

Abstract
MicroRNA (miR)-410 plays a potential role in the pathogenesis of atherosclerosis. The current study mainly focuses on the underlying mechanism of miR-410/histone deacetylase 1 (HDAC1)/KLF5/nuclear factor κB (NF-κB) inhibitor α (IKBα)/NF-κB axis in atherosclerosis. miR-410 expression was determined using quantitative real-time PCR in both mouse models of atherosclerosis and human umbilical endothelial cells (HUVECs) treated with oxidized low-density lipoprotein (ox-LDL). The study subsequently predicted regulators associated with miR-410 through bioinformatics, and their binding relation was further verified through dual luciferase reporter gene and RNA immunoprecipitation (RIP) assays, and how HDAC1 regulated KLF5 was tested through coimmunoprecipitation (coIP). In HUVECs, miR-410 and HDAC1 mRNA expression; HDAC1, KLF5, IKBα, p65, p-p65, VCAM-1, ICAM-1, and MCP-1 protein expression; and inflammatory cytokine expressions were detected using quantitative real-time PCR, western blot, and ELISA. The present study further tested cell functions by Cell Counting Kit-8 (CCK-8), flow cytometry, and the colony-formation assay. It was revealed that miR-410 could target HDAC1, whereas HDAC1 could target transcription factor KLF5, increasing IKBα expression, thus suppressing NF-κB in atherosclerosis. Furthermore, silencing miR-410 or overexpressing HDAC1 increased cell viability and suppressed apoptosis and an inflammatory reaction in HUVECs in atherosclerosis. Blocking miR-410 promotes HDAC1 expression and increases IKBα levels through KLF5 to suppress NF-κB, thus preventing development of atherosclerosis.
Keywords: miR-410, HDAC1, IKBα, NF-κB, atherosclerosis
Graphical abstract
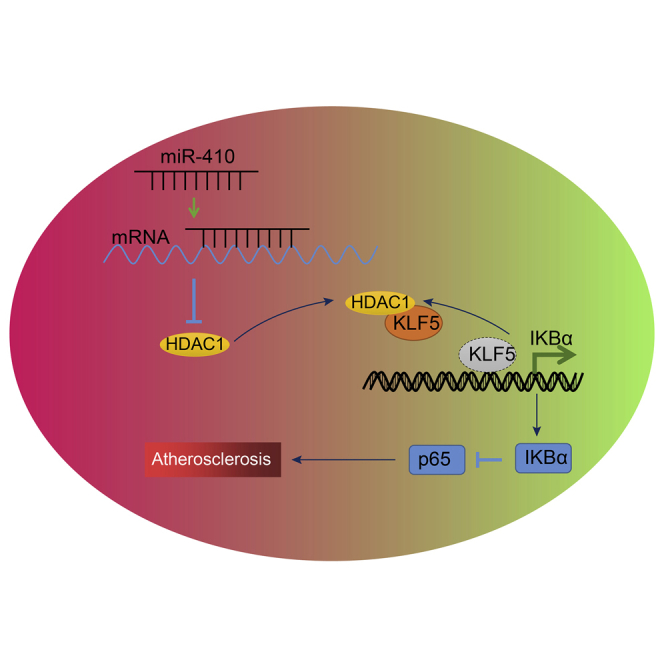
There has been emerging evidence of microRNAs (miRs) being important regulators in atherosclerosis. miR-410 plays a potential destructive role in the endothelial dysfunction of atherosclerosis, and our results revealed that miR-410 silencing promotes HDAC1 expression and increases IKBα levels through KLF5 to suppress NF-κB signaling, thus preventing development of atherosclerosis.
Introduction
Atherosclerosis is an ongoing chronic inflammatory disease, which occurs at the sites of disturbed blood flow.1 Unfortunately, atherosclerosis and its resultant clinical complication of cardiovascular diseases are major health problems in the modern world. The pathology of atherosclerosis is characterized by multiple monocyte-derived macrophages in the lipid core and vascular smooth muscle cells dominating in the fibrous cap with numerous lymphatic T cells.2 The plaques are generally covered by an intact endothelium until the eventual breakdown of the endothelial integrity, resulting in tragic progression and complications of the condition. One of the earliest onsets contributing to the occurrence of atherosclerosis is the impairment to the endothelial lining of the inner surface of the arterial vasculature. Mediated by a complex interplay of factors, stages, and molecular pathways, however, the endothelial dysfunction of atherosclerotic damage remains elusive.
microRNAs (miRNAs), as the endogenous, non-coding, evolutionarily conserved small RNAs, are estimated to regulate approximately 60% of human coding genes.3 The miRNAs have recently emerged as vital regulators of the cellular homeostasis and metabolic responses to stimuli in various human diseases including atherosclerosis.4,5 Previous studies have shown that miRNAs are involved either in a beneficial or harmful way in almost all molecular signaling of arterial remodeling and the genesis of atherosclerosis, including endothelial dysfunction, arterial wall invasion, platelet and vascular smooth muscle cell activation, as well as plaque formation.6,7 Additionally, microRNA (miR)-410 has been discovered to serve as oncogenes and tumor suppressors altering cell functions in numerous ways including proliferation, apoptosis, biochemistry metabolism, and inflammatory responses in different human malignancies, such as liver cancer, pancreatic cancer, colon cancer, and non-small cell lung cancer.8 It is unclear whether and how miR-410 is involved in the non-cancerous disorder processes such as endothelial dysfunction in atherosclerosis. There are studies that have reported that nuclear factor κB (NF-κB) is a crucial driver of endothelial dysfunction that is activated by multiple inflammatory cytokines.9 Furthermore, from the current study’s experimental results, it has been established that histone deacetylase 1 (HDAC1) was negatively regulated by miR-410. Therefore, the present study hypothesized that miR-410 participates in the initiation of atherosclerosis, targeting HDAC1, regulating NF-κB, resulting in endothelial dysfunction. To test this hypothesis, atherosclerosis of endothelial cell lines and mouse models was established, and the correlation among miR-410, downstream genes, NF-κB, inflammatory cytokines, and endothelial impairment was evaluated. Subsequently, we determined their respective roles in cholesterol instability-induced endothelial dysfunction in arterial vasculature.
Results
miR-40 is highly expressed in atherosclerosis
Through the hMDD database, we searched for atherosclerosis-related miRNAs and found that 29 miRNAs were considered as potential therapeutic targets for atherosclerosis (Table S1). At the same time, mouse atherosclerotic expression dataset GEO: GSE89858 was obtained through the GEO database. The expression of candidate miRNAs in the dataset was analyzed (Figure 1A). It was found that miR-410-3p showed significantly high expression in atherosclerotic samples (Figure 1B). Further sequence alignment analysis of miR-410 in human and mouse (Figure 1C) showed that their sequences were identical in human and mouse, suggesting that they may have similar regulatory mechanisms and functions.
Figure 1.

