

Supplemental Digital Content is available in the text.
Abstract
Ibrutinib is a covalently binding inhibitor of the B-cell receptor signaling-mediator Bruton’s tyrosine kinase (BTK) with great efficacy in chronic lymphocytic leukemia (CLL). Common side effects like atrial fibrillation (AF), bleeding and infections might be caused by ibrutinib’s inhibition of other kinases in non-B cells. Five-year follow-up of plasma biomarkers by proximity extension assay and immune cell numbers by flow cytometry during ibrutinib treatment revealed that 86 of the 265 investigated plasma biomarkers significantly changed during treatment, 74 of which decreased. Among the 12 markers that increased, 6 are associated with cardiovascular diseases and therefore potentially involved in ibrutinib-induced AF. Comparison between healthy donors and X-linked agammaglobulinemia (XLA) patients, who have nonfunctional BTK and essentially lack B cells, showed indicative changes in 53 of the 265 biomarkers while none differed significantly. Hence, neither B cells nor BTK-dependent pathways in other cells seem to influence the levels of the studied plasma biomarkers in healthy donors. Regarding immune cells, the absolute number of T cells, including subsets, decreased, paralleling the decreasing tumor burden. T helper 1 (Th1) cell numbers dropped strongly, while Th2 cells remained relatively stable, causing Th2-skewing. Thus, long-term ibrutinib treatment has a profound impact on the plasma proteome and immune cells in patients with CLL.
Introduction
Chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL) is associated with a broad immunodeficiency that causes frequent infections, autoimmune reactions and suboptimal antitumor immunity.1 Despite their ability to survive in the circulation, CLL cells expand in the secondary lymphoid organs and are highly dependent on the tumor microenvironment (TME).
Ibrutinib is a potent and irreversibly binding small-molecule inhibitor of Bruton’s tyrosine kinase (BTK).2 BTK inhibition blocks the transmission of important growth and survival signals through the B-cell receptor (BCR) in CLL cells. Ibrutinib therapy has substantially improved the prognosis of patients with relapsed or refractory (R/R) CLL, including those with tumor cells that carry del(17p) and/or TP53 mutations.
It has been clearly shown that ibrutinib interferes with the homing of B cells. BTK is essential for C-X-C motif chemokine ligand 12 (CXCL12)- and CXCL13-dependent integrin-mediated adhesion and migration of B cells into lymph nodes.3–5 Inhibition of BTK by ibrutinib reduces the expression of the CXCL12 receptor C-X-C motif chemokine receptor 4 (CXCR4) on leukemic B cells, which in turn leads to a rapid release of CLL cells from the lymphoid organs into the peripheral blood (PB).6–8 CLL cells with low CXCR4 surface expression do not migrate back to tissue niches and are therefore deprived of survival signals.7
Nonmalignant B cells that exist in the shadow of their transformed counterparts in CLL are presumably affected by ibrutinib as well, since BTK is an essential enzyme in physiological B-cell development.9 Moreover, the enzymatic activity of other kinases that have a similarly aligning cysteine residue to BTK is also inhibited by ibrutinib.2,10,11 Such “off-target” binding could potentially contribute to the clinical efficacy of ibrutinib, as well as to side effects.
Patients with X-linked agammaglobulinemia (XLA) have germline mutations in the gene that encodes BTK.12–14 Affected males essentially lack B-cell areas in lymphoid organs, with the ensuing loss of circulating mature B cells and serum immunoglobulins from birth. In the bone marrow, an increase in pro-B and pre-B cells and a reduction of all subsequent stages is observed.15,16 Patients with XLA are therefore an ideal reference to study BTK- or B cell-independent protein expression patterns.
Among the hallmarks of CLL are a compromised function of cytotoxic lymphocytes and an expansion of immunosuppressive immune cells. Circulating T-cell subsets have a terminally differentiated and pseudoexhausted phenotype, with less naïve and more effector and effector memory T cells.1,17 CD8+ T cells have reduced cytotoxic capabilities.1 CD4+ T cells are skewed toward a tumor growth-favoring T helper 2 (Th2) phenotype17,18 and the fraction and absolute number of regulatory T cells (Treg) cells is increased in the PB.17,19 Myeloid cells also add to the immunodeficient state of patients with CLL.20,21
The observation that infections are among the most frequent adverse events in ibrutinib clinical trials, especially during the first 6 months of treatment,22 has prompted investigations into the influence that this drug has on the profile and function of immune cells.23 Ibrutinib reduces the number of Th2 cells24 and of myeloid-derived suppressor cells,25 might potentiate T-cell therapies,26 improves dendritic cell activation,27 impedes natural killer (NK) cells in performing antibody-dependent cellular cytotoxicity28 and also affects granulocytes.23 T-cell numbers seem to normalize after 6–12 months of ibrutinib treatment,29–31 but data on the very long-term effects on immune cells are limited.
Ibrutinib treatment also results in rapid changes in plasma biomarkers, the majority of which are not B cell-derived.8,29 By investigating the effects on plasma biomarkers and cells other than tumor cells during long-term treatment, further insight can be gained not only into the mechanism of action of this drug, but also into CLL biology. To that end, we have analyzed the largest number of plasma biomarkers reported to date in both CLL and XLA and performed immunomonitoring.
Patients and methods
Patients and donors
Thirteen patients who started ibrutinib at the Hematology Department of the Karolinska University Hospital were included in this study. Seven of these patients were analyzed in our previous work, in which the baseline, 9 hours, 2 days, and 4 weeks timepoints have been reported.8 For 2 of these patients, follow-up was interrupted after 4 weeks. The baseline characteristics of the other 11 patients are listed in Table 1. PB samples were collected before the start of treatment, after 9 hours and 2 days in the 7 previously reported patients, at week 4, 10, 16, and 22, and after 8, 12, 24, 36, 48, and 60 months, or until treatment cessation. PB samples from 9 healthy donors were used as controls and PB samples from 8 patients with XLA harboring a verified mutation in BTK were acquired from the hospital’s immunodeficiency unit. The study was approved by the local ethics committee and all participating subjects gave written informed consent. Immunoglobulin heavy chain variable region mutational status was assessed as previously described.17 Chromosomal abnormalities, transformation-related protein 53 (TP53) mutations and absolute lymphocyte counts were determined by the hospital’s routine laboratories.
Table 1.
Patient Characteristics at Study Entry
| Patient Number | Age (Y)—Sex (Female/Male) | Diagnosis | Ann Arbor Stage (SLL)/Modified Rai Stage (CLL)/Binet Stage (CLL) | CIRS-G Score (Miller and Towers 1991) | Cytogenetic Abnormalities (FISH) | TP53 Mutation Status (Yes/No) | IGHV Mutation Status (UM/M) | Hypogammaglobulinemia (Yes/No) | Prior Treatment Lines (n) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| trisomy 12 (Yes/No) | del(13q) (Yes/No) | del(17p) (Yes/No) | del(11q) (Yes/No) | Complex Karyotypea (Yes/No) | |||||||||
| 01 | 77—M | SLL | IV/−/− | 6 | No | Yes | No | Yes | No | No | Mb | No | 1 |
| 02 | 57—M | CLL | −/high/C | 0 | No | Yes | No | Yes | No | No | UM | No | 2 |
| 03 | 73—M | CLL | −/inter./B | 8 | No | Yes | Yes | No | No | No | M* | Yes | 4 |
| 04 | 74—M | CLL | −/high/C | 4 | Yes | Yes | Yes | No | Yes | No | M | Yes | 1 |
| 05 | 73—M | CLL | −/inter./B | 3 | No | Yes | Yes | No | No | No | M | N/A | 1 |
| 06 | 75—F | CLL | −/high/C | 0 | No | Yes | Yes | No | No | Yes | M | Yes | 2 |
| 07 | 64—M | CLL | −/high/C | 4 | No | Yes | Yes | No | No | No | UM | Yes | 1 |
| 08 | 79—M | CLL | −/high/B | 4 | No | Yes | No | No | No | Yes | UM | N/A | 2 |
| 09 | 52—M | CLL | −/high/C | 0 | No | Yes | Yes | No | No | N/A | N/A | YES | 2 |
| 10 | 68—F | CLL | −/inter./B | 3 | No | Yes | Yes | No | No | N/A | N/A | YES | 1 |
| 11 | 66—F | CLL | −/high/C | 0 | Yes | Yes | Yes | No | Yes | N/A | N/A | Yes | 1 |
aComplex karyotype was defined as the presence of ≥3 cytogenetic abnormalities as measured by FISH.
bBorderline IGHV mutated (defined as 97%–98% sequence identity to germline).
