

Abstract
Background
Hailey–Hailey disease (HHD; OMIM: 169600) is an autosomal dominate genodermatosis, characterized by recurrent blisters and erosions clinically and remarkable acantholysis pathologically. The underlying pathogenic factor is the mutation of ATP2C1 gene (OMIM: 604384), which encodes secretory pathway Ca2+/Mn2+‐ATPase (SPCA1). Skin folds are the predilection site of HHD. Atypical cases with a generalized pattern have rarely been reported, making it prone to misdiagnosis.
Methods
In this study, we presented three Chinese pedigrees of Hailey–Hailey disease with generalized skin lesions. ATP2C1 mutations were screened by DNA sequencing and their transcripts were further confirmed by minigene assay. We also performed a literature review of previously published generalized HHD over past two decades together with our cases.
Results
Three splice‐site mutations were identified: c.2487+1G>A, c.2126+1G>A, and c.1891‐2A>G, which resulted in an exon 25‐truncated transcript, two exon 22‐truncated transcripts, and two exon 21‐truncated transcripts, respectively. The c.2487+1G>A and the c.1891‐2A>G mutations are novel mutations which have not been reported before. No clustered mutations of ATP2C1 gene were found in generalized HHD patients in literature along with our novel mutations.
Conclusion
We found no hot spot mutations in ATP2C1 correlated with the generalized pattern of HHD. Our study expanded the spectrum of ATP2C1 mutations, which would be useful for disease diagnosis and genetic counseling.
Keywords: ATP2C1 gene, DNA sequencing, generalized Hailey–Hailey disease, splice‐site mutation
The goal of our study was to explore the clinical and genetical features of generalized HHD. In our study, we identified three splice‐site mutations of ATP2C1 gene in three Chinese HHD pedigrees with generalized skin lesions, two of which have never been reported.
![]()
1. INTRODUCTION
Hailey–Hailey disease (HHD), also called familial benign chronic pemphigus (OMIM: 169600), is an uncommon autosomal dominant genodermatosis with complete penetrance. The estimated prevalence and incidence of this disease are 1:40,000 and 1:50,000, respectively (Burge, 1992; Deng & Xiao, 2017; Leducq et al., 2020; Szigeti & Kellermayer, 2006). HHD usually occurs after puberty in the second or third decade of life, with a subsequently fluctuating course. A positive family history was detected in around two‐thirds of HHD cases (Engin et al., 2015). HHD is characterized by erosive skin damage and recurrent blisters, with a predilection for intertriginous regions, such as the groins, axillae, neck, submammary, and perianal regions. The skin lesions often develop into malodorous vegetations and painful fissures, imposing physical and psychological pressure on the patient. Friction, heat, sweating, ultraviolet radiation, and microbial colonization may cause disease exacerbation and persistence (Deng & Xiao, 2017). Histopathologically, this disease is marked by widespread acantholysis starting at the suprabasal level, with mild hyperkeratosis, parakeratosis, and focal crusts, with moderate perivascular lymphocyte infiltration in the superficial dermis. Ultrastructural observations show disruption of the desmosome‐tonofilament complex and perinuclear tonofilament aggregation, leading to loss of cellular adhesion.
The ATPase calcium‐transporting type 2C member 1 gene (ATP2C1, OMIM: 604384) on chromosome 3q22 was first identified as the causative gene underlying HHD by two independent groups in 2000 (Hu et al., 2000; Sudbrak et al., 2000). This 27 exon gene encodes a secretory pathway Ca2+/Mn2+‐ATPase (SPCA1), which is important for maintaining cellular Ca2+ homeostasis. Misfolding or downregulation of this protein impedes Ca2+ sequestration, leading to depletion of Ca2+ in the Golgi lumen. HHD is predisposed to occur in skin folds, and generalized skin lesions spreading to the trunk and extremities are rare. In this study, we screened the ATP2C1 gene in patients in three Chinese HHD pedigrees with generalized skin lesions, and identified three splice‐site mutations of ATP2C1, of which two are novel mutations. Our study expanded the ATP2C1 mutation spectrum of HHD, especially for patients with generalized skin lesions, which would be useful for disease diagnosis and genetic counseling.
2. MATERIALS AND METHODS
2.1. Pedigrees and subjects
We studied three, four‐generation Chinese HHD families. Patients were diagnosed independently by two specialists at the Dermatology Department. Peripheral blood specimens were collected for genetic analysis. Written informed consent was obtained from each participant in line with the Declaration of Helsinki. The study was approved by the Institutional Review Board.
2.2. Mutation screening
Genomic DNA was extracted from the peripheral blood samples using a QIAamp DNA Blood Midi kit (Qiagen). Primers for all exons of the target gene ATP2C1 were designed using Primer3 Input (http://primer3.ut.ee/) (Hu et al., 2000; Sudbrak et al., 2000). All exons of ATP2C1 (NM_014382.3) were amplified by polymerase chain reaction (PCR) and the products were sequenced. The nucleotide sequences were viewed and variants were identified using CodonCode software (version 2.22, Technelysium). The variants were checked for presence in the GnomAD (http://gnomad.broadinstitute.org/), ExAC (http://exac.broadinstitute.org/) (Karczewski et al., 2017), 1000 Genomes (http://browser.1000genomes.org) (Genomes Project et al., 2012), ClinVar (http://www.ncbi.nlm.nih.gov/clinvar) (Landrum et al., 2014), HGMD Professional (http://www.hgmd.cf.ac.uk) (Stenson et al., 2017), and ATP2C1 LOVD v.3.0 databases (http://lovd.nl/ATP2C1) (Nellen et al., 2017).
