

ABSTRACT
Human innate cellular defence pathways have evolved to sense and eliminate pathogens, of which, viruses are considered one of the most dangerous. Their relatively simple structure makes the identification of viral invasion a difficult task for cells. In the course of evolution, viral nucleic acids have become one of the strongest and most reliable early identifiers of infection. When considering RNA virus recognition, RNA sensing is the central mechanism in human innate immunity, and effectiveness of this sensing is crucial for triggering an appropriate antiviral response. Although human cells are armed with a variety of highly specialized receptors designed to respond only to pathogenic viral RNA, RNA viruses have developed an array of mechanisms to avoid being recognized by human interferon-mediated cellular defence systems. The repertoire of viral evasion strategies is extremely wide, ranging from masking pathogenic RNA through end modification, to utilizing sophisticated techniques to deceive host cellular RNA degrading enzymes, and hijacking the most basic metabolic pathways in host cells. In this review, we aim to dissect human RNA sensing mechanisms crucial for antiviral immune defences, as well as the strategies adopted by RNA viruses to avoid detection and degradation by host cells. We believe that understanding the fate of viral RNA upon infection, and detailing the molecular mechanisms behind virus-host interactions, may be helpful for developing more effective antiviral strategies; which are urgently needed to prevent the far-reaching consequences of widespread, highly pathogenic viral infections.
KEWORDS: RNA viruses, viral RNA sensing, viral evasion, viral RNA degradation
Introduction
In light of recent outbreaks of severe acute respiratory syndrome (SARS) and chicken or pig flu, viruses have been observed to pose a threat, not only to the health of infected individuals, but more importantly, as a cause of pandemic. Such outbreaks may also result in serious problems for healthcare systems and economies worldwide.
Viruses are composed of a genetic material core, either single- (ss) or double-stranded (ds) DNA or RNA, and an outer shell. The genome of ssRNA viruses may be either positive-sense, the (+) strand, or negative-sense, the (-) strand. Upon infection, the genome of positive-strand RNA viruses, for example hepatitis C virus (HCV), SARS virus, and an alphavirus-like superfamily of viruses, is immediately ready to be translated into viral proteins. On the other hand, negative-strand RNA viruses, for example influenza or Ebola viruses, and dsRNA viruses, for example reoviruses, genomes are unreadable by ribosomes; thus requiring transcription by viral RNA (vRNA)-dependent RNA polymerase (RdRp) into positive-sense RNA strands prior to initiating viral gene expression [1].
Interestingly, highly pathogenic RNA viruses are responsible for zoonotic and epidemic diseases such as coronavirus disease (COVID-19) and flu, outbreaks of haemorrhagic fever such as yellow fever, Dengue fever, and Ebola disease, or encephalitis such as Japanese encephalitis and Zika fever. In this review, we will focus only on this particular group of viral pathogens, namely RNA viruses, and their interaction with human cell response pathways upon infection.
After only 24–48 hours after viral infection, up to 25% of all RNA molecules present in host cells are pathogenic vRNA [2], including viral messenger RNA (mRNA), encoding viral proteins, viral genomic RNA (gRNA), and ds replication intermediates (dsRNA). To prevent such a takeover of the host metabolism, the innate immune system protects cells, with adequate inflammatory and antiviral responses strongly relying on the proper detection of vRNAs. Viral nucleic acids are one of the strongest pathogen-associated molecular patterns (PAMPs), molecules causing particular immune system reactions. PAMPs are recognized by numerous pattern recognition receptors (PRRs) scattered across almost every part of the cell, where they sense the presence of viral particles. Among specific vRNA sensors, retinoic acid-inducible gene I (RIG-I)–like receptors (RLRs), Toll-like receptors (TLRs), 2′-5′-oligoadenylate synthetase (OAS), dsRNA-dependent protein kinase R (PKR), and interferon (IFN)-induced proteins with tetratricopeptide repeats (IFITs) can be found [3]. Upon detection of foreign RNA, type 1 IFN gene expression, as well as other inflammatory cytokines and chemokines, are stimulated to fight the infection. IFNs act in both an autocrine and paracrine manner, causing an effect on the cells from which the IFNs were produced or on nearby cells, respectively; thus activating IFN-alpha/beta receptors (IFNAR) and resulting in the expression of antiviral IFN-stimulated genes (ISGs) that have diverse antiviral and immunoregulatory functions.
In this review, we provide a comprehensive overview of the RNA sensing mechanisms involved in antiviral immune defence. Moreover, we discuss what countermeasures are implemented by RNA viruses to avoid being identified, as well as the cell strategies involved in diminishing the amount of vRNA upon its recognition. Understanding the RNA-sensing system and its detailed molecular mechanisms will help to better plan antiviral strategies, as more effective approaches are urgently needed to prevent the far-reaching consequences of highly pathogenic viral infections.
RNA sensing
During infection, vRNA is introduced into the cell. Foreign nucleic acid present in the cytosol is one of the most potent PAMPs; the host cell recognizes vRNA through the independent activation of several types of innate immune receptors in different cellular compartments (Fig. 1). In the cytoplasm, viral gRNA and its transcriptional intermediates are recognized mostly by RLRs, RIG-I, melanoma differentiation-associated gene 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2), which belong to the DExD/H-box helicase family. In endolysosomes, the major RNA sensors that recognize either ds or ss nucleic acid are TLRs 3, 7, and 8. Since newly RNA binding properties of IFITs were discovered, they have been started to be considered as the important ISGs responsible for foreign RNA recognition [2]. Moreover, very recently one member of NOD-like family of proteins, which is the best recognized as key players in activation of immune response during microbial infections [4], was identify as viral dsRNA sensor [5].
Figure 1.
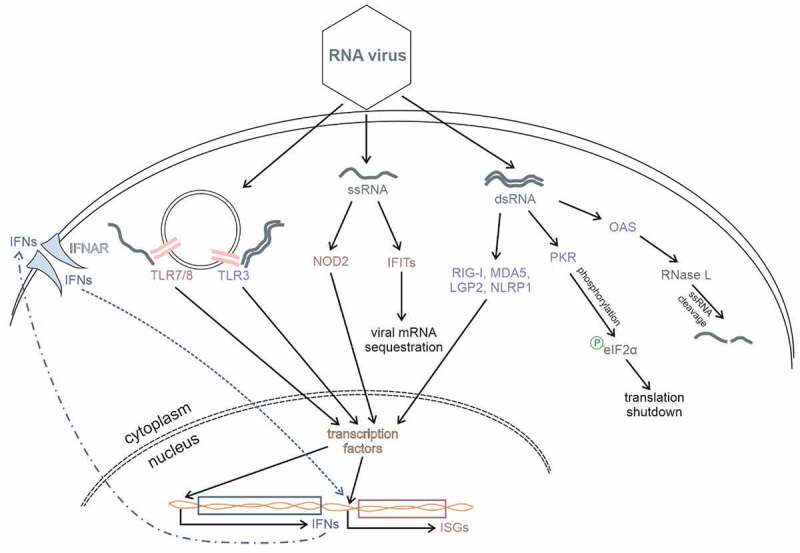
Cell signalling pathways that respond to viral RNA. During viral infection vRNA, single- or double-stranded, is introduced into the cell. This RNA is recognized by cellular sensors, RIG-I, MDA5, LGP2, PKR, OAS, TLR3/7/8, NLRP1, NOD2, and IFITs. Upon vRNA sensing, type I interferon response, as well as the production of antiviral IFN-stimulated genes (ISGs), is activated. Moreover, some vRNA sensors exert their function directly on viral RNA either by sequestrating viral transcripts from the pool of translationally active mRNAs (IFIT proteins) or by stimulating RNase L to degrade RNAs (OAS). Activation of PKR leads to phosphorylation of eukaryotic initiation factor eIF2α what subsequently results in global translation shutdown
For activation of any RNA sensor, detecting an abnormal molecular RNA pattern, not present under normal conditions, is obligatory. These patterns may be some chemical modification of RNA, or the absence of such one, specific secondary or tertiary RNA structure, particular sequence, or annealed dsRNA intermediates occurring during viral propagation [6].
Cellular RNA is usually unable to activate the immune response, with several modifications protecting self-RNA from being recognized by pattern recognition receptors in the cytoplasm, under the normal conditions described. Recent studies have revealed that discriminating between self and non-self RNA depends on the 5′ end of the molecule [7,8]. In the nucleus, RNA polymerases synthesize nascent transcripts possessing a 5′ triphosphate group (ppp-RNA) at the 5′ end. In the case of premature mRNA, 5′ ends of nascent transcripts undergo co-transcriptional modification – the addition of a cap structure, meaning N7-methylated guanosine is joined to the first transcribed nucleotide through a 5′-5′ triphosphate bridge, forming a cap-0 structure (Fig. 2). The roles of cap-0 in stability, splicing, polyadenylation, mRNA export, and translation have been well defined and characterized for many years [9,10]. Importantly, in humans, like in other higher eukaryotes, cap-0 is further modified by nuclear cap-specific 2′-O-RNA methyltransferase, where the 2′-O-position of the ribose of the first transcribed nucleotide is methylated. Furthermore, after mRNAs are exported to the cytoplasm, their cap-1 may be subjected to one more methylation, where the ribose of the second transcribed nucleotide may be methylated at position 2′-O, forming cap-210. Recent studies revealed that cap-1 plays a crucial role in distinguishing between self and non-self RNA [7,8], while the role of cap-2 is still elusive. Nevertheless, some other classes of cellular RNAs, for example transfer RNA (tRNA) and ribosomal RNA (rRNA), are subjected to specialized processing pathways through which mature 5′ monophosphorylated molecules are generated. Interestingly, a fraction of small nuclear RNAs (snRNAs) and small nucleolar RNAs (snoRNAs) undergo another modification in which a trimethylguanosine cap (TMG cap) is incorporated at their 5′ end [9]. Among the four nucleotides comprising cellular RNAs, namely adenosine triphosphate, cytidine triphosphate, uridine triphosphate, and guanosine triphosphate, some may be found chemically modified in a post-transcriptional manner. To date, more than 140 of these modifications have been characterized [11]. Interestingly, the presence of some post-transcriptionally modified nucleotides was shown to also modulate innate immune responses [11–13]. Finally, what also protects endogenous RNA from being recognized by RNA sensors is the association of nucleic acids with different proteins; as RNA rarely fulfils its function alone, rather operating as part of the ribonucleotide complex (RNP).
Figure 2.
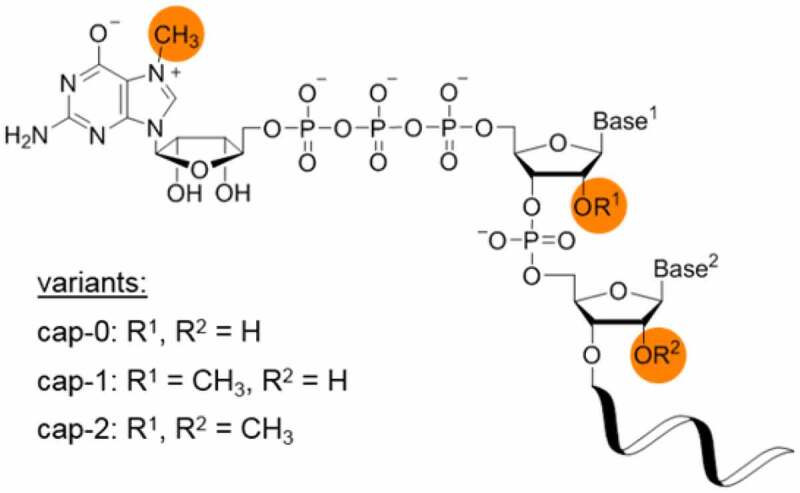
Structures of 7-methylguanosine RNA caps
RIG-I
RIG-I, with a multi-domain structure typical for proteins from the RLR group, is the first known member of the RLR family. RIG-I is composed of N- and C-terminal domains, with its central part known as a core. The core, a DExD/H-box helicase with an ATP-binding motif, is formed by subdomains Hel1 and Hel2, which are separated with a third subdomain, Hel2i. Hel1 and Hel2 bind ssRNA through several conserved motifs, named V and Ib. Hel2i has a regulatory function [31], also supposedly playing a role in dsRNA recognition [32,33]. Side RIG-I domains are the ss/dsRNA-binding C-terminal domain (CTD) and two N-terminal (caspase)-recruiting (CARD)-like domains. CARD domains stay repressed until the ligand is bound through the central DExD/H-box helicase core. Upon ligand binding, CTD releases the CARD domains, allowing them to bind to other CARD-containing proteins; this ability also makes CTD a repressor domain (RD) [34]. Interestingly, Hel2i can be mutated to produce constitutively active phenotypes, where the protein cannot be observed in its repressed form [31]. Interactions between the CARD domain and the Hel2i subdomain serve as receptor activation regulators, also governing ligand selection [35]. Activated RIG-I interacts with mitochondrial signalling protein (MAVS) at the cytosolic face of the outer mitochondrial membrane in order to trigger the signalling cascade [36]. However, to stimulate MAVS, CARD domain release is not sufficient; RIG-I must additionally undergo oligomerization [37].
The RIG-I CTD domain focuses on the 5′ end of RNA, recognizing molecules terminated with 5′-triphosphate or 5′-diphosphate groups [36,38,39] (Table 1). Therefore, distinguishing between self and non-self RNA occurs with 5′ end modification. Thanks to the presence of a cap structure on their 5′ end, endogenous mRNAs cannot be bound by RIG-I CTD [40]. Upon viral infection, short dsRNAs, 30–300 base pair (bp) molecules, bearing two to three phosphate groups at their 5′ ends, the most potent RIG-I activators, appear in the host cell cytoplasm. Nevertheless, RNAs up to 2 kbp have also been reported as RIG-I ligands [41,42]. Interestingly, dsRNA carrying cap-0 binds to RIG-I with similar affinity to ppp-dsRNA, with only the presence of cap-1 abrogating the interaction between dsRNA and this sensor protein [43]. Likewise, already mentioned RIG-I ligands, such as ssRNAs with a polyuridine-rich sequence and RNA bearing a 3′-phosphoryl group, can also activate this sensor protein [44,45]. Recently, Lu et al. described N6-methyladenosine (m6A) RNA modification as an additional marker for enabling discrimination between self and non-self RNAs. It was shown that human metapneumovirus RNAs are m6A methylated, guaranteeing deception of RIG-I [46].
Table 1.
