

Abstract
Shigellosis caused by Shigella dysenteriae is a major public health concern worldwide, particularly in developing countries. The bacterial genome is known, but there are many hypothetical proteins whose functions are yet to be discovered. A hypothetical protein (accession no. WP_128879999.1, 161 residues) of S. dysenteriae ATCC 12039 strain was selected in this study for comprehensive structural and functional analysis. Subcellular localization and different physicochemical properties of this hypothetical protein were estimated indicating it as a stable, soluble, and extracellular protein. Functional annotation tools, such as NCBI-CD Search, Pfam, and InterProScan, predicted our target protein to be an amidase effector protein 4 (Tae4) of type-VI secretion system (T6SS). Multiple sequence alignment of the homologous sequences coincided with previous findings. Random coil was found to be predominant in secondary structure. Three-dimensional (3D) structure of the protein was obtained using homology modeling method by SWISS-MODEL server using a template protein (PDB ID: 4J30) of 80.12% sequence identity. The 3D structure became more stable after YASARA energy minimization and was validated by several quality assessment tools like PROCHECK, QMEAN, Verify3D, and ERRAT. Superimposition of the target with the template protein by UCSF Chimera generated RMSD value of 0.115 Å, suggesting a reliable 3D structure. The active site of the modeled structure was predicted and visualized by CASTp server and PyMOL. Interestingly, similar binding affinity and key interacting residues were found for the target protein and a Salmonella enterica Tae4 protein with the ligand L-Ala D-Glu-mDAP by molecular docking analysis. Protein-protein docking was also performed between the target protein and hemolysin coregulated protein 1 of T6SS. Finally, the protein was found to be a unique protein of S. dysenteriae nonhomologous to human by comparative genomics approach indicating a potential therapeutic target. Most pathogens harboring T6SS in their system pose a significant threat to the human health. Many T6SSs and their effectors are associated with interbacterial competition, pathogenesis, and virulency; however, relationships between these effectors and pathogenicity of S. dysenteriae are yet to be determined. The study findings provide a lucrative platform for future antibacterial treatment.
Keywords: Shigella dysenteriae, in silico characterization, hypothetical protein, functional annotation, homology modeling, molecular docking, type-VI secretion system (T6SS), amidase effector protein 4, hcp1
Introduction
Next-generation sequencing (NGS) allows scientists to obtain huge amounts of data in a relatively short period of time. As more and more organisms are being sequenced, the challenge of assigning functions to genes is increasing.1,2 In many organisms, the molecular functions of more than 30% proteins are unknown termed as “Hypothetical Proteins (HP).”3 In silico characterization of hypothetical proteins help in determining 3-dimensional (3D) structures which may reveal new domains and motifs, pathways, protein networks, and so on.4-6 Furthermore, structural and functional annotation of HPs may also reveal potential biomarkers and pharmacological targets.7 Several bioinformatics databases and tools have been successfully used to annotate the functions of hypothetical proteins in different pathogenic micorganisms.8-14
A family of bacteria known as Shigella is responsible for nearly 700 000 deaths a year resulting from an intestinal disease, Shigellosis (Shigella infection).15 There are 4 known species of Shigella which are pathogenic: Shigella flexneri, Shigella boydii, Shigella sonnei, and Shigella dysenteriae.16 Among these 4 species, S. dysenteriae is prominently found in developing countries that can lead to deadly epidemics.17,18 It is a gram-negative, nonspore forming bacillus that survives as a facultative anaerobe. This organism is generally found in contaminated water supplies and in the stool of infected individuals.19
S. dysenteriae ATCC 12039 strain contains 4129 proteins of which nearly 8% are hypothetical.20 In silico analysis of these hypothetical proteins is essential because an understanding of the genome of this organism might contribute to the successful development of a drug or vaccine which is still under development in laboratories. A hypothetical protein (accession no. WP_128879999.1) of S. dysenteriae ATCC 12039 was selected in this study for comprehensive structural and functional analysis using various bioinformatics tools.
Materials and Methods
Sequence retrieval
Sixty five genomes of S. dysenteriae are available in NCBI (http://www.ncbi.nlm.nih.gov/)21 database. A hypothetical protein (accession no. WP_128879999.1) of S. dysenteriae ATCC 12039 strain containing 161 amino acid residues was selected for this study. Primary sequence of the protein was retrieved as FASTA format for subsequent analysis.
Analysis of physicochemical properties
Physical and chemical properties including molecular weight, aliphatic index (AI), extinction coefficients, GRAVY (grand average of hydropathy), and isoelectric point (pI) of the target protein were analyzed using ProtParam (http://web.expasy.org/protparam/)22 tool of ExPASy.
Subcellular localization and solubility prediction
The subcellular location of the hypothetical protein was predicted by CELLO (http://cello.life.nctu.edu.tw/).23 SOSUI (http://harrier.nagahama-i-bio.ac.jp/sosui/)24 calculates average hydrophobicity and determines the solubility of the protein. Any hydrophobic portion of the protein is labeled as transmembrane region.
Function prediction by domain and motif analysis
For domain analysis, NCBI Conserved Domain Search Service (CD Search) (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi),25 Pfam (https://pfam.xfam.org/),26 and InterProScan (http://www.ebi.ac.uk/Tools/services/web/toolform.ebi?tool=iprscan5)27 was used. CD Search identifies the conserved domains present in a protein sequence. It compares a query sequence by performing RPS-BLAST (Reverse Position-Specific BLAST) against position-specific score matrices resulting from conserved domain alignments present in the Conserved Domain Database (CDD). Pfam is a protein family database that includes annotations and multiple sequence alignments generated using hidden Markov models (HMMs).26 Protein sequence motif was analyzed using MOTIF (http://www.genome.jp/tools/motif/) server.