CLL = chronic lymphocytic leukemia; CIRS-G = Cumulative Illness Rating Scale for Geriatrics; FISH = fluorescence in situ hybridization; IGHV = immunoglobulin heavy chain variable region; inter. = intermediate; M = mutated; N/A = not available; SLL = small lymphocytic lymphoma; UM = unmutated.
Peripheral blood mononuclear cells (PBMCs) were isolated from whole heparinized blood by density gradient centrifugation using a Ficoll-Hypaque gradient (GE Healthcare, Uppsala, Sweden) and washed twice in Dulbecco’s Phosphate-Buffered Saline (Gibco, Life Technologies, Carlsbad, CA). Cells were stained freshly for flow cytometry analysis or vitally frozen in 10% dimethyl sulfoxide (Sigma-Aldrich, St. Louis, MO) and stored in liquid nitrogen until further use. Plasma was purified from whole heparinized blood by centrifugation and stored at −80°C until further use.
Proximity extension assay on plasma samples
Proximity extension assay (PEA; Olink Bioscience, Uppsala, Sweden)32 was performed on plasma from patients with CLL/SLL and XLA as well as healthy donors. By using 3 precision protein target panels (Olink Target 96 Inflammation, Immune Response and Oncology II), we have simultaneously studied 265 unique biomarkers. On samples from 7 patients from the earliest time points (ie, 9 hours, 2 days, and 4 weeks), we previously ran the Olink Target 96 Inflammation panel only, as reported.8 These data were added to the analyses in the current study. Here, we extended the data by performing PEA with 2 additional panels (ie, Olink Target 96 Immune response and Olink Target 96 Oncology II) on the earliest time points from the 7 previously reported patients and with all 3 panels on the samples from the 11 patients that were followed-up for longer time (up to 5 y). A complete list of all the biomarkers analyzed is provided in Supplemental Digital Table 1; http://links.lww.com/HS/A152. One microliter of plasma was used for each measurement and duplicates were run for each sample. The Wilcoxon signed-rank test was performed to compare paired measurements at baseline (pretreatment) with measurements during treatment. To correct for multiple-testing, the Benjamini-Hochberg procedure was applied and the molecules with an adjusted P ≤0.05 for one or more time points were considered significantly changed. For further details, see Supplemental Digital Methods; http://links.lww.com/HS/A152.
Validation of our data in a second cohort could strengthen our conclusions, but it would also delay the disclosure of our findings and the subsequent possibility of independent confirmation and was therefore omitted. Besides, a systems biology approach could in theory provide important complementary information and allow a deeper understanding of the integrated interactions between CLL and TME cells. However, it would also be challenging to perform computational modeling with only 265 selected biomarkers and we therefore chose to not pursue this strategy.
RNA sequencing of reference cells
The RNA sequencing data that is shown has been published previously.8,33 For further details see Supplemental Digital Methods; http://links.lww.com/HS/A152.
Flow cytometric characterization of immune cell subsets
For lymphocyte profiling, fresh whole blood cells from patients 01 to 08 and thawed PBMC from patients 09 to 11 were stained with antibodies from panel 1 in Supplemental Digital Table 2; http://links.lww.com/HS/A152, then subjected to red blood cell lysis and washed once before data acquisition. PBMC from patients 01 to 08 and thawed PBMC from patients 09 to 11 were stained with antibodies from panel 2 in Supplemental Digital Table 2; http://links.lww.com/HS/A152. PBMC from all CLL/SLL patients were thawed, washed, subsequently FcR blocked (Miltenyi, Bergisch Gladbach, Germany) and stained with antibodies from panel 3 in Supplemental Digital Table 2; http://links.lww.com/HS/A152. PBMC from patients 01 to 10 were thawed, washed and stained with antibodies from panel 4 in Supplemental Digital Table 2; http://links.lww.com/HS/A152. See Supplemental Digital Methods; http://links.lww.com/HS/A152 for full details of the staining procedures and Supplemental Digital Figure 1; http://links.lww.com/HS/A152 for gating strategies.
Results and discussion
Treatment outcome
For the 11 patients that were followed-up beyond 4 weeks of ibrutinib treatment, the objective response rate at 12 months was 100%, with 63% clinical complete responses according to iwCLL criteria (bone marrow aspirate and biopsy, as well as CT scan and minimal residual disease assessment were not always performed).34 A summary of the treatment duration and outcome, reasons for interruption, and of occurrence and severity of infections and atrial fibrillation (AF), is provided in Figure 1. In 2 patients, temporary dose reductions were applied due to toxicity.
Figure 1.
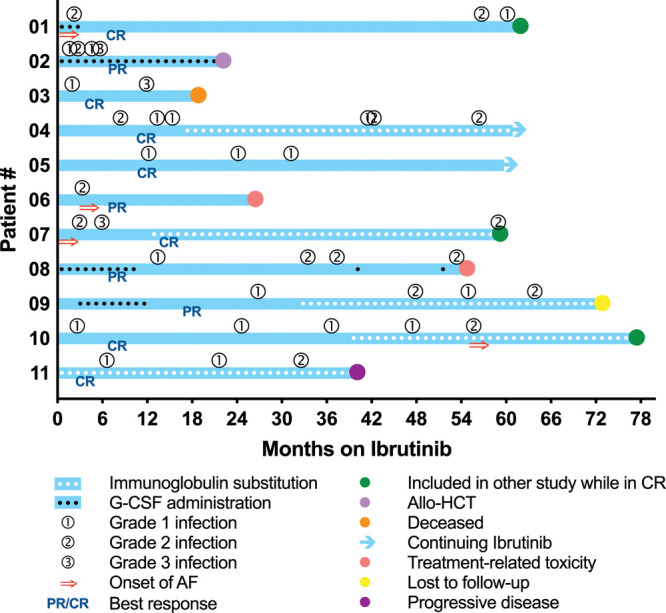
Treatment outcome and occurrence of infections and AF. Patients 01 and 07 already had AF before starting ibrutinib, while patients 06 and 10 developed AF after 3 and 55 mo of treatment, respectively. Colored dots indicate the timing and reason for discontinuation of follow-up. AF = atrial fibrillation; Allo-HCT = allogeneic hematopoietic cell transplantation; CR = complete response; G-CSF = granulocyte colony stimulating factor; PR = partial response.
Overview of the plasma biomarker analysis
The levels of 86 out of 265 (32%) analyzed plasma biomarkers were significantly different from baseline at ≥1 time point during treatment (Supplemental Digital Figure 2; http://links.lww.com/HS/A152). The majority (74/86) was reduced and an overview of how all biomarkers were categorized, as presented in the following sections, is shown in Figure 2. The biomarkers always remained either reduced or increased. RNA sequencing data from another study,33 which were used to cross-reference our data, showed that about one third of these 86 molecules are not normally produced by B cells. Some of these are known to originate from the CLL TME,20 others could be secreted by various other hematopoietic and non-hematopoietic cells. All 86 biomarkers that changed during treatment are listed with their alternative names in Supplemental Digital Table 3; http://links.lww.com/HS/A152.
Figure 2.
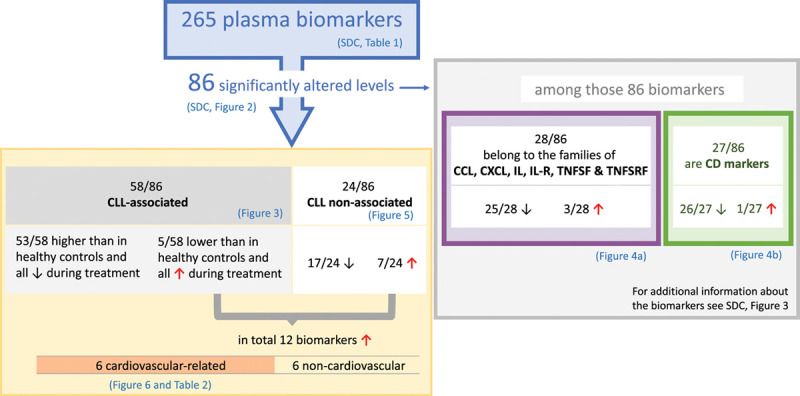
Schematic representation of the analysis 265 biomarkers in plasma from patients with CLL before and after treatment with ibrutinib. Biomarkers were subdivided into CLL-associated and CLL nonassociated (left) and were further assembled into CD markers and other families of proteins (right). Only 12/86 markers increased, 8 of which were cardiovascular-related, and indicative of “off-target” effects. Among the 86 biomarkers, 4 do not belong to either the CLL-associated, or CLL nonassociated biomarkers, IFNLR1, IL-12B, TXLNA, and SEZ6L, but are indicatively downregulated. Among the 2 categories CD-marker and other protein families (right) some of these biomarkers belong to both groups. CLL = chronic lymphocytic leukemia.
