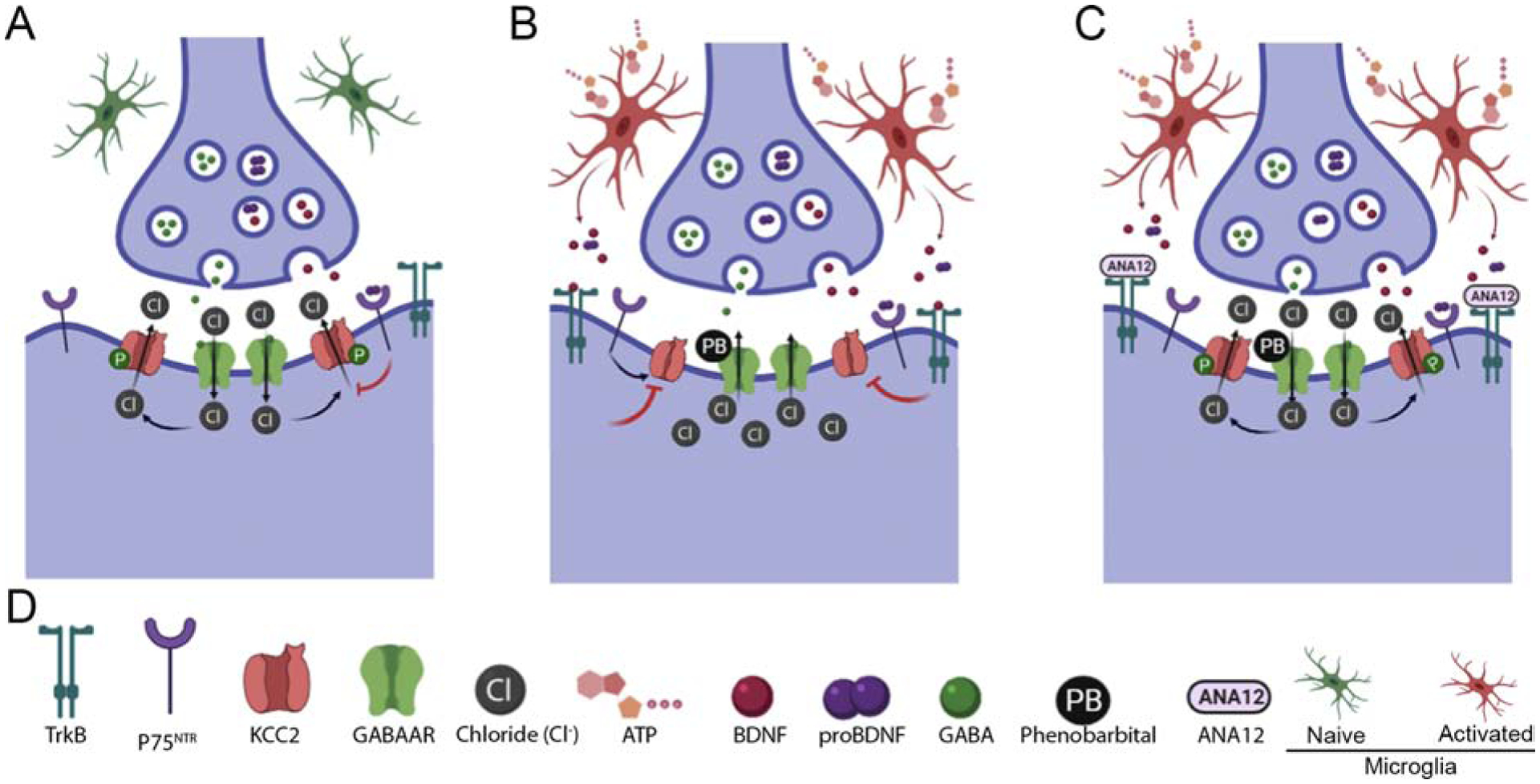
Post-ischemic BDNF-TrkB signaling is proposed to facilitate the emergence of phenobarbital unresponsive neonatal seizures. (A) Schematized inhibitory synapses in a non-pathological neonatal brain, (B) post-ischemic insult, and (C) ANA12 treated post-ischemic insult. (D) Legend for proteins, molecules, and cell types of interest. An improved understanding of the multiples levels of BDNF regulation in neonatal seizures is necessary.
