

Summary
Complex I (CI) is the largest enzyme of the mitochondrial respiratory chain, and its defects are the main cause of mitochondrial disease. To understand the mechanisms regulating the extremely intricate biogenesis of this fundamental bioenergetic machine, we analyze the structural and functional consequences of the ablation of NDUFS3, a non-catalytic core subunit. We show that, in diverse mammalian cell types, a small amount of functional CI can still be detected in the complete absence of NDUFS3. In addition, we determine the dynamics of CI disassembly when the amount of NDUFS3 is gradually decreased. The process of degradation of the complex occurs in a hierarchical and modular fashion in which the ND4 module remains stable and bound to TMEM126A. We, thus, uncover the function of TMEM126A, the product of a disease gene causing recessive optic atrophy as a factor necessary for the correct assembly and function of CI.
Keywords: respiratory complex I, CI, NDUFS3, CI modules, assembly factor, TMEM126A, SILAC, optic atrophy type 7
Graphical abstract
Highlights
-
•
A fraction of functional complex I assembles without the core subunit NDUFS3
-
•
Complex I disassembly differentially affects its submodules
-
•
With NDUFS3 absent, the ND4-module of the P-distal domain remains mostly stable
-
•
The TMEM126A/OPA7 factor interacts with the ND4-module and is necessary for CI maturation
D’Angelo et al. show that eliminating NDUFS3 does not completely abolish respiratory complex I maturation. Differential degradation of complex I subunits belonging to different structural/functional modules is triggered by NDUFS3 repression. The ND4 module remains stable and is bound to TMEM126A, which is, here, identified as a complex I assembly factor.
Introduction
Human complex I (CI; NADH:ubiquinone oxidoreductase), the largest multi-heteromeric enzyme of the mitochondrial respiratory chain (MRC), is composed of 45 subunits encoded by both the nuclear DNA (nDNA) and mitochondrial DNA (mtDNA) (Hirst, 2013; Sazanov, 2015). This enzyme couples the transfer of two electrons from NADH to ubiquinone with the pumping of four protons from the matrix to the intermembrane space, thus contributing to the generation of the proton-motive force exploited for ATP synthesis (Hirst, 2013; Sazanov, 2015). The essential catalytic core of the enzyme is constituted of 14 “core subunits” evolutionarily conserved from bacteria to the mitochondria of higher eukaryotes (Baradaran et al., 2013; Zhu et al., 2016; Agip et al., 2018). The remaining 30 supernumerary subunits lack direct catalytic function but provide stability and promote CI biogenesis (Stroud et al., 2016). Supernumerary subunits are all nDNA encoded, whereas seven core subunits (MT-ND1–6 and MT-ND4L) are encoded by mtDNA (Vinothkumar et al., 2014; Zhu et al., 2016). This gigantic complex has a distinctive L-shaped structure with a hydrophilic arm protruding into the matrix and a hydrophobic arm embedded in the inner mitochondrial membrane. It can be divided into three main functional modules: the NADH dehydrogenase module (N-module), the ubiquinone-binding module (Q-module), and the proton translocating module (P-module), which is located entirely in the membrane arm (Brandt, 2006). The transmembrane P-module contains all the mtDNA-encoded subunits and can be further divided into two proximal modules (the ND1- and ND2-modules) plus two distal modules (the ND4- and ND5-modules) (Guerrero-Castillo et al., 2017; Sánchez-Caballero et al., 2016a; Stroud et al., 2016). Such modular organization reflects the enzyme evolution (Friedrich and Scheide, 2000; Moparthi and Hägerhäll, 2011), structure (Agip et al., 2018) and function (Hirst, 2013), as well as the pathways involved in its assembly (Guerrero-Castillo et al., 2017). CI biogenesis is an intricate process that takes place in a modular fashion, in which each of the functional submodules are assembled and stabilized by a number of specific “assembly factors,” which can be chaperones or proteins involved in prosthetic group incorporation or in post-translational modification of the subunits (Formosa et al., 2018). Each of the modules are joined together in different stages, forming assembly intermediates, all eventually coming together at the end of the process to form the mature holoenzyme, releasing the assembly factors (Guerrero-Castillo et al., 2017; Sánchez-Caballero et al., 2016a). To further complicate this scenario, the near totality of CI is found associated with complexes III (CIII) and IV (CIV), forming the respiratory supercomplexes (SCs) or “respirasomes” (Schägger and Pfeiffer, 2000; Milenkovic et al., 2017). The biogenesis of CI has been proposed as either being completed before interacting with CIII and CIV (Acín-Pérez et al., 2008; Guerrero-Castillo et al., 2017) or occurring in the context of the SCs (Moreno-Lastres et al., 2012; Protasoni et al., 2020).
During CI assembly, the core subunit NDUFS3 is part of a subcomplex of about 89 kDa, containing NDUFS2 and NDUFA5, and appears in the early stages as the primary core for the generation of the Q-module (Vogel et al., 2007; Dieteren et al., 2012; Guerrero-Castillo et al., 2017). Hence, although it lacks a direct catalytic role, NDUFS3 is highly conserved and essential for CI assembly, and missense mutations in the NDUFS3 gene cause severe encephalomyopathy, including Leigh syndrome (LS) (Bénit et al., 2004; Jaokar et al., 2013; Lou et al., 2018; Pagniez-Mammeri et al., 2009). In addition, a recently developed conditional knockout (KO) mouse model showed that the ablation of Ndufs3 in skeletal muscle produces severe progressive myopathy and early mortality (Pereira et al., 2020).
Despite great advances in understanding mammalian MRC biogenesis in general and in the assembly of CI in particular, there are still many open questions concerning the step organization and mechanisms directing the different components of these pathways. These questions are relevant not only to understand basic mitochondrial biology but also to dissect the pathological mechanisms underlying CI deficiency in human disease.
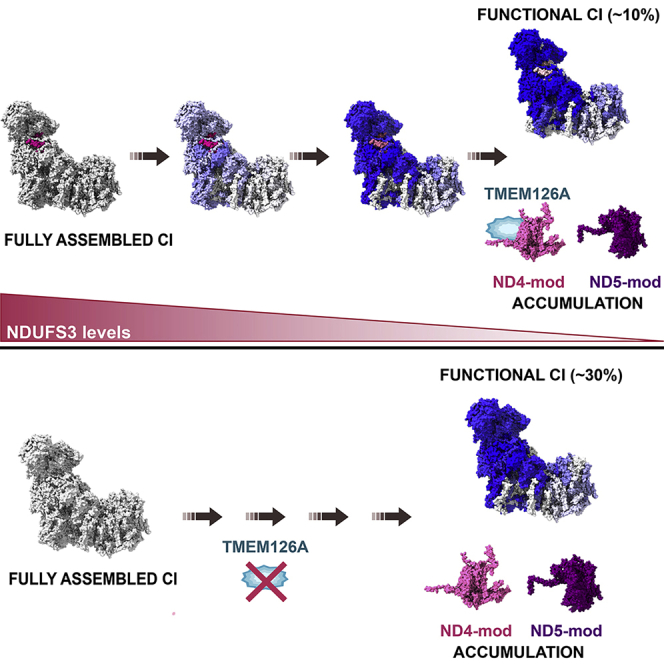
Here, we show that a progressive decrease or complete and permanent ablation of NDUFS3 severely affects CI stability, but a small fraction of the enzyme is still able to assemble and display its oxidoreductase activity. In addition, we have defined the dynamics of CI disassembly upon repression of NDUFS3 and gradual decrease of the protein levels, showing that the distal P-modules containing MT-ND4 and MT-ND5 remain mostly stable, whereas all the others are disassembled. Finally, we have identified TMEM126A/OPA7, whose function was thus far unknown, as a CI ND4-subcomplex interacting factor necessary for the correct maturation of the enzyme.
Results
A residual, fully assembled, and functional respiratory CI is present upon NDUFS3 ablation
We have previously reported that the genetic ablation of NDUFS3 in two human cell lines, from osteosarcoma 143B−/− and colorectal carcinoma HCT116−/−, induced a severe decrease of both CI NADH dehydrogenase activity and CI-driven ATP production (Kurelac et al., 2019). However, CI (rotenone-sensitive) activity was not completely abolished even if NDUFS3 was undetectable in 143B−/− and HCT116−/− (Figures 1A, S1A, and S1B; 13.62% and 20.31% of their isogenic controls, respectively). Such residual CI activity was evident in the two NDUFS3-null cell models by CI in-gel activity (CI-IGA) in both isolated, fully assembled CI and in SCs (Figures 1B, 1C, S1C, and S1D). This residual activity found in NDUFS3-null cells was undetectable in nDNA isogenic OS 143B cybrids carrying the homoplasmic frameshift mutation m.3571insC/MT-ND1, whereas some residual CI-IGA was detectable in OS-93 cells, in which the presence of 7% wild-type (WT) MT-ND1 was able to partially rescue the assembly of CI (Iommarini et al., 2018). The same activity was completely absent in mtDNA-depleted 143B Rho 0 cells (Figure 1B) or in blocked mitochondrial translation with 50 μg/mL chloramphenicol (CAP) (Figure S1E). This indicates the specific presence of a residual fully assembled and active CI, despite the complete lack of NDUFS3. NDUFS3 ablation was accompanied by marked reduction of several CI subunits belonging to all the structural modules of the enzyme, whereas subunits belonging to CIII and CIV were not affected (Figures 1D and S1F). Such reduction of CI subunits was associated with the accumulation of subassembly species reacting to NDUFB6-, NDUFB8-, and NDUFA9-specific antibodies. Nonetheless, two-dimensional (2D)-PAGE confirmed the presence of a residual, fully assembled CI in the SCs of 143B and HCT116 cells carrying genetic ablation of NDUFS3 (Figures 1E and S1G). On the other hand, SC CIII2+IV and fully assembled CIII and CIV were neither structurally affected nor showed defects in their redox activity (Figures 1E, S1G, and S2), and citrate synthase (CS) activity was not affected as well (Figure S2). We have previously shown that residual rotenone-sensitive CI-driven ATP synthesis was found in both 143B and HCT116 cells completely lacking NDUFS3, compared with controls, whereas CII- and CIII-mediated ATP synthesis was not significantly affected (Kurelac et al., 2019). We here show that such CI-driven ATP production is completely abolished in 143B−/− and HCT116−/− cells by treatment with 50 μg/mL CAP (Figure 1F). Furthermore, the presence of a residual, fully assembled, and functional CI was also evident in the mouse B16-F10 melanoma cell model lacking Ndufs3 (B16−/−), ruling out the possibility of a human-specific compensatory mechanism (Figures S3A–S3C).
Figure 1.

Detectable amounts of fully assembled and functional complex I are present in NDUFS3 KO cells
(A) Spectrophotometric measurements of complex I (CI) activity normalized to citrate synthase (CS). Data are means ± SD (n = 9 for 143B−/− and n = 10 for 143B+/+). ∗∗∗∗p < 0.0001 according to Mann-Whitney test.
(B) CI in-gel activity (CI-IGA) of 143B+/+, 143B−/−, OS93 (heteroplasmic 97% mutant m.3571insC/MT-ND1), OS (homoplasmic mutant m.3571insC/MT-ND1), and Rho 0 (mtDNA depleted) samples solubilized with n-dodecyl β-D-maltoside (DDM) and separated by high-resolution clear native PAGE (hrCNE).
(C) CI-IGA of 143B+/+ and 143B−/− samples solubilized with digitonin and separated by BN-PAGE. SCs include CI+III2 and respirasomes (I+III2+IV1−n).
(D) Immunodetection of CI, CIII, and CIV subunits on western blots of total lysates from 143B+/+ and 143B−/− cells lines resolved by SDS-PAGE. β-actin was used as loading control. Superfluous lanes were cropped in the UQCRC2 and relative loading control images.
