

PURPOSE
This phase I study (RAD1901-005; NCT02338349) evaluated elacestrant, an investigational oral selective estrogen receptor degrader (SERD), in heavily pretreated women with estrogen receptor–positive, human epidermal growth factor receptor 2–negative metastatic breast cancer, including those with estrogen receptor gene alpha (ESR1) mutation. The primary objective was to determine the maximum tolerated dose and/or recommended phase II dose (RP2D).
METHODS
The study consisted of a 3 + 3 design (elacestrant capsules) followed by expansion at RP2D (400-mg capsules, then 400-mg tablets) for the evaluation of safety and antitumor activity. Elacestrant was taken once daily until progression or intolerability.
RESULTS
Of 57 postmenopausal women enrolled, 50 received RP2D (400 mg once daily): median age, 63 years; median three prior anticancer therapies, including cyclin-dependent kinase 4,6 inhibitors (CDK4/6i; 52%), SERD (52%), and ESR1 mutation (circulating tumor DNA; 50%). No dose-limiting toxicities occurred; the most common adverse events at RP2D (400-mg tablet; n = 24) were nausea (33.3%) and increased blood triglycerides and decreased blood phosphorus (25.0% each). Most adverse events were grade 1-2 in severity. The objective response rate was 19.4% (n = 31 evaluable patients receiving RP2D), 15.0% in patients with prior SERD, 16.7% in patients with prior CDK4/6i, and 33.3% in patients with ESR1 mutation (n = 5/15). The clinical benefit rate (24-week) was 42.6% overall (n = 47 patients receiving RP2D), 56.5% (n = 23, ESR1 mutation), and 30.4% (n = 23, prior CDK4/6i). Elacestrant clinical benefit was associated with decline in ESR1 mutant allele fraction.
CONCLUSION
Elacestrant 400 mg orally once daily has an acceptable safety profile and demonstrated single-agent activity with confirmed partial responses in heavily pretreated patients with estrogen receptor–positive metastatic breast cancer. Notably, responses were observed in patients with ESR1 mutation as well as those with prior CDK4/6i and prior SERD. A phase III trial investigating elacestrant versus standard endocrine therapy is ongoing.
INTRODUCTION
Endocrine therapy (ET) is the mainstay treatment for patients with estrogen receptor–positive (ER+) metastatic breast cancer (mBC). However, the majority of patients with ER+ mBC experience disease progression, likely related to the development of resistance to ET.1,2 Notably, estrogen receptor gene alpha (ESR1) mutations are associated with acquired ET resistance and shorter progression-free survival (PFS) in patients receiving aromatase inhibitors (AIs),1,2 whereas PFS in patients treated with the selective estrogen receptor degrader (SERD) fulvestrant remains similar regardless of ESR1 mutation status.3,4 However, acquired ESR1 mutations also occur following fulvestrant treatment, possibly because of poor bioavailability and incomplete ER blockade by the required route of intramuscular administration.3 Fulvestrant is the only SERD approved for the treatment of postmenopausal women with hormone receptor–positive mBC. Thus, there is an unmet need for an SERD with activity in tumors harboring ESR1 mutations and improved bioavailability allowing oral administration, and therefore possibly improved activity.
CONTEXT
Key Objective
What is the recommended phase II dose of the novel oral selective estrogen receptor degrader elacestrant, and what is the preliminary efficacy and safety in postmenopausal women with advanced breast cancer with and without estrogen receptor gene alpha (ESR1) mutation?
Knowledge Generated
-
The recommended phase II dose of 400 mg once daily was safe and well-tolerated; adverse events were predominately grade 1-2 GI events.
In patients with a median of three prior lines of therapy for advanced breast cancer, 50% of whom had ≥ 1 ESR1 mutation, elacestrant 400 mg once daily demonstrated an objective response rate of 19.4%, including responses in patients with ESR1 mutation and overall clinical benefit rate (24 weeks) of 42.6%.
Relevance
These data demonstrated safety and preliminary antitumor activity of elacestrant, providing rationale for the phase III study comparing the efficacy and safety of elacestrant versus standard-of-care endocrine therapy (fulvestrant or aromatase inhibitor) in patients with estrogen receptor–positive, human epidermal growth factor receptor 2–negative advanced breast cancer (EMERALD; ClinicalTrials.gov identifier: NCT03778931).
Elacestrant is an investigational, nonsteroidal, oral SERD that degrades the ER in a dose-dependent manner and inhibits estradiol-dependent functions of ER target gene transcription induction and cell proliferation in ER+ BC cell lines.5-7 Estradiol-stimulated tumor growth was diminished by elacestrant in the ER+ MCF-7 cell line xenograft model and xenograft models derived from heavily pretreated patients,5-7 including models resistant to cyclin-dependent kinase 4,6 inhibitors (CDK4/6i) and fulvestrant and those harboring ESR1 mutations Y537S and D538G.5,8,9
RAD1901-005 (ClinicalTrials.gov identifier: NCT02338349) is a phase I study that evaluated elacestrant in heavily pretreated postmenopausal women with ER+, human epidermal growth factor receptor 2–negative (HER2−) mBC. This study was designed to determine the maximum tolerated dose and/or recommended phase II dose (RP2D) of elacestrant; assess safety, tolerability, and pharmacokinetics (PK); evaluate preliminary antitumor effect; and explore the relationship between ESR1 mutations identified in circulating tumor DNA (ctDNA) and clinical response to elacestrant.
METHODS
Study Design
RAD1901-005 was a multicenter, open-label, four-part, dose-escalation study conducted at 11 centers in the United States between April 2015 and October 2019. The study was originally designed as a two-part study evaluating safety, tolerability, and preliminary antitumor efficacy of elacestrant in a 3 + 3 dose-escalation phase (part A), followed by a safety expansion phase at the RP2D (part B)—both using a capsule formulation. Following the development of a tablet formulation, the design was amended to add two sequential cohorts: part C to evaluate the tablet administered at RP2D and part D to evaluate the tablet at RP2D in a more heavily pretreated patient population, including patients with prior CDK4/6i and fulvestrant and ≥ 2 lines of prior endocrine therapies for mBC (Data Supplement, online only).
The primary end point was the frequency of dose-limiting toxicities (DLTs) during the first 28 days of treatment (part A). Secondary end points included safety, PK, and investigator-assessed tumor response using RECIST v 1.1.10 Exploratory end points included correlation of tumor response with baseline ESR1 mutation status measured in ctDNA.
The study Protocol (online only) and supporting documents were approved by the institutional review board at each site. Written informed consent was obtained from all participants. The study was performed in accordance with ethical principles consistent with the Declaration of Helsinki and International Council of Harmonisation/Good Clinical Practice and applicable regulatory requirements.