2.3. In silico analysis
We predicted the pathogenicity of candidate mutations using Human Splicing Finder (http://www.umd.be/HSF3/) (Desmet et al., 2009), Mutation Taster (http://www.mutationtaster.org/) (Schwarz et al., 2014), and GeneSplicer (http://www.cbcb.umd.edu/software/GeneSplicer/gene_spl.shtml) (Pertea et al., 2001). The mutations were classified according to ACMG guidelines (Richards et al., 2015).
2.4. Minigene assay
We used a splicing reporter minigene assay to determine if the mRNA splicing was affected by the candidate mutations, using PCR amplification of wild‐type (WT) and mutant genomic DNA sequences (Cooper, 2005; Gaildrat et al., 2010). Specific primers were designed using In‐Fusion Cloning tools (TaKaRa; https://www.takarabio.com/learning‐centers/cloning/in‐fusion‐cloning‐tools), including the homologous arms of the restriction endonuclease BamH1 (New England Biolabs). The amplified target sequences contained the exon of ATP2C1 including the candidate mutations, together with several hundred base pairs (bp) of the 5′ and 3′ flanking intronic sequences, which were amplified by PCR. The pCAS2 reporter vector (provided by Mario Tosi, Rouen Institute for Biomedical Research) was digested with BamH1 for 3 h and separated by agarose gel electrophoresis to recover the target fragments (TIANgel Midi Purification Kit, TransGen Biotech). The recovered target fragments were then recombined into the digested pCAS2 reporter vector and the recombinant plasmids were transformed into competent Escherichia coli. Monoclonal E. coli were then picked and the plasmids were sequenced to confirm construction of the WT and mutant expression vectors. Finally, the recombinant plasmids were transfected into HeLa cells (Cell Resource Center, Peking Union Medical College) with Lipofectamine 2000 Transfection Reagent (Thermo Fisher Scientific) according to the manufacturer. Forty‐eight hours after transfection, cells were washed twice with PBS and total RNAs were extracted using TRIzol reagent (Life Technologies) and chloroform. The RNAs were used to create cDNAs by Reverse Transcription PCR (RT‐PCR), according to the Reverse Transcription System (Promega) instructions. cDNA sequences, including WT and MT, were amplified by PCR using a forward primer (5′‐GAATTCGTCCGCTGACGTCG‐3′) and a reverse primer (5′‐GCTCTACCACGCCTTCTCAG‐3′), which were located in the pCAS2 reporter vector. The resulting PCR products were visualized by gel electrophoresis and analyzed by further Sanger sequencing.
3. RESULTS
The proband in family 1 (P1, III:4) was a 49‐year‐old man with disease onset in his twenties. Lesions initially occurred at his bilateral groins with erosions and blisters on erythematous plaques, followed by generalized spreading to his bilateral axillae and abdomen (Figure 1a–c). Moist, malodorous vegetations, and fissures were also observed at his bilateral groins. The lesions were exacerbated by heat and sweating. The patient had been treated with topical corticosteroids, with minimal response. Histologic sections of lesions from the right groin showed mild to moderate hyperkeratosis, parakeratosis, acantholysis, intraepidermal cleft, and mild perivascular lymphocytic infiltration (Figure 1d). Dyskeratotic cells were scarce. The patient was diagnosed with HHD, and a detailed family history was obtained from this patient, which revealed similar clinical features in his mother (II:3) and uncle (II:2). In light of his poor response to topical corticosteroids, the patient was treated with oral corticosteroids combined with antibiotics. Sequencing of all exons of the ATP2C1 gene in family 1 identified a novel transition mutation: c.2487+1G>A at the splice donor site of intron 25 in P1 (Figure 2a). In silico analysis was used to predict the splice‐site mutation of this variant, which was further confirmed by minigene assay. Sanger sequencing of the minigene transcripts revealed that the c.2487+1G>A mutation in P1 resulted in skipping of exon 25 of the inserted ATP2C1 gene in the transfected HeLa cells, producing a truncated transcript that was 96 bp shorter than the WT transcript (Figure 2b–d), the corresponding protein change of which is p.Leu798_Gln829del (c.2392_2487del).
FIGURE 1.

Clinical presentation and histopathology of P1 (a) Pedigree of Family 1. Black arrow indicates proband (P1). (b–c) Skin lesions on the abdomen and groin in P1 (d) Histopathology of P1
FIGURE 2.