(E) Immunodetection of CI, CIII, and CIV subunits on western blots of mitochondrial fractions from 143B+/+ and 143B−/− cell lines solubilized with digitonin and separated by 2D BN-PAGE. All subunits were immunodetected on the same blot.
(F) CI-driven ATP synthesis rates (CI-ATP) for 143B+/+ (n = 6), 143B−/− (n = 6), 143B−/− treated with 50 μg/mL chloramphenicol (CAP) (n = 3), HCT116+/+ (n = 12), HCT116−/− (n = 8), and HCT116−/− treated with 50 μg/mL CAP (n = 4). Rotenone-sensitive ATP synthesis rates (nmol/min × mg) were normalized to CS activity and protein content. Values are means ± SD. ∗∗p = 0.0022, ∗∗∗∗p < 0.0001 according to Mann-Whitney test. ND, not detected.
See also Figures S1–S3.
To understand the dynamics of CI assembly upon ablation of NDUFS3, we investigated 143B cells in which the expression of NDUFS3 could be repressed by doxycycline (Dox) in a tetracycline (Tet)-off system; hereafter referred to as 143B−/−NDUFS3 (Kurelac et al., 2019). Treatment with 100 ng/mL Dox induced a progressive reduction of NDUFS3, which started promptly after 12 h and lasted until the subunit was virtually undetectable after 4 or 8 days of incubation (Figures 2A and S3D). These doses and treatment times did not affect mitochondrial translation, as shown by the normal levels of the mtDNA-encoded subunit MT-CO2 (Figure S3E). CI-IGA was still detectable in both isolated CI and in SCs at the same time points in which the presence of NDUFS3 was undetectable (Figures 2B and S3F). The gradual decrease of NDUFS3 levels was not always mirrored by a reduction in the steady-state levels of other CI subunits. In fact, the levels of subunits belonging to the N- and Q-modules (NDUFA12 and NDUFS6) promptly decreased after the progressive repression of NDUFS3, whereas NDUFB8 (ND5-module) and NDUFA9 showed a moderate reduction of their steady-state levels (Figure 2C). Notably, NDUFB6 levels remained consistently stable after virtually complete NDUFS3 repression (Figure 2C). Conversely, the reduction of NDUFS3 was followed by progressive decrease of fully assembled CI and accumulation of subcomplexes containing NDUFB8 and NDUFB6, whereas CIII and CIV were not affected (Figures 2D and 2E). These data are in agreement with the presence of subcomplexes with positive immunodetection for NDUFB8 and NDUFB6 found in NDUFS3-null 143B and HCT116 cells and may explain the high steady-state levels of these subunits after NDUFS3 repression. However, a residual CI was still found in the SCs after 8 days of Dox treatment with 143B−/−NDUFS3, as highlighted by positive staining for all tested CI, CIII, and CIV subunits (Figures 2D, 2E, and S3G), despite the virtually complete absence of NDUFS3. Moreover, fully assembled CI containing NDUFB8 was also evident in its isolated form after 8 days of incubation of 143B−/−NDUFS3 with 100 ng/mL Dox (Figure S3H). On the other hand, the incubation for 8 days with 100 ng/mL Dox together with 50 μg/mL CAP inhibited mitochondrial translation reducing the levels of mtDNA-encoded subunits (Figure S3E), resulting in the complete absence of CI (Figures 2B–2D).
Figure 2.

Persistence of functional respiratory CI during repression of NDUFS3
(A) Immunodetection of NDUFS3 on western blots of whole-cell lysates from 143B−/− NDUFS3 cells treated with 100 ng/mL doxycycline (Dox) for 0 (untreated), 1, 2, 4, and 8 days (n = 3). NDUFS3 band intensities were quantified by densitometry and normalized to the signal of Hsp70, used as loading control. The mean values of the treated cells were referred to those of untreated (control) set to 100%. Data are means ± SD. ∗∗∗∗p < 0.0001 treated versus untreated; one-way ANOVA with Sidak’s multiple comparisons test.
(B) CI-IGA of enriched mitochondrial fractions from repressed 143B−/−NDUFS3, solubilized either with digitonin or DDM and separated by BN-PAGE. SCs include CI+III2 and respirasomes (I+III2+IV1-n).
(C) Immunodetection of CI subunits on western blots of 143B−/−NDUFS3 cells, treated either with 100 ng/mL Dox for 0 (untreated), 1, 2, 4, and 8 days or simultaneously with 100 ng/mL Dox and 50 μg/mL CAP for 8 days, resolved by SDS-PAGE. β-actin was used as loading control.
(D) Immunodetection of CI subunits NDUFA12 (N-module), NDUFS2 (Q-module), NDUFS3 (Q-module), NDUFB8 (ND5-module), CIII subunit UQCRFS1, and CIV subunit MT-CO2, in western blots of mitochondrial fractions from 143B−/−NDUFS3, treated either with 100 ng/mL Dox for 0 (untreated), 1, 2, 4, and 8 days or simultaneously with 100 ng/mL Dox and 50 μg/mL CAP for 8 days, solubilized with digitonin and separated by BN-PAGE. SCs include CI+III2 and respirasomes (I+III2+IV1-n). SDHB and SDHA (CII) were used as loading controls.
(E) Immunodetection of CI, CIII, and CIV subunits on western blots of mitochondrial fractions from 143B−/−NDUFS3 cells treated with 100 ng/mL Dox for 0 (untreated), 2, 4, and 8 days, solubilized with digitonin and resolved by 2D BN-PAGE. All subunits were immunodetected on the same blot. One asterisk (∗) indicates a non-specific signal derived from an anti-NDUFA8 antibody with which the membrane was previously incubated. Two asterisks (∗∗) indicate a signal derived from anti-MT-CO1.
See also Figure S3.
Overall, these data demonstrate that, despite the complete absence of NDUFS3, either by genetic destruction or by strong expression inhibition, a residual but clearly detectable amount of active CI associated into SCs is present in both human and mouse cells, without affecting the composition and function of the remaining oxidative phosphorylation (OXPHOS) system, whereas mtDNA-encoded subunits are essential. The possibility that the detected CI is just a residual enzyme that has not been completely disassembled can be ruled out because treatment at the same time points with inhibitors of mitochondrial translation produced the complete disappearance of the complex (Guerrero-Castillo et al., 2017).
Progressive loss of NDUFS3 reveals the modular dynamics of CI disassembly and NDUFB6 as an ND4-module subunit
The Tet-off 143B−/−NDUFS3 model provided a dynamic system in which the kinetics of CI disassembly could be followed upon the progressive depletion of NDUFS3. Changes in mitochondrial protein levels relative to the untreated 143B−/−NDUFS3 cells were analyzed upon treatment with 100 ng/mL Dox after 2, 4, and 8 days by quantitative proteomics by using stable isotope labeling by amino acids in cell culture (SILAC) in duplicate experiments, in which the labeling was reversed, and was validated by western-blot and immunodetection (Figures 3A, S4A, and S4B). When the fold-change values for each protein in the two experiments were plotted (log2 H/L ratios of one experiment in the x axis; -log2 H/L ratios of the second experiment in the y axis), the protein points were distributed over a 45° diagonal line, reflecting the correlation between the two experiments (Figure 3A). Most of the detected proteins were unaffected by the progressive reduction of NDUFS3 levels because they clustered around the axis origin. In particular, subunits of CIII and CIV were found unaltered upon treatment with 100 ng/mL Dox up to 8 days (Figures 3A and S4B), confirming their stability when NDUFS3 is repressed. In contrast, significant changes in the relative abundance of CI subunits and assembly factors were detected (Figures 3A, 3B, S4A, and S4B). Immunoblotting data confirmed the observation that, upon NDUFS3 repression, the most affected subunits were those belonging to the N- and Q-modules (NDUFA12 and NDUFS8, respectively), whereas those belonging to the ND5- and, particularly, the ND4-modules remained stable (NDUFB8 and NDUFB11, respectively) (Figure S4A). Most of the CI subunits were detected in these time-course experiments, ranging from 77% to 82% of the 44 subunits. In agreement with western blotting data, NDUFS3 steady-state levels underwent a progressive, severe decrease upon treatment with Dox, which reached a logarithmic fold change of -3.73 ± 0.27 (approximately a 13-fold decrease) after 8 days (Figures 3A and 3B). Q-module subunits were the first to be downregulated after NDUFS3 depletion, being already significantly reduced after 2 days of Dox treatment (Figures 3A–3C). The levels of N-module subunits were less affected after 2 days of NDUFS3 suppression but were significantly decreased after 4 and 8 days (Figures 3A–3C). The stability of P-domain subunits was heterogeneous because some were not affected by NDUFS3 repression (Figures 3A–3C). In contrast, ND1-module subunits were markedly destabilized by NDUFS3 suppression and clustered together with Q- and N-modules after 8 days of Dox treatment. Among ND1-module subunits, NDUFA3 was the most stable with a log2 ratio of -1.89 ± 0.35. At the earliest time-point ND2-, ND4-, and ND5-modules were virtually unaffected by NDUFS3 repression. However, subunits belonging to the ND2-module became more unstable after 8 days of Dox treatment. Interestingly, NDUFA10 and NDUFS5 protein levels clustered with ND1-module subunits rather than those belonging to the ND2-module, with a logarithmic fold change of -2.86 ± 0.17 and -2.92 ± 0.15, respectively. Conversely, the ND4- and ND5-modules were particularly stable upon NDUFS3 depletion, especially those of the ND4-module, whose levels were practically unchanged (Figure 3B) with respect to those of the control, having their log2 H/L ratios clustered in the proximity the axes origin (Figure 3A). Interestingly, NDUFB6 relative levels after NDUFS3 depletion were much more similar to those of the ND4-module than those of the ND5-module subunits. The permanence of the distal membrane arm subunits was most likely due to the formation of stable CI subassemblies, as shown by the accumulation of NDUFB6- and NDUFB8-containing subassembly intermediates (Figures 1E, 2D, 2E, and S1G). It is important to note that NDUFB6 showed a different pattern of subassembly accumulation, compared with that of NDUFB8, having an extra band of a higher molecular weight that does not contain NDUFB8 (Figure 2E). NDUFB6, together with NDUFA9, NDUFA11, and NDUFB4, was part of a group of subunits whose module assignment was uncertain (Stroud et al., 2016). However, NDUFB6 was shown to interact in an early subassembly with NDUFB5, NDUFB6, NDUFB10, and NDUFB11 (Guerrero-Castillo et al., 2017), all subunits belonging to the ND4-module (Stroud et al., 2016). In view of our proteomics results, NDUFB6 can definitely be assigned to the ND4-module, confirming the most recent subunit assignment model (Guerrero-Castillo et al., 2017).
Figure 3.

Progressive loss of NDUFS3 reveals the modular dynamics of CI disassembly
(A) Scatterplots generated from the liquid chromatography-mass spectrometry (LC-MS) analysis of mitochondrial-enriched fractions corresponding to the duplicate SILAC experiments, comparing untreated 143B−/−NDUFS3 with cells treated with 100 ng/mL Dox for 2, 4, or 8 days. The values of the logarithmic fold change (log2 H/L) for each protein in experiment 1 (exp.1; light untreated, heavy Dox treated) are represented in the x axis. The logarithmic fold-change values (−log2 H/L) derived from experiment 2 (exp.2; heavy untreated, light Dox treated) are represented in the y axis. Each point represents the values for a specific protein. CI subunits and assembly factors are highlighted; CI subunits of the same module are the same color. The group termed “uncertain” includes the subunits with still-unclear domain assignments (Stroud et al., 2016).
(B) Heatmap generated from the mean of the duplicate SILAC experiments for the detected CI subunits at each of the Dox treatment points (2, 4, and 8 days). Grey, not detected either in one or both of the duplicates.