The BDNF protein
BDNF is initially translated in the endoplasmic reticulum as the precursor protein pre-proBDNF, which is then cleaved to form proBDNF and transported into the Golgi apparatus. proBDNF is sorted into either the regulated or constitutive secretory pathways. Intra-Golgi sortilin and intracellular huntingtin protein form a putative complex that determines the targeting of proBDNF to secretory granules and their transport along microtubules [3]. The conversion of proBDNF to mature BDNF (referred to as BDNF henceforth) is regulated by conversion enzymes and can occur intra- or extracellularly. proBDNF can be co-released with BDNF and/or conversion enzymes [3]. While the predominant cellular localization of BDNF is at pre-synaptic terminals, the synthesis and secretion of BDNF also occurs within post-synaptic dendrites, astrocytes, and microglia [3]. Furthermore, the secretion of BDNF depends on the elevation of intracellular Ca2+ and subsequent CaMKII activation [3]. Ca2+ accumulates in the cytoplasm after a diverse class of stimuli via ionotropic glutamate receptors, voltagegated calcium channels, and/or internal calcium stores. Thus, BDNF secretion is highly complex as it is governed by secretory pathways, conversion enzymes, the biophysics of Ca2+ diffusion, and the subsequent Ca2+-mediated intracellular signaling cascades. Currently, the age-dependent differences in these regulatory processes are unknown within the context of neonatal seizures (Figure 1).
BDNF-TrkB signaling
BDNF binds with high affinity to TrkB, a neurotrophin receptor that is enriched at the pre- and post-synapse. Upon binding to the ectodomain of TrkB, BDNF induces receptor dimerization, activation of the receptor kinase activity, and the subsequent trans-autophosphorylation of multiple tyrosine residues [3]. The autophosphorylation of TrkB creates docking sites for the adaptor proteins Shc and PLCγ1, that each initiate intracellular signaling cascades. Through these intracellular signaling cascades, BDNF-TrkB signaling influences neuronal Cl− extrusion capacity and GABA-mediated fast hyperpolarizing inhibition via its regulation of the neuronal specific K+-Cl− cotransporter 2 (KCC2) [4]. Following BDNF-TrkB binding, PLCγ1 binds to TrkB phosphotyrosine 816 (Y816) and inhibits the mRNA expression of the neuronal specific K+-Cl− cotransporter 2 (KCC2) [4]. In contrast, the PI3K and Ras/MAPK pathways result in Shc binding to TrkB phosphotyrosine 515 (Y515) and the subsequent promotion of Kcc2 transcription. However, when the Shc and PLCγ1 pathways are concurrently activated, KCC2 is downregulated [4]. In adult hippocampal preparations, the Trk inhibitor K252a (or scavenging endogenous BDNF with TrkB-Fc) can prevent the rapid BDNF-induced downregulation of KCC2 after the induction of continuous neuronal activity [4]. Therefore, BDNF-TrkB signaling is an important modulator of KCC2.
In contrast to BDNF, proBDNF preferentially binds to the pan-neurotrophin receptor p75 (p75NTR) and can form many different multimeric receptor complexes to produce diverse responses such as apoptosis, attenuated glutamatergic synaptic transmission, and KCC2 downregulation [5]. In the developing hippocampus, BDNF-TrkB signaling permits and strengthens the clustering of excitatory synapses, while proBDNF-p75NTR signaling facilitates the synaptic depression of unclustered excitatory synapses [6]. These results highlight the diversity of cellular responses mediated by either BDNF-TrkB or proBDNF-p75NTR signaling during development, suggesting that the conversion of proBDNF to BDNF can refine developing networks at single synapse precision [6]. The prevention of proBDNF to BDNF conversion via a cleavage resistant proBDNF rat model has demonstrated an increased susceptibility to chemoconvulsant induced seizures, increased KCC2 downregulation, and increased seizure frequency in the pilocarpine model of limbic epileptogenesis [5]. However, the influence of the BDNF-TrkB vs. proBDNF-p75NTR ratio during neonatal seizures is not well understood (Figure 1).
