

Abstract
The peroxisome proliferator activated receptor gamma (PPARG) nuclear receptor regulates energy metabolism and insulin sensitivity. In this study, we present novel evidence for an essential role of PPARG in the regulation of osteocyte function, and support for the emerging concept of the conjunction between regulation of energy metabolism and bone mass. We report that PPARG is essential for sclerostin production, a recently approved target to treat osteoporosis. Our mouse model of osteocyte-specific PPARG deletion (Dmp1CrePparγflfl or γOTKO) is characterized with increased bone mass and reduced bone marrow adiposity, which is consistent with upregulation of WNT signaling and increased bone forming activity of endosteal osteoblasts. An analysis of osteocytes derived from γOTKO and control mice showed an excellent correlation between PPARG and SOST/sclerostin at the transcript and protein levels. The 8 kb sequence upstream of Sost gene transcription start site possesses multiple PPARG binding elements (PPREs) with at least two of them binding PPARG with dynamics reflecting its activation with full agonist rosiglitazone and correlating with increased levels of Sost transcript and sclerostin protein expression (Pearson’s r=0.9910, p=0.001). Older γOTKO female mice are largely protected from TZD-induced bone loss providing proof of concept that PPARG in osteocytes can be pharmacologically targeted. These findings demonstrate that transcriptional activities of PPARG are essential for sclerostin expression in osteocytes and support consideration of targeting PPARG activities with selective modulators to treat osteoporosis.
Keywords: PPARG, SOST/sclerostin, PPRE, bone mass, marrow adipocytes
1. Introduction
Growing number of compelling evidences point to the mechanistic and functional connection between energy metabolism and bone mass [1–3]. Peroxisome proliferator-activated receptor gamma (PPARG) exemplifies this connection. PPARG is a transcription factor that regulates network of genes associated with glucose and lipid metabolism, insulin signaling, adipocyte differentiation, and inflammation. PPARG has been implicated in the pathology of numerous diseases like diabetes, obesity, cancer, and atherosclerosis [4–6]. In bone, PPARG controls osteoblasts and adipocytes differentiation from bone marrow stroma cells (BMSC) [7, 8], and osteoclasts differentiation from cells of hematopoietic lineage, monocytes [9]. Increased activity of PPARG in response to pharmacologic treatment with TZDs, PPARG full agonists and insulin sensitizers, results in bone loss associated with accumulation of fat in bone marrow, and up to two-fold increase in fracture rate in older women with diabetes [3, 10, 11]. Activated PPARG increases osteoclast resorptive activities, and is skewing BMSC lineage allocation toward adipocytes and away from osteoblasts, which at least in part explains PPARG negative effects on bone. Our previous studies have indicated that there is an in vivo relationship between PPARG activity and Sost expression in osteocytes, and most importantly that the bone anabolic effect of a new class of insulin sensitizers, which act as PPARG inverse agonists and block S112 dephosphorylation, is associated with decreased Sost expression [12]. These findings prompted us to explore the link between PPARG activity and sclerostin production with systematic and mechanistic approach.
Produced in osteocytes, the most abundant cells in bone, sclerostin protein regulates bone formation and resorption via inhibition of WNT signaling and production of RANKL [13]. The newly approved anti-osteoporotic therapy with romosozumab consists of a monoclonal antibody targeting sclerostin. Romosozumab is highly efficacious with respect to the anabolic effect on bone and fracture prevention in postmenopausal women; however, there are safety concerns caused by the growing number of reported serious cardiovascular (CV) effects among the users of this drug, especially among individuals with high risk for CV events [14–18]. Although the exact etiology of romosozumab-triggered CV effects is not determined, the off-target effects of the drug may include sclerostin produced in vascular endothelium and heart valves [19], and animal studies suggest that sclerostin produced by endothelium have a protective effect against vascular calcification [20]. The growing list of potential, although not yet clinically recognized off-target effects, includes also joint diseases. It has been shown that sclerostin improves posttraumatic osteoarthritis [21] and that synovial fibroblasts producing sclerostin have an inhibitory effect on progression of the TNFα-dependent rheumatoid arthritis, which is exacerbated with anti-sclerostin antibody therapy [22]. Based on the above concerns and the superior anti-osteoporotic effects of anti-sclerostin therapy, the transcriptional control of sclerostin production in osteocytes can be a great alternative approach to the antibody therapy, especially when the transcription factor has been already identified as a therapeutic target, which is a case for PPARG.
Thus, with our long-term objective to demonstrate the feasibility of therapeutic targeting of PPARG to treat simultaneously diabetes, diabetic bone disease, and osteoporosis we designed current study to understand the role of PPARG in regulation of osteocyte function. To achieve this goal, we have developed mouse model of osteocyte-specific deletion of PPARG using Dmp1Cre driver and Pparγ loxP system. Here, we report that PPARG in osteocytes is a viable target to regulate sclerostin levels specifically in bone, and as a consequence bone mass and marrow adiposity, and skeletal response to pharmacologic activators of PPARG. Sclerostin is transcriptionally regulated by PPARG and PPARG is essential for sclerostin expression. These findings support consideration of selective pharmacological targeting of PPARG in osteocytes to control sclerostin production.
2. Materials and Methods
2.1. Animals
Osteocyte-specific PPARG knock-out mice, γOTKO, were developed at the University of Toledo. The University of Toledo animal facility is operated as a pathogen-free, AAALAC approved facility and animal care and husbandry meet the requirements in the Guide for the Care and Use of Laboratory Animals. The animal breeding, care, and treatment were performed using a University of Toledo Health Science Campus Institutional Animal Care and Utilization Committee protocol. The γOTKO mice were constructed by crossing the 10kb Dmp1Cre mice (B6N.FVB-Tg (Dmp1-cre)1Jqfe/BwdJ) and Pparγfl/fl mice (B6.129-Ppargtm2Rev/J). Both mouse strains have C57BL/6 background and were obtained from the Jackson Laboratory (Bar Harbor, ME). Pparγfl/fl mice have loxP sites flanking exons 1 and 2 of Pparγ gene. The Cre-recombinase is under late osteoblast/osteocyte specific10kb Dmp1 promoter. The desired osteocyte-specific γOTKO and littermate control mice were obtained in the F3 progeny of 10 kb Dmp1Cre and Pparγfl/fl cross. As controls, mice with either Dmp1Cre only and/or Pparγfl/fl only genotypes were used after assurance that Pparγ and Sost expression were not affected by genotype. In addition, a post-hoc analysis of correlation between genotypes and experimental measures was performed for each experiment. Animals were housed under 12h dark-light cycle with unlimited access to chow (Teklad global 16% protein rodent diet; code 2916) and water. The special diet (Teklad global 19% protein extruded rodent diet; code 2919) was used during breeding.
For genotyping, tissue samples were collected at weaning using tail clipping and genomic DNA isolated after proteinase K (Sigma-Aldrich, Cat#P5568) digestion. PCR primers published by the Jackson Laboratory were used for genotyping. The Cre recombinase insertion was detected with following primers: F - TTG CCT TTC TCT CCA CAG GT and R - CAT GTC CAT CAG GTT CTT GC); while to differentiate between Pparγfl/fl (homozygous), Pparγfl/+ (heterozygous) and Pparγ+/+ (wildtype) strains following primers were used: F – TGGCTTCCAGTGCATAAGTT and R – TGTAATGGAAGGGCAAAAGG. Taq polymerase (Go Taq; Promega, Cat#M3005) based amplification was used for PCR followed by 1% agarose gel run for amplicon detection (167 bp for Cre; 200 bp for Pparγ wildtype, 230 bp for Pparγ homozygotes, both 200 bp and 230 bp for Pparγ heterozygotes).
Rosiglitazone supplemented diet experiment: Nine-month-old female mice (Ctrl and γOTKO) were randomly allocated to two groups and fed with either regular or rosiglitazone (Scripps Research Institute, Jupiter, FL) supplemented at the dose 25 mg/kg/day diet, as described [23]. DXA was performed at the beginning of the study and after 4 weeks of treatment to measure changes in BMD. After 6 weeks of treatment, animals were sacrificed, and tissues harvested. Right femur and vertebrae were scanned with mCT for bone parameters, then femur was decalcified, stained with OsO4 and scanned for marrow fat.
2.2. DXA
Dual Energy X-ray absorptiometry (DXA) was used to measure BMD values longitudinally as the mice age. Measurements were done using Lunar PIXImus II instrument (GE Lunar Corp., Madison, WI) at 4 mo and 6 mo of age and analyzed using LUNAR PIXImus v. 2.10 software provided by manufacturer. Head and metal tag region were excluded from region of interest (ROI).
2.3. mCT analysis of bone and marrow fat
Right tibiae and L4 vertebrae were collected from euthanized mice and stored in 10% buffered formalin for 48 hours to attain fixation plateau for all bones. Assessment of trabecular bone in proximal tibia and L4 vertebrae, and cortical bone at tibia midshaft was conducted by micro CT using μCT35 system (Scanco Medical AG, Bruettisellen, Switzerland). Bone scans were performed with the x-ray source operating at 70 kVp and 114 μA energy settings, and recording 500 projections/180° acquired at 300 ms integration time for proximal tibia, and 100 ms for vertebrae and tibia midshaft using 7 μm nominal resolution voxel for all bone locations. Scans of the proximal tibia consisted of 340 cross-sectional slices starting at the top of growth plate, and images of trabecular bone were segmented at optimized lower threshold value of 220 using per mille scale (approximately 3000 HU, or μ of 1.76) following manual contouring starting 10 slices down from the tip of intercondylar notch and extending for 200 more slices, excluding cortex. L4 vertebrae were scanned over the entire height of the vertebral body and trabecular bone was evaluated at lower threshold value of 220 starting 10 slices off both endplates, and excluding cortex. The analysis of the trabecular bone microstructure and cortical bone parameters was conducted using Evaluation Program V6.5–3 (Scanco Medical AG) and conformed to recommended guidelines [24].
To visualize marrow lipids, bones were decalcified using Formical 4 (Medline; Cat# STLB12145) and stained with freshly prepared 2% aqueous solution of osmium tetroxide (Electron Microscopy Sciences, Cat#19150) in 0.1 M sodium cacodylate buffer (pH 7.4), as described [25]. Staining was carried out in an exhaust hood and away from light due to osmium tetroxide toxicity and light sensitivity. Images of lipid depositions were acquired at 70 kVp and 114 μA settings and 12 μm nominal resolution voxel. Image segmentation was done under global threshold conditions by applying a gray scale threshold of 480–1000 using the per mille scale with Gauss filter set to sigma 1.2 and support 2.0. Lipid volumes were calculated directly from individual voxel volumes in three-dimensional reconstructions. All analyses were performed in a blinded manner.
2.4. Dynamic histomorphometry
Sterile filtered Calcein (Sigma, Cat#C0875–5G) solution (2.5 mg/ml) prepared in isotonic 2% NaHCO3 was administered intraperitonially (20mg/kg body weight) to mice, 10 days and 2 days before euthanasia. Left tibia was collected in 70% EtOH and embedded in methyl methacrylate (Indiana University Histology Core, Indianapolis, IN), and processed for slide preparation of coronal and transverse sections and staining. Coronal sections were used for static and dynamic histomorphometry. Images of the proximal tibia region adjacent to growth plate were taken using Nikon Eclipse 80i microscope and single and double calcein labelled surfaces were measured using Nikon NIS-Elements BR3.0 in a blinded manner by single operator. ROI included trabecular bone and endosteal bone surface in proximal tibia excluding growth plate and extending 2 mm downward the tibia. Measurements were done on two sections for each sample. Measurements of mineralizing surface (MS/BS), mineral apposition rate (MAR), and bone formation rate (BFR) were calculated, as recommended [26]. Transverse sections at diaphysis of the same tibia specimens were used for MS/BS measurements and calculations (2 sections per tibia).
2.5. Analysis of serum bone turnover markers
Bone specific alkaline phosphatase (BALP) was measured colorimetrically using the Alkaline Phosphatase Diagnostic Kit (Sigma-Aldrich, St. Louis, MO) in the presence of L-glucosamine to exclude muscle and liver enzymatic activity. The N-terminal propeptide of type I procollagen (P1NP) and tartrate-resistant acid phosphatase form 5b (TRAcP 5b) were measured using diagnostic kits provided by Immunodiagnostic Systems Inc. (IDS, Scottsdale, AZ).
2.6. Organ culture conditioned medium experiment
Conditioned media were collected from organ cultures of femora bone. Femora from six C57BL6 male mice were isolated, bone marrow was spun out and discarded, the outer surface of bone was cleaned off any remaining muscle fiber and periosteum, and one collagenase digestion followed by one EDTA digestion was performed, as described [27]. Using sterile bone-cutting scissors, the bones were then chopped into 1–2 mm fragments and distributed evenly on collagen coated 6-well plates with 3 ml culture medium/well (αMEM supplemented with 15% FBS and 10 mM glucose). Bone pieces were cultured for 6 days at 37C with 5% CO2 before conditioned medium (CM) was collected and pooled from all six bone organ cultures. Sclerostin was depleted from one half of the collected CM by overnight incubation with mouse SOST/sclerostin antibodies (R & D Systems, cat# AF1589) in the ratio of 20 μl antibodies per 9 ml CM, and pulled down with Protein A agarose beads (Invitrogen, cat#15918014). The other half of the CM was subjected to anti-mouse IgG pull-down step (2 μl for 9 ml CM) (Sigma-Aldrich, Cat#M9144) which was used as a negative control for this assay. Sclerostin levels after depletion were quantified using SOST ELISA kit (Quantikine ELISA, R&D Systems, Cat#MSST00).