Most of the CLL-associated plasma biomarkers that change during ibrutinib treatment decrease
Among the 86 biomarkers that significantly changed during ibrutinib treatment, 58 (67%) were classified as CLL-associated (Figure 3). Our arbitrary definition of “CLL-associated” is: biomarkers with levels that were significantly different between pretreatment and control samples. Thus, “CLL-associated” does not mean that these proteins originate from the tumor cells, but only that their levels differ between patients with CLL and healthy donors.
Figure 3.
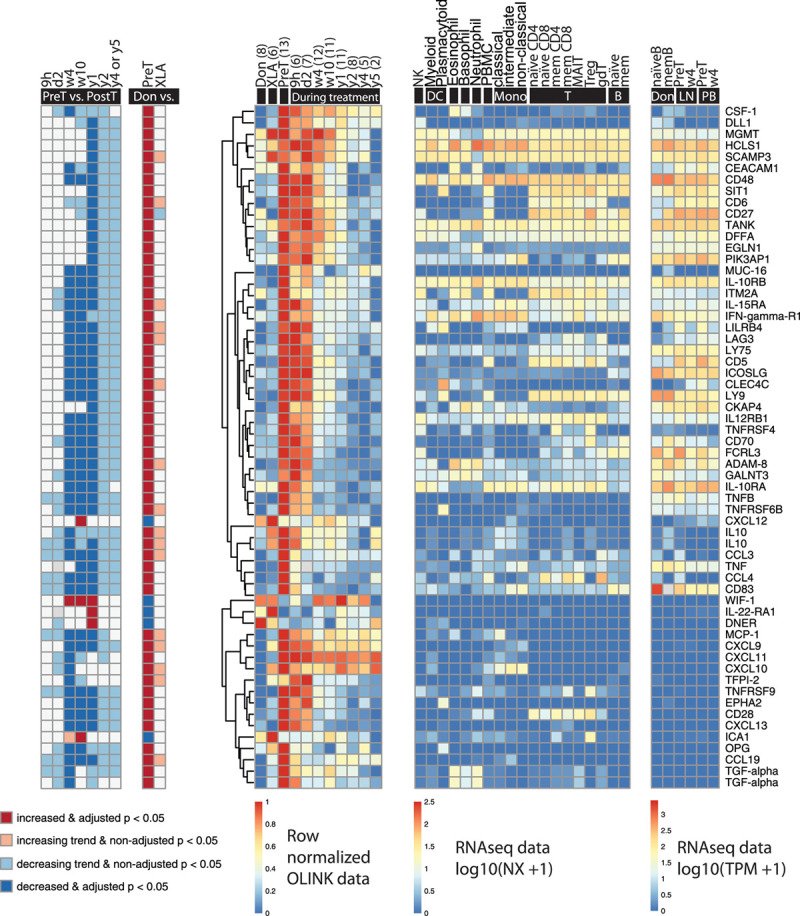
Significantly changed CLL-associated molecules during ibrutinib treatment (n = 58). Biomarkers that were different between healthy donors and patients with CLL at baseline were regarded as CLL-associated. (Left to right) The first heat-map shows significance and increase or decrease of the biomarkers at different time points during treatment compared to pretreatment; the second shows the comparison between healthy donors (6 males and 3 females, median age 65 y, range 45–79; Don), patients with CLL before the start of ibrutinib treatment (PreT) and patients with XLA (XLA); the third shows the row-normalized Olink data; the fourth shows mRNA expression in different cell types, from healthy individuals for comparison (data from Uhlen et al33); the fifth shows the mRNA expression in healthy donor and CLL B cells before and after 4 weeks of ibrutinib treatment in lymph nodes (LN) and peripheral blood (PB) (data from Palma et al8). All RNA sequencing data has been published previously.8,33 IL10 and TGF-alpha are depicted twice because 2 different Olink probes were used to assess them. The number of data points (n) is indicated per time point. B = B cells; CLL = chronic lymphocytic leukemia; DC = dendritic cells; gdT = gamma delta T cells; MAIT = mucosal-associated invariant T cells; mem = memory; Mono = monocytes; NK = natural killer cells; T = T cells; Treg = regulatory T cells.
At baseline, the levels of most of these biomarkers, namely 53 (91%) of them, were higher in patients with CLL than in healthy donors and decreased during treatment. The majority of these are produced, although not exclusively, by non-malignant B cells or CLL cells. However, 12 of them, including CD28, CD137 (TNFRSF) and 6 chemokines, are not. On the other hand, 5 of the CLL-associated biomarkers, that is, CXCL12, Delta and Notch-like epidermal growth factor-related receptor (DNER), islet cell autoantigen 1 (ICA1), interleukin (IL)-22 receptor α1 (IL-22Rα1) and wingless-type (Wnt) inhibitory factor 1 (WIF-1), were significantly lower at baseline and increased during treatment instead (Figure 3). A possible explanation could be that CLL cells repress the expression of these genes in the microenvironment, and hence their levels increase during effective ibrutinib therapy.
Here, we also confirm the reduction of all 23 markers that were reported in our previous study.8 As depicted in Figure 4 and elaborated on in the Supplemental Digital Results and Discussion; http://links.lww.com/HS/A152, the extended list contains 4 members of the chemokine ligand (CCL) family, 6 chemokine (C-X-C motif) ligand (CXCL) proteins, 9 IL and IL receptors, 8 of which decreased, and IL-22 receptor α1, which increased during treatment, as well as 9 members of the TNFSF and TNFRSF families. Moreover, altogether, 27 CD markers were significantly altered during treatment (Figure 4).
Figure 4.
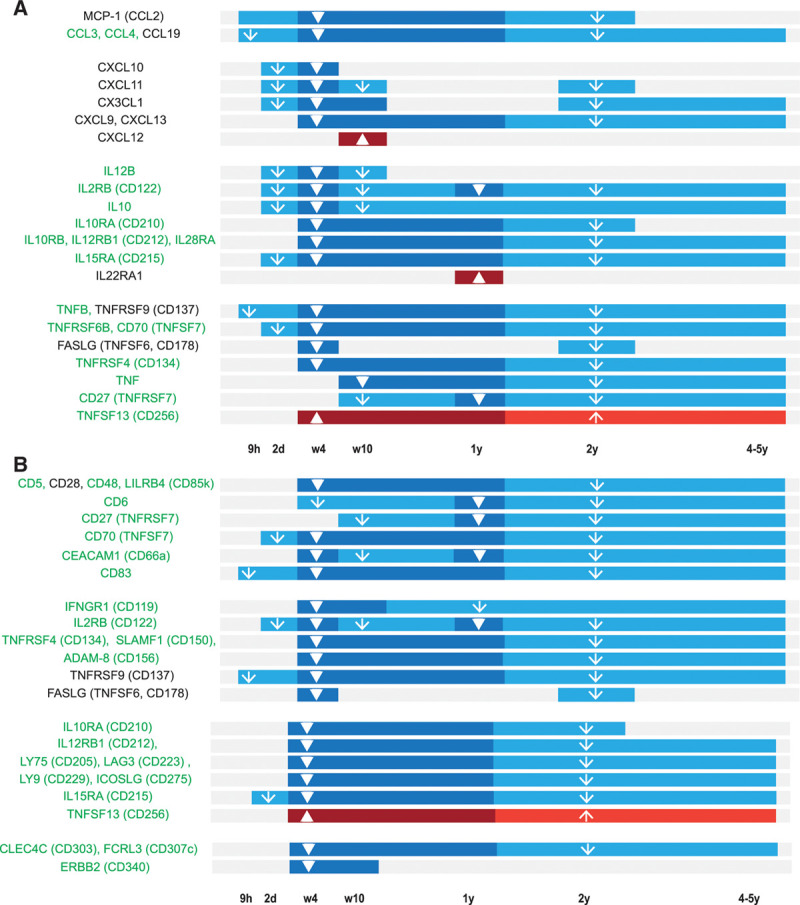
Timeline of plasma biomarkers that change during ibrutinib treatment. (A) Timeline for CCL proteins, CXCL proteins, ILs, IL-Rs, TNFSF, and TNFRSF members. (B) Timeline for CD molecules. Plasma biomarkers expressed by B cells are listed in green. Statistically significant changes in the plasma levels between the respective time points and pretreatment are depicted as a dark blue (decrease) or dark red (increase) bar with a thick arrow. Trends are depicted as a light blue (decreasing tendency) or light red (increasing tendency) bar with a thin arrow. CCL = chemokine ligand; CXCL12 = C-X-C motif chemokine ligand 12.