High miR-40 expression is observed in atherosclerosis
(A) The heatmap of candidate miRNA expression in GEO: GSE89858. The abscissa represents the sample number, the ordinate represents the miRNA name, each small square represents the expression of a miRNA in a sample, and the histogram on the right is the color scale. (B) The differential expression of miR-410 in GEO: GSE89858. The abscissa represents the sample type, the ordinate represents the expression, and the upper-left corner is the difference in p value. (C) The sequence alignment of miR-410 in human and mouse, in which Query is human miR-410 sequence, and Sbjct is mouse miR-410 sequence.
Downregulated miR-40 suppresses the development of atherosclerosis in mice
Subsequent to a high-fat diet (HFD), the current study detected the expression of miR-410 expression in ApoE−/− mice. Quantitative real-time PCR analysis showed that the miR-410 expression in the thoracic aorta tissues was significantly higher in the HFD group compared to the normal diet (ND) group (p < 0.05), and meanwhile, the HFD + miR-410 antagomir group demonstrated lower miR-410 levels than the HFD + negative control (NC) antagomir group (p < 0.05; Figure 2A). Subsequently, hematoxylin and eosin (H&E) and oil red O staining were performed on the aorta tissues (Figures 2B and 2C), which showed increased severity and area of inflammation and impairment in the HFD group in comparison to the ND group, whereas miR-410 antagomir rescued the damage.
Figure 2.

miR-40 inhibition suppresses the initiation of atherosclerosis in mice
(A) Detection of miR-410 expression level in aorta tissues. (B) Arterial impairment and inflammation. (C) The area of aorta impairment. (D) Serum TNF-α, IL-1, and IL-6 levels in mice. (E) VCAM-1, ICAM-1, and MCP-1 expression in aorta tissues; ∗p < 0.05. Measured data are described as mean ± standard deviation, analyzed using independent samples t test between two groups; n = 10.
The levels of serum inflammatory cytokines tumor necrosis factor (TNF)-α, interleukin (IL)-1, and IL-6 were found to be markedly elevated in the HFD group in comparison to the ND group as detected by the enzyme-linked immunosorbent assay (ELISA; p < 0.05), which was reversed by additional miR-410 antagomir treatment (Figure 2D). Furthermore, the western blot analysis revealed that exposure to an HFD could cause higher expression of endothelial cell glycoproteins VCAM-1, ICAM-1, and MCP-1 in the mice aorta tissues (p < 0.05), whereas miR-410 antagomir could minimize the change in those glycoproteins (Figure 2E). Cumulatively, the above-mentioned results demonstrated that an HFD significantly increased the miR-410 and inflammatory cytokine levels in the mice aorta tissues resulting in atherosclerosis, which could be rescued by miR-410 antagomir.
Interference of miR-410 expression can protect human umbilical endothelial cells (HUVECs) from oxidative damage
Aimed to determine whether the in vivo findings could be replicated in an in vitro cell line, HUVECs were treated with oxidized low-density lipoprotein (ox-LDL) to simulate HFD. Quantitative real-time PCR analysis detected higher miR-410 levels in cells treated with ox-LDL (Figure 3A). The cells were additionally treated with miR-410 antagomir or NC antagomir subsequent to ox-LDL induction, which revealed that miR-410 antagomir decreased the expression of miR-410 (p < 0.05; Figure 3B). Additionally, the ELISA and western blot analyses showed that the levels of TNF-α, IL-1, and IL-6 and the expressions of VCAM-1, ICAM-1, and MCP-1 were reduced in the supernatant of cells treated with miR-410 antagomir in comparison to those treated with NC antagomir (Figures 3F and 3G). Furthermore, a series of cell function experiments, including Cell Counting Kit-8 (CCK-8), colony-formation assay, and flow cytometry on cell viability and apoptosis, were performed. miR-410 antagomir increased the cell viability and suppressed apoptosis (Figures 3C−3E). The above-mentioned results showed that miR-410 promoted proliferation and decreased apoptosis in HUVECs.
Figure 3.

Blocking miR-410 prevents the development of atherosclerosis
(A) miR-410 expression in HUVECs treated with ox-LDL. (B) miR-410 expression in HUVECs after inhibiting miR-410. (C) HUVEC viability. (D) Colony formation to evaluate HUVEC viability. (E) HUVEC apoptosis. (F) The expression of TNF-α, IL-1, and IL-6 in HUVEC culture supernatant. (G) VCAM-1, ICAM-1, and MCP-1 levels in HUVECs; ∗p < 0.05. Measured data are described as mean ± standard deviation. Unpaired t test was used to analyze data between two groups and Bonferroni or Tukey’s test to analyze data among groups at different time points after repeated-measures ANOVA. All of the experiments were performed with technological triplicate.
Interfering miR-410 dampens development of atherosclerosis via regulation of HDAC1
The molecular mechanism underlying the miR-410 interference in atherosclerosis was studied. The bioinformatic prediction suggested that miR-410 potentially targeted genes HDAC1 and alpha-dystrobrevin (DTNA; Figure 4A). Additionally, microRNA.org suggested that miR-410 specifically bound to the HDAC1, which existed in both humans and mice (Figure 4B). Furthermore, it has been previously reported that HDAC1 can prevent the development of atherosclerosis, and its expression was decreased in atherosclerosis.10 We thus hypothesized that the regulatory mechanism of miR-410 on atherosclerosis might be through expression of HDAC1. Western blot analysis detected that the expression of HDAC1 was decreased in mice aorta tissues following an HFD compared to a ND (p < 0.05; Figure 4C). Moreover, in comparison to the HFD + NC antagomir group, the expression of HDAC1 was found to be elevated in the HFD + miR-410 antagomir group (p < 0.05).
Figure 4.

Interfering miR-410 inhibits development of atherosclerosis via regulation of HDAC1
(A) The bioinformatics websites StarBase, miRWalk, microT, RNAInter, and microRNA.org predicted the target genes of miR-410. (B) Prediction of targets of miR-410. (C) HDAC1 expression in mice aorta tissues (n = 10). (D) HDAC1 level in HUVECs treated with ox-LDL. (E) RIP-verified miR-410 binds to HDAC1. (F) The binding of miR-410 to HDAC1. (G) The expression of HDAC1 in HUVECs chose an optimal sequence for subsequent experiments. (H) miR-410 and HDAC1 expression in endothelial cells. (I) Cell viability. (J) Colony formation used evaluates the cell viability. (K) Cell apoptosis; ∗p < 0.05. Measured data are described as mean ± standard deviation. Unpaired t test was used to analyze data between two groups and Bonferroni or Tukey’s test to analyze data among groups at different time points after repeated-measures ANOVA. All of the experiments were performed with technological triplicate.