Recipient BMSC cultures were established from bone marrow isolated from femora of six C57BL6 male mice, pooled and plated on 6-well plates at the density 2×106 cells/well. After 7 days of growth, the culture medium was replaced with osteocyte CM either depleted of Sclerostin or IgG in triplicates, and cultured for three more days before processing for RNA isolation followed by analysis of adipocytic and osteoblastic markers using qPCR.
2.7. Isolation of osteocyte-enriched and endosteal osteoblast-enriched cell fractions from femora
An osteocyte-enriched fraction and endosteal osteoblast-enriched fraction were isolated after sequential collagenase digestion of femora [27, 28]. Briefly, femora were cleaned off muscles and bone marrow was spun-out after removal of epiphysis. The diaphysis-outer-surface was scrapped to remove periosteum and any remaining muscle fibers. Bones were then subjected to alternating digestion with collagenase and EDTA to retrieve endosteal osteoblasts and osteocytes. First two collagenase and EDTA digestions of femora were discarded. Cells from the 3rd digestion were collected and represented an endosteal osteoblast-enriched fraction 3 (F3). Alternating digestions continued and cells from the 6th digestion were collected and represented osteocyte-enriched fraction 6 (F6). The leftover bone pieces were homogenized and added to F6 osteocyte enriched fraction. Cell-type enrichment of isolated fractions was verified by analyzing levels of osteoblasts (Col1, Ocn) and osteocytes (Dmp1, Sost) gene markers expression (Suppl. Fig. S1).
2.8. Gene Expression Analysis
mRNA was isolated using Trizol (Thermo Fisher Scientific, Cat# 15596026). Half μg RNA was converted to cDNA using the Verso cDNA synthesis kit (Thermo Fisher Scientific, Cat#AB1453B). Gene transcripts were detected by quantitative real-time PCR using TrueAmp SYBR Green qPCR SuperMix (Smart Bioscience, Cat#7501–3) and processed with StepOne Plus System (Applied Biosystems, Carlsbad, CA). Relative gene expression was measured by the comparative ΔΔCT method using 18S RNA levels for normalization. Primers were designed using OligoPerfect Designer (Thermo Fisher Scientific) and are listed in the Suppl. Table S1.
2.9. Protein analysis using Western blots
Proteins were isolated from cell fraction enriched in osteocytes using TRIzol, following manufacturer’s protocol and quantified using BCA assay (Pierce BCA protein Assay Kit, ThermoScientific, Cat#23225). Ten μg of protein was loaded on each lane and resolved on 10% SDS-PAGE using BioRad system and electrophoretically transferred to Immobilon-FL membranes. Membranes were blocked at room temperature for 2h in Odyssey blocking buffer (LI-COR, cat#927–50000). Incubation with primary antibody was performed overnight at 4° with either mouse monoclonal PPARG antibodies at 1:500 dilution, (Santa Cruz Biotechnology, Santa Cruz, CA; cat # Sc-7273), or polyclonal SOST/sclerostin antibodies at 1:500 dilution, (R&D System, Minneapolis, MN; cat #AF1589), or mouse monoclonal β-actin antibodies at 1:10,000 dilution (Sigma-Aldrich Life Science, St. Louis, MO; cat #A1978). After incubation, membranes were washed 3 times in TBST (TBS plus 0.1% Tween 20) and incubated with appropriate secondary antibodies, either infrared anti-mouse (680RD; cat#925–68072) or infrared anti-goat (800CW; cat#925–32214) (LI-COR Bioscience, Lincoln, NE), at 1:10,000 dilution in TBS for 2h at room temperature. Immunoreactivity was visualized by infrared scanning in the Odyssey system (LI-COR Biosciences). Image J (version 1.52a) was used to calculate band intensity from Western blots and band intensity of β-actin was used to normalize PPARG and sclerostin signals.
2.10. Immunohistochemistry for sclerostin detection
Methyl methacrylate embedded specimens of tibia bone (coronal sections) were deplastinized and incubated with SOST antibodies (R&D System, Minneapolis, MN; cat #AF1589) at the dilution 1:100 for 8 hrs in 4C followed by incubation with secondary biotinylated antibodies from the Peroxidase VECTASTAIN® Elite ABC-HRP Kit (Goat IgG) (Vector Laboratories, Inc., Burlingame, CA; cat# PK-6105) and color development using peroxidase DAB substrate with nickel (Vector Laboratories, Inc.; cat# SK-4100), according to the manufacturers protocols. As negative controls, sections adjacent to those, which were used for sclerostin detection, were processed the same way except for primary antibody incubation.
2.11. In-silico mapping of PPREs in Sost regulatory region using JASPAR
The 8.85 kb sequence upstream to Sost gene transcription start site (TSS) was obtained from NCBI Genebank database. This sequence was divided into 3 equal fragments (2.95 kb each) and analyzed for mouse PPARG::RXRA heterodimer binding sites using JASPAR (version 2020), a database for curated, non-redundant set of transcription factor binding profiles. Position weight matrix (PWM) for mouse PPARG::RXRA binding site was run with relative profile score threshold set at 80%.
2.12. ChIP assay
Chromatin Immunoprecipitation (ChIP) assay to detect PPARG binding to selected PPRE sequences was performed on osteocyte like MLO-Y4 cells (kind gift from Dr. L. Bonewald). Cells were cultured on collagen coated 100 mm plates using 5% FBS, 5% CS and 1% P/S in alpha-MEM medium (at 37C with 5% CO2). Optimal passage time was 4–5 days before each splitting with 1:5 ratio after cultures reached 80% confluency. In each experiment, cells were treated in duplicates with either DMSO as vehicle, PPARG agonist rosiglitazone (1 μM), or a combination of rosiglitazone (1 μM) and antagonist GW9662 (10 μM). Sonicated only and Immunoglobulin G (IgG) pull-down samples were used as negative controls.
Chromatin Immunoprecipitation protocol was followed [29]. Briefly, after 24h treatment cells were fixed using 4% formaldehyde in PBS, lysed, and DNA was sonicated to fragments size 500 – 1000 bp (6 min total time per sample, 12 cycles of 30s pulse at 50% amplitude of Qsonica CL-18 sonicator followed by 90s incubation on ice after each cycle). After sonication, samples were incubated with Protein A agarose beads (Invitrogen, Cat#15918014) for 4h at 4C to reduce background. The beads were spun down and discarded, and either IgG (2 μl/sample; Sigma-Aldrich; Cat#M9144) or PPARG antibody (10 μl/sample; Santa Cruz Biotechnology;Cat#Sc-7273) were added and incubated overnight at 4C followed by adding fresh protein A agarose beads (60 μl/sample) and incubated for 4h at 4C. After spinning down the beads, antibody bound chromatin-PPARG fragments were eluted using elution buffer [29] followed by crosslink reversal (15h at 65C), Proteinase K digestion (1h at 50C) and purification of pulled-down DNA fragments using phenol-chloroform-isoamyl alcohol (25:24:1) extraction. The Taq polymerase (GoTaq) based PCR was performed with specifically designed primers for analyzed PPREs: PPRE3 F – TCTCCCAAGTCTGGAGCAAT and R - CTGAGGTGCAAAAGGAGGAG; PPRE14 & 15 F – TACCACGTCTCCCCTGTTTC and R – GGGCCTGTGTTTGCATAGTT. The presence of specific amplicons (532 bp for PPRE3 and 634 bp for PPRE14/15) was detected using 1% agarose gel.
2.13. Statistical analysis
Data are represented as means ± SD and were analyzed by statistical analysis software GraphPad Prism 8. Parametric, unpaired, two-tailed Student’s t-test was used for comparing means of two groups. Either One-way Anova or Two-way ANOVA followed by Tukey’s multiple comparison test were used to compare multiple groups. Pearson correlation was used to measure correlation between experimental variables. A p-value 0.05 and lower was considered as statistically significant.
3. Results
3.1. Pparγ is highly expressed in osteocytes, and its expression increases with aging and with rosiglitazone treatment.
Pparγ expression was analyzed in femora cortical bone fractions highly enriched in either osteocytes or endosteal osteoblasts, as validated at the level of cell type specific gene markers expression (Suppl. Fig.S1). As shown in Fig. 1A, an expression of Pparγ in osteocytes isolated from 6 mo old C57BL/6 males was 8-fold higher than the expression in endosteal osteoblasts isolated from the same femora cortical bone, whereas in 6 mo old females osteocyte expression of Pparγ was 40-fold higher than in osteoblasts. Interestingly, Pparγ mRNA expression increases with aging, as osteocytes derived from 10 mo old females have 3-fold higher Pparγ levels than osteocytes derived from 6 mo old females (Fig. 1B). Finally, Pparγ mRNA expression increases in osteocytes of mice treated with TZD rosiglitazone (Fig. 1C) indicating that PPARG protein in osteocytes is functional, because it responds to activation with the high affinity ligand by autoregulating its own expression, a response well documented in cells of adipocytic lineage [4].
Figure 1.
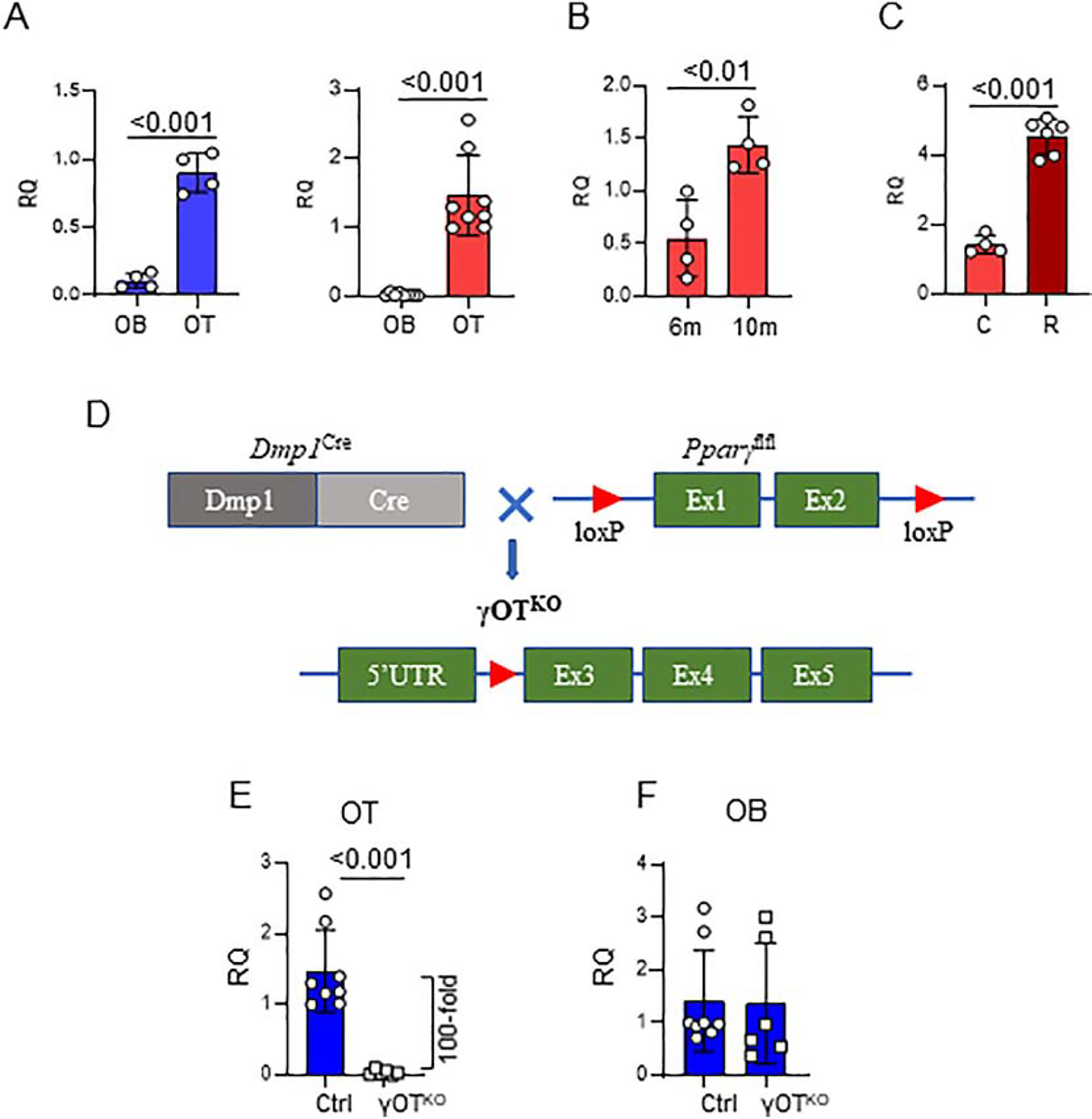
Development of γOTKO mice. A. Pparγ is highly expressed osteocytes (OT) as compared to osteoblasts (OB). OT and OB were isolated from femora cortical bone of 6.5 – 7 mo old C57BL/6 males (blue) (γOTKO n=4 and Ctrl n=4) and females (red) (γOTKO n=8 and Ctrl n=8) using a method of differential collagen digestion and immediately processed for RNA isolation. B. Pparγ expression in OT increases with aging. OT were isolated as above from femora of 6 mo and 10 mo old C57BL/6 female mice. C. Rosiglitazone treatment increases expression of osteocytic Pparγ. OT were isolated from 10 mo old C57BL/6 female mice treated with 25 mg/kg/d rosiglitazone (R) for 6 weeks (γOTKO n=6 and Ctrl n=4). D. Schematic of γOTKO mice development. γOTKO mice have deleted exon 1 and 2 from Pparγ gene, as a result of crossing Dmp1Cre and Pparγflfl mice. E. Pparγ expression in OT isolated from 6 mo old γOTKO (n=6) and Ctrl (n=8) male mice. F. Pparγ expression in OB isolated from the same mice as in E. Numbers above the horizontal bars indicate p values calculated with parametric unpaired Student t-test. RQ – relative quantity.