Patients
For all study parts, eligible patients were women of age ≥ 18 years and postmenopausal (defined in the Data Supplement) with ER+ (≥ 1% staining by immunohistochemistry)11 and HER2− locally advanced, inoperable, and/or metastatic breast adenocarcinoma and Eastern Cooperative Oncology Group performance status 0-1. Exclusion criteria included treatment with strong cytochrome 3A4 inducers or inhibitors; any ET within 14 days; chemotherapy within 28 days; luteinizing hormone-releasing hormone analogue within 12 months; and endometrial disorders, clinically significant cardiac disease, or history of thrombotic coagulopathy within prior 6 months. Prior anticancer therapy requirements differed between parts A-C and part D (Data Supplement). Briefly, parts A-C required ≤ 2 prior lines of chemotherapy for advanced or metastatic breast cancer and ≥ 6 months of prior ET in any setting with no limit on the number of lines of prior ET; part D required ≤ 1 prior line of chemotherapy for advanced or metastatic breast cancer, ≥ 2 prior lines of ET for advanced or metastatic breast cancer (as single agent or in combination, including one prior line of treatment with fulvestrant required with documented progression), and prior CDK4/6i. Full eligibility criteria are described in the Data Supplement.
Study Procedures
Patients were instructed to take elacestrant orally once daily approximately 30 minutes after a light meal. Elacestrant was provided as either multiple 100-mg capsules or a single 400-mg tablet. Elacestrant was continued until disease progression, intolerability, or withdrawn consent. Dosing interruptions < 7 days, but no dose reductions, were permitted for adverse events (AEs).
In part A, up to 18 patients were planned to be assigned sequentially to escalating doses of elacestrant with capsule formulation using a standard 3 + 3 design (200 mg-1,000 mg once daily in 200-mg dose increments), with a minimum of three to six evaluable patients at each dose. Dose escalation continued until the maximum tolerated dose (defined in the Data Supplement) was identified or an RP2D was selected based on safety and preliminary efficacy evaluations. In part B and part C, up to an additional 20 and 12 patients, respectively, were to be enrolled to evaluate the safety, tolerability, and preliminary efficacy of the RP2D of the capsule formulation (part B) and tablet formulation (part C). In part D, 36 patients with more specified prior anticancer therapies were to be enrolled to inform the planned phase II study design; change in corporate strategy led to early termination of enrollment after 10 patients in this cohort.
Assessments
Study visits occurred weekly for 1 month, then monthly, with a follow-up for 30 days after discontinuation of treatment or until resolution/stabilization of treatment-related AEs to grade ≤ 2. Physical examination, vital signs, hematology, chemistry and coagulation laboratory investigations, Eastern Cooperative Oncology Group assessment, and 12-lead ECG were performed monthly and at the end of treatment (EOT).
Tumor assessments (RECIST v1.1) were performed at baseline and then every 8 weeks. Responses were confirmed ≥ 4 weeks after the first documented response. Blood samples for ctDNA analysis were collected at screening, during treatment, and at the EOT. ESR1 mutations in ctDNA samples in parts A-C were analyzed using the OncoBEAM platform (Sysmex Inostics, Hamburg, Germany); samples in part D were analyzed using the Guardant360 assay (Guardant Health, Redwood City, CA); when residual samples were available, part A-C samples were retested using the Guardant assay (assay details in the Data Supplement). Blood samples for PK were collected pre- and postdose on day 8 and predose on day 28 and monthly (details in the Data Supplement). AEs were graded according to the National Cancer Institute Common Terminology Criteria for AEs v4.03 and coded using Medical Dictionary for Regulatory Activities, v17.1.
Statistical Methods
Data were summarized descriptively by dose cohort and the overall population treated at the RP2D. Safety data are presented separately for patients treated with capsule and tablet formulations. Efficacy data are presented for all parts combined, as well as for parts A-C and part D separately (Data Supplement) and patients with ESR1 mutation, prior SERD, and prior CDK4/6i.
Analysis populations are defined in the Data Supplement. Response analyses were performed on the response-evaluable (RE) population. Analyses of clinical benefit rate (CBR; defined as partial response plus stable disease for ≥ 24 weeks) were performed on the clinical benefit–evaluable (CBE) population. Analyses of PFS using the Kaplan-Meier method were performed on the intent-to-treat population.
RESULTS
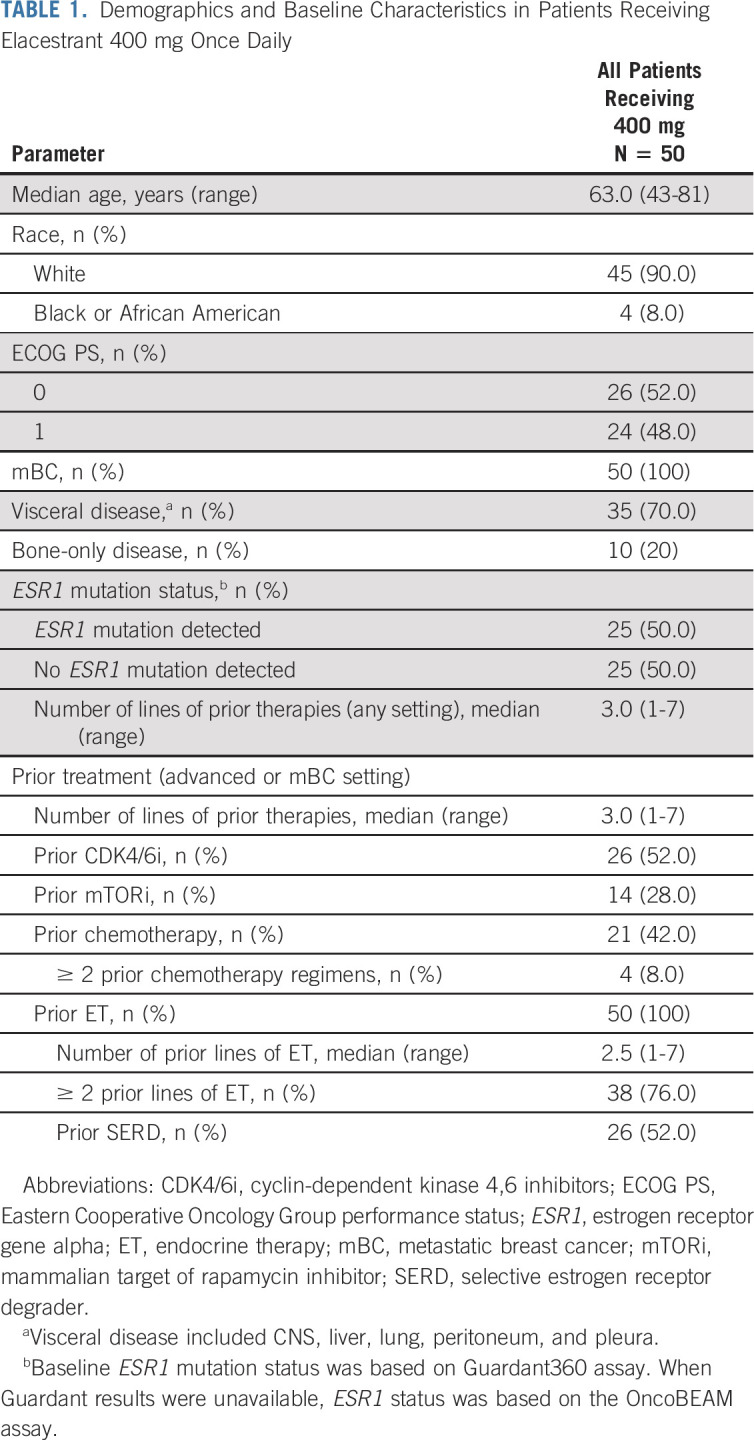
A total of 57 postmenopausal women with ER+, HER2− mBC were enrolled (Data Supplement), 50 at the RP2D of 400 mg once daily (26 with capsule and 24 with tablet). Baseline characteristics are presented for patients who received the RP2D (all patients, Table 1; by study part, Data Supplement) and for all dose cohorts (Data Supplement). In patients who received the RP2D, median age was 63 years; median number of prior lines of anticancer therapies in all settings and in the advanced setting was three; 52.0% of patients had prior CDK4/6i; and 52.0% had a prior SERD.