(C) CI subunit relative protein abundance changes induced by repression of NDUFS3 after 2 and 8 days of 100 ng/mL Dox treatment depicted on the structure of the active mouse CI (PDB: 6G2J) (Agip et al., 2018) using ChimeraX version 1.1 (Goddard et al., 2018).
See also Figure S4.
Among 14 detected CI assembly factors, 10 showed unchanged abundance upon NDUFS3 repression (Figure S4A and S4B). Interestingly, the levels of the Q-module assembly factors NDUFAF3 and NDUFAF4 had already declined significantly after 2 days of NDUFS3 repression and remained very low during the entire time-course. The levels of two assembly factors of the ND4-module FOXRED1 and ATP5SL (Fassone et al., 2010; Formosa et al., 2015; Guerrero-Castillo et al., 2017; Stroud et al., 2016) were also reduced in NDUFS3-repressed cells, although to a much lesser extent. This was in contrast with the amounts of the structural subunits of the same module, which were consistently unchanged. Therefore, these assembly factors may belong to other module(s) or act downstream, having a role in the joining of the ND4-module with the others. These data depict a dynamic scenario in which CI disassembly also happens in a modular fashion (Figures 3C and S4C). During this process, the Q-module is promptly affected by the reduced availability of NDUFS3, followed by the nearby N- and P-proximal (ND1- and ND2-) modules, whereas levels of the subunits belonging to the distal ND5-module are much less decreased and those of the ND4-module are virtually unaffected since they were found accumulated in stable assembly intermediates.
TMEM126A is an assembly factor of respiratory CI
The accumulation of such assembly intermediates prompted us to search for proteins that would be upregulated upon NDUFS3 repression and that were likely to interact with the accumulated P-distal (ND4 and ND5) modules. Such proteins are bona fide candidates to be CI assembly factors. To that end, CI was immunopurified from 143B−/−NDUFS3 cells harvested after 2, 4, and 8 days of NDUFS3 repression and quantified by SILAC (Figure 4A). The subunit coverage of detected subunits was very high in these experiments, ranging from 40 to 43 out of the 44 CI subunits. Among all the proteins preferentially enriched with the CI remnants upon NDUFS3 repression, TMEM126A/OPA7 was the only one showing significantly increased association with CI after 4 and 8 days of Dox treatment (Figure 4A). The accumulation of TMEM126A in CI-enriched fractions was not caused by a general increase of the protein in total lysates and mitochondrial fractions in the Tet-off model upon progressive repression of NDUFS3 (Figures 3A and 4B). Indeed, TMEM126A specifically co-migrated and was accumulated upon NDUFS3 repression in the higher molecular weight CI subassembly species containing NDUFB6, likely corresponding to the stable ND4-module (Figures 4C and 4D). The up-to-now uncharacterized mitochondrial protein TMEM126A is a paralog of the CI assembly factor TMEM126B (Andrews et al., 2013; Elurbe and Huynen, 2016; Heide et al., 2012) and mutations in TMEM126A (Online Mendelian Inheritance in Man [OMIM]: 612988) have been found in multiple families affected by autosomal recessive optic atrophy (arOA) (OMIM: 612989). However, its function and putative role in mitochondrial respiratory chain biogenesis was not clear, despite its possible role as a CI assembly factor candidate, being hypothesized to exert a redundant role to that of TMEM126B (Sánchez-Caballero et al., 2016b). To decipher the role of TMEM126A on respiratory chain biogenesis, we investigated CI assembly and function in commercially available HAP1 cells in which TMEM126A was knocked out by CRISPR-Cas9 (HAP1KO, Horizon Discovery, HZGHC05796c002). In these cells, the lack of TMEM126A was associated with a reduction of the steady-state levels of several subunits of CI belonging to different modules. The amounts of the tested CIII and CIV subunits were not affected by the loss of TMEM126A (Figures 5A and 5B). From a functional point of view, rotenone-sensitive CI activity was strongly reduced in HAP1KO compared with HAP1WT (Figures 5C and S5A), whereas CII, CIII, CIV, or CS activity were comparable to those of controls (Figures S5B and S5C), indicating that loss of TMEM126A induced an isolated CI deficiency. Moreover, the lack of TMEM126A was associated with a severe defect in the amount of active CI-containing SCs and isolated CI (Figure 5D), suggesting that TMEM126A is necessary for CI assembly or stability. This was confirmed by the observation that the decreased amount of fully assembled CI in the SCs in HAP1KO cells was concomitant with the accumulation of CI subassemblies detected with antibodies against NDUFB8 (ND5-module), NDUFB6, and NDUFB11 (ND4-module) (Figures 5D–5F). These biogenetic CI defects, however, did not affect basal respiration (Figure 5G), but a significant reduction in the maximal respiratory capacity was found in HAP1KO cells (Figure 5G). Moreover, ATP synthesis rates driven by CI and CII substrates were not affected by the isolated CI deficiency induced by the lack of TMEM126A (Figures S5D and S5E). These data indicate that the amount of CI able to assemble in the absence of TMEM126A, found in HAP1KO cells, is sufficient to sustain mitochondrial respiration and the generation of membrane potential under basal conditions, but not when the respiratory chain activity is forced to the maximum by uncoupling. Accordingly, HAP1KO were not able to survive under metabolic stress conditions, i.e., 5 mM galactose as carbon source (Figures 5H, S5F, and S5G). Overall, these data demonstrate that TMEM126A is a factor necessary to promote the assembly or the maintenance of stable and functional CI, likely through interaction with the ND4-module, allowing that module to join the other ones to complete the maturation of the enzyme.
Figure 4.

TMEM126A is associated with a stable CI subassembly of the ND4-module
(A) Scatterplots generated from the liquid chromatography-mass spectrometry (LC-MS) analysis of CI immunopurified fractions performed on the same mitochondrial-enriched fractions used for the proteomic analyses in Figure 3. The values of the logarithmic fold change (log2 H/L) for each protein in experiment 1 (exp.1; light untreated, heavy Dox treated) are represented on the x axis. The logarithmic fold change values (−log2 H/L) derived from experiment 2 (exp.2; heavy untreated, light Dox treated) are represented on the y axis. Each point represents a specific protein. CI subunits and assembly factors are highlighted; CI subunits of the same module are the same color. “Uncertain” subunits are those with still unclear assignment. TMEM126A is shown in red. Proteins with statistically significant changes are also labeled.
(B) Immunodetection of TMEM126A and NDUFS3 on western blots of whole-cell lysates and mitochondrial enriched fractions from 143B−/− NDUFS3 cells treated with 100 ng/mL Dox for 0 (untreated), 1, 2, 4, and 8 days (n = 2). TMEM126A band signal intensities were quantified by densitometry and normalized to the signal of either vinculin for whole-cell lysates, and CS or voltage-dependent anion channel (VDAC) for mitochondrial fractions. The mean values of the treated cells are the percentage of those of the untreated control. Data are means ± SD.
(C) Immunodetection of TMEM126A and NDUFB6 (ND4-module) in western blots of enriched-mitochondrial fractions from 143B−/−NDUFS3 cells treated with 100 ng/mL Dox for 0 (untreated), 1, 2, 4, and 8 days, solubilized using digitonin and separated by BN-PAGE. Two asterisks (∗∗) indicate a subassembly containing TMEM126A and co-migrating with NDUFB6 subassembly; one asterisk (∗) indicates a lower-molecular-sized subassembly with positive staining for TMEM126A.The two panels correspond to the same samples separated in the same BN-PAGE gel and transferred onto the same membrane, which was subsequently cut in two for immunodetection. SDHA (CII) was used as loading control in BN-PAGE blots. VDAC was used as loading control in western blots of the same samples separated by SDS-PAGE.
(D) Immunodetection of TMEM126A, NDUFB6 (ND4-module), UQCRC2 (CIII), and MT-CO2 (CIV) on western blots of mitochondrial enriched fractions from 143B−/−NDUFS3 cells treated with 100 ng/mL Dox for 0 (untreated) and 8 days, solubilized with 4 mg digitonin/mg protein and resolved by 2D BN-PAGE. All the subunits were immunodetected on the same blot. One asterisk (∗) indicates a signal derived from the anti-TMEM126A antibody with which the membrane was previously incubated.
Figure 5.

TMEM126A is required for respiratory CI biogenesis and function
(A) Immunodetection of TMEM126A on western blots of total lysates and mitochondrial fractions from HAP1WT and HAP1KO cells. β-actin and CS were used as loading controls.
(B) Immunodetection of CI, CIII, and CIV subunits on western blots of whole-cell lysates from HAP1WT and HAP1KO cells lines (n = 3–8) resolved by SDS-PAGE. β-actin or vinculin were used as loading controls. Band signal intensities were quantified by densitometry and normalized to the signal of the relative loading control. Data are means ± SD. ∗p < 0.0001 according to unpaired multiple t test, Holm-Sidak method, with α = 0.05.
(C) Spectrophotometric kinetic measurements of CI activity normalized to CS. Data are means ± SD (n = 3). ∗p = 0.0189 according to an unpaired Student’s t test.
(D) CI-IGA assay (left panel) and a western blot (right panel) in which NDUFB6 was immunodetected from HAP1WT and HAP1KO mitochondrial-enriched fractions solubilized with digitonin or DDM and separated by BN-PAGE. SDHA (CII) was the loading control.
(E) Immunodetection of CI, CIII, and CIV subunits on western blots of mitochondrial-enriched fractions from HAP1WT and HAP1KO cells, solubilized with digitonin and resolved by 2D BN-PAGE. All the subunits were immunodetected on the same blot. One asterisk (∗) indicates a signal derived from an anti-NDUFA9 antibody with which the membrane was previously incubated.
(F) Immunodetection of NDUFB11 and NDUFB6 (ND4-module) on western blots of mitochondrial-enriched fractions from HAP1WT and HAP1KO cells, solubilized with digitonin and resolved by 2D BN-PAGE. One asterisk (∗) indicates signals derived from anti-NDUFB11, with which the membrane was previously incubated.
(G) Oxygen consumption rate (OCR) profiles of HAP1WT and HAP1KO cells. Additions were 1 μM oligomycin (O), 0.25 μM FCCP (F), and 1 μM rotenone plus antimycin A (R/A) diluted in Seahorse medium, pH 7.4. Data are expressed as pmoles/min of O2 normalized on SRB absorbance and shown as means ± SEM of n = 4 independent replicates, ∗p < 0.01 according to unpaired multiple t test Holm-Sidak method, with α = 0.05.
(H) HAP1WT and HAP1KO cell viability determined by sulforhodamine B (SRB) assay after 72 h of incubation with either 25 mM glucose or glucose-free medium containing 5 mM galactose. Data are means ± SD of n = 4 independent replicates, ∗p = 0.0197 and ∗∗∗p < 0.0001 according to unpaired Student’s t test.
See also Figure S5.