These findings gave us strong bases for seeking the role of PPARG nuclear receptor in osteocytes. Mice with PPARG deficiency primarily in osteocytes, the γOTKO mice, were constructed by crossing Pparγflfl with 10kb Dmp1Cre strains to remove exon 1 and exon 2 from the Pparγ gene, as shown in Fig. 1D. Offspring with a desirable γOTKO genotype are viable and born with predicted Mendelian distribution with respect to sex and genotype. In all experiments, control mice (Ctrl) consist of littermates with either DmpCrePparγWT or DmpWTPparγfl/fl genotypes, as described in the Material and Methods.
The γOTKO model was validated on several levels. The expression of Pparγ in osteocytes isolated from femora of γOTKO mice was 100-fold lower than the expression in osteocytes isolated from Ctrl mice (Fig. 1E), while Pparγ expression in osteoblasts isolated from γOTKO mice was not affected, as compared to Ctrl mice (Fig. 1F). Since there is a concern that Cre recombinase under control of 10 kb Dmp1 promoter can be activated in other than osteocyte cell types, we analyzed the level of Pparγ expression in different tissues of adult γOTKO and Ctrl mice. As shown in the Suppl. Table S2, we did not detect significant differences in Pparγ expression in muscle, bone marrow, liver, WAT, BAT, kidney, cerebellum, and small intestine of Ctrl and γOTKO suggesting that even if 10 kb Dmp1Cre construct is “leaky”, the expression of Pparγ in other tissues of adult γOTKO mice is not obviously affected, which validates our study on the role of PPARG in osteocytes of mature bone.
3.2. PPARG transcription factor is essential for sclerostin production in osteocytes
The level of PPARG protein deletion in osteocytes is paralleled by the level of sclerostin protein produced in these cells (Fig. 2A and 2B). There is an excellent correlation (Pearson’s r=0.991, p=0.001) between PPARG and sclerostin levels, with both proteins being significantly decreased in osteocytes isolated from bone of γOTKO mice (Fig. 2C). Immunohistochemistry of tibia cortical bone confirmed major reduction of SOST protein in osteocytes deficient in PPARG (Fig. 2D) (note mouse #335 in Fig. 2A and 2D). Adjacent longitudinal section of tibia processed without primary antibody treatment was used as a negative control. Sclerostin levels in sera collected from γOTKO mice, including animals analyzed in Fig. 2A and 2B, show a tendency to decrease, however this decrease did not parallel a dramatic change in sclerostin protein levels in bone of the same animals (Fig. 2E). These suggest that sclerostin levels in circulation are not in direct correlation with the levels in bone and point to other sources for its production. Indeed, a number of studies indicates other organs such kidney, aorta and testes, as contributing to sclerostin levels in circulation [30–32].
Figure 2.
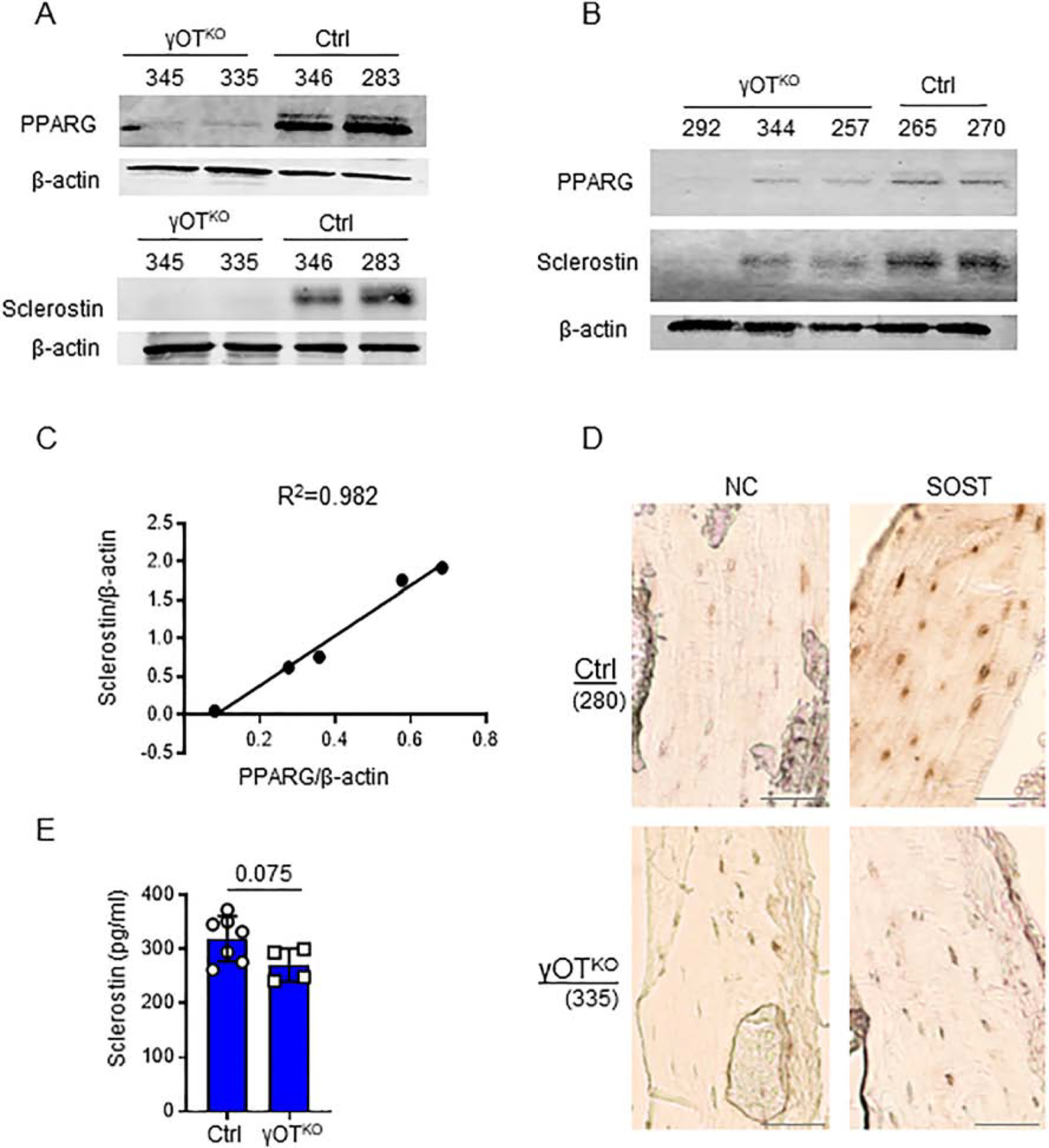
Correlation between PPARG and sclerostin levels in osteocytes. A. and B.Levels of PPARG and sclerostin proteins in OT isolated from femora of 6.5 mo old male γOTKO and Ctrl mice and either analyzed on separate (A) or the same (B) Western blot membrane. Numbers above the lanes indicate mice ID. (γOTKO n=5; Ctrl n=4). C. Pearson correlation analysis of PPARG and sclerostin protein levels shown in panels (A) and (B) (Pearson’s r=0.991, p=0.001) was calculated based on bands density assessed by Image J and normalized to β-actin. R2 represents coefficient of determination. D. Immunohistochemistry for sclerostin protein presence in osteocytes of tibia cortical bone. NC-negative control represents adjacent longitudinal sections processed without primary antibody incubation. Numbers in parenthesis indicate mice ID. Note that SOST is not detected in tibia osteocytes of mouse 335 (γOTKO) which correlates with an absence of detectable sclerostin levels in isolated femora osteocytes, as shown in panel (A). Scale bars correspond to 100 μm. NC – negative control consisted of adjacent sections being processed the same way as those for sclerostin detection, except for primary antibody incubation. E. Sclerostin levels measured by ELISA in sera of γOTKO (n=4) and Ctrl (n=7) 6.5 mo old male mice including animals analyzed in panels (A) – (D). Statistical significance was calculated using parametric unpaired Student’s t test.
3.3. Identification of PPREs located upstream of Sost transcription start site
The 8 kb DNA sequence upstream of transcription start site (TSS) of Sost gene was analyzed using a position weighted matrix (PWM) for prediction of binding sites for mouse PPARG::RXRA complex. The analysis was performed with assistance of the JASPAR database for curated, non-redundant set of transcription factor binding sites profiles. The position matrix algorithm is based on over 850 murine DNA sequences that were identified to bind the PPARG::RXRA heterodimer (Suppl. Fig. S2). Based on PWM, the JASPAR projected 15 PPARG response elements (PPRE) in the Sost gene promoter/enhancer region (Table 1 and Fig. 3A). The PPREs are located on both DNA strands. In the proximal 2 kb region, which represents Sost promoter, there are 4 PPREs, two of them with relatively low scores for PPARG binding consensus sequence are located within 1kb from TSS (PPRE1 and PPRE2) and the other two (PPRE3 and PPRE4) with higher scores at 1.8 – 1.9 kb distance from TSS. A cluster of 7 PPREs (PPRE5 to PPRE12) has been detected within 1 kb fragment at the 3.5 – 4.5 kb distance from TSS. Binding scores for PPREs in this cluster are high and varied from 8.1 to 12.5. The last cluster, with relatively high scores, consists of 3 PPREs (PPRE13 to PPRE15) and is located at the 7 – 8 kb upstream from TSS (Fig. 3A and Table 1). For further analysis, we selected two PPRE-containing regions based on their location and the highest scores for the consensus of PPARG binding sequence. The proximal PPRE3 located in the promoter region and two distal PPRE14 an PPRE15 located in the potential enhancer region. The consensus score for PPARG binding is 9.2 for PPRE3, and 15.3 and 9.6 for PPRE14 and PPRE15, respectively (Table 1).
Table 1.
Predicted PPRE sequences identified within 8.85 kb sequence upstream to Sost gene TSS using JASPAR position weighted matrix
| Start | End | DNA Strand | Predicted sequence | Score | |
|---|---|---|---|---|---|
| PPRE1 | 32 | 46 | + | ATGAGGCAGAGGGGG | 8.5 |
| PPRE2 | 666 | 680 | + | GTGGAGCTGGGGTCA | 8.9 |
| PPRE3 | 1829 | 1843 | + | CAGGGGAAAAGGAGA | 9.2 |
| PPRE4 | 1901 | 1915 | − | ACTTGGGAGAGGGCA | 8.7 |
| PPRE5 | 3435 | 3449 | + | CTGGGTGAAAAGAGA | 8.5 |
| PPRE6 | 3776 | 3790 | − | GGAGGCCAGAGGTCC | 12.5 |
| PPRE7 | 3984 | 3998 | − | GCAGGGGCAGGGGCA | 9.2 |
| PPRE8 | 3990 | 4004 | − | GCAGGGGCAGGGGCA | 9.2 |
| PPRE9 | 4184 | 4198 | − | GTAGACTAGAGGCTA | 8.1 |
| PPRE10 | 4277 | 4291 | + | GAGGCTCAGAGGGCA | 10.3 |
| PPRE11 | 4593 | 4607 | − | GTGTGCTAAAGGCCA | 10.6 |
| PPRE12 | 5331 | 5345 | − | TGGGGGCAGATTTCA | 8.4 |
| PPRE13 | 7229 | 7243 | + | CTGAAGCAGAGTGGA | 9.1 |
| PPRE14 | 7381 | 7395 | − | GTGGGGGAGGGGTCA | 15.3 |
| PPRE15 | 7726 | 7740 | + | GGAGGGCAAGGTGAA | 9.6 |
“Start” and “End” columns indicate PPRE bp position upstream of TSS. Bold text highlights PPREs chosen for further analysis.
Figure 3.