Types of viruses and viral PAMPs to be recognized by various RNA sensing mechanisms
| Sensor | Virus | Viral genome | Viral PAMP |
|---|---|---|---|
| RIG-I | Orthomyxoviruses (Influenza A virus – IAV) [37,38] Paramyxovirus (measles, mumps, Sendai virus) [14,37,38] |
Negative-strand RNA | ppp-ssRNA/dsRNA cap-0-ssRNA/dsRNA |
| Flaviviruses (Hepatitis C Virus – HCV, Japanese encephalitis virus) [15] | Positive-strand RNA | ||
| MDA5 | Picornaviruses (poliovirus, encephalomyocarditis virus – EMCV, Theiler′s virus, and Mengo virus and rotavirus) [16,17,18,19-21,37,38] Flaviviruses (Dengue virus) [74] |
Positive-strand RNA | dsRNAs |
| Paramyxoviruses (Sendai virus) [20] Rhabdoviruses (Rabies virus) [17] |
Negative-strand RNA | ||
| LGP2 | Orthomyxoviruses (IAV) [21] Paramyxoviruses (Sendai virus, Newcastle disease virus) [52] |
Negative-strand RNA | non direct binding |
| Flaviviruses (HCV) [34] | Positive-strand RNA | ||
| TLR3 | Pneumoviruses (Respiratory Syncytial Virus – RSV) [22] Flaviviruses (West Nile virus) [22] Orthomyxoviruses (IAV) [23] |
Negative-strand RNA | dsRNA |
| Picornaviruses (poliovirus) [59] | Positive-strand RNA | dsRNA | |
| TLR7 | Orthomyxoviruses (IAV) [24] Rhabdoviruses (Rabies virus, Vesicular stomatitis virus – VSV) [25] Paramyxoviruses (Sendai virus) [82] |
Negative-strand RNA | ssRNA |
| Flaviviruses (Dengue virus) [74] | Positive-strand RNA | ssRNA | |
| TLR8 | Flaviviruses (HCV) [76] | Positive-strand RNA | ssRNA |
| PKR | Rhabdoviruses (VSV) [26] | Negative-strand RNA | dsRNA |
| IFIT1 | Flaviviruses (Japanese encephalitis virus) [27] | Positive-strand RNA | ppp-ssRNA, cap-0-ssRNA |
| Rhabdoviruses (VSV) [28] | Negative-strand RNA | ||
| IFIT2 | Rhabdoviruses (Rabies virus, VSV) [29] | Negative-strand RNA | ppp-ssRNA, cap-0-ssRNA |
| IFIT3 | Orthomyxovirus (IAV) [28] | Negative-strand RNA | ppp-ssRNA, cap-0-ssRNA |
| Phenuiviruses (Rift Valley virus) [28] | Negative-strand RNA | ||
| IFIT5 | Orthomyxovirus (IAV) [30] Rhabdoviruses (VSV) [30] |
Negative-strand RNA | ppp-ssRNA |
| NLRP1 | Togaviruses (Semliki Forest virus) [5] | Negative-strand RNA | dsRNA |
| NOD2 | Orthomyxovirus (IAV) [106] Pneumoviruses (RSV) [106] |
Negative-strand RNA | ssRNA |
MDA5
Similarly to RIG-I, MDA5, the second member of the RLR family, is composed of CARD, CTD, and helicase domains. However, in contrast to RIG-I, MDA5 does not recognize specific chemical groups at the 5′ end of RNA, rather discriminating for dsRNA based on molecular length [37,38]. Polyriboinosinic:polyribocytidylic acid (poly I:C), a synthetic analogue of dsRNA, was shown to, when long enough, behave as an MDA5 agonist; however, when successively digested to a length below 2 kbp, poly I:C became a RIG-I agonist [38]. Viral dsRNAs in the range of ~0.5–7 kbp are also reportedly recognized by MDA553. Differences in substrate specificity between MDA5 and RIG-I result from discrepancies in their C-terminal domain structures. MDA5 CTD lacks a positive charge in its binding pocket, making it unable to recognize RNAs based on their negatively charged di/triphosphorylated 5′ ends. The RNA binding surface of MDA5 is also flatter than in RIG-I. Moreover, MDA5 CTD has a lower affinity for dsRNA, compared to the RIG-I C-terminal domain [47]. However, CTD is necessary for MDA5 to bind dsRNA with positive cooperativity (the longer the dsRNA, the more MDA5 particles can bind to the nucleic acid chain, with each attached MDA5 molecule making it easier for the next MDA5 protein to recognize the transcript), and when this domain is deleted [48] or replaced with RIG-I CTD, cooperative binding is abolished [47,48]. Structural studies have further revealed that, along dsRNA, MDA5 forms filaments consisting of ring-structured elements occupying ~14 bp each [48]. Consistent with these studies is the finding that the level of antiviral response positively correlates with the amount of MDA5 protein bound to dsRNA; the longer filaments are, the longer target is recognized [47,49]. Longer filaments formed by MDA5 are also more stable when bound to dsRNA, explaining the dsRNA-length-dependence of MDA5 signalling activity [49]. Similarly to RIG-I, after binding to target RNA, MDA5 exposes a CARD domain and initiates cytokine and type I IFN production via MAVS signalling[50]. Although some viruses can be recognized by both MDA5 and RIG-I, these receptors predominantly recognize nonoverlapping RNA targets, implying that they possess distinct virus specificities (Table 1).
LGP2
Another RLR family member, LGP2, was originally found as a highly expressed gene in mammary tumours[34], where it was considered to be a potential negative regulator of RLR signalling[51]. Later, LGP2 expression appeared to also be induced by the presence of dsRNA, IFN treatment, or viral infection [52]. For instance, LGP2 protein overproduction inhibits both Sendai virus and Newcastle disease virus signalling[52]. Compared to other members of the RLR family, LGP2 lacks functional CARD domains and therefore cannot induce downstream signalling on its own [53]. Historically, as it was shown to act as both an activator and repressor of the immune response, the role of LGP2 has been disputed. Three models describing LGP2 involvement in viral dsRNA recognition have been proposed: 1) The first assumes LGP2 direct binding to vRNA, preventing RIG-I- and MDA5-mediated recognition [52]; 2) In the second, LGP2 supposedly inhibits oligomerization of RIG-I and its subsequent interaction with MAVS via the LPG2 repressor domain [34]; 3) The last model describes LGP2 as a competing factor of Iκβ kinase I (IKK-I) in the recruitment of MAVS, thereby suppressing RLR signalling. However, in light of recent data, the aforementioned models must be questioned; since LGP2 appears to be an important regulator of RIG-I- and MDA5-mediated immune responses [54]. LGP2 facilitates the sensing of some viruses, greatly enhancing, or even enabling, viral dsRNA recognition [51] (Table 1). Most current data suggest that LGP2, through the DExD/H helicase domain, supports vRNA recognition via RIG-I and MDA557. Recently, Takahashi et al [55]. described a new LGP2 function, suggesting that RIG-I, but not MDA5, indirectly interacts with the TAR-RNA binding protein (TRBP) through LGP2, regulating endogenous microRNA-mediated RNA silencing. TRBP is a dsRNA binding protein that binds to the HIV type 1 trans-activation response (TAR) element and functions as an RNA silencing enhancer [56].
Other DEXD/H box helicases
Recently, several additional DEXD/H box helicases have been reported to sense non-self dsRNAs. Zhang et al. showed that DDX1, DDX21, and DHX36 form a complex, binding to foreign dsRNA, and activating the type I IFN response [57]. DDX60, an orthologue of the yeast exosome cofactor Ski2, whose expression increases during a viral infection, is another example [58]. Unlike the previously mentioned three helicases, DDX60 does not act independently to sense dsRNA; instead binding to RIG-I-like receptors and promoting an anti-viral response [58]. DDX60 can also act independent of RLRs, for example in HCV RNA degradation [59]. Furthermore, a growing body of evidence suggests that other DEXD/H box helicases may exhibit multiple functions in innate immunity. Just to mention that, for example DDX3 plays an antiviral role against a broad range of RNA viruses, including HCV [60], Dengue virus [61], Japanese encephalitis virus [62], and West Nile virus of the Flaviviridae family [63] and influenza of the Orthomyxoviridae family [64].
TLRs
TLRs are transmembrane glycoprotein receptors with an extracellular N-terminal part, responsible for PAMP-binding, and an intracellular C-terminal region, also known as a Toll/IL-1 R homology (TIR) domain, which takes part in downstream signalling. Characteristic features of this membrane-anchored receptor family include multiple leucine-rich repeats (LRRs), which form a horseshoe structure in the extracellular ectodomain, similarity in the TLR C-terminal region to the intracellular domain of the interleukin-1 receptor (IL-1 R) [65], and proteolytic pre-activation by specific digestion of the ectodomain in order to allow ligand binding [66,67]. In humans, this family consists of 10 members, of which four, TLR3, 7, 8, and 9, are responsible for sensing foreign nucleic acids. Sequence homology has revealed that TLR7, TLR8, and TLR9 exist in one evolutionarily conserved cluster within the TLR family [68]. TLR9 recognizes unmethylated deoxycytidyl-phosphate-deoxyguanosine (CpG) motifs commonly present in bacterial and viral genomes [69], whereas TLR3 and TLR7 and 8 sense ds- and ssRNAs, respectively [70,71]. In addition, TLR7 and 8 can recognize uridine-containing ssRNA [71] and synthetic guanosine-like compounds, such as resiquimod (R-848) [72], as well as, in the case of TLR7, short interfering RNAs (siRNAs) [73]. The most potent TLR7 agonists are vRNAs 0.8–2 kb long, while molecules exceeding 11 kb generally exhibit less potency [74]. Although TLR7 and TLR8 play a similar biological role, their expression varies among different cell types. TLR7 is present in plasmacytoid dendritic cells, while TLR8, which has been associated with enhanced human papillomavirus infection clearance and recognition of other viral ssRNA (Table 1) [75], is characteristic in monocytes and macrophages [76]. TLR7 and TLR8 [77] both possess two different binding sites. The first binding site, which is well conserved among TLR proteins, recognizes small chemical components and is localized in the sensor dimerization interface; upon ligand binding TLRs dimerize. The second binding site, which is responsible for ssRNA recognition, has different localization between the two TLRs; in TLR7 it is found in the dimerization region, while in TLR8 it localizes beyond the dimerization interface. The first binding site preferentially recognizes guanosine, while the second binds uridine moieties in ssRNA specifically. Additionally, this second binding site has slightly different sequence specificity for particular proteins; TLR7 preferentially binds polyU 3-mer, while TLR8 senses UG or UUG oligoribonucleotides [78]. In turn, it seems that TLR3 does not have any sequence specificity, with the minimum length of dsRNA required for its cooperative binding 40–45 bp [79,80]. It recognizes the dsRNA of rotaviruses, as well as viral replication intermediates of ssRNA viruses rich in guanosine and uridine [81] (Table 1). Availability of target RNA to TLRs depends largely on endocytosis of extracellular viral particles and on autophagy, which transports cytosolic viral replication intermediates into the lysosome [82]. Lysosomal localization is also beneficial as TLRs exhibit pH dependence of RNA binding [83]. Reduced pH of the endosome/lysosome promotes protonation of histidine residues, thereby enhancing RNA phosphate backbone binding affinity for TLRs. Interestingly, Wang et al. showed that the influenza virus activates TLRs at a lower pH than Dengue virus [74].
PKR
The antiviral response is also mediated through kinases, among which, PKR can be distinguished. PKR is a powerful factor that is activated upon foreign dsRNA binding, leading to a global translation shutdown [84]. It is composed of two distinct domains, an N-terminal regulatory domain for binding dsRNA (dsRBD), and a C-terminal catalytic domain (kinase domain). Under normal conditions, PKR is maintained as an inactive monomer. The primary function of dsRBD is to promote protein dimerization and activation [85]. This domain responds to both viral and synthetic dsRNA particles [86] (Table 1). PKR requires only 20–80 bp of dsRNA to optimally bind and activate an immune response [87]. Additionally, it may be activated directly or indirectly by other stimuli such as oxidative stress, growth factors, cytokines, and cellular proteins, such as PKR-associated activators (PACTs), or following TLR stimulation [88,89]. PKR activation disrupts the autoinhibitory state of this protein, enabling its homodimerization and autophosphorylation, thereby promoting phosphorylation of the eukaryotic initiation factor 2 alpha subunit (eIF2α) [89]; which results in blocked translation initiation and accumulation of stalled ribosomal pre-initiation complexes. Importantly, as most RNA viruses require the participation of eIF2α in mRNA translation, the ability to cause global arrest of not only cellular, but also viral protein synthesis, can lead to apoptosis in response to viral infection [84,88], making PKR a prominent antiviral agent. Viral infection and translation inhibition results in the appearance of specific structures called stress granules (SGs), which contain transiently repressed mRNAs, initiation factors, small ribosomal subunits, and RNA-binding proteins, of which, one is PKR [88].
Recently, Pham et al. reported an additional function of PKR, which is independent of translation inhibition [90]. In the aforementioned study, PKR was shown to be critical for IFN production downstream of MDA5, but not RIG-I. In addition, MDA5 was required for stimulation of PKR catalytic activity occurring in response to infection by an MDA5-restricted virus, but not in response to a RIG-I-sensed virus. These findings identified the previously uncharacterized role of PKR catalytic action; which cooperates with MDA5 signalling and highlights MDA5 unexpected role in stimulating PKR enzymatic activity [90]. Therefore, it is not surprising that viruses have developed mechanisms to counteract the antiviral functionality of PKR. To avoid the immune response, viral proteins either bind viral dsRNA to prevent PKR pathogenic nucleic acid recognition, for example non-structural protein 3 (NSP3) from rotaviruses [91], or interact directly with PKR to prevent its activation, for example NSP1 from influenza viruses [92]. Similarly, the Japanese encephalitis virus uses its NSP2A to modulate PKR activity [93]. Among the multitude of evasion mechanisms that the HCV has evolved, inhibition of PKR through internal ribosome entry sites (IRESs) of gRNA has been distinguished. The HCV IRES has a complex secondary structure that efficiently competes with dsRNA for PKR binding, thus preventing kinase activation [94]. Moreover, HCV uses its NSP5A and E2 [95] proteins to directly bind and inhibit PKR [96], although the exact role of these proteins in this mechanism is not well established [97]. Interestingly, other viruses such as poliovirus and enterovirus induce rapid degradation of PKR [98,99].
IFITs
Among ISGs, the most important contributors to facilitating the immune response to exogenous RNA are IFITs [2] (Table 1). The human genome encodes five different IFIT proteins, namely IFIT1, IFIT1B, IFIT2, IFIT3, and IFIT5[100,101,102]. Although IFITs, in contrast to RLRs, recognize ssRNA and bind directly to non-self RNA to inhibit their translation or replication, only IFIT1 and IFIT5 are able to interact directly with RNA 5′ end. IFIT1 and IFIT5 discriminate RNA on the basis of the 5′ end of the transcript, with IFIT5 binding only ssRNAs with a triphosphate group at this position, while ligands of IFIT1 may possess a triphosphate group or a cap-0 [2,101,102]. Although crystallographic studies have revealed the presence of a similar positively charged tunnel through which IFIT1 and IFIT5 interact with the phosphate backbone of bound RNA at the 5′ end, only the former possesses a large hydrophobic cavity, accommodating the cap structure, at the rear of the tunnel [103]. The finding that IFIT1 could directly bind capped RNA supports a model in which it competes with eukaryotic translation initiation factor 4E (eIF4E) to bind to the N7-methylguanosine triphosphate cap at the 5′ end of vRNA [2]. Endogenous mRNAs are protected from IFIT1 recognition via the presence of cap-1. Interestingly, with the exception of IFIT5 which exists exclusively as a monomer, IFIT proteins have the propensity to homo- and heterooligomerize [100]. IFIT1 is able to form complexes of different composition, interacting with both, or one, of IFIT2 and IFIT3 [102,103]. In each case, complex formation strengthens the interaction between IFIT1 and RNA. Furthermore, it occurred that IFIT2 specifically binds to AU-rich RNAs irrespective of modifications present at transcript 5′ end [86]. Tran et al. revelled that IFIT2 binding enhances translation efficiency of cellular mRNAs including interferon stimulated genes in order to stimulate antiviral response [104]. Interestingly, influenza mRNAs also contain AU-rich sequences and thus influenza exploits IFIT2 to stimulate its own gene expression and replication.