While the onset of the decrease varied profoundly among the biomarkers (Supplemental Digital Results and Discussion; http://links.lww.com/HS/A152), for most of them the plasma level changes seemed to be maintained throughout 4–5 years of treatment.
Ibrutinib reduces CLL nonassociated plasma biomarkers
Interestingly, among the 86 biomarkers that were significantly altered over time, 24 (28%) were classified as “CLL nonassociated.” These correspond to the plasma biomarkers that neither differed significantly nor showed trends between healthy donors and patients with CLL before start of treatment (Figure 5). Moreover, only 2 of them were modestly altered in XLA as compared to pretreatment samples (Figure 5), suggesting that the levels of most of these biomarkers are unrelated to BTK activity in non-B cells and instead represent “off-target” effects. In this group, we mainly observed decreasing levels during treatment as well. Interestingly, 7 CLL non-associated biomarkers, that is, amphiregulin (AREG), cystatin D (CST5), ectodysplasin A receptor (EDAR), epidermal growth factor (EGF), plexin-A4 (PLXNA4), R-spondin-3 (RSPO3), and TNFSF13, increased during treatment (Figure 5). See Supplemental Digital Results and Discussion; http://links.lww.com/HS/A152, for other findings.
Figure 5.
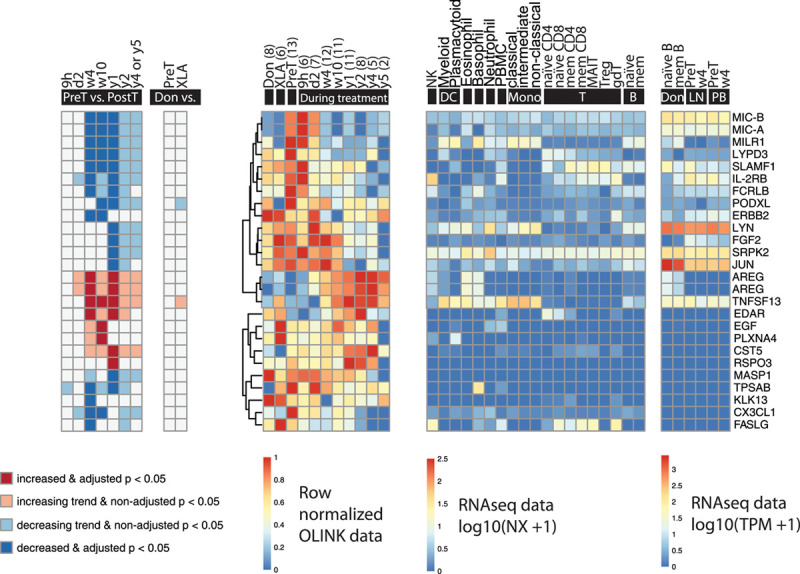
Significantly changed CLL nonassociated molecules during ibrutinib treatment (n = 24). Biomarkers that were not significantly different nor showed trends between healthy donors and patients at baseline were regarded as CLL nonassociated. (Left to right) The first heat-map shows significance and increase or decrease of the biomarkers at different time points during treatment compared to pretreatment; the second shows the comparison between healthy donors (6 males and 3 females, median age 65 y, range 45–79; Don), patients with CLL before the start of ibrutinib treatment (PreT) and patients with XLA (XLA); the third shows the row-normalized Olink data; the fourth shows mRNA expression in different cell types (data from Uhlen et al33); the fifth shows the mRNA expression in healthy donor and CLL B cells before and after 4 wks of ibrutinib treatment in lymph nodes (LN) and peripheral blood (PB) (data from Palma et al8). All RNA sequencing data has been published previously.8,33 MIC-A and MIC-B were assessed using the same Olink probe and therefore counted as one unique protein. AREG is depicted twice because 2 different Olink probes were used to assess it. The number of data points (n) is indicated per time point. B = B cells; CLL = chronic lymphocytic leukemia; DC = dendritic cells; gdT = gamma delta T cells; MAIT = mucosal-associated invariant T cells; mem = memory; Mono = monocytes; NK = natural killer cells; T = T cells; Treg = regulatory T cells.
Six cardiovascular disease-related plasma biomarkers increase during ibrutinib treatment
We observed that the levels of 12 of all 86 changing biomarkers significantly increased during treatment, 9 of which are not derived from B cells (Figures 2 and 6).
Figure 6.
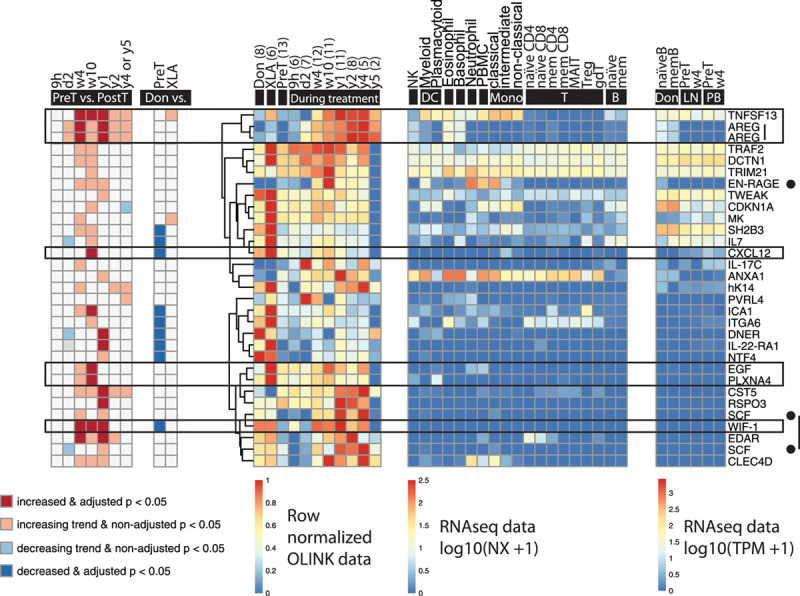
Plasma biomarkers that increase during ibrutinib treatment (n = 29). Boxed biomarkers (n = 6) indicate those implicated in cardiovascular diseases. Biomarkers indicated by a black dot (n = 2) have also been related to cardiovascular diseases, but only show a trend toward increase. (Left to right) The first heat-map shows significance and increase or decrease of the biomarkers at different time points during treatment compared to pretreatment; the second shows the comparison between healthy donors (6 males and 3 females, median age 65 y, range 45–79; Don), patients with CLL before the start of ibrutinib treatment (PreT) and patients with XLA (XLA); the third shows the row-normalized Olink data; the fourth shows mRNA expression in different cell types (data from Uhlen et al33); the fifth shows the mRNA expression in healthy donor and CLL B cells before and after 4 weeks of ibrutinib treatment in lymph nodes (LN) and peripheral blood (PB) (data from Palma et al8). All RNA sequencing data have been published previously.8,33 AREG and SCF are depicted twice because two different Olink probes were used to assess them. The number of data points (n) is indicated per time point. B = B cells; CLL = chronic lymphocytic leukemia; DC = dendritic cells; gdT = gamma delta T cells; MAIT = mucosal-associated invariant T cells; mem = memory; Mono = monocytes; NK = natural killer cells; T = T cells; Treg = regulatory T cells.