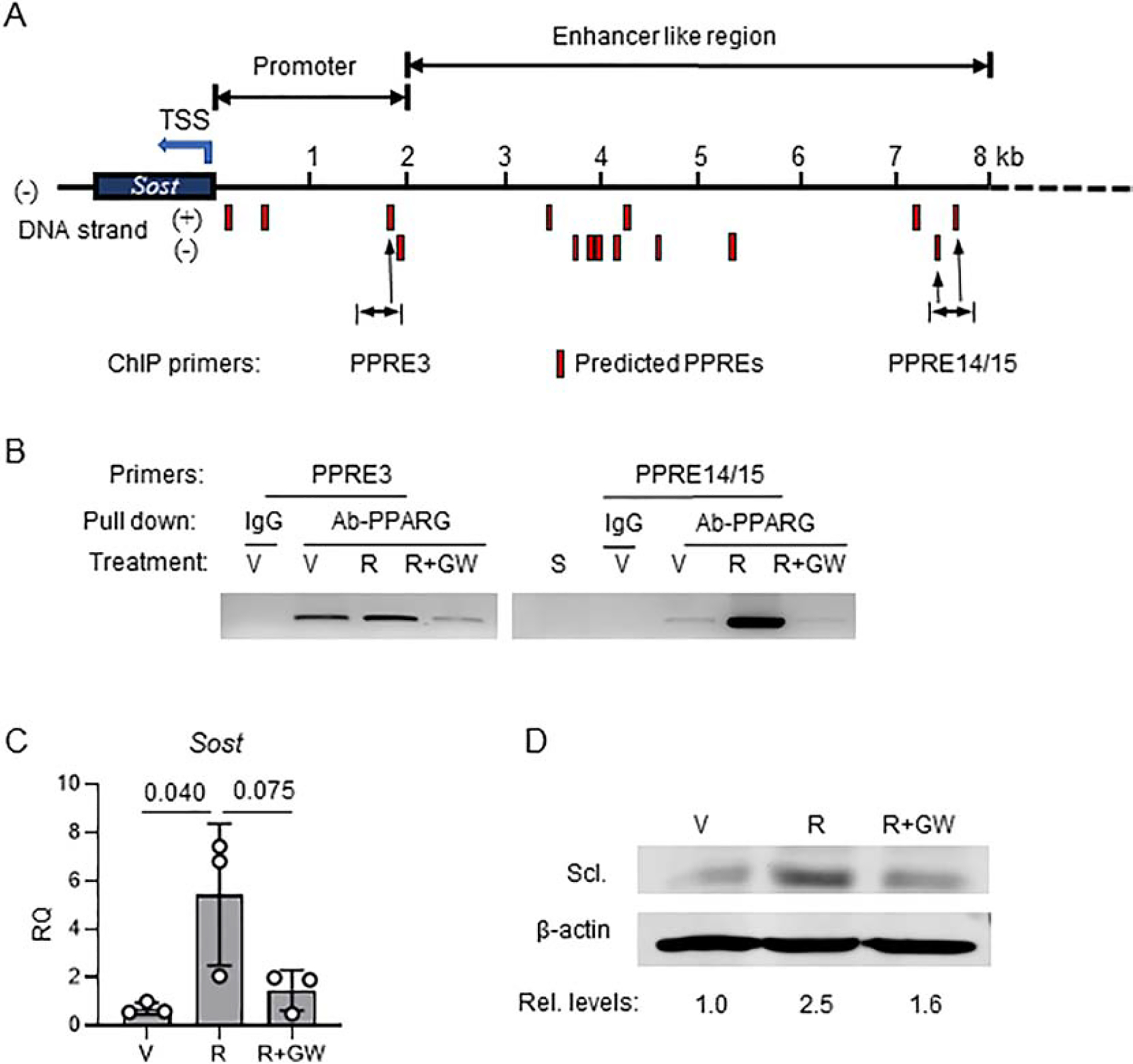
PPARG positively regulates sclerostin expression. A. Schematic positioning of JASPAR projected PPARG response elements (PPREs) (red bars) upstream of sclerostin transcription start site (TSS). B. Chromatin immunoprecipitation (ChIP) assay end point agarose gel image (full image of the gel is presented in Suppl. Fig. 1). ChIP was performed on osteocyte-like MLO-Y4 cells targeting PPRE3 and PPRE14/15. Cells were treated for 24h with either vehicle (V), or 1μM rosiglitazone (R) or combination of 1μM R and 10μM GW9662 antagonist (R+GW) followed by ChIP assay, as described in Material and Methods. S – sonicated lysate (no antibody pulldown). C. Expression of Sost mRNA in MLO-Y4 cells treated for 3 days with either vehicle (V), or rosiglitazone (R), or combination of rosiglitazone and GW9662 (R+GW) at the same concentration as in (B). One-way ANOVA followed by Tukey’s post hoc analysis was performed for significance calculation. D. Western blot of sclerostin protein levels in MLO-Y4 cells treated as in (C). Sclerostin protein levels were normalized to β-actin levels measured as band density using Image J and relative levels of expression had been calculated and shown below Western blot images.
Using the chromatin immunoprecipitation assays (ChIP), we tested selected PPREs for their binding PPARG in basal conditions and upon activation with rosiglitazone. The assay was performed on osteocyte-like MLO-Y4 cells and showed that PPARG binds to both locations of the PPRE motifs (Fig. 3B). In basal conditions, PPARG protein occupies proximal PPRE3 and its binding to the PPRE14/15 location is rather weak. Activation with rosiglitazone slightly increases binding to PPRE3, while treatment with the covalent antagonist GW9662 significantly decreases PPARG binding to this site (Fig. 3B and Suppl. Fig. S3). In contrast, treatment with rosiglitazone induces robust recruitment of PPARG to PPRE14/15 binding sites, which is completely abrogated in the presence of GW9662 antagonist (Fig. 3B).
The pattern of dynamic changes of PPARG interactions with PPREs was paralleled by regulation of Sost mRNA expression in the same MLO-Y4 cells. Treatment with rosiglitazone upregulated Sost expression, which in the presence of GW9662 antagonist was attenuated (Fig. 3C). This was followed by changes in the sclerostin protein levels. Treatment with rosiglitazone increased sclerostin by 2.5-fold, which was then reduced to 1.6-fold in the presence of GW9662 antagonist (Fig. 3D). These results demonstrate that PPARG is a positive regulator of Sost transcription and sclerostin protein production. Most importantly, the PPARG transcriptional activity regulating sclerostin can be modulated pharmacologically.
3.4. Expression of WNT signaling and bone formation markers is elevated in endosteal osteoblasts of γOTKO mice, as compared to controls
Sclerostin acts as an inhibitor of WNT and BMP signaling in osteoblasts. Consistent with low levels of sclerostin in γOTKO osteocytes, the expression of members of WNT signaling is upregulated in endosteal osteoblasts. Osteoblasts isolated from γOTKO mice have increased expression of WNT ligands, Wnt10b and Wnt16, and β-catenin, the transcriptional mediator of canonical WNT pathway activity (Fig. 4A). This is followed by increased expression of genes, which are positively regulated by this signaling, including Axin2, Connexin43 and Cyclin D (Fig. 4B). We have also analyzed expression of osteoblast-specific BMPs, Bmp2 and Bmp4, and found that Bmp4 expression is significantly increased in endosteal osteoblasts derived from γOTKO mice (Fig. 4C). Finally, an increase in activity of WNT and BMP pathways is reflected in upregulation of osteoblast-specific transcription factors (Runx2 and Dlx5) and alkaline phosphatase (Alp), a regulator of bone matrix mineralization (Fig. 4D).
Figure 4.
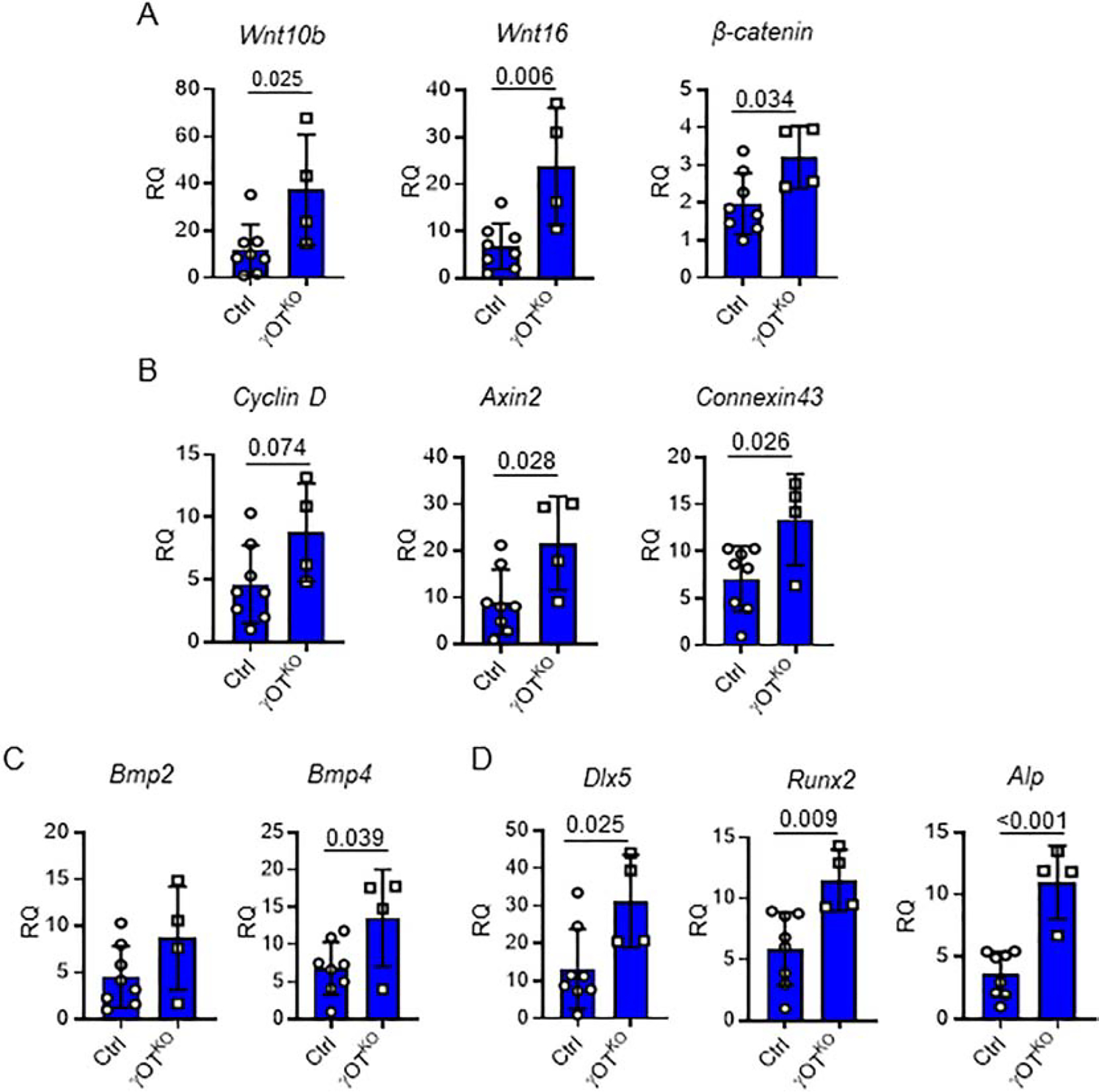
Relative expression of signaling pathways and osteoblast gene markers analyzed in fraction of endosteal osteoblasts freshly isolated by differential collagen digestion of femora cortical bone of 6.5 mo old males. A. WNT pathway signaling gene markers. B. Expression of genes positively regulated by WNT signaling. C. BMP signaling gene markers. D. Expression of genes positively regulated by WNT and BMP signaling and representing markers of bone forming osteoblasts. Ctrl (n=8) and γOTKO (n=4). Statistical significance was calculated using parametric unpaired Student’s t test.
3.5. γOTKO mice have higher BMD and increased bone formation in trabecular and cortical compartment
In support of increased WNT signaling and osteoblast activity, γOTKO mice, both males and females, have high bone mass. In males, an increased global bone mineral density (BMD) is noticed as early as 4 mo of age, while in females it develops later in life (Fig. 5A). An analysis of bone formation rate in calcein double-labeled trabecular bone of proximal tibia showed that both γOTKO males and females at the age of 6 mo have higher bone formation rate (BFR) as compared to Ctrl mice with intact PPARG in osteocytes (Fig. 5B). An increased BFR correlates with increase in fraction of bone surface undergoing process of mineralization (MS/BS), but not a mineral apposition rate (MAR) (Fig.5B).
Figure 5.