TABLE 1.
Demographics and Baseline Characteristics in Patients Receiving Elacestrant 400 mg Once Daily
ESR1 mutations were detected at baseline in 50% of patients treated at RP2D. The most common ESR1 mutations were D538G and Y537S; 44.0% of patients had > 1 ESR1 mutation (Fig 1A and Data Supplement). The frequency of ESR1 mutations increased with increasing lines of prior ET and was highest among patients with prior AI therapy (Fig 1B).
FIG 1.

ESR1 mutation distribution and correlation with therapy in the intent-to-treat population receiving elacestrant 400 mg (n = 50). (A) Distribution and prevalence of baseline ESR1 mutations/indels. (B) Alteration frequency of ESR1 at baseline versus prior treatment history in correlation with the number of lines of prior ET in the mBC setting or type of prior ET (AI, SERM, or SERD) in any setting. Baseline ESR1 mutation status is based on Guardant360 assay; when Guardant results were unavailable, ESR1 status was based on Sysmex OncoBEAM assay. AI, aromatase inhibitor; ESR1, estrogen receptor gene alpha; ET, endocrine therapy; mBC, metastatic breast cancer; SERD, selective estrogen receptor degrader; SERM, selective estrogen receptor modulator.
Safety
Dose escalation proceeded to 600 mg once daily. No DLTs were reported; however, upper GI events (grade 1-2 nausea, vomiting, dyspepsia, esophageal pain, gastroesophageal reflux disease, and eructation) were concerning for long-term tolerability at 600 mg. Therefore, 400 mg once daily was selected as the RP2D. Expansion cohorts in parts B, C, and D confirmed acceptability of the safety profile.
For all patients receiving elacestrant 400 mg (parts A-D), the most common treatment-emergent AEs (TEAEs) were nausea (50.0%), dyspepsia (32.0%), vomiting (30.0%), and fatigue (28.0%; Table 2). These TEAEs all occurred with lower frequency among the 24 patients receiving one 400-mg tablet compared with the 26 patients receiving four 100-mg capsules: nausea (33.3% v 65.4%, respectively), dyspepsia (20.8% v 42.3%), vomiting (16.7% v 42.3%), and fatigue (20.8% v 34.6%). The majority of these events were of grade 1-2 severity. At the 400-mg dose level, grade 3-4 TEAEs occurred in 10 patients (41.7%) receiving the tablet and 12 patients (46.2%) receiving capsules. The most common grade 3-4 TEAEs in patients receiving the tablet were syncope (clinical event detailed in the Data Supplement) and decreased blood phosphorus, occurring in two patients each (8.3%). TEAEs leading to dose interruption and discontinuation and serious TEAEs are summarized in Table 2 and the Data Supplement. Dose interruptions for any reason occurred in 23 patients (46.0%).
TABLE 2.
TEAEs in Patients Receiving Elacestrant 400-mg Tablets or Capsules