Multiple sequence alignment
A BLASTp search from NCBI (http://www.ncbi.nlm.nih.gov/) against the nonredundant database with default parameters was performed to find the homologues of the protein. Multiple sequence alignment and phylogenetic tree was constructed using Jalview.28
Secondary structure determination
Secondary structure was predicted using the self-optimized prediction method with alignment (SOPMA) (https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_sopma.html).29 We also used PSIPRED (http://bioinf.cs.ucl.ac.uk/psipred/)30 and ENDscript (http://endscript.ibcp.fr/ESPript/ENDscript/)31 to validate the results obtained from SOPMA.
Homology modeling
The 3D structure of the target protein was determined using SWISS-MODEL32 server based on homology modeling. The server automatically performs BLASTp search to identify templates for each protein sequence. From the query result, template protein 4j30.1.A was selected for homology modeling. This is an X-ray diffraction model of a Salmonella typhimurium putative cytoplasmic protein with 80.12% sequence identity which was a reliable score to initiate modeling. The 3D model structure was visualized by BIOVIA Discovery Studio Visualizer (version 20.1.0.19295).
Energy minimization of the model structure
Three-dimensional model structure from SWISS-MODEL server was energy minimized using YASARA force field minimizer.33 It minimizes the energy and gives a more accurate and stable 3D structure of the desired protein.
Quality assessment
The quality of the model structure was evaluated by PROCHECK (https://www.ebi.ac.uk/thornton-srv/software/PROCHECK/),34 Verify3D (https://servicesn.mbi.ucla.edu/Verify3D/),35 QMEAN (https://swissmodel.expasy.org/qmean/)36 programs of ExPASy server of SWISS-MODEL Workspace and ERRAT (https://servicesn.mbi.ucla.edu/ERRAT/).37 Furthermore, the model and the template structure was superimposed and visualized by UCSF Chimera software.38 The Z scores for both the proteins were also estimated by ProSA-web server.39
Active site determination
Computed atlas of surface topography of proteins (CASTp) (http://sts.bioe.uic.edu/castp/) server was used to determine the active site of the protein. The topographical features of a protein are obtained in a detailed, comprehensive, and quantitative manner by CASTp. Active pockets located on protein surfaces and in the interior site of the 3D structure can be precisely located and be measured. Thus, it has become an indispensable platform for prediction of the regions and key residues of protein which interact with ligands.40 The CASTp result was also visualized by PyMOL software.41
Molecular docking analysis
Docking analysis was performed using Autodock Vina (http://vina.scripps.edu/download.html) software.42 It helps study and predict how ligands interact with macromolecules. The ligand used for the docking was L-Alanyl-gamma-D-glutamyl-meso-diaminopimelic acid (a peptidoglycan fragment). The binding affinity of the target protein and a Salmonella enterica Tae4 protein WP_129397493.1 (3D structure was developed by HHpred MODELER)43 with the ligand was obtained using Autodock Vina. Protein-protein docking between the target protein and hemolysin coregulated protein-1 of T6SS was performed by ClusPro 2.0 server.44 Docking result was analyzed using PyMOL and Discovery Studio Visualizer.
Comparative genomics approach
To know if our target hypothetical protein WP_128879999.1 has any resemblance to human, a BLASTp (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins)45 search against Homo sapiens proteome was performed. A threshold E-value (expected value) of .005 and a minimum bit score of 100 was set to filter the hits.
Results
Physicochemical properties and subcellular localization
Several physicochemical properties of the hypothetical protein WP_128879999.1 were estimated by ProtParam tool which is shown in Table 1. The protein was predicted to contain 161 amino acids, possess a molecular weight of 17 589.04, theoretical pI of 7.72, and grand average of hydropathicity (GRAVY) of −0.072. The instability index (II) of the target protein was predicted to be 31.46 classifying the protein as stable.
Table 1.
Physicochemical properties estimated by ProtParam tool.
| No. of amino. acids. | Molecular Weight | Half life | pI | (Asp + Glu) | (Arg + Lys) | Aliphatic index (AI) | Instabiliy index (II) | Grand average of hydropathicity (GRAVY) |
|---|---|---|---|---|---|---|---|---|
| 161 | 17 589.04 | 30 hr | 7.72 | 12 | 13 | 80.43 | 31.46 | −0.072 |
Subcellular localization of a hypothetical protein would be useful to have insight into their function, as different cellular locations represent different functions. This knowledge can also be used to design a drug against the target protein.46 The subcellular localization of our target protein was predicted as “extracellular” by CELLO program. SOSUI server predicted the protein to be a soluble protein.
Protein family and phylogeny analysis
Several annotation tools were used to identify conserved domains and potential function of our target protein. Based on predictions made by NCBI-CD Search, Pfam and InterProScan, the target protein was suggested to contain domain of Tae4 superfamily and is classified as amidase effector protein 4 (Tae4) of type-VI secretion system (T6SS). NCBI-CDD server predicted the Tae4 superfamily domain at 40-139 amino acid residues with an E-value of 5.62e−21. Pfam also predicted the Tae4 superfamily domain at 40-151 amino acid residues. MOTIF found Tae4 superfamily at 41-138 position with an E-value of 1.1e−16.