Six of the 12 significantly increased plasma biomarkers have been related to AF or other cardiovascular diseases. These are: AREG, CXCL12, EGF, PLXNA4, TNFSF13, and WIF-1 (Supplemental Digital Figure 3; http://links.lww.com/HS/A152). Extracellular newly identified RAGE-binding protein (EN-RAGE; also known as S100A12) and stem cell factor (SCF; also known as Kit ligand) are two additional cardiovascular disease-related biomarkers that showed an increasing trend. TNFSF13, also known as “a proliferation-inducing ligand” (APRIL), is a growth factor for B cells.
Apart from CXCL12 and WIF-1, all cardiovascular disease-related biomarkers whose levels increased during treatment were CLL nonassociated. CXCL12 and WIF-1 levels were lower in the pretreated patients compared to healthy donors, and both increased, essentially normalizing. Considering this, and because they are CLL-associated, we regard them as less likely candidates for reflecting cardiovascular toxicity. Moreover, AREG, EGF, EN-RAGE, PLXNA4, and WIF-1 are not expressed by B cells nor CLL cells. Importantly, these biomarkers seem to be BTK-independent, since their plasma levels do not differ between healthy donors and patients with XLA (Figure 6). References to publications that show the expression of these biomarkers in human cardiac tissue and their association with AF are listed in Table 2 and Supplemental Digital Table 4; http://links.lww.com/HS/A152.
Table 2.
Eight Cardiovascular Disease-related Biomarkers That Increase During Ibrutinib Treatment
| Protein Name | Expression in Human Cardiac Tissue | Association with AF |
|---|---|---|
| AREG | 35 | 36,37 |
| CXCL12 (SDF-1)a | 38 | 39 |
| EGF | 40 | 41 |
| Plexin-A4 | see Supplemental Digital Table 4; http://links.lww.com/HS/A152 | see Supplemental Digital Table 4; http://links.lww.com/HS/A152 |
| TNFSF13 (APRIL) | see Supplemental Digital Table 4;http://links.lww.com/HS/A152 | 42 |
| WIF-1 | 43,44 | 43,45 |
| SCF (Kit ligand)b | 46,47 | 48 |
| EN-RAGE (S100A12)b | 49,50 | 51,52 |
aCLL-associated markers normalizing during ibrutinib treatment.
bIncreasing trend (indicative but not statistically significant) in this study.
CCL = chemokine ligand; CXCL12 = C-X-C motif chemokine ligand 12.
Ibrutinib-treated patients have a higher risk of developing AF than age-matched healthy individuals or patients with CLL who do not take the drug.53 Other cardiovascular events, such as ventricular arrhythmias, conduction disorders, and hypertension, have also been observed under ibrutinib.53
Here, we hypothesize that the plasma levels of 6 of the biomarkers which increase, 4 statistically significant, 2 trending (ie, AREG, EGF, EN-RAGE, PLXNA4, TNFSF13, and SCF) may be related to AF. Their characteristics are detailed in the Supplemental Digital Results and Discussion; http://links.lww.com/HS/A152. Five of them are mainly expressed in cardiac tissue (Table 2 and Supplemental Digital Table 4; http://links.lww.com/HS/A152), while TNFSF13 is also synthesized by CLL cells (Figure 6). It may seem contradictory that TNFSF13 is categorized as a non-associated marker, since it can be produced by B cells as well. However, as discussed in the next section, the TNFSF13 level is increased in XLA patients who essentially lack B cells and, moreover, the level increases in patients with CLL during ibrutinib treatment.
Plasma biomarker levels in patients with XLA
To further investigate the origin of the biomarkers, we compared their levels in patients with XLA with those in healthy donors. Because the CD5+ cells represent the B lymphocyte population most sensitive to BTK deficiency, while, conversely, CLL cells are known to carry CD5, B cells in these disorders could be regarded as representing opposite ends of the spectrum. Interestingly, this population, at least in mice, is autoreactive,54 similar to the malignant clone in many patients with CLL. Moreover, CD5 marks both CLL and mantle cell lymphoma, which both respond to BTK inhibitors, while other B cell malignancies lack, or show low levels, of this surface marker,55 suggesting that there may be a functional correlation.
BTK is expressed in hematopoietic cells such as B cells and myeloid cells, but not in T cells and plasma cells.56,57 No significant changes were observed when comparing the 265 plasma biomarker levels, when P values were corrected for the number of comparisons. However, 53 of the biomarkers show indicative changes and, unexpectedly, 46 (87%) of these were increased in patients with XLA (Supplemental Digital Table 5; http://links.lww.com/HS/A152). Among these, CXCL10, IL-6, IL-10, and TNFSF13 have been previously reported as elevated in patients with XLA.58–61 This verifies our set of data, suggesting that our findings might have biological relevance in spite of not reaching statistical significance.
Two conclusions can be made based on these findings. First, while nonmalignant B and plasma cells contribute to the large immunoglobulin fraction among plasma proteins, other plasma biomarkers seem to originate only marginally from nonmalignant B cells. Second, while many other hematopoietic cells express BTK,56,57 the plasma proteins that derive from these cells seem to be essentially independent of BTK. This is compatible with the fact that the signs and symptoms of XLA are mainly caused by the B-cell defect.14
Among the molecules that showed a tendency toward reduction in patients with XLA, T-cell leukemia/lymphoma 1 (TCL1) was observed (Supplemental Digital Table 5; http://links.lww.com/HS/A152). TCL1 is a predominantly cytoplasmic, 114 amino acid, lymphocyte-expressed protein, whose activity causes CLL in IgH-Eμ-TCL1-transgenic mice,62 a leading model for experimental CLL. See SDC, Results and Discussion for further details.
Reduction of tumor burden drives changes in T- and NK-cell numbers
In patients with CLL, the CD8+ T-cell population consists of dominant clones and grows in association with the size of the CLL clone.63–65 Moreover, different patients with CLL with the same stereotypical BCR on their tumor cells share identical dominant T-cell clones, suggesting expansion of tumor-reactive T cells.66
In our cohort of patients responding to ibrutinib treatment, we found that the total T-cell, CD8+ and CD4+ T-cell numbers decreased and normalized (Figure 7C, E, G), and that the CD3-CD56+ population (NK cells) also decreased (Figure 7I), correlating positively with CD19+ cells numbers (Figure 7D, F, H, J), that is, with the shrinking of the tumor burden.
Figure 7.