Neonatal seizures and their management
Neonatal seizures occur in an estimated 1 to 3.5 per 1000 live births in the term infant [7]. Acute seizures occur more often during the neonatal period than at any other age. The most common etiologies of acute symptomatic neonatal seizures are hypoxic-ischemic encephalopathy (HIE), ischemic stroke, intracranial hemorrhage, and intracranial infections [7]. Neonatal seizures are a major clinical challenge in the neonatal intensive care unit. Their subtle semiology frequently requires electroencephalography (EEG) recordings to accurately identify and quantify seizure burdens [7]. These technical challenges are coupled with neonatal seizures’ poor response to anti-seizure medication (ASM) [7]. Uncontrolled neonatal seizures may exacerbate secondary brain damage as they are associated with unfavorable outcomes [8]. Neonatal HIE is the most common cause of neonatal seizures, it results from inadequate cerebral blood flow that deprives the fetal brain of oxygen and glucose [7]. The outcomes of prolonged neonatal seizures after HIE can include mortality, intellectual disability, developmental delay, and epilepsy [7].
Development of evidence-based guidelines for the management of neonatal seizures requires randomized controlled trials that include neonatal pharmacokinetic studies, standardized EEG recording durations, primary anti-seizure outcome measures, and secondary long-term neurological outcome measures [7]. Treatment algorithms vary between institutions (e.g. doses, therapeutic hypothermia protocols, and second-line therapies) resulting in physician preference and institutional traditions determining clinical management [7]. However, the current standard of care for EEG confirmed seizures in a high-risk scenario (e.g. HIE) is a loading dose of phenobarbital (PB: a positive allosteric modulator for GABAA receptors) as a first-line therapy [7,8]. While regular EEG monitoring and earlier ASM administration have been shown to improve the efficacy of PB for neonatal seizures, the current consensus is that standard PB therapy is often ineffective for the treatment of neonatal seizures [7]. Therefore, there is an urgent need for the identification of new anti-seizure therapies specifically developed for this population of patients.
Targeting BDNF-TrkB-KCC2 signaling in neonatal seizures
One mechanism potentially underlying neonatal hyperexcitability is the high concentration of neuronal intracellular Cl− ([Cl−]i) mediated by the lower expression of KCC2 during development [9]. The tight regulation of [Cl−]i is necessary for GABAA receptor mediated fast synaptic inhibition. Further, the anti-seizure efficacy of phenobarbital may be dependent upon intracellular Cl− regulation. Therefore, KCC2 is a focus of preclinical neonatal seizure research as it may underlie seizures unresponsive to phenobarbital [9]. BDNF-TrkB signaling is one major regulator of KCC2 and it is significantly modulated by seizures [2,9]. However, investigations of BDNF-TrkB within the broad context of seizure disorders have almost entirely been performed in adult models of limbic epileptogenesis (Table 1).
Table 1.