Pharmacokinetics
Limited PK data were collected. The elacestrant plasma concentrations were similar between the tablet and capsule formulations at the two observation time points (predose and 4-hour postdose) (Data Supplement). Although this comparison does not reach the level of a bioequivalence assessment, these plasma concentration data supported the use of the tablet formulation in part D.
Antitumor Activity
Among the 50 patients receiving 400 mg once daily, 31 patients were in the RE population and 47 patients were in the CBE population. Eleven patients (22.0%) remained on treatment for ≥ 12 months; the longest treatment duration was 43 months (Fig 2A).
FIG 2.
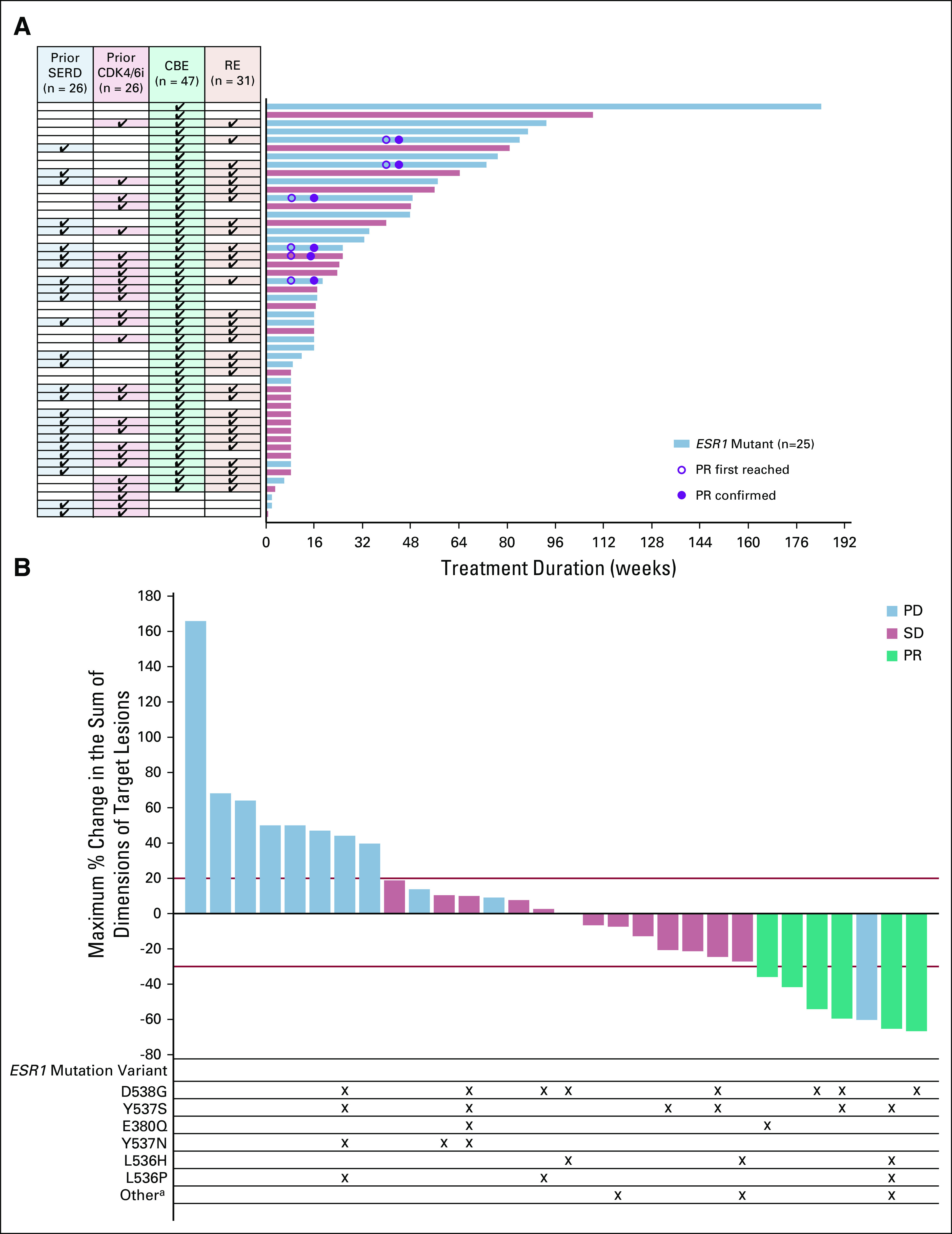
Duration of treatment in patients treated with 400 mg of elacestrant in parts A-D (A) and maximum percent change in sum of diameters of all target lesions (B) in response-evaluable patients treated with 400 mg of elacestrant in parts A-D. Baseline ESR1 mutation status is based on Guardant360 assay. When Guardant results were unavailable, ESR1 status was based on Sysmex OncoBEAM assay. aOther includes Y537C, E542D, L536_Y537del, S463P, and V534L. CBE, clinical benefit–evaluable; CDK4/6i, cyclin-dependent kinase 4,6 inhibitors; ESR1, estrogen receptor gene alpha; PD, progressive disease; PR, partial response; RE, response-evaluable; SD, stable disease; SERD, selective estrogen receptor degrader.
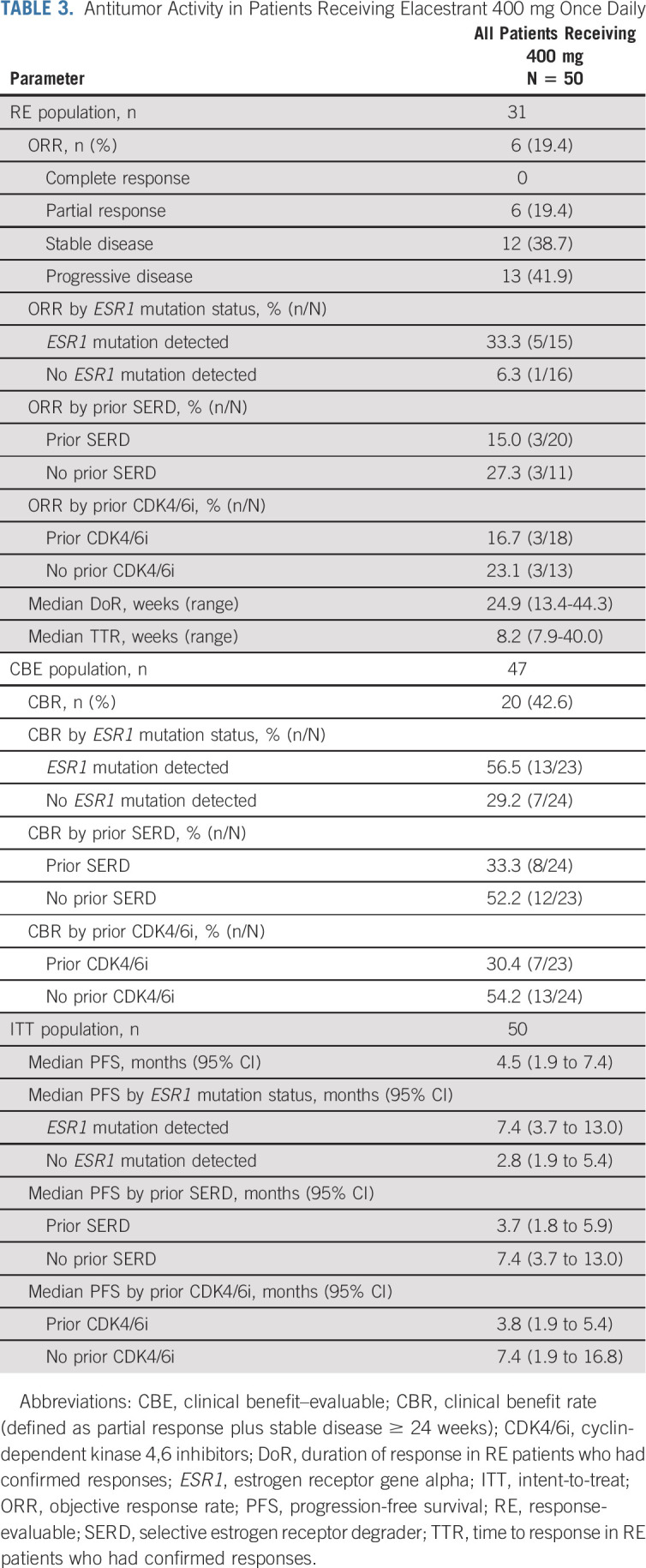
A partial response was observed in six patients for an objective response rate (ORR) of 19.4% among all 31 patients receiving 400 mg once daily (Table 3, Fig 2B). Responses were observed in patients with prior SERD (15.0%) and prior CDK4/6i (16.7%). Median time to response was 1.9 months (range, 1.8-9.3); median duration of response was 5.8 months (range, 3.1-10.3). The CBR was 42.6% among the 47 patients who received 400 mg once daily. The overall median PFS was 4.5 months (Data Supplement). Data for parts A-C, part D, and the intent-to-treat population (all doses studied) are presented in the Data Supplement.
TABLE 3.
Antitumor Activity in Patients Receiving Elacestrant 400 mg Once Daily
Relationship Between ESR1 Mutation and Response
Responses were observed in patients whose tumors harbored ESR1 mutation, including those commonly associated with ET resistance, that is, Y537S and D538G (Data Supplement). The ORR was 33.3% in patients with ESR1 mutation (n = 5/15) and 6.3% in patients with no ESR1 mutation (n = 1/16; Table 3). The CBR was 56.5% in patients with ESR1 mutation (n = 13/23) and 29.2% in patients with no ESR1 mutation (n = 7/24). Median PFS was 7.4 months in patients with ESR1 mutation and 2.8 months in patients with no ESR1 mutation.