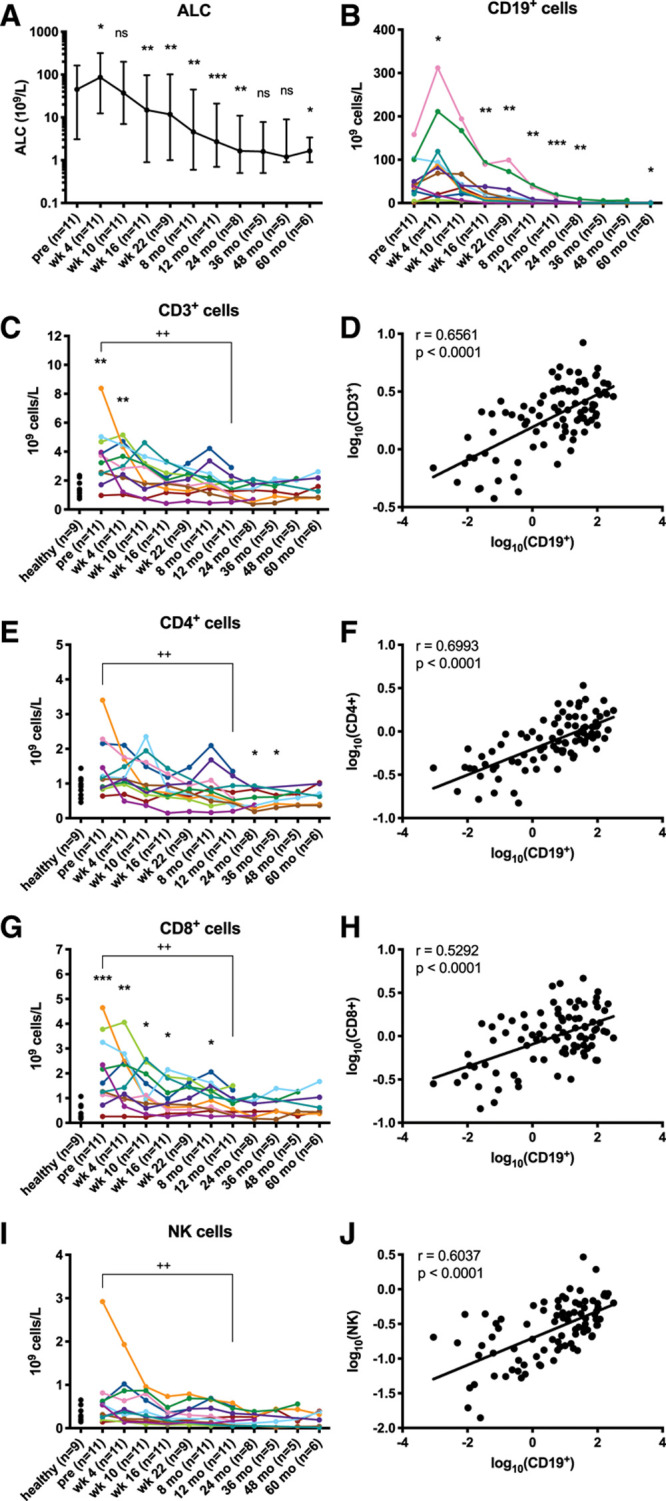
Lymphocytes and subsets decrease along with a reduction in tumor burden. (A) ALC is depicted as median and range on a log10 scale and (B) absolute numbers of CD19+ cells are depicted on a linear scale. Stars indicate a statistically significant difference between pre-treatment and the respective time point as analyzed by Wilcoxon signed-rank tests. (C, E, G, and I) Absolute numbers of cells are depicted per patient. Plus signs indicate a statistically significant difference between pretreatment and the respective time point as analyzed by Wilcoxon signed-rank tests. Stars indicate a statistically significant difference between healthy donors (6 males and 3 females, median age 65 y, range 45–79) and the respective time point as analyzed by Mann-Whitney U tests. (D, F, H, and J) Log10-transformed absolute cell numbers from all individual patient time points are depicted and lines indicate the linear regression of the X and Y variables. r indicates the Pearson correlation coefficient. *P < 0.05, **/++P < 0.005, ***P < 0.0005. ALC = absolute lymphocyte count; ns = not significant; pre = pretreatment.
Even if an increase in T-cell numbers after 6 months of ibrutinib treatment has been reported,67 probably related to a still partial reduction of the CLL clone, our results are consistent with data from other previous reports with longer follow-up.29–31 See Supplemental Digital Results and Discussion; http://links.lww.com/HS/A152 for further details.
T helper cells decrease along with the receding tumor burden
Ibrutinib has the potential to selectively decrease Th2 cell numbers by blocking ITK, causing Th1 skewing in patients24,68 and it has recently been demonstrated that ibrutinib’s effects on T cells are completely BTK-independent.69 Several studies have confirmed the reduction of some Th2-cytokines in ibrutinib-treated patients.29,30,67,70
However, we observed a steady decline in Th1 cell numbers, while Th2 cells did not decrease significantly. This led to a significant decrease of the Th1/Th2 ratio (Figure 8E). No statistically significant changes were observed in signature Th-cytokines (Supplemental Digital Results and Discussion; http://links.lww.com/HS/A152 and Supplemental Digital Figure 4; http://links.lww.com/HS/A152). However, a strong reduction in Th1 cells, which facilitates CD8+ T cell-mediated anti-tumor immunity, might have been triggered by the decreasing tumor burden in our cohort, which is not mutually exclusive with a selective reduction of Th2 cell numbers through ITK inhibition. Our observations from Th17 cells and Tregs are described in the Supplemental Digital Results and Discussion; http://links.lww.com/HS/A152 and displayed in Supplemental Digital Figure 5; http://links.lww.com/HS/A152.
Figure 8.
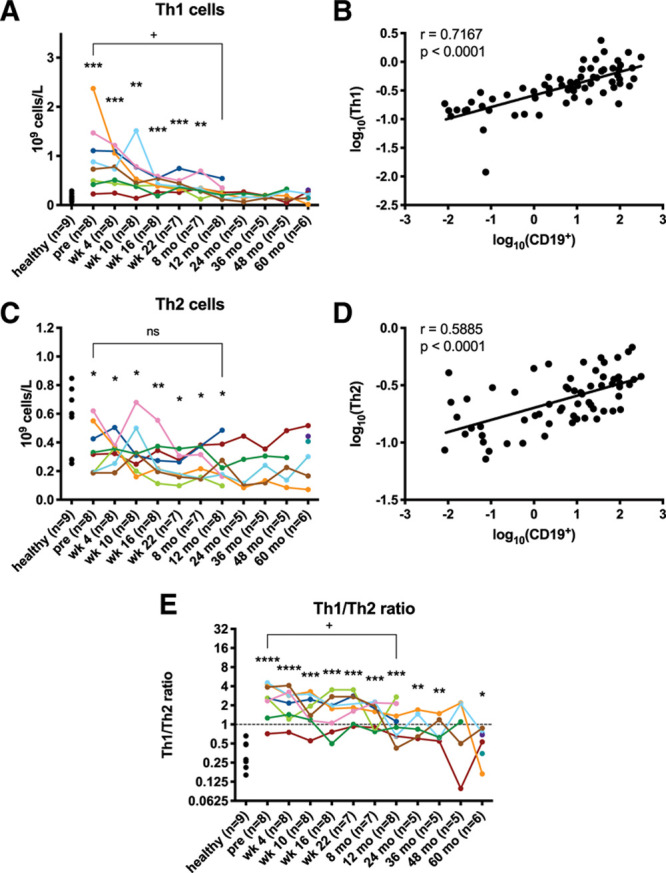
Th1 cell numbers decrease, causing Th2-skewing. (A, C, and E) Absolute numbers of cells are depicted per patient. Plus signs indicate a statistically significant difference between pretreatment and the respective time point as analyzed by Wilcoxon signed-rank tests. Stars indicate a statistically significant difference between healthy donors (6 males and 3 females, median age 65 y, range 45–79) and the respective time point as analyzed by Mann-Whitney U tests. (B and D) Log10-transformed absolute cell numbers from all individual patient time points are depicted and lines indicate the linear regression of the X and Y variables. r indicates the Pearson correlation coefficient. */+P < 0.05, **P < 0.005, ***P < 0.0005, ****P < 0.0001.
Chronically activated antigen-experienced T-cell populations subside
T cells are functionally suppressed in CLL1 and ibrutinib might reduce the expression of suppressive molecules on T cells.29,67,71 We observed a reduction in CTLA-4 and PD-1 expressing T cells as well (Supplemental Digital Figure 6; http://links.lww.com/HS/A152). Moreover, plasma levels of lymphocyte-activation gene 3 (LAG-3), another member of the immune checkpoint family of receptors, were significantly reduced (Figure 3). Furthermore, CD4+ effector memory (EM), CD8+ EM and CD8+ T effector memory re-expressing CD45RA (TEMRA) cells decreased after 12 months as well (Supplemental Digital Results and Discussion; http://links.lww.com/HS/A152; Supplemental Digital Figure 7; http://links.lww.com/HS/A152; Supplemental Digital Figure 8; http://links.lww.com/HS/A152).
Effects on non-malignant B cells, monocyte subsets and dendritic cell in peripheral blood
Long-term ibrutinib treatment also seemed to affect other immune cells (Supplemental Digital Results and Discussion; http://links.lww.com/HS/A152; Supplemental Digital Figure 9; http://links.lww.com/HS/A152; Supplemental Digital Figure 10; http://links.lww.com/HS/A152).
In conclusion, these plasma protein and immune monitoring data confirm that ibrutinib has evident immunomodulatory characteristics and reveal potential mediators of ibrutinib-induced AF. While it cannot be excluded that the disease severity, prior treatment history and the relatively low number of patients examined might have had an impact on our observations, we believe that these could serve as a starting point for more in-depth functional studies, for example, to characterize the effect of BTK inhibitors with a narrower effect on other kinases using the biomarker assay system.
Disclosures
The authors have no conflicts of interest to disclose.
Sources of funding
This work was supported by grants from The Swedish Cancer Society, The Cancer Society in Stockholm, Swedish Research Council, King Gustav V Jubilee Fund, The Cancer and Allergy Foundation, The Karolinska Institutet Foundations, The Stockholm County Council, AFA Insurance, Dr Ake Olsson’s Foundation for Hematology research, The Knut and Alice Wallenberg Foundation and a generous donation from Björn and Lena Ulvaeus. In addition, the Karolinska Institutet doctoral education program (KID) and the Minciencias-Colombia scholarship program 756 (2016) supported the doctoral studies of LPP. and HYE., respectively.