Targeting TrkB in Seizure Disorders
| Citation (PMID) | Date | Model | Method | Age | Results |
|---|---|---|---|---|---|
| Isackson et al. (2054188) | 1991 | DG focal lesion in male rats | Behavioral seizure scoring | Adult | BDNF mRNA expression is increased after limbic seizures |
| Kokaia et al. (7649227) | 1995 | Electrical kindling in male mice | Behavioral seizure scoring | Adult | The development of electrical kindling is suppressed in BDNF+/− mice |
| Binder et al. (9952416) | 1999 | Electrical kindling in male rats | Behavioral seizure scoring | Adult | Chronic intraventricular infusion of TrkB receptor bodies inhibited the development of electrical kindling |
| Croll et al. (10501474) | 1999 | Intraperitoneal KA injection in mice | Behavioral seizure scoring | Adult | BDNF overexpression increased KA seizure severity |
| Lahteinen et al. (11886452) | 2002 | Intraperitoneal KA injection in male mice | Video-EEG monitoring | Adult | Transgenic mice that overexpress truncated TrkB had significantly less spontaneous recurrent seizures and interictal spikes after KA induced epilepsy |
| Scharfman et al. (11922662) | 2002 | Chronic BDNF intrahippocamp al infusion in male rats | Behavioral seizure scoring and acute scalp recordings | Adult | Chronic intrahippocampal infusion of BDNF induced spontaneous seizures and decreased latency to pilocarpine induced status epilepticus |
| He et al. (15233915) | 2004 | Electrical kindling in mice | Behavioral seizure scoring | Adult | Conditional deletion of TrkB prevented the development of electrical kindling |
| He et al. (20445044) | 2010 | Electrical kindling in TrkBPLC/PLC mice | Behavioral seizure scoring | Adult | Genetic uncoupling of TrkB (Y816F) from PLCƴ/1 inhibited the development of electrical kindling |
| Liu et al. (23790754) | 2013 | Right amygdala KA infusion in adult TrkBF616A mice | Video-EEG monitoring | Adult | Chemical-genetic inhibition of TrkB (two weeks) in combination with diazepam (40 minutes) and lorazepam (1 hour) prevented spontaneous recurrent seizures after KA induced SE |
| Gu et al. (26481038) | 2015 | Electrical kindling in mice | Video-EEG monitoring | Adult | Peptide that prevents the coupling of TrkB pY816 to PLCγ1 prevents kindling |
| Kang et al. (26452067) | 2015 | Unilateral carotid ligation in P7 CD-1 mice | Video-EEG monitoring | Neonatal | Acute TrkB-inhibition with ANA-12 prevented phenobarbital resistant neonatal seizures in P7 CD- 1 pups |
| Carter et al. (30097625) | 2018 | Unilateral carotid ligation in P7 and P10 CD-1 mice | Video-EEG monitoring | Neonatal | Acute TrkB-inhibition with ANA-12 prevented phenobarbital resistant P7 neonatal seizures in a dose dependent manner |
| Gu et al. (29408225) | 2018 | Partial neocortical isolation model in rats | Video-EEG monitoring | Adult | Chronic administration of the TrkB agonist PTX BD4–3 after UC decreased PTZ induced electrographic and behavioral ictal events |
| Riffault et al. (27913431) | 2018 | PTZ injection in CR-proBDNF mice | Behavioral seizure scoring | Adult | Cleavage resistant proBDNF increased the severity of PTZ induced seizures and the frequency of seizures in the pilocarpine model of temporal lobe epilepsy |
| Krishnamurthy et al. (31525273) | 2019 | Electrical kindling in mice | Video-EEG monitoring | Adult | Acute chemical genetic TrkB inhibition prevented the development of electrical kindling |
| Kang et al. (31864171) | 2020 | Unilateral carotid ligation in P7 CD-1 pups | Video-EEG monitoring | Neonatal and Adult | Acute TrkB inhibition with ANA-12 prevented phenobarbital resistant neonatal seizures a P7 and has positive long-term outcomes in adulthood on vEEG |
| Kipnis et al. (32427585) | 2020 | Unilateral carotid ligation in P7 CD-1, P10 CD-1, and P7 TrkBF616A pups | Video-EEG monitoring | Neonatal | Acute administration of with partial TrkB agonist LM22A-4, TrkB agonist HIOC, or the chemical-genetic inactivation of TrkB all prevented P7 phenobarbital resistant neonatal seizures |
To investigate the pathophysiology of neonatal seizures, a CD-1 mouse model of unilateral carotid ligation without transection was characterized using continuous video-electroencephalogram (vEEG) recordings after ligation at P7 or P10 [10]. The seizures at P7 had a significant proportion of subclinical electrographic seizures after ligation, and did not demonstrate focal strokes when evaluated at P18 with histopathology [10]. Neonatal seizure models that depend on behavioral seizure scoring in the absence of vEEG are likely inaccurate as electrographic seizures were present in the CD-1 model. The Racine scale quantifies seizure behavioral phenotypes in adult rodent models of epilepsy. In neonatal seizure models, a modified Racine scale has commonly been used to quantify the frequency of convulsive vs. non-convulsive seizures on video when EEG is also available for confirmation [10]. The quantification of these behavioral phenotypes in neonates is limited by the motor manifestations of those seizures; as they differ significantly between neonatal and adult ages. In this CD-1 model, unilateral carotid ligation induced the activation of TrkB at both ages [10]. At P7, post ischemic TrkB activation was associated with PB refractory neonatal seizures and reduced KCC2. In contrast, post ischemic neonatal seizures responded to PB at P10 when KCC2 expression is greater than P7 [10].