NOD-like receptors
NOD-like receptor (NLR) is a large family of pattern recognition receptors responsible for immune system activation during microbial infection, cellular injury or stress [4]. NLR members share common structural features, they all are composed of N-terminal effector, central nucleotide binding domain (NBD) and C-terminal leucine-rich repeat (LRR) domain. Some of them, e.g. NLRP1 and NLRP3, were acknowledged to form multi-protein complexes known as inflammasomes, which are molecular platform responsible for signal transduction. However, other NLR members, e.g NOD1 and NOD2, can act in immune response in an inflammasome-independent manner. Interestingly, besides their well-established role in immune response to bacterial infection, it occurred that some NLR members are engaged in antiviral response, among them NOD2 and NLRP3 could be distinguished. It is known that NOD2 is involved in the restriction of respiratory syncytial virus and influenza virus [105], whereas NLRP3 is important for immune response upon Sendai virus and influenza infection [5,106]. Both proteins induce interferon production via MAVS signalling, however only NOD2 was described as direct RNA sensor (Table 1). Very recently, unexpectedly Bauernfried et al. presented that another member of NLR, namely NLRP1, which antiviral activity was well documented in rodents [107], is engaged in immune response in human cells [5]. It occurred that NLRP1 is a direct sensor for RNA binding double-stranded molecules [5], while NOD2 recognizes ssRNAs [105]. Moreover, NLRP3, like MDA5, activates immune response upon recognition long dsRNAs [5,37,38]. This observation puts NLRP1 together with other pattern recognition receptors such as MDA5 and RIG-I as genuine dsRNA sensor.
Viral evasion and host shutoff
Although human cells have formed various pathways for the detection of viral particles, the multitude of our defence systems have forced viruses to develop sophisticated mechanisms through which to deceive host cells (Fig. 3). Several methods through which RNA viruses hide, shield, or modify their own genetic material to avoid recognition are discussed below. We also present some clever methods that viruses utilize to modulate cell RNA metabolism, and finally lead to host shutoff.
Figure 3.
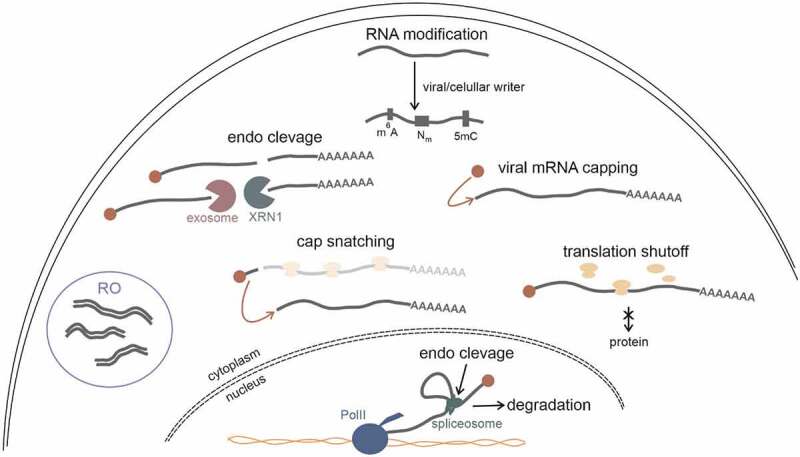
RNA viruses strategies either to avoid recognition by host cell immune system or to modulate cellular translation. Viruses use several methods to hide their genetic material from cellular RNA sensors. In order to do that viral RNA can undergo post-transcriptional modifications such as N6-methylation of adenosine (m6A), internal 2′-O-methylation (Nm), and cytosine-5-methylation (5mC). Moreover, to evade recognition by anti-viral immune system, on 5′ end of viral mRNA cap structure can be installed either by viral capping enzyme or through the ‘cap snatching’. RNA intermediates of viral replication can be hidden from host RNA sensors in specialized bodies called replication organelles (ROs). RNA viruses also learned how to lower the translation rate of cellular transcripts either by directly interfere with the biosynthesis of host proteins or by modulating RNA metabolism what in consequence leads to degradation of cellular transcripts
Replication in specialized bodies
During replication, RNA viruses produce several RNA species normally absent in uninfected cells, such as dsRNAs and ppp-RNAs [108]. Typically in host cells, RNA is not copied from RNA templates, thus, these unusual intermediate RNA molecules are recognized by innate immune sensors; however, viruses have learned how to circumvent and/or suppress these intracellular antiviral responses. One of the primary strategies that (+)ssRNA viruses utilize is to ‘hide’ highly recognizable replication intermediates from host sensors e.g. RIG-I and MDA5. Upon entry, coronaviruses and rhinoviruses elaborately modify intracellular membrane organelles, such as endoplasmic reticulum (ER) and mitochondria, to form cytosol isolated organ-like spaces for vRNA replication, also called replication organelles (ROs) [109–111]. RO structures concentrate the viral replication machinery and replication byproducts inside membrane-bound vesicles or invaginations, making them undetectable by host cell cytosolic innate immune sensors. ROs can be divided into two types: the invaginated vesicle/spherule, and the double-membrane vesicle [112]. Although the exact method of RO formation is not known, invaginated vesicle/spherule ROs are usually formed upon infection with Flaviviridae, such as Dengue or Zika virus, while double-membrane vesicle ROs are established during infection with Picornaviridae, such as poliovirus or coxsackievirus B3, or Coronaviridae, such as severe acute respiratory syndrome coronavirus (SARS-CoV, also named SARS-1) or middle east respiratory syndrome coronavirus (MERS-CoV). Aside from the number of layers composing the specific types of replication organelles, the main difference between ROs is whether their content is connected to the cytosol. Only invaginated vesicle/spherule ROs can freely exchange their content with the cytosol. Interestingly, infection of cells with HCV leads to formation of membranous web, structure which is combination of single layer invaginated vesicle/spherule and double layer double-membrane vesicle [113]. Very little is known about how the number of ROs during infection correlates with infection severity in animal models or real hosts. Al-Mulla et al. showed that during infection with coronavirus mutants, those able to produce only half of the original number of ROs, no effect on replication or fitness of these viruses was observed [114]; although it may be arguable that viral replication occurs outside ROs in the case of these mutants. Interestingly, the presence of ROs in (-)ssRNA viruses infected cells is questionable; although, respiratory syncytial virus reportedly has replication enzymes associated with cytosolic occluded structures, here named inclusion bodies [115].
Capping of vRNA
Although replication organelles serve as a spatial barrier enabling viral replication intermediates to hide from cell immune sensors, in some cases, this is not sufficient protection against host RNA receptor recognition. As mentioned above, some sensors rely on recognizing 5′ end RNA structures; therefore, many RNA viruses have developed additional protection strategies, including protection of vRNA 5′ ends through cap addition or structure imitation. A 5′ cap not only masks viral molecules from detection, but also provides efficient translation of invader proteins [116]. Poliovirus encodes a specialized cap mimicking the VPq protein, attaching it to the 5′ end of vRNAs, thus protecting these nucleic acids from recognition [117,118]. Interestingly, although these viruses do not require a cap structure for translation, since their vRNAs undergo cap-independent IRES-mediated translation [119,120], most viruses with an ssRNA genome synthesize a 5′ cap using their own set of enzymes [121,122]. After adding guanosine to the 5′ end of vRNA, through a triphosphate bridge, this newly formed cap is methylated at positions N7 and 2′-O by either one bi-functional N7/2′-O methyltransferase, like in flaviviruses [123], or by two separate enzymes, like in coronaviruses [124]. These two methylation steps enable formation of the vRNA cap-1 structure, a molecular signature of endogenous ‘self’ RNA in human cells. Lack of 2′-O-methylation, and in consequence formation of cap-0, leads to activation of the host defence system. Viral mRNAs bearing only cap-0 are sensed by IFIT proteins and excluded from the pool of translationally active mRNAs [8]. Daffis et al. demonstrated that IFIT proteins recognized vRNAs produced by a mutated strain of the flavivirus, West Nile virus, which lacked 2′-O-methyltransferase activity [101]. Interestingly, cap 2′-O-methylation not only masks vRNAs from IFIT protein recognition, but also impacts MDA5 and TLR7 viral transcripts sensing. Züst et al. showed that in wild-type mice, 2′-O-methyltransferase deficient coronaviruses were non-pathogenic, although their replication and spread was restored in murine strains lacking MDA5 and TLR7 to the same level as in IFNAR mice [125].
Interestingly, viruses from the Togaviridae family, such as alphaviruses and rubiviruses, synthesize RNA 5′ cap structure lacking 2′-O-methylation at the first transcribed nucleotide [121,122,126,127]. However, despite bearing only a cap-0, alphavirus transcripts evade host recognition as non-self RNAs. This is possibly due to secondary structures within the 5′ untranslated region (UTR) of alphavirus RNA, which protects it from being sensed by IFIT proteins [128].
A different approach to protecting vRNAs is presented by (-)ssRNA viruses belonging to Arenaviridae, Bunyaviridae, and Orthomyxoviridae families [121,122]. Although these pathogens do not have their own cap synthesizing machinery, they are able to steal caps from endogenous cellular mRNAs to prime replication of their own vRNAs, a process known as ‘cap snatching’. Upon recognizing a hosts mRNA cap, the transcript is cleaved by viral endonucleases usually 10–20 bases downstream of this structure [129]. These short-capped molecules are then used as primers for the synthesis of vRNAs via RNA-dependent RNA polymerase (RdRp). Interestingly, influenza carries out ‘cap snatching’ in the host nucleus, while most other viruses perform this action in the cell cytoplasm [129–131]. Following endonucleolytic cleavage, the remaining host mRNA is subjected to degradation via cellular RNA degradation machinery, for example enzyme XRN1. Limited studies investigate how ‘cap snatching’ affects host cell functioning, however this phenomenon likely plays a role in viral-host interplay.
Post-transcriptional viral RNA modifications
To date, more than 140 RNA post-transcriptional modifications have been reported in humans [11]. Inserting, deleting, substituting, or chemically modifying nucleotides within nascent RNA are not the only ways through which to manipulate protein expression, although these are convenient tactics to avoid activating the immune response via self-RNA molecules. Several major RNA modifications and editing events also occur in the case of vRNA, which have an influence on viral gene expression similarly to cellular RNA [40,46,132,133]. These include N6-methylation of adenosine, internal 2′-O-methylation, cytosine-5-methylation, isomerization of uridine to pseudo-uridine, and deamination of adenosine to inosine (A-to-I). These RNA modification/editing events have distinct consequences on viral infection, with the presence of post-transcriptional modifications in viral genomes potentially an evolutionary adaptation for immune evasion.
The best-studied, and likely the most abundant vRNA modification, post-transcriptional modifications of cellular RNAs is m6A [134,135]. Three types of enzymes are related to m6A modification processing, namely writer, eraser, and reader enzymes. Writers, methyltransferase-like enzymes, such as METTL3 and METTL14, together with Wilms tumour 1-associated protein (WTAP) and KIAA1429, deposit this modification onto RNA [136]. Erasers, such as the demethylase fat mass and obesity-associated protein (FTO) and α‐ketoglutarate-dependent dioxygenase AlkB homolog 5 (ALKBH5), are responsible for removing the methyl group from adenosine [137,138]. The YTH domain family of reader proteins, including YTHDC1, YTHDC2, YTHDF1, YTHDF3, and YTHDF3, direct m6A post-transcriptional functions [136]. Upon binding, YTHDF1 promotes translation of mRNAs, while YTHDF2 targets transcripts for degradation. YTHDF3 has been found to play a dual role, promoting protein biosynthesis in synergy with YTHDF1, and also affecting methylated mRNA decay mediated by YTHDF2 [139]. RNA genomes of HCV, Zika virus, Dengue virus, yellow fever virus, and West Nil virus of the Flaviviridae family contain such m6A modifications [140–142]. The main reported role of m6A refers to its actions in viral gene expression [134,135,143]. This type of methylation is considered a negative regulator of both HCV and Zika virus life cycles [140,142]. Manipulating writer and eraser amounts results in significant changes to intracellular HCV protein levels, as well as the amount of HCV RNA in the extracellular environment [140]. Most recently, human metapneumovirus RNA was shown to be m6A methylated in order to avoid RIG-I recognition [46].
For cellular RNA, pseudouridine is the most abundant post-transcriptional modifications; although there are only a few reports that confirm its presence in vRNAs [144,145]. In human cells, as in other eukaryotes, isomerization of uridine to form 5-ribosyl uracil (pseudouridine) is catalysed by pseudouridine synthases (PUS) [145]. Although, in yeast RNA, pseudouridine is suggested to play a role on transcript stability [133], its function in regulating viral gene expression remains largely unexplored. Recently, PUS proteins were identified in a CRISPR-based screen designed to reveal host factors facilitating flavivirus replication [146]. Interestingly, pseudouridine, when incorporated into a synthetic transcript for therapeutic purposes, guaranteed both higher mRNA stability and protein biosynthesis yield, also helping modified molecules to escape immune recognition [147,148].
The next most common modification found in different human RNA species is cytosine-5-methylation (5mC). This is present in highly abundant tRNA and rRNA, supposedly having an impact on nucleic acid structure and function [132]. Although this modification was also identified in mRNA, its role in messenger molecule metabolism is still enigmatic. Currently, it is known that the position of 5mC within mRNA has differential effects on transcript fate [149–152]. Enzymes responsible for 5mC belong either to DNMT2 or to NOL1/NOP2/sun (Nsun) methyltransferase subgroups [153,154]. Unfortunately, far less is known about 5mC effector proteins. Recently, 5mC was shown to be specifically recognized by the mRNA export adaptor ALYREF, whose downregulation in HeLa cells resulted in increased nuclear retention of 5mC-containing transcripts [150]. Infection with Dengue virus, Zika virus, HCV, or poliovirus changes the 5mC RNA landscape in infected cells, suggesting its potential role in the viral replication cycle. Moreover, data show that 5mC is present in both vRNA isolated from cells and in released viral particles [155,156].
Interestingly, some viruses that cap their own viral mRNA with cap-1, using 2′-O- methyltransferase, can utilize the same enzyme to introduce internal modifications, for example, Dengue virus employs its NSP5 protein to modify internal adenosines in vRNA [157]. 2′-O-methylated nucleotides, present in short vRNA sequences, repress cytokine and IFN production; a result of activating the endosomal immune sensor TLR7 [158]. It is possible that internal 2′-O-methylated adenosines also limit innate sensing during viral entry, although such a conclusion requires verification [159].
In human cells, enzymes called adenosine deaminases act on RNA (ADAR) 1 and 2, catalysing the deamination of position C6 in adenine (A) to produce inosine (I) [160]. These A-to-I editing events occur in dsRNA. This conversion may have significant biological implications such as changing the coding capacity of ribosomes or viral RNA dependent RNA polymerases, and altering the RNA structure, as I behaves similarly to guanosine (G). Interestingly, in human cells, ADARs have been shown to edit genomes of paramyxoviruses, such as human respiratory syncytial virus, parainfluenza virus 5, and human metapneumovirus [111,161,162]. Additionally, I has also been found in (+)ssRNA flaviviruses, for example HCV, Zika virus, and Dengue virus, and picornaviruses, namely poliovirus [155,163]. Taylor et al. showed that this type of post-transcriptional modifications successfully reduced the presence of HCV proteins, most likely due to IFN‐mediated decreases in cap-independent translation [163]. In contrast, installation of I also masks vRNA from MDA5 sensor detection, and prevents IFN and other inflammatory protein synthesis [164,165]. Thus, A-to-I editing in vRNA may potentially suppress the innate immune response [166].
Viral endonucleases
One of the most intriguing ways through which several viruses avoid recognition is endonuclease action on their own vRNA. Though it may seem counter-intuitive to destroy one’s own RNA, this has proven to be a useful strategy in evading immune recognition. For instance, the SARS-CoV genome encodes for NSP15, an endoribonuclease [167]. Although NSP15 does not degrade viral or host RNA in a non-specific manner, it is responsible for cleaving the poly(U) tail, generated at the 3′ end of transcripts during viral replication, from vRNAs. Eliminating this tail limits PAMP formation, hampering MDA5s ability to activate the innate immune response to viral infection [168].