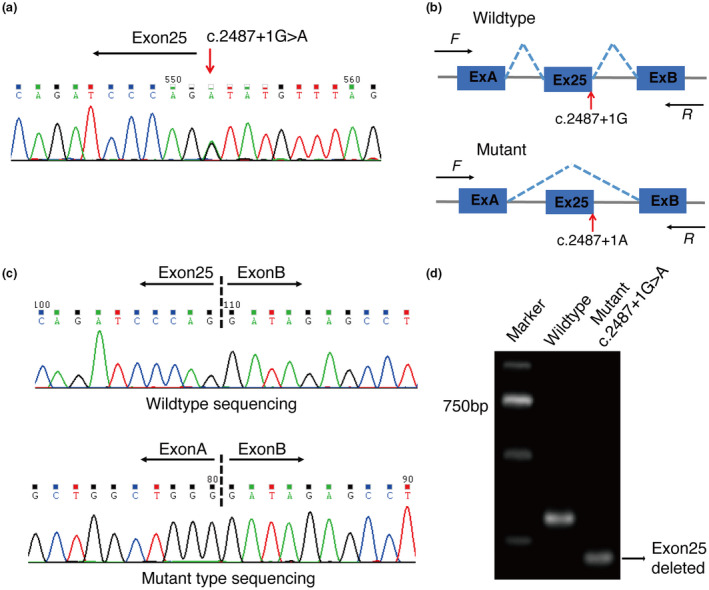
Genetic findings in HHD family 1 (a) A splice‐site mutation c.2487+1G>A of ATP2C1 gene in P1 found by PCR and sequencing. (b) Schematic diagram of the abnormal splicing process caused by c.2487+1G>A mutation. (c) Sanger sequencing of RT‐PCR products of minigene assay identified exon 25‐truncated transcript in the mutant type. (d) RT‐PCR products were separated by electrophoresis
The proband in family 2 (P2, II:2) was a 55‐year‐old man who developed erythema, and macerated and crusted erosions at his bilateral groins, axillae, neck, and backs of his hands at the age of 23 years (Figure 3a–h). Atypical maculopapular lesions were also seen on his lower extremities (Figure 3e,g). The lesions were often aggravated in summer and remitted in winter. Topical corticosteroids combined with antibiotics transiently relieved his symptoms, but frequent recurrence severely limited the patient's quality of life. Histopathological sections obtained from his neck and right thigh showed prominent hyperkeratosis, parakeratosis, acantholysis likened to a “dilapidated brick wall,” and lymphocyte infiltration in the superficial dermis (Figure 4a,b). Hematoxylin and eosin staining also revealed papillomatous proliferation in the lesion on his right thigh, which is an uncommon finding only reported in a few cases of HDD (Figure 4a) (Chauhan et al., 2019; Lu et al., 2019). The patient was diagnosed with HHD with a generalized, symmetrical pattern. Sequencing of ATP2C1 of P2 identified a transition mutation: c.2126+1G>A at the splice donor site of intron 22 (Figure 5a). Sanger sequencing of the minigene transcripts revealed that the c.2126+1G>A mutation in P2 produced two skipped variants: one variant in which the inserted exon 22 of ATP2C1 was skipped, resulting in a 69 bp shorter transcript, and the consequent protein change of p.Ala688_Ser710del (c.2058_2126del), and another variant including a new splice acceptor site generated on exon 23, causing deletion of exon 22, and a partial sequence of the 5′ end of exon 23, resulting in a 142 bp shorter transcript than the WT and the resultant protein change of p.Met686Ilefs*19 (c.2058_2199del) (Figure 5b–d). To the best of our knowledge, two aberrant splice pattern variants from a single mutation in ATP2C1 have rarely been reported (Kono et al., 2018). Four other relatives (I:2, II:3, III:2, and III:5) in this four‐generation family also demonstrated similar clinical characteristics and harbored the same ATP2C1 mutation (Figure 5e).
FIGURE 3.

Clinical presentation of P2 (a) Pedigree of Family 2. Black arrow indicates proband (P2). (b–h) Generalized skin lesions in P2
FIGURE 4.

Histopathology of P2 (a) Histopathology of skin lesion on right thigh of P2. Enlarged view indicates acantholysis likened to a “dilapidated brick wall.” (b) Histopathology of skin lesion on the neck of P2
FIGURE 5.

Genetic findings in HHD family 2 (a) A splice‐site mutation c.2126+1G>A of ATP2C1 gene in family 2 found by PCR and sequencing. (b) Schematic diagram of the abnormal splicing process caused by c.2126+1G>A mutation. (c) Sanger sequencing of RT‐PCR products identified exon 22‐truncated and deletion of exon 22 plus a partial sequence of exon 23's 5′ end in the mutant type. (d) RT‐PCR products were separated by electrophoresis. (e) Sequencing result of I:2, II:3, III:2, and III:5 of family 2
The proband in family 3 (P3, III:3) was a 52‐year‐old man who presented with multiple blisters, erosions, crusts, and pigmentation on his chest, abdomen, perianal region, and bilateral axilla and groins (Figure 6a–f). Histopathological examination of a lesion from his right axilla showed hyperkeratosis, focal crusts, and suprabasal clefting with plenty of acantholytic cells likened to a “dilapidated brick wall.” Dyskeratotic cells could also be seen. Prominent lymphocyte infiltration was observed in the superficial dermis (Figure 6g). The patient was diagnosed with generalized HHD and treated with oral minocycline and nicotinamide. A review of his family history showed that seven other relatives in his four‐generation family (I:2, II:2, II:3, II:5, II:6, III:1, and III:5) had similar clinical characteristics with varying severities. Sanger sequencing of P3 and his unaffected nephew revealed a novel mutation: c.1891‐2A>G at the splice acceptor site of intron 20 in P3 (Figure 7a), which was confirmed to generate two exon 21‐truncated transcripts by minigene assay (Figure 7b–d): one variant in which the inserted exon 21 was skipped, causing a 167 bp shorter abnormal transcript and the corresponding protein change of p.Ser631Valfs*7 (c.1891_2057del), and another variant including a new splice acceptor site generated on exon 21, causing 9‐base‐pair deletion of 5′ end of exon 21, and the resultant protein change, p.Ser631_Gln633del (c.1891_1899del), is an in‐frame 3 amino acids deletion.
