

ABSTRACT
P-glycoprotein (P-gp/ABCB1) and breast cancer resistance protein (BCRP/ABCG2) modulate the distribution of drugs and toxins across the blood-brain barrier (BBB). Animal studies reported that infection-induced disruption of these transporters in the developing BBB impairs fetal brain protection. However, the impact of infection mimics on P-gp/BCRP function in human brain endothelium is less well understood. We hypothesized that Toll-like receptor ligands mimicking bacterial and viral infection would modify the expression and function of P-gp and BCRP in human brain endothelial cells (BECs). Human cerebral microvascular endothelial cells (hCMEC/D3) were challenged with bacterial [Lipopolysaccharide (LPS)] and viral-mimics [polyinosinic:polycytidylic acid (PolyI:C) or single-stranded RNA (ssRNA)], or pro-inflammatory cytokines interleukin (IL)-6, tumor necrosis factor (TNF)-α and interferon gamma (IFN)-ɣ. P-gp and BCRP function was assessed after 4 or 24 h, using Calcein-AM and Chlorin-6 assays, respectively. Western blot and qPCR quantified P-gp/ABCB1 and BCRP/ABCG2 expression following treatments. Infection mimics are potent modulators of drug transporters in human BECs in vitro. LPS and PolyI:C increased, while ssRNA exposure reduced P-gp activity. In contrast, LPS and PolyI:C decreased, while ssRNA increased BCRP activity (P < .05). There was little correlation between drug transporter function, gene expression and total protein level. Altered plasma membrane BCRP may suggest modified intracellular trafficking induced by infection in human BECs. Bacterial and viral infection mimics modify P-gp and BCRP transport function in human BECs, in vitro. This knowledge may contribute and have important implications for human brain protection and possible altered biodistribution of drugs and xenobiotics in the brain following exposure to TLR agonists.
KEYWORDS: Infection mimics, lps, polyi:C, ssRNA, cytokines, blood-brain barrier (BBB), hCMEC/D3, p-glycoprotein (P-gp/ABCB1), breast Cancer Resistance Protein (BCRP/ABCG2)
Introduction
The blood-brain barrier (BBB) is a selective barrier formed by specialized brain endothelial cells (BECs) that protect the brain from invasive pathogens and penetration of toxic compounds from the bloodstream.1 The multidrug-resistance transporters, P-glycoprotein (P-gp; encoded by ABCB1) and breast cancer resistance protein (BCRP; ABCG2), belong to the ATP-binding cassette (ABC) transporters superfamily of efflux transporters and are central protective components of the BBB.2 They are present at the luminal surface of the cerebral microvascular endothelium, where they actively efflux endogenous (steroid hormones, cytokines, chemokines) and exogenous substrates (analgesics, antiretrovirals, antiepileptics, antibiotics, synthetic glucocorticoids, selective serotonin reuptake inhibitor (SSRIs), agrochemicals), back into the bloodstream. As such, they act to control accumulation of their substrates in the central nervous system (CNS).2
The development of the mammalian brain is highly vulnerable to xenobiotics and environmental toxins.1,3 Using animal models, our group demonstrated that BBB drug transport function is regulated by early exposure to several factors, including the pro-inflammatory cytokines interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α. These cytokines inhibit P-gp function in BECs, while exposure to glucocorticoids and transforming growth factor-β1 (TGF-β1) up-regulates P-gp function.1,4–7 Therefore, P-gp activity in BECs (the major component of the BBB) may either be reduced or increased depending on the nature of stimuli, resulting in altered accumulation of xenobiotics in the brain.
No studies have investigated the impact of bacterial or viral infection on drug transport function in BECs in adulthood or during development in humans. In the present study, we sought to investigate the impact of infection mimics on P-gp and BCRP transport function using an in vitro human brain endothelium model. We hypothesized that bacterial and viral mimics alter P-gp and BCRP function in the human brain endothelium. We challenged human cerebral microvascular endothelial cells (hCMEC/D3) with bacterial- and viral-mimics and key infection-associated cytokines and demonstrated that control (i.e. inhibition or activation) of human BEC function is dependent on the nature of infective/inflammatory stimuli.
Material and methods
Cell culture and reagents
The human cerebral microvascular endothelial cell line (hCMEC/D3, Cedarlane Labs #CLU512, Burlington, ON, Canada) was selected as a model to investigate drug transporters function since it has been extensively used and characterized to have a brain endothelial phenotype and is, thus a widely used model to investigate human BBB function.8–10 The cells have similar morphology and protein expression levels to primary human cerebral microvascular endothelial cells, and they are well adapted for drug transport studies.11 The cells were purchased between passages 25 and 26 (P25/26), and cultured at 37°C in 5% CO2 with EndoGROTM-MV Complete Culture Media Kit® (Millipore, ON, Canada, #SCME004), consisting of EndoGRO Basal Medium and EndoGRO Supplement kit containing recombinant human epidermal growth factor (5 ng/mL), L-glutamine (10 mM), hydrocortisone hemisuccinate (1.0 µg/mL), heparin sulfate (0.75 U/mL), ascorbic acid (50 µg/mL) and fetal bovine serum (5%; FBS). The media was supplemented with human basic fibroblast growth factor (1 ng/mL; Sigma, #F0291) and penicillin-streptomycin (1%; 10000 units – 10000 μg/mL, Life Technologies, #15140-122). Cells were expanded through P29, frozen in 5% DMSO in FBS for 24 h (−80°C), and then stored in liquid nitrogen. All procedures, treatments, and analyses were performed with hCMEC/D3 cells at P30. Lipopolysaccharide (LPS, #L4391), polyinosinic:polycytidylic acid (PolyI:C, #P9582), and calcein-acetoxymethyl ester (Ca-AM, #177831) were purchased from Sigma-Aldrich (Oakville, ON, Canada). Single-stranded RNA (ssRNA, #tlr-lrna40) and its vehicle Lyovec (#lyec-12) were purchased from InvivoGen (San Diego, California, USA), and the cytokines IL-6 (#PHC0065), TNF-α (#PHC3015) and interferon-gamma (IFN)-ɣ (#PHC4031) were purchased from Gibco (Grand Island, New York, USA).
