

Abstract
Myeloid derived suppressor cells (MDSCs) can be subset into monocytic (M-), granulocytic (G-) or polymorphonuclear (PMN-), and immature (i-) or early MDSCs and have a role in many disease states. In cancer patients, the frequencies of MDSCs can positively correlate with stage, grade, and survival. Most clinical studies into MDSCs have been undertaken with peripheral blood (PB); however, in the present studies, we uniquely examined MDSCs in the spleens and PB from patients with gastrointestinal cancers. In our studies, MDSCs were rigorously subset using the following markers: Lineage (LIN) (CD3, CD19 and CD56), human leukocyte antigen (HLA)-DR, CD11b, CD14, CD15, CD33, CD34, CD45, and CD16. We observed a significantly higher frequency of PMN- and M-MDSCs in the PB of cancer patients as compared to their spleens. Expression of the T-cell suppressive enzymes arginase (ARG1) and inducible nitric oxide synthase (i-NOS) were higher on all MDSC subsets for both cancer patients PB and spleen cells as compared to MDSCs from the PB of normal donors. Similar findings for the activation markers lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1), program death ligand 1 (PD-L1) and program cell death protein 1 (PD-1) were observed. Interestingly, the total MDSC cell number exported to clustering analyses was similar between all sample types; however, clustering analyses of these MDSCs, using these markers, uniquely documented novel subsets of PMN-, M- and i-MDSCs. In summary, we report a comparison of splenic MDSC frequency, subtypes, and functionality in cancer patients to their PB by clustering and cytometric analyses.
Keywords: MDSC, SPADE, Flow cytometry, Cancer patient spleen, Cancer patient peripheral blood
1. Introduction
In cancer patients the frequency of MDSCs positively correlate with cancer stage [1, 2] and grade [3], and in some instances, overall survival [3]. Furthermore, the increased MDSC frequency in the PB of cancer patients is associated with tumor secretion of hematopoietic growth factors [4]. Initial studies defined suppressor cells based on their functionality [5, 6]. In the early studies, phenotypic markers for macrophages, T-cells, B-cells, and NK cells were not yet fully available and so these cells were termed null cells [5]. Initially, these cells were phenotypically defined as CD14+ monocytes [6], and later with the availability of more extensive antibody markers, suppressive myeloid cells in head and neck cancer patients were reported to express the progenitor cell marker, CD34 [6]. Later, these myeloid cells were also described as LIN−, lacking markers for CD3, CD19, CD56, and CD13, as well as HLA-DR [7]. In other studies, MDSC subsets were shown to express the myeloid markers CD33 and/or CD11b in addition to being LIN− and HLA-DR−/low [7].
Currently, most researchers define the MDSC phenotype in humans as LIN−HLA-DR−/low and express CD33+ and CD11b+ [8] together with the expression of either the monocytic marker CD14 or a granulocytic marker (CD15 or CD66b) to identify subsets of M- and PMN-MDSCs respectively [7, 9, 10]. Additional markers to assess MDSC differentiation have included CD16 and CD62L [9]. Recent studies have suggested that the expression of LOX-1 can identify PMN-MDSCs [11]; however, we report herein that LOX-1 is expressed in other MDSC subtypes. Presently, in addition to PMN-MDSCs and M-MDSCs an i-MDSC subset can be identified as CD14−CD15− and CD33+ [8].
MDSCs can further be functionally assessed by staining for ARG1 [12], reactive oxygen species (ROS) [13], and i-NOS [12, 13]; whose expression has been directly correlated to T-cell suppression [12, 13]. The substrate for both ARG1 and i-NOS is L-arginine. Hydrolysis of L-arginine by ARG1 decreases T-cell activation [14], proliferation [14] and signaling by the zeta protein of the T-cell receptor [14, 15]. Metabolism of L-arginine by i-NOS produces high levels of nitric oxide (NO) which is cytotoxic to lymphocytes [16]. MDSCs also produce superoxides that rapidly react with many molecules to form ROS such as hydrogen peroxide, peroxynitrate and hydroxyl radicals [17]. ROS’s are additional contributors to the suppressive activity of MDSCs and regulate the immune system by suppressing T-cell function [18],[17].
MDSCs are activated myelopoietic progenitor cells and in some studies have been reported to express PD-L1 [19], which can suppress T-cell function and inhibits T-cell activation via PD-1 binding [20, 21]. Previous studies on myeloid cells suggested that they do not express PD-1; however, PD-1 expression was recently reported on M-MDSCs [22] in patients with treatment-refractory non-Hodgkin’s lymphoma who had a higher frequency of PD-1+M-MDSCs in their PB as compared to patients who responded to treatment or normal donors [22]. This suggest that PD-1 expression on MDSCs could be clinically relevant, as this may reverse therapy resistance to checkpoint inhibitors [22].
Flow cytometric analyses can identify MDSCs as confirmed by functional analyses [23]; however, no biological clustering analyses have been used to further subset MDSC population beyond PMN-, M- or i-MDSCs. Clustering algorithms such as, spanning tree progression analysis (SPADE) [24] and CytoBackBone [25] can be used to identify rare cellular subsets [24]. The SPADE algorithm analyzes multidimensional data acquired from flow cytometry and/or CyTOF and clusters phenotypically similar cell populations into a hierarchically organized branched tree structure [24]. However, SPADE does not allow the inclusion of markers from multiple staining panels, which is possible using CytoBackBone [25]. Both clustering algorithms were used in our studies to assess if additional novel human MDSC subpopulations could be identified.
We report herein a lower frequency of MDSCs in the spleens of cancer patients as compared to their PB. Further, expression of the Fc receptor, CD16 [26, 27], can act as a maturation marker differentiating mature PMNs from immature myeloid cells. In addition, the clustering analyses identified both common and unique MDSCs subpopulations based on the expression of maturational and functional markers. Thus, in addition to extending the MDSC phenotypic studies to include the spleen cells of cancer patients our studies also used both SPADE and CytoBackBone analysis of human MDSCs in the PB and spleens of cancer patients; provided unique insight into potential novel MDSC subsets.
2. Materials and Methods
2.1. Tissue Samples
All samples from patients and donors were obtained after receiving the approval of the Institutional Review Board at the University of Nebraska Medical Center; informed consents were obtained from all donors. Venous blood samples from healthy adult donors were collected in sodium heparin tubes (Becton Dickinson, Franklin Lakes, NJ). Cancer patient PB samples were collected by venous blood draw prior to surgery. Blood samples were kept at 4 °C and stained within two hours of collection. To ensure that all micrometastases are cleared the regional lymph nodes are removed and assessed and a splenectomy is routinely done with a distal pancreatectomy as the spleen and body/tail of the pancreas share the same blood supply. Spleens from cancer patients, eight of whom received neoadjuvant chemotherapy six weeks prior to surgery, were collected from 14 males and 7 females with a mean age of 64 years (Table 1). Patient samples were collected from 11 pancreatic adenocarcinomas, 4 pancreatic neuroendocrine tumors, 1 intraductal papillary mucinous neoplasm, 1 neuroendocrine gastric mucosa, 1 colorectal cancer, 1 colon cancer, 1 lymphoepithelial cyst, and 1 bland, hypocellular, spindle cell lesion. Spleens were removed as early as possible to limit ischemia and cell loss following ligature of the splenic vasculature. Following removal from the patient, the spleen was sterilely macerated into approximately 1-cm3 fragments in the operating room and placed into a sterile 500 mL easy grip polystyrene container (Corning, Corning, NY) with 100 mLs of Hanks Balanced Salt Solution (HBSS) (Gibco, Gaithersburg, MD). Specimen weight was obtained by weighing the container and HBSS prior to and following spleen addition. To analyze total cell populations, processing of splenic tissue was conducted in a Stomacher 80 Biomaster (Seward, Bohemia, NY), a laboratory paddle blender [28–30]. The spleen fragments were placed into a stomacher bag (Seward, Bohemia, NY) with HBSS, and processed into a single cell suspension using a Stomacher 80 Biomaster (Seward, Bohemia, NY). All samples were stained for flow cytometry analysis within 2 hours of collection.
Table 1.
Characteristics of both cancer patients and normal donors (controls).
| Cancer Patient | Controls | |
|---|---|---|
| Age | 64±3 | 35±12 |
| Gender (Male:Female) | 14/7 | 5/5 |
| Platelet Count (103/mm3) | 248±21 | 159±59 |
| Neutrophils | 68±3 | 62±9 |
| Monocytes | 9.4±0.6 | 7.6±1.9 |
| Lymphocytes | 19±2 | 22±9.9 |
| Neoadjuvant (Yes/No) | 8/13 | |
| Grade | ||
| 1 | 4/21 | |
| 2 | 3/21 | |
| 3 | 3/21 | |
| 4 | 2/21 | |
| Not Graded | 9/21 | |
2.2. Multicolor Staining and Acquisition
Membrane phenotypic staining was undertaken on all samples following ammonium-chloride-potassium lysis of red blood cells. Post lyse, cells were washed and blocked with intravenous immunoglobulin G (IVIG) (Grifols, Research Triangle Park, NC) for 30 minutes at 4°C. Post incubation cells were washed, aliquoted at 1×106 cells/ml/tube and stained with monoclonal antibodies (Supplemental Table (ST) 1) per manufacturer recommendation and incubated on ice for 30 minutes in the dark. These staining panels and appropriate fluorochrome-labeled CD45 compensation controls for each panel are shown in ST 2. The samples were stained using the following flow panels as follows: Enzyme Panel - Arginase and i-NOS expression, Panel 2- expression of CTLA-4, CD33, CD34 and LOX-1 and Panel 3- expression of PD-L1 and PD-1 on MDSCs. Post antibody incubation the stained cells were washed and fixed with BD stabilizing fixative per manufacturer’s instructions. Intracellular flow staining was similar to the above procedure; however, after 15 minutes of the 30-minute antibody incubation intracellular solution A (Invitrogen, Camarillo, CA) was added to the intracellular tubes. Post 30-minute antibody incubation, cells were washed, and intracellular solution B (Invitrogen) and the intracellular antibody of interest was added to the intracellular tubes for an additional 20 minutes. Post intracellular staining cells were again washed and fixed. Cells were analyzed on an LSRII Green Flow Cytometer configured with 355-nm, 405-nm, 532-nm, 561-nm, 488-nm, and 633-nm lasers (BD). Internal quality assurance procedures included cytometer setup using tracking beads (BD), that were used according to the manufacturer’s instructions.