In HUVECs treated with ox-LDL, the expression of HDAC1 was also found to be increased when miR-410 was silenced by antagomir (Figure 4D). Moreover, the RNA immunoprecipitation (RIP) showed that the amount of miR-410 binding to HDAC1 in the anti-AGO2 group was significantly increased in comparison to that in the anti-immunoglobulin G (IgG) group (Figure 4E). Additionally, in comparison to the NC mimic group, the co-transfection with wild-type (WT)-HDAC1-3′ untranslated region (UTR) in the miR-410 mimic group resulted in a significant decrease in the fluorescence intensity (p < 0.05), and meanwhile, there was no significant difference noted in the fluorescence intensity subsequent to co-transfection with mutant (mut)-HDAC1-3′ UTR (p > 0.05; Figure 4F), which suggested that miR-410 could directly bind to HDAC1.
The sequences of short hairpin (sh)-HDAC1-1 (shRNA targeting HDAC1-1) and sh-HDAC1-2 were designed as previously described.11 Western blot analysis showed that the HDAC1 level was markedly decreased in the sh-HDAC1-1 and sh-HDAC1 groups in comparison to the sh-NC group and was the lowest in the sh-HDAC1-2 group; therefore, it was selected for subsequent experiments (Figure 4G). Aimed to investigate the effect of miR-410 binding with HDAC1 on HUVEC proliferation and apoptosis, miR-410 and HDAC1 were blocked simultaneously in the HUVECs previously treated with ox-LDL. Quantitative real-time PCR analysis revealed that the expression of miR-410 was decreased, and the HDAC1 level was increased in the miR-410 antagomir + sh-NC group in comparison to the NC antagomir + sh-NC group (Figure 4H). The miR-410 expression showed no significant change in the miR-410 antagomir + sh-NC group and miR-410 antagomir + sh-HDAC1 group, whereas the expression of HDAC1 was found to be significantly reduced in the sh-HDAC1 group (p < 0.05; Figure 4H). Furthermore, the cell function tests, including CCK-8 assay, colony-formation assay, and flow cytometry, demonstrated that the cell viability was improved, and apoptosis was reduced in the miR-410 antagomir + sh-NC group in comparison to the NC antagomir + sh-NC group, whereas the miR-410 antagomir + sh-HDAC1 group had less cell viability and enhanced apoptosis than the group of miR-410 antagomir + sh-NC (Figures 4I−4K). The aforementioned data suggested that HDAC1 is a downstream target of miR-410, and the inhibition of miR-410 promoted the development of atherosclerosis through HDAC1.
HDAC1 binds KLF5 to induce the expression of NF-κB inhibitor α (IKBα) in HUVECs
Although it has been previously reported that HDAC1 can inhibit KLF5,12 the underlying mechanism remains elusive. The bioinformatic website STRING predicted HDAC1 could regulate KLF5, and the coimmunoprecipitation (coIP) assay verified that HDAC1 could bind to KLF5 and that HDAC1 inhibited its transcriptional activity by targeting and directly binding to KLF5 (Figures 5A and 5B). It has been established that KLF5 reduces the expression of IKBα.13 Additionally, the expression of IKBα has been found to be decreased in atherosclerosis, whereas the upregulation of IKBα was revealed to attenuate the pathogenesis.14 The present study observed that the expression of KLF5 was increased when KLF5 was overexpressed in the HUVECs treated with ox-LDL, and it was also observed that IKBα expression was markedly decreased in the oe-KLF5 (overexpressed KLF5) group in comparison to the oe-NC (NC of oe-KLF5) group (Figure 5C). Furthermore, the dual luciferase reporter gene assay showed the luciferase activity in the IKBα promoter + oe-KLF5 group was significantly reduced compared to the IKBα promoter + oe-NC group (p < 0.05). The luciferase activity in the IKBα promoter + oe-KLF5 + oe-HDAC1 (overexpressed HDAC1) group was found to be higher than that in the IKBα promoter + oe-KLF5 group but showed no significant change in comparison to the IKBα promoter + oe-NC group (Figure 5D). These results implied that HDAC1 binds directly to KLF5 in order to inhibit its transcription, thus increasing the expression of IKBα.
Figure 5.

HDAC1 binds KLF5 to suppress its transcription and induce the expression of IKBα in HUVECs
(A) Bioinformatics website STRING (https://string-db.org/cgi/network.pl?taskId=LIdtFKtm9mBY) predicted that HDAC1 targeting regulates KLF5. (B) The binding of HDAC1 and KLF5 in HUVECs treated with ox-LDL. (C) KLF5 and IKBα levels in HUVECs. (D) HDAC1 can inhibit IKBα through KLF5; ∗p < 0.05. Measured data are described as mean ± standard deviation. Unpaired t test was used to analyze data between two groups and ANOVA test to analyze data among multiple groups. All of the experiments were performed with technological triplicate.
HDAC1 prevents atherosclerosis in mice via promoting IKBα expression
On the basis of HDAC1 regulating IKBα through the inhibition of KLF5, this study explored the effect of HDAC1 promoting the expression of IKBα on the development of atherosclerosis, and thus, HDAC1 was overexpressed in the HFD mice. The western blot revealed that the HDAC1 and IKBα levels in the aorta tissue were remarkably elevated in the HFD + oe-HDAC1 group in comparison to the HFD + oe-NC group (Figure 6A). The area of aorta impairment was revealed to be reduced in the HFD + oe-HDAC1 group (Figure 6B). Meanwhile, HDAC1 overexpression decreased serum TNF-α, IL-1, and IL-6 levels in mice (Figure 6D). Additionally, the western blot analysis showed HDAC1 overexpression could also suppress the expression of VCAM-1, ICAM-1, and MCP-1 in mice aorta tissues (p < 0.05; Figure 6C). Cumulatively, the above-mentioned results suggested that HDAC1 prevented the pathogenesis of atherosclerosis in mice through the upregulation of IKBα expression.
Figure 6.

HDAC1 prevents atherosclerosis in mice via promoting IKBα expression
(A) HDAC1 and IKBα levels in mice aorta tissues. (B) The area of aortic impairment (×400). (C) VCAM-1, ICAM-1, and MCP-1 levels in aorta tissues. (D) Serum TNF-α, IL-1, and IL-6 expression in mice; ∗p < 0.05. Measured data are described as mean ± standard deviation. Unpaired t test was used to analyze data between two groups; n = 10.
HDAC1 prevents atherosclerosis in HUVECs through upregulation of IKBα
To investigate whether HDAC1 suppressed the development of atherosclerosis in vitro via IKBα, ox-LDL-treated HUVECs were co-transfected with plasmids of HDAC1 overexpression and IKBα silencing. The results of the western blot analysis demonstrated that HDAC1 and IKBα levels were elevated in the oe-HDAC1 + sh-NC group in comparison to the control group, whereas the level of IKBα was increased without the HDAC1 level significantly changed in the oe-HDAC1 + sh-IKBα group compared to the oe-HDAC1 + sh-NC group (Figure 7A). Additionally, the cell function experiments showed that the oe-HDAC1 + sh-NC group had improved cell viability and reduced apoptosis more than the oe-NC + sh-NC group (p < 0.05), whereas it was opposite in the oe-HDAC1 + sh-IKBα groups in comparison to the oe-HDAC1 + sh-NC group (Figures 7B−7D). The expression of VCAM-1, ICAM-1, and MCP-1 was lower in the oe-HDAC1 + sh-NC group than the oe-NC + sh-NC group but higher in the oe-HDAC1 + sh-IKBα group than the oe-HDAC1 + sh-NC group (Figure 7E). The results manifest that HDAC1 prevents the pathogenesis of atherosclerosis in HUVECs via upregulation of IKBα expression.