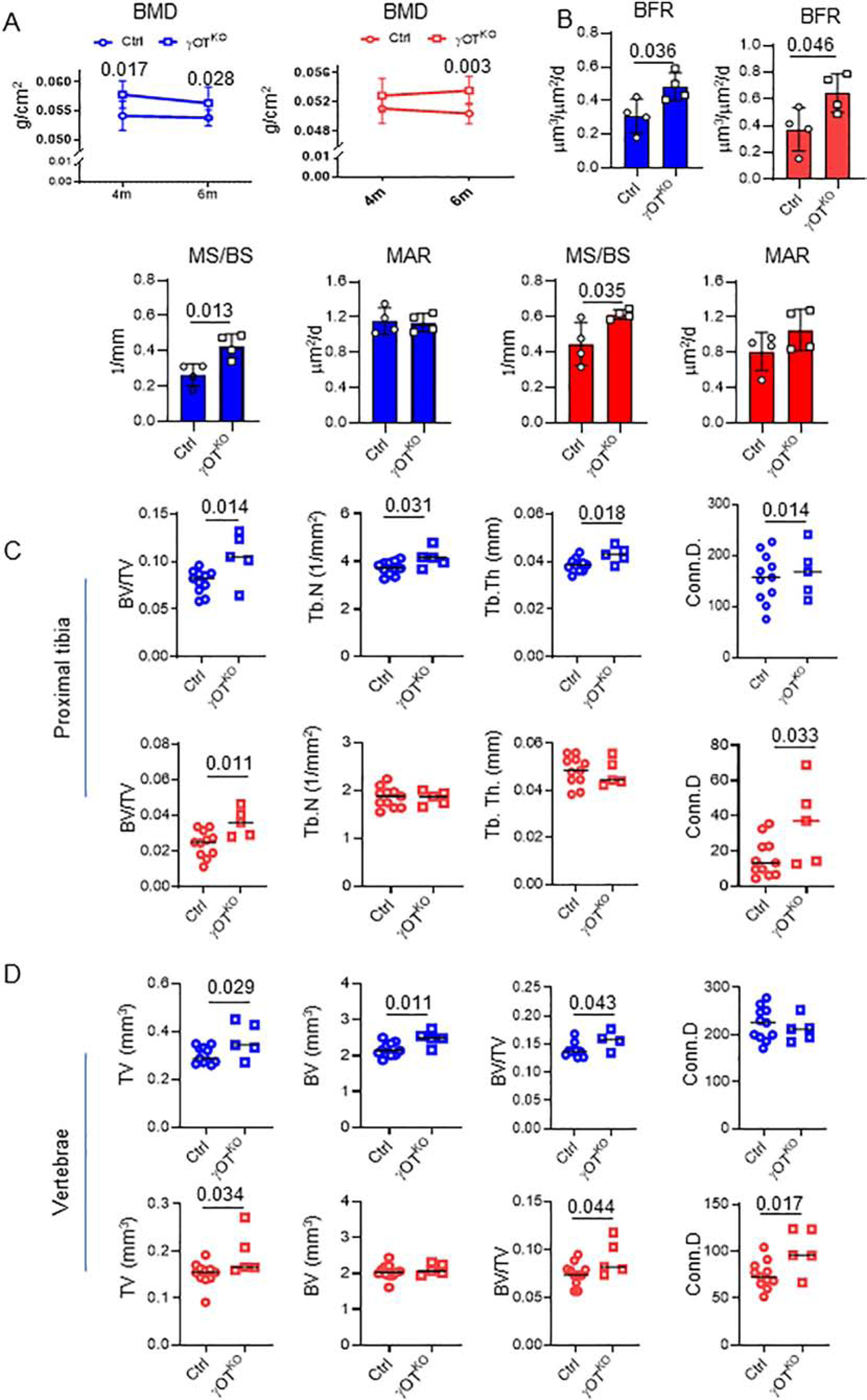
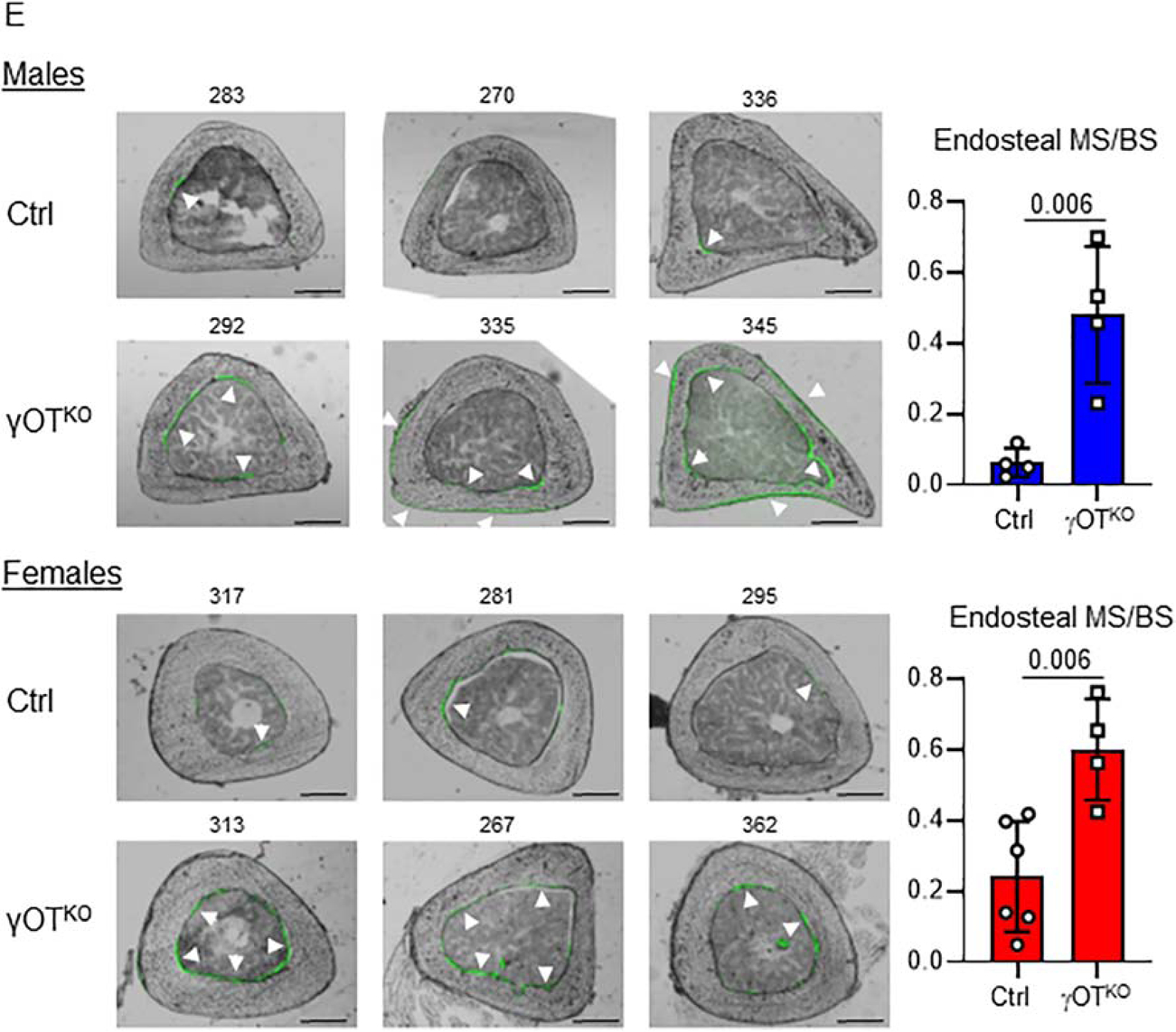
Both males (blue) and females (red) γOTKO mice exhibit high bone mass and high bone formation. A. Measurements of global BMD using DXA at 4 mo and 6 mo of age. Ctrl (n=11) and γOTKO (n=5). B. Bone formation rate (BFR), fraction of mineralizing bone surface (MS/BS), and mineral apposition rate (MAR) in 6.5 mo old males and 7 mo old females measured in calcein double-labeled tibia. Ctrl (n=4) and γOTKO (n=4). C. μCT measurements of trabecular bone in proximal tibia. D. μCT measurements of trabecular bone in L4 vertebrae. C and D. Ctrl (n=11) and γOTKO (n=5). TV – tissue volume, BV – bone volume, BV/TV – trabecular bone mass, Tb.N – trabecular number, Tb.Th – trabecular thickness, Conn.D – connectivity density. Statistical significance was calculated using parametric unpaired Student’s t test. E. Fluorescence and bright fields overlayed images of cross-sectioned tibia midshaft of the same animals as in (B). Numbers above the images indicate mice IDs. Note the correlation between very low sclerostin levels in OT of γOTKO mice (#335, 345, 292) in Fig. 2A and 2B and high calcein labeling on endosteal and periosteal tibia bone surface of the same mice. In contrast, very low calcein labeling of cortical bone corresponds to normal sclerostin levels in Ctrl mice (#283 and 270) as shown in Fig. 2A and 2B. White arrows point to calcein labeled bone surface. Scale bars correspond to 500 μm. Graphs represent quantification of endosteal mineralized surface of males (blue) (Ctrl n=4; γOTKO n=4) and females (red) (Ctrl n=6; γOTKO n=4). Statistical significance was calculated using parametric unpaired Student’s t test.
An analyses of bone microarchitecture using micro-computed tomography (mCT), showed that both sexes have increased trabecular bone mass in appendicular and axial skeleton (Fig. 5C and 5D). In males at 6 mo of age, high trabecular bone mass in proximal tibia was consistent with high trabecular number and thickness, whereas in females at the same age high trabecular bone mass correlated rather with structural changes at the level of connectivity density (Fig. 5C). Similarly, both males and females have high trabecular bone mass in L4 vertebra which correlated with increased connectivity density (Fig. 5D). Interestingly, in both sexes the vertebra body is larger in γOTKO, as compared to Ctrl mice (Fig. 5D). Remarkably, the changes in global BMD and trabecular bone mass were not followed by changes in properties of tibia cortical bone.
In γOTKO mice, a fraction of calcein labeled endosteal bone surface is much higher than in Ctrl animals (Fig. 5E) indicating increased bone formation. There is also increase in periosteal bone formation, especially in γOTKO males.
Serum parameters of bone turnover support increase in bone formation (Fig. 6). Bone specific alkaline phosphatase (BALP) activity is significantly higher and levels of N-terminal propeptide of type I procollagen (P1NP) have tendency to increase in sera of γOTKO, as compared to Ctrl mice. At the same time, there is no difference in the levels of tartrate resistant acid phosphatase (TRAcP 5b), a marker of osteoclast activities (Fig. 6). However, the cortical thickness, bone area, marrow area, bone material properties (BMD and TMD), and bone geometry did not differ between γOTKO and Ctrl mice (Table 2). These results suggest that PPARG absence in osteocytes alters, besides sclerostin suppression, other activities which neutralize the expected anabolic effect [31] and result in “no gain” in cortical bone parameters.
Figure 6.

Levels of bone turnover markers in sera of 6.5 mo old males (blue) (Ctrl n=6–9; γOTKO n=5–6) and 7 mo old females (red) (Ctrl n=4; γOTKO n=5). BALP – bone specific alkaline phosphatase, P1NP - N-terminal propeptide of type I procollagen (P1NP), TRAcP 5b - tartrate-resistant acid phosphatase form 5b. Statistical significance was calculated using parametric unpaired Student’s t test.
Table 2.
mCT cortical bone analysis
| Males | Females | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| γOTKO | Ctrl | γOTKO | Ctrl | |||||||
| Mean | SD | Mean | SD | p value | Mean | SD | Mean | SD | p value | |
| T.Ar (mm2) | 2.033 | 0.259 | 2.066 | 0.131 | 0.730 | 1.558 | 0.120 | 1.617 | 0.102 | 0.327 |
| B.Ar (mm2) | 0.722 | 0.116 | 0.691 | 0.031 | 0.411 | 0.553 | 0.046 | 0.570 | 0.031 | 0.415 |
| M.Ar (mm2) | 1.311 | 0.154 | 1.375 | 0.119 | 0.374 | 1.005 | 0.076 | 1.048 | 0.087 | 0.363 |
| Ct.Th. (mm) | 0.194 | 0.011 | 0.190 | 0.008 | 0.460 | 0.182 | 0.011 | 0.185 | 0.009 | 0.523 |
| BMD (mg HA/cm3) | 583.4 | 19.1 | 565.4 | 22.7 | 0.147 | 607.9 | 27.2 | 610.7 | 40.6 | 0.892 |
| TMD (mg HA/cm3) | 1048.9 | 12.5 | 1047.8 | 7.8 | 0.828 | 1048.5 | 9.1 | 1055.2 | 25.4 | 0.583 |
| pMOI (mm4) | 0.222 | 0.045 | 0.224 | 0.018 | 0.879 | 0.128 | 0.016 | 0.136 | 0.017 | 0.381 |
| Imax/Cmax (mm3) | 0.180 | 0.027 | 0.176 | 0.011 | 0.670 | 0.111 | 0.014 | 0.115 | 0.012 | 0.623 |
| Imin/Cmin (mm3) | 0.133 | 0.020 | 0.135 | 0.009 | 0.781 | 0.094 | 0.008 | 0.099 | 0.009 | 0.309 |
T.Ar – total area, B.Ar – bone area, M.Ar – marrow area, Ct.Th – cortical thickness, BMD – bone mineral density, TMD – tissue mineral density, pMOI – polar moment of inertia or torsional strength, Imax/Cmax - bending strength across the bone along the maximal centroid-to-edge distance, Imin/Cmin - bending strength across the bone along the minimal centroid-to-edge distance
3.6. PPARG in osteocytes regulates marrow fat volume at least in part via sclerostin axes
Besides regulation of WNT signaling in osteoblasts, sclerostin is also implicated in regulation of marrow adipocyte differentiation, probably through the inhibitory effect on WNT pathway activity in cells of adipocytic lineage [33]. In general, marrow fat content in tibia of γOTKO mice is decreased. In males, the decrease in total volume of bone marrow adipose tissue (BMAT) is mostly due to its decrease in distal part since the proximal tibia contains very little fat in 6 mo old animals (Fig. 7A). However, in females, which have much more BMAT in proximal tibia than males, PPARG ablation in osteocytes resulted in substantial reduction of BMAT in this location (Fig. 7B and 7C). Similarly to males, females have decreased BMAT volume in distal tibia. Pearson correlational analysis of BMAT volume and osteocyte levels of sclerostin protein in the same animals resulted in the partial but statistically significant coefficient of determination, with the value R2 = 0.695 (Fig. 7D).
Figure 7.