Baseline immunohistochemical characterization
Immunohistochemical analysis was performed to confirm the phenotype of the human BEC cell line. Cells were rinsed twice with cold PBS, fixed with ice-cold Methanol for 10 min, and then permeabilized with 0.2% Triton X-100 (5 min; room temperature). Cells were then blocked with 0.1% BSA in PBS (1 h), and incubated with primary antibodies anti-P-gp (Santa Cruz Biotechnology, Dallas, Texas, USA, #SC-55510, 1:200), anti-BCRP (Millipore, #MAB146, 1:200), anti-Claudin-5 (Invitrogen #352588, 1:200), anti-PECAM-1 (Dako, Burloak, ON, Canada, #M0823, 1:200) and anti-GFAP (Cell Signaling Technology, Whitby, ON, Canada, #3670, 1:300) overnight at 4°C. The primary antibodies anti-rabbit IgG (Abcam, Toronto, ON, Canada, #ab171870), anti-mouse IgG1 (Dako, #X0931), anti-mouse IgG2a (Dako, #X0943), and anti-mouse IgG2b (Dako, #X0944) were used as isotype controls. Subsequently, cells were washed (3X) and incubated with fluorochrome-labeled secondary antibody Alexa Fluor 488 (Molecular Probes Inc., Eugene, OR, USA, 1:300) and counterstained with DAPI (1 mg/mL, 60 min). The area and intensity of antibody staining were analyzed, and representative images were obtained using a spinning disc confocal fluorescent microscope (Leica DMI6000 B, Concord, ON, Canada) at 40X magnification.
Experimental design
hCMEC/D3 cells (P30) were plated in clear flat bottom 6-well or 96-well TC-treated polystyrene culture plates (Costar, Kennebunk, ME, USA, #3516; or Falcon, Mississauga, ON, Canada, #353072, respectively) at a density of 20000 cells/cm2 with EndoGROTM, and 5% charcoal-stripped (CS)-FBS in DMEM. After 24 h of incubation, cells were challenged with the Toll-like receptor (TLR)-4 ligand, LPS (103−6 pg/mL bacterial mimic); the TLR-3 ligand PolyI:C (103−6 pg/mL; double-stranded viral mimic) or the TLR-7/8 ligand ssRNA (103−6 pg/mL; single-stranded viral mimic), to mimic bacterial and viral infection on human BECs, in vitro. The cytokines IL-6 (101−4 pg/mL), TNF-α (101−4 pg/mL), and IFN-ɣ (101−4 pg/mL) were used to determine the action of downstream mediators induced by infection/inflammation. Functional drug transport assays were undertaken or cells were harvested for total/plasma membrane protein or mRNA analysis 4- and 24 h after treatments. Dose and time of exposure regimens were selected based on previous studies from our group.4,6
Gene expression analyses
Total RNA was extracted using the RNeasy Mini Kit (Qiagen #74104, Toronto, ON, Canada), according to the manufacturer’s instructions. RNA concentration and purity were determined using a NanoDrop1000 Spectrophotometer (Thermo Scientific, Wilmington, DE, USA) and total RNA (1 μg) was reverse transcribed to cDNA using iScriptTM Reverse Transcription Supermix (Bio-Rad #1708840, Mississauga, ON, Canada). The sequences of the primers used are listed in Table 1. SYBR Green reagent (Sigma #S9194) was used to run the qPCR, using the CFX 380 Real-Time system C1000 TM Thermal Cycler (Bio-Rad). The parameters used were: one cycle (95°C, 2 min), followed by 40 cycles (95°C, 5s) and 60°C for 20s. RNA (10 ng) was used per reaction and each sample was run in triplicate. Hypoxanthine-guanine phosphoribosyltransferase (HPRT), TATA-box binding protein (TBP), and tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein zeta (YWHAZ) were used as reference genes and their geometric mean was used to normalize expression of our genes of interest. The relative mRNA expression was calculated by comparing the PCR cycle threshold (CT) between groups using the 2−ΔΔCT Method, 14 and the control group was normalized to 1.
Table 1.
Sequence of the primers used to run the qPCR in this study
| Primer | |||
|---|---|---|---|
| Target | Forward | Reverse | Reference |
| ABCB1 | agc aga ggc cgc cgc tgt tcg tt | cca ttc cga cct cgc gct cc | 12 |
| ABCG2 | tgg aat cca gaa cag agct ggg gt | aga gtt cca cgg ctg aaa cac tgc | 12 |
| HPRT | tga cac tgg caa aac aat gca | ggt cct ttt cac cag caa gct | 13 |
| IL-6 | tca atg agg aga ctt gcc tg | tgg ctt gtt cct cac tac tct | * |
| YWHAZ | act ttt ggt aca ttg tgg ctt caa | ccg cca gga caa acc agt at | 12 |
| TLR-3 | tta cga aga ggc tgg aat gg | agg aac tcc ttt gcc ttg gt | 12 |
| TLR-4 | aca gga aac ccc atc cag ag | att tgt ctc cac agc cac ca | 12 |
| TLR-8 | tta tgt gtt cca gga act cag aga a | taa tac cca agt tga tag tcg ata agt ttg | * |
| TBP | tgc aca gga gcc aag agt gaa | cac atc aca gct ccc cac ca | 13 |
*Primers designed using Primer-Blast tool at the National Center for Biotechnology Information website <https://www.ncbi.nlm.nih.gov/tools/primer-blast/>
P-gp and BCRP functional assays
P-gp activity was assessed as described previously by our group with adaptations.4–7,15 Briefly, charcoal-stripped (CS)-FBS (5%) was withdrawn and cells were washed twice with warm Tyrode salts’ solution (Sigma, #T2145) supplemented with sodium bicarbonate (1 g/L; Sigma, #S6014). Cells were incubated with 10−6 M Ca-AM at 37°C and 5% CO2 (1 h). Ca-AM is intracellularly cleaved by endogenous esterases, producing fluorescent calcein that can be used as an indirect measure of P-gp activity, since it is transported out of cells by P-gp. After incubation with Ca-AM, the plates were placed on ice and the cells were washed twice with ice-cold PBS, followed by cell lysis with 1% Triton X-100 (Sigma #X100) lysis buffer. The BCRP activity assay was adapted from a previous study16 using Chlorin 6 (Ce6, Santa Cruz Biotechnology, #SC-263067) as a specific BCRP substrate.17 Cells were incubated with 2 nM Ce6 (37°C; 5% CO2; 1 h). Plates were placed on ice, cells were washed (2X) with cold Tyrode salt solution and lysis buffer added. Calcein and Ce6 accumulation were quantified using a fluorescent microplate reader, at excitation/emission wavelengths of 485/510 nm and 407/667 nm, respectively. The uptake of Ca-AM and Ce6 by P-gp and BCRP was confirmed under the exposure to their specific inhibitors, Verapamil and Ko143, respectively, to validate the assays (see Supplementary Figure 1). N = 6–8 was used per group; assays were repeated 2–4 times. Mean background readings were subtracted and P-gp and BCRP activity following treatment were expressed as a percentage of controls.