FIGURE 6.

Clinical presentation, histopathology, and sequencing in P3 (a) Pedigree of Family 3. Black arrow indicates proband (P3). (b–f) Generalized skin lesions in P3. (g) Histopathology of lesions in P3
FIGURE 7.

Genetic findings in HHD family 3 (a) A splice‐site mutation c.1891‐2A>G of ATP2C1 gene in family 3 found by PCR and sequencing. (b) Schematic diagram of the abnormal splicing process caused by c.1891‐2A>G mutation. (c) Sanger sequencing of RT‐PCR product identified exon 21‐truncated and 9‐base‐pair deletion of exon 21’s 5′ end in the mutant type. (d) RT‐PCR products were separated by electrophoresis
4. DISCUSSION
HHD is caused by mutation of the ATP2C1 gene, encoding SPCA1 (UniProt ID P98194). SPCA1 belongs to the type II phosphorylation (P)‐type Ca2+ transport ATPase family. It is a large transmembrane protein of 919 amino acids, located on the Golgi apparatus. It includes 10 transmembrane domains (M1–10) and three cytosolic domains (actuator domain, phosphorylation domain, and nucleotide‐binding domain) (Deng & Xiao, 2017). The normal abundance and functioning of SPCA1 are critical for maintaining the Golgi ribbon and sequestering Ca2+ in the Golgi lumen (Micaroni et al., 2010). Golgi Ca2+ is known to be important for protein processing (Dürr et al., 1998), and its depletion may impair the processing of junctional proteins necessary for cell‐to‐cell adhesion. ATP2C1 mutations have been reported to decrease SPCA1 protein levels in HHD patients through nonsense mRNA decay or protein misfolding and instability (Fairclough et al., 2004). This further supports the haploinsufficiency theory accounting for the dominant inheritance pattern of HHD. At least 210 ATP2C1 mutations have been reported in the literature to date (Maruyama et al., 2020; Wang et al., 2019). These mutations are distributed uniformly across the gene, with no obvious clusters. Furthermore, no correlations between clinical phenotype and the underlying ATP2C1 mutations have been identified (Deng & Xiao, 2017; Dobson‐Stone et al., 2002; Fairclough et al., 2004; Nellen et al., 2017). Furthermore, the severity of HDD may be exacerbated by external effects, such as drugs, comorbid dermatitis, and bacterial infection.
In this study, we presented three HHD probands from three different Chinese HHD pedigrees, who presented with atypical clinical features involving generalized, rather than intertriginous‐restricted lesions. The examined lesion in the proband from family 2 also showed papillomatous proliferation, which has rarely been reported in the literature (Chauhan et al., 2019; Lu et al., 2019). The lesion was initially confounded by warty dyskeratosis, but a definite diagnosis was made based on ATP2C1 gene mutation. The differential diagnoses of HHD include Darier's disease, pemphigus vulgaris, relapsing linear acantholytic dermatosis, and many types of dermatitis. A review of family history, clinical features, and histological and immunological examinations may be sufficient to diagnose typical HDD skin lesions; however, the presence of atypical lesions may increase the diagnostic difficulty, and screening for pathogenic mutations may be helpful in these cases. In the current study, we identified three splice‐site mutations of the ATP2C1 gene in three Chinese pedigrees: c.2487+1G>A mutation, c.2126+1G>A mutation, and c.1891‐2A>G mutation, all of which were assigned as “Pathogenic” (PVS1, PS3, PM2, PP1, and PP4) based on ACMG classification criteria. Of the three variants, c.2487+1G>A and c.1891‐2A>G mutation were two novel mutations identified in our study. The c.2487+1G>A mutation generated an exon 25‐truncated transcript, c.2126+1G>A mutation generated two exon 22‐truncated transcripts, and c.1891‐2A>G mutation generated two exon 21‐truncated transcripts. Exon 25 has previously been reported to encode the transmembrane 8 (M8) domain of the SPCA1 protein (Deng & Xiao, 2017). Truncation of exon 25 caused by the c.2487+1G>A transition might impair the normal localization of SPCA1 to the Golgi apparatus, resulting in loss of cell‐to‐cell adhesion in the HHD patient described above. Furthermore, given that exon 22 encodes the N‐terminal of the transmembrane 6 (M6) domain and the C‐terminal of the Mn2+‐binding domain (Deng & Xiao, 2017), deletion of exon 22 due to the c.2126+1G>A mutation may impede Mn2+ transport from the cytoplasm to the Golgi apparatus. The same abnormality may result from the c.1891‐2A>G mutation, which caused the deletion of exon 21, given that exon 21 also encodes a Mn2+‐binding domain (Deng & Xiao, 2017). An imbalance of Mn2+ in the cytoplasm and Golgi impairs the detoxification of Mn2+ (Table 1). We reviewed 20 previous cases of generalized HHD reported since the ATP2C1 mutation was first reported in 2000, together with the three current cases, with the aim of identifying genotype–phenotype correlations or mutation hotspots (Databases: Embase, Web of Science, PubMed; search terms: “Hailey‐Hailey disease” OR “HHD” OR “familial benign chronic pemphigus”). The mutations included seven missense mutations, six splice mutations, four frameshift mutations, and five nonsense mutations, and one in‐frame mutation (Figure 8, Table 2) (Chao et al., 2012; Ding et al., 2009; Dobson‐Stone et al., 2002; Hamada et al., 2008; Ikeda et al., 2001; Kono et al., 2018; Leducq et al., 2020; Li et al., 2016; Lu et al., 2019; Mizuno et al., 2014; Nellen et al., 2017; Rácz et al., 2005; Tian et al., 2010; Yasuda et al., 2017; Zhang et al., 2007, 2012). All the mutations were reported or verified to be pathogenic. The missense mutation may not affect the cellular localization of SPCA1, but may reduce its expression level and activity, which would in turn affecting the Ca2+ and Mn2+ transport rates. Partial splice, frameshift, and nonsense mutations may result in a premature stop codon and an abnormally truncated SPCA1. Nonsense‐mediated RNA decay or endoplasmic reticulum‐mediated protein degradation may significantly reduce protein expression levels, while partial splice and frameshift mutations may damage the structural and functional domains of SPCA1, affecting its cellular localization or function. However, this review of the current and previous cases of generalized HHD failed to find any correlation between specific mutations and the generalized phenotype; the mutations were scattered throughout the ATP2C1 gene, and no mutation hotspots were identified (Figure 8). This suggests that external factors rather than intrinsic mutations may be responsible for the generalized skin lesions observed in some patients with HHD.