The BLASTp search against the nonredundant database showed homology (up to 96% sequence similarity) with other known T6SS amidase effector protein 4 from different enterobacteriaceae (Table 2). We also retrieved 2 hypothetical proteins containing Tae4 domain from S. flexneri (Accession No. OUZ56033.1) and S. sonnei (Accession No. PBP02122.1). Multiple sequence alignment of 5 sequences from BLASTp result along with these 2 sequences was completed using Jalview 2.11.1.3 and shown in Figure 1. It is interesting to see that the sequences are also conserved in other Shigella species (WP_094105598.1 and OUZ56033.1 from S. flexneri; PBP02122.1 from S. sonnei) along with the target protein. Russell et al. 2012, described the distribution of Tae4 in bacteria; however, they were unable to discover it in Shigella.47 To address this, we performed a tblastn search using our target hypothetical protein as query against the nonredundant nucleotide database and RefSeq Genome database. Interestingly, we found Tae4 homologs in some Proteobacteria (Serratia marcescens, Pseudomonas fluorescens, Burkholderia multivorans, Burkholderia cepacia, etc.) including Shigella species that could not be identified by Russell et al. (Supplementary files 1 and 2). Zhang et al,48 Plos One 2013, referred to the residues Cys44-His126-Asp137 as the catalytic triad of the Tae4 amidase. However, our target protein lacks His126 and Asp137 and contains Tyr126 and Glu137 instead. Glu137 is also present in another S. enterica amidase WP_023213614.1. Both of the substitutions are conservative which might retain protein’s catalytic activity. The His126 is used for deprotonation of the Cys44, whereas Asp 137 forms hydrogen bond with His126 in the catalytic triad which might be possible by the substituted amino acids in our target protein.49,50 We also compared the 3D conformation of the catalytic triad of an S. enterica Tae4 protein (WP_129397493.1) with our target protein and found similarity between them (Figure 2). Nevertheless, whether these substitutions retain the protein’s catalytic activity or result in loss of function needs to be experimentally validated.
Table 2.
BLASTp result showing similarity between proteins.
| Accession No. | Organism | Protein name | Score | Percent identity | E-value |
|---|---|---|---|---|---|
| |WP_128879999.1| | Shigella dysenteriae | Hypothetical protein | 332 | 100% | 3e−115 |
| |WP_000533466.1| | Enterobacteriaceae | MULTISPECIES: type-VI secretion system amidase effector protein Tae4 | 322 | 96.89% | 4e−111 |
| |WP_001558594.1| | Escherichia coli | type-VI secretion system amidase effector protein Tae4 | 321 | 96.27% | 1e−110 |
| |WP_096858990.1| | Escherichia coli | type-VI secretion system amidase effector protein Tae4 | 321 | 96.27% | 1e−110 |
| |WP_094105598.1| | Shigella flexneri | type-VI secretion system amidase effector protein Tae4 | 321 | 96.27% | 2e−110 |
Figure 1.

MSA among different amidase effector 4 (Tae4) proteins using ClustalOmega algorithm by Jalview software. (Top row—target protein, Rows 2 and 3—Shigella flexneri, Row 4—Shigella sonnei, Row 5—Enterobacteriaceae, Rows 6 and 7—Escherichia coli, and Row 8—Salmonella enterica). Marked white boxes indicate conserved catalytic cysteine (C) and histidine (H) residues typical of amidases. MSA indicate multiple sequence alignment.
Figure 2.

Three-dimensional conformation of the catalytic triad of Salmonella enterica Tae4 protein (WP_129397493.1) (A) and the target hypothetical protein (B) generated by PyMOL software.
A phylogenic tree was constructed using many Tae4 protein sequences by Jalview software. The Tae4 proteins used for phylogenetic tree construction were retrieved from BLASTp results which were mostly from Escherichia coli and S. enterica species. The target protein along with 3 other Shigella Tae4 amidases seems to share the most recent common ancestor with E. coli amidases rather than S. enterica amidases (Figure 3).
Figure 3.

A phylogenetic tree showing evolutionary relationship of the target protein (yellow marked) with other Tae4 proteins. The tree was generated using neighbor joining method based on BLOSUM62 scoring matrix by Jalview software. The values indicate percentage mismatches between 2 nodes (branch length). The target protein along with 3 other Shigella Tae4 amidases (shaded) seems to share the most recent common ancestor with E. coli amidases (blue color) rather than Salmonella enterica amidases (black color).
Structure analysis and model quality assessment
The secondary structure of the protein was predicted by PSIPRED, SOPMA, and ENDscript server. According to SOPMA estimation, the random coil was found to be the most predominant (42.24%) one followed by alpha helix (27.33%), extended strand (19.88%), and beta turn (10.56%). In case of 3 conformational states prediction by SOPMA, the results were found to be random coil (54.66%), alpha helix (26.09%), and extended strand (19.25%). Similar results were obtained from ENDscript (not shown here) and PSIPRED (random coil: 58.38%, alpha helix: 24.84%, and extended strand: 16.77%). Secondary structure of the protein predicted by PSIPRED is shown in Figure 4. Tertiary structure of the protein was obtained from SWISS-MODEL server using the template 4j30.1.A which shows 80.12% sequence identity with the target protein. The structure obtained through SWISS-MODEL is depicted in Figure 5.
Figure 4.

Predicted secondary structure of the target protein using PSI-PRED server.
Figure 5.
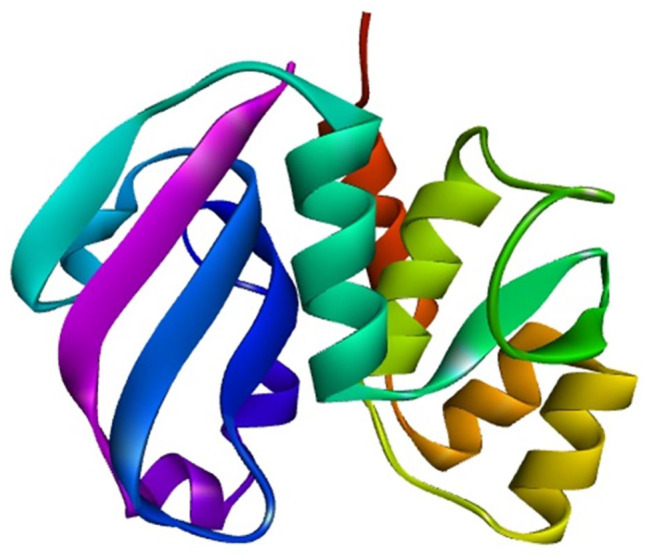
Predicted 3-dimensional structure of the target protein through SWISS-MODEL server after YASARA energy minimization (visualized by BIOVIA Discovery Studio Visualizer version 20.1.0.19295).