Discussion
In this study, we demonstrate that a small portion of fully assembled and functional respiratory CI is still present at the level of SCs when NDUFS3 is absent. NDUFS3 is a core subunit, highly conserved across evolution and believed to be essential for CI assembly, being an early entry of the Q-module (Formosa et al., 2018; Sánchez-Caballero et al., 2016a; Vogel et al., 2007). Even so, lack of NDUFS3 concurred with the presence of a small amount of CI that is apparently intact at the levels of structure (same electrophoretic mobility as in the WT) and function (detectable NADH dehydrogenase activity and CI-linked ATP synthesis). If these two parameters correlate, as they seem to do in our cell models, it would mean that the small amounts of CI that are assembled in the absence of NDUFS3 retain full catalytic capacity. This phenomenon is not exclusive to a single cell line because residual CI activity was also evident in independent NDUFS3 KO models derived from different human and murine cancer cell lines. It is important to note that the existence of residual CI was also evident in differentiated mouse skeletal muscle tissue of a conditional Ndufs3-KO model (Pereira et al., 2020), indicating that such phenotype is not a prerogative of cancer cells. In particular, the presence of the N-module subunits at the levels of SCs indicated completion of active CI assembly. Our hypothesis is that an alternative, although not prominent, route for CI assembly can be followed when NDUFS3 is missing. SCs may help stabilize and activate the NDUFS3-less CI, providing a platform for the stabilization of mature CI as suggested previously (Moreno-Lastres et al., 2012; Protasoni et al., 2020). Whether this alternative route can also be followed when other CI subunits are missing or is exclusive to NDUFS3 warrants future work. Indeed, NDUFS3 has the peculiarity of being a core subunit without real catalytic activity and when it is missing, several other subunits are reduced but not completely absent, supporting the possibility for some residual CI to complete the assembly and become functionally active. Unsurprisingly, we have shown that mtDNA-encoded subunits are pivotal for the assembly of the enzyme and no residual dehydrogenase activity was detected in the presence of a truncating mutation of MT-ND1, when mtDNA was depleted (Rho 0 cells) or when mitochondrial translation was inhibited with CAP. On the other hand, the ablation of accessory subunits was shown to affect CI assembly to a different extent depending on the role and topology of the subunit (Stroud et al., 2016).
Upon NDUFS3 repression, an intact CI remains stable for about 2 days and then starts to decrease, indicating that CI turnover takes place within that time frame (Dieteren et al., 2012). The kinetics of reduction of the amount of CI subunits reported here is in agreement with the current organization and modular assembly models (Guerrero-Castillo et al., 2017; Stroud et al., 2016). Q-module subunits rapidly reduced upon NDUFS3 repression, followed by N-module subunits, confirming that the assembly of the N-module is independent from the Q-module, but also that their interaction is necessary to build the whole CI. Regarding the membrane arm modules, the most affected subunits are those belonging to the proximal P-module, whereas the distal P-module subunits remained stable forming readily detectable subcomplexes containing NDUFB6 and NDUFB8, which progressively accumulate with the disassembly of the fully assembled enzyme. The fact that the turnover of the different CI modules varies greatly was also evidenced when the mitochondrial matrix protease CLPxP was knocked out (Szczepanowska et al., 2020). In this case, the subunits of the N-module accumulated to much greater amounts than those of the Q-module, which were moderately accumulated, or the P-module, which were unchanged with respect to the WT. This study also revealed that the degradation and replacement of the N-module happens much faster than the rest, which is in accordance with our observations in the repressible NDUFS3 model where, after the Q-module, the N-module subunits are the first ones to disappear. The elevated stability of ND4- and ND5-modules is not specific to the lack of NDUFS3 but is also evident when other subunits belonging to the Q- and ND1-module are knocked out (Stroud et al., 2016). This is probably due to the fact that ND4- and ND5-modules are not in direct contact with the Q-module and again supports the idea of a modular subassembly process.
The time-course quantitative proteomics analysis performed upon NDUFS3 repression has allowed us to reliably assign a module for a few subunits whose localization was still uncertain. Our data support the assignment of both NDUFA6 and NDUFA7 to the N-module, as described in Stroud et al. (2016), even if they were found to assemble independently (Guerrero-Castillo et al., 2017). On the other hand, abundance changes of NDUFB6 clustered with those of ND4-module subunits and remained consistently stable across the different time points, strongly suggesting its assignment to this module. This fact was evidenced in the analysis of CI assembly dynamics (Guerrero-Castillo et al., 2017) but remained unclear in the proteomic studies of residual CI after ablation of supernumerary subunits (Stroud et al., 2016). Likewise, the 2D blue native (BN)-PAGE pattern, of two distinct subassemblies of around 350 kDa and 200 kDa containing NDUFB6, was distinct to that of NDUFB8 of the ND5-module, where only one species of around 200 kDa was accumulated. Interestingly, the ND2-module subunits NDUFA10 and NDUFS5 behaved differently relative to the other module subunits, being markedly affected by NDUFS3 repression. This discrepancy was observed across the time points, and their fold changes were similar to those of N-module subunits. Being that NDUFA10 and NDUFS5 form extensive interactions with the Q- and ND1-modules, one possibility is that the assembly impairment of such modules could be transferred to these subunits in case they are added to a preformed ND2-module.
The lack of NDUFS3 and the consequent decrease in CI amount and activity did not affect the stability or activity of other MRC complexes, in particular CIII2 and CIV. This is a common phenomenon for mutations in structural or ancillary CI proteins, where the biochemical manifestation is isolated CI deficiency (Ghezzi and Zeviani, 2018). Contrariwise, mutations causing a strong impairment in CIII2 biogenesis are often associated with secondary loss of CI and CIV activities (Acin-Perez and Enriquez, 2014; Carossa et al., 2014; Protasoni et al., 2020). In some cases, severe defects in CIV biogenesis also have an effect on CI assembly or stability (Čunátová et al., 2021; Diaz et al., 2006). In human cells, the near totality (90%–95%) of CI appears associated with SCs when the mitochondria are solubilized with a mild detergent, such as digitonin, whereas approximately 50% of CIII2 is inside SCs and 90% of CIV is in the “free” monomeric form (Lobo-Jarne and Ugalde, 2018). There is growing evidence in the literature of the existence of alternative maturation routes for CIII2 and CIV, depending on whether they are in the free or in the SC-associated forms (Lobo-Jarne et al., 2020; Moreno-Lastres et al., 2012; Timón-Gómez et al., 2020). Moreover, only a residual amount of CI is able to mature when CIII2 is absent (Protasoni et al., 2020); therefore, the efficient incorporation of the N-module of CI only occurs in the context of the SCs in human cells (Moreno-Lastres et al., 2012; Protasoni et al., 2020). The dependent assembly of CI, on one hand, and independent biogenesis of CIII2 and CIV, on the other, would explain why defects in the latter induce secondary defects in the CI assembly but not the opposite. However, this phenomenon was also viewed as the consequence of an oxidative-damage-triggered degradation of fully assembled CI, by yet undescribed mechanisms, when it is not associated with CIII2 (Acín-Pérez et al., 2004; Guarás et al., 2016).
An important finding described herein is that the mitochondrial protein TMEM126A/OPA7 is consistently accumulated with distal P-module subcomplexes upon NDUFS3 repression. TMEM126A encodes an evolutionary conserved transmembrane mitochondrial protein of yet unknown function. TMEM126A shows high sequence homology with the assembly factor TMEM126B, which is part of the mitochondrial CI intermediate assembly (MCIA) complex, necessary for the assembly of the ND2-module (Formosa et al., 2020; Heide et al., 2012). Homozygous missense and nonsense mutations (c.163C > T; c.107C > T; c.86+2T > C; and c.497A > G) in TMEM126A were described as pathogenic in both syndromic and non-syndromic forms of arOA (Désir et al., 2012; Hanein et al., 2009; Kloth et al., 2019; La Morgia et al., 2019; Meyer et al., 2010). Partial CI deficiency and elevated lactic acid levels in blood, a typical sign of OXPHOS dysfunction, have been reported in patients (Hanein et al., 2009; La Morgia et al., 2019). Despite the considerable involvement in human pathology and the proposal of a role redundant to that of TMEM126B (Sánchez-Caballero et al., 2016a), the TMEM126A function has so far remained unexplored. We demonstrate here that this protein is a factor necessary for the correct biogenesis and function of CI, through the binding of the ND4-module, containing NDUFB6, within the distal P-module. Three main lines of evidence support the fact that TMEM126A is involved in this specific aspect within the CI assembly pathway. First, there is an accumulation of subunits belonging to this module inside stable subassembly species in the cells lacking NDUFS3, whereas other topological distinct subunits are strongly reduced. Second, TMEM126A is also accumulated upon NDUFS3 repression, physically interacts with the remaining CI subunits, and clearly co-migrates with the NDUFB6-containing subcomplex. Third, the lack of TMEM126A produces a clear defect in CI function and biogenesis, which translates into a reduced respiratory capacity and the inability of the cells to adapt to metabolic stress conditions. In particular, because the lack of TMEM126A induces the accumulation of CI subassembly intermediates containing NDUFB6, we propose that this assembly factor is responsible for the interaction of preformed ND4-module with other submodules already preassembled and, consequently, to its correct incorporation during the process of CI biogenesis. In this frame, we identify TMEM126A as a novel assembly factor specifically involved in the incorporation of the pre-assembled ND4-module, expanding the list of mitochondrial proteins required for the maturation of respiratory CI.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-TMEM126A | Atlas Antibodies | Cat#HPA046648; RRID:AB_2679737 |
| Mouse monoclonal anti-MTCO1 | Abcam | Cat#ab14705; RRID:AB_2084810 |
| Mouse monoclonal anti-MTCO2 | Abcam | Cat#ab110258; RRID:AB_10887758 |
| Mouse monoclonal anti-UQCRC2 | Abcam | Cat#ab14745; RRID:AB_2213640 |
| Rabbit polyclonal anti-CYC1 | Proteintech | Cat#10242-1-AP; RRID:AB_2090144 |
| Mouse monoclonal anti-UQCRFS1 | Abcam | Cat#ab14746; RRID:AB_301445 |