2.3. Cytometric Data Analysis
Post acquisition, data was exported and stored using the flow cytometry standard (FCS) 3.1 format and analyzed using FlowJo software version 10.6 (FlowJo, Ashland, OR). Cells were compensated using FlowJo with the CD45+ stained compensation tubes. A representative dot plot analysis of the gating strategy for panel 1 is shown in Supplemental Figure (SF) 1. For flow analysis, all panels were first gated as forward scatter-area (FSC-A) x side scatter-A (SSC-A) to omit dead or apoptotic cells. This was followed by two single-cell gates to omit doublets (FSC-A x FSC-height (H) and SSC-A x SSC-H), followed by a CD45 x SSC-A gate to remove any additional apoptotic cells.
The CD45+ cells were placed on a LIN x SSC-A gate to select LIN− lymphocytes (to remove CD3+ T-cells, CD19+ B-cells, and CD56+ NK cells). A HLA-DR x SSC-A gate was applied to the LIN− cell population to remove macrophages. HLA-DR−/low cells were gated by a CD11b x SSC-A to collect CD11b+ myeloid cells. The CD11b+ cell population was then gated on a CD16 x SSC-A gate to differentiate mature PMN’s (CD16+) from immature MDSCs (CD16−). CD16− cells were separated into CD15+ PMN-MDSC and CD14+ M-MDSC populations. Cells that didn’t express CD15 or CD14 were gated using a CD33 x SSC-A gate to collect CD33+ i-MDSCs. In summary, M-MDSC were defined as CD45+LIN− HLA-DR−/low CD11b+CD16−CD14+CD15−, PMN-MDSC as CD45+LIN− HLA-DR−/lowCD11b+CD16−CD14−CD15+, and i-MDSC as CD45+LIN− HLA-DR−/lowCD11b+CD16−CD14−CD15−CD33+. Monocytes and granulocytes were defined as LIN−CD11b+HLA-DR+CD14+ and LIN−CD11b+HLA-DR+CD15+ respectively. Median fluorescence intensity (MFI) was used to report the extent of marker expression on the cell membrane. MFI was collected using FlowJo software on positive populations.
2.4. Clustering Analysis
In our studies, a SPADE tree consists of two main components which are: nodes and clusters. Each node on a SPADE tree is represented as a circle that varies in size and color, representing both cell frequency (the larger the node the higher the cell frequency) and intensity (MFI) of marker expression (ranging from low to high). Nodes that are phenotypically similar are grouped together to form a cluster that is identified with a number (annotation). Thus, clusters reflect the results of all parameters assessed. The SPADE algorithm clusters the data into a hierarchy such that cell clusters located on the outer aspects of the SPADE tree are less differentiated as compared to the inner clusters.
SPADE analysis was performed using MATLAB (Natick, MA) with the SPADE 3.0 source code and parameters recommended and described by Qui Peng [24]. Each of the three sample types (cancer patients PB and spleen, and normal donors PB) and sources were assessed for each flow tube with images of the corresponding SPADE trees captured. Parameters used for each SPADE analysis were as follows: 1) The target number of nodes was = 200; 2) arcsin transformation was set to 150; and 3) percent down sampling = 3%. Cellular phenotypes were assigned to SPADE clusters based upon the expression of the chosen phenotypic markers. The relative marker expression for each annotation was exported from SPADE and used to generate a heatmap in Excel based upon first through third median quartiles. The fluorochromes from each flow cytometry panel had varying expression levels dependent upon sample type and source (ST 3).
In our initial studies, the total CD45+ cell populations for each sample and panel were analyzed in SPADE; however, the low frequency of MDSCs precluded an in-depth clustering analysis of unique subpopulations. To achieve our objective, the total MDSC cell populations (CD45+LIN− HLA-DR−/lowCD11b+CD16− ) were exported from FlowJo and analyzed using SPADE. This allowed further insight and identification of discrete, unique MDSC subpopulations (clusters). This approach is similar to a study by Anchang et al [31] who applied SPADE to an manually gated population of B-cells to assess B-cell lymphopoiesis.
To further aid in the identification of novel MDSC subsets and to incorporate markers from multiple staining panels, we utilized the CytoBackBone algorithm. CytoBackBone is run in R and merges overlapping markers from multiple staining panels [25]; the source code is available at: https://github.com/tchitchek-lab/CytoBackBone. Unfortunately, the Enzyme panel could not be included as it incorporated intracellular staining, resulting in variable FSC-A and SSC-A. A distance threshold of 3 was used for each merge, FCS files were exported from R and analyzed in SPADE.
2.5. Statistics
All statistics were calculated using SPSS (Statistical Package for Social Sciences, Armonk, NY). Data was first tested for normality using a Shapiro-Wilk test. An independent T-test was used to determine significant difference between unpaired data. To determine significant differences between paired cancer patient PB and spleen cells a paired T-test was conducted. Results are presented as means ± standard error of the mean (SEM). To correct for multiple comparisons, the Benjamini-Hochberg [32] (BH) procedure was used with a false discovery rate of 0.05. An asterisk or other marker is used to designate statistical significance with a BH adjusted P-value ≦ 0.05.
3. Results
3.1. Physical Characterization of MDSC Subpopulations
The frequency (percent), size, and granularity of PMN-, M-, and i-MDSCs from the PB and spleen cells of cancer patients are compared to the PB of normal donors as shown in Fig. 1. PMN-MDSCs, due to their larger size (FSC) and granularity (SSC) have similar light scattering properties to granulocytes. Furthermore, M-MDSC are smaller and less granular than PMN-MDSCs and have a similar FSC and SSC profile similar to monocytes. The smallest and least granular MDSCs are i-MDSCs, which can be subset based on their FSC and SSC profile that is similar to hematopoietic progenitor cells [33]. The frequency of total MDSCs (based on CD45+ cells) in the PB of cancer patients (9.5±1.6), was significantly higher than observed in their spleens (3.3±0.8) or in the PB of normal donors (2.2±0.26) (Fig. 1). Furthermore, PMN-MDSCs as a frequency of CD45+ cells was significantly higher than M-MDSCs; with an increase in cancer patients PB (45.5 fold), cancer patients spleen cells (39 fold) and normal donors PB (8.5 fold) respectively (Fig. 1). Similarly, as a frequency of CD45+ cells we observed a significantly higher frequency of M-MDSCs, but not PMN- or i-MDSCs in the PB of cancer patients as compared to their spleens (Fig. 1).
Figure 1. Comparison of MDSC frequency, size and granularity.

A graphical representation of granulocytic (G), monocytic (M), and immature (i) myeloid derived suppressor cells (MDSC) on an FSC-A x SSC-A plot for normal donor peripheral blood (PB), cancer patient PB, and cancer patient spleen cells. PMN-, M- and i-MDSCs are colored black, red, and green respectively. In the lower table MDSC frequencies are reported as the frequency of CD45+ cells The Benjamini-Hochberg (BH) procedure was used with a false discover rate of 0.05 to correct for multiple comparisons. Significance was determined by an independent T-test with BH adjusted p-value<0.05; * significant difference to cancer spleen, # significant difference to cancer PB.
3.2. Frequency and Absolute Number of Myeloid Cell Subsets
Herein, we report the relative frequencies of myeloid cells and MDSCs, as a frequency of CD45+ cells, in the PB and spleens of cancer patients as compared to normal donors (Fig. 2). The frequency of granulocytes (LIN−CD11b+HLA-DR+CD15+) in the PB of cancer patients was observed to be statistically identical to the PB of normal donors. In addition, we observed a significantly higher frequency of granulocytes in the PB of cancer patients as compared to their spleens. Interestingly a significantly higher frequency of PMN-MDSCs (reported as a frequency of CD45+ cells) was observed in the PB of cancer patients as compared to their spleens. The frequency of PMN-MDSCs and total MDSCs in the PB of cancer patients was significantly higher as compared to the PB of normal donors. Furthermore, the absolute numbers of total MDSCs and PMN-MDSCs were significantly higher in the PB of cancer patients as compared to the PB of normal donors (8.8- and 10.9-fold, respectively). However, we were unable to compare the absolute numbers of myeloid cells and MDSCs for spleens because a section of splenic tissue was used for pathology. Interestingly, the frequency of monocytes, PMN-, and i-MDSC subpopulations were significantly higher in the PB of normal donors and cancer patients as compared to the spleen cells of cancer patients. Indeed, the frequencies of all myeloid cells, including M-MDSCs, was significantly higher in the PB of cancer patients as compared to their spleens; with the exception of hematopoietic progenitor cells (LIN−CD14−CD15−CDllb−CD34+), which were significantly higher in the spleens of cancer patients.
Figure 2. Frequency of CD45+ Cell Types.

A comparison of cellular phenotypes reported as the frequency of CD45+ cells in cancer patients’ peripheral blood (PB), normal donors PB, and cancer patients’ spleens. Absolute numbers of the above cell populations for cancer and normal donor PB samples are shown in the lower table. Hematopoietic progenitor cells are defined as Lin−HLA-Dr−/lowCD11b−CD14−CD15−CD16−CD34+. The Benjamini-Hochberg (BH) procedure was used with a false discover rate of 0.05 to correct for multiple comparisons. Significance was determined by an independent T-test with BH adjusted p-value<0.05; * significant difference to cancer spleen, # significant difference to cancer PB.
3.3. Functional and Maturational Markers on MDSC Subsets
In our studies, we observed a lower frequency of CD33+ PMN-MDSCs (as a percent of PMN-MDSCs) in cancers patients’ PB (82%) and spleens (78%) as compared to the PB of normal donors (87% respectively) (Fig. 3A). Similarly, a lower frequency of CD33+ M-MDSCs (as a percent of M-MDSCs) was observed in cancers patients’ PB (77%) and spleens (74%) as compared to the PB of normal donors (96.9%) respectively (Fig. 3B). CD33 expression could not be assessed on i-MDSCs as they’re phenotypically defined as being CD33+ (Fig. 3C). Reported as the percent of PMN-MDSCs, the frequency of CD34+ PMN-MDSCs in the PB and spleens of cancer patients was statistically identical to normal PB (Fig. 3A). The same was observed for CD34+ M-MDSCs (as a percent of M-MDSCs) for all sample types and sources (Fig. 3B). Reported as a frequency of CD45+ cells, the PB of cancer patients had a higher frequency of i-MDSCs as compared to their spleens (Fig. 2). However, when reported as the frequency of i-MDSCs, the PB of cancer patients had lower frequency of CD34+ i-MDSCs as compared to their spleens (Fig. 3C). In addition, the frequency of CD34+ i- and M-MDSC for each sample type and source was 3.4–5.7-fold and 2.2–3.2-fold lower than PMN-MDSCs respectively. Interestingly, the frequency of CD34+CD33+ M- and i-MDSCs were 2.7–3.3 and 3.3–8.9-fold lower as compared to CD34+CD33+ PMN-MDSCs for each sample type and source. Furthermore, the frequency of CD33+CD34+ PMN-MDSCs (Fig. 3A), M-MDSCs (Fig. 3B), and i-MDSCs (Fig. 3C) (reported as the frequency of PMN-, M-, and i-MDSCs respectively) for any sample type or source was statistically the same.