YASARA Energy Minimization Server minimized the energy of the model protein structure from −64 076.4 to −85 175.9 kJ/mol. The preliminary score was −0.56 but after energy minimization, the final score turned to be 0.47 indicating a more stable form.
The quality of our modeled 3D structure was assessed by PROCHECK, Verify 3D, QMEAN, and ERRAT program. According to PROCHECK result, 92.3% amino acid residues fell within the most favored region in “Ramachandran plot” (Table 3 and Figure 6A). The model structure successfully passed the Verify 3D server where 93.75% of the residues have averaged 3D-1D score ⩾0.2. QMEAN tool placed the model inside the dark gray zone with QMEAN4 value of 0.49 which is considered as good (Figure 6B). ERRAT also predicted the protein structure to be of good quality with a quality factor of 99.3243.
Table 3.
Ramachandran plot statistics of target protein.
| Statistics | Number of AA residues | Percentage (%) |
|---|---|---|
| Residues in the most favored regions [A, B, L] | 120 | 92.3 |
| Residues in the additional allowed regions [a, b, l, p] | 10 | 7.7 |
| Residues in the generously allowed regions [~a, ~b, ~l, ~p] | 0 | 0.0 |
| Residues in disallowed regions | 0 | 0.0 Total: 100 |
| Number of nonglycine and nonproline residues | 130 | |
| Number of end-residues (excl. Gly and Pro) | 0 | |
| Number of glycine residues (shown as triangles) | 20 | |
| Number of proline residues | 10 | |
| Total number of residues | 160 |
Figure 6.

Quality assessment of the model: (A) Ramachandran plot of model structure validated by PROCHECK program, (B) graphical representation of QMEAN result of the model structure (indicates good agreement between the model structure and experimental structures of similar size).
Superimposition between the model and the template protein (PDB ID: 4J30) is shown in Figure 7. The RMSD (root-mean-square deviation of atomic positions) value obtained from the superimposition in UCSF Chimera was found to be 0.115 Å, suggesting a reliable 3D model. The Z score of the model indicates overall model quality and is used to check whether the input structure is within the range of scores usually found for native proteins of similar size.39 The Z score for the model obtained from ProSA was −6.29 (Figure 8A) and for the template was −5.08 (Figure 8B), proposing the homology between the template and the model.
Figure 7.
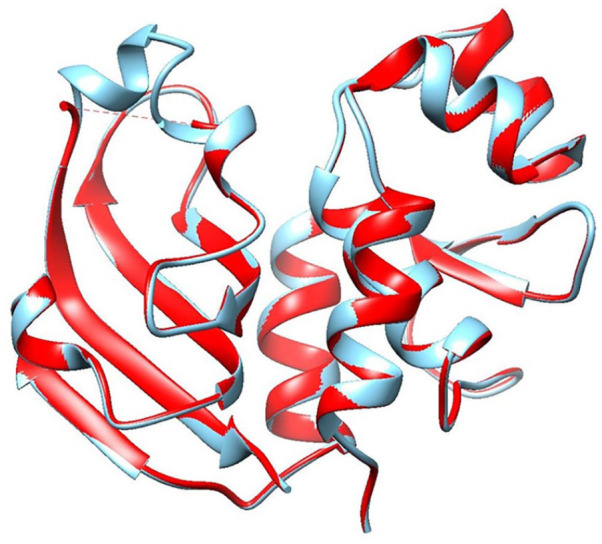
Superimposition of the model (red color) and the template (cyan color) protein using UCSF Chimera software.
Figure 8.

Z scores of the target (A) and template (B) protein using ProSA server. Both of the structure fell in the region typically found for experimentally determined (NMR and X-ray) native proteins of similar size.
Active site determination and molecular docking analysis
The active site of the model structure was analyzed using the CASTp server and the amino acid residues of the active site were also determined. The result was then visualized using PyMOL (Figure 9). Identification and characterization of active site residues is the key step toward the design of a drug or an inhibitor. According to CASTp prediction, active residues of the model protein (of 2 largest active pockets with solvent accessible [SA] area of 229.931 and 108.012, respectively) were found to be Val20, Glu41, Asn42, Ala43, Cys44, Arg47, Val80, Pro101, Thr102, Pro103, Phe106, Ile113, Val115, Trp120, Asn122, Ala123, Gly125, Tyr126, Val127, Thr128, Trp130, Cys135, Glu137, Gln138, His140, Leu141, Leu142, Asp144, Asp146, Asn147, Phe150, and Pro152. These residues lie on the Tae4 superfamily domain of the target protein consistent with the prediction made by NCBI-CD Search, Pfam, and InterProScan (discussed in “Protein family and phylogeny analysis” section). The predicted active residues by CASTp also included the catalytic triad Cys44-Tyr126-Glu137 (Tyr126 instead of His126 and Glu137 instead of Asp137 as discussed earlier) and some conserved Cys and His residues typical of amidases.51
Figure 9.

Determination of active site using CASTp server and visualized (2 largest pockets) in PyMOL (left). Active amino acid residues are highlighted in the right figure.