Acknowledgments
The authors would like to thank the patients who consented to the use of their samples for this study. Thanks are due to Richard Rosenquist and Emma Young for performing the immunoglobulin heavy chain variable region mutational analysis; to Aleksandra Krstic, Barbro Näsman-Glaser and Ann Svensson for technical assistance; to Qing Wang for helping with patient sample collection.
Supplementary Material
Footnotes
CIES and MP contributed equally.
Supplemental digital content is available for this article.
References
- 1.Riches JC, Davies JK, McClanahan F, et al. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood. 2013; 121:1612–1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Honigberg LA, Smith AM, Sirisawad M, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci U S A. 2010; 107:13075–13080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ortolano S, Hwang IY, Han SB, et al. Roles for phosphoinositide 3-kinases, Bruton’s tyrosine kinase, and Jun kinases in B lymphocyte chemotaxis and homing. Eur J Immunol. 2006; 36:1285–1295 [DOI] [PubMed] [Google Scholar]
- 4.de Gorter DJ, Beuling EA, Kersseboom R, et al. Bruton’s tyrosine kinase and phospholipase Cgamma2 mediate chemokine-controlled B cell migration and homing. Immunity. 2007; 26:93–104 [DOI] [PubMed] [Google Scholar]
- 5.Herman SE, Mustafa RZ, Jones J, et al. Treatment with ibrutinib inhibits BTK- and VLA-4-dependent adhesion of chronic lymphocytic leukemia cells in vivo. Clin Cancer Res. 2015; 21:4642–4651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Rooij MF, Kuil A, Geest CR, et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood. 2012; 119:2590–2594 [DOI] [PubMed] [Google Scholar]
- 7.Chen SS, Chang BY, Chang S, et al. BTK inhibition results in impaired CXCR4 chemokine receptor surface expression, signaling and function in chronic lymphocytic leukemia. Leukemia. 2016; 30:833–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palma M, Krstic A, Peña Perez L, et al. Ibrutinib induces rapid down-regulation of inflammatory markers and altered transcription of chronic lymphocytic leukaemia-related genes in blood and lymph nodes. Br J Haematol. 2018; 183:212–224 [DOI] [PubMed] [Google Scholar]
- 9.Douglas AP, Trubiano JA, Barr I, et al. Ibrutinib may impair serological responses to influenza vaccination. Haematologica. 2017; 102:e397–e399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan Z, Scheerens H, Li SJ, et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem. 2007; 2:58–61 [DOI] [PubMed] [Google Scholar]
- 11.Berglöf A, Hamasy A, Meinke S, et al. Targets for ibrutinib beyond B cell malignancies. Scand J Immunol. 2015; 82:208–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsukada S, Saffran DC, Rawlings DJ, et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell. 1993; 72:279–290 [DOI] [PubMed] [Google Scholar]
- 13.Vetrie D, Vorechovský I, Sideras P, et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature. 1993; 361:226–233 [DOI] [PubMed] [Google Scholar]
- 14.Sideras P, Smith CI. Molecular and cellular aspects of X-linked agammaglobulinemia. Adv Immunol. 1995; 59:135–223 [DOI] [PubMed] [Google Scholar]
- 15.Noordzij JG, de Bruin-Versteeg S, Comans-Bitter WM, et al. Composition of precursor B-cell compartment in bone marrow from patients with X-linked agammaglobulinemia compared with healthy children. Pediatr Res. 2002; 51:159–168 [DOI] [PubMed] [Google Scholar]
- 16.Del Pino Molina L, Wentink M, van Deuren M, et al. Precursor B-cell development in bone marrow of Good syndrome patients. Clin Immunol. 2019; 200:39–42 [DOI] [PubMed] [Google Scholar]
- 17.Palma M, Gentilcore G, Heimersson K, et al. T cells in chronic lymphocytic leukemia display dysregulated expression of immune checkpoints and activation markers. Haematologica. 2017; 102:562–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Podhorecka M, Dmoszynska A, Rolinski J, et al. T type 1/type 2 subsets balance in B-cell chronic lymphocytic leukemia–the three-color flow cytometry analysis. Leuk Res. 2002; 26:657–660 [DOI] [PubMed] [Google Scholar]
- 19.Beyer M, Kochanek M, Darabi K, et al. Reduced frequencies and suppressive function of CD4+CD25hi regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood. 2005; 106:2018–2025 [DOI] [PubMed] [Google Scholar]
- 20.ten Hacken E, Burger JA. Molecular pathways: targeting the microenvironment in chronic lymphocytic leukemia–focus on the B-cell receptor. Clin Cancer Res. 2014; 20:548–556 [DOI] [PubMed] [Google Scholar]
- 21.Jitschin R, Braun M, Büttner M, et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood. 2014; 124:750–760 [DOI] [PubMed] [Google Scholar]
- 22.Sun C, Tian X, Lee YS, et al. Partial reconstitution of humoral immunity and fewer infections in patients with chronic lymphocytic leukemia treated with ibrutinib. Blood. 2015; 126:2213–2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prezzo A, Cavaliere FM, Bilotta C, et al. Ibrutinib-based therapy impaired neutrophils microbicidal activity in patients with chronic lymphocytic leukemia during the early phases of treatment. Leuk Res. 2019; 87:106233. [DOI] [PubMed] [Google Scholar]
- 24.Dubovsky JA, Beckwith KA, Natarajan G, et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood. 2013; 122:2539–2549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stiff A, Trikha P, Wesolowski R, et al. Myeloid-derived suppressor cells express bruton’s tyrosine kinase and can be depleted in tumor-bearing hosts by ibrutinib treatment. Cancer Res. 2016; 76:2125–2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mhibik M, Wiestner A, Sun C. Harnessing the effects of BTKi on T cells for effective immunotherapy against CLL. Int J Mol Sci. 2019; 21:E68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Natarajan G, Oghumu S, Terrazas C, et al. A Tec kinase BTK inhibitor ibrutinib promotes maturation and activation of dendritic cells. Oncoimmunology. 2016; 5:e1151592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kohrt HE, Sagiv-Barfi I, Rafiq S, et al. Ibrutinib antagonizes rituximab-dependent NK cell-mediated cytotoxicity. Blood. 2014; 123:1957–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niemann CU, Herman SE, Maric I, et al. Disruption of in vivo chronic lymphocytic leukemia tumor-microenvironment interactions by ibrutinib–findings from an investigator-initiated phase II study. Clin Cancer Res. 2016; 22:1572–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yin Q, Sivina M, Robins H, et al. Ibrutinib therapy increases T cell repertoire diversity in patients with chronic lymphocytic leukemia. J Immunol. 2017; 198:1740–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burger JA, Sivina M, Jain N, et al. Randomized trial of ibrutinib vs ibrutinib plus rituximab in patients with chronic lymphocytic leukemia. Blood. 2019; 133:1011–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Assarsson E, Lundberg M, Holmquist G, et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS One. 2014; 9:e95192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uhlen M, Karlsson MJ, Zhong W, et al. A genome-wide transcriptomic analysis of protein-coding genes in human blood cells. Science. 2019; 366:eaax9198. [DOI] [PubMed] [Google Scholar]
- 34.Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018; 131:2745–2760 [DOI] [PubMed] [Google Scholar]
- 35.Koeppen M, Lee JW, Seo SW, et al. Hypoxia-inducible factor 2-alpha-dependent induction of amphiregulin dampens myocardial ischemia-reperfusion injury. Nat Commun. 2018; 9:816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu L, Song S, Zhang YP, et al. Amphiregulin promotes cardiac fibrosis post myocardial infarction by inducing the endothelial-mesenchymal transition via the EGFR pathway in endothelial cells. Exp Cell Res. 2020; 390:111950. [DOI] [PubMed] [Google Scholar]
- 37.Sohns C, Marrouche NF. Atrial fibrillation and cardiac fibrosis. Eur Heart J. 2020; 41:1123–1131 [DOI] [PubMed] [Google Scholar]