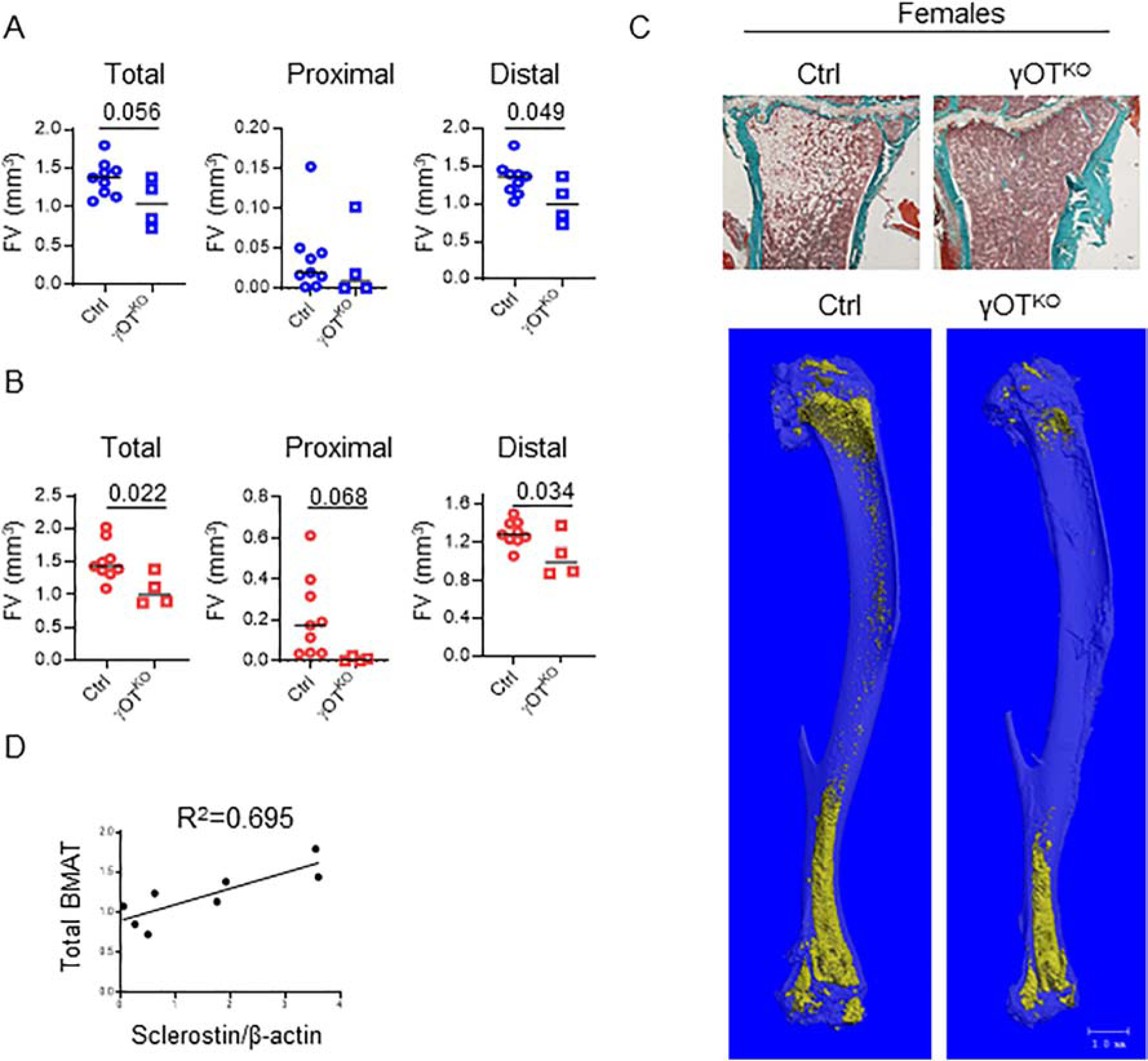
PPARG in osteocytes regulates marrow fat content. A. BMAT content in tibia of 6.5 mo old males γOTKO (n=4) and Ctrl (n=9) mice. Measurements were done using μCT after decalcification of tibia followed by OsO4 staining. B. BMAT measurements in tibia of 7 mo old females γOTKO (n=4) and Ctrl (n=9) mice. C. Representative Goldner’s Trichrome stained proximal tibia of female Ctrl and γOTKO mice and renderings of OsO4 stained entire tibia (below) μCT images of female Ctrl and γOTKO mice. Bar on mCT renderings indicates 1 mm. D. Pearson correlation of marrow fat volume (determined by mCT) and sclerostin protein levels (determined by Western blot) in osteocytes of Ctrl and γOTKO male mice (n=8), the same animals which were analyzed in Fig. 7A and Fig.2A and 2B (Pearson’s r=0.883, p=0.010). R2 – represents coefficient of determination. Statistical significance was calculated using parametric unpaired Student’s t test.
The conditioned medium (CM) experiment assessed sclerostin contribution to the regulation of BMSC lineage allocation toward adipocytes (Fig. 8A). CM, collected from organ culture of cortical femora bone of intact C57BL/6 mice and depleted from sclerostin with specific antibodies (Fig. 8B), decreased expression of Adiponectin and Fabp4, and have a tendency to decrease an expression of adipocyte-specific Pparγ2, in recipient BMSC isolated from the same mice (Fig. 8C).
Figure 8.
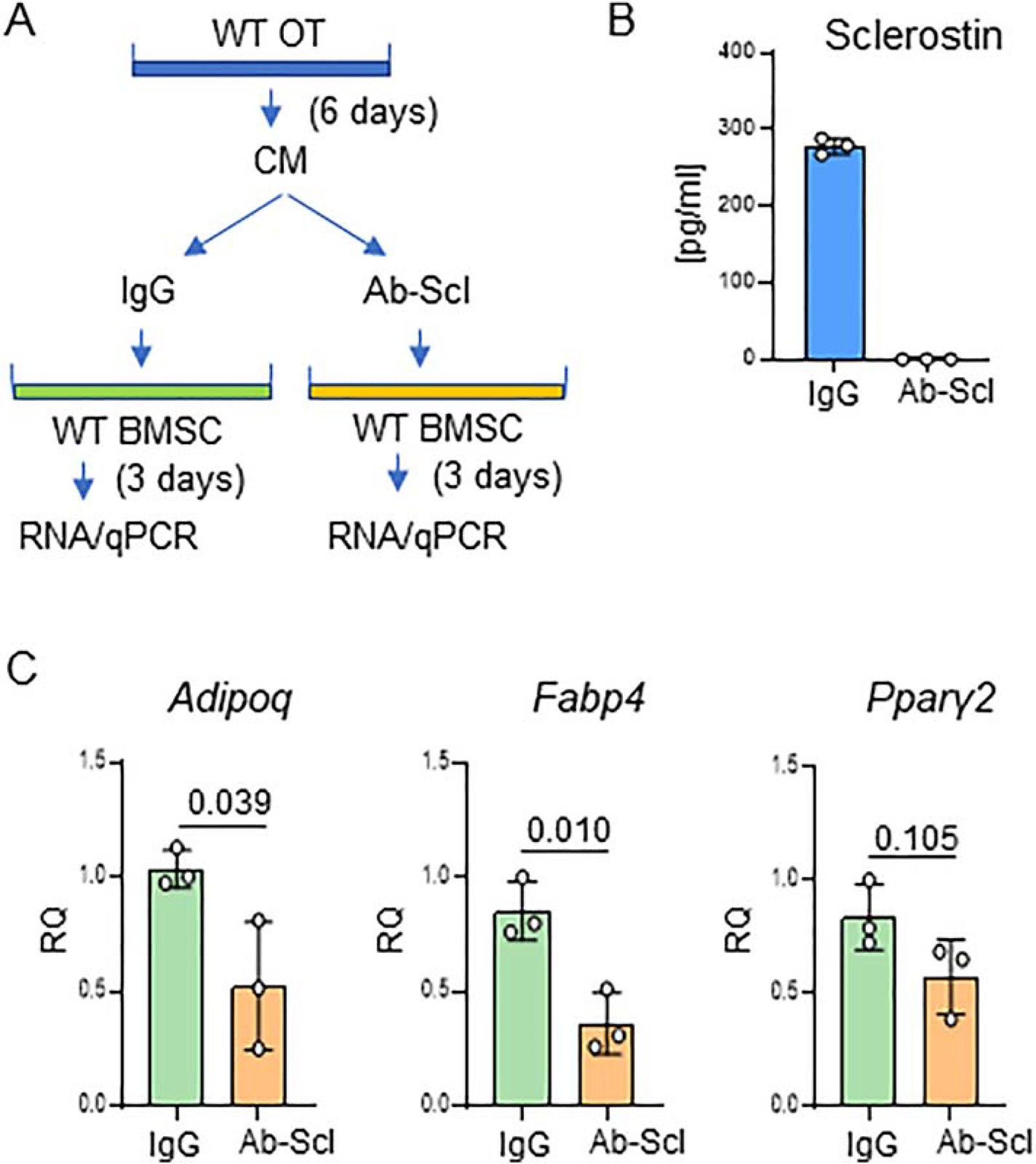
Osteocyte derived sclerostin positively contributes to marrow adipocyte differentiation. A. Schematic showing experimental design of co-culture of BMSC with intact or sclerostin depleted conditioned medium (CM). One group of adherent BMSC culture received IgG depleted CM from primary osteocytes (control group) while the other group received sclerostin depleted CM (group of interest). B. ELISA measurements of sclerostin level in CM after anti-SOST antibody mediated depletion. C. Expression of adipocytic and osteoblastic gene markers in adherent BMSCs treated with CM from primary osteocytes IgG or sclerostin (Ab-Scl) depleted. Statistical significance was calculated using parametric unpaired Student’s t test.
A significant decrease in BMAT volume in γOTKO mice, together with a partial correlation between sclerostin levels and BMAT volume, and a decrease in adipocytic phenotype of BMSC subjected to CM depleted from sclerostin, indicate that PPARG in osteocytes is essential for physiological BMAT formation and that sclerostin contributes to this process.
3.7. γOTKO mice are largely resistant to rosiglitazone-induced bone loss
The evidence presented in Fig. 1C and Fig. 3, collectively imply that PPARG activated with rosiglitazone increases sclerostin production. It has been documented extensively by our lab and others that rosiglitazone decreases bone mass in mice and humans. Therefore, we asked a question whether PPARG in osteocytes contributes to the rosiglitazone-induced bone loss. Female mice at the age of 10 mo were fed a diet supplemented with rosiglitazone for 4 weeks, which resulted in substantial decrease in a global bone mineral density (BMD) in Ctrl mice, but not in γOTKO mice (Fig. 9A). The mCT examination of trabecular bone mass in vertebra body confirmed that γOTKO mice are spared of the negative effect of rosiglitazone. While the vertebra BV/TV of γOTKO mice was unaffected by rosiglitazone, the Ctrl mice BV/TV decreased significantly by 34.4% (Fig. 9B). Consistently, the trabeculae number (Tb.N) were decreased and spacing between trabeculae (Tb.Sp) were increased in Ctrl mice, whereas the γOTKO mice were largely protected from these effects (Fig. 9B).
Figure 9.
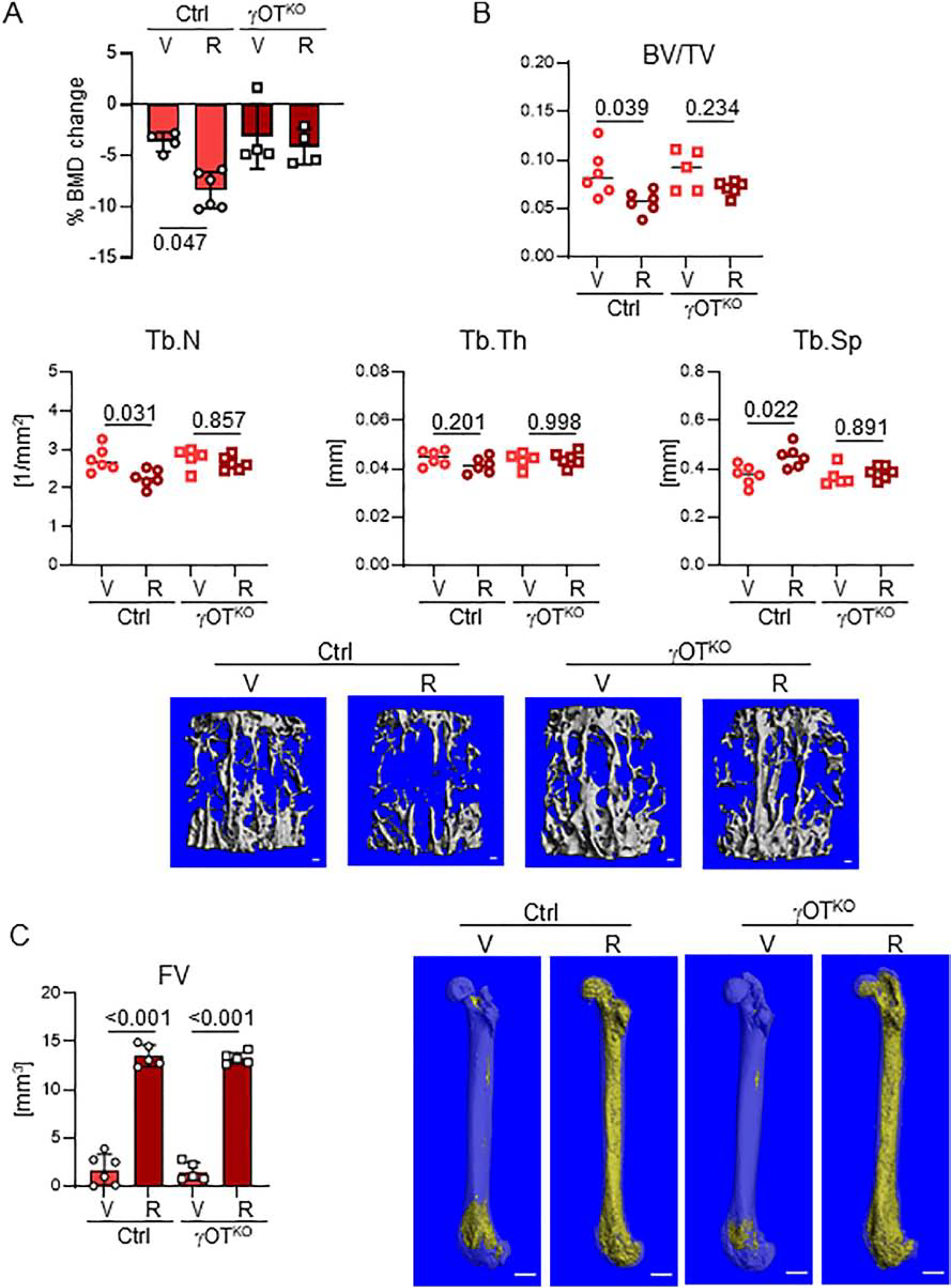
Bone of γOTKO mice is partially protected from the detrimental effects of rosiglitazone. A. Percent change in global BMD after 4 wks treatment of 10 mo old females with rosiglitazone (25 mg/kg/d). n=4–5 per group. B. μCT measurements of trabecular bone in L4 vertebrae and representative renderings. n=5–6/group. BV/TV – trabecular bone mass, Tb.N – trabecular number, Tb.Th – trabecular thickness, Tb.Sp – trabecular spacing. Bar on renderings indicates 0.1 mm. C. Left - Total fat volume in femora measured with μCT after decalcification and staining with OsO4 (n=5/group). Right - Representative mCT renderings. Bar on renderings indicates 1 mm. Statistical analysis was performed using Two-way Anova followed by Tukey’s multiple comparison test and p values are indicated over horizontal lines on graphs.