TABLE 1.
Three novel mutations of ATP2C1 gene identified in three Chinese HHD pedigrees
| Pedigree ID | Mutation site | Mutation type | Putative SPCA1 domain | Transcript 1 | Transcript 2 | Classification ACMG | ||
|---|---|---|---|---|---|---|---|---|
| cDNA change | AA change | cDNA change | AA change | |||||
| Family 1 | c.2487+1G>A | Splice‐site mutation | M8 | c.2392_2487del | p.Leu798_Gln829del | — | — | Pathogenic (PVS1, PS3, PM2, PP1, PP4) |
| Family 2 | c.2126+1G>A | Splice‐site mutation | M6 | c.2058_2126del | p.Ala688_Ser710del | c.2058_2199del | p.Met686Ilefs*19 | Pathogenic (PVS1, PS3, PM2, PP1, PP4) |
| Family 3 | c.1891‐2A>G | Splice‐site mutation | Mn2+‐binding domain | c.1891_2057del | p.Ser631Valfs*7 | c.1891_1899del | p.Ser631_Gln633del | Pathogenic (PVS1, PS3, PM2, PP1, PP4) |
FIGURE 8.

Schematic representation of ATP2C1 gene mutations found in reported Hailey–Hailey disease patients with generalized skin lesions
TABLE 2.
Pathogenic ATP2C1 mutations and phenotypes in patients with generalized HHD
| No | Sex | Sporadic/familiar | Clinical characteristics | Nucleotide change | Amino acid change | Location | Protein domain | Mutation type | Novel or reference | |
|---|---|---|---|---|---|---|---|---|---|---|
| Age of onset | Lesion distribution | |||||||||
| 1 | m | F | 29 | Back, axillae, groin | c.366T>A | p. Tyr122* | Exon 6 | M2 | Nonsense | Tian et al. (2010) |
| 2 | m | S | 20 | Axillae, back, and groin | c.457C>T | p. Arg153* | Exon 7 | A | Nonsense | Hamada et al. (2008) |
| 3 | m | F | 20 | Groin, neck, back, abdomen, perianal regions, cubital area, popliteal space, axillae, and trunk | c.506T>A | p. Val169Glu | Exon 7 | A | Missense | Lu et al. (2019) |
| 4 | f | S | 22 | Extending to chest and back | c.659G>A | p. Gly220Glu | Exon 8 | A | Missense | Nellen et al. (2017) |
| 5 | f | F | 35 | Scalp, neck, axillae, groin, finger, abdomen, back, extremities, and trunk | c.832G>A | p. Ile313Lysfs*25 | Exon 10 | M4 | Frameshift | Chao et al. (2012) |
| 6 | m | S | Childhood | Groin, intertriginous areas, occipital areas, and upper back | c.832+2T>C | p. Ala253Glufs*22 | Intron10 | M3 | Splice site | Ikeda et al. (2001) |
| ,7 | f | F | 30 | Extremities and trunk | c.920C>T | p. Pro307Leu | Exon 12 | M4 | Missense | Zhang et al. (2007) |
| 8 | f | F | 45 | Extensive flexural vegetating hyperkeratosis | c.926G>T | p. Gly309Val | Exon 12 | M4 | Missense | Nellen et al. (2017) |
| 9 | m | F | 53 | General keratotic papules and erythema at intertriginous areas | c.951_959delins24 | p. L318‐L320delinsTMCWCYEN | Exon 12 | P | In‐frame | Ikeda et al. (2001) |
| 10 | m | S | 24 | Neck, trunk, and intertriginous areas | c.1085insA | p. Trp363Tyrfs*11 | Exon 13 | P | Frameshift | Rácz et al. (2005) |
| 11 | m | F | 21 | Axillae, chelidon, wrist, finger, popliteal space, groin, and midriff areas | c.1330delC | p. Gln444Lysfs*36 | Exon 16 | P | Frameshift | Li et al. (2016) |
| 12 | m | S | 80 | Itchy generalized rash, recurrent erosions located in flexural areas | c.1535_1536dup | p. Glu513Lysfs*25 | Exon 17 | N | Frameshift | Leducq et al. (2020) |
| 13 | m | S | 48 | Posterior neck, popliteal fossae, back, abdomen, axillae, and thigh | c.1627G>T | p. Gly543* | Exon 18 | N | Nonsense | Yasuda et al. (2017) |
| 14 | m | F | 52 | Chest, abdomen, perianal regions, axillae, and groin | c.1891‐2A>G | — | intron 20 | S5 | Splice site | Novel |
| 15 | f | 4 | Axillae, groin, trunk, arm, and thigh | c.1891‐1G>T | — | Intron 20 | S5 | Splice site | Mizuno et al. (2014) | |
| 16 | m | F | 17 | Axillae, groin, neck, and back | c.1982T>G | p. Met661Arg | Exon 21 | S5 | Missense | Ding et al. (2009) |
| 17 | m | S | 66 | Neck and trunk | c.2126+1G>A | — | Intron 22 | M5 | Splice site | Kono et al. (2018) |
| 18 | F | 29 | Head, periocular, submammary, and perianal regions | c.2132T>G | p. Ile711Arg | Exon 23 | M5 | Missense | Zhang et al. (2012) | |
| 19 | F | 27 | Head, neck, chelidon, popliteal space, axillae, and groin | c.2198A>G | p. Gln733Arg | Exon 23 | M6 | Missense | Zhang et al. (2012) | |
| 20 | f | S | 54 | Knee and forearm | c.2385G>A | p. Trp795* | Exon 24 | I4 | Nonsense | Hanamura et al. (2018) |