Figure 3. Expression of activation, myeloid and functional markers on MDSCs.

Reported as the frequency of each individual MDSC subset, based on the expression of activation (D-F), myeloid (A-C), and functional markers (G-I). This is shown for PMN- (A,D,G), M- (B,E,H) and i-(C,F,I) MDSCs in normal peripheral blood (PB), cancer PB, and cancer patient’s spleen cells. The Benjamini-Hochberg (BH) procedure was used with a false discover rate of 0.05 to correct for multiple comparisons. Significance was determined by an independent T-test with BH adjusted p-value<0.05; * significant difference to cancer spleen, # significant difference to cancer PB.
The expression of checkpoint markers (PD-L1 and PD-1) and the scavenger receptor, LOX-1, on MDSCs were also analyzed (Fig. 3D–F). The frequency of PD-L1+ PMN-MDSCs (Fig. 3D), M-MDSCs (Fig. 3E), and i-MDSCs (Fig. 3F) (as a percent of PMN-, M-, and i-MDSCs respectively) were statistically identical for each sample type and source. Furthermore, no significant difference was observed in the frequency of PD-L1+ MDSCs, as a percent of total MDSCs, from any sample type or source (data not shown). The frequency of PD-1+ PMN-MDSCs (as a percent of PMN-MDSCs) in the spleens (2.6±1.3) of cancer patients, but not their PB (1.84±0.73), were significantly higher as compared to the PB of normal donors (0.05±0.02) (Fig. 3D). However, no significant difference was observed in the frequency of PD-1 expression on M- (Fig. 3E) or i-MDSCs (Fig. 3F) for any sample type or source examined.
The frequency of LOX-1+ PMN-MDSCs (as a percent of PMN-MDSCs) in the PB (4.7±0.79) and spleen cells (8.8±2.0) of cancer patients were significantly higher as compared to the PB of normal donors (0.37±0.35) (Fig. 3D). Similarly, the frequency of LOX-1+ M-MDSCs (as a percent of M-MDSCs) in the PB (1.98 ± 0.67) and spleens (1.9±0.58) of cancer patients was significantly higher as compared the PB of normal donors (0±0) (Fig. 3E). As reported previously (Fig. 2), the frequency of i-MDSCs (as a percent of CD45+ cells) was lower in the PB and spleens of cancer patients as compared to the PB of normal donors. However, when reported as a percent of i-MDSCs, a higher frequency of LOX-1+ i-MDSCs in the PB of cancer patients was observed as compared to the PB of normal donors (Fig. 3F). In contrast, no significant difference was observed in the frequency of CTLA-4+ PMN-, M-, or i-MDSCs for any sample type or source (Fig. 3D–F).
We also examined the frequency of MDSCs, which express the functional mediators ARG1 and i-NOS. ARG1+ PMN-MDSCs (as a percent of PMN-MDSCs) in the PB (70.3±6.1) and spleen cells (77.8±16.1) of cancer patients were significantly higher as compared to the PB of normal donors (38.6±10.3) (Fig. 3G). Conversely, the frequency of ARG1+ M- (Fig. 3H) and i-MDSCs (Fig. 3I) (as a percent of M- and i-MDSCs respectively) were identical between each sample type or source. In contrast to the identical frequencies in M-MDSCs between the different PB sources (Fig. 2), there were significantly higher frequencies of i-NOS+ M-MDSCs (as a percent of M-MDSCs) in the PB and spleens of cancer patients as compared to the PB of normal donors (Fig. 3H). Similarly, a significantly higher frequency of i-NOS+ M-MDSCs was observed (as a percent of M-MDSCs) in the spleen cells of cancer patients as compared to their PB (Fig. 3H). Interestingly, as a percent of i-MDSCs, a significantly higher frequency of i-NOS+ i-MDSCs was observed in the PB of cancer patients as compared to the PB of normal donors (Fig. 3I). Despite these differences, no significant differences were observed in ARG1+i-NOS+ PMN-, M-, or i-MDSCs for any sample type or source examined.
3.4. MFI of Functional Markers ARG1 and i-NOS
The MFI of ARG1 expression in the PB and spleen cells of PMN-MDSCs (610±64 and 690±68 respectively) and granulocytes (642±39 and 722±59 respectively) of cancer patients was significantly higher as compared to the PB of normal donors (321±43 and 502±12 respectively) (Table 2). The MFI of ARG1 expression in PMN-MDSCs was statistically similar to granulocytes in the PB and spleen cells of cancer patients. Similarly, the MFI of ARG1 expression for all cellular population studied in the PB and spleen cells of cancer patients was statistically similar. The MFI of i-NOS expression in PMN-MDSCs was significantly higher in the PB of cancer patients (455±140) and normal donors (447±234) as compared to spleens (80±80). Similarly, the MFI of i-NOS expression in granulocytes in the PB of cancer patients (2,729±288) was significantly higher as compared to their spleens (1,924±206), but not the PB of normal donors (2,177±67). However, no significant difference was observed in the MFI of i-NOS in M- or i-MDSCs or monocytes for any sample type or source (Table 2).
Table 2.
The relative median fluorescence intensity (MFI) of Arginase (ARG1+) and i-NOS+ on granulocytes (Grans), monocytes (Mono), and granulocytic (G), monocytic (M), and immature (i)- myeloid derived suppressor cells (MDSCs) in normal peripheral blood (PB), cancer PB, and cancer patient spleen.
| MFI | |||||
|---|---|---|---|---|---|
| Arginase | |||||
| PMN-MDSC | M-MDSC | i-MDSC | Grans | Mono | |
| Normal PB | 321±43*# | 192±88 | 380±29 | 502±12*# | 447±61 |
| Cancer PB | 610±64 | 525±102 | 340±67 | 642±39 | 420±70 |
| Cancer Spleen | 690±68 | 326±131 | 403±87 | 722±59 | 373±65 |
| i-NOS | |||||
| Normal PB | 447±234# | 778±168 | 457±182 | 2,177±67 | 1,252±214 |
| Cancer PB | 455±140# | 1,167±218 | 727±217 | 2,729±288# | 1,859±953 |
| Cancer Spleen | 80±80* | 783±132 | 439±183 | 1,924±206* | 1,394±742 |
The Benjamini-Hochberg (BH) procedure was used with a false discover rate of 0.05 to correct for multiple comparisons. Significance was determined by an independent T-test with BH adjusted p-value<0.05
significant difference to cancer spleen
significant difference to cancer PB.
3.5. Clustering Analysis (SPADE) of Total Cells
In our initial studies, the SPADE algorithm was used to identify the major cellular subsets in total (CD45+) PB leukocytes (Fig 4A). However, because total leukocytes include granulocytes, monocytes, and lymphocytes, SPADE was limited in its ability to extensively differentiate subpopulations within the low frequency of MDSCs. While this approach identified three additional MDSC subsets, it was unable to further subset them (Fig. 4A). However, when we selected total MDSCs using FlowJo for analysis in SPADE, numerous additional MDSC clusters were identified (Fig. 4B).
Figure 4. SPADE tree analysis comparison in the peripheral blood.

SPADE tree comparisons of CD45+ cells versus total MDSC populations. The comparison used total CD45+ cells from a normal donor as a standard cellular source, while the MDSCs were from the peripheral blood (PB) of a cancer patient as a source rich in MDSCs. Annotations of the selected cell populations is shown for SPADE trees from the PB of normal donors and cancer patients. Annotations 1–16 are the CD45+ cells from normal PB, identifying 6 MDSC subpopulations. Annotations 17–28 are representative of the 11 MDSC subpopulations identified in the total MDSC population in the PB of cancer patients.
3.6. Clustering Analyses Identification of Novel MDSC Subsets
SPADE trees were established (Fig. 5 and SF 2–4) for each flow panel. Although, the cell number of MDSCs exported into the clustering analyses were similar between sample types and sources, we noted differences in the frequency of each type of MDSC (Fig. 5). In Fig. 5, a series of SPADE trees are shown demonstrating the results from cell frequency. The SPADE trees indicates the cell frequency ranging from low (blue) to high (red) and follows a hierarchical organization with immature cells on the outer branches and differentiated cells on the inner branches. Consistent with the hierarchical organization of the SPADE trees, progenitor cells are found at a low frequency (i.e. blue) on the outer branches as compared to the more frequency (i.e. red) and differentiated cellular populations on the inner branches (Fig. 5). A comparison of these SPADE trees demonstrate that the subsets of MDSCs differ based upon the sample type, sample source, and flow panel. The numbers (annotations) in the SPADE trees shown in Fig. 5 identify cellular frequencies for cohorts of similar cells, which SPADE identifies as clusters. SPADE trees for each fluorophore, specimen type, source and flow panel were also analyzed (SF 2–4). Furthermore, the MFI of the cells in every annotation in the SPADE tree can also be expressed as a heatmap (ranging from low (blue) to high (red) MFI); providing a more constrained, in-depth analysis (Tables 3–4). The MDSC subpopulations in annotations 1–5 were consistently observed for each sample type, source, and flow panel (Table 3). Annotations 1–5 included two PMN-, two M-, and one i-MDSC population. Consistent with our flow cytometric results (Fig. 1), the heatmap shown in Fig. 5 demonstrates that both PMN-MDSC subpopulations (LOX-1+ and LOX-1Br) have a higher SSC-A and FSC-A value, as compared to either M- or i-MDSCs subpopulations. Similarly, the M-MDSC subpopulations (CD33+ and CD33Dull) have a lower FSC-A and SSC-A value as compared to the PMN-MDSC subpopulations but a greater FSC-A and SSC-A as compared to the i-MDSC subpopulation (Table 3). The MFI expression levels differ across flow panels for each sample type and source preventing a comparison between flow panels, because the relative MFI’s for each fluorophore differ due to the varying antibody markers in each flow panel, sample types and sources.
Figure 5. SPADE analysis of each sample type and panel.