Protein expression
Cells were detached and lysed using lysis buffer (1 mol/L Tris-HCL pH 6.8, 2% SDS, 10% glycerol, containing protease and phosphatase inhibitor cocktail from Thermo Scientific (Mississauga, ON, Canada, #78440) and extracted by sonication. Protein concentration was determined using the Pierce BCA Protein assay kit (Thermo Scientific). Plasma membrane fractions were obtained using the Minute™ Plasma Membrane Protein Isolation and Cell Fractionation kit (Invent Biotechnologies, Plymouth, MN, USA, #SM-005), following the manufacturers’ instructions. Total (30 µg) or plasma membrane (10 µg) proteins were separated by electrophoresis (100 V, 1 h) using SDS polyacrylamide gels (8%) and then transferred to polyvinylidene fluoride membranes using the Trans-Blot® Turbo™ Transfer System (Bio-Rad; 10 min). Membranes for BCRP analysis were blocked with skim milk (5%), while those for P-gp analysis were blocked with bovine serum albumin (BSA; 5%), in Tris-buffered saline containing 0.1% Tween (TBS-T; 1 h). Blots were incubated overnight (4°C) with the following primary antibodies: anti-P-gp (Abcam #ab170903, 1:1000), anti-BCRP (Abcam #ab108312, 1:2000), anti-ERK2 (Santa Cruz Biotechnology #SC-292838, 1:2000), and anti-Na+/K+-ATPase (Abcam, #ab76020, 1:20000). Membranes were then washed (3X) with TBS-T, and incubated (1 h) with HRP-linked anti-rabbit secondary antibody (1:10000; GE Healthcare Bio-Science, Baie d’Urfe, QC, Canada). Protein-antibody complexes were detected by incubating the membranes with Laminate Crescendo Western HRP Substrate (5 mins; Millipore). Chemiluminescence was detected under UV using ChemiDoc™ MP Imaging system (Bio-Rad). BCRP and P-gp protein band intensity were quantified using Image Lab™ software and normalized against ERK2 or Na+/K+-ATPase signals for total or plasma membrane protein levels assessment, respectively.
Statistical analyses
Statistical analysis was performed using Prism (version 7.0; GraphPad Software, Inc., San Diego, CA). Data were assessed for normal distribution using D’Agostino and Pearson or the Shapiro-Wilk test; any outliers were excluded using Grubbs’ test. Gene and protein expression were analyzed using unpaired Student’s t-test. Functional assays for P-gp and BCRP were analyzed using the Kruskal-Wallis test, followed by Dunn’s multiple comparison test, comparing the different treatments to the control (vehicle) group. Number of samples (n) varied from 3 to 8 per group, depending on the experiment. Specific n values are identified in each table/figure legend. Differences were considered significant when p-value<0.05. Data are presented as mean ± standard error of the mean (SEM).
Results
Characterization of human BECs
Immunohistochemical analysis confirmed the presence of P-gp and BCRP in hCMEC/D3 cells Figure 1. hCMEC/D3 cells also expressed claudin-5 and PECAM protein, consistent with a BEC phenotype. The astrocyte marker GFAP was not present. Mouse IgG1 was used as a negative control for GFAP and PECAM, and mouse IgG2a and 2b (insets) in Figure 1 were used as negative controls for BCRP, P-gp and Claudin-5, respectively. No staining in negative controls was detected.
Figure 1.
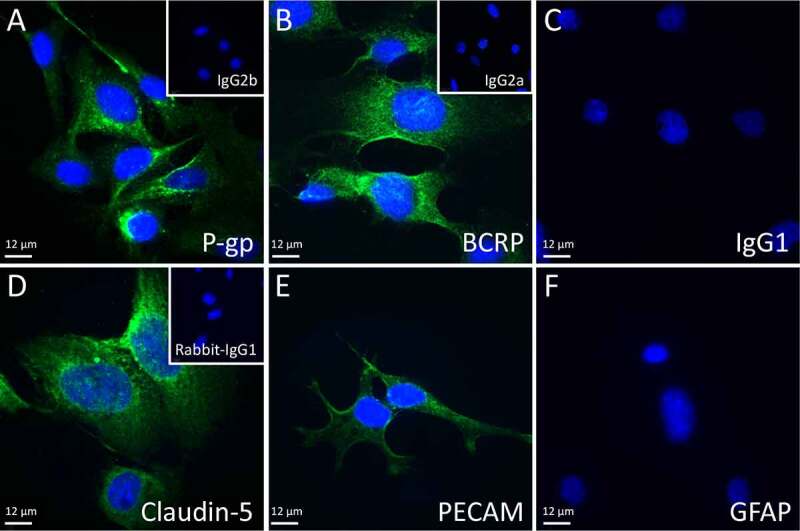
Representative fluorescent immunohistochemical images used for hCMEC/D3 characterization. Blue indicates DAPI staining, green represents specific antibody staining: A) P-gp, B) BCRP, C) IgG1, D) Claudin-5, E) PECAM, and F) GFAP. Insets show IgG2b (a), IgG2a (b) and rabbit IgG (d), 40X objective. Scale bars = 12 µm
Bacterial- and viral-mimics and pro-inflammatory cytokine responses
Bacterial- and viral-mimics evoked a pro-inflammatory response in human BECs (Supplementary Figure 2). LPS induced a dramatic increase in IL-6 mRNA expression, at 4 h and 24 h (p < .001 and p < .05). PolyI:C increased IL-6 mRNA 24 h (p < .05) after exposure. ssRNA induced a near significant increase in IL-6 mRNA at 4 h (p = .061) with a similar trend at 24 h. The responsiveness of TLR to their respective ligand, as well as potential cross-regulation, was investigated in human BEC 4 h after ligand stimulation (Supplementary Figure 2). LPS increased TLR-4 (p < .01), with concomitant up-regulation of TLR-3 (p < .001) mRNA. PolyI:C increased TLR-3 mRNA (p < .05), with no change in TLR-4 or TLR-8 mRNA. ssRNA exposure had no effect of TLRs mRNA levels.
The responses of cytoplasmic recognition receptors retinoid-acid-inducible gene I (RIG-I) and melanoma differentiation associated gene 5 (MDA5) to bacterial and viral mimics are presented in Supplementary Figure 3. RIG-I and MDA5 recognize ssRNA and PolyI:C in the cytoplasm, 18 activating an antiviral response mediated by nuclear factor-kB (NF-kB) and IFN regulatory factors (IRFs).19 LPS increased RIG-I (p < .001) and MDA5 (p < .05 and p < .001) mRNA levels 4 h and 24 h after exposure. PolyI: C increased MDA5 (p < .05) mRNA expression 24 h post-treatment, while ssRNA had no effect on RIG-I or MDA5 mRNA.
P-gp and BCRP function
As expected, exposure of hCMEC/D3 cells to Verapamil and Ko143 significantly increased Ca-AM and Ce6 accumulation, respectively. This confirms correct functionof the activity assays (Supplementary Figure 1). LPS (105 pg/mL) treatment increased (p < .01) P-gp activity at 24 h, whereas BCRP activity was decreased in response to LPS (106 pg/mL, p < .05) at 24 h Figure 2. PolyI:C treatment (106 pg/mL) increased (p < .05) P-gp activity at 24 h, while PolyI:C (104 and 105 pg/mL) decreased (p < .05, p < .01) BCRP activity 24 h after exposure. ssRNA treatment led to a marked decrease in P-gp function 4 h (104 pg/mL, p < .01; 105 pg/mL, p < .05) and 24 h (104 pg/mL, p < .05; 105 and 106 pg/mL, p < .01) after treatment. In contrast, ssRNA exposure resulted in a pronounced increase in BCRP function at 24 h (104 pg/mL, p < .01; 105 pg/mL, p < .05; Figure 2).