The polymerase acidic protein (PA) of influenza also possesses endoribonuclease activity. PA, together with subunits PB1 and PB2, forms a viral polymerase complex [130]. The PA factor participates in endonucleolytic cleavage of host mRNA during the ‘cap-snatching’ process [131]. However, the alternative products of the PA gene, called PA-X, and its truncated variant PAXΔC20, were recently shown to be responsible for host shutoff in influenza infected cells [169–172]. Although PA-X preferentially targets ssRNA transcribed by host RNA polymerase II (Pol II) only, it also efficiently cleaves dsRNA [173]. Interestingly, the PAXΔC20 protein is characterized by severely reduced endonuclease activity. Recently, Gaucherand et al. revealed that PA-X targets majority of host cell mRNA for degradation [170]. Following endonucleolytic cleavage, resulting RNA fragments are eliminated via host RNA degradation machinery. Additionally, PA-X selective cleavage of endogenous Pol II transcripts is tightly connected with RNA splicing. These data suggest distinct cellular roles for both PA-X and PAXΔC20 proteins in viral infection. Taken together, influenza viruses possess a unique host shutoff mechanism that contributes to viral infection efficiency. Moreover, this is a remarkable example of how viruses hinder host cell immunity while promoting the expression of their own viral mRNA.
Hijacking host translation
General host shutoff (GHS) is a broadly termed consequence of all viral actions, eventually halting overall protein expression in the infected cell. Such a strategy allows viruses to take full capacity advantage of cellular translation machinery. The lower total protein expression rate means that fewer RNA receptors are produced by the host cell, allowing more viral transcripts to be efficiently processed into viral proteins. Various viruses handle expression hijacking differently. Coronaviruses, for example SARS-CoV and MERS-CoV, cause host shutoff at the post-transcriptional level; where the process is mediated by NSP1, which binds to cellular translation factors, preventing mRNA translation. SARS-CoV NSP1 blocks the 40S ribosomal subunit, effectively repressing the translation of both viral and host transcripts [174–176]. Data by Lokugamage et al. revealed that, depending on the mechanism of initiation operating on the mRNA template, NSP1 is able to inhibit translation at this stage [175]. Furthermore, MERS-CoV NSP1 utilizes yet another mechanism, through which it is able to differentiate between cellular mRNA produced in the nucleus and viral mRNA synthesized in the cytosol. SARS-CoV NSP1 is exclusively localized in the cytoplasm, stably binding to the 40S ribosomal subunit, while MERS-CoV NSP1 is present not only in the cytoplasm, but also in the cell nucleus, with a less stable interaction occurring with 40S. These data suggest that MERS-CoV NSP1 targets host mRNA in the nucleus, exporting it together with host transcripts to the cytoplasm, thus sparing viral mRNAs of cytoplasmic origin [177].
Viruses of the family Picornaviridae, including poliovirus, human rhinoviruses, and coxsackievirus, use a yet another tactic to inhibit cap-dependent translation of host mRNA [178–180]. Although picornaviruses use 2A proteinase to cleave the translation initiation factor elF4G, viral protein biosynthesis is unaffected since translation from viral mRNA depends on IRES [181]. Recent work by Aumayr et al. indicated that, in order to efficiently cleave eIF4G, human rhinovirus 2A proteases must bind to another translation initiation factor, elF4E [182].
In the case of human respiratory syncytial virus, from the Paramyxoviridae family, it is difficult to identify a single mechanism responsible for host shutoff. Bruce et al. suggested that human respiratory syncytial virus may specifically alter the expression of surfactant protein A (SP-A) in human pulmonary epithelial cells by interfering with protein translation [183]. The role of SP-A proteins is to bind directly to human respiratory syncytial virus, marking the virus for degradation by the host defence system. Moreover, the human respiratory syncytial virus genome encodes two unique nonstructural proteins, NSP1 and NSP2; the presence of which should lead to an effective induction of host shutoff. Production of these proteins strongly reduces IFN response activation upon infection with human respiratory syncytial virus [184]. Furthermore, both NSP1 and 2 individually and cooperatively target proteins from the IFN signalling cascade, including RNA sensors, such as RIG-I, MDA5 and OAS, and ISGs, such as IFIT1 [185–188]. Therefore, to better understand the impact of NS1 and NS2 on the innate immune system, Goswami et al. performed a series of experiments suggesting that these proteins are part of a multiprotein complex known as the NS degradosome [189]. Another layer of complexity to how human respiratory syncytial virus may gain advantage over host cells is through stress granule formation, a phenomenon benefited by viral replication [190,191].
Viral RNA degradation
Recognition of vRNA is a crucial step in the host cells defence against invading viruses. To efficiently eradicate an infection, prompt degradation of recognized vRNA is vital. Regular RNA decay enzymes digest vRNA from the 5′ to 3′ end, and vice versa; however, cells have also developed specialized factors dedicated to eliminating vRNA particles [192–194]. To combat this, viruses have again gained specific mechanisms to counteract these cellular RNA degradation systems.
XRN1
The 5′-3′ exonuclease, XRN1, is a key player in RNA turnover and surveillance pathways, catalysing mRNA degradation [195]. XRN1-dependent mRNA degradation regulates the proteome by controlling the mRNA population available for translation, which also serves a significant role in shaping the cell response to viral infection. RNA molecules are protected from XRN1 digestion when their 5′ end is capped; with the decapping process strictly controlled by a complex net of interacting proteins. Elimination of this cap structure predestinates endogenous mRNAs, and vRNA alike, for degradation by XRN1. Thus, viral pathogens have evolved means through which to manipulate the fate of their mRNA [195]. RNA viruses present different strategies via which to circumvent XRN1 action, thereby protecting their genomic integrity. Those which have m7GTP-capped mRNA simply avoid the exonucleolytic activity of XRN1196. As mentioned above, capped host transcripts must first undergo decapping, a reaction performed by the DCP1-DCP2 complex, which leads to generation of 7-methylguanosine diphosphate (m7GDP) and a 5′-monophosphorylated mRNA exposed to 5′-exonucleolytic degradation. Additionally, viral capped RNAs are the perfect substrates for this decapping complex, the action of which would leave pathogenic molecules exposed to XRN1 degradation.
Therefore, it is not surprising that viruses attempt to reduce XRN1 availability in the host cytoplasm in order to accumulate their own RNAs. Rotaviruses decrease the level of cytoplasmic XRN1, along with DCP1, by relocating these proteins to the cell nucleus [196]. Moreover, rotavirus NSP1 protein has been shown to target another component of the RNA degradation machinery, PAN3, a component of the deadenylation complex [196]. Furthermore, poliovirus also targets this same set of enzymes, although upon viral infection, all three RNA decay factors, XRN1, DCP1, and PAN3, undergo accelerated degradation [197].
A different approach to XRN1 targeting is exploited by flaviviruses such as Zika or Dengue virus. Although, initially, XRN1 appears to act as an antiviral agent, degrading pathogenic gRNA, presence of highly structured sequences in its 3′ UTR causes XRN1 stalling. This results in accumulation of subgenomic flavivirus RNA (sfRNA), which is toxic to the cell [198–200]. Accumulated sfRNAs facilitate infection with flavivirus by interacting with many cellular proteins responsible for the modulation of host RNA interference, RNA decay, and IFN activation [201]. Moreover, XRN1 stalling on sfRNAs results in repression of XRN1 activity and accumulation of uncapped cellular mRNAs [200,202]. A slightly different strategy to repress XRN1 is applied by other members of the Flaviviridae family, such as HCV and bovine viral diarrhoea virus. In this case, the region where XRN1 stalls is located in the 5′ UTR of gRNA, leading to repression of XRN1 activity [203]. This dysregulation causes stabilization of normally short-lived mRNAs, including mRNAs encoding immune regulators and oncogenes. Therefore, this finding contributes a new layer of evidence to HCV pathogenesis, including for hepatocellular carcinoma.
The mechanisms through which host RNA degradation machinery exerts its function in order to eliminate vRNAs remain elusive. Recently, Ng et al. shed some light on this process, suggesting that XRN1 and DCP2 repress replication of the Newcastle disease virus, from the Paramyxoviridae family, as well as the encephalomyocarditis virus, from the Picornaviridae family, inside discrete cytoplasmic foci, distinct from SGs and processing bodies (P-bodies) [204]. Interestingly, this finding indicates that the function of both XRN1 and DCP2 could be tightly regulated to specifically target vRNAs, separate to host transcripts.
Exosome complex
Another major component of RNA degradation machinery is a multisubunit exosome complex, which predominantly behaves as a 3′-5′ exoribonuclease [205]. It is composed of catalytic proteins and nine core subunits, six of which, Rrp41, Rrp45, Mtr3, Rrp42, Rrp43, and Rrp46, form a ring-like structure capped by a trimer of Rrp4, Rrp40, and Csl4. Interestingly, although each hexameric ring subunit possesses an RNase PH-like domain, the exosome core is catalytically inactive. Therefore, to fulfill its duties, the exosome core in the cytoplasm of human cells associates with other subunits such as RRP44, a 3′-5′ exoribonuclease also named DIS3, which can also act as an endonuclease [206], or the 3′-5′ exoribonuclease DIS3L. Additionally, the RRP6 exoribonuclease supports the exosome with its 3′-5′ activity [207,208]. Moreover, productive exosome action requires interactions with several cofactors, which target the exosome to different RNA substrates. Many of these cofactors were described in budding yeast, including the Ski (super killing) complex, composed of Ski2/Ski3/Ski8, which is responsible for feeding RNAs to the exosome in the cytoplasm [209,210]. Historically, Ski genes were named for their dsRNA virus ‘super killing’ ability; pathogens which would have otherwise been lethal for yeast strains deficient in these genes [209]. Importantly, orthologues of all three Ski genes are also present in higher eukaryotes, although their substrate specificity is still unknown [211,212]. Recently, Eckard et al. showed that SKIV2L, the human orthologue of yeast Ski2, engaged in the regulatory mechanism responsible for limiting RIG-I activation. Cells depleted in SKIV2L were found to accumulate cleavage products of the inositol-requiring enzyme 1 (IRE-1) endonuclease, leading to RIG-I activation. Moreover, patients with SKIV2L deficiency show a potent IFN signature in their peripheral blood cells [213].
In humans, the nuclear exosome associates with at least three different cofactors, including NEXT (MTR4, ZCCHC8, and RBM7), PAX (MTR4, ZFC3H1, and PABPN1), and hTRAMP (MTR4, ZCCHC7 and PAPD5 [205]), in which only RNA helicase MTR4 is a common component. Recently, it has been demonstrated that, upon infection with an ssRNA virus, normally nuclear proteins, MTR4 and ZCCHC7, shuttle to the cytoplasm to form cytoplasmic complexes that specifically recognize and induce degradation of viral mRNA [214]; however, the exact mechanism of viral transcript degradation is yet to be established.
OAS and RNase L
RNase L is a ubiquitous cellular endoribonuclease whose expression in humans, as in most vertebrates, is induced upon recognition of vRNA. IFNs transcriptionally induce OAS proteins that, by binding dsRNA, are activated to produce, from cellular ATP, higher oligomers of 2ʹ-5ʹ-oligoadenylates, which activate RNase L. Upon binding these 2ʹ-5ʹ-oligoadenylates, RNase L forms either homo- or oligomers, thus becoming an active enzyme that cuts, without distinguishing, both viral and cellular ssRNA [45,89]. RNase L cleavage products are recognized by RIG-I and MDA5, further increasing IFN production and the antiviral response. It is still unclear how cells coordinate RNA sensing via signalling and IFN production. Recently, RNase L was shown to participate in the formation of antiviral SGs (avSGs) through its dsRNA cleavage products. These dsRNAs activate PKR and recruit other antiviral proteins, including RIG-I, PKR, and avSGs [89,215–217].
RNP assemblies formed in the cytoplasm in response to diverse stress signals are called SGs [215]. As the main role of SGs is to prevent unnecessary protein generation by pausing translation upon cellular stress, SG composition depends on the nature of the stress signal. Different proteins are recruited to non-membranous complexes, for example protein kinases such as PKR, general control non-derepressible 2 (GCN2), or PKR-like ER kinase (PERK), in response to specific types of stimuli, including heat, oxidation, hypoxia, and osmotic pressure [215,218,219]. Transcripts accumulated in SGs are either subjected to degradation, or translation is re-initiated.
avSGs are formed in response to viral infections and can be compared to typical SGs formed as a response to canonical stress signals. SGs, whose main role is signalling, are RNP assemblies in the cytoplasm [89,220]. The production of IFNs during several viral infections was shown to require avSGs for formation. Thus, avSGs have been described as an important platform for sensing vRNAs during viral infection.
ISG20
The protein of the IFN-stimulated gene of 20 kDa (ISG20) was first described as an IFN-induced protein associated with pathological structures of promyelocytic leukaemia (PML) [221,222]. ISG20 is a member of the DnaQ-like 3′-5′ exonuclease superfamily, with 3′-5′ activity towards RNA. Its catalytic property has been associated with replication inhibition in a broad range of RNA viruses, including Flaviviridae (yellow fever virus, West Nile virus, Dengue virus, and HCV) [223–226] and Orthomyxoviridae (influenza) [227].
Supposedly, the main antiviral mechanism of ISG20 is direct 3′-5′ exonucleolytic degradation of vRNA; as suggested by its potent exonuclease function in vitro and the observed loss of antiviral effects upon its mutation at residues of the death effector domain (DEDD) [222,228,229]. However, several studies also reported ISG20 viral inhibition in the absence of vRNA degradation [225,230], raising the question whether alternative mechanisms may be involved. ISG20 was recently showed to inhibit the vesicular stomatitis virus through translational blockage. Data by Wu et al. revealed that ISG20 exhibited the ability to act as a general translational modulator of exogenous transcripts [231]. Interestingly, the authors also showed that it appears to spare self RNA [231], although its exact mechanism of RNA discrimination remains unknown. Nonetheless, researchers excluded the idea that ISG20 affects mRNA nucleocytoplasmic transport and translation initiation [231]. Furthermore, specific co- or post-transcriptional modifications, such as m6A, may contribute to RNA discrimination.
ZAP
Identification of vRNA by the host innate immune system could be performed not only based on vRNAs characteristic features or the presence of a triphosphate group at the 5′ end, but also on more genome-wide attributes of RNA viruses, such as nucleotide composition. The zinc-finger antiviral protein (ZAP) eliminates cytoplasmic RNAs recognized as non-self, due to their elevated CpG dinucleotide content, compared with endogenous mRNAs. Therefore, it is not surprising that the replication of high CpG content-RNA viruses, such as alphaviruses, influenza viruses, and flaviviruses, is controlled by ZAP [226,232–236]. The ZAP protein contains several domains, among which the N-terminal RNA-binding domain, composed of four CCCH zinc fingers, can be distinguished. This RNB domain has the ability to directly bind to CpG motifs in the RNA sequence, making it necessary for the protein to fulfill its function [237]. Interestingly, RNA viruses with an increased frequency of UpA also undergo ZAP-mediated attenuation [238,239]. In line with these results is the observation that a reduction in CpG or UpA frequencies leads to an increase in viral replication rate [239].