SPADE trees and annotations for the peripheral blood (PB) of normal donors and cancer patients, and cancer patients spleens for all three flow panels (CTLA-4, PD-L1, and enzyme); reported as cell frequency from exported total MDSC population. Annotations 1–5 are representative of cell clusters consistently found for all sample types (Table 3). Annotations 6–12, 13–18, and 19–24 (Table 4) are representative of MDSC subpopulations identified in each sample type and source for flow panels CTLA-4, PD-L1, and enzyme respectively.
Table 3.
Numerically denoted phenotypic marker expression from SPADE analysis of the consistently observed myeloid derived suppressor cell (MDSC) subpopulations (annotations 1–5) in the peripheral blood (PB) of normal donors and cancer patients, and cancer patients’ spleens (Fig. 5). Relative antibody marker expression is shown for all three flow panels (CTLA-4, PD-L1, and enzyme) and ranges from low (blue) to high (red). The median and range of the phenotypic marker expression can be referenced in ST 3.
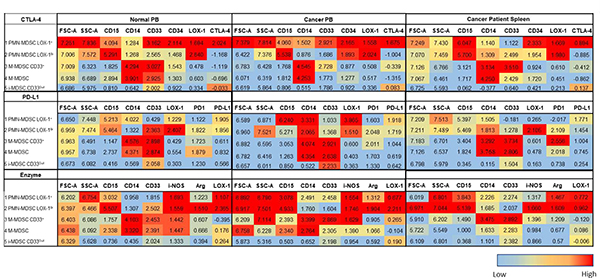 |
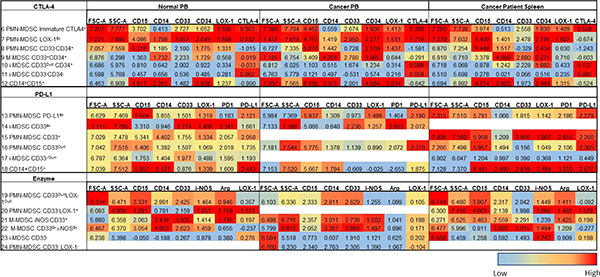
Table 4.
Numerically denoted phenotypic marker expression from SPADE analysis of the myeloid derived suppressor cell (MDSC) subpopulations (annotations 6–24) in the peripheral blood (PB) of normal donors and cancer patients, and cancer patients’ spleens (Fig. 5). Relative phenotypic marker expression is shown for all three flow panels (CTLA-4, PD-L1, and enzyme) and ranges from low (blue) to high (red). The median and range of the phenotypic marker expression can be references in ST 3.
 |
Because CD14 and CD15 are the primary differentiation markers for MDSC subsets, in the SPADE analyses we did not emphasize them instead focusing on the MFI expression of activation, maturation, and functional parameters. Thus, the two consistent PMN-MDSC subpopulations differ in their MFI levels across the flow panels (Table 3). The relative MFI ranges (ST 3) differ for each marker and between sample types and sources, contributing to the ability to differentiate between subpopulations. In the CTLA-4 flow panel, differences were observed between the two PMN-MDSC subpopulations based on the MFI of CD33, CD34 and CTLA-4 for each sample type and source (Table 3). Similar results were also observed in the PD-L1 flow panel for the MFI of PD-1, CD33, and LOX-1 expression (Table 3). In the enzyme flow panel, the two PMN-MDSC subpopulations differed in the MFI expression of CD33, ARG1 and i-NOS (Table 3). In the CTLA-4 flow panel, the CD33 MFI was lower for one PMN-MDSC subpopulation in the PB of normal donors as compared to the PB and spleen cells of cancer patients (Table 3). In contrast, we observed a higher CD34 MFI in the PB and spleen cells of cancer patients as compared to the PB of normal donors (Table 3). Similarly, one PMN-MDSC subpopulation had a higher MFI for PD-1, ARG1, and i-NOS as compared to the second PMN-MDSC subpopulation (Table 3). As shown in Table 3, additional differences in the MFI were observed between the two PMN-MDSC subpopulations but are not discussed.
In the CTLA-4 flow panel, one of the two M-MDSC subpopulations had a higher CD33 MFI for each sample type and source. In contrast, the CD33Dull M-MDSC subpopulation had a higher CD34 MFI expression as compared to the other M-MDSC subpopulation (Table 3). Furthermore, one of the two M-MDSC subpopulations in the PD-L1 flow panel had a higher PD-1 MFI for each sample type and sample source (Table 3). The M-MDSC subpopulations in the enzyme panel also differed in the ARG1 and i-NOS MFI expression levels (Table 3). For each flow panel analyzed, one M-MDSC subpopulation had a higher CTLA-4, PD-1 and i-NOS MFI in the PB and spleen cells of cancer patients as compared to the PB of normal donors (Table 3). In contrast to the differences observed in the PMN- and M-MDSC subpopulations there were few differences in the MFI of these markers in the i-MDSC subpopulation (Table 3).
The results in Table 4 (annotations 6–24) document varying differences in MDSC subpopulations between each sample type, sample source, and flow panels. In the CTLA-4 flow staining panel three PMN-, one M-, and two i-MDSC subpopulations (annotations 6–12) were identified (Table 4). The identity of annotations 6–12 were delineated based upon the MFI expression of CD33 and CD34 (Table 4). Interestingly, in the CTLA-4 panel, immature PMN-MDSCs (CD33−CD34+) for all sample types and sources expressed LOX-1 with low MFI levels of CTLA-4. However, CTLA-4 expression was observed on the mature, CD33+ PMN- and M-MDSC subpopulations for all sample types and sources (Table 4). In contrast to the PMN- and M-MDSC subpopulations, the CTLA-4 MFI was higher on both i-MDSC subpopulations (Table 4). Interestingly, the MFI of CD34 was higher on the spleen cells as compared to the PB of cancer patients and normal donors (Table 4). In the PD-L1 flow staining panel, annotations 13–18 included four PMN-MDSC and one i-MDSC subpopulations, which are differentiated based on the MFI of CD33, LOX-1, PD-1, and PD-L1. One of these PMN-MDSC subpopulations is unique to normal PB, and expresses the immature myeloid marker CD33, a high PD-1 MFI and a dull LOX-1 MFI. The other three PMN-MDSC subpopulations were found in all sample types and had a high PD-L1 MFI. The one i-MDSC subpopulation identified was specific to the PB of normal donors and the spleens of cancer patients (Table 4). Furthermore, the i-MDSC subpopulation had higher MFI levels of CD33, LOX-1, PD-L1 and PD-1 in the PB of normal donors as compared to the spleens of cancer patients (Table 4). Within the three sample types analyzed, all the MDSC subpopulations in the spleen cells had a high MFI levels of PD-L1, while the normal PB had the lowest. In the enzyme flow panel, annotations 19–24 included three PMN-MDSCs, two M-MDSCs, and one i-MDSC subpopulations (Table 4). The MDSC subpopulations were differentiated based on the MFI levels of CD33, ARG1, and i-NOS (Table 4). However, the presence of the various PMN-MDSC subpopulations differed between sample types and sources (Table 4). One PMN-MDSC subpopulation was observed only in the spleen cells of cancer patients and the PB of normal donors; with a high MFI levels of ARG1, i-NOS and LOX-1 (Table 4). The other PMN-MDSC subpopulations were observed only in the PB of cancer patients and had high levels of FSC-A and LOX-1 MFI (Table 4).
The CytoBackBone algorithm allowed us to merge the CTLA-4 and PD-L1 flow panel results, combining unique markers with replicated markers. A CytoBackBone based SPADE tree is shown for each sample type and source in Fig. 6. SPADE trees (SF 5) are color coded according to the MFI levels of the specific marker and flow panel. Each SPADE tree represents one PMN- and three i-MDSC subpopulations (annotations 1–4) that were observed in all sample types and sources (Table 5). The PMN-MDSC subpopulation varied in its MFI expression of CD33, CD34, PD-L1, PD-1 and CTLA-4 but not LOX-1 between sample types and sources (Table 5). The PMN-MDSC subpopulation in the spleen cells from cancer patients had lower CD33 and PD-1 and higher CD34, PD-L1, and CTLA-4 MFI levels as compared to the PB of either cancer patients or normal donors. The i-MDSC subpopulations were differentiated by the MFI levels of LOX-1, CTLA-4, PD-1, CD33, and CD34. Normal PB i-MDSC subpopulations had a higher CTLA-4 MFI as compared to the PB or spleens of cancer patients. Furthermore, the i-MDSC subpopulations in the PB of normal donors had similar LOX-1 MFI levels relative to the spleen cells of cancer patients. The i-MDSC subpopulations in the spleens of cancer patients had a higher CD33 and CD34 MFI levels as compared to the PB of cancer patients or normal donors.
Figure 6. CytoBackBone Analysis.

SPADE trees generated post merging of the CTLA-4 and PD-L1 flow panels using the CytoBackBone algorithm for the peripheral blood (PB) of normal donors, and the spleen cells and PB of cancer patients; reported as cell frequency of the exported total MDSC populations. Annotations 1–4 are representative of clusters consistently identified in all sample types and sources (Table 5). Annotations 5–12, 13–20, and 21–30 are representative of MDSC subpopulations identified in the PB of normal donors, and the PB and spleen cells of cancer patients, respectively (Table 5).
Table 5.
Numerically denoted phenotypic marker expression from CytoBackBone analysis of the myeloid derived suppressor cell (MDSC) subpopulations in the peripheral blood (PB) of normal donors and cancer patients, and cancer patients’ spleens (Fig. 6). Annotations 1–4 are representative of clusters consistently identified in all sample types and sources (Table 5). Annotations 5–12, 13–20, and 21–30 are representative of MDSC subpopulations identified in the PB of normal donors, and the PB and spleen cells of cancer patients, respectively (Table 5). Relative phenotypic marker expression is shown for the merged CTLA-4 and PD-L1 flow panels and ranges from low (blue) to high (red). The median and range of the phenotypic marker expression can be references in ST 4.