Figure 2.
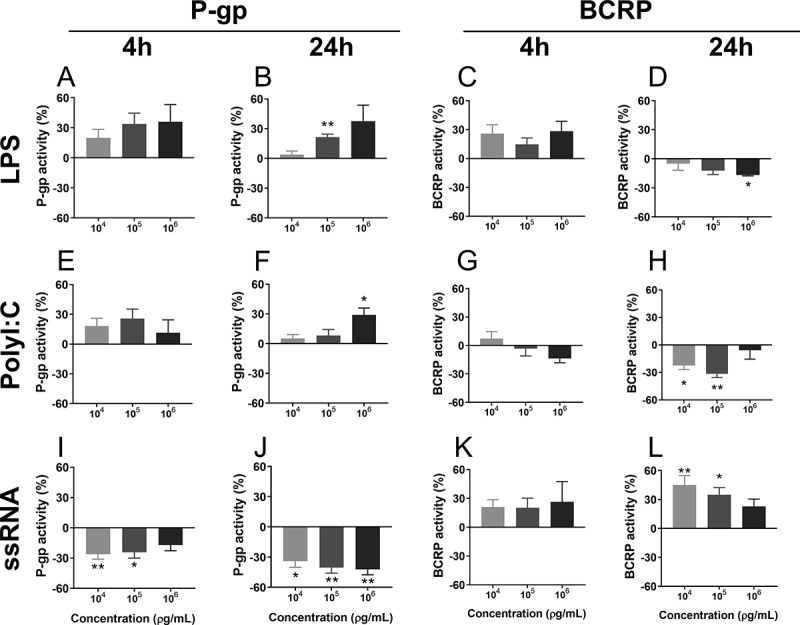
P-gp and BCRP function 4 h or 24 h after treatment with LPS (a-d), PolyI:C (e-h) or ssRNA (i-l), measured using the Ca-AM and Ce6 assays. Doses: 103–106 pg/mL. N = 6/group. Values are expressed as a percentage of control (vehicle) groups. Data are presented as mean ± SEM. Statistical analysis: Kruskal-Wallis test, followed by Dunn’s multiple comparison test. *p < .05, and **p < .01
Expression of P-gp/ABCB1 and BCRP/ABCG2
The effects of LPS on ABCB1 and ABCG2 mRNA levels and P-gp and BCRP total protein are illustrated in Table 2 and Figure 3, respectively. LPS decreased both ABCB1 and ABCG2 mRNA levels at 24 h (p < .05; Table 2), and reduced P-gp protein expression at 4 h (p < .05), however BCRP protein levels were increased by LPS 4 h post-treatment (p < .05). PolyI:C had no effect on ABCB1 or ABCG2 mRNA or P-gp protein at either time-point, but increased BCRP expression at 4 h of treatment (p < .05). ssRNA exposure up-regulated ABCB1 mRNA at 24 h, and ABCG2 at 4 h post-treatment, respectively (p < .01). While there was no effect of ssRNA on total P-gp, total BCRP protein levels were increased at 4 h (p < .05).
Table 2.
ABCB1 and ABCG2 mRNA levels following LPS, PolyI:C or ssRNA (106 pg/mL) treatment (4- or 24 h), measured using qPCR
| |
ABCB1 |
ABCG2 |
||
|---|---|---|---|---|
| 4 h | 24 h | 4 h | 24 h | |
| Ctrl | 1.00 ± 0.05 | 1.00 ± 0.05 | 1.00 ± 0.22 | 1.00 ± 0.12 |
| LPS | 0.88 ± 0.04 | 0.77 ± 0.02* | 1.03 ± 0.04 | 0.55 ± 0.16* |
| Ctrl | 1.00 ± 0.21 | 1.00 ± 0.11 | 1.00 ± 0.17 | 1.00 ± 0.12 |
| PolyI:C | 1.01 ± 0.12 | 1.10 ± 0.27 | 1.16 ± 0.10 | 0.91 ± 0.21 |
| Ctrl | 1.00 ± 0.12 | 1.00 ± 0.24 | 1.00 ± 0.02 | 1.00 ± 0.11 |
| ssRNA | 1.51 ± 0.38 | 1.96 ± 0.13** | 1.45 ± 0.19* | 0.98 ± 0.08 |
Statistical analysis: unpaired Student’s t-test. N = 4–6/group. Mean ± SEM. *p < 0.05 and **p < 0.01 versus respective controls.
Figure 3.
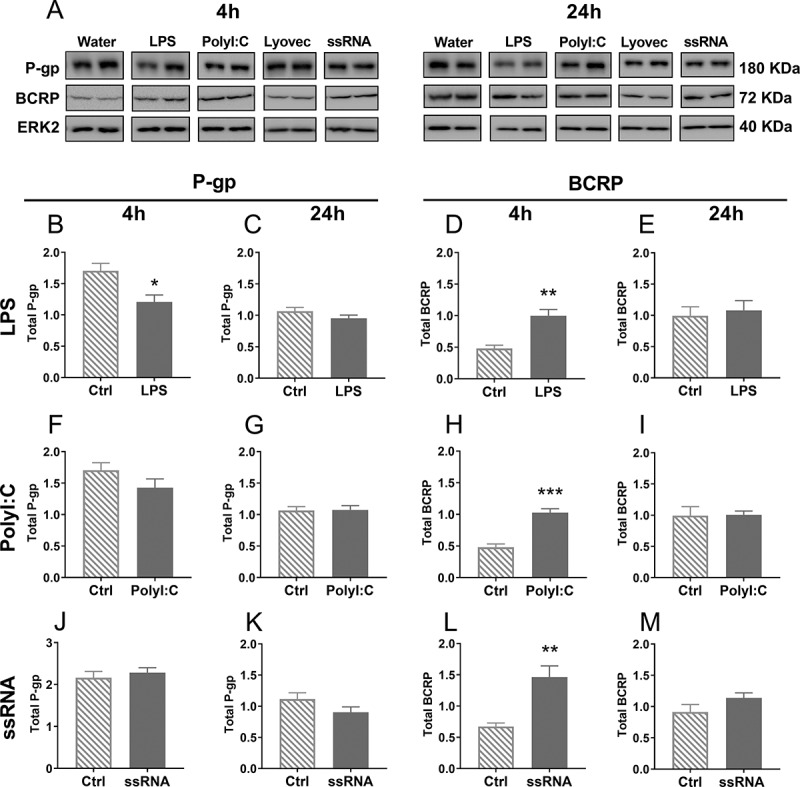
Total P-gp and BCRP protein expression in BECs, 4 h or 24 h after treatment with LPS (b-e), PolyI:C (f-i) or ssRNA (j-m) and their respective controls, measured using Western blot. Representative images of P-gp, BCRP and ERK2 (loading control) bands were cropped and are shown in A. N = 4–5/group. Dose: 106 pg/mL. Statistical analysis: unpaired Student’s t-test. Data are presented as mean ± SEM. *p < .05, **p < .01 and ***p < .001
The inconsistency between total cellular P-gp and BCRP protein levels and P-gp and BCRP function following exposure to bacterial and viral mimics, led us to undertake analysis of P-gp and BCRP levels in the plasma membrane of human BEC cells following treatment Figure 4. LPS had no effect on plasma membrane P-gp at 4 or 24 h; however, it reduced plasma membrane BCRP at 24 h post-treatment (p < .05). PolyI:C treatment increased P-gp levels in the plasma membrane at 4 h of exposure (p < .001), with no changes after 24 h. PolyI:C did not affect BCRP levels in the plasma membrane. ssRNA had no effect on P-gp in the plasma membrane but increased BCRP 4 h after treatment (p < .05).