Among the 25 patients with any baseline ESR1 mutation, 16 had at least one paired baseline and postbaseline sample, tested with the same assay, to assess the change in ESR1 mutant allele fraction (MAF). Among the 16 patients with any ESR1 mutation at baseline and at least one postbaseline sample (28 specific ESR1 mutations), reduction in MAF at cycle (C) 1 day (D) 28, C2D28, C3D28, and EOT occurred in 81.8%, 81.8%, 100%, and 73.1% of mutations, respectively (Fig 3A and Data Supplement). Among the eight patients with the most frequent ESR1 mutation, D538G, reduction in MAF at C1D28, C2D28, C3D28, and EOT occurred in 100%, 100%, 100%, and 75.0% of patients, respectively (Fig 3B). Patients with partial response tended to have decline in MAF on treatment, with rise in MAF at EOT (ie, disease progression) (Figs 3A and 3C).
FIG 3.
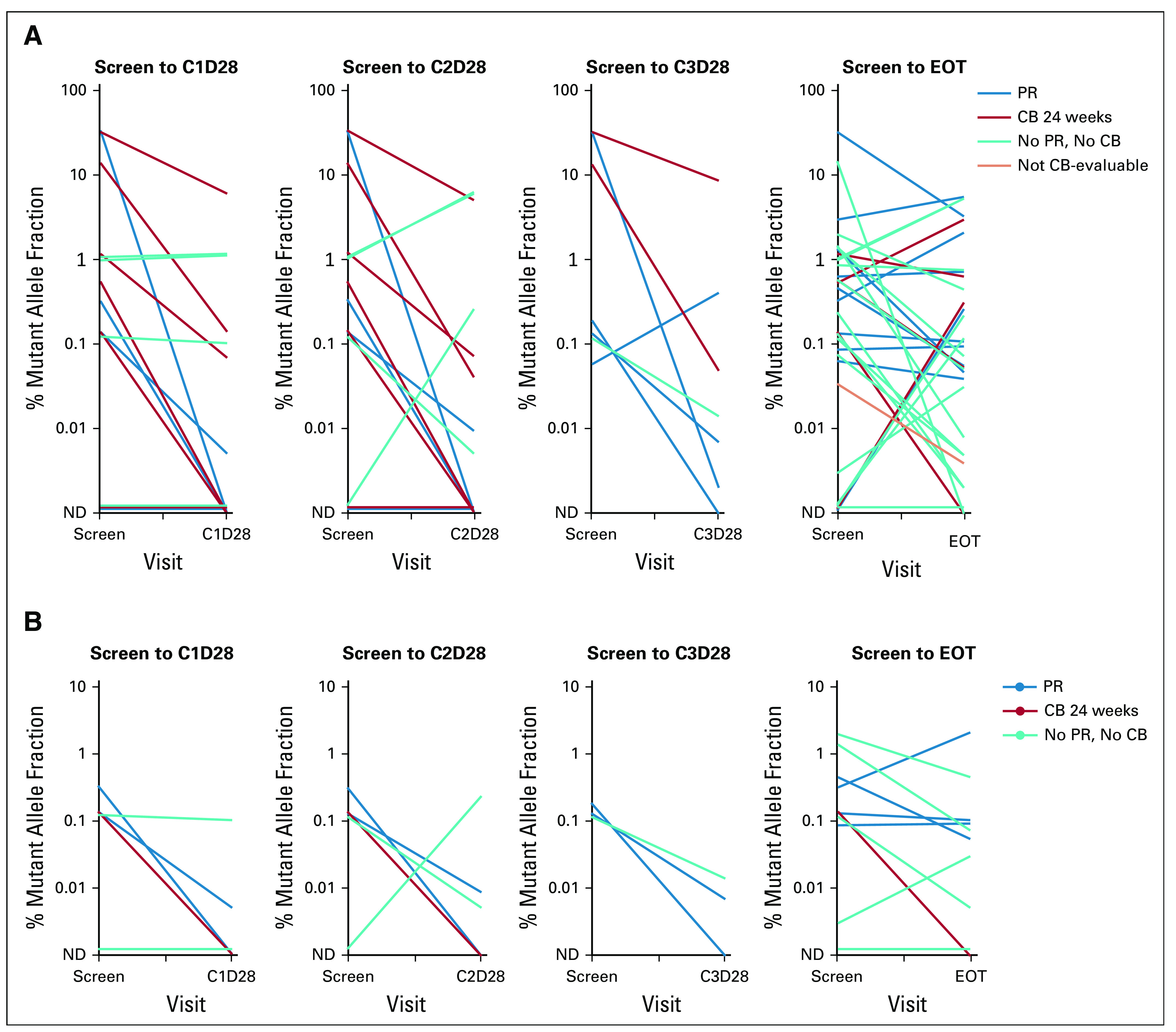
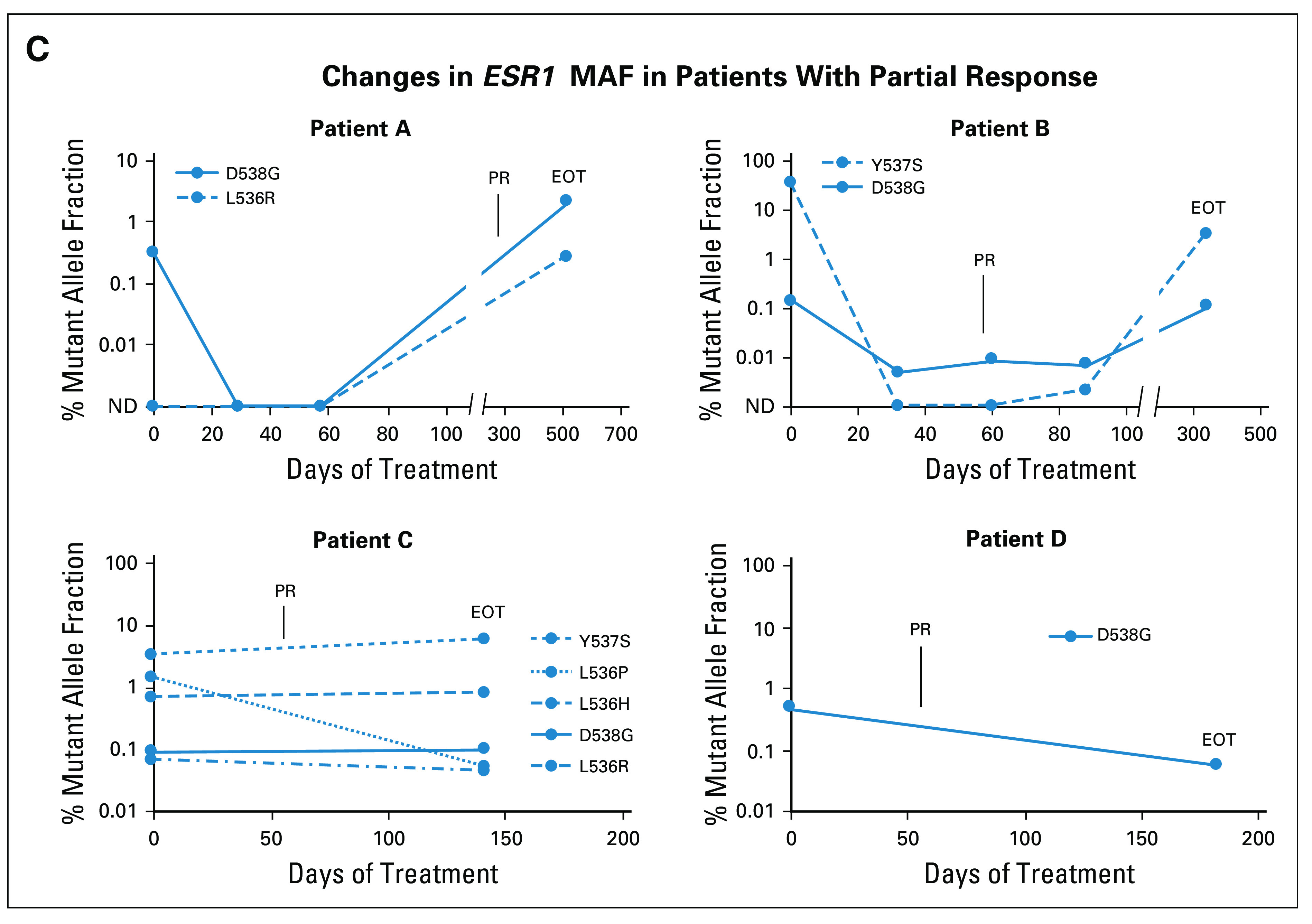
Change in ESR1 MAF for patients who received elacestrant 400 mg. Change in ESR1 MAF for all mutations in all patients with any baseline (screen) ESR1 mutation and at least one baseline and postbaseline sample tested with the same assay (A). Change in ESR1 D538G MAF for all patients with baseline D538G mutation and at least one baseline and postbaseline sample tested with the same assay (B). Change in ESR1 MAF for all ESR1 mutations in four patients with PR who had paired ESR1 data (C). C, cycle; CB, clinical benefit; EOT, end of treatment; ESR1, estrogen receptor gene alpha; MAF, mutant allele fraction; ND, mutation not detected; PR, partial response.
DISCUSSION
This phase I study of elacestrant demonstrated no DLTs at doses ranging from 200 mg to 600 mg once daily, a tolerable safety profile with the RP2D of 400 mg once daily, and reduced GI toxicity with the tablet formulation. At the 400-mg once-daily dose, antitumor activity was observed in patients with ER+, HER2− mBC, including those with prior fulvestrant and/or prior CDK4/6i and those patients whose tumors harbored ESR1 mutations.
Patients were heavily pretreated with a median of three prior lines of therapy in the advanced or metastatic breast cancer setting, 76% having received ≥ 2 prior ET, 52% having received a prior SERD (ie, fulvestrant), and 52% having received a CDK4/6i. Additionally, 50% of patients had ≥ 1 ESR1 mutation, consistent with extensive prior ET.2,12 In this poor prognostic setting, elacestrant demonstrated an ORR of 19.4%, CBR of 42.6%, and median PFS of 4.5 months. Only nine RE patients were enrolled in part D, and they had received more prior ET (median, three lines v two lines in parts A-C), which may explain the lack of responses in part D.