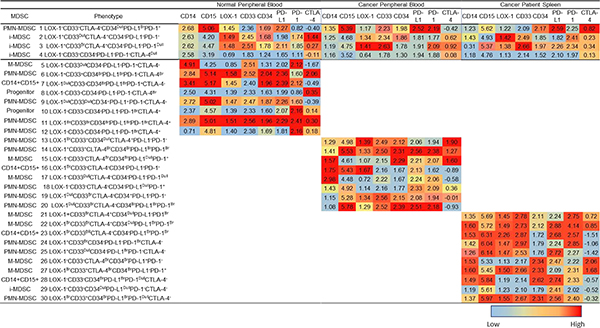 |
MDSC subpopulations which varied between each sample type and source are shown in Table 5. These subpopulations were identified by differences in the MFI levels of CD33, CD34, PD-1, PD-L1 and LOX-1 (annotations 5–30). In the PB of normal donors, MFI expression of CTLA-4 was observed on both PMN- and i-MDSCs, but not M-MDSCs. Conversely, in the PB of cancer patients CTLA-4 was expressed on some of the M-MDSC subpopulations and on all the splenic M-MDSC subpopulations. Consistent with the SPADE analyses, the highest levels of CD33 and CD34 MFI was observed on the spleen cells as compared to the PB of normal donors and cancer patients. Interestingly, two subpopulations of CD14+CD15+ MDSCs are observed in the spleens of cancer patients as compared to one CD14+CD15+ MDSCs in the PB of cancer patients and normal donors (Table 5). Furthermore, the two CD14+CD15+ MDSC subpopulations in the spleens of cancer patients can be differentiated based on the MFI levels of PD-L1 and PD-1 (Table 5). Higher MFI levels of PD-L1 and PD-1 was observed on the MDSC subpopulations in the PB and spleens of cancer patients as compared to the PB of normal donors.
4. Discussion
The phenotype of human MDSCs and their subsets are controversial [10, 34], with few studies focused on the frequency and phenotypes of myeloid cells in secondary lymphoid organs. To date, four papers have reported the frequency of myeloid cells in human spleens [35–38], albeit with limitations. The studies by Aggarwal et al [37] examined spleens from patients who received an urgent splenectomy, resulting in a suboptimal viability (average of 82% ranging from 54–99%). Jordan et al [36] examined spleen cells following a Ficoll-Hypaque gradient separation and cryopreservation. The Ficoll-Hypaque gradient separation results in the removal of myeloid cells, while cryopreservation negatively impacts cellular viability and morphology of granulocytes and MDSCs [39, 40]. Basso et al [35] examined total spleen cells and PB from patients with benign and malignant disease; however, no information was given regarding sample processing. Furthermore, the flow cytometric panel for MDSCs in their study was limited to antibodies against CD33, HLA-DR, CD45, CD14, and PD-L1 or CTLA-4 [35]; omitting the identification of i-MDSCs or more immature (CD16−) phenotype. Lastly, Tavukcuoglu et al [38] studied spleen and PB samples from newly diagnosed gastric and pancreatic cancer patients; however, cellular viability was not reported and the samples underwent density gradient separation prior to analysis [38].
These studies contrast with ours, which analyzed the total leukocyte populations from rapidly procured, unmanipulated spleen and PB samples resulting in a high cell viability (average of 97% ranging from 90–99%). We also utilized a multiparametric flow panel (ST2), that allowed the differentiation of granulocytes, monocytes, and PMN-, M-, and i-MDSCs (Fig. 2). Our analyses incorporated more rigorous flow panels (ST2) than are frequently used [41–43], as well as, maturational and functional markers together with multiple clustering algorithms to identify novel MDSC subsets. We note that our study is not without its limitations, as our control population was not age matched to the cancer patient population studied.
4.1. Myeloid Cells in the Spleens of Cancer Patients
Patients with high tumor burdens are frequently neutrophilic [44, 45] and occasionally present with splenomegaly [46] in part due to extramedullary myelopoiesis [47]. However, we observed significantly fewer granulocytes in the spleens of cancer patients as compared to their PB or the PB of normal donors. These studies are similar to the study by Aggarwal et al [37] that observed a lower frequency of granulocytes in the spleens of cancer patients as compared to their PB, but unfortunately did not conduct a statistical analysis. Tavukcuoglu et al [38] compared the percentage of normal density (ND) and low density (LD) granulocytes, a cell population which are phenotypically consistent with a PMN-MDSC or activated granulocyte [48] in the PB and spleens of cancer patients. However, in their studies the cells underwent a density gradient separation obscuring myeloid cell frequencies. Perhaps because of this experimental design they observed a lower (though not significant) percentage of ND granulocytes in the spleens of cancer patients as compared to their PB. In our studies, a significantly lower frequency of splenic monocytes (5.0±0.79) was observed as compared to the PB of cancer patients and normal donors. This splenic monocyte frequency contrasts with the results from Aggarwal et al’s [37] studies, which observed a lower frequency of splenic monocytes (1.6±1). This difference is likely associated with our focus on spleens from cancer patients versus the analysis of traumatic injury patients in Aggarwal’s [37] studies. In our studies, the frequency of granulocytes and monocytes in the PB of normal donors (45.3% and 8.7% respectively) were statistically identical to the PB of cancer patients (46.8% and 8.4% respectively). The normal leukocyte levels is consistent with a low grade / stage patient population as patients with advanced disease typically present with neutrophilia [44, 49]. Tumor induced neutrophilia is associated with the tumor secretion of hematopoietic growth factors [49–51], which results in extramedullary myelopoiesis [50]. This supports our observation of a significantly higher frequency of hematopoietic progenitor cells in the spleens of cancer patients as compared to their PB; although the spleens of cancer patients had a significantly lower frequency of granulocytes (10.8±2.2%) relative to their PB (46.8±3.9%). To our knowledge, this is the first comparison of the frequency of granulocytes in spleens as compared to the PB. Taken together these hematologic parameters are consistent with the tumors being low grade and/or stage.
We observed a significantly higher frequency of PMN-MDSCs in the PB of cancer patients (9.1±1.6) as compared to their spleens (3.1±0.8) (Fig. 1). In contrast, Jordan et al [36, 38], observed a significantly higher frequency of PMN-MDSCs in the spleens of cancer patients (0.90±0.24) as compared to their PB (0.21±0.41). This might be due to differences in the phenotypic definition of PMN-MDSCs between the studies. Further, parameters in Jorden’s study that might contribute to these differences include Ficoll-Hypaque processing of cells and flow analysis following freeze/thaw storage. The frequency of PMN-MDSCs in the spleen versus the PB in the studies by Tavukcuoglu et al [38] were variable and dependent on the tumor phenotype. Further, in this study [34] gastric cancer patients had a significantly higher frequency of PMN-MDSCs in their spleen (16.51±3.19) as compared to their PB (12.35±3.65). In contrast, pancreatic cancer patients had a similar frequency of PMN-MDSCs in their spleen and PB (13.12±2.55 and 14.31±3.93 respectively), reported in the absence of a statistical analysis [38]. Similarly, no statistical analysis was undertaken for the comparison of the frequency of LD granulocytes in the spleens (25%) of cancer patients as compared to their PB (5%) [34]. Furthermore, the majority of the LD granulocytes in the spleens of cancer patients were band cells, supporting their immaturity [38]. The median frequency of PMN-MDSCs in the PB of cancer patients in our study (7.54%) is similar to results from Choi et al [27] (4.69%) and Khaled et al [51] (8.1%). The differences in our study versus Tavukcuoglu et al [38] may be due to our analysis using whole blood as compared PBMCs isolated by density gradient separation. Tavukcuoglu et al [38] also collected data on newly diagnosed, chemotherapy naïve patients unlike patients in our studies who received neoadjuvant chemotherapy (38%). Furthermore, our flow panel incorporated CD16, which is expressed on mature myeloid cells [26, 52], but not myeloid progenitor cells [53]; improving the discrimination between granulocytes and PMN-MDSCs.
In our studies, a significantly higher frequency of M-MDSCs (2.5-fold) was also observed in the PB of cancer patients as compared to their spleens, which was similar to the results of Jordan et al [36] (2.9-fold). The studies by Tavukcuoglu et al [38] and Basso et al [35] also reported a non-significantly higher frequency of M-MDSCs in the PB of cancer patients as compared to their spleens. In the studies by Basso et al [35], the frequency of M-MDSCs were variable in the PB (0.04 – 21.9%) and spleens (0 – 2.2%) of pancreatic cancer patients and dependent on the extent of “vascular invasion”. We suggest that the differences between these five studies in the frequencies of M- and PMN-MDSCs is attributed to sample processing and flow panel design (CD16 inclusion).
In our studies, a significantly higher frequency of i-MDSCs were observed in the PB of cancer patients versus their spleens. This finding contrasts with a report that examined the frequency of i-MDSCs, [38] in the PB of gastric cancer patients versus their spleens and observed no significant difference. Differing variables between these studies included the patient population (i.e. grade, stage, and chemotherapy regimen), sample processing, and flow panel design. The decreased frequency of i-MDSCs in the spleens of cancer patients observed in our studies is consistent with the decreased frequency of PMN- and M-MDSCs that may be associated with higher CSF levels in cancer patients, driving the extravasation of MDSCs from the spleen into the PB [54].
4.2. Maturation, Activation and Functional Marker Expression on Splenic MDSCs
Immature myeloid cells vary in their expression of myeloid progenitor / stem cell markers, CD33 [10, 55, 56] and CD34 [55, 57]; both expressed on subsets of MDSCs early in their differentiation [10, 55–57]. Furthermore, murine studies have documented that MDSC differentiation is blocked due to tumor secretion of inflammatory mediators [58, 59], limiting apoptosis [60] and allowing proliferation in the absence of terminal differentiation [61, 62]. In our studies, a significantly higher frequency of CD34+ i-MDSCs was observed in the spleens of cancer patients as compared to their PB supporting the spleen in humans as a potential site of MDSC proliferation.
To date, there are no reports on the frequency of PD-L1, PD-1, or LOX-1 expression in the spleens of cancer patients. Indeed, little data has been published regarding activation marker expression on MDSCs in the PB of cancer patients and normal donors [63, 64]. The studies by Iwata et al [63] reported a higher frequency of MDSCs that express PD-L1 in the PB of head and neck cancer patients as compared to the PB of normal donors. In contrast, we observed a higher frequency of MDSCs that expressed PD-L1 in the PB of normal donors as compared to cancer patients. These differences are perhaps due to variations in sample processing, cancer diagnosis or flow panel design. In our studies, the frequency of PMN-MDSCs that were PD-L1+ was higher in the PB of cancer patients as compared to normal donors. Tavukcuoglu et al [38] reported histograms of PD-L1 expression on spleen cells and PB of cancer patients, but did not report their frequency. They also reported a histogram showing PD-L1 expression on PMN-MDSCs in both the spleen and PB of cancer patients; however, this was not quantified [38].