Figure 4.
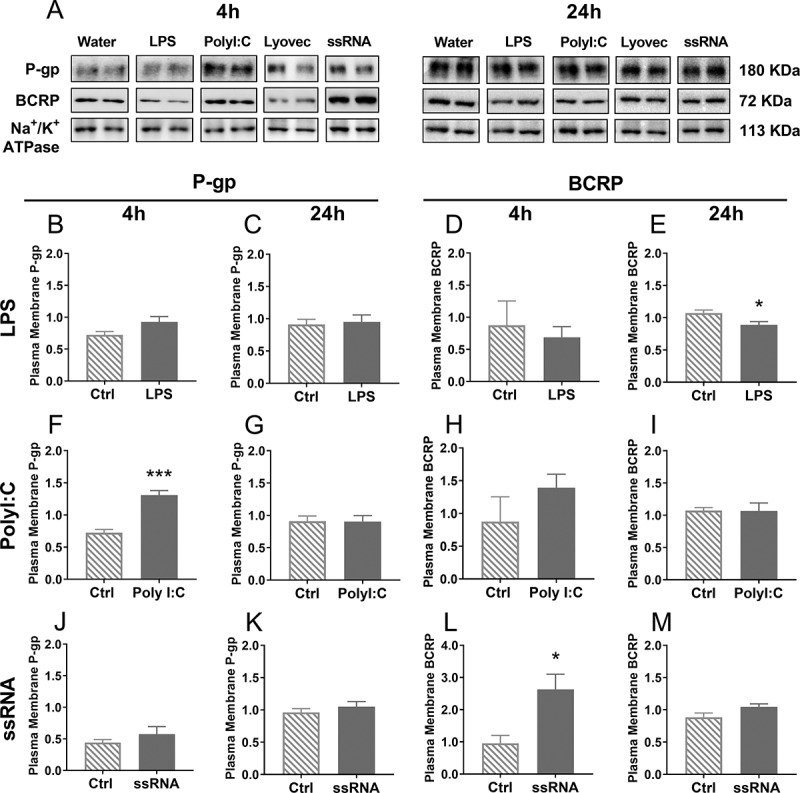
P-gp and BCRP protein expression in the plasma membrane 4 h or 24 h after treatment with LPS (b-e), PolyI:C (f-i) or ssRNA (j-m) and their respective controls, measured using Western blot. Representative images of P-gp, BCRP and Na+/K+-ATPase (loading control) bands were cropped and are shown in A. N = 4–5/group. Dose: 106 pg/mL. Statistical analysis: unpaired Student’s t-test. Data are presented as mean ± SEM. *p < .05 and ***p < .001
Cytokine regulation of P-gp/ABCB1 and BCRP/ABCG2 mRNA expression and functionP-gp and BCRP function and ABCB1 and ABCG2 mRNA levels were assessed in BECs following 4 h and 24 h exposure to key infection-associated pro-inflammatory cytokines, IL-6, TNF-α and IFN-ɣ Figure 5 and Table 3. Exposure to IL-6 inhibited P-gp activity after 24 h at all concentrations investigated (101−4 pg/mL; p < .05), though no effects were observed at 4 h. Similarly, IL-6 (103 pg/mL) down-regulated ABCB1 mRNA levels (p < .001) 24 h after treatment. IL-6 also decreased BCRP function (103 and 104 pg/mL; p < .01) and ABCG2 mRNA levels (103 pg/mL, p < .05) 24 h after treatment Figure 5d and Table 3. There were no effects of IL-6 on BCRP function or ABCG2 mRNA after 4 h of treatment. There were also no effects of TNF-α treatment on P-gp activity or ABCB1 mRNA levels. However, TNF-α treatment decreased BCRP activity (103 and 104 pg/mL; p < .01) 4 h after treatment, though there were no effects on ABCG2 mRNA levels. IFN-ɣ increased P-gp function (103 and 104 pg/mL; p < .01), but down-regulated ABCB1 mRNA levels (p < .05) 24 h after treatment Figure 5j and Table 2. In contrast, IFN-ɣ decreased BCRP function both 4 h (103 pg/mL; p < .05) and 24 h (103 and 104 pg/mL; p < .01) after treatment Figure 5(k, L), though there were no effects on ABCG2 mRNA levels.
Figure 5.
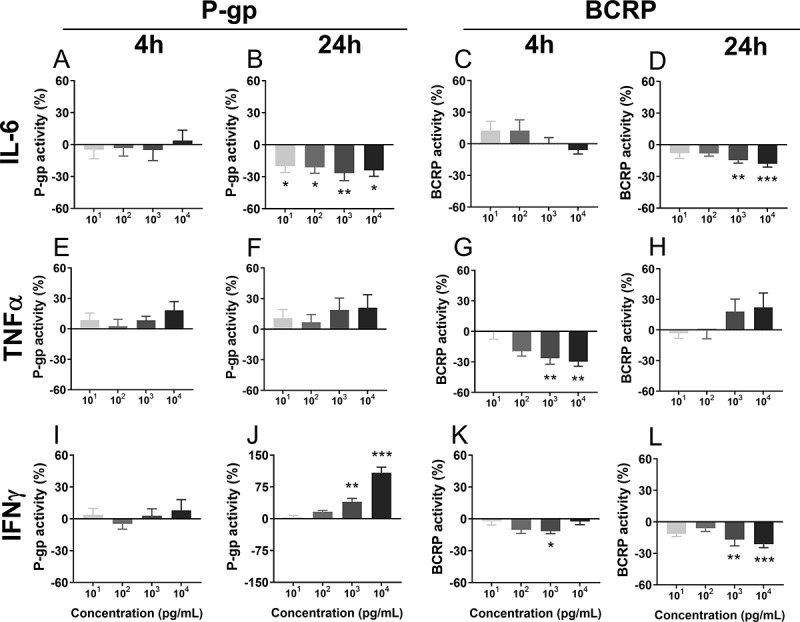
P-gp and BCRP function 4 h or 24 h after treatment with the pro-inflammatory factors IL-6 (a-d), TNF-α (e-h) or IFN-ɣ (i-l), measured using the Ca-AM and Ce6 assays. Dose: 101–104 pg/mL. N = 6–8/group. Values are expressed as a percentage of controls (vehicle). Data are presented as mean ± SEM. Statistical analysis: Kruskal-Wallis test, followed by Dunn’s multiple comparison test. *p < .05, **p < .01 and ***p < .001
Table 3.