As shown in Figs.7 and 8, PPARG in osteocytes regulates marrow adiposity and its deletion decreases BMAT content. Thus, we assessed whether PPARG activity in osteocytes contributes to marrow BMAT accumulation after rosiglitazone treatment. As shown in Fig. 9C, PPARG deficiency in osteocytes with parallel sclerostin deficiency did not protect from accumulation of large volume of marrow fat, comparable to that of Ctrl mice, in response to rosiglitazone treatment. Thus, TZDs-related bone loss in female mice relies to large degree on PPARG function in osteocytes and may include sclerostin upregulation, while proadipocytic effect of TZDs consists of direct effect on BMSC, which in γOTKO model have unaffected expression of PPARG protein.
4. Discussion
Presented studies provide compelling evidence that PPARG in osteocytes plays an important role in transcriptional regulation of sclerostin expression. First, we have shown that there is an excellent positive correlation between protein levels of PPARG and sclerostin in osteocytes. It is notable that in the absence of PPARG there is an absence of sclerostin, which points to this nuclear receptor as essential for sclerostin production. Consequently, PPARG activation with full agonist rosiglitazone increases Sost transcript expression and sclerostin protein levels. Second, we have identified multiple PPRE sequences upstream of transcription start site, and demonstrated that at least two of them bind PPARG protein with dynamics reflecting status of PPARG activation. Thus, PPARG on molecular levels actively interacts with Sost gene DNA regulatory elements located in promoter/enhancer fragment upstream of TSS. Third, an ablation of PPARG in osteocytes leads to increased bone mass and decreased physiological levels of BMAT. Increases in the global BMD and trabecular bone mass, which are associated with increased WNT signaling in osteoblasts, are expected outcomes of sclerostin deficiency in osteocytes [13, 31]. Interestingly however, we did not observe increase in cortical bone mass despite activation of endosteal osteoblasts and active bone formation on up to 60% of endosteal surface, as measured by calcein labeling. This suggests that there are other factors in osteocytes under the PPARG control that mitigate the anabolic effect of sclerostin absence on bone. It is also possible that mechanical loading will reveal full potential of PPARG-regulated cortical bone response, however we did not address this aspect in presented study. Finally, PPARG deficiency in osteocytes protects trabecular bone from the negative effect of rosiglitazone indicating that osteocytes are the major contributors to the TZD-induced bone loss. Although, we cannot conclude with certainty that the skeletal resistance to rosiglitazone treatment is entirely mediated by a lack of sclerostin, however presented results provide proof of concept that pharmacologic modulators of PPARG activity reach osteocytes in their lacunae and exert the function including differential regulation of sclerostin protein.
Finding the functional link between PPARG and sclerostin is of great importance, because both proteins are pharmacological targets for existing therapies to treat diabetes and osteoporosis, respectively. This finding provides means to explore therapeutic overlap between anti-diabetic and anti-osteoporotic therapies. In fact, our previous study showed that anabolic effect on bone of a novel type of insulin sensitizers, which act as inverse agonists in respect to the PPARG pro-adipocytic and anti-osteoblastic activity, was associated with down-regulation of Sost expression in osteocytes [12]. Here, we have demonstrated that PPARG is a positive regulator of sclerostin protein expression by showing that its ablation from osteocytes shuts down, while its activation increases, sclerostin production.
A potential prospect of pharmacological targeting PPARG to reduce sclerostin in bone have several caveats which need to be corroborated including sclerostin expression in aorta, kidney and joints. These are potential off-targets for therapies with sclerostin antibodies [14–18, 20, 22]. Thus, it needs to be addressed whether regulation of sclerostin expression in other organs is also under the control of PPARG. An existence of multiple PPRE sequences in the Sost promoter region may indicate tissue-specific regulation, and may hold a premise for selective modulation of PPARG activities controlling sclerostin production specifically in osteocytes. Another caveat consists of the PPARG function in osteocytes beside sclerostin expression. Bone phenotype of γOTKO mice is rather mild and does not include increase in cortical bone mass despite significant activation of endosteal and even periosteal bone formation. This suggests that PPARG plays a larger role in regulation of osteocyte activities, which needs to be characterized in order to precise modulation of its activity to selectively invoke those which are beneficial for bone.
Since discovery of sclerostin and its encoding gene Sost, there have been great efforts to identify bone-specific transcriptional mechanisms regulating its expression. The transcriptional control of Sost is complex and implicates number of signaling pathways and only few identified transcription factors, which directly bind to the promoter region and regulate its activity. The most prominent is PTH signaling, which acts on ECR5 enhancer via MEF2c/HDAC5 transcriptional regulators, and negatively regulates Sost expression in osteocytes [34–36]. However, there is ample of evidence for other signaling mechanisms acting either in a positive (BMP, TNFα, Vitamin D, IL-1α, IL-1β, TWEAK, RA, and calcitonin), or in a negative (PGE2, Oncostatin M, Cardiotrophin, and Sirtuin1), or in both manners determined by biological context (TGFβ, glucocorticoids, estrogen, and hypoxia), and interacting with either an immediate promoter region or more distal enhancers [37, 38].
Mechanistically, activities of these signaling are channeled to the interaction of specific transcriptional regulators with cognate regulatory elements in the Sost promoter/enhancer region. Up to date, few of them have been identified for their binding to the promoter including RUNX2, OSX/SP7, ZFP467, and TIEG [39–42]. An array of loss- and gain-of-function experiments determined that RUNX2, OSX/SP7 and ZFP467 act as positive regulators of Sost promoter activity and that at least between RUNX2 and OSX/SP7, which binding sites are located in the proximal promoter region between −106 bp to −260 bp upstream of TSS, there is a great degree of coordination in this regulation [43]. In contrast, TIEG, or TGFβ inducible early gene-1, has been identified as a negative regulator of sclerostin expression acting downstream of estrogen signaling [42]. Interestingly, two identified KLF/SP1 binding sites for TIEG, which are located at −1864 kb and −1956 kb upstream of TSS, are in close proximity to the PPRE3 (−1829 kb), identified here as binding PPARG. It is well recognized that PPARG and estrogen signaling oppose each other during adipose tissue expansion, including BMAT expansion in conditions of estrogen deficiency [44–46]. It also has been shown that estrogen is a negative regulator of sclerostin [42]. Thus, there is a prospect that PPARG interaction with Sost promoter may be affected by, or may be subjected to, the modulatory effect of neighboring regulatory elements, such as KLF/SP1 perhaps in estrogen dependent manner. On the other hand, two distal PPRE14 and PPRE15, which have very high scores for PPRE consensus sequence and bind PPARG with dynamics coresponding to the status of PPARG activition, are located approximately in the 1 kb distance upstream to the region identified as enhancer responsive to Vitamin D and possessing a putative VDR binding element [47], which creates a possibility of PPREs interaction with a modulatory element under control of another nuclear receptor, VDR.
On note, we have also identified up to 7 PPREs with relatively good scores, which are clustered within a 1 kb fragment at the 4 kb distance from TSS; however, in this study we did not analyze the functional significance of this region to the regulation of Sost expression. Nevertheless, the pattern of such close clustering is known to facilitating strong interaction with binding proteins, even if the particular consensus sequences are not at the highest scores [48]. We have previously identified similar pattern of PPREs distribution in the promoter of genes known to be directly and positively regulated by PPARG including adipocyte-specific gene Fabp4 (Suppl. Fig. 4 in ref. [8]). The significance of this region to Sost gene expression remains to be established for both PPARG basal activity and the activity modulated pharmacologically by either full or inverse agonists. Taking into account the PPRE number, pattern of their distribution, and binding activity of those which were analyzed, it concludes in a strong support for PPARG directly regulating Sost expression via interaction with promoter and enhancer region.
Besides regulating bone remodeling, osteocytes also modulate marrow environment including marrow adiposity with sclerostin having a prominent role in this process. By the fact that sclerostin acts as an inhibitor of WNT pathway activity, it has a role in skewing BMSC lineage allocation toward adipocyte and away from osteoblast differentiation. The proadipocytic activity of sclerostin has been reported in several systems of adipocyte differentiation including 3T3–L1 cell line representing progenitors for white adipose tissue [49], ear mesenchymal stem cells, and BMSC [50]. Consistent with these findings, a decrease in sclerostin protein levels in bone of γOTKO mice correlates with a reduction in BMAT volume. However, there are few interesting aspects of this phenotype including different manifestation in respect to skeletal location and a degree of correlation.
Several research groups have shown that BMAT represents heterogenous tissue consisting of adipocytes originating from different progenitors and having different phenotype and function depending on skeletal location and exposure to environmental or hormonal signaling [46, 51, 52]. Based on the model of murine tibia, it has been postulated that BMAT located in proximal, in contrast to distal tibia, is highly sensitive to a variety of stimuli. Here we show that proximal BMAT is dependent on osteocyte signaling to a much larger degree than distal BMAT. Specifically, in females PPARG ablation in osteocytes almost completely prevents BMAT development in proximal tibia while in distal location BMAT volume is reduced by only 20%. The coculture experiments with intact or sclerostin depleted conditioned media confirm that sclerostin is partially, but not entirely, responsible for this phenotype. The degree of inhibition of adipocyte gene markers in recipient cells in vitro together with a rather modest correlation of BMAT volume in tibia with sclerostin levels in the same bone, suggest that there are other factors under PPARG control in osteocytes, which regulate BMAT specifically in the proximal tibia location. Another explanation of different response of proximal vs. distal BMAT to PPARG deficiency in osteocytes may include difference in the osteocyte-specific signaling gradients, which are determined by different ratio of bone area to marrow volume in these two locations.
The significance of presented study is reinforced by the fact that there is a scarcity of information on the role of PPARγ in osteocytes. To our knowledge, there are only two reports that previously addressed this issue. The first consists of development a model of PPARG deficiency in osteocytes constructed from the same Dmp1Cre driver and Pparγflfl mice, which was characterized with high bone mass including increased cortical bone mass and periosteal bone formation; the phenotype that we partially confirmed in our model [53]. This study was limited to male mice and did not focus on the mechanism by which PPARG in osteocytes controls bone mass including regulation of sclerostin expression and bone marrow composition in respect to BMAT [53]. The second model, demonstrated that sclerostin expression is increased in osteocyte-like MLO-Y4 in response to treatment with PPARG agonist rosiglitazone, which is consistent with our results; however, the mechanism of this effect was not provided [54].
4.1. Conclusion
In presented study, we have shown that PPARG nuclear receptor regulates osteocyte function supporting bone formation and marrow adipose tissue development with substantial fraction of these activities being channeled through sclerostin activities. We have demonstrated that sclerostin is transcriptionally regulated by PPARG and that PPARG is essential for sclerostin expression. We have focused specifically on the PPARG-sclerostin connection; however, we recognize that this relationship is a part of a larger role which PPARG plays in regulation of osteocyte function supporting bone homeostasis; the role which is pursued in another ongoing study. Presented findings support consideration of selective pharmacological targeting of PPARG in osteocytes to control sclerostin production and bone mass.
Supplementary Material
Highlights.
PPARG nuclear receptor in osteocytes is essential for sclerostin expression
Osteocytic PPARG regulates bone mass and marrow adiposity
Mice deficient in PPARG in osteocytes are protected from rosiglitazone-induced bone loss
5. Acknowledgements
This work was supported by grants from the National Institute of Diabetes, Digestive and Kidney Diseases (NIDDK) (R01 DK105825) to PRG and BLC, and the American Diabetes Association Innovative Basic Science Award (1-19-IBS-029) to BLC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure
All authors state that they have no conflicts of interest.