Docking analysis between the target protein and the ligand was performed using Autodock Vina software. Tae4 protein hydrolyzes peptide crosslinks of the peptidoglycan at the γ-D-glutamyl-mDAP (meso-diaminopimelic acid) LD-bond.47 Hence, the ligand L-Alanyl-gamma-D-glutamyl-meso-diaminopimelic acid was docked with both the target protein and an S. enterica Tae4 protein (WP_129397493.1). A strong binding affinity was found for the ligand with both of the proteins. The binding affinity of the ligand for the model and the S. enterica Tae4 protein was −6.2 kcal/mol and −5.9 kcal/mol with grid box center X: 33.642; Y: 34.998; Z: 10.456 and X: 31.516; Y: 34.278; Z: 3.073, respectively. Many interacting residues in the active site were found to be similar for both of the proteins. The result is also consistent with active site prediction by CASTp. So far, to our knowledge, no crystal structure of Tae4 with its substrate is available till now. Structure of Tae4 with the ligand is needed to further investigate and compare our results. However, the findings were similar to previous studies which strengthen our prediction.49,52 Docking analysis results are shown in Table 4 and Figure 10.
Table 4.
Summary of docking analysis results from Autodock Vina.
| Protein | Ligand | Binding affinity (Kcal/Mol) | Key interacting residues |
|---|---|---|---|
| Hypothetical Protein (WP_128879999.1) | L-Ala D-Glu-mDAP | −6.2 | Phe18, Val20, Asn42, Ala43, Cys44, Arg47, Trp120, Asn122, Tyr126, Glu137, His140, Leu141, Asp144, Asn147 |
| Salmonella enterica Tae4 (WP_129397493.1) | L-Ala D-Glu-mDAP | −5.9 | His18, Val20, Ala43, Cys44, Arg47, Trp120, Asp137, His140, Leu142, Asn143 |
Figure 10.

L-Ala D-Glu-mDAP ligand (red stick) docked in the active site of proteins: (A) ligand-bound hypothetical protein (WP_128879999.1), (B) ligand-bound S. enterica Tae4 protein (WP_129397493.1) (analyzed by PyMOL), (C) key interacting residues of WP_129397493.1 with ligand, and (D) key interacting residues of hypothetical protein with ligand (analyzed by Discovery Studio Visualyzer).
Sana et al53 found that, the Hcp1 (hemolysis coregulated protein) of T6SS in S. typhimurium selectively binds to the Tae4 antibacterial toxin and helps stabilize the effector and allow for proper delivery. However, no hcp1 protein of S. dysenteriae or any other Shigella species is reported so far. Hence, we built a 3D structure of an S. typhimurium hcp1 protein (NP_459274.1) based on homology modeling method by HHpred MODELER43 using a template (PDB ID: 5XHH). The protein was then docked with both the target hypothetical protein and S. enterica Tae4 protein (Accession No. WP_129397493.1) using ClusPro 2.0 server. Docking results are summarized in Table 5. The interactions were then analyzed using PyMOL software (Figure 11). It is to be noted that many interacting residues varied between S. typhimurium and S. dysenteriae probably because the protein-ligand complex conformation from ClusPro server was chosen based on highest cluster members in each case. However, the specific interaction between hcp1 and Tae4 protein is yet to be discovered experimentally.
Table 5.
Summary of docking analysis results from ClusPro server.
| Receptor | Ligand | Cluster members | Weighted energy score of the center |
|---|---|---|---|
| S. typhimurium Hcp1 (NP_459274.1) | Hypothetical protein (WP_128879999.1) | 80 | −720.8 |
| S. typhimurium Hcp1 (NP_459274.1) | S. enterica Tae4 (WP_129397493.1) | 227 | −869.5 |
Figure 11.

Hcp1-Tae4 interaction analysis by PyMOL software resulted from ClusPro server: (A) interaction of Salmonella typhimurium Hcp1 (red) with Salmonella enterica Tae4 (teal), (B) interaction of Salmonella typhimurium Hcp1 (red) with the target hypothetical protein (teal). The interacting residues of Hcp1 and Tae4 are marked in black and blue color, respectively.
Structural and functional annotation of the hypothetical protein being successfully performed, comparative genomics strategy was applied to further characterize our target protein. A BLASTp search against human proteome was performed to identify whether the target protein has any human homologue. The result showed no homology of the target protein to any of the known human protein and was identified as a unique protein of S. dysenteriae. Targeting microbial proteins that are nonhomologous to human proteins would be a suitable drug candidate avoiding any side effect.