| Mouse monoclonal anti-NDUFA9 | Abcam | Cat#ab14713; RRID:AB_301431 |
| Rabbit polyclonal anti-NDUFS2 | Thermo Fisher Scientific | Cat#PA5-22364; RRID:AB_11152516 |
| Mouse monoclonal anti-NDUFS3 | Abcam | Cat#ab110246; RRID:AB_10861972 |
| Mouse monoclonal anti-NDUFS8 | Santa Cruz Biotechnology | Cat#SC-515527 |
| Mouse monoclonal anti-NDUFB8 | Abcam | Cat#ab110242; RRID:AB_10859122 |
| Rabbit polyclonal anti-NDUFB11 | Proteintech | Cat#16720-1-AP; RRID:AB_2298378 |
| Rabbit polyclonal anti-NDUFA12 | Sigma-Aldrich | Cat#SAB2701046 |
| Mouse monoclonal anti-NDUFB6 | Abcam | Cat#ab110244; RRID:AB_10865349 |
| Rabbit monoclonal anti-NDUFS6 | Abcam | Cat#ab195807 |
| Mouse monoclonal anti-SDHB | Abcam | Cat#ab14714; RRID:AB_301432 |
| Mouse monoclonal anti-SDHA | Abcam | Cat#ab14715; RRID:AB_301433 |
| Mouse monoclonal anti-Hsp70 | Abcam | Cat#ab2787; RRID:AB_303300 |
| Mouse monoclonal anti-β-actin | Sigma-Aldrich | Cat#A1978; RRID:AB_476692 |
| Mouse monoclonal anti-β-tubulin | Sigma-Aldrich | Cat#T5201; RRID:AB_609915 |
| Rabbit polyclonal anti-vinculin | GeneTex | Cat#GTX113294; RRID:AB_10732545 |
| Rabbit polyclonal anti-CS | Abcam | Cat#ab96600; RRID:AB_10678258 |
| Rabbit monoclonal anti-VDAC | Abcam | Cat#ab154856; RRID:AB_2687466 |
| Chicken polyclonal anti-TIMMDC1 | Prof. John E. Walker’s group | N/A |
| Goat anti-mouse IgG | Promega | Cat#W4021; RRID:AB_430834 |
| Goat anti-rabbit IgG | Promega | Cat#W4011; RRID:AB_430833 |
| Anti-chicken IgY | Promega | Cat#G1351; RRID:AB_430845 |
| Chemicals, peptides, and recombinant proteins | ||
| DMEM | GIBCO Thermo Fisher Scientific | Cat#31966 |
| IMDM | GIBCO Thermo Fisher Scientific | Cat#12440046 |
| DMEM for SILAC | GIBCO Thermo Fisher Scientific | Cat#88364 |
| L-Lysine-13C6,15N2 hydrochloride | Sigma-Aldrich | Cat#608041 |
| L-Arginine-13C6,15N4 hydrochloride | Sigma-Aldrich | Cat#608033 |
| L-Lysine monohydrochloride | Sigma-Aldrich | Cat#L8662 |
| L-Arginine | Sigma-Aldrich | Cat#A8094 |
| L-Proline | Sigma-Aldrich | Cat#P5607 |
| Uridine | Sigma-Aldrich | Cat#U3003 |
| FBS | GIBCO Thermo Fisher Scientific | Cat#10270 |
| Dialyzed FBS | GIBCO Thermo Fisher Scientific | Cat#26400 |
| Digitonin, High Purity | Calbiochem | Cat#300410 |
| DDM | Thermo Fisher Scientific | Cat#89903 |
| Doxycycline | Sigma-Aldrich | Cat#D9891 |
| Chloramphenicol | Fisiopharma | Cat#J01BA01 |
| G418 | Sigma-Aldrich | Cat# A1720 |
| Puromycin | Thermo Fisher Scientific | Cat#A1113803 |
| XF DMEM Medium | Agilent | Cat#103575-100 |
| Critical commercial assays | ||
| Complex I Immunocapture Kit | Abcam | Cat#ab109711 |
| ATP assay mix | Sigma-Aldrich | Cat#FLAAM |
| XFe Cell Mito Stress Test Kit | Agilent | Cat#103015-100 |
| Experimental models: Cell lines | ||
| Human 143B+/+ (NDUFS3+/+) | Kurelac et al., 2019 | N/A |
| Human 143B−/− (NDUFS3−/−; c.9_10insCGGCG/NDUFS3) | Kurelac et al., 2019 | N/A |
| Human HCT116+/+ (NDUFS3+/+) | Kurelac et al., 2019 | N/A |
| Human HCT116−/− (NDUFS3−/−; c.9_10insCGGCG/NDUFS3) | Kurelac et al., 2019 | N/A |
| Human 143B−/− NDUFS3 (NDUFS3−/−; c.9_10insCGGCG/NDUFS3) | Kurelac et al., 2019 | N/A |
| Human OS-93 (m.3571insC/MT-ND1 93% heteroplasmic) | Iommarini et al., 2018 | N/A |
| Human OS (m.3571insC/MT-ND1 100% homoplasmic) | Iommarini et al., 2018 | N/A |
| Human Rho 0 (mtDNA depleted) | King and Attardi, 1989 | N/A |
| Mouse B16-F10 | ATCC | Cat#CRL-6475; RRID:CVCL_0159 |
| Mouse B16+/+ (Ndufs3+/+) | This paper | N/A |
| Mouse B16−/− (Ndufs3−/−; c.148A > G/Ndufs3 and c.150_151InsCT/Ndufs3) | This paper | N/A |
| Human HAP1WT | Horizon Discovery | Cat#C631; RRID:CVCL_Y019 |
| Human: HAP1KO: TMEM126AKO; 79bp insertion | Horizon Discovery | Cat#HZGHC005796c002; RRID:CVCL_XU39 |
| Oligonucleotides | ||
| Forward primer for Sanger sequencing: CTGTAACTCCAGTCTCAGGGA | This paper | N/A |
| Reverse primer for Sanger sequencing CACACTGCAGGGATCACTTG | This paper | N/A |
| gRNA: TTGTGGGTCACATCACTCCG PAM sequence: GGG | This paper | N/A |
| Software and algorithms | ||
| GraphPad Prism v.7 | GraphPad Software Inc. | https://www.graphpad.com:443/scientificsoftware/prism/ |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| ChimeraX v1.1 | Goddard et al., 2018 | https://www.cgl.ucsf.edu/chimerax/ |
| MaxQuant v1.6.17.0 | Cox and Mann, 2008 | https://www.maxquant.org/ |
| Perseus v1.6.15.0 | Cox and Mann, 2011 | https://www.maxquant.org/perseus/ |
Resource availability
Lead contacts
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Luisa Iommarini (luisa.iommarini2@unibo.it).
Materials availability
In this work, the sole newly generated material is represented by murine melanoma cells B16-F10 knocked out for Ndufs3, generated by Dr. Kurelac and Prof. Gasparre. According to University of Bologna regulations, cells can be shared only upon the subscription of a Material Transfer Agreement (MTA).
Data and code availability
The datasets supporting the current study related to Figures 3, 4, and S4 are available as Tables S1 and S2. Source data for figures in the paper are available from the corresponding authors on request.
Experimental model and subject details
Cell lines
Human female osteosarcoma 143B TK- and male colorectal cancer HCT116 cell lines bearing the homozygous c.9_10insCGGCG/NDUFS3 frameshift mutation, which resulted in a premature stop codon and the truncation of the protein (p.Val6Alafs∗3), were analyzed relative to their isogenic controls (143B+/+ and HCT116+/+). These NDUFS3 knockout cells, referred as 143B−/− and HCT116−/−, were generated as previously reported and recently underwent authentication by sequence tandem repeats (microsatellites) analysis (Kurelac et al., 2019). Cell origin was authenticated using AMPFISTRIdentifiler kit (Applied Biosystems #4322288) and their STR profile corresponded to their putative background. The results are available upon request. 143B TK- cybrid cells carrying the truncative frameshift m.3571insC/MT-ND1 mutation were also used. Specifically, the previously characterized homoplasmic cells (OS) and heteroplasmic cells with 93% of mutated MT-ND1 (OS-93) were used (Porcelli et al., 2010; Gasparre et al., 2011; Iommarini et al., 2018). 143B TK- mtDNA-depleted cells (Rho 0) (King and Attardi, 1989) provided CI-negative control. Doxycycline-inducible NDUFS3 knockout 143B cells (referred as 143B−/− NDUFS3) were also used. They were generated re-expressing NDUFS3 using a Tet-Off inducible system ensuring the transcriptional repression of the complemented gene in the presence of doxycycline as previously described (Kurelac et al., 2019). B16-F10 murine male melanoma cell line was purchased from ATCC (#CRL-6475) and Ndufs3 knockout cell line (B16−/−) was generated by genome editing (see Genome editing). Human male chronic myeloid leukemia HAP1 cell line with a 79 bp-insertion in TMEM126A exon 4 (HAP1KO; Horizon Discovery, #HZGHC005796c002) and its relative wild-type control (HAP1WT; Horizon Discovery, #C631) were used to demonstrate the role of TMEM126A. B16-F10 and HAP1 cells were purchased in 2020, thus they do not underwent cell authentication analyses.
Genome editing
CRISPR/Cas9 system was used to introduce a frameshift mutation in Ndufs3 gene in B16-F10 murine melanoma cell line. In detail, Cas9 protein (Invitrogen #A36497) was transfected following manufacturer’s instructions using Lipofectamine CRISPRMAX Cas9 Transfection Reagent (Invitrogen #CMAX00015) together with synthetic RNA guides designed by Deskgen and purchased from Synthego. Exon 3 targeting guide TTGTGGGTCACATCACTCCG with PAM sequence GGG was used. Cells were split 48 hours after transfection and DNA was extracted using Mammalian Genomic DNA Miniprep Kit (Sigma-Aldrich #G1N350). Non-homologous repair efficiency was evaluated by Sanger Sequencing using KAPA2G Taq polymerase (Kapa Biosystems #KK5601) and Big Dye protocol (Life Technologies #4337451). In particular, 61°C annealing temperature was used for the PCR reaction, with primers forward CTGTAACTCCAGTCTCAGGGA and reverse CACACTGCAGGGATCACTTG. Manual clonal selection was performed in order to identify the cells with frameshift Ndufs3 mutations, leading to the generation of a pool of clones carrying the homozygous c.148A>G and c.150_151insCT mutations. DNA extraction from 96-well plates was performed using 8 μL of Lysis Solution (Sigma-Aldrich #L3289) and 80 μL of Neutralization Buffer (Sigma-Aldrich #N9784) per sample, following manufacturer’s instructions.
Cell maintenance and treatments
143B, HCT116 and B16-F10 cell lines were grown in high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM) plus Glutamax and sodium pyruvate (GIBCO Thermo Fisher Scientific, #31966) supplemented with 10% fetal bovine serum (GIBCO Thermo Fisher Scientific, #10270), 1% Pen/Strep and 50 μg/mL uridine (Sigma-Aldrich, #U3003), in an incubator at 37°C with a humidified atmosphere at 5% CO2. 143B−/−NDUFS3 cells were grown in the same medium but with addition of 100 μg/mL G418 (Sigma-Aldrich, # A1720) and 0.25 μg/mL puromycin (Thermo Fisher Scientific, #A1113803) to sustain the selective pressure. In this cell model, expression of NDUFS3 was repressed by addition of 100 ng/mL doxycycline (Sigma-Aldrich, #D9891). For the inhibition of mitochondrial translation 50 μg/mL chloramphenicol (Fisiopharma, #J01BA01) was used. HAP1 cell lines were cultured in Iscove’s Modified Dulbecco’s Medium (IMDM; GIBCO Thermo Fisher Scientific, #12440046) with 10% fetal bovine serum, 1% Pen/Strep, 1% L-glutamine and 50 μg/mL uridine. For SILAC experiments, 143B−/−NDUFS3 cells were grown in SILAC DMEM (GIBCO Thermo Fisher Scientific, #88364) added with either isotopically labeled 152.1 mg/L L-Lysine-15N213C6 (Sigma-Aldrich, #608041) plus 87.8 mg/L L-Arginine-15N413C6 (Sigma-Aldrich, #608033) (“Heavy” medium), or unlabelled 143.8 mg/L L-Lysine (Sigma-Aldrich, #L8662) and 69.3 mg/L L-Arginine (Sigma-Aldrich, #A8094) (“Light” medium). Both differentially labeled media were supplemented also with 10% dialyzed serum (GIBCO Thermo Fisher Scientific, #26400-044), 50 μg/mL uridine, 100 μg/mL G418, 0.25 μg/mL puromycin and 200 mg/mL L-Proline (Sigma-Aldrich, #P5607). Incorporation of the “Heavy” amino acids into the cells proteins was completed after approximately 9 passages (Ong et al., 2002). Cell lines were regularly tested for mycoplasma.
Method details
SDS-PAGE
Whole-cell lysates were obtained incubating cell pellets with a Lysis Buffer (2% n-dodecyl β-d-maltoside (DDM) (Thermo Scientific, #89903) plus 1X protease inhibitor cocktail (Roche, #11697498001 in PBS) for 20 min at 4°C. Protein concentration in the extracts was measured with a Nanodrop UV-visible spectrophotometer at λ = 280nm. 25 μg of total protein were combined with 2X Laemmli Sample Buffer (2% sodium dodecyl sulfate (SDS), 4.6% β-mercaptoethanol, 20% glycerol, 0.025% bromophenol blue and 62.5 mM Tris/HCl at pH 6.8) in one-to-one ratio, loaded on pre-cast NuPAGE™ 4%–12% or 12% Bis-Tris gels (Invitrogen, #NP0321, #NP0322, #NP0341, #NP0342) and resolved in a electrophoresis cell at 100 V for approximately 2 hours at RT. Either NuPAGE™ MOPS or MES SDS Running Buffers were used (Invitrogen, #NP0001, #NP0002). Precision Plus Protein™ Standard (Bio-Rad, #1610374) was used for molecular weight (MW) estimation.