The ZAP protein binds directly to specific vRNA, recruiting the exosome complex and degrading targeted transcripts [234,240,241]. On the other hand, vRNAs recognized by ZAP could undergo decapping and would thus be subjected to XRN1 degradation [240,241]; however, whether viral transcripts are eliminated via this alternative pathway is still unclear [234]. Upon alphavirus infection, ZAP was also shown to repress viral mRNA cap-dependent translation by interfering with translational initiation factors eIF4G and eIF4A [242]. Moreover, Odon et al. unexpectedly revealed that sequences with a high CpG or UpA content may be recognized via OAS and be further degraded by RNase L [238]. However, further work is needed to investigate the potential crosstalk between ZAP-, OAS- and RNase L-mediated antiviral pathways.
RNA interference
In canonical RNAi pathway dsRNA is cleaved by Dicer endoribonuclease producing 20–24 nt long RNA duplexes – short interfering RNAs (siRNAs) [243]. When separated, one strand from siRNA molecule is incorporated into RNA-Induced Silencing Complex (RISC) and serves as a guide RNA for finding a complementary mRNA. Target mRNA and RISC binding induces transcript cleavage mediated by Argonaute protein, component of RISC, what leads to particular gene silencing. In course of the studies it occurred that RNAi pathway is also an important antiviral mechanism. Dicer could digest viral dsRNA intermediates what would lead to production of virus-derived siRNAs (vsiRNAs) that facilitate antiviral response. Presence of vsiRNAs in infected cells was well documented in plants and in invertebrates, such as fruit fly or nematode. Thus, it is also possible that the same antiviral mechanism could be present in mammalian cells. Several studies dedicated to find vsiRNAs in mammalian cells failed what put into question possibility of generating these siRNA species upon viral infection in mammals [244,245]. Interestingly, an exception arises in the case of murine oocytes and embryonic stem cells [246–248]. These observations led to conclusion that RNAi pathway could play a role in antiviral response only in cells that are undifferentiated somatic cells. This discrepancy between undifferentiated and differentiated cells could be due to differences in Dicer domain composition, somatic Dicer is full length protein, while in embryonic stem cells truncated form is present [249]. It was shown that only amino-terminally truncated Dicer was able to process dsRNAs and turn these into functional siRNAs in murine and human cells [249,250]. However, situation changed completely in 2013, when Mallard et al. and Li et al. proved independently that vsiRNAs are also present in virus infected somatic mammalian cells [251,252]. Previous inability to capture the presence of vsiRNAs in viral infected cells could be explained in several ways. Differentiated cells are able to induce potent interferon response upon virus infection, which inhibits Dicer activity [55,253,254]. Moreover, many RNA viruses, like Nodamura virus, influenza, and Dengue virus, actively antagonize host immunity by the production of viral suppressors of RNAi: B2, NS1 and 2A protein, respectively, to hamper Dicer activity [251,252,255]. Thus, infection of mammalian cells with viral strains deficient in aforementioned proteins enabled vsiRNA detection.
Based on already carried out studies it is certain that RNAi pathway helps to eradicate viruses from mammalian cells but from the other hand this strategy is not the main mechanism to fight viral infection. Further studies are needed to learn to what extent RNAi contributes to antiviral response.
Concluding remarks and future perspectives
Significant progress has been made in elucidating which mechanisms lead to induction of host cell antiviral defences. Although much is known about the main molecular features of virally-derived RNAs responsible for inducing innate immune system receptors, significantly less is known about how viruses countermeasure a host defence mechanisms. Recent work has highlighted the importance of masking vRNAs through various 5′ end modifications, as well as processes of mimicking cellular RNAs via ‘cap snatching’ or even direct blocking of receptor proteins, thus preventing recognition. Among the vast repertoire of viral tricks are m6A methylation to avoid RIG-I recognition, 2′-O-methylation preventing IFIT, MDA5, and TLR7 recognition, forming complex secondary RNA structures which interfere with PKR sensing. Interestingly, majority of RNA viruses use multiple mechanisms to avoid recognition by the host cell. For instance, coronaviruses instal a cap-1 structure on their mRNA, as well as hide their replication in cytosol isolated organ-like structures, known as ROs. In addition to reducing vRNA visibility, viruses are able to harness those host cellular mechanisms dedicated to eliminating them, for their own benefit. Flaviviruses exploit the 5′-3′ exonuclease, XRN1, to generate, from their genomic nucleic acids, sfRNAs, which are toxic to the host cell.
Despite our knowledge of well-known viral evasion mechanisms, our understanding of exactly how host RNA degradation machinery exerts its function to eliminate vRNAs remains unclear. Thus, further studies are needed to fully understand the interplay between viruses and host cells. Moreover, an in-depth understanding of the molecular mechanisms between viral-host relationships would allow for the design of appropriate antiviral drugs.
Acknowledgments
This work was financially supported by the National Science Centre (Poland, grants UMO-2018/31/D/NZ1/03526 to PJS). PJS designed the manuscript outline and coordinated work on the paper. LM and PJS performed the literature search and wrote first draft of the manuscript. KD prepared the figures. KD and PJS contributed to writing and editing of the final manuscript version. All authors have given approval to the final version of the manuscript.
Funding Statement
This work was supported by the Narodowe Centrum Nauki [UMO-2018/31/D/NZ1/03526].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Ahlquist P. Parallels among Positive-Strand RNA Viruses, Reverse-Transcribing Viruses and Double-Stranded RNA Viruses. Nature Reviews Microbiology. 2006;4(5):371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pichlmair A, Lassnig C, Eberle C-A, et al. IFIT1 Is an Antiviral Protein That Recognizes 5′-Triphosphate RNA. Nat Immunol. 2011;12(7):624–630. [DOI] [PubMed] [Google Scholar]
- [3].Chi H, Flavell RA.. Innate Recognition of Non-Self Nucleic Acids. Genome Biol. 2008;9(3):211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Platnich JM, Muruve DA. NOD-like Receptors and Inflammasomes: A Review of Their Canonical and Non-Canonical Signaling Pathways. Arch Biochem Biophys. 2019;670:4–14. [DOI] [PubMed] [Google Scholar]
- [5].Bauernfried S, Scherr MJ, Pichlmair A, et al. Human NLRP1 Is a Sensor for Double-Stranded RNA. Science. 2020;eabd0811. 10.1126/science.abd0811 [DOI] [PubMed] [Google Scholar]
- [6].Rehwinkel J, Reis E Sousa C. RIGorous Detection: exposing Virus Through RNA Sensing. Science. 2010;327(5963):284–286. [DOI] [PubMed] [Google Scholar]
- [7].Habjan M, Pichlmair A. Cytoplasmic Sensing of Viral Nucleic Acids. Curr Opin Virol. 2015;11:31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hyde JL, Diamond MS. Innate Immune Restriction and Antagonism of Viral RNA Lacking 2׳-O Methylation. Virology. 2015;479–480:66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Warminski M, Sikorski PJ, Kowalska J, et al. Applications of Phosphate Modification and Labeling to Study (m)RNA Caps. Top Curr Chem Cham. 2017;375(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Galloway A, Cowling VH. mRNA cap regulation in mammalian cell function and fate. Biochimica Et Biophysica Acta (BBA) - Gene Regulatory Mechanisms. 2019;1862(3):270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nachtergaele S, He C. Chemical Modifications in the Life of an MRNA Transcript. Annu Rev Genet. 2018;52(1):349–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Weissman D, Pardi N, Muramatsu H, et al. Purification of In Vitro Transcribed Long RNA. InSynthetic Messenger RNA and Cell Metabolism Modulation. Rabinovich PM, editor. Methods in Molecular Biology Humana Press:Totowa, NJ. 2013. Vol. 969. 43–54. [DOI] [PubMed] [Google Scholar]
- [13].Karikó K, Muramatsu H, Ludwig J, et al. Generating the Optimal MRNA for Therapy: HPLC Purification Eliminates Immune Activation and Improves Translation of Nucleoside-Modified, Protein-Encoding MRNA. Nucleic Acids Res. 2011;39(21):e142–e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yoneyama M, Kikuchi M, Matsumoto K, et al. Shared and Unique Functions of the DExD/H-Box Helicases RIG-I, MDA5, and LGP2 in Antiviral Innate Immunity. J Immunol. 2005;175(5):2851–2858. [DOI] [PubMed] [Google Scholar]
- [15].Imran M, Waheed Y, Manzoor S, et al. Interaction of Hepatitis C Virus Proteins with Pattern Recognition Receptors. Virol J. 2012;9(1):126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gitlin L, Barchet W, Gilfillan S, et al. Essential Role of Mda-5 in Type I IFN Responses to Polyriboinosinic:Polyribocytidylic Acid and Encephalomyocarditis Picornavirus. Proc Natl Acad Sci. 2006;103(22):8459–8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Faul EJ, Wanjalla CN, Suthar MS, et al. Rabies Virus Infection Induces Type I Interferon Production in an IPS-1 Dependent Manner While Dendritic Cell Activation Relies on IFNAR Signaling. PLoS Pathog. 2010;6(7):e1001016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fredericksen BL, Keller BC, Fornek J, et al. Establishment and Maintenance of the Innate Antiviral Response to West Nile Virus Involves Both RIG-I and MDA5 Signaling through IPS-1. J Virol. 2008;82(2):609–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sen A, Pruijssers AJ, Dermody TS, et al. The Early Interferon Response to Rotavirus Is Regulated by PKR and Depends on MAVS/IPS-1, RIG-I, MDA-5, and IRF3. J Virol. 2011;85(8):3717–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gitlin L, Benoit L, Song C, et al. Melanoma Differentiation-Associated Gene 5 (MDA5) Is Involved in the Innate Immune Response to Paramyxoviridae Infection In Vivo. PLoS Pathog. 2010;6(1):e1000734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Si-Tahar M, Blanc F, Furio L, et al. Protective Role of LGP2 in Influenza Virus Pathogenesis. J Infect Dis. 2014;210(2):214–223. [DOI] [PubMed] [Google Scholar]
- [22].Kawai T, Akira S. Toll-like Receptor and RIG-1-like Receptor Signaling. Ann N Y Acad Sci. 2008;1143(1):1–20. [DOI] [PubMed] [Google Scholar]
- [23].Guillot L, Le Goffic R, Bloch S, et al. Involvement of Toll-like Receptor 3 in the Immune Response of Lung Epithelial Cells to Double-Stranded RNA and Influenza A Virus. J Biol Chem. 2005;280(7):5571–5580. [DOI] [PubMed] [Google Scholar]
- [24].Diebold SS. Innate Antiviral Responses by Means of TLR7-Mediated Recognition of Single-Stranded RNA. Science. 2004;303(5663):1529–1531. [DOI] [PubMed] [Google Scholar]
- [25].Lund JM, Alexopoulou L, Sato A, et al. Recognition of Single-Stranded RNA Viruses by Toll-like Receptor 7. Proc Natl Acad Sci. 2004;101(15):5598–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Amici C, La Frazia S, Brunelli C, et al. Inhibition of Viral Protein Translation by Indomethacin in Vesicular Stomatitis Virus Infection: role of EIF2α Kinase PKR. Cell Microbiol. 2015;17(9):1391–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kimura T, Katoh H, Kayama H, et al. Ifit1 Inhibits Japanese Encephalitis Virus Replication through Binding to 5ʹ Capped 2ʹ-O Unmethylated RNA. J Virol. 2013;87(18):9997–10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Johnson B, VanBlargan LA, Xu W, et al. Human IFIT3 Modulates IFIT1 RNA Binding Specificity and Protein Stability. Immunity. 2018;48(3):487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Davis BM, Fensterl V, Lawrence TM, et al. Ifit2 Is a Restriction Factor in Rabies Virus Pathogenicity. J Virol. 2017;91(17):e00889–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Diamond MS. IFIT1: A Dual Sensor and Effector Molecule That Detects Non-2′-O Methylated Viral RNA and Inhibits Its Translation. Cytokine Growth Factor Rev. 2014;25(5):543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gee P, Chua PK, Gevorkyan J, et al. Essential Role of the N-Terminal Domain in the Regulation of RIG-I ATPase Activity. J Biol Chem. 2008;283(14):9488–9496. [DOI] [PubMed] [Google Scholar]
- [32].Kowalinski E, Lunardi T, McCarthy AA, et al. Structural Basis for the Activation of Innate Immune Pattern-Recognition Receptor RIG-I by Viral RNA. Cell. 2011;147(2):423–435. [DOI] [PubMed] [Google Scholar]
- [33].Jiang F, Ramanathan A, Miller MT, et al. Structural Basis of RNA Recognition and Activation by Innate Immune Receptor RIG-I. Nature. 2011;479(7373):423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Saito T, Hirai R, Loo Y-M, et al. Regulation of Innate Antiviral Defenses through a Shared Repressor Domain in RIG-I and LGP2. Proc Natl Acad Sci. 2007;104(2):582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ramanathan A, Devarkar SC, Jiang F, et al. The Autoinhibitory CARD2-Hel2i Interface of RIG-I Governs RNA Selection. Nucleic Acids Res. 2016;44(2):896–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cui S, Eisenächer K, Kirchhofer A, et al. Terminal Regulatory Domain Is the RNA 5′-Triphosphate Sensor of RIG-I. Mol Cell. 2008;29(2):169–179. [DOI] [PubMed] [Google Scholar]
- [37].Loo Y-M, Fornek J, Crochet N, et al. RIG-I and MDA5 Signaling by RNA Viruses in Innate Immunity. J Virol. 2008;82(1):335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kato H, Takeuchi O, Sato S, et al. Differential Roles of MDA5 and RIG-I Helicases in the Recognition of RNA Viruses. Nature. 2006;441(7089):101–105. [DOI] [PubMed] [Google Scholar]