In our studies, the frequency of M-MDSCs that were PD-L1+ was higher in the PB of normal donors as compared to cancer patients. This differs from a report by Okla et al [64], which noted a higher frequency of M-MDSCs that expressed PD-L1 in the PB of cancer patients as compared to normal donors. This was an unexpected observation, as a higher frequency of M-MDSCs expressing PD-L1 in the PB of cancer patients would be expected due to their inflammatory microenvironment. The differences between our studies and Okla et al’s [64] may be due to differences in cancer diagnosis, phenotypic definition of M-MDSC and/or flow panel design. Another important observation in our studies was the higher frequency of i-MDSCs that expressed PD-L1 in the spleens of cancer patients as compared to the PB of normal donors and cancer patients. This higher frequency in the spleens of cancer patients as compared to the PB may be due to the proximity of the spleen to the primary tumor and it’s secretion of inflammatory cytokines.
Similar to the PD-L1 studies, few studies have reported the frequency of PD-1+ expression on myeloid cells in the PB of cancer patients [22,70]. However, there is currently no data on the frequency of PD-1+ expression on myeloid cells in the spleens of cancer patients. In a study of patients with refractory non-Hodgkin’s Lymphoma [22], a significantly higher frequency of M-MDSCs that were PD-1+ was reported in the PB as compared to healthy donors [22]. Similarly, we observed a higher frequency of PMN-, M-, and i-MDSCs that were PD-1+ in the PB and spleen cells of cancer patients as compared to the PB of normal donors, but no differences in PD-1 expression on MDSCs was observed when comparing the PB and spleen of cancer patients. Further, in our studies we did not observe any difference in the MFI of PD-1 expression on granulocytes and monocytes in the PB and spleen cells of normal donors and cancer patients. This differed from the study by MacFarlane et al [65], that reported the MFI of PD-1 expression was decreased on monocytes and neutrophils in the PB of cancer patients.
Previous reports observed a higher frequency of myeloid cells that express LOX-1 in the PB of cancer patients as compared to normal donors [11, 66, 67], such that LOX-1+ expression can discriminate PMN-MDSCs [11, 68]. We note, in these studies [11, 68] that LOX-1+ expression was not assessed on either M- or i-MDSCs. To our knowledge no studies have reported the frequency of LOX-1+ M- or i-MDSCs in cancer patients. In contrast, we report that all MDSC subsets (PMN-, M- and i-MDSCs) and not just PMN-MDSCs, expressed LOX-1. Furthermore, the frequency of PMN- and M-MDSCs that were LOX-1+ was significantly lower in the PB of normal donors as compared to the PB or spleen cells of cancer patients.
The expression of the immunosuppressive mediators ARG1 and i-NOS have been reported to positively correlate with the suppressive function of MDSCs [69–71]. Building on this finding we note that the frequency of ARG1+ PMN-MDSCs was significantly higher in the spleen cells of cancer patients as compared to the PB of normal donors but not the PB of cancer patients. However, the PB and spleens of cancer patients had similar expression of ARG1 on PMN- and M-MDSCs. This suggests that the MDSCs in the PB and spleens of cancer patients are more suppressive as compared to MDSCs in the PB of normal donors. Consistent with this finding, the frequency of M-MDSCs that were i-NOS+ was significantly higher in the spleens and PB of cancer patients as compared to normal PB. These data suggest that despite a significantly lower frequency of MDSCs in the spleens of cancer patients, as compared to their PB, the higher expression of the immunosuppressive mediators (ARG1 and i-NOS) support an immunosuppressive functionality. The higher frequency of PMN-, M-, and i-MDSCs that express ARG1+ and i-NOS+ in the spleens of cancer patients, as compared to their PB, may be due to the proximity of the spleen to the pro-inflammatory tumor environment.
4.3. Clustering Analyses
In addition to being subset based on the myeloid progenitor (CD33) and stem cell marker (CD34), MDSCs can be further subset on their expression of activation (PD-L1, PD-1 and LOX-1) and functional markers (i-NOS and ARG1) using clustering analyses. Although there are published SPADE studies reporting the conventional MDSC subsets there are no SPADE or CytoBackBone analyses that have further subset MDSCs. The sequential use of CytoBackBone following SPADE analysis allows the merger of multiple flow panels; increasing the number of parameters analyzed. Furthermore, no study has reported the use of FlowJo to select and export cells with the MDSC phenotype for further analysis. The one SPADE analysis of human MDSCs was by Roussel et al [72], who analyzed total MDSCs that were differentiated in vitro with GM-CSF and/or IL-6 from human PB and bone marrow and may not be an accurate representation of circulating MDSCs [72].
Of the novel MDSC subtypes identified by SPADE, five were common to each specimen type and source. In addition to these consistently observed novel MDSC subsets, there were nineteen that varied depending on the sample type, source, and panel. The spleen cells of cancer patients had a higher frequency of CD34 expression on PMN-, M- and i-MDSCs as compared to the PB of cancer patients and normal donors. This supports the spleen as a site of EMM and MDSC proliferation in humans, similar to mice [73]. The frequency of CD33 expression, a marker of myeloid committed progenitor cells was lower on M-MDSCs but not PMN- or i-MDSCs in the PB and spleens of cancer patients as compared to normal PB; a finding supported by Roussel et al’s study [72]. The expression of all other markers studied was similar on the i-MDSCs between specimens and tissue types. Consistent with the higher frequency of functional marker expression on all MDSC subsets, the activation and immaturity markers were also increased in the spleen and PB of cancer patients as compared to the PB of normal donors. The increase in functional marker expression could be due to the higher serum level of inflammatory mediators in cancer PB [74, 75].
We also used CytoBackBone, to combine the results from two different flow panel tubes to increase our N and markers for analysis. These studies again supported that multiple, novel subsets of MDSCs can be identified based upon tissue source and markers of maturity and activation. In our CytoBackBone analysis, the overlapping novel MDSC subsets identified in each specimen and tissue type were limited to PMN- and i-MDSCs. This contrasts with our SPADE analysis, where there were overlapping PMN-, M-, and i-MDSC subpopulations. The lack of overlapping M-MDSC subpopulations in CytoBackBone may be associated with increased variability in the activation and functional marker expression by M-MDSCs. However, similar to our SPADE analysis we observed higher frequency of CD34 expression on PMN-, M-, and i-MDSCs in the spleen cells of cancer patients as compared to their PB and the PB of normal donors. Furthermore, CytoBackBone allowed us to differentiate the i-MDSC subpopulations based on their expression of activation and maturation markers. This approach of combining multiple sets of staining parameters and CytoBackBone analysis allowed us to further discriminate between the M- and i-MDSC populations.
In conclusion, the frequency of MDSCs and their expression of both maturational and activation markers differ between individuals and tissue types. We used unique staining panels and clustering analyses, in our studies, to identify novel MDSC subsets based upon maturational, functional, and activation marker expression. Some of these differences were independent of tissue type or source with others dependent on the markers used in each staining panel. Although, the number of MDSCs in our clustering analyses were similar between each sample type and source, we observed multiple novel MDSC subsets. This observation warrants future investigation to determine if there is a correlation between these subsets and patient outcomes or clinical response Further, using flow cytometry, we observed a higher frequency of MDSCs in the PB of cancer patients as compared to their spleens; with MDSCs in the spleens of cancer patients having a more immature, although highly functional, phenotype as compared to their PB. These data provide an in-depth and unique analysis of novel MDSCs in both the PB and spleens of cancer patients as compared to the PB of normal donors.
Supplementary Material
Supplemental Figure 1. Flow gating strategy. Our flow gating strategy for differentiating granulocytic (G), monocytic (M), and immature (i) myeloid derived suppressor cells (MDSC).
Supplemental Figure 2. CTLA-4 flow panel SPADE analysis. SPADE trees and the relative phenotypic marker intensities for each antibody marker in the CTLA-4 flow panel for the total exported MDSC populations from the peripheral blood (PB) of normal donors and the PB and spleen cells of cancer patients.
Supplemental Figure 3. PD-L1 flow panel SPADE analysis. SPADE trees and the relative phenotypic marker intensities for each antibody marker in the PD-L1 flow panel for the total exported MDSC populations from the peripheral blood (PB) of normal donors and the PB and spleen cells of cancer patients.
Supplemental Figure 4. Enzyme flow panel SPADE analysis. SPADE trees and the relative phenotypic marker intensities for each antibody marker in the enzyme flow panel for the total exported MDSC populations from the peripheral blood (PB) of normal donors and the PB and spleen cells of cancer patients.
Supplemental Figure 5. CytoBackBone SPADE analysis. SPADE trees and the relative phenotypic marker intensities for each antibody marker for the total exported MDSC populations of the merged flow panels CTLA-4 and PD-L1. These studies were from the peripheral blood (PB) of normal donors and the PB and spleen cells of cancer patients.
Cancer patient spleen a source of immature myeloid cells
Higher frequency of MDSCs in cancer patient’s peripheral blood versus their spleen
Clustering analysis reveals novel MDSC subsets
Checkpoint protein expression is higher in peripheral blood versus spleen
Acknowledgements
This work was supported, in whole or in part, by National Institutes of Health Grants for Specialized Programs of Research Excellence, Grant P50 CA127297 and by the Fred & Pamela Buffett Cancer Center Support Grant P30 CA036727.We also greatly appreciate the insightful commentary and support provided by Peng Qiu, regarding the use of SPADE.