Blue-Native PAGE
For the preparation of samples for Blue-Native PAGE, mitochondrial-enriched fractions were obtained by digitonin treatment (Nijtmans et al., 2002). Briefly, cells were harvested by trypsinization, 5-8x106 cell pellets were washed twice and resuspended in cold DPBS and incubated in ice for 10 minutes with 2 mg/mL digitonin (Calbiochem, #3000410). Next, cold DPBS was added and a centrifugation at 10,000 g for 10 min and 4°C was performed. The mitochondrial-enriched fractions obtained were solubilized in a Solubilization Buffer (1.5 M aminocaproic acid, 50 mM Bis-Tris/HCl at pH 7). Proteins were quantified by a detergent-compatible Lowry-based method (Bio-Rad DC Protein Assay), solubilized either with 1.6 μg DDM/μg total proteins or 4 μg digitonin/μg total proteins and incubated at 4°C for 5 min (Wittig et al., 2006). Insoluble material was removed by a centrifugation (18,000 g for 30 min at 4°C) and Sample Buffer (750 mM aminocaproic acid, 50 mM Bis-Tris/HCl pH 7, 0.5 mM EDTA and 5% Coomassie Blue G250) was added. For 1D-BN PAGE, 50-75 μg of proteins were loaded on Pre-cast Native PAGE 3%–12% Bis-Tris gels (Invitrogen, #BN2011BX10) at 150 V and 4°C for 4 hours. Cathode Buffer A containing 1x NativePAGE Running Buffer (Invitrogen, #BN2001) plus 1x NativePAGE Cathode Buffer Additive (Invitrogen, #BN2002); Cathode Buffer B containing 1x NativePAGE Running Buffer and 0.1x NativePAGE Cathode Buffer Additive; and Anode Buffer consisting of 1x NativePAGE Running Buffer were used.
2D BN-PAGE
1D BN-PAGE lanes were excised from the gel and incubated with a Denaturing Solution (1% 2-mercaptoethanol, 1% SDS) at RT for 1 hour. Then, the lanes were inserted in denaturing Pre-cast NuPAGE™ 4%–12% Bis-Tris Gels (Invitrogen, #NP0326) in a 90° orientation and separated in either MES or MOPS SDS Running Buffer at 100 V for approximately 2 hours at RT.
Western blotting and immunodetection
Proteins resolved by either SDS-PAGE or 2D BN-PAGE were transferred onto a polyvinylidene difluoride (PVDF) membrane. The blotting was performed at 100 V for 1 hour at 4°C in Tris-Glycine Transfer Buffer (12.5 mM Tris, 96 mM glycine, 20% Methanol, 0.025% SDS). Membranes were blocked 1 hour at RT with 5% powdered skimmed milk in PBS containing 0.1% Tween 20 (PBS-T), washed three times for 10 min in PBS-T and incubated overnight at 4°C with the following primary antibodies diluted in a blocking solution containing 3% BSA in PBS-T: anti-NDUFS2 (Thermo Fisher Scientific, #PA522364) 1:2000; anti-NDUFA9 (Abcam, #ab14713) 1:1000; anti-NDUFS3 (Abcam, #ab110246) 1:1000 or 1:500; anti-NDUFS8 (Santa Cruz Biotechnology, #SC-515527) 1:1000; anti-NDUFB8 (Abcam, #ab110242) 1:1000; anti-NDUFB11 (Proteintech, #16720-1-AP) 1:700; anti-NDUFA12 (Sigma-Aldrich, #SAB2701046) 1:1000; anti-NDUFB6 (Abcam, #ab110244) 1:1000; anti-NDUFS6 (Abcam, #ab195807) 1:1000; anti-SDHA (Abcam, #ab14715) 1:1000; anti-SDHB (Abcam, #ab14714) 1:1000; anti-UQCRC2 (Abcam, #ab14745) 1:1000; anti-CYC1 (Proteintech, #10242-1-AP) 1:1000; anti-UQCRFS1 (Abcam, #ab14746) 1:1000; anti-MT-CO1 (Abcam, #ab14705) 1:1000; anti-MT-CO2 (Abcam, #ab110258) 1:1000; anti-TIMMDC1 (homemade, a generous gift from Prof. John E. Walker’s group) 1:2500; anti-TMEM126A (Sigma-Aldrich, #HPA046648); anti-CS (Abcam, #ab96600) 1:1000; anti-VDAC (Abcam, #ab154856) 1:1000; anti-Hsp70 (Abcam, #ab2787) 1:1000; anti-β-actin (Sigma-Aldrich, #AI978); 1:1000; anti-β-tubulin (Sigma-Aldrich, #T5201) 1:1000; anti-vinculin (GeneTex #GTX113294) 1:2000. Then, three washes of 10 minutes in PBS-T were performed and membranes were incubated 1 hour at RT with the following 1:5000 or 1:2500 horseradish peroxidase conjugated secondary antibodies in 1% milk in PBS-T: anti-mouse (Promega, #W4021), anti-rabbit (Promega, #4011) and anti-chicken (Promega, #G1351). Proteins resolved by 1D BN-PAGE were electroblotted as above, except that the blotting was performed at 300 mA for 1 hour at 4°C in Bicarbonate Transfer Buffer (10 mM NaHCO3 and 3 mM Na2CO3). The immunoreactive bands were visualized with ECL Western Blotting Reagents (GE Healthcare, #RPN2106). When performed, band signal intensities were quantified by densitometry using ImageJ (Schneider et al., 2012) and 2-way ANOVA with Sidak’s post hoc test was used for multiple comparison statistical analysis.
Enzymatic activity assays
Crude mitochondria were used to assess respiratory chain enzymatic activities. A pellet of 15-20 × 106 cells was suspended in ice-cold Sucrose-Mannitol Buffer (200 mM mannitol, 70 mM sucrose, 1 mM EGTA and 10 mM Tris-HCl at pH 7.6) and homogenized using a glass/teflon Potter homogenizer. The obtained sample was centrifuged at 600 g for 10 min at 4°C to discard unbroken cells and nuclei. The resulting supernatant was centrifuged at 10,000 g for 20 min at 4°C to separate crude mitochondria from the remaining sub-cellular fractions of the sample. The pellet was resuspended in Sucrose-Mannitol Buffer and stored in aliquotes at −80°C. Kinetic measurements of respiratory chain complex activities were assessed at 37°C in a spectrophotometer (V550 Jasco Europe, Modena, Italy) as previously detailed (Ghelli et al., 2013). CI activity (NADH:DB:DCIP oxidoreductase) was assessed incubating 5 μL of mitochondria with 0.3 mM KCN, 1 μM antimycin A (Aa), 200 μM NADH, 60 μM dichlorophenol indophenol (DCIP; λ: 600 nm; εDCIP: 19.1 mM-1cm-1), 70 μM decylbenzoquinone (DB), and subtracting the rotenone-insensitive activity. CII activity (succinate/DCIP) was determined incubating 5 μL of mitochondria with 1 μM rotenone, 0.3 mM KCN, 1 μM Aa, 80 μM DCIP, 200 μM ATP, 50 μM decylbenzoquinone (DB), 10 mM Na+ succinate, and subtracting the K+ malonate-insensitive activity. CIII activity (cytochrome c/DBH2) was measured incubating 5 μL mitochondria with 0.3 mM KCN, 20 μM oxidized cytochrome c (λ: 550 nm; εcytc: 19 mM-1cm-1), 50 μM decylbenzoquinol (DBH2), and subtracting the Aa-insensitive activity. CIV activity (cytochrome c oxidase) was assessed following the oxidation of 20 μM reduced cytochrome c and subtracting the KCN-insensitive activity. The measurements were performed in a buffer containing 50 mM KH2PO4 (pH 7.6), 1 mM EDTA, 2.5 mg/mL BSA (3.5 mg/mL for CI activity assessment). Each specific activity was normalized to protein content and citrate synthase (DTNB:oxaloacetate) activity. The latter was measured incubating 10 μL mitochondria with 100 μM dithionitrobenzoic acid (DTNB), 300 μM acetyl-CoA, 500 μM oxalacetate, 100 mM Tris (pH 8.1) and 0.1% Triton X-100. CI-IGA assays were performed incubating native gels either with 2 mM Tris-Cl (pH 7.4), 0.15 mM NADH, and 2.5 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) or with 0.5 M Tris/HCl (pH 7.4), 0.1 mg/mL NADH, 1 mg/mL nitro tetrazolium blue (NTB) at RT overnight.
ATP synthesis rate assay
The rate of CI-driven ATP synthesis was measured in digitonin-permeabilized cells treated or not with 50 μg/mL chloramphenicol. Cells were harvested by trypsinization, pellets were washed in PBS and the cells (10 × 106 /mL) were suspended in a buffer containing 150 mM KCl, 25 mM Tris-HCl, 2 mM EDTA, 0.1% BSA, 10 mM potassium phosphate, 0.1 mM MgCl2, pH 7.4. Then, cellular suspensions were incubated with 50 μg/mL digitonin for 1 minute at RT with gentle agitation. As a result of the permeabilization 90%–100% of cells were positive to Trypan Blue staining. Aliquots of 3 × 105 permeabilized cells were incubated in the same buffer with 0.9 mM Na+ pyruvate, 0.9 mM K+ malate, and 10 μL of ATP assay mix (Sigma-Aldrich; #FLAAM) in the presence of 0.1 mM adenylate kinase inhibitor P1,P5-di(adenosine-5′) pentaphosphate. After the addition of 0.09 mM ADP, chemiluminescence was determined as a function of time with Sirius L Tube luminometer (Titertek-Berthold, Pforzheim, Germany). The chemiluminescence signal was calibrated with 11 μM internal ATP standard (Sigma-Aldrich; #FLAAS) after the addition of 1 μM rotenone The rates of the ATP synthesis were normalized to protein content and citrate synthase (CS) activity.
Quantitative proteomics
Proteomic analyses were performed on mitochondrial enriched fractions and quantified using SILAC (Andrews et al., 2013). Each proteomic analysis was performed in duplicate: in one experiment, the heavy-labeled Dox-treated cells were mixed, in a one-to-one ratio, with the light-labeled untreated ones; in the second experiment, the labeling was inverted. Accordingly, mitochondrial enriched fractions obtained from two reciprocal cellular pellets (15 mg total protein each) were solubilized in 500 μL of 10% glycerol, 1X protease inhibitor cocktail, 1.5% DDM, and a mixture of Avanti® polar lipids (Avanti; #850457C, #8407575C, #840457C) and incubated in ice for 30 minutes. Then, samples were centrifuged at 18,000 g for 10 minutes at 4°C to remove insoluble materials. In the present study, portions from each sample were retained before performing CI immunoprecipitation and directly analyzed by liquid chromatography-mass spectrometry (LC-MS). For CI co-immunopurification, solubilized mitochondrial-enriched fractions were incubated overnight at 4°C with Complex I Immunocapture Kit (Abcam; #ab109711) suspension. Both immunopurified and non-immunopurified samples were reduced, alkylated, and fractioned by SDS-PAGE on 10%–20% Tris-glycine gels (Invitrogen; #XP10200) for 2 hours at 100 V. Tryptic peptide mixtures from gel slices were resolved by Nanoscale Ultra High Performance Liquid Chromatography (nano-UPLC) in a C18 reverse phase column (75 μm x 100 mm) with a gradient of acetonitrile in 0.1% formic acid and a flow rate of 300 nL/min. The eluate was analyzed with Q Exactive Plus Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Fisher Scientific) operating in mass tandem (MS/MS) mode. Protein identification and quantification were performed with the MaxQuant computational platform (Cox and Mann, 2008; Cox et al., 2011). For the relative quantification, MaxQuant calculated the ratios between the peak intensities of heavy- and light-labeled peptide pairs (H/L ratio). Fold changes for each protein were plotted in scatterplots with the log2H/L ratios of one experiment in the x axis and -log2H/L ratios of the second experiment in the y axis. Correlation between the two duplicate experiments was reflected by the distribution of the protein over a 45° diagonal line of the scatterplots. Statistical significances of the differences for enriched and depleted proteins in each reciprocal experiment were determined by applying the B-significance test with Perseus (Cox and Mann, 2011; Tyanova et al., 2016).