- [39].Goubau D, Schlee M, Deddouche S, et al. Antiviral Immunity via RIG-I-Mediated Recognition of RNA Bearing 5′-Diphosphates. Nature. 2014;514(7522):372–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Picard-Jean F, Brand C, Tremblay-Létourneau M, et al. 2ʹ-O-Methylation of the MRNA Cap Protects RNAs from Decapping and Degradation by DXO. PloS One. 2018;13(3):e0193804–e0193804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kato H, Takeuchi O, Mikamo-Satoh E, et al. Recognition of Double-Stranded Ribonucleic Acids by Retinoic Acid–Inducible Gene-I and Melanoma Differentiation–Associated Gene 5. J Exp Med. 2008;205(7):1601–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Baum A, Sachidanandam R, García-Sastre A. Preference of RIG-I for Short Viral RNA Molecules in Infected Cells Revealed by next-Generation Sequencing. Proc Natl Acad Sci. 2010;107(37):16303–16308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Devarkar SC, Wang C, Miller MT, et al. Structural Basis for M7G Recognition and 2ʹ-O-Methyl Discrimination in Capped RNAs by the Innate Immune Receptor RIG-I. Proc Natl Acad Sci. 2016;113(3):596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Saito T, Owen DM, Jiang F, et al. Innate Immunity Induced by Composition-Dependent RIG-I Recognition of Hepatitis C Virus RNA. Nature. 2008;454(7203):523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Malathi K, Saito T, Crochet N, et al. RNase L Releases a Small RNA from HCV RNA That Refolds into a Potent PAMP. RNA. 2010;16(11):2108–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lu M, Zhang Z, Xue M, et al. Enables Viral RNA to Escape Recognition by RNA Sensor RIG-I. Nat Microbiol. 2020;5(4):584–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Peisley A, Lin C, Wu B, et al. Cooperative Assembly and Dynamic Disassembly of MDA5 Filaments for Viral DsRNA Recognition. Proc Natl Acad Sci. 2011;108(52):21010–21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Berke IC, Modis Y. MDA5 Cooperatively Forms Dimers and ATP-Sensitive Filaments upon Binding Double-Stranded RNA: MDA5 Forms Dimers and Filaments on Binding DsRNA. Embo J. 2012;31(7):1714–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Peisley A, Jo MH, Lin C, et al. Kinetic Mechanism for Viral DsRNA Length Discrimination by MDA5 Filaments. Proc Natl Acad Sci. 2012;109(49):E3340–E3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kang DC, Gopalkrishnan RV, Wu Q, et al. Mda-5: an Interferon-Inducible Putative RNA Helicase with Double-Stranded RNA-Dependent ATPase Activity and Melanoma Growth-Suppressive Properties. Proc Natl Acad Sci. 2002;99(2):637–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Satoh T, Kato H, Kumagai Y, et al. LGP2 Is a Positive Regulator of RIG-I- and MDA5-Mediated Antiviral Responses. Proc Natl Acad Sci. 2010;107(4):1512–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Rothenfusser S, Goutagny N, DiPerna G, et al. The RNA Helicase Lgp2 Inhibits TLR-Independent Sensing of Viral Replication by Retinoic Acid-Inducible Gene-I. J Immunol. 2005;175(8):5260–5268. [DOI] [PubMed] [Google Scholar]
- [53].Murali A, Li X, Ranjith-Kumar CT, et al. Structure and Function of LGP2, a DE X (D/H) Helicase That Regulates the Innate Immunity Response. J Biol Chem. 2008;283(23):15825–15833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Sanchez David RY, Combredet C, Najburg V, et al. LGP2 Binds to PACT to Regulate RIG-I– and MDA5-Mediated Antiviral Responses. Sci Signal. 2019;12(601):eaar3993. [DOI] [PubMed] [Google Scholar]
- [55].Takahashi T, Nakano Y, Onomoto K, et al. Virus Sensor RIG-I Represses RNA Interference by Interacting with TRBP through LGP2 in Mammalian Cells. Genes (Basel). 2018;9(10):511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Dorin D, Bonnet MC, Bannwarth S, et al. The TAR RNA-Binding Protein, TRBP, Stimulates the Expression of TAR-Containing RNAs in Vitro and in Vivo Independently of Its Ability to Inhibit the DsRNA-Dependent Kinase PKR. J Biol Chem. 2003;278(7):4440–4448. [DOI] [PubMed] [Google Scholar]
- [57].Zhang Z, Kim T, Bao M, et al. DDX1, DDX21, and DHX36 Helicases Form a Complex with the Adaptor Molecule TRIF to Sense DsRNA in Dendritic Cells. Immunity. 2011;34(6):866–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Miyashita M, Oshiumi H, Matsumoto M, et al. DDX60, a DEXD/H Box Helicase, Is a Novel Antiviral Factor Promoting RIG-I-Like Receptor-Mediated Signaling. Mol Cell Biol. 2011;31(18):3802–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Oshiumi H, Okamoto M, Fujii K, et al. TLR3/TICAM-1 Pathway Is Mandatory for Innate Immune Responses to Poliovirus Infection. J Immunol. 2011;187(10):5320–5327. [DOI] [PubMed] [Google Scholar]
- [60].Ariumi Y, Kuroki M, Abe K, et al. DDX3 DEAD-Box RNA Helicase Is Required for Hepatitis C Virus RNA Replication. J Virol. 2007;81(24):13922–13926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Li G, Feng T, Pan W, et al. Box RNA Helicase DDX3X Inhibits DENV Replication via Regulating Type One Interferon Pathway. Biochem Biophys Res Commun. 2015;456(1):327–332. [DOI] [PubMed] [Google Scholar]
- [62].Li C, Ge L, Li P, et al. DDX3 Regulates Japanese Encephalitis Virus Replication by Interacting with Viral Un-Translated Regions. Virology. 2014;449:70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Brai A, Martelli F, Riva V, et al. DDX3X Helicase Inhibitors as a New Strategy To Fight the West Nile Virus Infection. J Med Chem. 2019;62(5):2333–2347. [DOI] [PubMed] [Google Scholar]
- [64].Thulasi Raman SN, Liu G, Pyo HM, et al. DDX3 Interacts with Influenza A Virus NS1 and NP Proteins and Exerts Antiviral Function through Regulation of Stress Granule Formation. J Virol. 2016;90(7):3661–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Bowie A, O’Neill LAJ. The Interleukin-1 Receptor/Toll-like Receptor Superfamily: signal Generators for pro-Inflammatory Interleukins and Microbial Products. J Leukoc Biol. 2000;67(4):508–514. [DOI] [PubMed] [Google Scholar]
- [66].Park B, Brinkmann MM, Spooner E, et al. Proteolytic Cleavage in an Endolysosomal Compartment Is Required for Activation of Toll-like Receptor 9. Nat Immunol. 2008;9(12):1407–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ewald SE, Lee BL, Lau L, et al. The Ectodomain of Toll-like Receptor 9 Is Cleaved to Generate a Functional Receptor. Nature. 2008;456(7222):658–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Du X, Poltorak A, Wei Y, et al. Three Novel Mammalian Toll-like Receptors: gene Structure, Expression, and Evolution. Eur Cytokine Netw. 2000;11(3):362–371. [PubMed] [Google Scholar]
- [69].Hemmi H, Takeuchi O, Kawai T, et al. Recognizes Bacterial DNA. Nature. 2000;408(6813):740–745. [DOI] [PubMed] [Google Scholar]
- [70].Alexopoulou L, Holt AC, Medzhitov R, et al. Recognition of Double-Stranded RNA and Activation of NF-ΚB by Toll-like Receptor 3. Nature. 2001;413(6857):732–738. [DOI] [PubMed] [Google Scholar]
- [71].Heil F. Species-Specific Recognition of Single-Stranded RNA via Toll-like Receptor 7 and 8. Science. 2004;303(5663):1526–1529. [DOI] [PubMed] [Google Scholar]
- [72].Heil F, Ahmad-Nejad P, Hemmi H, et al. Receptor 7 (TLR7)-Specific Stimulus Loxoribine Uncovers a Strong Relationship within the TLR7, 8 and 9 Subfamily. Eur J Immunol. 2003;33(11):2987–2997. [DOI] [PubMed] [Google Scholar]
- [73].Hornung V, Guenthner-Biller M, Bourquin C, et al. Sequence-Specific Potent Induction of IFN-α by Short Interfering RNA in Plasmacytoid Dendritic Cells through TLR7. Nat Med. 2005;11(3):263–270. [DOI] [PubMed] [Google Scholar]
- [74].Wang JP, Liu P, Latz E, et al. Flavivirus Activation of Plasmacytoid Dendritic Cells Delineates Key Elements of TLR7 Signaling beyond Endosomal Recognition. J Immunol. 2006;177(10):7114–7121. [DOI] [PubMed] [Google Scholar]
- [75].Daud II, Scott ME, Ma Y, et al. Association between Toll-like Receptor Expression and Human Papillomavirus Type 16 Persistence. Int J Cancer. 2011;128(4):879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Zhang Y, El-Far M, Dupuy FP, et al. HCV RNA Activates APCs via TLR7/TLR8 While Virus Selectively Stimulates Macrophages Without Inducing Antiviral Responses. Sci Rep. 2016;6:29447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Colak E, Leslie A, Zausmer K, et al. RNA and Imidazoquinolines Are Sensed by Distinct TLR7/8 Ectodomain Sites Resulting in Functionally Disparate Signaling Events. J Immunol. 2014;192(12):5963–5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Krüger A, Oldenburg M, Chebrolu C, et al. Human TLR8 Senses UR/URR Motifs in Bacterial and Mitochondrial RNA. EMBO Rep. 2015;16(12):1656–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Leonard JN, Ghirlando R, Askins J, et al. The TLR3 Signaling Complex Forms by Cooperative Receptor Dimerization. Proc Natl Acad Sci. 2008;105(1):258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Gosu V, Son S, Shin D, et al. Insights into the Dynamic Nature of the DsRNA-Bound TLR3 Complex. Sci Rep. 2019;9(1):3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Bell JK, Askins J, Hall PR, et al. The DsRNA Binding Site of Human Toll-like Receptor 3. Proc Natl Acad Sci. 2006;103(23):8792–8797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Lee HK, Lund JM, Ramanathan B, et al. Autophagy-Dependent Viral Recognition by Plasmacytoid Dendritic Cells. Science. 2007;315(5817):1398–1401. [DOI] [PubMed] [Google Scholar]
- [83].Akira S, Uematsu S, Takeuchi O. Pathogen Recognition and Innate Immunity. Cell. 2006;124(4):783–801. [DOI] [PubMed] [Google Scholar]
- [84].Balachandran S, Roberts PC, Brown LE, et al. Essential Role for the DsRNA-Dependent Protein Kinase PKR in Innate Immunity to Viral Infection. Immunity. 2000;13(1):129–141. [DOI] [PubMed] [Google Scholar]
- [85].Ung TL, Cao C, Lu J, et al. Heterologous Dimerization Domains Functionally Substitute for the Double-Stranded RNA Binding Domains of the Kinase PKR. Embo J. 2001;20(14):3728–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Feng F, Yuan L, Wang YE, et al. Crystal Structure and Nucleotide Selectivity of Human IFIT5/ISG58. Cell Res. 2013;23(8):1055–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Manche L, Green SR, Schmedt C, et al. Interactions between Double-Stranded RNA Regulators and the Protein Kinase DAI. Mol Cell Biol. 1992;12(11):5238–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].García MA, Gil J, Ventoso I, et al. Impact of Protein Kinase PKR in Cell Biology: from Antiviral to Antiproliferative Action. Microbiol Mol Biol Rev MMBR. 2006;70(4):1032–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Manivannan P, Siddiqui MA, Malathi K. RNase L Amplifies Interferon Signaling by Inducing PKR-Mediated Antiviral Stress Granules. J Virol. 2020. DOI: 10.1128/JVI.00205-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Pham AM, Santa Maria FG, Lahiri T, et al. PKR Transduces MDA5-Dependent Signals for Type I IFN Induction. PLOS Pathog. 2016;12(3):1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Langland JO, Pettiford S, Jiang B, et al. Products of the Porcine Group C Rotavirus NSP3 Gene Bind Specifically to Double-Stranded RNA and Inhibit Activation of the Interferon-Induced Protein Kinase PKR. J Virol. 1994;68(6):3821–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Schierhorn KL, Jolmes F, Bespalowa J, et al. Influenza A Virus Virulence Depends on Two Amino Acids in the N-Terminal Domain of Its NS1 Protein To Facilitate Inhibition of the RNA-Dependent Protein Kinase PKR. J Virol. 2017;91(10). DOI: 10.1128/jvi.00198-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Tu Y-C, Yu C-Y, Liang -J-J, et al. Blocking Double-Stranded RNA-Activated Protein Kinase PKR by Japanese Encephalitis Virus Nonstructural Protein 2A. J Virol. 2012;86(19):10347–10358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Vyas J, Elia A, Clemens MJ. Inhibition of the Protein Kinase PKR by the Internal Ribosome Entry Site of Hepatitis C Virus Genomic RNA. Rna N Y N. 2003;9(7):858–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Taylor DR. Inhibition of the Interferon- Inducible Protein Kinase PKR by HCV E2 Protein. Science. 1999;285(5424):107–110. [DOI] [PubMed] [Google Scholar]
- [96].François C, Duverlie G, Rebouillat D, et al. Expression of Hepatitis C Virus Proteins Interferes with the Antiviral Action of Interferon Independently of PKR-Mediated Control of Protein Synthesis. J Virol. 2000;74(12):5587–5596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Frese M, Interferon-Induced Effector DE. Proteins and Hepatitis C Virus Replication. Jirillo E editor. Hepatitis C Virus Disease. Springer New York: New York. 2008. 106–129. 10.1007/978-0-387-71376-2_6. [DOI] [Google Scholar]
- [98].Black TL, Safer B, Hovanessian A, et al. 68,000-Mr Protein Kinase Is Highly Autophosphorylated and Activated yet Significantly Degraded during Poliovirus Infection: implications for Translational Regulation. J Virol. 1989;63(5):2244–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Chang Y-H, Lau KS, Kuo R-L, et al. DsRNA Binding Domain of PKR Is Proteolytically Released by Enterovirus A71 to Facilitate Viral Replication. Frontiers in Cellular and Infection Microbiology. 2017;7:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Mears HV, Sweeney TR. Better Together: the Role of IFIT Protein–Protein Interactions in the Antiviral Response. Journal of General Virology. 2018;99(11):1463–1477. [DOI] [PubMed] [Google Scholar]
- [101].Daffis S, Szretter KJ, Schriewer J, et al. 2′-O Methylation of the Viral MRNA Cap Evades Host Restriction by IFIT Family Members. Nature. 2010;468(7322):452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Abbas YM, Pichlmair A, Górna MW, et al. Structural Basis for Viral 5′-PPP-RNA Recognition by Human IFIT Proteins. Nature. 2013;494(7435):60–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Sen GC, Fensterl V. Crystal Structure of IFIT2 (ISG54) Predicts Functional Properties of IFITs. Cell Res. 2012;22(10):1407–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Tran V, Ledwith MP, Thamamongood T, et al. Influenza Virus Repurposes the Antiviral Protein IFIT2 to Promote Translation of Viral MRNAs. . Nature Microbiology. 2020;5(12):1490–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Sabbah A, Chang TH, Harnack R, et al. Activation of Innate Immune Antiviral Responses by Nod2. Nat Immunol. 2009;10(10):1073–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Allen IC, Scull MA, Moore CB, et al. The NLRP3 Inflammasome Mediates In Vivo Innate Immunity to Influenza A Virus through Recognition of Viral RNA. Immunity. 2009;30(4):556–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Yu C-H, Moecking J, Geyer M, et al. Mechanisms of NLRP1-Mediated Autoinflammatory Disease in Humans and Mice. Journal of Molecular Biology. 2018;430(2):142–152. [DOI] [PubMed] [Google Scholar]
- [108].Feng Q, Hato SV, Langereis MA, et al. MDA5 Detects the Double-Stranded RNA Replicative Form in Picornavirus-Infected Cells. Cell Rep. 2012;2(5):1187–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].den Boon JA, Organelle-Like Membrane AP. Compartmentalization of Positive-Strand RNA Virus Replication Factories. Annu Rev Microbiol. 2010;64(1):241–256. [DOI] [PubMed] [Google Scholar]