Figure 7.

HDAC1 protects against atherosclerosis in HUVECs through upregulation of IKBα
(A) HDAC1 and IKBα levels in HUVECs. (B) Cell viability. (C) Colony-formation assay of cell viability (×400). (D) Cell apoptosis. (E) VCAM-1, ICAM-1, and MCP-1 expression in HUVECs; ∗p < 0.05. Measured data are described as mean ± standard deviation. ANOVA test was used to analyze data among multiple groups. All of the experiments were performed with technological triplicate.
miR-410 inhibits NF-κB signaling by upregulation of IKBα through HDAC1
The NF-κB signaling accelerates the development of atherosclerosis by activating the inflammatory response.14 The current study further blocked the expression of miR-410 in mice fed with HFD. The result of the western blot analysis showed that the level of IKBα was increased, and the p-p65 level was decreased in the HFD + miR-410 antagomir group in comparison to the HFD + NC antagomir group, whereas there was no significant change in the expression of p65 (Figure 8A). Subsequently, miR-410 and IKBα were silenced in the ox-LDL-treated HUVECs. Additionally, the western blot also revealed that the level of IKBα was increased, and the level of p-p65 was decreased in the miR-410 antagomir + sh-NC group in comparison to the NC antagomir + sh-NC group, without any significant change in the expression of p65 (Figure 8B). Meanwhile, the level of IKBα was decreased, and the p-p65 level was increased in the miR-410 antagomir + sh-IKBα group compared with the miR-410 antagomir + sh-NC group, without any significant change in the expression of p65. Furthermore, the present study also examined the effect of miR-410 antagomir treatment on p-p65 nuclear translocation (Figure S1). It was observed that miR-410 antagomir treatment increased the nuclear translocation of p-p65.
Figure 8.

miR-410 inhibits NF-κB by upregulation of IKBα through HDAC1
(A) The IKBα, p65, and p-p65 levels in aorta tissues (n = 10). (B) IKBα, p65, and p-p65 expression in HUVECs. (C) HDAC1, p65, and p-p65 levels in HUVECs. (D) Cell viability. (E) Colony-formation assay results of cell viability (×400). (F) Apoptosis in HUVECs. (G) VCAM-1, ICAM-1, and MCP-1 expression in cells; ∗p < 0.05. Measured data are described as mean ± standard deviation. Unpaired t test was used to analyze data between two groups and ANOVA test to analyze data among multiple groups. All of the experiments were performed with technological triplicate.
Aimed to further explore the effect of inhibiting the NF-κB signaling on atherosclerosis, HDAC1 was blocked in the HUVECs with additional IKB JSH-23 (HY-13982; MedChemExpress; 20 μm).15 The expression of HDAC1 was found to be decreased, whereas the expression of p-p65 was found to be increased with no significant change in the level of p65 in the sh-HDAC1 + DMSO group compared to the sh-NC + DMSO group, as detected by the western blot (Figure 8C). Additionally, in comparison to the sh-HDAC1 + DMSO group, there was no significant change noted in the HDAC1 and p65 levels in the sh-HDAC1 + JSH-23 group (p > 0.05), whereas the p-p65 expression was found to be decreased (p < 0.05).
The cell viability was inhibited, and apoptosis was enhanced in the sh-HDAC1 + DMSO group compared to the sh-NC + DMSO group, whereas it was opposite in the sh-HDAC1 + JSH-23 group in comparison to the sh-NC + DMSO group (Figures 8D−8F). Furthermore, the western blot analysis demonstrated that the expression levels of VCAM-1, ICAM-1, and MCP-1 in the sh-HDAC1 + DMSO group were elevated compared to the sh-NC + DMSO group, whereas they were found to be reduced in the sh-HDAC1 + JSH-23 group compared to the sh-HDAC1 + DMSO group. Cumulatively, the aforementioned results suggested that miR-410 inhibits the development of atherosclerosis by promoting IKBα expression through HDAC1 and inhibiting the NF-κB signaling.
Discussion
Atherosclerosis and its complications of cardiovascular disease are the leading cause of mortality and morbidity in the world.16 It is of great necessity to identify the target to counteract atherosclerosis. The present study showed that the cholesterol exposure remarkably increased miR-410 levels in cultured HUVECs and in mouse thoracic aorta. The cell line treated with ox-LDL and mouse models treated with HFDs were used to mimic cholesterol-induced vascular endothelial dysfunction in atherosclerosis patients. Furthermore, it was discovered that HDAC1 was a downstream gene regulated by miR-410 and could bind directly to KLF5 to suppress its transcription and promote IKBα expression, thus facilitating cell proliferation of HUVECs and inhibiting NF-κB to alleviate endothelial cell activation and dysfunction. Therefore, blocking miR-410 protected against atherosclerosis through the miR-410/HDAC1/KLF5/IKBα/NF-κB axis, which presented itself as a potential target in atherosclerosis prophylaxis and treatment.