| 21 | m | F | 47 | Groin, axillae, and chest | c.2416C>T | p. Arg806* | Exon 25 | I4 | Nonsense | Dobson‐Stone et al. (2002) |
| 22 | m | F | 20 | Groin, axillae, and abdomen | c.2487+1G>A | — | Intron 25 | M8 | Splice site | Novel |
| 23 | f | F | 42 | Groin and axillae, extension to trunk | c.2630‐3C>A | — | Intron 26 | M10 | Splice site | Nellen et al. (2017) |
These findings increase the repertoire of known ATP2C1 mutations and expand the clinical pattern of HHD, with useful implications for disease diagnosis and genetic counseling.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
ETHICS APPROVAL
Written informed consent was obtained from each participant in line with the Declaration of Helsinki. The study was approved by the Institutional Review Board of Peking Union Medical College Hospital, Beijing, China.
CONSENT TO PARTICIPATE
Each participant provided their written informed consent to participate in this study.
CONSENT FOR PUBLICATION
Written informed consent was obtained from each participant for publication.
CODE AVAILABILITY
Not applicable.
ACKNOWLEDGMENT
We thank Susan Furness, PhD, from Liwen Bianji, Edanz Editing China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript. We give the financial credit for the support of Fundamental Research Funds for the Central Universities (3332018025), the National Scientific Data Sharing Platform for Population and Health ‐ Clinical Centre (NCMI‐ABD02‐201709), Beijing Dongcheng District Excellent Talent Support Training project (2019JGM‐5), the National Key Research and Development Program of China Grant (2016YFC0901500), and the Center for Rare Diseases Research, Chinese Academy of Medical Sciences, Beijing, China.
Lu Yang and Qianli Zhang have equal contributions.
Funding informationThis work was supported by grants from the Fundamental Research Funds for the Central Universities (3332018025), the National Scientific Data Sharing Platform for Population and Health ‐ Clinical Centre (NCMI‐ABD02‐201709), Beijing Dongcheng District Excellent Talent Support Training project (2019JGM‐5), the National Key Research and Development Program of China Grant (2016YFC0901500), and the Center for Rare Diseases Research, Chinese Academy of Medical Sciences, Beijing, China.
Contributor Information
Yaping Liu, Email: ypliu@ibms.pumc.edu.cn.
Tao Wang, Email: wangtaopumch@126.com.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- Burge, S. M. (1992). Hailey‐Hailey disease: The clinical features, response to treatment and prognosis. The British Journal of Dermatology, 126, 275–282. [DOI] [PubMed] [Google Scholar]
- Chao, S.‐C. , Lee, J.‐Y.‐Y. , Wu, M.‐C. , & Hsu, M.‐M.‐L. (2012). A novel splice mutation in the ATP2C1 gene in a woman with concomitant psoriasis vulgaris and disseminated Hailey‐Hailey disease. International Journal of Dermatology, 51, 947–951. [DOI] [PubMed] [Google Scholar]
- Chauhan, P. , Meena, D. , Hazarika, N. , Mrigpuri, S. , & Parsad, D. (2019). Generalized Hailey‐Hailey disease with flexural keratotic papules: An interesting presentation and remarkable response with minocycline. Dermatologic Therapy, 32, e12945. [DOI] [PubMed] [Google Scholar]
- Cooper, T. A. (2005). Use of minigene systems to dissect alternative splicing elements. Methods, 37, 331–340. [DOI] [PubMed] [Google Scholar]
- Deng, H. , & Xiao, H. (2017). The role of the ATP2C1 gene in Hailey‐Hailey disease. Cellular and Molecular Life Sciences, 74, 3687–3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet, F.‐O. , Hamroun, D. , Lalande, M. , Collod‐Béroud, G. , Claustres, M. , & Béroud, C. (2009). Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Research, 37, e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, Y. G. , Fang, H. , Lao, L. M. , Jiang, X. J. , & Chen, H. C. (2009). Genetic diagnosis of Hailey‐Hailey disease in two Chinese families: Novel mutations in the ATP2C1 gene. Clinical and Experimental Dermatology, 34, e968–e971. [DOI] [PubMed] [Google Scholar]
- Dobson‐Stone, C. , Fairclough, R. , Dunne, E. , Brown, J. , Dissanayake, M. , Munro, C. S. , Strachan, T. , Burge, S. , Sudbrak, R. , Monaco, A. P. , & Hovnanian, A. (2002). Hailey‐Hailey disease: Molecular and clinical characterization of novel mutations in the ATP2C1 gene. The Journal of Investigative Dermatology, 118, 338–343. [DOI] [PubMed] [Google Scholar]