Abbreviations:
- PB
peripheral blood
- MDSC
myeloid derived suppressor cells
- PMN
polymorphonuclear cells
- LIN
lineage
- ARG1
arginase
- i-NOS
inducible nitric oxide synthase
- LOX-1
low-density lipoprotein receptor −1
- PD-L1
program death ligand 1
- PD-1
program cell death protein 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Wang L, Chang EW, Wong SC, Ong SM, Chong DQ, Ling KL, Increased myeloid-derived suppressor cells in gastric cancer correlate with cancer stage and plasma S100A8/A9 proinflammatory proteins, Journal of immunology, 190 (2013) 794–804. [DOI] [PubMed] [Google Scholar]
- [2].Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ, Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy, Cancer Immunol Immunother, 58 (2009) 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW, Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13, Cancer Immunol Immunother, 60 (2011) 1419–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Young MRI, Wright MA, Lozano Y, Prechel MM, Benefield J, Leonetti JP, Collins SL, Petruzzelli GJ, Increased recurrence and metastasis in patients whose primary head and neck squamous cell carcinomas secreted granulocyte-macrophage colony-stimulating factor and contained CD34+ natural suppressor cells, International Journal of Cancer, 74 (1997) 69–74. [DOI] [PubMed] [Google Scholar]
- [5].Slavin S, Strober S, Induction of allograft tolerance after total lymphoid irradiation (TLI): development of suppressor cells of the mixed leukocyte reaction (MLR) Journal of immunology, 123 (1979) 942–946. [PubMed] [Google Scholar]
- [6].Pak AS, Wright MA, Matthews JP, Collins SL, Petruzzelli GJ, Young MR, Mechanisms of immune suppression in patients with head and neck cancer: presence of CD34(+) cells which suppress immune functions within cancers that secrete granulocyte-macrophage colony-stimulating factor, Clin Cancer Res, 1 (1995) 95–103. [PubMed] [Google Scholar]
- [7].Poschke I, Mougiakakos D, Hansson J, Masucci GV, Kiessling R, Immature immunosuppressive CD14+HLA-DR-/low cells in melanoma patients are Stat3hi and overexpress CD80, CD83, and DC-sign, Cancer Res, 70 (2010) 4335–4345. [DOI] [PubMed] [Google Scholar]
- [8].Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI, Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer, Journal of immunology, 166 (2001) 678–689. [DOI] [PubMed] [Google Scholar]
- [9].Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R, Ochoa AC, Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes, Cancer Res, 69 (2009) 1553–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dumitru CA, Moses K, Trellakis S, Lang S, Brandau S, Neutrophils and granulocytic myeloid-derived suppressor cells: immunophenotyping, cell biology and clinical relevance in human oncology, Cancer Immunol Immunother, 61 (2012) 1155–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, Tcyganov E, Hashimoto A, Nefedova Y, Lin C, Partlova S, Garfall A, Vogl DT, Xu X, Knight SC, Malietzis G, Lee GH, Eruslanov E, Albelda SM, Wang X, Mehta JL, Bewtra M, Rustgi A, Hockstein N, Witt R, Masters G, Nam B, Smirnov D, Sepulveda MA, Gabrilovich DI, Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients, Sci Immunol, 1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhang N, Deng J, Wu F, Lu X, Huang L, Zhao M, Expression of arginase I and inducible nitric oxide synthase in the peripheral blood and lymph nodes of HIVpositive patients, Mol Med Rep, 13 (2016) 731–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI, Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species, Journal of immunology, 172 (2004) 989–999. [DOI] [PubMed] [Google Scholar]
- [14].Zea AH, Rodriguez PC, Culotta KS, Hernandez CP, DeSalvo J, Ochoa JB, Park H-J, Zabaleta J, Ochoa AC, l-Arginine modulates CD3ζ expression and T cell function in activated human T lymphocytes, Cellular Immunology, 232 (2004) 21–31. [DOI] [PubMed] [Google Scholar]
- [15].Rodriguez PC, Zea AH, Culotta KS, Zabaleta J, Ochoa JB, Ochoa AC, Regulation of T cell receptor CD3zeta chain expression by L-arginine, J Biol Chem, 277 (2002) 21123–21129. [DOI] [PubMed] [Google Scholar]
- [16].Brzozowski T, Konturek SJ, Sliwowski Z, Drozdowicz D, Zaczek M, Kedra D, Role of L-arginine, a substrate for nitric oxide-synthase, in gastroprotection and ulcer healing, J Gastroenterol, 32 (1997) 442–452. [DOI] [PubMed] [Google Scholar]
- [17].Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI, Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells, Journal of immunology, 182 (2009) 5693–5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, Padhya T, McCaffrey TV, McCaffrey JC, Gabrilovich DI, Mechanism Regulating Reactive Oxygen Species in Tumor-Induced Myeloid-Derived Suppressor Cells, The Journal of Immunology, 182 (2009) 5693–5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lu C, Redd PS, Lee JR, Savage N, Liu K, The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells, Oncoimmunology, 5 (2016) e1247135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ostrand-Rosenberg S, Horn LA, Haile ST, The programmed death-1 immune-suppressive pathway: barrier to antitumor immunity, Journal of immunology, 193 (2014) 3835–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, Riley JL, CTLA-4 and PD-1 Receptors Inhibit T-Cell Activation by Distinct Mechanisms, Molecular and Cellular Biology, 25 (2005) 9543–9553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, Pal R, Yuan M, Asara J, Patsoukis N, Boussiotis VA, Targeted deletion of PD-1 in myeloid cells induces antitumor immunity, Sci Immunol, 5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gros A, Turcotte S, Wunderlich JR, Ahmadzadeh M, Dudley ME, Rosenberg SA, Myeloid cells obtained from the blood but not from the tumor can suppress T-cell proliferation in patients with melanoma, Clin Cancer Res, 18 (2012) 5212–5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Qiu P, Simonds EF, Bendall SC, Gibbs KD Jr., Bruggner RV, Linderman MD, Sachs K, Nolan GP, Plevritis SK, Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE, Nat Biotechnol, 29 (2011) 886–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Leite Pereira A, Lambotte O, Le Grand R, Cosma A, Tchitchek N, CytoBackBone: an algorithm for merging of phenotypic information from different cytometric profiles, Bioinformatics, 35 (2019) 4187–4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Carulli G, Gianfaldoni ML, Azzara A, Papineschi F, Vanacore R, Minnucci S, Testi R, Ambrogi F, FcRIII (CD16) expression on neutrophils from chronic myeloid leukemia. A flow cytometric study, Leuk Res, 16 (1992) 1203–1209. [DOI] [PubMed] [Google Scholar]
- [27].Choi J, Suh B, Ahn YO, Kim TM, Lee JO, Lee SH, Heo DS, CD15+/CD16low human granulocytes from terminal cancer patients: granulocytic myeloid-derived suppressor cells that have suppressive function, Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine, 33 (2012) 121–129. [DOI] [PubMed] [Google Scholar]
- [28].Hata K, Zhang XR, Iwatsuki S, Van Thiel DH, Herberman RB, Whiteside TL, Isolation, phenotyping, and functional analysis of lymphocytes from human liver, Clinical Immunology and Immunopathology, 56 (1990) 401–419. [DOI] [PubMed] [Google Scholar]
- [29].Fogler WE, Volker K, McCormick KL, Watanabe M, Ortaldo JR, Wiltrout RH, NK cell infiltration into lung, liver, and subcutaneous B16 melanoma is mediated by VCAM-1/VLA-4 interaction, Journal of immunology, 156 (1996) 4707–4714. [PubMed] [Google Scholar]
- [30].Twilley TA, Mason L, Talmadge JE, Wiltrout RH, Increase in liver-associated natural killer activity by polyribonucleotides, Nat Immun Cell Growth Regul, 6 (1987) 279–290. [PubMed] [Google Scholar]
- [31].Anchang B, Hart TDP, Bendall SC, Qiu P, Bjornson Z, Linderman M, Nolan GP, Plevritis SK, Visualization and cellular hierarchy inference of single-cell data using SPADE, Nature Protocols, 11 (2016) 1264–1279. [DOI] [PubMed] [Google Scholar]
- [32].Benjamini Y, Hochberg Y, Controlling the false discovery rate: a practical and powerful approach to multiple testing, Journal of the Royal statistical society: series B (Methodological), 57 (1995) 289–300. [Google Scholar]
- [33].Venditti A, Battaglia A, Del Poeta G, Buccisano F, Maurillo L, Tamburini A, Del Moro B, Epiceno AM, Martiradonna M, Caravita T, Santinelli S, Adorno G, Picardi A, Zinno F, Lanti A, Bruno A, Suppo G, Franchi A, Franconi G, Amadori S, Enumeration of CD34+ hematopoietic progenitor cells for clinical transplantation: comparison of three different methods, Bone Marrow Transplant, 24 (1999) 1019–1027. [DOI] [PubMed] [Google Scholar]
- [34].Veglia F, Perego M, Gabrilovich D, Myeloid-derived suppressor cells coming of age, Nat Immunol, 19 (2018) 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Basso D, Fogar P, Falconi M, Fadi E, Sperti C, Frasson C, Greco E, Tamburrino D, Teolato S, Moz S, Bozzato D, Pelloso M, Padoan A, De Franchis G, Gnatta E, Facco M, Zambon CF, Navaglia F, Pasquali C, Basso G, Semenzato G, Pedrazzoli S, Pederzoli P, Plebani M, Pancreatic tumors and immature immunosuppressive myeloid cells in blood and spleen: role of inhibitory co-stimulatory molecules PDL1 and CTLA4. An in vivo and in vitro study, PLoS One, 8 (2013) e54824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jordan KR, Kapoor P, Spongberg E, Tobin RP, Gao D, Borges VF, McCarter MD, Immunosuppressive myeloid-derived suppressor cells are increased in splenocytes from cancer patients, Cancer Immunol Immunother, 66 (2017) 503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Aggarwal N, Fischer J, Swerdlow SH, Craig FE, Splenic Lymphoid Subsets With Less Well-Recognized Phenotypes Mimic Aberrant Antigen Expression, American Journal of Clinical Pathology, 140 (2013) 787–794. [DOI] [PubMed] [Google Scholar]
- [38].Tavukcuoglu E, Horzum U, Yanik H, Uner A, Yoyen-Ermis D, Nural SK, Aydin B, Sokmensuer C, Karakoc D, Yilmaz KB, Hamaloglu E, Esendagli G, Human splenic polymorphonuclear myeloid-derived suppressor cells (PMN-MDSC) are strategically located immune regulatory cells in cancer, European Journal of Immunology, n/a. [DOI] [PubMed] [Google Scholar]
- [39].Kotsakis A, Harasymczuk M, Schilling B, Georgoulias V, Argiris A, Whiteside TL, Myeloid-derived suppressor cell measurements in fresh and cryopreserved blood samples, J Immunol Methods, 381 (2012) 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Trellakis S, Bruderek K, Hutte J, Elian M, Hoffmann TK, Lang S, Brandau S, Granulocytic myeloid-derived suppressor cells are cryosensitive and their frequency does not correlate with serum concentrations of colony-stimulating factors in head and neck cancer, Innate Immun, 19 (2013) 328–336. [DOI] [PubMed] [Google Scholar]