Cell viability assay
HAP1 cell viability was evaluated by sulforhodamine B (SRB) assay. Briefly, 30 × 103 cells/well were seeded in 24-well plates and incubated overnight. Then, IMDM was replaced with either glucose-free and galactose-containing (5 mM) DMEM or glucose-containing (25 mM) DMEM. At the end of incubation time points (24, 48 and 72 hours), medium was replacedwith a fresh one to eliminate detached cells, 10% trichloroacetic acid (TCA) was added, and the plates were incubated for 1 hour at 4°C to fix the cells. 5 washes in water were performed. Then, fixed cells were incubated with 0.4% SRB in 1% acetic acid for 30 minutes at RT. SRB was removed and 5 washes with 1% acetic acid were carried out to eliminate the residual dye. Finally, the SRB bound to the proteins was solubilized in 10 mM Tris and the absorbance at 560 nm was read in a Victor2 plate reader (Perkin-Elmer, Turku; Finland). Measurements were blank corrected.
Oxygen consumption rate
Mitochondrial respiration was evaluated using the Seahorse XFe Cell Mito Stress Test Kit (Agilent #103015-100) following the manufacturer instructions. Cells were seeded (40 × 103 cells/well) in 80 μL of IMDM medium into XFe96 cell culture plate and allowed to attach for 24 h. Cell culture media was replaced with Seahorse XF DMEM Medium, pH 7.4 (Agilent #103575-100). OCR was measured over a 3 min period, followed by 3 min of mixing and re-oxygenation of the medium. To perform Mito Stress Test, complete growth medium was replaced with 180 μL of unbuffered XF medium supplemented with 10 mM glucose pH 7.4 pre-warmed at 37 °C. Cells were incubated at 37 °C for 30 min to allow temperature and pH equilibration. After 3 OCR baseline measurements, 1 μM oligomycin, 0.25 μM carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), and 0.5 μM rotenone plus 0.5 μM antimycin A were sequentially added to each. Three measurements of OCR were obtained following injection of each drug. FCCP concentration was optimized on cell lines prior to experiments. At the end of each experiment, the medium was removed and SRB assay was performed to determine the amount of total cell proteins as described above. OCR data were normalized to total protein levels (SRB protein assay) in each well. Each cell line was represented at least four wells per experiment (n = 4 replicate experiments). Data are normalized on SRB absorbance and expressed as pmoles of O2 per minute.
Quantification and statistical analysis
GraphPad Prism v.7 (GraphPad Software Inc., San Diego, CA, USA) was used to perform statistical tests and create bar plots and graphs. Quantitative data are mean ± SD or ± SEM as indicated in the figure legends. Unless stated otherwise, a two-tailed unpaired Student’s t tests assuming equal variance were performed to compare averages. For each experiment, at least three biological replicates were analyzed. Statistical significance was defined by p value < 0.05.
Acknowledgments
Luisa Iommarini and Erika Fernandez-Vizarra want to dedicate this work to the memory of their friend and colleague Prof. Francisca Diaz, University of Miami, Department of Neurology, Miller School of Medicine. This work was supported by University of Bologna AlmaIdea Junior grant INTACT to L.I.; EU H2020 ITN Marie Curie project TRANSMIT (grant no. GA722605) to A.M.P.; Italian Ministry of Health grant DISCO TRIP (grant no. GR-2013-02356666) to G.G.; and by a Core Grant from the Medical Research Council (grant MC_UU_00015/5) to M.Z. and E.F.-V. M.D.L. was supported by an Associazione Italiana Ricerca sul Cancro (AIRC) fellowship. We thank Fondazione Dal Monte (Bologna, Italy) and Centro Studi della Barbariga (Padua, Italy) for the financial support finalized to the acquisition of instruments Seahorse XFe96 and Jasco V550 spectrophotometer, respectively. We thank Dr. Joe Carroll and Prof. John E. Walker (Mitochondrial Biology Unit, Cambridge) for the insightful discussion on the role of TMEM126A and the kind gift of the anti-TIMMDC1 antibody. We are grateful to Prof. Francesco Musiani (University of Bologna) for technical suggestions on the production of complex I structure pictures.
Author contributions
Conceptualization, M.Z., A.M.P., E.F.-V., and L.I.; methodology, L.D.A., E.A., M.D.L., I.K., N.U.-G., S.D., I.M.F., E.F.-V., and L.I.; investigation, L.D.A., E.A., M.D.L., S.D., and L.I.; formal analysis, L.D.A., E.A., S.D., and L.I.; writing – original draft, L.D.A., E.A., G.G., A.M.P., E.F.-V., and L.I.; writing – review & editing, L.D.A., G.G., M.Z., I.M.F, A.M.P., E.F.-V., L.I.; funding acquisition, M.Z., G.G., A.M.P., E.F.-V., and L.I.; supervision, E.F.V. and L.I.
Declaration of interest
The authors declare no competing interests.
Published: April 20, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109002.
Contributor Information
Erika Fernandez-Vizarra, Email: erika.fernandez-vizarra@glasgow.ac.uk.
Luisa Iommarini, Email: luisa.iommarini2@unibo.it.
Supplemental information
References
- Acin-Perez R., Enriquez J.A. The function of the respiratory supercomplexes: the plasticity model. Biochim. Biophys. Acta. 2014;1837:444–450. doi: 10.1016/j.bbabio.2013.12.009. [DOI] [PubMed] [Google Scholar]
- Acín-Pérez R., Bayona-Bafaluy M.P., Fernández-Silva P., Moreno-Loshuertos R., Pérez-Martos A., Bruno C., Moraes C.T., Enríquez J.A. Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol. Cell. 2004;13:805–815. doi: 10.1016/s1097-2765(04)00124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acín-Pérez R., Fernández-Silva P., Peleato M.L., Pérez-Martos A., Enriquez J.A. Respiratory active mitochondrial supercomplexes. Mol. Cell. 2008;32:529–539. doi: 10.1016/j.molcel.2008.10.021. [DOI] [PubMed] [Google Scholar]
- Agip A.A., Blaza J.N., Bridges H.R., Viscomi C., Rawson S., Muench S.P., Hirst J. Cryo-EM structures of complex I from mouse heart mitochondria in two biochemically defined states. Nat. Struct. Mol. Biol. 2018;25:548–556. doi: 10.1038/s41594-018-0073-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews B., Carroll J., Ding S., Fearnley I.M., Walker J.E. Assembly factors for the membrane arm of human complex I. Proc. Natl. Acad. Sci. USA. 2013;110:18934–18939. doi: 10.1073/pnas.1319247110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baradaran R., Berrisford J.M., Minhas G.S., Sazanov L.A. Crystal structure of the entire respiratory complex I. Nature. 2013;494:443–448. doi: 10.1038/nature11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bénit P., Slama A., Cartault F., Giurgea I., Chretien D., Lebon S., Marsac C., Munnich A., Rötig A., Rustin P. Mutant NDUFS3 subunit of mitochondrial complex I causes Leigh syndrome. J. Med. Genet. 2004;41:14–17. doi: 10.1136/jmg.2003.014316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt U. Energy converting NADH:quinone oxidoreductase (complex I) Annu. Rev. Biochem. 2006;75:69–92. doi: 10.1146/annurev.biochem.75.103004.142539. [DOI] [PubMed] [Google Scholar]
- Carossa V., Ghelli A., Tropeano C.V., Valentino M.L., Iommarini L., Maresca A., Caporali L., La Morgia C., Liguori R., Barboni P. A novel in-frame 18-bp microdeletion in MT-CYB causes a multisystem disorder with prominent exercise intolerance. Hum. Mutat. 2014;35:954–958. doi: 10.1002/humu.22596. [DOI] [PubMed] [Google Scholar]
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Cox J., Mann M. Quantitative, high-resolution proteomics for data-driven systems biology. Annu. Rev. Biochem. 2011;80:273–299. doi: 10.1146/annurev-biochem-061308-093216. [DOI] [PubMed] [Google Scholar]
- Cox J., Neuhauser N., Michalski A., Scheltema R.A., Olsen J.V., Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- Čunátová K., Reguera D.P., Vrbacký M., Fernández-Vizarra E., Ding S., Fearnley I.M., Zeviani M., Houštěk J., Mráček T., Pecina P. Loss of COX4I1 leads to combined respiratory chain deficiency and impaired mitochondrial protein synthesis. Cells. 2021;10:369. doi: 10.3390/cells10020369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Désir J., Coppieters F., Van Regemorter N., De Baere E., Abramowicz M., Cordonnier M. TMEM126A mutation in a Moroccan family with autosomal recessive optic atrophy. Mol. Vis. 2012;18:1849–1857. [PMC free article] [PubMed] [Google Scholar]
- Diaz F., Fukui H., Garcia S., Moraes C.T. Cytochrome c oxidase is required for the assembly/stability of respiratory complex I in mouse fibroblasts. Mol. Cell. Biol. 2006;26:4872–4881. doi: 10.1128/MCB.01767-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieteren C.E.J., Koopman W.J.H., Swarts H.G., Peters J.G.P., Maczuga P., van Gemst J.J., Masereeuw R., Smeitink J.A.M., Nijtmans L.G.J., Willems P.H.G.M. Subunit-specific incorporation efficiency and kinetics in mitochondrial complex I homeostasis. J. Biol. Chem. 2012;287:41851–41860. doi: 10.1074/jbc.M112.391151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elurbe D.M., Huynen M.A. The origin of the supernumerary subunits and assembly factors of complex I: A treasure trove of pathway evolution. Biochim. Biophys. Acta. 2016;1857:971–979. doi: 10.1016/j.bbabio.2016.03.027. [DOI] [PubMed] [Google Scholar]
- Fassone E., Duncan A.J., Taanman J.-W., Pagnamenta A.T., Sadowski M.I., Holand T., Qasim W., Rutland P., Calvo S.E., Mootha V.K. FOXRED1, encoding an FAD-dependent oxidoreductase complex-I-specific molecular chaperone, is mutated in infantile-onset mitochondrial encephalopathy. Hum. Mol. Genet. 2010;19:4837–4847. doi: 10.1093/hmg/ddq414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formosa L.E., Mimaki M., Frazier A.E., McKenzie M., Stait T.L., Thorburn D.R., Stroud D.A., Ryan M.T. Characterization of mitochondrial FOXRED1 in the assembly of respiratory chain complex I. Hum. Mol. Genet. 2015;24:2952–2965. doi: 10.1093/hmg/ddv058. [DOI] [PubMed] [Google Scholar]
- Formosa L.E., Dibley M.G., Stroud D.A., Ryan M.T. Building a complex complex: Assembly of mitochondrial respiratory chain complex I. Semin. Cell Dev. Biol. 2018;76:154–162. doi: 10.1016/j.semcdb.2017.08.011. [DOI] [PubMed] [Google Scholar]
- Formosa L.E., Muellner-Wong L., Reljic B., Sharpe A.J., Jackson T.D., Beilharz T.H., Stojanovski D., Lazarou M., Stroud D.A., Ryan M.T. Dissecting the roles of mitochondrial complex I intermediate assembly complex factors in the biogenesis of complex I. Cell Rep. 2020;31:107541. doi: 10.1016/j.celrep.2020.107541. [DOI] [PubMed] [Google Scholar]
- Friedrich T., Scheide D. The respiratory complex I of bacteria, archaea and eukarya and its module common with membrane-bound multisubunit hydrogenases. FEBS Lett. 2000;479:1–5. doi: 10.1016/s0014-5793(00)01867-6. [DOI] [PubMed] [Google Scholar]