- Dürr, G. , Strayle, J. , Plemper, R. , Elbs, S. , Klee, S. K. , Catty, P. , Wolf, D. H. , & Rudolph, H. K. (1998). The medial‐Golgi ion pump Pmr1 supplies the yeast secretory pathway with Ca2+ and Mn2+ required for glycosylation, sorting, and endoplasmic reticulum‐associated protein degradation. Molecular Biology of the Cell, 9, 1149–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin, B. , Kutlubay, Z. , Çelik, U. , Serdaroğlu, S. , & Tüzün, Y. (2015). Hailey‐Hailey disease: A fold (intertriginous) dermatosis. Clinics in Dermatology, 33, 452–455. [DOI] [PubMed] [Google Scholar]
- Fairclough, R. J. , Lonie, L. , Van Baelen, K. , Haftek, M. , Munro, C. S. , Burge, S. M. , & Hovnanian, A. (2004). Hailey‐Hailey disease: Identification of novel mutations in ATP2C1 and effect of missense mutation A528P on protein expression levels. Journal of Investigative Dermatology, 123, 67–71. [DOI] [PubMed] [Google Scholar]
- Gaildrat, P. , Killian, A. , Martins, A. , Tournier, I. , Frébourg, T. , & Tosi, M. (2010). Use of splicing reporter minigene assay to evaluate the effect on splicing of unclassified genetic variants. Methods in Molecular Biology, 653, 249–257. [DOI] [PubMed] [Google Scholar]
- Genomes Project, C. , Abecasis, G. R. , Auton, A. , Brooks, L. D. , DePristo, M. A. , Durbin, R. M. , Handsaker, R. E. , Kang, H. M. , Marth, G. T. , & McVean, G. A. (2012). An integrated map of genetic variation from 1,092 human genomes. Nature, 491, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada, T. , Fukuda, S. , Sakaguchi, S. , Yasumoto, S. , Kim, S.‐C. , & Hashimoto, T. (2008). Molecular and clinical characterization in Japanese and Korean patients with Hailey‐Hailey disease: Six new mutations in the ATP2C1 gene. Journal of Dermatological Science, 51, 31–36. [DOI] [PubMed] [Google Scholar]
- Hanamura, T. , Takeichi, T. , Okuno, Y. , Ichikawa, D. , Kono, M. , & Akiyama, M. (2018). Mild case of Hailey‐Hailey disease caused by a novel ATP2C1 mutation. The Journal of Dermatology., 45, e207–e208. [DOI] [PubMed] [Google Scholar]
- Hu, Z. , Bonifas, J. M. , Beech, J. , Bench, G. , Shigihara, T. , Ogawa, H. , Ikeda, S. , Mauro, T. , & Epstein, E. H. (2000). Mutations in ATP2C1, encoding a calcium pump, cause Hailey‐Hailey disease. Nature Genetics, 24, 61–65. [DOI] [PubMed] [Google Scholar]
- Ikeda, S. , Shigihara, T. , Mayuzumi, N. , Yu, X. , & Ogawa, H. (2001). Mutations of ATP2C1 in Japanese patients with Hailey‐Hailey disease: Intrafamilial and interfamilial phenotype variations and lack of correlation with mutation patterns. The Journal of Investigative Dermatology, 117, 1654–1656. [DOI] [PubMed] [Google Scholar]
- Karczewski, K. J. , Weisburd, B. , Thomas, B. , Solomonson, M. , Ruderfer, D. M. , Kavanagh, D. , Hamamsy, T. , Lek, M. , Samocha, K. E. , Cummings, B. B. , Birnbaum, D. , The Exome Aggregation, C. , Daly, M. J. , & MacArthur, D. G. (2017). The ExAC browser: Displaying reference data information from over 60 000 exomes. Nucleic Acids Research, 45, D840–D845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono, M. , Niizawa, M. , Takeichi, T. , Muro, Y. , & Akiyama, M. (2018). Mild Hailey‐Hailey disease cases with aberrant splicing variants of ATP 2 C1 successfully controlled with excimer light. Journal of the European Academy of Dermatology and Venereology, 32, e413–e416. [DOI] [PubMed] [Google Scholar]
- Landrum, M. J. , Lee, J. M. , Riley, G. R. , Jang, W. , Rubinstein, W. S. , Church, D. M. , & Maglott, D. R. (2014). ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Research, 42, D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leducq, S. , Duchatelet, S. , Zaragoza, J. , Ventéjou, S. , de Muret, A. , Eymieux, S. , Blanchard, E. , Machet, L. , Hovnanian, A. , & Kervarrec, T. (2020). A previously unreported frameshift ATP 2C1 mutation in a generalized Hailey–Hailey disease. Journal of the European Academy of Dermatology and Venereology, 34, e118–e120. [DOI] [PubMed] [Google Scholar]
- Li, H. , Chen, L. , Mei, A. , Chen, L. , Xu, Y. , Hu, W. , Dong, Y. , Zhang, Y. , Wang, T. , Liu, D. , & Deng, Y. (2016). Four novel ATP2C1 mutations in Chinese patients with Hailey‐Hailey disease. The Journal of Dermatology, 43, 1197–1200. [DOI] [PubMed] [Google Scholar]