Inflammation plays a crucial role at all stages of the atherosclerosis formation and controls the development and the destabilization of the plaques. It has been widely accepted that the formation of atherosclerosis is triggered by endothelial dysfunction, causing disturbed aggregation of platelets, vascular smooth muscle growth, inflammation, blood flow, as well as the vascular tone.17 Additionally, it has been established that endothelial cells and leukocytes use the NF-κB signaling to enhance the expression of inflammatory genes, including various adhesion molecules and cytokines, which induce the inflammatory activities of the arterial wall.18 miRNAs serve as silencers of their target genes by modulating their expression via binding to 3′ UTR.19 It has been observed that several miRNAs regulate the proliferation and differentiation of endothelial cells and may affect cell apoptosis, such as miR-34, miR-217, and miR-146.20,21 Furthermore, some miRNAs can also alter the inflammatory state of the vasculature on pro- or anti-inflammatory cytokines,22 whereas other miRNAs present different expression patterns depending on the disease pathogenesis. According to previously conducted studies, miR-410 can be either an oncogene or a tumor suppressor gene in different neoplasms, and it is a component of the regulatory network of cell apoptosis.23,24 In the current study, our findings showed that hyperlipidemia, more specifically ox-LDL, increased the level of miR-410 with elevated inflammatory cytokines and adhesion molecules in both cultured endothelial cell HUVECs and mouse aorta, whereas the inhibition of miR-410 could reverse the changes. These findings are consistent with the result of a previous study that miR-410 silencing can inhibit the ox-LDL-induced cell proliferation of HUVECs and rescue cell apoptosis.25
The protection against atherosclerosis relies on cell proliferation, and the current study suggests the involvement of miR-410 in this process. It was found that the miR-410 expression was increased in atherosclerosis and was associated with the elevation of NF-κB, and the bioinformatic prediction of HDAC1 being the target gene fulfills the circle. HDAC1 is considered as a positive regulator of cell proliferation, and the silencing of HDAC1 in mice would cause growth disorders, and the p21 cyclin-dependent inhibitor was increased accordingly.10,26 As a further prediction of KLF being the target gene of HDAC1 and IKBα being the downstream component, we verified existence of miR-410/HDAC1/KLF5/IKBα/NF-κB axis. Additionally, KLF5, a zinc-finger-structured transcription factor, plays an essential role in cardiovascular remodeling through acceleration of the phenotypic transformation of vascular smooth muscle cells from a contractile phenotype to a disadvantageous proliferative phenotype and then accelerates the formation of atherosclerotic plaques.27 It has been shown that promoting KLF5 downregulation protects from vascular injury.28 Furthermore, the activation of NF-κB was demonstrated to be triggered by signal-induced degradation of IKBα protein. As IKBα protein is degraded, the complex of NF-κB is subsequently released into the cell nucleus, where it can “turn on” the expression of specific genes with a nearby DNA binding site of NF-κB causing the cascade of endothelial dysfunction.29,30 This highlights the therapeutic potential of targeting miR-410 or its downstream molecules in order to provide a holistic approach for the prevention of atherosclerosis.31,32 Further investigation on the expansive role of miR-410, as well as miR-410/HDAC1/KLF5/IKBα/NF-κB axis, will provide insight on the most effective way to fully capitalize on their properties in a therapeutic manner.
Conclusively, miR-410 silencing promotes HDAC1 expression and increases IKBα levels through KLF5 to suppress NF-κB, thus preventing the endothelial dysfunction of atherosclerosis. This provides a novel potential target to counteract atherosclerosis.
Materials and methods
Screening of atherosclerosis-related miRNAs
The miRNAs related to atherosclerosis were searched through HMDD version (v.)3.2 database (http://www.cuilab.cn/hmdd), and the miRNAs labeled as therapeutic target in the search results were selected as candidate miRNAs. Through GEO database (https://www.ncbi.nlm.nih.gov/geo/), the mouse atherosclerosis expression dataset GEO: GSE89858 was obtained. The selected samples were 6-week samples, including 3 normal samples and 3 disease model samples. By taking normal samples as control, wilcox.test was used to measure differential expression of miR-410 in normal samples and disease samples.
shRNA screening
The shRNA sequence was designed on the basis of HDAC1 gene sequence obtained from the GenBank database. Two optimal shRNAs sequences targeting different regions within the HDAC1 gene that have the fewest BLAST matches were selected (Table S2) and constructed with the pGMLV-SC6 vector (component sequence: hU6-MCS-CMV-RFP-PGK-puromycin; GenePharma, Shanghai, China). The plasmids were successfully created after enzyme digestion and sequencing, named sh-HDAC1-1 and sh-HDAC1-2, and built into the HDAC1-shRNA vectors and the NC vectors for subsequent transfection into human ovarian surface epithelial cells (HOSEpiC cells). Western blot was employed to detect the levels of HDAC1 in the cells to determine the most effective shRNA sequence.
Mouse model of atherosclerosis
Sixty 4- to 6-week-old C57BL/6J ApoE−/− male specific pathogen-free (SPF) mice (16−21 g; Peking University Health Science Center, Beijing, China) were raised in a SPF animal lab under the controlled conditions of 40%–60% humidity and 18°C−23°C temperature. The mice were randomly divided into six groups (n = 10 in each group): the ND group (as controls, as they were fed with regular mouse food); HFD group (fed with high fat food with 0.25% cholesterol and 15% fat); HFD + NC antagomir group; HFD + miR-410 antagomir group; HFD + oe-NC group (lentivirus oe-NC was injected 2 × 107 TU/mouse into tail veins); and HFD + oe-HDAC1 group (lentivirus oe-HDAC1 was injected 2 × 107 TU/mouse into tail veins). (For the last 4 groups, mice were also fed with high fat food for 16 weeks, whereas the lentivirus injection started at week 4.). The mice were anesthetized subsequent to a 16-week diet for the collection of 0.7 mL blood through renal artery centesis, and the thoracic aortas were harvested and stored in liquid nitrogen following euthanasia.
H&E staining
The paraffin-embedded mice aorta tissue sections were dewaxed using xylene and further hydrated with gradient ethanol. Subsequent to staining with hematoxylin for the duration of 10 min, the sections were rinsed with tap water and dipped in 0.5% hydrochloric acid ethanol to remove any excessive staining. After being rinsed with running water, the sections were stained with hematoxylin for the duration of 5 min and then with eosin for the duration of 10 min. Following the ethanol dehydration and xylene dewaxing, the sections were sealed with neutral gum and observed under an XSP-8CA upright microscope (Shanghai Optical Instrument Factory, Shanghai, China) for micrographs.
Oil red O staining
The mice aorta tissue sections were incubated in distilled water for 1 min and subsequently rinsed with 100% propylene glycol (Polyscientific, Bayshore, NY, USA) for the duration of 2 min. Part of the tissue sections were stained with oil red O working solution for 36 h, and the remainder was rinsed with 85% propylene glycol and then stained with hematoxylin. The slides were then mounted on glycerin jelly.
Cell culture and transfection
HUVECs (ATCC CRL-1730) were purchased from American Type Culture Collection (Manassas, VA, USA) and cultured in DMEM (Thermo Fisher Scientific, Waltham, MA, USA) with 10% fetal bovine serum (FBS; Thermo Fisher Scientific). The cells were cultured at the controlled temperature of 37°C in a 5% CO2 atmosphere. The cells in the logarithmic growth phase were extracted and seeded into a 6-well plate (3 × 105 cells/well). When the cell confluence reached 50%, the cells were transfected using Lipofectamine 2000 (11668019; Invitrogen, Carlsbad, CA, USA). Subsequent to 24 h of transfection, the cells were further treated with 50 μg/mL ox-LDL (Beijing Xiesheng Bio-Technology, Beijing, China) for an additional 24 h.
To verify the effect of NF-κB signaling on atherosclerosis, HDAC1 was inhibited in HUVECs, and IKB JSH-23 (HY-13982; MedChemExpress, NJ, USA; 20 μm) was added to the cells.15 The experiment was divided into three groups: sh-NC + DMSO, sh-HDAC1 + DMSO, and sh-HDAC1 + JSH-23. Subsequent to the transfection, the expressions of HDAC1, p65, and p-p65 were detected using the western blot analysis, the cell activity was detected by the CCK-8, HUVEC survival was detected by clone formation, and the apoptosis was detected using flow cytometry.