- [110].Romero-Brey I, Bartenschlager R. Membranous Replication Factories Induced by Plus-Strand RNA Viruses. Viruses. 2014;6(7):2826–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].van der Hoeven B, Oudshoorn D, Koster AJ, et al. Architecture of Arterivirus Replication Organelles. Virus Res. 2016;220:70–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Scutigliani EM, Kikkert M. Interaction of the Innate Immune System with Positive-Strand RNA Virus Replication Organelles. Cytokine Growth Factor Rev. 2017;37:17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Neufeldt CJ, Joyce MA, Van Buuren N, et al. The Hepatitis C Virus-Induced Membranous Web and Associated Nuclear Transport Machinery Limit Access of Pattern Recognition Receptors to Viral Replication Sites. PLOS Pathog. 2016;12(2):1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Al-Mulla HMN, Turrell L, Smith NM, et al. Competitive Fitness in Coronaviruses Is Not Correlated with Size or Number of Double-Membrane Vesicles under Reduced-Temperature Growth Conditions. mBio. 2014;5(2):e01107–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Collins PL, Fearns R, Graham BS. Respiratory Syncytial Virus: virology, Reverse Genetics, and Pathogenesis of Disease. In:Challenges and Opportunities for Respiratory Syncytial Virus Vaccines. Anderson LJ, Graham BS, editors. Current Topics in Microbiology and Immunology Springer Berlin Heidelberg:Berlin, Heidelberg. 2013. Vol. 372. 3–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Jaafar ZA, Kieft JS. Viral RNA Structure-Based Strategies to Manipulate Translation. Nat Rev Microbiol. 2019;17(2):110–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Flanegan JB, Petterson RF, Ambros V, et al. Covalent Linkage of a Protein to a Defined Nucleotide Sequence at the 5ʹ-Terminus of Virion and Replicative Intermediate RNAs of Poliovirus. Proc Natl Acad Sci. 1977;74(3):961–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Lee YF, Nomoto A, Detjen BM, et al. A Protein Covalently Linked to Poliovirus Genome RNA. Proc Natl Acad Sci. 1977;74(1):59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Jang SK, Kräusslich HG, Nicklin MJ, et al. Segment of the 5ʹ Nontranslated Region of Encephalomyocarditis Virus RNA Directs Internal Entry of Ribosomes during in Vitro Translation. J Virol. 1988;62(8):2636–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Pelletier J, Sonenberg N. Internal Initiation of Translation of Eukaryotic MRNA Directed by a Sequence Derived from Poliovirus RNA. Nature. 1988;334(6180):320–325. [DOI] [PubMed] [Google Scholar]
- [121].Decroly E, Biochemical Principles CB. Inhibitors to Interfere with Viral Capping Pathways. Curr Opin Virol. 2017;24:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Decroly E, Ferron F, Lescar J, et al. Unconventional Mechanisms for Capping Viral MRNA. Nat Rev Microbiol. 2011;10(1):51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Liu L, Dong H, Chen H, et al. Flavivirus RNA Cap Methyltransferase: structure, Function, and Inhibition. Front Biol. 2010;5(4):286–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Bouvet M, Debarnot C, Imbert I, et al. In Vitro Reconstitution of SARS-Coronavirus MRNA Cap Methylation. PLOS Pathog. 2010;6(4):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Züst R, Cervantes-Barragan L, Habjan M, et al. 2′-O-Methylation Provides a Molecular Signature for the Distinction of Self and Non-Self MRNA Dependent on the RNA Sensor Mda5. Nat Immunol. 2011;12(2):137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].PETTERSSON RF, SÖDERLUND H, KÁÁRIÁINEN L. The Nucleotide Sequences of the 5′-Terminal T1 Oligonucleotides of Semliki-Forest-Virus 42-S and 26-S RNAs Are Different. Eur J Biochem. 1980;105(3):435–443. [DOI] [PubMed] [Google Scholar]
- [127].Hefti E, Bishop DHL, Dubin DT, et al. 5ʹ Nucleotide Sequence of Sindbis Viral RNA. J Virol. 1976;17(1):149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Hyde JL, Gardner CL, Kimura T, et al. RNA Structural Element Alters Host Recognition of Nonself RNA. Science. 2014;343(6172):783–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Reguera J, Gerlach P, Rosenthal M, et al. Functional Analysis of Bunyavirus and Arenavirus Cap-Snatching Endonucleases. PLOS Pathog. 2016;12(6):1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Dias A, Bouvier D, Crépin T, et al. The Cap-Snatching Endonuclease of Influenza Virus Polymerase Resides in the PA Subunit. Nature. 2009;458(7240):914–918. [DOI] [PubMed] [Google Scholar]
- [131].De Vlugt C, Sikora D, Pelchat M. Insight into Influenza: A Virus Cap-Snatching. Viruses. 2018;10(11):641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Trixl L, Lusser A. The Dynamic RNA Modification 5-Methylcytosine and Its Emerging Role as an Epitranscriptomic Mark. WIREs RNA. 2019;10(1):e1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Schwartz S, Bernstein DA, Mumbach MR, et al. Reveals Widespread Dynamic-Regulated Pseudouridylation of NcRNA and MRNA. Cell. 2014;159(1):148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Gokhale NS, Horner SM, Modifications RNA. Go Viral. PLOS Pathog. 2017;13(3):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Gonzales-van Horn SR, Sarnow P. Making the Mark: the Role of Adenosine Modifications in the Life Cycle of RNA Viruses. Cell Host Microbe. 2017;21(6):661–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Shi H, Wei J, Where HC. When, and How: context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol Cell. 2019;74(4):640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Jia G, Fu Y, Zhao X, et al. N6-Methyladenosine in Nuclear RNA Is a Major Substrate of the Obesity-Associated FTO. Nat Chem Biol. 2011;7(12):885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Zheng G, Dahl JA, Niu Y, et al. ALKBH5 Is a Mammalian RNA Demethylase That Impacts RNA Metabolism and Mouse Fertility. Mol Cell. 2013;49(1):18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Shi H, Wang X, Lu Z, et al. YTHDF3 Facilitates Translation and Decay of N6-Methyladenosine-Modified RNA. Cell Res. 2017;27(3):315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Gokhale NS, McIntyre ABR, McFadden MJ, et al. N6-Methyladenosine in Flaviviridae Viral RNA Genomes Regulates Infection. Cell Host Microbe. 2016;20(5):654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Hao H, Hao S, Chen H, et al. N6-Methyladenosine Modification and METTL3 Modulate Enterovirus 71 Replication. Nucleic Acids Res. 2018;47(1):362–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Lichinchi G, Zhao BS, Wu Y, et al. Dynamics of Human and Viral RNA Methylation during Zika Virus Infection. Cell Host Microbe. 2016;20(5):666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Kennedy EM, Courtney DG, Tsai K, et al. Viral Epitranscriptomics. J Virol. 2017;91(9). DOI: 10.1128/JVI.02263-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].COHN WE, VOLKIN E. Nucleoside-5′-Phosphates from Ribonucleic Acid. Nature. 1951;167(4247):483–484. [Google Scholar]
- [145].Cohn WE. 5-Ribosyl Uracil, a Carbon-Carbon Ribofuranosyl Nucleoside in Ribonucleic Acids. Biochim Biophys Acta. 1959;32:569–571. [DOI] [PubMed] [Google Scholar]
- [146].Marceau CD, Puschnik AS, Majzoub K, et al. Genetic Dissection of Flaviviridae Host Factors through Genome-Scale CRISPR Screens. Nature. 2016;535(7610):159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Karikó K, Muramatsu H, Welsh FA, et al. Incorporation of Pseudouridine Into MRNA Yields Superior Nonimmunogenic Vector With Increased Translational Capacity and Biological Stability. Mol Ther. 2008;16(11):1833–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Aspden JL, Jackson RJ. Differential Effects of Nucleotide Analogs on Scanning-Dependent Initiation and Elongation of Mammalian MRNA Translation in Vitro. RNA. 2010;16(6):1130–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Xing J, Yi J, Cai X, et al. NSun2 Promotes Cell Growth via Elevating Cyclin-Dependent Kinase 1 Translation. Mol Cell Biol. 2015;35(23):4043–4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Yang X, Yang Y, Sun B-F, et al. 5-Methylcytosine Promotes MRNA Export — NSUN2 as the Methyltransferase and ALYREF as an M5C Reader. Cell Res. 2017;27(5):606–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Tang H, Fan X, Xing J, et al. NSun2 Delays Replicative Senescence by Repressing P27 (KIP1) Translation and Elevating CDK1 Translation. Aging (Albany NY). 2015;7(12):1143–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Hoernes TP, Clementi N, Faserl K, et al. Nucleotide Modifications within Bacterial Messenger RNAs Regulate Their Translation and Are Able to Rewire the Genetic Code. Nucleic Acids Res. 2016;44(2):852–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Bujnicki JM, Feder M, Ayres CL, et al. Sequence-Structure-Function Studies of TRNA:M5C Methyltransferase Trm4p and Its Relationship to DNA:M5C and RNA:M5U Methyltransferases. Nucleic Acids Res. 2004;32(8):2453–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Motorin Y, Lyko F, Helm M. 5-Methylcytosine in RNA: detection, Enzymatic Formation and Biological Functions. Nucleic Acids Res. 2010;38(5):1415–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].McIntyre W, Netzband R, Bonenfant G, et al. Positive-Sense RNA Viruses Reveal the Complexity and Dynamics of the Cellular and Viral Epitranscriptomes during Infection. Nucleic Acids Res. 2018;46(11):5776–5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Courtney DG, Chalem A, Bogerd HP, et al. Extensive Epitranscriptomic Methylation of A and C Residues on Murine Leukemia Virus Transcripts Enhances Viral Gene Expression. mBio. 2019;10(3). DOI: 10.1128/mBio.01209-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Dong H, Chang DC, Hua MHC, et al. 2′-O Methylation of Internal Adenosine by Flavivirus NS5 Methyltransferase. PLOS Pathog. 2012;8(4):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Petes C, Odoardi N, Gee K. The Toll for Trafficking: toll-Like Receptor 7 Delivery to the Endosome. Front Immunol. 2017;8:1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Cruz-Oliveira C, Freire JM, Conceição TM, et al. Receptors and Routes of Dengue Virus Entry into the Host Cells. FEMS Microbiol Rev. 2015;39(2):155–170. [DOI] [PubMed] [Google Scholar]
- [160].Samuel CE. Adenosine Deaminases Acting on RNA (ADARs) Are Both Antiviral and Proviral. Virology. 2011;411(2):180–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Martínez I, Melero JAA. Model for the Generation of Multiple A to G Transitions in the Human Respiratory Syncytial Virus Genome: predicted RNA Secondary Structures as Substrates for Adenosine Deaminases That Act on RNA. J Gen Virol. 2002;83:1445–1455. [DOI] [PubMed] [Google Scholar]
- [162].Rima BK, Gatherer D, Young DF, et al. Stability of the Parainfluenza Virus 5 Genome Revealed by Deep Sequencing of Strains Isolated from Different Hosts and Following Passage in Cell Culture. J Virol. 2014;88(7):3826–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Taylor DR, Puig M, Darnell MER, et al. New Antiviral Pathway That Mediates Hepatitis C Virus Replicon Interferon Sensitivity through ADAR1. J Virol. 2005;79(10):6291–6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Mannion NM, Greenwood SM, Young R, et al. Editing Enzyme ADAR1 Controls Innate Immune Responses to RNA. Cell Rep. 2014;9(4):1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Liddicoat BJ, Piskol R, Chalk AM, et al. RNA Editing by ADAR1 Prevents MDA5 Sensing of Endogenous DsRNA as Nonself. Science. 2015;349(6252):1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Pfaller CK, Donohue RC, Nersisyan S, et al. Extensive Editing of Cellular and Viral Double-Stranded RNA Structures Accounts for Innate Immunity Suppression and the Proviral Activity of ADAR1p150. PLOS Biol. 2018;16(11):1–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Deng X, Hackbart M, Mettelman RC, et al. Coronavirus Nonstructural Protein 15 Mediates Evasion of DsRNA Sensors and Limits Apoptosis in Macrophages. Proc Natl Acad Sci. 2017;114(21):E4251–E4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Hackbart M, Deng X, Baker SC. Coronavirus Endoribonuclease Targets Viral Polyuridine Sequences to Evade Activating Host Sensors. Proc Natl Acad Sci. 2020;117(14):8094–8103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Jagger BW, Wise HM, Kash JC, et al. An Overlapping Protein-Coding Region in Influenza A Virus Segment 3 Modulates the Host Response. Science. 2012;337(6091):199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Gaucherand L, Porter BK, Levene RE, et al. The Influenza A Virus Endoribonuclease PA-X Usurps Host MRNA Processing Machinery to Limit Host Gene Expression. Cell Rep. 2019;27(3):776–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Hayashi T, MacDonald LA, Takimoto T. Influenza A Virus Protein PA-X Contributes to Viral Growth and Suppression of the Host Antiviral and Immune Responses. J Virol. 2015;89(12):6442–6452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Hayashi T, Chaimayo C, McGuinness J, et al. Critical Role of the PA-X C-Terminal Domain of Influenza A Virus in Its Subcellular Localization and Shutoff Activity. J Virol. 2016;90(16):7131–7141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Khaperskyy DA, Schmaling S, Larkins-Ford J, et al. Selective Degradation of Host RNA Polymerase II Transcripts by Influenza A Virus PA-X Host Shutoff Protein. PLOS Pathog. 2016;12(2):1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Tanaka T, Kamitani W, DeDiego ML, et al. Severe Acute Respiratory Syndrome Coronavirus Nsp1 Facilitates Efficient Propagation in Cells through a Specific Translational Shutoff of Host MRNA. J Virol. 2012;86(20):11128–11137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Lokugamage KG, Narayanan K, Huang C, et al. Severe Acute Respiratory Syndrome Coronavirus Protein Nsp1 Is a Novel Eukaryotic Translation Inhibitor That Represses Multiple Steps of Translation Initiation. J Virol. 2012;86(24):13598–13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Kamitani W, Huang C, Narayanan K, et al. Strategy to Suppress Host Protein Synthesis by SARS Coronavirus Nsp1 Protein. Nat Struct Mol Biol. 2009;16(11):1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Lokugamage KG, Narayanan K, Nakagawa K, et al. Middle East Respiratory Syndrome Coronavirus Nsp1 Inhibits Host Gene Expression by Selectively Targeting MRNAs Transcribed in the Nucleus While Sparing MRNAs of Cytoplasmic Origin. J Virol. 2015;89(21):10970–10981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Foeger N, Schmid EM, Human Rhinovirus ST. 2 2A pro Recognition of Eukaryotic Initiation Factor 4GI: INVOLVEMENT OF AN EXOSITE. J Biol Chem. 2003;278(35):33200–33207. [DOI] [PubMed] [Google Scholar]
- [179].Etchison D, Ederys I, Sonenberggl N (1982) Inhibition of HeLa Cell ProteinSynthesis Following Poliovirus Infection Correlates with the Proteolysis of a 220,000-Dalton Polypeptide Associated with Eucaryotic Initiation Facto3rand a Cap Binding Protein Complex. 5. [PubMed]
- [180].Song -Q-Q, Lu M-Z, Song J, et al. Coxsackievirus B3 2A Protease Promotes Encephalomyocarditis Virus Replication. Virus Res. 2015;208:22–29. [DOI] [PubMed] [Google Scholar]
- [181].Martínez-Salas E, Ryan MD. Translation and Protein Processing. In: The Picornaviruses. American Society of Microbiology Press, Washington DC, 2010. 140–161. [Google Scholar]
- [182].Aumayr M, Schrempf A, Üzülmez Ö, et al. Interaction of 2A Proteinase of Human Rhinovirus Genetic Group A with EIF4E Is Required for EIF4G Cleavage during Infection. Virology. 2017;511:123–134. [DOI] [PubMed] [Google Scholar]