Responses were observed with elacestrant in patients whose tumors harbored ESR1 mutations that confer endocrine resistance, including Y537S and D538G. These data are consistent with results from a pharmacodynamic study (RAD1901-106) where elacestrant demonstrated an ORR and CBR of 11.1% and 30.8% among nine RE and 13 CBE patients, respectively, and a PFS of 5.3 months in a heavily pretreated population, of which 56% had ESR1 mutations.13 Study RAD1901-106 also demonstrated that elacestrant greatly reduced ER availability, as measured by 16α-18F-fluoro-17β-estradiol positron emission tomography with low-dose computed tomography imaging, showing a median reduction of 88% in tumor 16α-18F-fluoro-17β-estradiol uptake from baseline to day 14 that was consistent regardless of ESR1 mutation status.13 The decline in MAF of different ESR1 mutations with elacestrant in the present study also provides pharmacodynamic proof of principle of the RP2D and highlights broad activity across different ESR1 mutations. Additionally, it suggests the potential to use ESR1 MAF using ctDNA to monitor response to elacestrant therapy.
Elacestrant efficacy in ESR1-mutated tumors may indicate that ESR1 mutation identifies an ER-dependent tumor that is more likely to respond to an ER antagonist in the endocrine-refractory setting. Endocrine resistance is conferred by various genetic mutations in addition to and mutually exclusive from ESR1 mutation, including genes involved in mitogen-activated protein kinase signaling that may confer ER independence and therapeutic refractoriness to ER inhibition.14
An unmet need exists for an oral SERD with improved activity, including tumors harboring ESR1 mutations. In trials evaluating fulvestrant monotherapy in predominantly second or later lines of therapy, response rates are approximately 10% or less.15-18 In BELLE-3, patients with AI pretreatment and endocrine/mTOR inhibitor combination therapy resistance demonstrated a 3% ORR to fulvestrant.15
Although elacestrant is the most advanced in clinical development, several investigational oral SERDs and an oral selective estrogen receptor covalent antagonist are in early development, including SAR439859, AZD9833, LSZ102, GDC-9545, G-1T48, and H3B-6545. Response rates for studies performed in a population of patients similar to the population used in this study have ranged from 1.3% for LSZ102 to 16% for AZD9833, with activity noted in patients with ESR1 mutations for most agents.19-23 Of note, direct cross-trial comparisons should be done with caution due to differences in eligibility and small patient numbers at different dose levels in these phase I studies.
GI side effects of elacestrant were improved with the tablet formulation compared with the capsule formulation, possibly due to reduced number of pills required with the tablet formulation and/or dissolution of the tablet lower in the GI tract. All GI events were approximately halved in frequency with the tablet compared with the capsule: nausea, 33.3% versus 65.4%; dyspepsia, 20.8% versus 42.3%; vomiting, 16.7% versus 42.3%; and diarrhea, 12.5% versus 34.6%. As such, all subsequent studies of elacestrant are performed with the tablet formulation.