Careful interpretations are recommended for models that utilize chemoconvulsants to induce neonatal seizures. Specifically, the acute administration of pentylenetetrazol in P7 CD-1 pups induced seizures that were both responsive to PB and were associated with an increase in KCC2 [11]. After PTZ induced seizures or ischemic seizures in P7 CD-1 mice the chloride imported Na-K-2Cl cotransporter 1 does not undergo significant changes in expression [10,11]. Therefore, chemoconvulsant models of neonatal seizures present with high seizure burdens that respond to first line anti-seizure therapies and lack reductions in KCC2.
TrkB mediated KCC2 hypofunction after ischemia in P7-CD1 mice was necessary for the emergence of pharmacoresistant neonatal seizures. A single dose of the small-molecule TrkB antagonist ANA12 administered to P7 CD-1 pups after unilateral carotid ligation [12] prevented the emergence of neonatal seizures unresponsive to phenobarbital in a dose dependent manner [12,13]. Neonatal pups treated with combination of ANA12 and PB improved the long-term outcomes in adulthood, in comparison to pups that only received PB [14]. Therefore, acutely and transiently preventing TrkB activation in post-ischemic immature brains with ANA12 was sufficient to prevent the emergence of refractory neonatal seizures and KCC2 hypofunction. The subsequent long-term developmental benefits further highlight the importance of curbing refractory seizures in neonatal mice.
In vivo evidence for the net effect of exogenous BDNF in the neonatal brain is not available as BDNF has poor blood-brain barrier penetrance. The recently developed BDNF loop II mimetic LM22A-4 (LM) provides one potential solution for evaluating the net effect of increased BDNF-TrkB signaling post ischemia. When low doses of the BDNF mimetic LM were administered to CD-1 P7 pups after unilateral carotid ligation, LM significantly exacerbated post-ischemic neonatal seizures [15]. In contrast, at higher doses LM prevented the emergence of PB-refractory neonatal seizures and KCC2 hypofunction [15]. These results indicate that TrkB agonists can also prevent the emergence of refractory seizures, potentially as functional antagonists of BDNF-TrkB signaling [15]. This effect did not seem to be loop II specific since the full TrkB agonists HIOC and deoxygedunin both prevented the emergence of refractory neonatal seizures [15]. These data suggest that BDNF-TrkB signaling may facilitate post-ischemic seizures in the neonatal brain. However, future studies are required to understand the role of BDNF-TrkB in other etiologies of neonatal seizures and after treatments such as therapeutic hypothermia.
Conclusion
Targeting BDNF-TrkB signaling is a novel therapeutic strategy for neonatal seizures. An improved understanding of the multiple levels of BDNF regulation during neonatal seizures will provide insights for the development of new therapies.
Acknowledgements
This review was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health under Award Number R01HD090884 (SDK). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations:
- BDNF
Brain derived neurotrophic factor
- TrkB
tyrosine receptor kinase B
- KCC2
K+-Cl− cotransporter 2
- ASM
anti-seizure medicine
- PB
phenobarbital
- proBDNF
probrain derived neurotrophic factor
- LM
LM22A-4
- vEEG
ANA-12, video-electroencephalogram
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest statement
SDK is listed as an author on US patent 10525024B2, “Methods for rescuing phenobarbital-resistance of seizures by ANA-12 or ANA-12 in combination with CLP290.”