- 38.Kucia M, Ratajczak J, Reca R, et al. Tissue-specific muscle, neural and liver stem/progenitor cells reside in the bone marrow, respond to an SDF-1 gradient and are mobilized into peripheral blood during stress and tissue injury. Blood Cells Mol Dis. 2004; 32:52–57 [DOI] [PubMed] [Google Scholar]
- 39.Li D, Bjørnager L, Langkilde A, et al. Stromal cell-derived factor 1α (SDF-1α): a marker of disease burden in patients with atrial fibrillation. Scand Cardiovasc J. 2016; 50:36–41 [DOI] [PubMed] [Google Scholar]
- 40.Munk M, Memon AA, Goetze JP, et al. Hypoxia changes the expression of the epidermal growth factor (EGF) system in human hearts and cultured cardiomyocytes. PLoS One. 2012; 7:e40243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Büttner P, Werner S, Sommer P, et al. EGF (epidermal growth factor) receptor ligands in atrial fibrillation: from genomic evidence to the identification of new players. Circ Arrhythm Electrophysiol. 2019; 12:e007212. [DOI] [PubMed] [Google Scholar]
- 42.Yamada Y, Sakuma J, Takeuchi I, et al. Identification of TNFSF13, SPATC1L, SLC22A25 and SALL4 as novel susceptibility loci for atrial fibrillation by an exome-wide association study. Mol Med Rep. 2017; 16:5823–5832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu D, Dong W, Zhang X, et al. WIF1 causes dysfunction of heart in transgenic mice. Transgenic Res. 2013; 22:1179–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meyer IS, Jungmann A, Dieterich C, et al. The cardiac microenvironment uses non-canonical WNT signaling to activate monocytes after myocardial infarction. EMBO Mol Med. 2017; 9:1279–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ress C, Paulweber M, Goebel G, et al. Circulating Wnt inhibitory factor 1 levels are associated with development of cardiovascular disease. Atherosclerosis. 2018; 273:1–7 [DOI] [PubMed] [Google Scholar]
- 46.Vandervelde S, van Luyn MJ, Rozenbaum MH, et al. Stem cell-related cardiac gene expression early after murine myocardial infarction. Cardiovasc Res. 2007; 73:783–793 [DOI] [PubMed] [Google Scholar]
- 47.Jahanyar J, Youker KA, Torre-Amione G, et al. Increased expression of stem cell factor and its receptor after left ventricular assist device support: a potential novel target for therapeutic interventions in heart failure. J Heart Lung Transplant. 2008; 27:701–709 [DOI] [PubMed] [Google Scholar]
- 48.Chua W, Purmah Y, Cardoso VR, et al. Data-driven discovery and validation of circulating blood-based biomarkers associated with prevalent atrial fibrillation. Eur Heart J. 2019; 40:1268–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ligthart S, Sedaghat S, Ikram MA, et al. EN-RAGE: a novel inflammatory marker for incident coronary heart disease. Arterioscler Thromb Vasc Biol. 2014; 34:2695–2699 [DOI] [PubMed] [Google Scholar]
- 50.Buyukterzi Z, Can U, Alpaydin S, et al. Enhanced S100A9 and S100A12 expression in acute coronary syndrome. Biomark Med. 2017; 11:229–237 [DOI] [PubMed] [Google Scholar]
- 51.Oesterle A, Bowman MA. S100A12 and the S100/calgranulins: emerging biomarkers for atherosclerosis and possibly therapeutic targets. Arterioscler Thromb Vasc Biol. 2015; 35:2496–2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nazari A, Khorramdelazad H, Hassanshahi G, et al. S100A12 in renal and cardiovascular diseases. Life Sci. 2017; 191:253–258 [DOI] [PubMed] [Google Scholar]
- 53.Salem JE, Manouchehri A, Bretagne M, et al. Cardiovascular toxicities associated with ibrutinib. J Am Coll Cardiol. 2019; 74:1667–1678 [DOI] [PubMed] [Google Scholar]
- 54.Graf R, Seagal J, Otipoby KL, et al. BCR-dependent lineage plasticity in mature B cells. Science. 2019; 363:748–753 [DOI] [PubMed] [Google Scholar]
- 55.Debord C, Wuillème S, Eveillard M, et al. Flow cytometry in the diagnosis of mature B-cell lymphoproliferative disorders. Int J Lab Hematol. 2020; 42Suppl 1113–120 [DOI] [PubMed] [Google Scholar]
- 56.de Weers M, Verschuren MC, Kraakman ME, et al. The Bruton’s tyrosine kinase gene is expressed throughout B cell differentiation, from early precursor B cell stages preceding immunoglobulin gene rearrangement up to mature B cell stages. Eur J Immunol. 1993; 23:3109–3114 [DOI] [PubMed] [Google Scholar]
- 57.Smith CI, Baskin B, Humire-Greiff P, et al. Expression of Bruton’s agammaglobulinemia tyrosine kinase gene, BTK, is selectively down-regulated in T lymphocytes and plasma cells. J Immunol. 1994; 152:557–565 [PubMed] [Google Scholar]
- 58.Amedei A, Romagnani C, Benagiano M, et al. Preferential Th1 profile of T helper cell responses in X-linked (Bruton’s) agammaglobulinemia. Eur J Immunol. 2001; 31:1927–1934 [DOI] [PubMed] [Google Scholar]
- 59.Gagliardi MC, Finocchi A, Orlandi P, et al. Bruton’s tyrosine kinase defect in dendritic cells from X-linked agammaglobulinaemia patients does not influence their differentiation, maturation and antigen-presenting cell function. Clin Exp Immunol. 2003; 133:115–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jin R, Kaneko H, Suzuki H, et al. Age-related changes in BAFF and APRIL profiles and upregulation of BAFF and APRIL expression in patients with primary antibody deficiency. Int J Mol Med. 2008; 21:233–238 [PubMed] [Google Scholar]
- 61.González-Serrano ME, Estrada-García I, Mogica-Martínez D, et al. Increased pro-inflammatory cytokine production after lipopolysaccharide stimulation in patients with X-linked agammaglobulinemia. J Clin Immunol. 2012; 32:967–974 [DOI] [PubMed] [Google Scholar]
- 62.Bichi R, Shinton SA, Martin ES, et al. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc Natl Acad Sci U S A. 2002; 99:6955–6960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wen T, Mellstedt H, Jondal M. Presence of clonal T cell populations in chronic B lymphocytic leukemia and smoldering myeloma. J Exp Med. 1990; 171:659–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Goolsby CL, Kuchnio M, Finn WG, et al. Expansions of clonal and oligoclonal T cells in B-cell chronic lymphocytic leukemia are primarily restricted to the CD3(+)CD8(+) T-cell population. Cytometry. 2000; 42:188–195 [PubMed] [Google Scholar]
- 65.Blanco G, Vardi A, Puiggros A, et al. Restricted T cell receptor repertoire in CLL-like monoclonal B cell lymphocytosis and early stage CLL. Oncoimmunology. 2018; 7:e1432328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vardi A, Vlachonikola E, Karypidou M, et al. Restrictions in the T-cell repertoire of chronic lymphocytic leukemia: high-throughput immunoprofiling supports selection by shared antigenic elements. Leukemia. 2017; 31:1555–1561 [DOI] [PubMed] [Google Scholar]
- 67.Long M, Beckwith K, Do P, et al. Ibrutinib treatment improves T cell number and function in CLL patients. J Clin Invest. 2017; 127:3052–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ryan CE, Sahaf B, Logan AC, et al. Ibrutinib efficacy and tolerability in patients with relapsed chronic lymphocytic leukemia following allogeneic HCT. Blood. 2016; 128:2899–2908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Estupiñán HY, Bouderlique T, He C, et al. Novel mouse model resistant to irreversible BTK inhibitors: a tool identifying new therapeutic targets and side effects. Blood Adv. 2020; 4:2439–2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Podhorecka M, Goracy A, Szymczyk A, et al. Changes in T-cell subpopulations and cytokine network during early period of ibrutinib therapy in chronic lymphocytic leukemia patients: the significant decrease in T regulatory cells number. Oncotarget. 2017; 8:34661–34669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kondo K, Shaim H, Thompson PA, et al. Ibrutinib modulates the immunosuppressive CLL microenvironment through STAT3-mediated suppression of regulatory B-cell function and inhibition of the PD-1/PD-L1 pathway. Leukemia. 2018; 32:960–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.