6. References
- [1].Dirckx N, Moorer MC, Clemens TL, Riddle RC, The role of osteoblasts in energy homeostasis, Nat Rev Endocrinol 15(11) (2019) 651–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Schwartz AV, Diabetes, bone and glucose-lowering agents: clinical outcomes, Diabetologia 60(7) (2017) 1170–1179. [DOI] [PubMed] [Google Scholar]
- [3].Lecka-Czernik B, Diabetes, bone and glucose-lowering agents: basic biology, Diabetologia 60(7) (2017) 1163–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, Evans RM, PPARγ signaling and metabolism: the good, the bad and the future, Nature medicine 19(5) (2013) 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Elrod HA, Sun SY, PPARgamma and Apoptosis in Cancer, PPAR research 2008 (2008) 704165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Staels B, PPARγ and atherosclerosis, Current Medical Research and Opinion 21(sup1) (2005) S13–S20. [DOI] [PubMed] [Google Scholar]
- [7].Lecka-Czernik B, Gubrij I, Moerman EJ, Kajkenova O, Lipschitz DA, Manolagas SC, Jilka RL, Inhibition of Osf2/Cbfa1 expression and terminal osteoblast differentiation by PPARgamma2, J Cell Biochem 74(3) (1999) 357–71. [PubMed] [Google Scholar]
- [8].Shockley KR, Lazarenko OP, Czernik PJ, Rosen CJ, Churchill GA, Lecka-Czernik B, PPARgamma2 nuclear receptor controls multiple regulatory pathways of osteoblast differentiation from marrow mesenchymal stem cells, J Cell Biochem 106(2) (2009) 232–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wan Y, Chong L-W, Evans RM, PPAR-γ regulates osteoclastogenesis in mice, Nature Medicine 13(12) (2007) 1496–1503. [DOI] [PubMed] [Google Scholar]
- [10].Kahn SE, Zinman B, Lachin JM, Haffner SM, Herman WH, Holman RR, Kravitz BG, Yu D, Heise MA, Aftring RP, Viberti G, Rosiglitazone-associated fractures in type 2 diabetes: an Analysis from A Diabetes Outcome Progression Trial (ADOPT), Diabetes care 31(5) (2008) 845–51. [DOI] [PubMed] [Google Scholar]
- [11].Zou W, Rohatgi N, Chen TH, Schilling J, Abu-Amer Y, Teitelbaum SL, PPAR-gamma regulates pharmacological but not physiological or pathological osteoclast formation, Nat Med 22(11) (2016) 1203–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Stechschulte LA, Czernik PJ, Rotter ZC, Tausif FN, Corzo CA, Marciano DP, Asteian A, Zheng J, Bruning JB, Kamenecka TM, Rosen CJ, Griffin PR, Lecka-Czernik B, PPARG Post-translational Modifications Regulate Bone Formation and Bone Resorption, EBioMedicine 10 (2016) 174–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Delgado-Calle J, Sato AY, Bellido T, Role and mechanism of action of sclerostin in bone, Bone 96 (2017) 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Saag KG, Petersen J, Brandi ML, Karaplis AC, Lorentzon M, Thomas T, Maddox J, Fan M, Meisner PD, Grauer A, Romosozumab or Alendronate for Fracture Prevention in Women with Osteoporosis, N Engl J Med 377(15) (2017) 1417–1427. [DOI] [PubMed] [Google Scholar]
- [15].Koos R, Brandenburg V, Mahnken AH, Schneider R, Dohmen G, Autschbach R, Marx N, Kramann R, Sclerostin as a potential novel biomarker for aortic valve calcification: an in-vivo and ex-vivo study, J Heart Valve Dis 22(3) (2013) 317–25. [PubMed] [Google Scholar]
- [16].Brandenburg VM, Kramann R, Koos R, Kruger T, Schurgers L, Muhlenbruch G, Hubner S, Gladziwa U, Drechsler C, Ketteler M, Relationship between sclerostin and cardiovascular calcification in hemodialysis patients: a cross-sectional study, BMC Nephrol 14 (2013) 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kawaguchi H, Serious Adverse Events With Romosozumab Use in Japanese Patients: Need for Clear Formulation of Contraindications Worldwide, J Bone Miner Res 35(5) (2020) 994–995. [DOI] [PubMed] [Google Scholar]
- [18].Saag KG, Curtis JR, Reid IR, Reply to Serious Adverse Events With Romosozumab Use in Japanese Patients: Need for Clear Formulation of Contraindications Worldwide, J Bone Miner Res 35(5) (2020) 996–997. [DOI] [PubMed] [Google Scholar]
- [19].Zhu D, Mackenzie NC, Millan JL, Farquharson C, MacRae VE, The appearance and modulation of osteocyte marker expression during calcification of vascular smooth muscle cells, PLoS One 6(5) (2011) e19595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Krishna SM, Seto SW, Jose RJ, Li J, Morton SK, Biros E, Wang Y, Nsengiyumva V, Lindeman JH, Loots GG, Rush CM, Craig JM, Golledge J, Wnt Signaling Pathway Inhibitor Sclerostin Inhibits Angiotensin II-Induced Aortic Aneurysm and Atherosclerosis, Arterioscler Thromb Vasc Biol 37(3) (2017) 553–566. [DOI] [PubMed] [Google Scholar]
- [21].Chang JC, Christiansen BA, Murugesh DK, Sebastian A, Hum NR, Collette NM, Hatsell S, Economides AN, Blanchette CD, Loots GG, SOST/Sclerostin Improves Posttraumatic Osteoarthritis and Inhibits MMP2/3 Expression After Injury, J Bone Miner Res 33(6) (2018) 1105–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wehmeyer C, Frank S, Beckmann D, Bottcher M, Cromme C, Konig U, Fennen M, Held A, Paruzel P, Hartmann C, Stratis A, Korb-Pap A, Kamradt T, Kramer I, van den Berg W, Kneissel M, Pap T, Dankbar B, Sclerostin inhibition promotes TNF-dependent inflammatory joint destruction, Sci Transl Med 8(330) (2016) 330ra35. [DOI] [PubMed] [Google Scholar]
- [23].Lazarenko OP, Rzonca SO, Hogue WR, Swain FL, Suva LJ, Lecka-Czernik B, Rosiglitazone induces decreases in bone mass and strength that are reminiscent of aged bone, Endocrinology 148(6) (2007) 2669–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R, Guidelines for assessment of bone microstructure in rodents using micro-computed tomography, J Bone Miner Res 25(7) (2010) 1468–86. [DOI] [PubMed] [Google Scholar]
- [25].Pai VM, Kozlowski M, Donahue D, Miller E, Xiao X, Chen MY, Yu Z-X, Connelly P, Jeffries K, Wen H, Coronary artery wall imaging in mice using osmium tetroxide and micro-computed tomography (micro-CT), J Anat 220(5) (2012) 514–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR, Parfitt AM, Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee, J Bone Miner Res 28(1) (2013) 2–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Stern AR, Stern MM, Van Dyke ME, Jähn K, Prideaux M, Bonewald LF, Isolation and culture of primary osteocytes from the long bones of skeletally mature and aged mice, Biotechniques 52(6) (2012) 361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kramer I, Halleux C, Keller H, Pegurri M, Gooi JH, Weber PB, Feng JQ, Bonewald LF, Kneissel M, Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis, Mol Cell Biol 30(12) (2010) 3071–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lee TI, Johnstone SE, Young RA, Chromatin immunoprecipitation and microarray-based analysis of protein location, Nat Protoc 1(2) (2006) 729–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Weivoda MM, Youssef SJ, Oursler MJ, Sclerostin expression and functions beyond the osteocyte, Bone 96 (2017) 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yee CS, Manilay JO, Chang JC, Hum NR, Murugesh DK, Bajwa J, Mendez ME, Economides AE, Horan DJ, Robling AG, Loots GG, Conditional Deletion of Sost in MSCDerived Lineages Identifies Specific Cell-Type Contributions to Bone Mass and B-Cell Development, Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 33(10) (2018) 1748–1759. [DOI] [PubMed] [Google Scholar]
- [32].Jastrzebski S, Kalinowski J, Stolina M, Mirza F, Torreggiani E, Kalajzic I, Won HY, Lee SK, Lorenzo J, Changes in bone sclerostin levels in mice after ovariectomy vary independently of changes in serum sclerostin levels, J Bone Miner Res 28(3) (2013) 618–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cawthorn WP, Bree AJ, Yao Y, Du B, Hemati N, Martinez-Santibanez G, Macdougald OA, Wnt6, Wnt10a and Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a beta-catenin-dependent mechanism, Bone 50(2) (2012) 477–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bellido T, Ali AA, Gubrij I, Plotkin LI, Fu Q, O’Brien CA, Manolagas SC, Jilka RL, Chronic Elevation of Parathyroid Hormone in Mice Reduces Expression of Sclerostin by Osteocytes: A Novel Mechanism for Hormonal Control of Osteoblastogenesis, Endocrinology 146(11) (2005) 4577–4583. [DOI] [PubMed] [Google Scholar]
- [35].Leupin O, Kramer I, Collette NM, Loots GG, Natt F, Kneissel M, Keller H, Control of the SOST Bone Enhancer by PTH Using MEF2 Transcription Factors, Journal of Bone and Mineral Research 22(12) (2007) 1957–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wein MN, Spatz J, Nishimori S, Doench J, Root D, Babij P, Nagano K, Baron R, Brooks D, Bouxsein M, Pajevic PD, Kronenberg HM, HDAC5 controls MEF2C-driven sclerostin expression in osteocytes, J Bone Miner Res 30(3) (2015) 400–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].St John HC, Hansen SJ, Pike JW, Analysis of SOST expression using large minigenes reveals the MEF2C binding site in the evolutionarily conserved region (ECR5) enhancer mediates forskolin, but not 1,25-dihydroxyvitamin D3 or TGFbeta1 responsiveness, J Steroid Biochem Mol Biol 164 (2016) 277–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sebastian A, Loots GG, Transcriptional control of Sost in bone, Bone 96 (2017) 76–84. [DOI] [PubMed] [Google Scholar]
- [39].Sevetson B, Taylor S, Pan Y, Cbfa1/RUNX2 directs specific expression of the sclerosteosis gene (SOST), J Biol Chem 279(14) (2004) 13849–58. [DOI] [PubMed] [Google Scholar]
- [40].Yang F, Tang W, So S, de Crombrugghe B, Zhang C, Sclerostin is a direct target of osteoblast-specific transcription factor osterix, Biochem Biophys Res Commun 400(4) (2010) 684–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].You L, Chen L, Pan L, Gu WS, Chen JY, Zinc finger protein 467 regulates Wnt signaling by modulating the expression of sclerostin in adipose derived stem cells, Biochem Biophys Res Commun 456(2) (2015) 598–604. [DOI] [PubMed] [Google Scholar]
- [42].Subramaniam M, Pitel KS, Bruinsma ES, Monroe DG, Hawse JR, TIEG and estrogen modulate SOST expression in the murine skeleton, J Cell Physiol 233(4) (2018) 3540–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Perez-Campo FM, Santurtun A, Garcia-Ibarbia C, Pascual MA, Valero C, Garces C, Sanudo C, Zarrabeitia MT, Riancho JA, Osterix and RUNX2 are Transcriptional Regulators of Sclerostin in Human Bone, Calcif Tissue Int 99(3) (2016) 302–9. [DOI] [PubMed] [Google Scholar]
- [44].Newell-Fugate AE, The role of sex steroids in white adipose tissue adipocyte function, Reproduction 153(4) (2017) R133–R149. [DOI] [PubMed] [Google Scholar]
- [45].Syed FA, Oursler MJ, Hefferanm TE, Peterson JM, Riggs BL, Khosla S, Effects of estrogen therapy on bone marrow adipocytes in postmenopausal osteoporotic women, Osteoporos Int 19(9) (2008) 1323–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lecka-Czernik B, Stechschulte LA, Czernik PJ, Sherman SB, Huang S, Krings A, Marrow Adipose Tissue: Skeletal Location, Sexual Dimorphism, and Response to Sex Steroid Deficiency, Front Endocrinol (Lausanne) 8 (2017) 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wijenayaka AR, Yang D, Prideaux M, Ito N, Kogawa M, Anderson PH, Morris HA, Solomon LB, Loots GG, Findlay DM, Atkins GJ, 1α,25-dihydroxyvitamin D3 stimulates human SOST gene expression and sclerostin secretion, Molecular and Cellular Endocrinology 413 (2015) 157–167. [DOI] [PubMed] [Google Scholar]
- [48].Czernik PJ, Peterson CA, Hurlburt BK, Preferential binding of MyoD-E12 versus myogenin-E12 to the murine sarcoma virus enhancer in vitro, J Biol Chem 271(15) (1996) 9141–9. [DOI] [PubMed] [Google Scholar]
- [49].Ukita M, Yamaguchi T, Ohata N, Tamura M, Sclerostin Enhances Adipocyte Differentiation in 3T3–L1 Cells, J Cell Biochem 117(6) (2016) 1419–1428. [DOI] [PubMed] [Google Scholar]
- [50].Fairfield H, Falank C, Harris E, Demambro V, McDonald M, Pettitt JA, Mohanty ST, Croucher P, Kramer I, Kneissel M, Rosen CJ, Reagan MR, The skeletal cell-derived molecule sclerostin drives bone marrow adipogenesis, Journal of cellular physiology 233(2) (2018) 1156–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Scheller EL, Doucette CR, Learman BS, Cawthorn WP, Khandaker S, Schell B, Wu B, Ding SY, Bredella MA, Fazeli PK, Khoury B, Jepsen KJ, Pilch PF, Klibanski A, Rosen CJ, MacDougald OA, Region-specific variation in the properties of skeletal adipocytes reveals regulated and constitutive marrow adipose tissues, Nat Commun 6 (2015) 7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Horowitz MC, Berry R, Holtrup B, Sebo Z, Nelson T, Fretz JA, Lindskog D, Kaplan JL, Ables G, Rodeheffer MS, Rosen CJ, Bone marrow adipocytes, Adipocyte 6(3) (2017) 193–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Brun J, Berthou F, Trajkovski M, Maechler P, Foti M, Bonnet N, Bone Regulates Browning and Energy Metabolism Through Mature Osteoblast/Osteocyte PPARγ Expression, Diabetes 66(10) (2017) 2541–2554. [DOI] [PubMed] [Google Scholar]
- [54].Mabilleau G, Mieczkowska A, Edmonds ME, Thiazolidinediones induce osteocyte apoptosis and increase sclerostin expression, Diabet Med 27(8) (2010) 925–32. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.