ELISA
Taking IL-6, for example, the kit (mouse: RAB0287, Sigma-Aldrich, St. Louis, MO, USA; human: EHC007.48, NeoBioscience, Shenzhen, China) was coated with specific IL-6 antibody at the bottom. Subsequently, plasma was added, and then the IL-6-specific biotinylated antibodies were further added to bind the IL-6 in the samples. The unbound biotinylated antibodies were washed away following incubation at room temperature, and then the streptavidin-peroxidase (SP) conjugate was added for the SP-conjugation reaction. Furthermore, the tetramethyl benzidine (TMB) contained in the color-developing solution could be catalyzed by SP to produce a blue conjugate, which would turn yellow when acidic-terminating solution was added. The ratio of the yellow color density and the content of IL-6 combined at the bottom of the kit and the absorbance (optical density [OD]) value of the yellow solution could be measured; thus, the content of IL-6 in the samples was calculated according to the standard curve. The detection of TNF-α (mouse: Sigma-Aldrich; human: E-EL-H0109c, Elabscience, Wuhan, China) and IL-1 (mouse: ab100704, Abcam, UK; human: 583311-96, Cayman, Beijing, China) was conducted following the above-mentioned procedure.
Quantitative real-time PCR
The total RNA was extracted by the TRIzol reagent (15596026; Invitrogen). The RNA samples were quantified using the TaqMan MicroRNA Assays (Applied Biosystems, Foster City, CA, USA). The synthesis of cDNA from mRNA was generated using a commercially available kit (RR047A; Takara, Japan). Additionally, the synthesis of cDNA of miR-410 was carried out using miRNA First Strand cDNA Synthesis (Tailing Reaction) kit (B532451-0020) in accordance with the instructions provided by the manufacturer (Sangon Biotech, Shanghai, China). Subsequently, the cDNA was subjected to quantitative real-time PCR using the SYBR Premix Ex Taq II (Perfect Real Time) Kit (DRR081; Takara) with the Applied Biosystems (ABI) 7500 instrument (ABI, Foster City, CA, USA), and each reaction was run in technological triplicate. The universal RT primer of miRNA and forward primer of U6 were provided by the miRNA First Strand cDNA Synthesis Kit. Moreover, individual miRNA-specific forward primer and mRNA primer information is listed in Table S3. The miR-410 level was normalized to U6 and the target mRNA level to GAPDH. The results of the experiment were calculated using the 2−△△CT method.
Western blot
Protein extraction was performed using the protease inhibitor-contained radioimmunoprecipitation assay (RIPA) buffer (R0010; Solarbio, Beijing, China), and the concentration was measured using the BCA protein assay kit (Thermo Fisher Scientific). The protein sample was separated using freshly prepared SDS-PAGE, electrotransferred onto polyvinylidene fluoride (PVDF) membranes, and then probed with primary antibodies. Next, the membrane was re-probed with goat anti-rabbit IgG (1:10,000, ab6721; Abcam), and the immunoblots were visualized with enhanced chemiluminescence detection reagents. Additionally, the gray values of the target protein bands were quantified by employing the Image Pro Plus 6.0 software (Media Cybernetics, USA), with GAPDH used for normalization. The primary antibodies used were the following: anti-HDAC1 antibody (1:2,000, ab7028; Abcam), anti-KLF5 antibody (1:2,000, ab137676; Abcam), anti-IKBα antibody (1:1,000, ab32518; Abcam), anti-p65 antibody (1:1,000, ab16502; Abcam), anti-p-p65 antibody (1:1,000, ab97726; Abcam), anti-VCAM-1 antibody (1:2,000, ab134047; Abcam), anti-ICAM-1 antibody (1:1,000, ab7815; Abcam), and anti-MCP-1 antibody (1:1,000, DF7577; Affinity Biosciences, USA). All western blot tests were conducted with technological triplicate to obtain sufficient data for further analysis.
CCK-8
The cells were seeded into 96-well plates at a density of 2 × 103 cells/well, leaving the blank control group with only cell medium but no cells for zero adjustment. Subsequent to 24-h transfection, 10 μL of CCK-8 solution was added to each well to co-incubate for an additional duration of 4 h at 37°C at 0, 24, 48, 72, and 96 h. The absorbance at the wavelength of 450 nm was measured using a microplate reader (Bio-Rad, Hercules, CA, USA). The ratio of the absorbance of the experimental group to the control group was calculated and made into a curve of cell viability. The average result was obtained with technological triplicates to ensure variable data for further analysis.
Colony-formation assay
The cells in the logarithmic growth phase were treated with 0.25% trypsin and gently dissociated into a single cell suspension. The cells in each group were then inoculated into dishes containing 10 mL preheated culture medium at the temperature of 37°C at a gradient density of 50,100 and 200 cells/10 cm well, respectively. The cells were then evenly dispersed by gentle rotation, which was followed by further incubation at 37°C and 5% CO2 for 1 week, with the culture medium changed once every other day. The culture was halted once the clone in the dish was visible to the naked eyes. Subsequent to the removal of the supernatant, the cells were washed twice with PBS and then fixed with 2 mL 4% paraformaldehyde for the duration of 20 min. The fixed cells were stained with 2 mL Giemsa’s solution (G4640; Solarbio Science & Technology, Beijing, China) for 40 min. Subsequently, the number of cell clones was counted under an inverted IX-50 microscope (Olympus, Japan), and the number of colonies with >10 cells/colony was counted.
Evaluation of cell apoptosis by flow cytometry
The day following transfection, the cells in each group were digested with 0.25% trypsin, and the digestion was terminated using the RPMI-1640 medium containing 10% FBS (Thermo Fisher Scientific). The cells were then centrifuged at 1,000 rpm for the duration of 5 min, and the resultant supernatant was discarded. Subsequently, the cells were fixed with 70% ethanol, precooled at 4°C, adjusted to the concentration of 1 × 106 cells/mL, and stained with 10 mL Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI; 556547; Shanghai Shuojia Biotechnology, Shanghai, China) for the duration of 15−30 min at the controlled temperature of 4°C. The cell apoptosis ratio was evaluated employing the EPICS XL flow cytometer (Beckman Coulter, CA, USA) at excitation wavelength of 480 nm, FITC at 530 nm, and PI at greater than 575 nm.
RIP chromatin immunoprecipitation (ChIP) assay
The RIP kit (Millipore, MA, USA) was employed to detect the binding of HDAC1 to AGO2 protein. The supernatant was discarded after the cells were rinsed with precooled PBS. Subsequently, the resultant cells were lysed with the same volume of RIP lysate in an ice bath for the duration of 5 min and centrifuged at 14,000 rpm for 10 min at the controlled temperature of 4°C to collect the supernatant. Partial cell extract was taken as input, and the remainder was incubated with antibodies for co-precipitation. Consequently, 50 μL of magnetic beads was taken from each co-precipitation reaction system, resuspended in 100 μL RIP wash buffer, and then added with 5 μg antibody, according to the experimental group. Following this, the magnetic bead-antibody complexes were resuspended in 900 μL RIP wash buffer, added with 100 μL cell extract, and incubated overnight at 4°C. The sample was then placed on a magnetic base to collect the magnetic bead-protein complexes. The samples and the input were digested using proteinase K to extract RNA for subsequent PCR detection. The antibodies used in RIP were AGO2 (ab32381, 1:50; Abcam) mixed at room temperature for the duration of 30 min, and IgG (1:100, ab109489; Abcam) was used as the NC. HDAC1 was eventually detected by quantitative real-time PCR analysis.