- Gasparre G., Kurelac I., Capristo M., Iommarini L., Ghelli A., Ceccarelli C., Nicoletti G., Nanni P., De Giovanni C., Scotlandi K. A mutation threshold distinguishes the antitumorigenic effects of the mitochondrial gene MTND1, an oncojanus function. Cancer Res. 2011;71:6220–6229. doi: 10.1158/0008-5472.CAN-11-1042. [DOI] [PubMed] [Google Scholar]
- Ghelli A., Tropeano C.V., Calvaruso M.A., Marchesini A., Iommarini L., Porcelli A.M., Zanna C., De Nardo V., Martinuzzi A., Wibrand F. The cytochrome b p.278Y>C mutation causative of a multisystem disorder enhances superoxide production and alters supramolecular interactions of respiratory chain complexes. Hum. Mol. Genet. 2013;22:2141–2151. doi: 10.1093/hmg/ddt067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezzi D., Zeviani M. Human diseases associated with defects in assembly of OXPHOS complexes. Essays Biochem. 2018;62:271–286. doi: 10.1042/EBC20170099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard T.D., Huang C.C., Meng E.C., Pettersen E.F., Couch G.S., Morris J.H., Ferrin T.E. UCSF chimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. doi: 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarás A., Perales-Clemente E., Calvo E., Acín-Pérez R., Loureiro-Lopez M., Pujol C., Martínez-Carrascoso I., Nuñez E., García-Marqués F., Rodríguez-Hernández M.A. The CoQH2/CoQ ratio serves as a sensor of respiratory chain efficiency. Cell Rep. 2016;15:197–209. doi: 10.1016/j.celrep.2016.03.009. [DOI] [PubMed] [Google Scholar]
- Guerrero-Castillo S., Baertling F., Kownatzki D., Wessels H.J., Arnold S., Brandt U., Nijtmans L. The assembly pathway of mitochondrial respiratory chain complex I. Cell Metab. 2017;25:128–139. doi: 10.1016/j.cmet.2016.09.002. [DOI] [PubMed] [Google Scholar]
- Hanein S., Perrault I., Roche O., Gerber S., Khadom N., Rio M., Boddaert N., Jean-Pierre M., Brahimi N., Serre V. TMEM126A, encoding a mitochondrial protein, is mutated in autosomal-recessive nonsyndromic optic atrophy. Am. J. Hum. Genet. 2009;84:493–498. doi: 10.1016/j.ajhg.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heide H., Bleier L., Steger M., Ackermann J., Dröse S., Schwamb B., Zörnig M., Reichert A.S., Koch I., Wittig I., Brandt U. Complexome profiling identifies TMEM126B as a component of the mitochondrial complex I assembly complex. Cell Metab. 2012;16:538–549. doi: 10.1016/j.cmet.2012.08.009. [DOI] [PubMed] [Google Scholar]
- Hirst J. Mitochondrial complex I. Annu. Rev. Biochem. 2013;82:551–575. doi: 10.1146/annurev-biochem-070511-103700. [DOI] [PubMed] [Google Scholar]
- Iommarini L., Ghelli A., Tropeano C.V., Kurelac I., Leone G., Vidoni S., Lombes A., Zeviani M., Gasparre G., Porcelli A.M. Unravelling the effects of the mutation m.3571insC/MT-ND1 on respiratory complexes structural organization. Int. J. Mol. Sci. 2018;19:764. doi: 10.3390/ijms19030764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaokar T.M., Patil D.P., Shouche Y.S., Gaikwad S.M., Suresh C.G. Human mitochondrial NDUFS3 protein bearing Leigh syndrome mutation is more prone to aggregation than its wild-type. Biochimie. 2013;95:2392–2403. doi: 10.1016/j.biochi.2013.08.032. [DOI] [PubMed] [Google Scholar]
- King M.P., Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- Kloth K., Synofzik M., Kernstock C., Schimpf-Linzenbold S., Schuettauf F., Neu A., Wissinger B., Weisschuh N. Novel likely pathogenic variants in TMEM126A identified in non-syndromic autosomal recessive optic atrophy: two case reports. BMC Med. Genet. 2019;20:62. doi: 10.1186/s12881-019-0795-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurelac I., Iommarini L., Vatrinet R., Amato L.B., De Luise M., Leone G., Girolimetti G., Umesh Ganesh N., Bridgeman V.L., Ombrato L. Inducing cancer indolence by targeting mitochondrial Complex I is potentiated by blocking macrophage-mediated adaptive responses. Nat. Commun. 2019;10:903. doi: 10.1038/s41467-019-08839-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Morgia C., Caporali L., Tagliavini F., Palombo F., Carbonelli M., Liguori R., Barboni P., Carelli V. First TMEM126A missense mutation in an Italian proband with optic atrophy and deafness. Neurol. Genet. 2019;5:e329. doi: 10.1212/NXG.0000000000000329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo-Jarne T., Ugalde C. Respiratory chain supercomplexes: Structures, function and biogenesis. Semin. Cell Dev. Biol. 2018;76:179–190. doi: 10.1016/j.semcdb.2017.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo-Jarne T., Pérez-Pérez R., Fontanesi F., Timón-Gómez A., Wittig I., Peñas A., Serrano-Lorenzo P., García-Consuegra I., Arenas J., Martín M.A. Multiple pathways coordinate assembly of human mitochondrial complex IV and stabilization of respiratory supercomplexes. EMBO J. 2020;39:e103912. doi: 10.15252/embj.2019103912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X., Shi H., Wen S., Li Y., Wei X., Xie J., Ma L., Yang Y., Fang H., Lyu J. A Novel NDUFS3 mutation in a Chinese patient with severe Leigh syndrome. J. Hum. Genet. 2018;63:1269–1272. doi: 10.1038/s10038-018-0505-0. [DOI] [PubMed] [Google Scholar]
- Meyer E., Michaelides M., Tee L.J., Robson A.G., Rahman F., Pasha S., Luxon L.M., Moore A.T., Maher E.R. Nonsense mutation in TMEM126A causing autosomal recessive optic atrophy and auditory neuropathy. Mol. Vis. 2010;16:650–664. [PMC free article] [PubMed] [Google Scholar]
- Milenkovic D., Blaza J.N., Larsson N.-G., Hirst J. The enigma of the respiratory chain supercomplex. Cell Metab. 2017;25:765–776. doi: 10.1016/j.cmet.2017.03.009. [DOI] [PubMed] [Google Scholar]
- Moparthi V.K., Hägerhäll C. The evolution of respiratory chain complex I from a smaller last common ancestor consisting of 11 protein subunits. J. Mol. Evol. 2011;72:484–497. doi: 10.1007/s00239-011-9447-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Lastres D., Fontanesi F., García-Consuegra I., Martín M.A., Arenas J., Barrientos A., Ugalde C. Mitochondrial complex I plays an essential role in human respirasome assembly. Cell Metab. 2012;15:324–335. doi: 10.1016/j.cmet.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijtmans L.G.J., Henderson N.S., Holt I.J. Blue Native electrophoresis to study mitochondrial and other protein complexes. Methods. 2002;26:327–334. doi: 10.1016/S1046-2023(02)00038-5. [DOI] [PubMed] [Google Scholar]
- Ong S.E., Blagoev B., Kratchmarova I., Kristensen D.B., Steen H., Pandey A., Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- Pagniez-Mammeri H., Lombes A., Brivet M., Ogier-de Baulny H., Landrieu P., Legrand A., Slama A. Rapid screening for nuclear genes mutations in isolated respiratory chain complex I defects. Mol. Genet. Metab. 2009;96:196–200. doi: 10.1016/j.ymgme.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Pereira C.V., Peralta S., Arguello T., Bacman S.R., Diaz F., Moraes C.T. Myopathy reversion in mice after restauration of mitochondrial complex I. EMBO Mol. Med. 2020;12:e10674. doi: 10.15252/emmm.201910674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcelli A.M., Ghelli A., Ceccarelli C., Lang M., Cenacchi G., Capristo M., Pennisi L.F., Morra I., Ciccarelli E., Melcarne A. The genetic and metabolic signature of oncocytic transformation implicates HIF1α destabilization. Hum. Mol. Genet. 2010;19:1019–1032. doi: 10.1093/hmg/ddp566. [DOI] [PubMed] [Google Scholar]
- Protasoni M., Pérez-Pérez R., Lobo-Jarne T., Harbour M.E., Ding S., Peñas A., Diaz F., Moraes C.T., Fearnley I.M., Zeviani M. Respiratory supercomplexes act as a platform for complex III-mediated maturation of human mitochondrial complexes I and IV. EMBO J. 2020;39:e102817. doi: 10.15252/embj.2019102817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Caballero L., Guerrero-Castillo S., Nijtmans L. Unraveling the complexity of mitochondrial complex I assembly: a dynamic process. Biochim. Biophys. Acta. 2016;1857:980–990. doi: 10.1016/j.bbabio.2016.03.031. [DOI] [PubMed] [Google Scholar]
- Sánchez-Caballero L., Ruzzenente B., Bianchi L., Assouline Z., Barcia G., Metodiev M.D., Rio M., Funalot B., van den Brand M.A.M., Guerrero-Castillo S. Mutations in complex I assembly factor TMEM126B result in muscle weakness and isolated complex I deficiency. Am. J. Hum. Genet. 2016;99:208–216. doi: 10.1016/j.ajhg.2016.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sazanov L.A. A giant molecular proton pump: structure and mechanism of respiratory complex I. Nat. Rev. Mol. Cell Biol. 2015;16:375–388. doi: 10.1038/nrm3997. [DOI] [PubMed] [Google Scholar]
- Schägger H., Pfeiffer K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000;19:1777–1783. doi: 10.1093/emboj/19.8.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud D.A., Surgenor E.E., Formosa L.E., Reljic B., Frazier A.E., Dibley M.G., Osellame L.D., Stait T., Beilharz T.H., Thorburn D.R. Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature. 2016;538:123–126. doi: 10.1038/nature19754. [DOI] [PubMed] [Google Scholar]
- Szczepanowska K., Senft K., Heidler J., Herholz M., Kukat A., Höhne M.N., Hofsetz E., Becker C., Kaspar S., Giese H. A salvage pathway maintains highly functional respiratory complex I. Nat. Commun. 2020;11:1643. doi: 10.1038/s41467-020-15467-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timón-Gómez A., Garlich J., Stuart R.A., Ugalde C., Barrientos A. Distinct roles of mitochondrial HIGD1A and HIGD2A in respiratory complex and supercomplex biogenesis. Cell Rep. 2020;31:107607. doi: 10.1016/j.celrep.2020.107607. [DOI] [PubMed] [Google Scholar]
- Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- Vinothkumar K.R., Zhu J., Hirst J. Architecture of mammalian respiratory complex I. Nature. 2014;515:80–84. doi: 10.1038/nature13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel R.O., Dieteren C.E.J., van den Heuvel L.P.W.J., Willems P.H.G.M., Smeitink J.A.M., Koopman W.J.H., Nijtmans L.G.J. Identification of mitochondrial complex I assembly intermediates by tracing tagged NDUFS3 demonstrates the entry point of mitochondrial subunits. J. Biol. Chem. 2007;282:7582–7590. doi: 10.1074/jbc.M609410200. [DOI] [PubMed] [Google Scholar]
- Wittig I., Braun H.-P., Schägger H. Blue native PAGE. Nat. Protoc. 2006;1:418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- Zhu J., Vinothkumar K.R., Hirst J. Structure of mammalian respiratory complex I. Nature. 2016;536:354–358. doi: 10.1038/nature19095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the current study related to Figures 3, 4, and S4 are available as Tables S1 and S2. Source data for figures in the paper are available from the corresponding authors on request.