- [41].Porembka MR, Mitchem JB, Belt BA, Hsieh CS, Lee HM, Herndon J, Gillanders WE, Linehan DC, Goedegebuure P, Pancreatic adenocarcinoma induces bone marrow mobilization of myeloid-derived suppressor cells which promote primary tumor growth, Cancer Immunol Immunother, 61 (2012) 1373–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ostrand-Rosenberg S, Fenselau C, Myeloid-Derived Suppressor Cells: Immune-Suppressive Cells That Impair Antitumor Immunity and Are Sculpted by Their Environment, The Journal of Immunology, 200 (2018) 422–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, Rodriguez PC, Sica A, Umansky V, Vonderheide RH, Gabrilovich DI, Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards, Nature Communications, 7 (2016) 12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hocking W, Goodman J, Golde D, Granulocytosis associated with tumor cell production of colony-stimulating activity, Blood, 61 (1983) 600–603. [PubMed] [Google Scholar]
- [45].Fredeau L, Bohelay G, Shourick J, Piver D, Guyot A, Schlageter MH, Caux F, Maubec E, Paraneoplastic neutrophilic leukaemoid reaction in a patient with melanoma: association between tumour volume and leucocytosis, Br J Dermatol, (2020). [DOI] [PubMed] [Google Scholar]
- [46].Klein B, Stein M, Kuten A, Steiner M, Barshalom D, Robinson E, Gal D, Splenomegaly and solitary spleen metastasis in solid tumors, Cancer, 60 (1987) 100–102. [DOI] [PubMed] [Google Scholar]
- [47].Wang X, Cho SY, Hu CS, Chen D, Roboz J, Hoffman R, C-X-C motif chemokine 12 influences the development of extramedullary hematopoiesis in the spleens of myelofibrosis patients, Exp Hematol, 43 (2015) 100–109.e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Schmielau J, Finn OJ, Activated Granulocytes and Granulocyte-derived Hydrogen Peroxide Are the Underlying Mechanism of Suppression of T-Cell Function in Advanced Cancer Patients, Cancer Research, 61 (2001) 4756–4760. [PubMed] [Google Scholar]
- [49].Hirashima M, Higuchi S, Sakamoto K, Nishiyama T, Okada H, The ratio of neutrophils to lymphocytes and the phenotypes of neutrophils in patients with early gastric cancer, Journal of Cancer Research and Clinical Oncology, 124 (1998) 329–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bussolino F, Wang JM, Defilippi P, Turrini F, Sanavio F, Edgell CJ, Aglietta M, Arese P, Mantovani A, Granulocyte- and granulocyte-macrophage-colony stimulating factors induce human endothelial cells to migrate and proliferate, Nature, 337 (1989) 471–473. [DOI] [PubMed] [Google Scholar]
- [51].Khaled YS, Ammori BJ, Elkord E, Increased levels of granulocytic myeloid-derived suppressor cells in peripheral blood and tumour tissue of pancreatic cancer patients, J Immunol Res, 2014 (2014) 879897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Terstappen LW, Safford M, Loken MR, Flow cytometric analysis of human bone marrow. III. Neutrophil maturation, Leukemia, 4 (1990) 657–663. [PubMed] [Google Scholar]
- [53].Baum CM, Weissman IL, Tsukamoto AS, Buckle AM, Peault B, Isolation of a candidate human hematopoietic stem-cell population, Proc Natl Acad Sci U S A, 89 (1992) 2804–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Groblewska M, Mroczko B, Wereszczyńska-Siemiatkowska U, Myśliwiec P, Kedra B, Szmitkowski M, Serum levels of granulocyte colony-stimulating factor (G-CSF) and macrophage colony-stimulating factor (M-CSF) in pancreatic cancer patients, Clin Chem Lab Med, 45 (2007) 30–34. [DOI] [PubMed] [Google Scholar]
- [55].Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, Carbone DP, Gabrilovich DI, Increased Production of Immature Myeloid Cells in Cancer Patients: A Mechanism of Immunosuppression in Cancer, The Journal of Immunology, 166 (2001) 678–689. [DOI] [PubMed] [Google Scholar]
- [56].Mandruzzato S, Brandau S, Britten CM, Bronte V, Damuzzo V, Gouttefangeas C, Maurer D, Ottensmeier C, van der Burg SH, Welters MJ, Walter S, Toward harmonized phenotyping of human myeloid-derived suppressor cells by flow cytometry: results from an interim study, Cancer Immunol Immunother, 65 (2016) 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Young MR, Young ME, Wright MA, Stimulation of immune-suppressive bone marrow cells by colony-stimulating factors, Exp Hematol, 18 (1990) 806–811. [PubMed] [Google Scholar]
- [58].Sade-Feldman M, Kanterman J, Ish-Shalom E, Elnekave M, Horwitz E, Baniyash M, Tumor necrosis factor-α blocks differentiation and enhances suppressive activity of immature myeloid cells during chronic inflammation, Immunity, 38 (2013) 541–554. [DOI] [PubMed] [Google Scholar]
- [59].Condamine T, Gabrilovich DI, Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function, Trends Immunol, 32 (2011) 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Chornoguz O, Grmai L, Sinha P, Artemenko KA, Zubarev RA, Ostrand-Rosenberg S, Proteomic pathway analysis reveals inflammation increases myeloid-derived suppressor cell resistance to apoptosis, Mol Cell Proteomics, 10 (2011) M110.002980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, Kavanaugh D, Carbone DP, Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells, Nat Med, 2 (1996) 1096–1103. [DOI] [PubMed] [Google Scholar]
- [62].Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, Ortiz M, Nacken W, Sorg C, Vogl T, Roth J, Gabrilovich DI, Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein, J Exp Med, 205 (2008) 2235–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Iwata T, Kondo Y, Kimura O, Morosawa T, Fujisaka Y, Umetsu T, Kogure T, Inoue J, Nakagome Y, Shimosegawa T, PD-L1+MDSCs are increased in HCC patients and induced by soluble factor in the tumor microenvironment, Scientific Reports, 6 (2016) 39296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Okla K, Rajtak A, Czerwonka A, Bobinski M, Wawruszak A, Tarkowski R, Bednarek W, Szumilo J, Kotarski J, Accumulation of blood-circulating PD-L1-expressing M-MDSCs and monocytes/macrophages in pretreatment ovarian cancer patients is associated with soluble PD-L1, J Transl Med, 18 (2020) 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].MacFarlane A.W.t., Jillab M, Plimack ER, Hudes GR, Uzzo RG, Litwin S, Dulaimi E, Al-Saleem T, Campbell KS, PD-1 expression on peripheral blood cells increases with stage in renal cell carcinoma patients and is rapidly reduced after surgical tumor resection, Cancer Immunol Res, 2 (2014) 320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Wu CF, Wang CC, Tai TS, Su YC, ER stress of cancer cell SCC25 induces LOX-1-expressed immunosuppressive neutrophils, Annals of Oncology, 30 (2019) xi55. [Google Scholar]
- [67].Nan J, Xing YF, Hu B, Tang JX, Dong HM, He YM, Ruan DY, Ye QJ, Cai JR, Ma XK, Chen J, Cai XR, Lin ZX, Wu XY, Li X, Endoplasmic reticulum stress induced LOX-1(+ ) CD15(+) polymorphonuclear myeloid-derived suppressor cells in hepatocellular carcinoma, Immunology, 154 (2018) 144–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Chai E, Zhang L, Li C, LOX-1+ PMN-MDSC enhances immune suppression which promotes glioblastoma multiforme progression, Cancer Manag Res, 11 (2019) 7307–7315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, McDermott D, Quiceno D, Youmans A, O’Neill A, Mier J, Ochoa AC, Arginase-Producing Myeloid Suppressor Cells in Renal Cell Carcinoma Patients: A Mechanism of Tumor Evasion, Cancer Research, 65 (2005) 3044–3048. [DOI] [PubMed] [Google Scholar]
- [70].Mao F.-y., Zhao Y.-l., Lv Y.-p., Teng Y.-s., Kong H, Liu Y.-g., Wu X.-l., Hao C.-j., Chen W, Duan M.-b., Han B, Ma Q, Wang T.-t., Peng L.-s., Zhang J.-y., Cheng P, Su C.-y., Fu X.-l., Zou Q.-m., Guo G, Guo X.-l., Zhuang Y, CD45+CD33lowCD11bdim myeloid-derived suppressor cells suppress CD8+ T cell activity via the IL-6/IL-8-arginase I axis in human gastric cancer, Cell Death & Disease, 9 (2018) 763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Zhang H, Li Z-L, Ye S-B, Ouyang L-Y, Chen Y-S, He J, Huang H-Q, Zeng Y-X, Zhang X-S, Li J, Myeloid-derived suppressor cells inhibit T cell proliferation in human extranodal NK/T cell lymphoma: a novel prognostic indicator, Cancer Immunology, Immunotherapy, 64 (2015) 1587–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Roussel M, Ferrell PB Jr., Greenplate AR, Lhomme F, Le Gallou S, Diggins KE, Johnson DB, Irish JM, Mass cytometry deep phenotyping of human mononuclear phagocytes and myeloid-derived suppressor cells from human blood and bone marrow, J Leukoc Biol, 102 (2017) 437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Younos IH, Dafferner AJ, Gulen D, Britton HC, Talmadge JE, Tumor regulation of myeloid-derived suppressor cell proliferation and trafficking, Int Immunopharmacol, 13 (2012) 245–256. [DOI] [PubMed] [Google Scholar]
- [74].Groblewska M, Mroczko B, Wereszczyńska-Siemiątkowska U, Myśliwiec P, Kędra B, Szmitkowski M, Serum levels of granulocyte colony-stimulating factor (G-CSF) and macrophage colony-stimulating factor (M-CSF) in pancreatic cancer patients, Clinical Chemistry and Laboratory Medicine (CCLM), 45 (2007) 30. [DOI] [PubMed] [Google Scholar]
- [75].Ławicki S, Będkowska GE, Wojtukiewicz M, Szmitkowski M, Hematopoietic cytokines as tumor markers in breast malignancies. A multivariate analysis with ROC curve in breast cancer patients, Adv Med Sci, 58 (2013) 207–215. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Flow gating strategy. Our flow gating strategy for differentiating granulocytic (G), monocytic (M), and immature (i) myeloid derived suppressor cells (MDSC).
Supplemental Figure 2. CTLA-4 flow panel SPADE analysis. SPADE trees and the relative phenotypic marker intensities for each antibody marker in the CTLA-4 flow panel for the total exported MDSC populations from the peripheral blood (PB) of normal donors and the PB and spleen cells of cancer patients.
Supplemental Figure 3. PD-L1 flow panel SPADE analysis. SPADE trees and the relative phenotypic marker intensities for each antibody marker in the PD-L1 flow panel for the total exported MDSC populations from the peripheral blood (PB) of normal donors and the PB and spleen cells of cancer patients.
Supplemental Figure 4. Enzyme flow panel SPADE analysis. SPADE trees and the relative phenotypic marker intensities for each antibody marker in the enzyme flow panel for the total exported MDSC populations from the peripheral blood (PB) of normal donors and the PB and spleen cells of cancer patients.
Supplemental Figure 5. CytoBackBone SPADE analysis. SPADE trees and the relative phenotypic marker intensities for each antibody marker for the total exported MDSC populations of the merged flow panels CTLA-4 and PD-L1. These studies were from the peripheral blood (PB) of normal donors and the PB and spleen cells of cancer patients.