CoIP assay
The HUVECs were placed in RIPA lysate, added with 1% cocktail (1:100; Sigma-Aldrich), and then lysed on ice at 4°C following ultrasound treatment. The cell debris was removed by centrifugation, and the resultant cell lysate was incubated with 1 μg of anti-HDAC1 antibody (1:2,000, ab7028; Abcam), IgG (1:2,000, ab6721; Abcam), and 15 μL protein A/G beads (Santa Cruz Biotechnology, TX, USA) for the duration of 2 h. Subsequent to extensive washing, the beads were boiled at the temperature of 100°C for 5 min. The proteins were then separated using SDS-PAGE, transferred to a nitrocellulose membrane (Millipore), and then subjected to immunoblotting.
Dual luciferase reporter gene assay
The biological prediction websites StarBase (starbase.sysu.edu.cn/), miRWalk (mirwalk.umm.uni-heidelberg.de/), microT (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r%20=%20microT_CDS), RNAInter (www.rna-society.org/rnainter/), and microRNA.org (www.microrna.org/microrna/home.do) were employed to predict the target genes of miR-410, and the prediction results of the above-mentioned five websites were tested using the Venn diagram online tool (bioinformatics.psb.ugent.be/webtools/Venn/). Additionally, the STRING website (https://string-db.org/cgi/network.pl?taskId=LIdtFKtm9mBY) was used to analyze the binding correlation between HDAC1 and KLF5. Furthermore, the luciferase report method verified the effect of miR-410 on HDAC1 and that HDAC1 could regulate the transcription of KLF5. The full length of HDAC1 was synthesized with additional sequences of SpeI and HindIII cleavage sites so that it could be introduced into pMIR-reporter (Promega, WI, USA). In the WT HDAC, we designed the complementary sequence mutation position of the seed and inserted the target fragment into the pMIR-reporter plasmid using the T4 DNA ligase. Subsequently, the sequenced luciferase reporter plasmids WT and mut were co-transfected with NC mimic and miR-410 mimic, respectively, into the HEK293T cell line (Shanghai Institutes for Biological Sciences, Shanghai, China). Following 48 h of transfection, the cells were collected and lysed, and the luciferase activity was detected using the luciferase detection kit (K801-200; BioVision, CA, USA) and the Glomax 20/20 luminometer fluorescence detector (Promega) and further calculated as Luc/Rel of the relative fluorescence value with Renilla being the internal reference.
Additionally, the IKBα promoter region was amplified, and the target fragment was inserted into the pGL3-basic reporter plasmid co-transfection system using the T4 DNA ligase. The correctly sequenced luciferase reporter plasmid promoter was co-transfected with oe-NC, oe-HDAC1, and oe-KLF5 into the HEK293T cell line. The luciferase activity was detected and calculated as previously described. All of the experiments were performed with technological triplicate to obtain sufficient and variable data for further analysis.
Statistical analysis
All statistical analyses were completed employing the SPSS 21.0 software (IBM, Armonk, NY, USA). The results were expressed as the mean ± standard deviation. Unless otherwise noted, the statistical comparisons were performed using the unpaired t test when only two groups were compared or by Tukey’s test-corrected one-way analysis of variance (ANOVA) when more than two groups were compared. The variables were analyzed at different time points using the Bonferroni-corrected repeated-measures ANOVA. Statistical significance was considered existing when two-tailed p <0.05.
Study approval
All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee of The Second Hospital of Jilin University. Extensive measures were taken to minimize the suffering of all of the included animals.
Acknowledgments
We would like to give our sincere appreciation to the reviewers for their helpful comments on this article.
Author contributions
S.N. and C.X. conducted the experiments. Y.W. and H.W. designed the experiments and wrote the paper. S.N. and H.W. performed the statistical analysis and preparation of figures. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.03.009.
Supplemental information
References
- 1.Hansson G.K., Libby P., Tabas I. Inflammation and plaque vulnerability. J. Intern. Med. 2015;278:483–493. doi: 10.1111/joim.12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jonasson L., Holm J., Skalli O., Bondjers G., Hansson G.K. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis. 1986;6:131–138. doi: 10.1161/01.atv.6.2.131. [DOI] [PubMed] [Google Scholar]
- 3.Bajan S., Hutvagner G. Regulation of miRNA processing and miRNA mediated gene repression in cancer. MicroRNA. 2014;3:10–17. doi: 10.2174/2211536602666140110234046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 5.Siasos G., Bletsa E., Stampouloglou P.K., Oikonomou E., Tsigkou V., Paschou S.A., Vlasis K., Marinos G., Vavuranakis M., Stefanadis C., Tousoulis D. MicroRNAs in cardiovascular disease. Hellenic J. Cardiol. 2020;61:165–173. doi: 10.1016/j.hjc.2020.03.003. [DOI] [PubMed] [Google Scholar]
- 6.Papageorgiou N., Tousoulis D., Androulakis E., Siasos G., Briasoulis A., Vogiatzi G., Kampoli A.M., Tsiamis E., Tentolouris C., Stefanadis C. The role of microRNAs in cardiovascular disease. Curr. Med. Chem. 2012;19:2605–2610. doi: 10.2174/092986712800493048. [DOI] [PubMed] [Google Scholar]
- 7.Feinberg M.W., Moore K.J. MicroRNA Regulation of Atherosclerosis. Circ. Res. 2016;118:703–720. doi: 10.1161/CIRCRESAHA.115.306300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wen R., Umeano A.C., Essegian D.J., Sabitaliyevich U.Y., Wang K., Farooqi A.A. Role of microRNA-410 in molecular oncology: A double edged sword. J. Cell. Biochem. 2018;119:8737–8742. doi: 10.1002/jcb.27251. [DOI] [PubMed] [Google Scholar]
- 9.Sun X., Belkin N., Feinberg M.W. Endothelial microRNAs and atherosclerosis. Curr. Atheroscler. Rep. 2013;15:372. doi: 10.1007/s11883-013-0372-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y., Zhang K., Mao W. Inhibition of miR-34a prevents endothelial cell apoptosis by directly targeting HDAC1 in the setting of atherosclerosis. Mol. Med. Rep. 2018;17:4645–4650. doi: 10.3892/mmr.2018.8411. [DOI] [PubMed] [Google Scholar]