- [183].Bruce SR, Atkins CL, Colasurdo GN, et al. Respiratory Syncytial Virus Infection Alters Surfactant Protein A Expression in Human Pulmonary Epithelial Cells by Reducing Translation Efficiency. Am J Physiol-Lung Cell Mol Physiol. 2009;297(4):L559–L567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Spann KM, Tran KC, Collins PL. Effects of Nonstructural Proteins NS1 and NS2 of Human Respiratory Syncytial Virus on Interferon Regulatory Factor 3, NF-B, and Proinflammatory Cytokines. J Virol. 2005;79:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Lifland AW, Jung J, Alonas E, et al. Human Respiratory Syncytial Virus Nucleoprotein and Inclusion Bodies Antagonize the Innate Immune Response Mediated by MDA5 and MAVS. J Virol. 2012;86(15):8245–8258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Ling Z, Tran KC, Teng MN. Human Respiratory Syncytial Virus Nonstructural Protein NS2 Antagonizes the Activation of Beta Interferon Transcription by Interacting with RIG-I. J Virol. 2009;83(8):3734–3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Ribaudo M, Barik S. The Nonstructural Proteins of Pneumoviruses Are Remarkably Distinct in Substrate Diversity and Specificity. Virol J. 2017;14(1):215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Dhar J, Cuevas RA, Goswami R, et al. 2ʹ-5ʹ-Oligoadenylate Synthetase-Like Protein Inhibits Respiratory Syncytial Virus Replication and Is Targeted by the Viral Nonstructural Protein 1. J Virol. 2015;89(19):10115–10119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Goswami R, Majumdar T, Dhar J, et al. Viral Degradasome Hijacks Mitochondria to Suppress Innate Immunity. Cell Res. 2013;23(8):1025–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Lindquist ME, Lifland AW, Utley TJ, et al. Respiratory Syncytial Virus Induces Host RNA Stress Granules to Facilitate Viral Replication. J Virol. 2010;84(23):12274–12284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Lindquist ME, Mainou BA, Dermody TS, et al. Activation of Protein Kinase R Is Required for Induction of Stress Granules by Respiratory Syncytial Virus but Dispensable for Viral Replication. Virology. 2011;413(1):103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Moon SL, Barnhart MD, Inhibition WJ. Avoidance of MRNA Degradation by RNA Viruses. Curr Opin Microbiol. 2012;15(4):500–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Dickson AM, Wilusz J. Strategies for Viral RNA Stability: live Long and Prosper. Trends Genet. 2011;27(7):286–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Abernathy E, Glaunsinger B. Emerging Roles for RNA Degradation in Viral Replication and Antiviral Defense. Virology. 2015;479–480:600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Burgess HM, Cellular MI. 5′-3′ MRNA Exonuclease Xrn1 Controls Double-Stranded RNA Accumulation and Anti-Viral Responses. Cell Host Microbe. 2015;17(3):332–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Bhowmick R, Mukherjee A, Patra U, et al. Cytoplasmic P Bodies during Infection. Virus Res. 2015;210:344–354. [DOI] [PubMed] [Google Scholar]
- [197].Dougherty JD, White JP, Lloyd RE. Poliovirus-Mediated Disruption of Cytoplasmic Processing Bodies. J Virol. 2011;85(1):64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Silva PAGC, Pereira CF, Dalebout TJ, et al. An RNA Pseudoknot Is Required for Production of Yellow Fever Virus Subgenomic RNA by the Host Nuclease XRN1. J Virol. 2010;84(21):11395–11406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Chapman EG, Costantino DA, Rabe JL, et al. The Structural Basis of Pathogenic Subgenomic Flavivirus RNA (SfRNA) Production. Science. 2014;344(6181):307–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Pijlman GP, Funk A, Kondratieva N, et al. A Highly Structured, Nuclease-Resistant, Noncoding RNA Produced by Flaviviruses Is Required for Pathogenicity. Cell Host Microbe. 2008;4(6):579–591. [DOI] [PubMed] [Google Scholar]
- [201].Michalski D, Ontiveros JG, Russo J, et al. Zika Virus Noncoding SfRNAs Sequester Multiple Host-Derived RNA-Binding Proteins and Modulate MRNA Decay and Splicing during Infection. J Biol Chem. 2019;294(44):16282–16296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Moon SL, Anderson JR, Kumagai Y, et al. RNA Produced by Arthropod-Borne Flaviviruses Inhibits the Cellular Exoribonuclease XRN1 and Alters Host MRNA Stability. RNA. 2012;18(11):2029–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Moon SL, Blackinton JG, Anderson JR, et al. XRN1 Stalling in the 5ʹ UTR of Hepatitis C Virus and Bovine Viral Diarrhea Virus Is Associated with Dysregulated Host MRNA Stability. PLOS Pathog. 2015;11(3):1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Ng CS, Kasumba DM, Fujita T, et al. Characterization of the Antiviral Activity of the XRN1-DCP1/2 Aggregation against Cytoplasmic RNA Viruses to Prevent Cell Death. Cell Death Differ. 2020. DOI: 10.1038/s41418-020-0509-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Schmid M, Jensen T.The Nuclear RNA Exosome and Its Cofactors. In: Advances in experimental medicine and biology. Vol. 1203; 2019. p. 113–132. [DOI] [PubMed] [Google Scholar]
- [206].Lebreton A, Tomecki R, Dziembowski A, et al. RNA Cleavage by a Eukaryotic Exosome. Nature. 2008;456(7224):993–996. [DOI] [PubMed] [Google Scholar]
- [207].Tomecki R, Kristiansen MS, Lykke-Andersen S, et al. The Human Core Exosome Interacts with Differentially Localized Processive RNases: HDIS3 and HDIS3L. Embo J. 2010;29(14):2342–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Staals RHJ, Bronkhorst AW, Schilders G, et al. Dis3-like 1: A Novel Exoribonuclease Associated with the Human Exosome. Embo J. 2010;29(14):2358–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Toh-E A, Guerry P, Wickner RB. Chromosomal Superkiller Mutants of Saccharomyces Cerevisiae. J Bacteriol. 1978;136(3):1002–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Ridley SP, Sommer SS, Wickner RB. Superkiller Mutations in Saccharomyces Cerevisiae Suppress Exclusion of M2 Double-Stranded RNA by L-A-HN and Confer Cold Sensitivity in the Presence of M and L-A-HN. Mol Cell Biol. 1984;4(4):761–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Orban TI, Izaurralde E. Decay of MRNAs Targeted by RISC Requires XRN1, the Ski Complex, and the Exosome. Rna N Y N. 2005;11(4):459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Zhu B, Mandal SS, Pham A-D, et al. The Human PAF Complex Coordinates Transcription with Events Downstream of RNA Synthesis. Genes Dev. 2005;19(14):1668–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Eckard SC, Rice GI, Fabre A, et al. The SKIV2L RNA Exosome Limits Activation of the RIG-I-like Receptors. Nat Immunol. 2014;15(9):839–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Molleston JM, Sabin LR, Moy RH, et al. A Conserved Virus-Induced Cytoplasmic TRAMP-like Complex Recruits the Exosome to Target Viral RNA for Degradation. Genes Dev. 2016;30(14):1658–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Onomoto K, Yoneyama M, Fung G, et al. Antiviral Innate Immunity and Stress Granule Responses. Trends Immunol. 2014;35(9):420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Onomoto K, Jogi M, Yoo J-S, et al. Critical Role of an Antiviral Stress Granule Containing RIG-I and PKR in Viral Detection and Innate Immunity. PLoS ONE. 2012;7(8):e43031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Reineke LC, Kedersha N, Langereis MA, et al. Stress Granules Regulate Double-Stranded RNA-Dependent Protein Kinase Activation through a Complex Containing G3BP1 and Caprin1. mBio. 2015;6(2). DOI: 10.1128/mBio.02486-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Nover L, Scharf KD, Neumann D. Cytoplasmic Heat Shock Granules Are Formed from Precursor Particles and Are Associated with a Specific Set of MRNAs. Mol Cell Biol. 1989;9(3):1298–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Williams BRG. Signal Integration via PKR. Sci Signal. 2001;2001(89):re2. [DOI] [PubMed] [Google Scholar]
- [220].Hu S, Sun H, Yin L, et al. Dependent Cytosolic CGAS Foci Are Necessary for Intracellular DNA Sensing. Sci Signal. 2019;12:609. [DOI] [PubMed] [Google Scholar]
- [221].Gongora C, David G, Pintard L, et al. Molecular Cloning of a New Interferon-Induced PML Nuclear Body-Associated Protein. J Biol Chem. 1997;272(31):19457–19463. [DOI] [PubMed] [Google Scholar]
- [222].Espert L, Degols G, Gongora C, et al. ISG20, a New Interferon-Induced RNase Specific for Single-Stranded RNA, Defines an Alternative Antiviral Pathway against RNA Genomic Viruses. J Biol Chem. 2003;278(18):16151–16158. [DOI] [PubMed] [Google Scholar]
- [223].Jiang D, Weidner JM, Qing M, et al. Identification of Five Interferon-Induced Cellular Proteins That Inhibit West Nile Virus and Dengue Virus Infections. J Virol. 2010;84(16):8332–8341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Jiang D, Guo H, Xu C, et al. Identification of Three Interferon-Inducible Cellular Enzymes That Inhibit the Replication of Hepatitis C Virus. J Virol. 2008;82(4):1665–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Zhou Z, Wang N, Woodson SE, et al. Antiviral Activities of ISG20 in Positive-Strand RNA Virus Infections. Virology. 2011;409(2):175–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Zhang Y, Burke CW, Ryman KD, et al. Identification and Characterization of Interferon-Induced Proteins That Inhibit Alphavirus Replication. J Virol. 2007;81(20):11246–11255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Qu H, Li J, Yang L, et al. Influenza A Virus-Induced Expression of ISG20 Inhibits Viral Replication by Interacting with Nucleoprotein. Virus Genes. 2016;52(6):759–767. [DOI] [PubMed] [Google Scholar]
- [228].Espert L, Degols G, Lin Y-L, et al. ISG20 Exhibits an Antiviral Activity against Human Immunodeficiency Virus Type 1. J Gen Virol. 2005;86:2221–2229. [DOI] [PubMed] [Google Scholar]
- [229].Nguyen LH, Espert L, Mechti N, et al. The Human Interferon- and Estrogen-Regulated ISG20/HEM45 Gene Product Degrades Single-Stranded RNA and DNA in Vitro. Biochemistry. 2001;40(24):7174–7179. [DOI] [PubMed] [Google Scholar]
- [230].Weiss CM, Trobaugh DW, Sun C, et al. The Interferon-Induced Exonuclease ISG20 Exerts Antiviral Activity through Upregulation of Type I Interferon Response Proteins. mSphere. 2018;3(5):e00209–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Wu N, Nguyen X-N, Wang L, et al. The Interferon Stimulated Gene 20 Protein (ISG20) Is an Innate Defense Antiviral Factor That Discriminates Self versus Non-Self Translation. PLOS Pathog. 2019;15(10):1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Liu C-H, Zhou L, Chen G, et al. Battle between Influenza A Virus and a Newly Identified Antiviral Activity of the PARP-Containing ZAPL Protein. Proc Natl Acad Sci. 2015;112(45):14048–14053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Tang Q, Wang X, Gao G. The Short Form of the Zinc Finger Antiviral Protein Inhibits Influenza A Virus Protein Expression and Is Antagonized by the Virus-Encoded NS1. J Virol. 2017;91(2). DOI: 10.1128/JVI.01909-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Chiu H-P, Chiu H, Yang C-F, et al. Inhibition of Japanese Encephalitis Virus Infection by the Host Zinc-Finger Antiviral Protein. PLoS Pathog. 2018;14(7):e1007166–e1007166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Takata MA, Gonçalves-Carneiro D, Zang TM, et al. Enables Antiviral Defence Targeting Non-Self RNA. Nature. 2017;550(7674):124–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Bick MJ, Carroll J-WN, Gao G, et al. Expression of the Zinc-Finger Antiviral Protein Inhibits Alphavirus Replication. J Virol. 2003;77(21):11555–11562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Gao G, Guo X, Goff SP. Inhibition of Retroviral RNA Production by ZAP, a CCCH-Type Zinc Finger Protein. Science. 2002;297(5587):1703–1706. [DOI] [PubMed] [Google Scholar]
- [238].Odon V, Fros JJ, Goonawardane N, et al. The Role of ZAP and OAS3/RNAseL Pathways in the Attenuation of an RNA Virus with Elevated Frequencies of CpG and UpA Dinucleotides. Nucleic Acids Res. 2019;47(15):8061–8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Atkinson NJ, Witteveldt J, Evans DJ, et al. The Influence of CpG and UpA Dinucleotide Frequencies on RNA Virus Replication and Characterization of the Innate Cellular Pathways Underlying Virus Attenuation and Enhanced Replication. Nucleic Acids Res. 2014;42(7):4527–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Guo X, Ma J, Sun J, et al. Antiviral Protein Recruits the RNA Processing Exosome to Degrade the Target MRNA. Proc Natl Acad Sci. 2007;104(1):151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Zhu Y, Chen G, Lv F, et al. Zinc-Finger Antiviral Protein Inhibits HIV-1 Infection by Selectively Targeting Multiply Spliced Viral MRNAs for Degradation. Proc Natl Acad Sci U S A. 2011;108(38):15834–15839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Zhu Y, Wang X, Goff SP, et al. Translational Repression Precedes and Is Required for ZAP-Mediated MRNA Decay. Embo J. 2012;31(21):4236–4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Li M-L, Weng K-F, Shih S-R, et al. The Evolving World of Small RNAs from RNA Viruses: the Evolving World of Small RNAs from RNA Viruses. Wiley Interdiscip Rev RNA. 2016;7(5):575–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Parameswaran P, Sklan E, Wilkins C, et al. Six RNA Viruses and Forty-One Hosts: viral Small RNAs and Modulation of Small RNA Repertoires in Vertebrate and Invertebrate Systems. PLoS Pathog. 2010;6(2):e1000764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Scott JC, Brackney DE, Campbell CL, et al. Comparison of Dengue Virus Type 2-Specific Small RNAs from RNA Interference-Competent and –Incompetent Mosquito Cells. PLoS Negl Trop Dis. 2010;4(10):e848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Tam OH, Aravin AA, Stein P, et al. Pseudogene-Derived Small Interfering RNAs Regulate Gene Expression in Mouse Oocytes. Nature. 2008;453(7194):534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Babiarz JE, Ruby JG, Wang Y, et al. Mouse ES Cells Express Endogenous ShRNAs, SiRNAs, and Other Microprocessor-Independent, Dicer-Dependent Small RNAs. Genes Dev. 2008;22(20):2773–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Watanabe T, Totoki Y, Toyoda A, et al. Endogenous SiRNAs from Naturally Formed DsRNAs Regulate Transcripts in Mouse Oocytes. Nature. 2008;453(7194):539–543. [DOI] [PubMed] [Google Scholar]
- [249].Flemr M, Malik R, Franke V, et al. Isoform Directs Endogenous Small Interfering RNA Production in Mouse Oocytes. Cell. 2013;155(4):807–816. [DOI] [PubMed] [Google Scholar]
- [250].Kennedy EM, Whisnant AW, Kornepati AVR, et al. Production of Functional Small Interfering RNAs by an Amino-Terminal Deletion Mutant of Human Dicer. Proc Natl Acad Sci. 2015;112(50):E6945–E6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Maillard PV, Ciaudo C, Marchais A, et al. Antiviral RNA Interference in Mammalian Cells. Science. 2013;342(6155):235–238.24115438 [Google Scholar]
- [252].Li Y, Lu J, Han Y, et al. Functions as an Antiviral Immunity Mechanism in Mammals. Science. 2013;342(6155):231–234.24115437 [Google Scholar]
- [253].Maillard PV, Van der Veen AG, Deddouche‐Grass S, et al. Inactivation of the Type I Interferon Pathway Reveals Long Double‐stranded RNA ‐mediated RNA Interference in Mammalian Cells. Embo J. 2016;35(23):2505–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Veen AG, Maillard PV, Schmidt JM, et al. The RIG‐I‐like Receptor LGP2 Inhibits Dicer‐dependent Processing of Long Double‐stranded RNA and Blocks RNA Interference in Mammalian Cells. Embo J. 2018;37(4). DOI: 10.15252/embj.201797479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Qiu Y, Xu Y-P, Wang M, et al. Flavivirus Induces and Antagonizes Antiviral RNA Interference in Both Mammals and Mosquitoes. Sci Adv. 2020;6(6):eaax7989. [DOI] [PMC free article] [PubMed] [Google Scholar]