Preliminary reports of safety data for other investigational SERDs and selective estrogen receptor covalent antagonist have reported diarrhea rates as high as 27% to > 50% with some SERDs19,20 and other AEs including bradycardia, ranging from 8% to 45%, and visual disturbances in 53% of patients.21,22,24
In conclusion, elacestrant at the RP2D of 400 mg orally once daily has an acceptable safety profile, suitability for administration as monotherapy or in combination with other targeted agents, and improved tolerability with the tablet formulation as compared with the initial capsule formulation with a safety profile characterized predominately by grade 1-2 GI events. Elacestrant demonstrated single-agent activity with confirmed partial responses in heavily pretreated postmenopausal women with advanced ER+ breast cancer, including those with prior CDK4/6i and prior fulvestrant as well as those whose tumors harbored ESR1 mutations that confer resistance to ET. These data provided the rationale for the phase III study comparing the efficacy and safety of elacestrant versus standard-of-care endocrine treatment (fulvestrant or AI) in patients with ER+, HER2− advanced breast cancer (EMERALD; NCT03778931), with the primary end point of PFS, assessed in both the overall population and the population of patients with ESR1 mutation.23 Elacestrant is the first oral SERD to be studied in a phase III clinical trial (activated in 2018), and this phase I study provides preliminary evidence of clinical activity as a potential new therapeutic class in breast cancer.
ACKNOWLEDGMENT
The authors thank Mark Phillips, PharmD, and Laura Evans, PharmD of Phillips Gilmore Oncology Communications Inc, for professional assistance with manuscript preparation.
Aditya Bardia
Consulting or Advisory Role: Novartis, Genentech, Pfizer, Spectrum Pharmaceuticals, BioTheranostics, Merck, Radius Health, Inc., Immunomedics, Genentech/Roche, Innocrin Pharma, Sanofi, Puma Biotechnology, Daiichi Sankyo/AstraZeneca
Research Funding: BioTheranostics
Virginia Kaklamani
Honoraria: Celgene, Eisai, Genentech, Novartis, Pfizer, Genomic Health, Puma Biotechnology, AstraZeneca, Seattle Genetics, Daiichi Sankyo
Consulting or Advisory Role: Amgen, Eisai, Puma Biotechnology, Celldex, AstraZeneca, Athenex
Speakers' Bureau: Eisai, Genentech, Celgene, Novartis, Genomic Health, Puma Biotechnology, Pfizer
Research Funding: Eisai
Sharon Wilks
Honoraria: Seattle Genetics
Consulting or Advisory Role: Seattle Genetics
Research Funding: Seattle Genetics
Donald Richards
Consulting or Advisory Role: Ipsen, Taiho Pharmaceutical, Seattle Genetics/Astellas, Mirati Therapeutics
Wael Harb
Research Funding: AI Therapeutics
Cynthia Osborne
Honoraria: Agendia, Guardant Health, Breast Cancer Index, Seattle Genetics, Immunomedics
Speakers' Bureau: Novartis, Lilly
Travel, Accommodations, Expenses: Novartis, Lilly
Robert Wesolowski
Consulting or Advisory Role: Puma Biotechnology, Pfizer, Roche Diagnostics
Research Funding: Acerta Pharma
Travel, Accommodations, Expenses: Puma Biotechnology, Pfizer, Roche Diagnostics
Meghan Karuturi
Consulting or Advisory Role: Pfizer, MD Anderson Physician's Network
Paul Conkling
Employment: Virginia Oncology Associates
Research Funding: Bristol-Myers Squibb, EMD Serono, Arcus Biosciences, Aptose Biosciences, Janssen, Pfizer, Bayer, Astex Pharmaceuticals
Rebecca G. Bagley
Employment: Radius Health, Inc.
Yamei Wang
Employment: Radius Health, Inc.
Stock and Other Ownership Interests: RDUS
Maureen G. Conlan
Employment: Radius Health, Inc.
Stock and Other Ownership Interests: Radius Health, Inc.
Peter Kabos
Consulting or Advisory Role: Lilly
Research Funding: Radius Health, Inc., Lilly, Pfizer, AstraZeneca, Sanofi, Genentech, Angiochem
No other potential conflicts of interest were reported.
See accompanying article on page 1383
SUPPORT
Supported by Radius Health, Inc.
CLINICAL TRIAL INFORMATION
AUTHOR CONTRIBUTIONS
Conception and design: Aditya Bardia, Maureen G. Conlan
Financial support: Maureen G. Conlan, Peter Kabos
Administrative support: Maureen G. Conlan
Provision of study materials or patients: Aditya Bardia, Sharon Wilks, Donald Richards, Wael Harb, Robert Wesolowski, Paul Conkling, Maureen G. Conlan, Peter Kabos
Collection and assembly of data: Aditya Bardia, Virginia Kaklamani, Sharon Wilks, Amy Weise, Donald Richards, Wael Harb, Cynthia Osborne, Robert Wesolowski, Paul Conkling, Rebecca G. Bagley, Maureen G. Conlan, Peter Kabos
Data analysis and interpretation: Aditya Bardia, Virginia Kaklamani, Sharon Wilks, Amy Weise, Wael Harb, Cynthia Osborne, Robert Wesolowski, Meghan Karuturi, Paul Conkling, Rebecca G. Bagley, Yamei Wang, Maureen G. Conlan, Peter Kabos
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Phase I Study of Elacestrant (RAD1901), a Novel Selective Estrogen Receptor Degrader, in ER-Positive, HER2-Negative Advanced Breast Cancer
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Aditya Bardia
Consulting or Advisory Role: Novartis, Genentech, Pfizer, Spectrum Pharmaceuticals, BioTheranostics, Merck, Radius Health, Inc., Immunomedics, Genentech/Roche, Innocrin Pharma, Sanofi, Puma Biotechnology, Daiichi Sankyo/AstraZeneca
Research Funding: BioTheranostics
Virginia Kaklamani
Honoraria: Celgene, Eisai, Genentech, Novartis, Pfizer, Genomic Health, Puma Biotechnology, AstraZeneca, Seattle Genetics, Daiichi Sankyo
Consulting or Advisory Role: Amgen, Eisai, Puma Biotechnology, Celldex, AstraZeneca, Athenex
Speakers' Bureau: Eisai, Genentech, Celgene, Novartis, Genomic Health, Puma Biotechnology, Pfizer
Research Funding: Eisai
Sharon Wilks
Honoraria: Seattle Genetics
Consulting or Advisory Role: Seattle Genetics
Research Funding: Seattle Genetics
Donald Richards
Consulting or Advisory Role: Ipsen, Taiho Pharmaceutical, Seattle Genetics/Astellas, Mirati Therapeutics
Wael Harb
Research Funding: AI Therapeutics
Cynthia Osborne
Honoraria: Agendia, Guardant Health, Breast Cancer Index, Seattle Genetics, Immunomedics
Speakers' Bureau: Novartis, Lilly
Travel, Accommodations, Expenses: Novartis, Lilly
Robert Wesolowski
Consulting or Advisory Role: Puma Biotechnology, Pfizer, Roche Diagnostics
Research Funding: Acerta Pharma
Travel, Accommodations, Expenses: Puma Biotechnology, Pfizer, Roche Diagnostics
Meghan Karuturi
Consulting or Advisory Role: Pfizer, MD Anderson Physician's Network
Paul Conkling
Employment: Virginia Oncology Associates
Research Funding: Bristol-Myers Squibb, EMD Serono, Arcus Biosciences, Aptose Biosciences, Janssen, Pfizer, Bayer, Astex Pharmaceuticals
Rebecca G. Bagley
Employment: Radius Health, Inc.