Discussion
Researchers are striving to develop a Shigella vaccine, but there are no licensed vaccines available still now. Rapid development of low-cost sequencing technologies has generated vast amount of genomic and proteomic data, although research on hypothetical proteins is yet to keep pace with. Characterization of HPs can help better understand bacterial metabolic pathways, disease progression, drug development, and disease control strategies.54 In this study, various bioinformatics resources were used for structural and functional characterization of the hypothetical protein WP_128879999.1 of S. dysenteriae ATCC 12039 strain. By analyzing physicochemical properties, the protein was estimated to contain 161 amino acids with a molecular weight of 17 589.04, theoretical pI of 7.72, and grand average of hydropathicity (GRAVY) of −0.072 (Table 1). CELLO server predicted this soluble protein to be extracellular. Secondary structure of the protein consists of random coil, alpha helix, beta turn, and extended strand with random coil being the predominant one. Domain and motif analysis predicted our target hypothetical protein to be an amidase effector protein 4 (Tae4) of T6SS by all the annotation tools with high confidence. BLASTp result against the nonredundant database showed up to 96% sequence similarity with other known T6SS amidase effector proteins validating the prediction (Table 2). Tae4 is a new form of toxin-antitoxin system protein of T6SS which is a specialized secretion system recently identified in gram-negative bacteria. Type-VI secretion system is mostly involved in bacterial competition by using it to kill neighboring nonimmune bacteria.55 Bacteria encode cognate immunity proteins (e.g. Tai4) that neutralize the toxic activities of T6SS effectors (e.g. Tae4) to protect themselves from self-intoxication. These antibacterial proteins are secreted directly into the periplasm of the target bacterial cell in a contact-dependent manner. A few T6SSs have been found to play role in pathogenesis, biofilm formation, and macrophage survival.56-58 Several T6SSs have been discovered in many pathogenic E. coli strains that cause persistent diarrhea in children, infants, and immunocompromised individuals.59,60 Recently, T6SS has been found in many E. coli strains with extensive drug resistance (XDR) properties.61 Previously, it was reported that virulent species of Shigella contained T6SS orthologs but not the avirulent ones suggesting its crucial role in imparting pathogenicity to an organism.62 However, relationship between T6SS effectors and pathogenecity of S. dysenteriae is still unknown. Toxic activities of T6SS are mediated by deployment of different effectors including amidases into a neighboring cell.63 These amidases exert antibacterial activities by hydrolyzing the cell wall peptidoglycan.57 Russell et al47 reported the first superfamily of Tae (type-VI amidase effector) consisting 4 families, named Tae1-4. All 4 families contained the conserved catalytic cysteine and histidine residues typical of amidases.51 This also coincides with our findings from the multiple sequence alignments of different amidase effector 4 proteins with the target protein (Figure 1). However, Russell et al. could not report Tae4 in Shigella which might be due to less available genomic sequences at that time or a possible recent emergence of Tae4 protein in those species urging further study. Three-dimensional structure of the protein obtained using SWISS-MODEL server successfully passed all of the model quality assessment tools like PROCHECK, Verify 3D, QMEAN and ERRAT. The 3D structure became more stable after YASARA energy minimization process. Superimposition of the model protein with the template protein (S. typhimurium putative cytoplasmic protein, PDB ID: 4J30) by UCSF chimera also suggested the 3D structure to be reliable with RMSD value of 0.115 Å (discussed in “Structure analysis & model quality assessment” section). The active site amino acid residues computed by CASTp server were consistent with the prediction of functional annotation tools and lie in the Tae4 superfamily domain region. However, 2 conservative substitutions were found in the protein’s catalytic triad (His126Tyr and Asp137Glu) typical of Tae4 amidases. Further experimentations are needed to confirm whether these substitutions retain the protein’s catalytic activity or result in loss of function. Molecular docking was performed by Autodock Vina tool to know the interaction between the target protein with the ligand L-Ala D-Glu-mDAP (peptidoglycan fragment). A strong binding affinity was found for the ligand with the target protein and an S. enterica Tae4 protein further confirming our findings. Many interacting residues in the active site of the proteins were found to be similar (Table 4, Figure 10). A component of T6SS, hemolysin coregulated protein (hcp1) selectively binds Tae4 protein and helps stabilize the effector.53 Protein-protein docking was also performed by S. typhimurium hcp1 protein with the target protein and an S. enterica Tae4 protein (Table 5, Figure 11). Comparative genomics study revealed the protein to be a unique S. dysenteriae protein nonhomologous to human indicating a potential therapeutic target. Interestingly, the sequences were also found to be conserved in other Shigella species, including S. sonnei and S. flexneri, which reinforces the potential of Shigella Tae4 to be used as a treatment target. However, we did not find any homolog in the avirulent species S. boydii which is consistent with previous findings.62 Further research and experimental validations are needed to confirm our findings about this crucial protein. Most pathogens containing a T6SS in their system are an important threat to the human health. While there has been considerable progress in recent years toward understanding the roles of T6SSs, many structural and functional features of T6SSs and their effectors remain unknown. So far, to our knowledge, this is the first study to characterize a T6SS amidase effector of S. dysenteriae from both structural and functional aspects. Annotation of the hypothetical protein like this may help in designing an effective drug/vaccine. The study of individual effectors will be useful to understand antibacterial mechanisms. We underscore the importance of continued research into T6SSs and their effectors not only in Shigella but also in other pathogenic microorganisms to develop future treatment strategies.
Supplemental Material
Supplemental material, sj-xlsx-1-bbi-10.1177_11779322211011140 for In Silico Characterization of a Hypothetical Protein from Shigella dysenteriae ATCC 12039 Reveals a Pathogenesis-Related Protein of the Type-VI Secretion System by Md. Fazley Rabbi, Saiwda Asma Akter, Md. Jaimol Hasan and Al Amin in Bioinformatics and Biology Insights
Supplemental material, sj-xlsx-2-bbi-10.1177_11779322211011140 for In Silico Characterization of a Hypothetical Protein from Shigella dysenteriae ATCC 12039 Reveals a Pathogenesis-Related Protein of the Type-VI Secretion System by Md. Fazley Rabbi, Saiwda Asma Akter, Md. Jaimol Hasan and Al Amin in Bioinformatics and Biology Insights
Acknowledgments
The authors acknowledge the Department of Biotechnology and Genetic Engineering, Noakhali Science and Technology University for providing support to conduct the research work. They also acknowledge the book “Gobeshonay Bioinformatics—1st Part.”
Footnotes
Funding:The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: M.F.R. conceived and designed the experiments, made critical revisions, and approved the final version. M.F.R. and S.A.A. analyzed the data and wrote the first draft of the manuscript. M.F.R., S.A.A., M.J.H., and A.A. reviewed the analysis and contributed to the preparation of the manuscript. All authors reviewed and approved the final manuscript.
ORCID iD: Md. Fazley Rabbi  https://orcid.org/0000-0002-7545-9065
https://orcid.org/0000-0002-7545-9065
Supplemental Material: Supplemental material for this article is available online.