ABCB1 and ABCG2 mRNA levels following IL-6, TNF-α or IFN-ɣ (103 pg/mL) treatment (4- or 24 h), measured using qPCR
| |
ABCB1 |
ABCG2 |
||
|---|---|---|---|---|
| 4 h | 24 h | 4 h | 24 h | |
| Ctrl | 1.00 ± 0.13 | 1.00 ± 0.06 | 1.00 ± 0.44 | 1.00 ± 0.04 |
| IL-6 | 1.17 ± 0.06 | 0.63 ± 0.05*** | 1.40 ± 0.55 | 0.77 ± 0.04* |
| Ctrl | 1.00 ± 0.13 | 1.00 ± 0.06 | 1.00 ± 0.22 | 1.00 ± 0.04 |
| TNF-α | 1.29 ± 0.12 | 0.85 ± 0.06 | 1.26 ± 0.09 | 1.09 ± 0.05 |
| Ctrl | 1.00 ± 0.13 | 1.00 ± 0.06 | 1.00 ± 0.22 | 1.00 ± 0.04 |
| IFN-ɣ | 0.95 ± 0.09 | 0.54 ± 0.08* | 1.00 ± 0.07 | 1.05 ± 0.15 |
Statistical analysis: unpaired Student’s t-test. N = 4–5/group. Mean ± SEM. *p < 0.05 and ***p < 0.001 versus respective controls.
Discussion
The current study has demonstrated, for the first time, that bacterial and viral infection-mimics can impact drug transporter activity in human brain endothelial cells in vitro. hCMEC/D3 cells exposed to TLR ligands and pro-inflammatory cytokines exhibit altered P-gp and BCRP activity and expression. Interestingly, we also showed that these transporters were regulated in opposite directions by all infection mimics, which may indicate a compensatory regulation between P-gp and BCRP function in human BECs. These findings may have considerable implications for biodistribution of drugs in the CNS during bacterial and viral infection or inflammation.
LPS, a TLR-4 agonist, caused transporter-specific time-dependant changes in function. LPS induced a long-term (24 h) increase of P-gp activity, which, in vivo, would likely be associated with reduced concentrations of P-gp substrates in the brain, as shown previously using brain capillaries.20,21 Conversely, LPS exposure (24 h) decreased BCRP activity. Considering that altered drug transporter function can profoundly impact the accumulation and efficacy of CNS-targeting drugs, findings in the present study may be extremely important in the context of CNS infection-related therapeutics.
Chikungunya (CHIKV), Dengue virus (DENV), Human Immunodeficiency virus (HIV), Japanese Encephalitis (JEV), SARS-CoV-2, Zika virus (ZIKV) and West Nile virus (WNV) all belong to the class of ssRNA viruses.22,23 In the present study, we have shown that the viral-mimic ssRNA inhibits P-gp function both in the short (4 h) and longer-term (24 h). This may indicate potential implications in drug transfer across the BBB in vivo, in infections induced by ssRNA viruses. To our surprise, BCRP function was increased by ssRNA exposure at 24 h. Further studies are clearly required to determine the impact of ssRNA exposure on drug and xenobiotics accumulation in animal and human brain.
Cytomegalovirus (CMV), DENV and ZIKV activate the host’ immune system through TLR-3 stimulation, 24–26 therefore we used the TLR-3 agonist PolyI:C as an alternative viral mimic. We demonstrated that drug transporter function is regulated in a TLR type-specific manner, since ssRNA and PolyI:C induced opposite effects on P-gp and BCRP function. Human endometrial endothelial cells also present distinct responses to TLR-3 and TLR-7 agonists, based on the distinct inflammatory and antiviral immune pathways that they activate.27 Nevertheless, infections mimicked by PolyI:C may profoundly influence the accumulation of potentially toxic compounds by reducing BCRP activity. Taken together, our results provide evidence that there may be viral-specific effects on human brain protection through modification of P-gp and BCRP function in the brain vascular endothelium.
All infection-mimics had opposite effects on P-gp and BCRP function. LPS and PolyI:C stimulated P-gp function, but inhibited BCRP activity. In contrast, ssRNA decreased P-gp function whilst increasing BCRP activity. P-gp and BCRP can modulate the transfer of cytokines and other inflammatory mediators from the brain parenchyma to the blood, suggesting that infection itself might alter the accumulation of these compounds in the brain. While P-gp and BCRP share common inhibitors and substrates, their functional relationship is not well understood. In this regard, Abcb1a−/- mutant mice exhibit threefold higher levels of Abcg2 mRNA in brain capillaries compared to wild type animals, 28 suggesting a BCRP compensation for the loss of P-gp. Results from the present study, provide the first evidence of a potential compensatory relationship between P-gp and BCRP at the level of transporter function in human BECs. Further studies are required to better understand this important functional relationship at the level of brain protection.
Another interesting finding of this study is that modification of transporter function in BECs by bacterial and viral mimics seems to occur independent of total protein or mRNA abundance. Studies have reported several post-translational mechanisms regulating P-gp and BCRP activity in BECs, with no changes in gene or total protein expression. They include the insertion/removal of P-gp into/from the cell surface by Caveolin-1, 29,30 ERM (ezrin, radixin, and moesin) proteins,31 or small GTPases.32 In addition, phosphorylation33,34 or glycosylation35 can also promote changes in drug transporter function, independent of changes in absolute transporter levels.
There were no observable changes in plasma membrane P-gp associated with altered P-gp function in human BECs after infection. In this case, other regulatory mechanisms such as changes in the lipid microenvironment or protein-membrane composition36,37 may be contributing to the observed changes in P-gp function. Furthermore, indirect regulation of drug transporter activity mediated by the actin filament-associated protein (AFAP)-1 has been recently shown to inhibit P-gp function without changing total cellular or plasma membrane levels of P-gp protein in BECs.38 Further studies are required to elucidate which signaling pathways are involved in the regulation of P-gp activity following infection.