Yamei Wang
Employment: Radius Health, Inc.
Stock and Other Ownership Interests: RDUS
Maureen G. Conlan
Employment: Radius Health, Inc.
Stock and Other Ownership Interests: Radius Health, Inc.
Peter Kabos
Consulting or Advisory Role: Lilly
Research Funding: Radius Health, Inc., Lilly, Pfizer, AstraZeneca, Sanofi, Genentech, Angiochem
No other potential conflicts of interest were reported.
REFERENCES
- 1.Fribbens C O'Leary B Kilburn L, et al. : Plasma ESR1 mutations and the treatment of estrogen receptor-positive advanced breast cancer. J Clin Oncol 34:2961-2968, 2016 [DOI] [PubMed] [Google Scholar]
- 2.Chandarlapaty S Chen D He W, et al. : Prevalence of ESR1 mutations in cell-free DNA and outcomes in metastatic breast cancer: A secondary analysis of the BOLERO-2 clinical trial. JAMA Oncol 2:1310-1315, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O'Leary B Cutts RJ Liu Y, et al. : The genetic landscape and clonal evolution of breast cancer resistance to palbociclib plus fulvestrant in the PALOMA-3 trial. Cancer Discov 8:1390-1403, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spoerke JM Gendreau S Walter K, et al. : Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nature Comm 7:11579, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bihani T Patel HK Arlt H, et al. : Elacestrant (RAD1901), a selective estrogen receptor degrader (SERD), has antitumor activity in multiple ER+ breast cancer patient-derived xenograft models. Clin Cancer Res 23:4793-4804, 2017 [DOI] [PubMed] [Google Scholar]
- 6.Wardell SE Nelson ER Chao CA, et al. : Evaluation of the pharmacological activities of RAD1901, a selective estrogen receptor degrader. Endocr Relat Cancer 22:713-724, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garner F Shomali M Paquin D, et al. : RAD1901: A novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models. Anticancer Drugs 26:948-956, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patel HK Tao N Lee K-M, et al. : Elacestrant (RAD1901) exhibits anti-tumor activity in multiple ER+ breast cancer models resistant to CDK4/6 inhibitors. Breast Cancer Res 21:146, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel H Tao N Arlt H, et al. : Anti-tumor activity of elacestrant (RAD1901) in models harboring ESR1 mutations resistant to standard of care therapies. San Antonio Breast Cancer Symposium, San Antonio, TX, December 4-8, 2018 (abstr P6-20-08)
- 10.Eisenhauer EA Therasse P Bogaerts J, et al. : New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer 45:228-247, 2009 [DOI] [PubMed] [Google Scholar]
- 11.Hammond MEH Hayes DF Dowsett M, et al. : American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol 28:2784-3543, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeselsohn R Yelensky R Buchwalter G, et al. : Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor–positive breast cancer. Clin Cancer Res 20:1757-1767, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jager A de Vries EGE Menke-van der Houven van Oordt CW, et al. : A phase 1b study evaluating the effect of elacestrant treatment on estrogen receptor expression and estradiol binding to the estrogen receptor in metastatic breast cancer lesions using 18F-FES PET/CT imaging. Breast Cancer Res 22:97, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamilton EP Patel MR Armstrong AC, et al. : A first-in-human study of the new oral selective estrogen receptor degrader AZD9496 for ER+/HER2- advanced breast cancer. Clin Cancer Res 24:3510-3518, 2018 [DOI] [PubMed] [Google Scholar]
- 15.Di Leo A Johnston S Lee KS, et al. : Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 19:87-100, 2018 [DOI] [PubMed] [Google Scholar]
- 16.Johnston SR Kilburn LS Ellis P, et al. : Fulvestrant plus anastrozole or placebo versus exemestane alone after progression on non-steroidal aromatase inhibitors in postmenopausal patients with hormone-receptor-positive locally advanced or metastatic breast cancer (SoFEA): A composite, multicentre, phase 3 randomised trial. Lancet Oncol 14:989-998, 2013 [DOI] [PubMed] [Google Scholar]
- 17.Cristofanilli M Turner NC Bondarenko I, et al. : Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol 17:425-439, 2016 [DOI] [PubMed] [Google Scholar]
- 18.Baselga J Im S-A Iwata H, et al. : Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 18:904-916, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jhaveri K Juric D Yap Y-S, et al. : Interim results of a phase 1/1b study of LSZ102, an oral selective estrogen receptor degrader, in combination with ribociclib or alpelisib in patients with ER+ breast cancer who had progressed after endocrine therapy. Ann Oncol 31:S62-S82, 2020(suppl 1; abstr LBA1) [Google Scholar]
- 20.Hamilton EP Dees EC Wang JS-Z, et al. : Phase I dose escalation of H3B-6545, a first-in-class highly selective ERα covalent antagonist (SERCA), in women with ER-positive, HER2-negative breast cancer (HR+ BC). J Clin Oncol 37:1059, 2019(suppl; abstr 1059) [Google Scholar]
- 21.Hamilton EP Oliveira M Banerji U, et al. : A phase I dose escalation and expansion study of the next generation oral SERD AZD9833 in women with ER-positive, HER2-negative advanced breast cancer. J Clin Oncol 38:1024, 2020(suppl; abstr 1024) [DOI] [PubMed] [Google Scholar]
- 22.Lim E Jhaveri KL Perez-Fidalgo JA, et al. : A phase Ib study to evaluate the oral selective estrogen receptor degrader GDC-9545 alone or combined with palbociclib in metastatic ER-positive HER2-negative breast cancer. J Clin Oncol 38:1023, 2020(suppl; abstr 1023) [Google Scholar]
- 23.Bardia A Aftimos P Bihani T, et al. : EMERALD: Phase III trial of elacestrant (RAD1901) vs endocrine therapy for previously treated ER+ advanced breast cancer. Future Oncol 15:3209-3218, 2019 [DOI] [PubMed] [Google Scholar]
- 24.Campone M Bardia A Ulaner GA, et al. : Phase 1/2 study of SAR439859, an oral selective estrogen receptor (ER) degrader (SERD), in ER-positive (ER+)/human epidermal growth factor receptor 2-negative (HER2-) metastatic breast cancer (mBC). J Clin Oncol 38:1070, 2020(suppl; abstr 1070)32058846 [Google Scholar]