References
- 1. Choi HP, Juarez S, Ciordia S, et al. Biochemical characterization of hypothetical proteins from helicobacter pylori. PLoS ONE. 2013;8:e66605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Morozova O, Marra MA. Applications of next-generation sequencing technologies in functional genomics. Genomics. 2008;92:255-264. [DOI] [PubMed] [Google Scholar]
- 3. Shahbaaz M, Bisetty K, Ahmad F, Hassan MI. Current advances in the identification and characterization of putative drug and vaccine targets in the bacterial genomes. Curr Top Med Chem. 2016;16:1040-1069. [DOI] [PubMed] [Google Scholar]
- 4. Nimrod G, Schushan M, Steinberg DM, Ben-Tal N. Detection of functionally important regions in “hypothetical proteins” of known structure. Structure (London, England: 1993). 2008;16:1755-1763. [DOI] [PubMed] [Google Scholar]
- 5. Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. PNAS. 1999;96:4285-4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Idrees SNS, Kanwal S, Ehsan B, Yousaf A, Nadeem SMIR. In silico sequence analysis, homologymodeling and function annotation of Ocimum basilicum hypothetical protein G1CT28_OCIBA. Int J Bioautomation. 2012;16:111-118. [Google Scholar]
- 7. Lubec G, Afjehi-Sadat L, Yang JW, John JP. Searching for hypothetical proteins: theory and practice based upon original data and literature. Prog Neurobiol. 2005;77:90-127. [DOI] [PubMed] [Google Scholar]
- 8. Turab Naqvi AA, Rahman S, Zeya F, et al. Genome analysis of Chlamydia trachomatis for functional characterization of hypothetical proteins to discover novel drug targets. Int J Biol Macromol. 2017;96:234-240. [DOI] [PubMed] [Google Scholar]
- 9. Naqvi AA, Anjum F, Khan FI, Islam A, Ahmad F, Hassan MI. Sequence analysis of hypothetical proteins from Helicobacter pylori 26695 to identify potential virulence factors. Genomics Inform. 2016;14:125-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shahbaaz M, Hassan MI, Ahmad F. Functional annotation of conserved hypothetical proteins from Haemophilus influenzae Rd KW20. PLoS ONE. 2013;8:e84263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang Z, Zeng X, Tsui SK. Investigating function roles of hypothetical proteins encoded by the Mycobacterium tuberculosis H37Rv genome. BMC Genomics. 2019;20:394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Prava J, G P, Pan A. Functional assignment for essential hypothetical proteins of Staphylococcus aureus N315. Int J Biol Macromol. 2018;108:765-774. [DOI] [PubMed] [Google Scholar]
- 13. Islam MS, Shahik SM, Sohel M, Patwary NI, Hasan MA. In silico structural and functional annotation of hypothetical proteins of Vibrio cholerae O139. Genomics Inform. 2015;13:53-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ferdous N, Reza MN, Emon MTH, Islam MS, Mohiuddin AKM, Hossain MU. Molecular characterization and functional annotation of a hypothetical protein (SCO0618) of Streptomyces coelicolor A3(2). Genomics Inform. 2020;18:e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guidelines for the control of shigellosis, including epidemics due to Shigella dysenteriae type 1.WHO; 2005:2. https://www.who.int/cholera/publications/shigellosis/en/. [Google Scholar]
- 16. Taneja N, Mewara A. Shigellosis: epidemiology in India. Indian J Med Res. 2016;143:565-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Faruque SM, Chowdhury N, Khan R, et al. Shigella dysenteriae type 1-specific bacteriophage from environmental waters in Bangladesh. Appl Environ Microbiol. 2003;69:7028-7031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Levine MM, Kotloff KL, Barry EM, Pasetti MF, Sztein MB. Clinical trials of Shigella vaccines: two steps forward and one step back on a long, hard road. Nat Rev Microbiol. 2007;5:540-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hale TLK, Gerald T. Shigella: structure, classification, and antigenic types. In: Baron S, ed. Medical Microbiology, 4th ed. Galveston, TX: University of Texas Medical Branch; 1996. https://www.ncbi.nlm.nih.gov/books/NBK8038/. [Google Scholar]
- 20. Schroeder MR, Juieng P, Batra D, et al. High-quality complete and draft genome sequences for three Escherichia spp. and three Shigella spp. generated with Pacific Biosciences and Illumina sequencing and optical mapping. Genome Announc. 2018;6:e01384-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Rapp BA, Wheeler DL. GenBank. Nucleic Acids Res. 2002;30:17-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gasteiger E, Hoogland C, Gattiker A, et al. Protein identification and analysis tools on the ExPASy server. In: Walker JM, ed. The Proteomics Protocols Handbook. Totowa, NJ: Humana Press; 2005:571-607. [Google Scholar]
- 23. Yu CS, Chen YC, Lu CH, Hwang JK. Prediction of protein subcellular localization. Proteins. 2006;64:643-651. [DOI] [PubMed] [Google Scholar]
- 24. Hirokawa T, Boon-Chieng S, Mitaku S. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics (Oxford, England). 1998;14:378-379. [DOI] [PubMed] [Google Scholar]
- 25. Lu S, Wang J, Chitsaz F, et al. CDD/SPARCLE: the Conserved Domain Database in 2020. Nucleic Acids Res. 2020;48:D265-D268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. El-Gebali S, Mistry J, Bateman A, et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019;47:D427-D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Quevillon E, Silventoinen V, Pillai S, et al. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33:W116-W120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics (Oxford, England). 2009;25:1189-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Combet C, Blanchet C, Geourjon C, Deléage G. NPS@: network protein sequence analysis. Trends Biochem Sci. 2000;25:147-150. [DOI] [PubMed] [Google Scholar]
- 30. Buchan DWA, Jones DT. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019;47:W402-W407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gouet P, Robert X, Courcelle E. ESPript/ENDscript: extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 2003;31:3320-3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Waterhouse A, Bertoni M, Bienert S, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296-W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Krieger E, Joo K, Lee J, et al. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins. 2009;77:114-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK—a program to check the stereochemical quality of protein structures. J App Cryst. 1993;26:283-291. [Google Scholar]