In the case of BCRP, LPS (bacterial infection mimic) decreased transporter activity and plasma membrane BCRP protein at 24 h while total cellular BCRP levels remained unchanged, suggesting a possible change in BCRP intracellular translocation induced by LPS. Similar regulation of BCRP has been previously reported in human embryonic stem cells39 and vascular endothelial cells.40
Pro-inflammatory cytokines, including IL-6, TNF-α, and IFN-ɣ, represents a critical component of the response to bacterial and viral infection.41 We found in the present study that exposure to IL-6 decreased P-gp function and ABCB1 mRNA levels in BECs, consistent with previous findings in BECs derived from guinea pigs at various stages of development.6 IL-6 (24 h) also down-regulated BCRP function and ABCG2 mRNA levels in human BECs in vitro, which was consistent with a previous study that utilized 72 h exposure in the same cell line (72 h).42 TNF-α is a critical mediator of viral neurotrophic infections43 and in the current study, there were no effects of TNF-α on P-gp function or ABCB1 mRNA levels. However, BCRP activity was acutely (4 h) reduced after TNF-α treatment, consistent with previous reports in human, porcine, and guinea pig BECs.42,44
IFN-ɣ plays a central role in viral replication and pathogenesis such as the ZIKV, 45 and disrupts the BBB, inducing the migration of inflammatory cells into the brain.46 Therefore, we hypothesized IFN-ɣ would impact BBB function by downregulating drug transporter activity in human BECs. Indeed, IFN-ɣ exposure resulted in a significant reduction in BCRP function at 4 h and 24 h. In contrast, we found increased P-gp activity 24 h after exposure to IFN-ɣ. IFN-ɣ mediates BBB disruption following JEV infection in mice.47 Thus, we can speculate that increased P-gp activity after exposure to this cytokine represents an adaptive mechanism in response to increased BBB permeability. Further studies are needed to address this possibility in human BECs. Notwithstanding, this study has identified the potential of IFN-ɣ to modulate both P-gp and BCRP at the human BBB, in vitro.
Although the current study focused on drug transporter activity and expression, infection/inflammation also changes other aspects of the BBB structure. Indeed, tight junctions (TJ), represent important modulators of BBB integrity following exposure to infective/inflammatory stimuli. Studies have demonstrated that the presence of Claudin-5, and its ratio to other claudins, are crucial to normal BBB function.48–50 BBB breakdown induced by downregulation/degradation of TJ is a common feature of virus-induced neuropathogenesis. In vivo and in vitro studies have reported increased BBB permeability with concomitant downregulation of TJ proteins following different models of viral encephalitis, as well as exposure to infective agents, infection mimics, and pro-inflammatory mediators.51–61 Clearly, further studies are required to investigate the relationship of TJ and drug transporter function during infection.
Although the calcein-AM activity assay is a commonly used method for in vitro assessment of P-gp function and that calcein has been used as P-gp substrate for several years by different research groups, 7,62–69 some studies report that calcein could also be substrate for other ABC transporters, including MRP1 and MRP2.70–72 There is no evidence in the literature of ABCC1/MRP1 or ABCC2/MRP2 responsiveness (altered expression or function) following infection in human BBB. However, considering hCMEC/D3 plasma membrane expression levels of MRP1 (1.65 ± 0.23 fmol/µg protein) compared to P-gp (3.87 ± 0.39 fmol/µg protein), 69 it is possible that the response we found may be driven by a combination of P-gp and MRP1 efflux. We recognize this is a potential limitation and highlight the importance of future studies to dissect the specific roles of P-gp and MRP1 in brain protection during infection.
In conclusion, we have shown, for the first time, that bacterial and viral mimics influence the expression and function of major drug transporter systems in hCMEC/D3 cells. The current findings are important in the context of bacterial/viral CNS insult, that include Streptococcus pneumoniae, Haemophilus influenzae, Neisseria meningitidis, arboviruses, coronavirus, enterovirus, herpesvirus, poliovirus and rotavirus .26,73–75 These pathogens can induce the onset of meningitis/encephalitis, leading to severe cerebral injury and long-lasting neurological dysfunction. We provide in vitro evidence that stimulation of TLRs is associated with disrupted P-gp and BCRP function in human BECs. Should a similar effect occur in vivo brain protection against xenobiotics would be compromised and there altered biodistribution of drugs in the CNS could result. Further studies with co-culture models would be extremely relevant to highlight the possible influence of astrocytes and microglia on brain endothelial drug transporter response to TLRs ligands. Also, given the transporter-specific sensitivity of P-gp and BCRP in BECs to different infection-mimics, polymicrobial infections may result in diverse patterns of P-gp and BCRP dysfunction in the brain endothelium, and this concept should be further investigated. Our results highlight the need to assess the effects of infection-mimics, and of important human pathogens on the exposure of xenobiotics and accumulation of therapeutic drugs at multiple times across the life course, in vivo.
Supplementary Material
Acknowledgments
We are grateful to Alisa Kostaki for her technical support in this research.
Funding Statement
EB is supported by Coordenação de Aperfeiçoamento Pessoal de Nível Superior (CAPES, finance code 001, Capes-Print fellowship); SGM is funded by the Canadian Institutes of Health Research (SGM; FDN-148368).