References
- [1].Maynard KR, Hill JL, Calcaterra NE, Palko ME, Kardian A, Paredes D, et al. Functional Role of BDNF Production from Unique Promoters in Aggression and Serotonin Signaling. Neuropsychopharmacology 2016;41:1943–55. 10.1038/npp.2015.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].McNamara JO, Scharfman HE. Temporal Lobe Epilepsy and the BDNF Receptor, TrkB. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. 4th ed., Bethesda (MD): National Center for Biotechnology Information (US); 2012. [PubMed] [Google Scholar]
- [3].Gottmann K, Mittmann T, Lessmann V. BDNF signaling in the formation, maturation and plasticity of glutamatergic and GABAergic synapses. Exp Brain Res 2009;199:203–34. 10.1007/s00221-009-1994-z. [DOI] [PubMed] [Google Scholar]
- [4].Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipilä S, et al. Mechanism of Activity-Dependent Downregulation of the Neuron-Specific K-Cl Cotransporter KCC2. J Neurosci 2004;24:4683–91. 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Riffault B, Kourdougli N, Dumon C, Ferrand N, Buhler E, Schaller F, et al. Pro-Brain-Derived Neurotrophic Factor (proBDNF)-Mediated p75NTR Activation Promotes Depolarizing Actions of GABA and Increases Susceptibility to Epileptic Seizures. Cereb Cortex 2018;28:510–27. 10.1093/cercor/bhw385. [DOI] [PubMed] [Google Scholar]
- [6].Niculescu D, Michaelsen-Preusse K, Güner Ü, van Dorland R, Wierenga CJ, Lohmann C. A BDNF-Mediated Push-Pull Plasticity Mechanism for Synaptic Clustering. Cell Reports 2018;24:2063–74. 10.1016/j.celrep.2018.07.073. [DOI] [PubMed] [Google Scholar]
- [7].Volpe J, Inder T, Darras B, de Vries L, du Plessis A, Neill J, et al. Volpe’s Neurology of the Newborn - 6th Edition. 6th ed. Elsevier; 2017. [Google Scholar]
- [8].Glass HC, Glidden D, Jeremy RJ, Barkovich AJ, Ferriero DM, Miller SP. Clinical Neonatal Seizures are Independently Associated with Outcome in Infants at Risk for Hypoxic-Ischemic Brain Injury. J Pediatr 2009;155:318–23. 10.1016/j.jpeds.2009.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sullivan BJ, Kadam SD. The involvement of neuronal chloride transporter deficienceis in epilepsy. In: Tang X, editor. Neuronal Chloride Transporters in Health and Disease. 1st ed., Elsevier; 2020, p. 650. [Google Scholar]
- [10].Kang SK, Markowitz GJ, Kim ST, Johnston MV, Kadam SD. Age- and sex-dependent susceptibility to phenobarbital-resistant neonatal seizures: role of chloride co-transporters. Front Cell Neurosci 2015;9:173-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kharod SC, Carter BM, Kadam SD. Pharmaco-resistant Neonatal Seizures: Critical Mechanistic Insights from a Chemoconvulsant Model. Dev Neurobiol 2018;78:1117–30. 10.1002/dneu.22634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kang SK, Johnston MV, Kadam SD. Acute TrkB inhibition rescues phenobarbital-resistant seizures in a mouse model of neonatal ischemia. Eur J Neurosci 2015;42:2792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Carter BM, Sullivan BJ, Landers JR, Kadam SD. Dose-dependent reversal of KCC2 hypofunction and phenobarbital-resistant neonatal seizures by ANA12. Scientific Reports 2018;8:11987. 10.1038/s41598-018-30486-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kang SK, Ammanuel S, Adler DA, Kadam SD. Rescue of PB-resistant neonatal seizures with single-dose of small-molecule TrkB antagonist show long-term benefits. Epilepsy Research 2020;159:106249. 10.1016/j.eplepsyres.2019.106249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kipnis PA, Sullivan BJ, Carter BM, Kadam SD. TrkB agonists prevent postischemic emergence of refractory neonatal seizures in mice. JCI Insight 2020;5. 10.1172/jci.insight.136007. [DOI] [PMC free article] [PubMed] [Google Scholar]