- Lu, Y. , Li, Q. , & Li, R. (2019). Generalized Familial benign chronic pemphigoid. Journal of Clinical Dermatology, 48, 494–497. [Google Scholar]
- Maruyama, A. , Wada, M. , Kondo, Y. , Kira, M. , Nakano, H. , & Katoh, N. (2020). Case of bullous pemphigoid following Hailey‐Hailey disease with novel mutation of the ATP2C1 gene. The Journal of Dermatology, 47, e79–e80. [DOI] [PubMed] [Google Scholar]
- Micaroni, M. , Perinetti, G. , Berrie, C. P. , & Mironov, A. A. (2010). The SPCA1 Ca2+ pump and intracellular membrane trafficking. Traffic, 11, 1315–1333. [DOI] [PubMed] [Google Scholar]
- Mizuno, K. , Hamada, T. , Hashimoto, T. , & Okamoto, H. (2014). Successful treatment with narrow‐band UVB therapy for a case of generalized Hailey‐Hailey disease with a novel splice‐site mutation in ATP2C1 gene. Dermatologic Therapy, 27, 233–235. [DOI] [PubMed] [Google Scholar]
- Nellen, R. G. , Steijlen, P. M. , van Steensel, M. A. , Vreeburg, M. , European Professional, C. , Frank, J. , & van Geel, M. (2017). Mendelian disorders of cornification caused by defects in intracellular calcium pumps: Mutation update and database for variants in ATP2A2 and ATP2C1 associated with Darier disease and Hailey‐Hailey disease. Human Mutation, 38, 343–356. [DOI] [PubMed] [Google Scholar]
- Pertea, M. , Lin, X. , & Salzberg, S. L. (2001). GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Research, 29, 1185–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rácz, E. , Csikós, M. , & Kárpáti, S. (2005). Novel mutations in the ATP2C1 gene in two patients with Hailey‐Hailey disease. Clinical and Experimental Dermatology, 30, 575–577. [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , & Rehm, H. L. ; Committee, A. L. Q. A. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, J. M. , Cooper, D. N. , Schuelke, M. , & Seelow, D. (2014). MutationTaster2: Mutation prediction for the deep‐sequencing age. Nature Methods, 11, 361–362. [DOI] [PubMed] [Google Scholar]
- Stenson, P. D. , Mort, M. , Ball, E. V. , Evans, K. , Hayden, M. , Heywood, S. , Hussain, M. , Phillips, A. D. , & Cooper, D. N. (2017). The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next‐generation sequencing studies. Human Genetics, 136, 665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudbrak, R. , Brown, J. , Dobson‐Stone, C. , Carter, S. , Ramser, J. , White, J. , Healy, E. , Dissanayake, M. , Larrègue, M. , Perrussel, M. , Lehrach, H. , Munro, C. S. , Strachan, T. , Burge, S. , Hovnanian, A. , & Monaco, A. P. (2000). Hailey‐Hailey disease is caused by mutations in ATP2C1 encoding a novel Ca(2+) pump. Human Molecular Genetics, 9, 1131–1140. [DOI] [PubMed] [Google Scholar]
- Szigeti, R. , & Kellermayer, R. (2006). Autosomal‐dominant calcium ATPase disorders. The Journal of Investigative Dermatology, 126, 2370–2376. [DOI] [PubMed] [Google Scholar]
- Tian, H. , Yan, X. , Liu, H. , Yu, Y. , & Zhang, F. (2010). Six novel ATP2C1 mutations identified in Chinese patients with Hailey‐Hailey disease. Journal of Dermatological Science, 58, 80–82. [DOI] [PubMed] [Google Scholar]
- Wang, Z. , Li, L. , Sun, L. , Mi, Z. , Fu, F. , Yu, G. , Fu, X. , Liu, H. , & Zhang, F. (2019). Review of 52 cases with Hailey‐Hailey disease identified 25 novel mutations in Chinese Han population. The Journal of Dermatology, 46, 1024–1026. [DOI] [PubMed] [Google Scholar]
- Yasuda, H. , Kanazawa, N. , Matsuda, M. , Hamada, T. , Furumura, M. , Hashimoto, T. , Nakama, T. , & Furukawa, F. (2017). A case of Hailey‐Hailey disease with a novel nonsense mutation in the ATP2C1 gene. Annals of Dermatology, 29, 642–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, F. , Yan, X. , Jiang, D. , Tian, H. , Wang, C. , & Yu, L. (2007). Eight novel mutations of ATP2C1 identified in 17 Chinese families with Hailey‐Hailey disease. Dermatology, 215, 277–283. [DOI] [PubMed] [Google Scholar]
- Zhang, H. Z. , Tian, H. Q. , Du, D. H. , Wang, G. J. , Yan, X. X. , Liu, H. , Zhou, G. Z. , Fu, X. A. , Yu, Y. X. , Yu, G. Q. , Liu, H. X. , & Zhang, F. R. (2012). Analysis of ATP2C1 gene mutations in Chinese patients with Hailey‐Hailey disease. Clinical and Experimental Dermatology, 37, 190–193. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.