- 11.Moore C.B., Guthrie E.H., Huang M.T., Taxman D.J. Short hairpin RNA (shRNA): design, delivery, and assessment of gene knockdown. Methods Mol. Biol. 2010;629:141–158. doi: 10.1007/978-1-60761-657-3_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsumura T., Suzuki T., Aizawa K., Munemasa Y., Muto S., Horikoshi M., Nagai R. The deacetylase HDAC1 negatively regulates the cardiovascular transcription factor Krüppel-like factor 5 through direct interaction. J. Biol. Chem. 2005;280:12123–12129. doi: 10.1074/jbc.M410578200. [DOI] [PubMed] [Google Scholar]
- 13.Liu F.F., Dong L., Yang X., Li D.J., Shen Y.Y., Liu Z.L. KLF5 silence attenuates proliferation and epithelial-mesenchymal transition induction in Hep-2 cells through NF-κB signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2019;23:3867–3875. doi: 10.26355/eurrev_201905_17814. [DOI] [PubMed] [Google Scholar]
- 14.Tang Z.H., Peng J., Ren Z., Yang J., Li T.T., Li T.H., Wang Z., Wei D.H., Liu L.S., Zheng X.L., Jiang Z.S. New role of PCSK9 in atherosclerotic inflammation promotion involving the TLR4/NF-κB pathway. Atherosclerosis. 2017;262:113–122. doi: 10.1016/j.atherosclerosis.2017.04.023. [DOI] [PubMed] [Google Scholar]
- 15.Kasparkova J., Thibault T., Kostrhunova H., Stepankova J., Vojtiskova M., Muchova T., Midoux P., Malinge J.M., Brabec V. Different affinity of nuclear factor-kappa B proteins to DNA modified by antitumor cisplatin and its clinically ineffective trans isomer. FEBS J. 2014;281:1393–1408. doi: 10.1111/febs.12711. [DOI] [PubMed] [Google Scholar]
- 16.White H.D., Chew D.P. Acute myocardial infarction. Lancet. 2008;372:570–584. doi: 10.1016/S0140-6736(08)61237-4. [DOI] [PubMed] [Google Scholar]
- 17.Lu Y., Thavarajah T., Gu W., Cai J., Xu Q. Impact of miRNA in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2018;38:e159–e170. doi: 10.1161/ATVBAHA.118.310227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ait-Oufella H., Taleb S., Mallat Z., Tedgui A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011;31:969–979. doi: 10.1161/ATVBAHA.110.207415. [DOI] [PubMed] [Google Scholar]
- 19.Lu T.X., Rothenberg M.E. MicroRNA. J. Allergy Clin. Immunol. 2018;141:1202–1207. doi: 10.1016/j.jaci.2017.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao W.J., Rosenblat J.D., Roth N.C., Kuliszewski M.A., Matkar P.N., Rudenko D., Liao C., Lee P.J., Leong-Poi H. Therapeutic Angiogenesis by Ultrasound-Mediated MicroRNA-126-3p Delivery. Arterioscler. Thromb. Vasc. Biol. 2015;35:2401–2411. doi: 10.1161/ATVBAHA.115.306506. [DOI] [PubMed] [Google Scholar]
- 21.Incalza M.A., D’Oria R., Natalicchio A., Perrini S., Laviola L., Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 2018;100:1–19. doi: 10.1016/j.vph.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 22.Chen J., Zhu R.F., Li F.F., Liang Y.L., Wang C., Qin Y.W., Huang S., Zhao X.X., Jing Q. MicroRNA-126a Directs Lymphangiogenesis Through Interacting With Chemokine and Flt4 Signaling in Zebrafish. Arterioscler. Thromb. Vasc. Biol. 2016;36:2381–2393. doi: 10.1161/ATVBAHA.116.308120. [DOI] [PubMed] [Google Scholar]
- 23.Liu C., Zhang A., Cheng L., Gao Y. miR-410 regulates apoptosis by targeting Bak1 in human colorectal cancer cells. Mol. Med. Rep. 2016;14:467–473. doi: 10.3892/mmr.2016.5271. [DOI] [PubMed] [Google Scholar]
- 24.Li M., Zheng R., Yuan F.L. MiR-410 affects the proliferation and apoptosis of lung cancer A549 cells through regulation of SOCS3/JAK-STAT signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2018;22:5987–5993. doi: 10.26355/eurrev_201809_15933. [DOI] [PubMed] [Google Scholar]
- 25.Hu M.Y., Du X.B., Hu H.B., Shi Y., Chen G., Wang Y.Y. MiR-410 inhibition induces HUVECs proliferation and represses ox-LDL-triggered apoptosis through activating STAT3. Biomed. Pharmacother. 2018;101:585–590. doi: 10.1016/j.biopha.2018.02.111. [DOI] [PubMed] [Google Scholar]
- 26.Jurkin J., Zupkovitz G., Lagger S., Grausenburger R., Hagelkruys A., Kenner L., Seiser C. Distinct and redundant functions of histone deacetylases HDAC1 and HDAC2 in proliferation and tumorigenesis. Cell Cycle. 2011;10:406–412. doi: 10.4161/cc.10.3.14712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang W., Zhang Y., Wang L., Li J., Li Y., Yang X., Wu Y. mircroRNA-152 prevents the malignant progression of atherosclerosis via down-regulation of KLF5. Biomed. Pharmacother. 2019;109:2409–2414. doi: 10.1016/j.biopha.2018.08.014. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y.N., Xie B.D., Sun L., Chen W., Jiang S.L., Liu W., Bian F., Tian H., Li R.K. Phenotypic switching of vascular smooth muscle cells in the ‘normal region’ of aorta from atherosclerosis patients is regulated by miR-145. J. Cell. Mol. Med. 2016;20:1049–1061. doi: 10.1111/jcmm.12825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang F.Z., Zhou J., Li W.W., Wang F., Wen P.Y., Zhou L., Wang J.G., Zheng X.X. [Nuclear factor κB and IKB expression and calcium deposition of atherosclerotic plaques in apolipoprotein E and low density lipoprotein receptor knockout mice] Zhonghua Xin Xue Guan Bing Za Zhi. 2012;40:684–689. [PubMed] [Google Scholar]
- 30.Liang W.J., Yang H.W., Liu H.N., Qian W., Chen X.L. HMGB1 upregulates NF-kB by inhibiting IKB-α and associates with diabetic retinopathy. Life Sci. 2020;241:117146. doi: 10.1016/j.lfs.2019.117146. [DOI] [PubMed] [Google Scholar]
- 31.Solly E.L., Dimasi C.G., Bursill C.A., Psaltis P.J., Tan J.T.M. MicroRNAs as Therapeutic Targets and Clinical Biomarkers in Atherosclerosis. J. Clin. Med. 2019;8:2199. doi: 10.3390/jcm8122199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bao Y., Li S., Ding Y., Du X., Zhang M., Tang W., Zhou S. MiRNA: a potential target for gene diagnosis and treatment of atherosclerotic stroke. Int. J. Neurosci. 2021;131:283–288. doi: 10.1080/00207454.2020.1738428. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.