- 35. Eisenberg D, Lüthy R, Bowie JU. VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997;277:396-404. [DOI] [PubMed] [Google Scholar]
- 36. Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics (Oxford, England). 2011;27:343-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511-1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605-1612. [DOI] [PubMed] [Google Scholar]
- 39. Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007;35:W407-W410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tian W, Chen C, Lei X, Zhao J, Liang J. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res. 2018;46:W363-W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rigsby RE, Parker AB. Using the PyMOL application to reinforce visual understanding of protein structure. Biochem Mol Biol Educ. 2016;44:433-437. [DOI] [PubMed] [Google Scholar]
- 42. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455-461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zimmermann L, Stephens A, Nam SZ, et al. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J Mol Biol. 2018;430:2237-2243. [DOI] [PubMed] [Google Scholar]
- 44. Kozakov D, Hall DR, Xia B, et al. The ClusPro web server for protein-protein docking. Nat Protoc. 2017;12:255-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403-410. [DOI] [PubMed] [Google Scholar]
- 46. Wang J, Sung WK, Krishnan A, Li KB. Protein subcellular localization prediction for Gram-negative bacteria using amino acid subalphabets and a combination of multiple support vector machines. BMC Bioinformatics. 2005;6:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Russell AB, Singh P, Brittnacher M, et al. A widespread bacterial type VI secretion effector superfamily identified using a heuristic approach. Cell Host Microbe. 2012;11:538-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang H, Gao ZQ, Wei Y, Xu JH, Dong YH. Insights into the cross-immunity mechanism within effector families of bacteria type VI secretion system from the structure of StTae4-EcTai4 complex. PLoS ONE. 2013;8:e73782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Benz J, Reinstein J, Meinhart A. Structural insights into the effector—immunity system Tae4/Tai4 from Salmonella typhimurium. PLoS ONE. 2013;8:e67362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Aramini JM, Rossi P, Huang YJ, et al. Solution NMR structure of the NlpC/P60 domain of lipoprotein Spr from Escherichia coli: structural evidence for a novel cysteine peptidase catalytic triad. Biochemistry. 2008;47:9715-9717. [DOI] [PubMed] [Google Scholar]
- 51. Rigden DJ, Jedrzejas MJ, Galperin MY. Amidase domains from bacterial and phage autolysins define a family of gamma-D, L-glutamate-specific amidohydrolases. Trends Biochem Sci. 2003;28:230-234. [DOI] [PubMed] [Google Scholar]
- 52. Zhang H, Zhang H, Gao ZQ, et al. Structure of the type VI effector-immunity complex (Tae4-Tai4) provides novel insights into the inhibition mechanism of the effector by its immunity protein. J Biol Chem. 2013;288:5928-5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sana TG, Flaugnatti N, Lugo KA, et al. Salmonella typhimurium utilizes a T6SS-mediated antibacterial weapon to establish in the host gut. PNAS. 2016;113:E5044-E5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sen T, Verma NK. Functional annotation and curation of hypothetical proteins present in a newly emerged serotype 1c of Shigella flexneri: emphasis on selecting targets for virulence and vaccine design studies. Genes. 2020;11:340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Anderson MC, Vonaesch P, Saffarian A, Marteyn BS, Sansonetti PJ. Shigella sonnei encodes a functional T6SS used for interbacterial competition and niche occupancy. Cell Host Microbe. 2017;21:769-776.e3. [DOI] [PubMed] [Google Scholar]
- 56. Bingle LE, Bailey CM, Pallen MJ. Type VI secretion: a beginner’s guide. Curr Opin Microbiol. 2008;11:3-8. [DOI] [PubMed] [Google Scholar]
- 57. Journet L, Cascales E. The type VI secretion system in Escherichia coli and related species. EcoSal Plus. 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Navarro-Garcia F, Ruiz-Perez F, Cataldi Larzábal ÁM. Type VI secretion system in pathogenic Escherichia coli: structure, role in virulence, and acquisition. Front Microbiol. 2019;10:1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Harrington SM, Dudley EG, Nataro JP. Pathogenesis of enteroaggregative Escherichia coli infection. FEMS Microbiol Lett. 2006;254:12-18. [DOI] [PubMed] [Google Scholar]
- 60. Estrada-Garcia T, Navarro-Garcia F. Enteroaggregative Escherichia coli pathotype: a genetically heterogeneous emerging foodborne enteropathogen. FEMS Immunol Med Microbiol. 2012;66:281-298. [DOI] [PubMed] [Google Scholar]
- 61. Boisen N, Melton-Celsa AR, Scheutz F, O’Brien AD, Nataro JP. Shiga toxin 2a and enteroaggregative Escherichia coli—a deadly combination. Gut Microbes. 2015;6:272-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Shrivastava S, Mande SS. Identification and functional characterization of gene components of Type VI secretion system in bacterial genomes. PLoS ONE. 2008;3:e2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jana B, Salomon D. Type VI secretion system: a modular toolkit for bacterial dominance. Future Microbiol. 2019;14:1451-1463. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, sj-xlsx-1-bbi-10.1177_11779322211011140 for In Silico Characterization of a Hypothetical Protein from Shigella dysenteriae ATCC 12039 Reveals a Pathogenesis-Related Protein of the Type-VI Secretion System by Md. Fazley Rabbi, Saiwda Asma Akter, Md. Jaimol Hasan and Al Amin in Bioinformatics and Biology Insights
Supplemental material, sj-xlsx-2-bbi-10.1177_11779322211011140 for In Silico Characterization of a Hypothetical Protein from Shigella dysenteriae ATCC 12039 Reveals a Pathogenesis-Related Protein of the Type-VI Secretion System by Md. Fazley Rabbi, Saiwda Asma Akter, Md. Jaimol Hasan and Al Amin in Bioinformatics and Biology Insights