Disclosure of interest
The authors report no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Bloise E, Petropoulos S, Iqbal M, Kostaki A, Ortiga-Carvalho TM, Gibb W, Matthews SG.. Acute effects of viral exposure on p-glycoprotein function in the mouse fetal blood-brain barrier. Cell Physiol Biochem. 2017;41:1–14. doi: 10.1159/000461569. [DOI] [PubMed] [Google Scholar]
- 2.Bloise E, Ortiga-Carvalho TM, Reis FM, Lye SJ, Gibb W, Matthews SG. ATP-binding cassette transporters in reproduction: a new frontier. Hum Reprod Update. 2015;dmv049. doi: 10.1093/humupd/dmv049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tohyama C. Developmental neurotoxicity test guidelines: problems and perspectives. J Toxicol Sci. 2016;41:SP69–SP79. doi: 10.2131/jts.41.SP69. [DOI] [PubMed] [Google Scholar]
- 4.Iqbal M, Baello S, Javam M, Audette MC, Gibb W, Matthews SG. Regulation of multidrug resistance p-glycoprotein in the developing blood-brain barrier: interplay between glucocorticoids and cytokines. J Neuroendocrinol. 2016;28:12360. doi: 10.1111/jne.12360. [DOI] [PubMed] [Google Scholar]
- 5.Baello S, Iqbal M, Bloise E, Javam M, Gibb W, Matthews SG. TGF-β1 regulation of multidrug resistance P-glycoprotein in the developing male blood-brain barrier. Endocrinology. 2014;155:475–484. doi: 10.1210/en.2013-1472. [DOI] [PubMed] [Google Scholar]
- 6.Iqbal M, Ho HL, Petropoulos S, Moisiadis VG, Gibb W, Matthews SG, Frasch M. Pro-inflammatory cytokine regulation of P-glycoprotein in the developing blood-brain barrier. PLoS One. 2012;7:e43022. doi: 10.1371/journal.pone.0043022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baello S, Iqbal M, Kearney S, Kuthiala S, Bloise E, Gibb W, Matthews SG. Glucocorticoids modify effects of TGF-β1 on multidrug resistance in the fetal blood-brain barrier. Growth Factors. 2016;34:33–41. doi: 10.3109/08977194.2016.1162163. [DOI] [PubMed] [Google Scholar]
- 8.Ohtsuki S, Ikeda C, Uchida Y, Sakamoto Y, Miller F, Glacial F, Decleves X, Scherrmann JM, Couraud PO, Kubo Y, et al. Quantitative targeted absolute proteomic analysis of transporters, receptors and junction proteins for validation of human cerebral microvascular endothelial cell line hCMEC/D3 as a human blood-brain barrier model. Mol Pharm. 2013;10:289–296. doi: 10.1021/mp3004308. [DOI] [PubMed] [Google Scholar]
- 9.Dubey SK, Ram MS, Krishna KV, Saha RN, Singhvi G, Agrawal M, Ajazuddin SS, Alexander A. Recent expansions on cellular models to uncover the scientific barriers towards drug development for Alzheimer’s Disease. Cell Mol Neurobiol. 2019;39:181–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maherally Z, Fillmore HL, Tan SL, Tan SF, Jassam SA, Quack FI, Hatherell KE, Pilkington GJ. Real-time acquisition of transendothelial electrical resistance in an all-human, in vitro, 3-dimensional, blood-brain barrier model exemplifies tight-junction integrity. Faseb J. 2018;32:168–182. doi: 10.1096/fj.201700162R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weksler B, Romero IA, Couraud PO. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS. 2013;10:16. doi: 10.1186/2045-8118-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lye P, Bloise E, Dunk C, Javam M, Gibb W, Lye SJ, Matthews SG. Effect of oxygen on multidrug resistance in the first trimester human placenta. Placenta. 2013;34:817–823. doi: 10.1016/j.placenta.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 13.Audette MC, Greenwood SL, Sibley CP, Jones CJ, Challis JR, Matthews SG, Jones RL. Dexamethasone stimulates placental system A transport and trophoblast differentiation in term villous explants. Placenta. 2010;31:97–105. doi: 10.1016/j.placenta.2009.11.016. [DOI] [PubMed] [Google Scholar]
- 14.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 15.Baello S, Iqbal M, Gibb W, Matthews SG. Astrocyte-mediated regulation of multidrug resistance p-glycoprotein in fetal and neonatal brain endothelial cells: age-dependent effects. Physiol Rep. 2016;4(16):e12853. doi: 10.14814/phy2.12853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paturi DK, Kwatra D, Ananthula HK, Pal D, Mitra AK. Identification and functional characterization of breast cancer resistance protein in human bronchial epithelial cells (Calu-3). Int J Pharm. 2010;384:32–38. doi: 10.1016/j.ijpharm.2009.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bakhsheshian J, Hall MD, Robey RW, Herrmann MA, Chen JQ, Bates SE, Gottesman MM. Overlapping substrate and inhibitor specificity of human and murine ABCG2. Drug Metab Dispos. 2013;41:1805–1812. doi: 10.1124/dmd.113.053140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 19.Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol. 2006;6:644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- 20.Cen J, Liu L, Li MS, He L, Wang LJ, Liu YQ, Liu M, Ji BS. Alteration in P-glycoprotein at the blood-brain barrier in the early period of MCAO in rats. J Pharm Pharmacol. 2013;65:665–672. doi: 10.1111/jphp.12033. [DOI] [PubMed] [Google Scholar]
- 21.Spudich A, Kilic E, Xing H, Kilic U, Rentsch KM, Wunderli-Allenspach H, Bassetti CL, Hermann DM. Inhibition of multidrug resistance transporter-1 facilitates neuroprotective therapies after focal cerebral ischemia. Nat Neurosci. 2006;9:487–488. doi: 10.1038/nn1676. [DOI] [PubMed] [Google Scholar]
- 22.Nazmi A, Mukherjee S, Kundu K, Dutta K, Mahadevan A, Shankar SK, Basu A. TLR7 is a key regulator of innate immunity against Japanese encephalitis virus infection. Neurobiol Dis. 2014;69:235–247. doi: 10.1016/j.nbd.2014.05.036. [DOI] [PubMed] [Google Scholar]
- 23.Yang P, Wang X. COVID-19: a new challenge for human beings. Cell Mol Immunol. 2020;17:555–557. doi: 10.1038/s41423-020-0407-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dang J, Tiwari SK, Lichinchi G, Qin Y, Patil VS, Eroshkin AM, Rana TM. Zika virus depletes neural progenitors in human cerebral organoids through activation of the innate immune receptor TLR3. Cell Stem Cell. 2016;19:258–265. doi: 10.1016/j.stem.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsai YT, Chang SY, Lee CN, Kao CL. Human TLR3 recognizes dengue virus and modulates viral replication in vitro. Cell Microbiol. 2009;11:604–615. [DOI] [PubMed] [Google Scholar]
- 26.Yew KH, Harrison CJ. Blockade of Lyn kinase upregulates both canonical and non-canonical TLR-3 pathways in THP-1 monocytes exposed to human cytomegalovirus. Acta Virol. 2011;55:243–253. doi: 10.4149/av_2011_03_243. [DOI] [PubMed] [Google Scholar]
- 27.Krikun G, Potter JA, Abrahams VM. Human endometrial endothelial cells generate distinct inflammatory and antiviral responses to the TLR3 agonist, Poly(I:C) and the TLR8 agonist, viral ssRNA. Am J Reprod Immunol. 2013;70:190–198. doi: 10.1111/aji.12128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cisternino S, Mercier C, Bourasset F, Roux F, Scherrmann JM. Expression, up-regulation, and transport activity of the multidrug-resistance protein Abcg2 at the mouse blood-brain barrier. Cancer Res. 2004;64:3296–3301. doi: 10.1158/0008-5472.CAN-03-2033. [DOI] [PubMed] [Google Scholar]
- 29.Barakat S, Demeule M, Pilorget A, Regina A, Gingras D, Baggetto LG, Beliveau R. Modulation of p-glycoprotein function by caveolin-1 phosphorylation. J Neurochem. 2007;101:1–8. doi: 10.1111/j.1471-4159.2006.04410.x. [DOI] [PubMed] [Google Scholar]
- 30.Hawkins BT, Sykes DB, Miller DS. Rapid, reversible modulation of blood-brain barrier P-glycoprotein transport activity by vascular endothelial growth factor. J Neurosci. 2010;30:1417–1425. doi: 10.1523/JNEUROSCI.5103-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pokharel D, Roseblade A, Oenarto V, Lu JF, Bebawy M. Proteins regulating the intercellular transfer and function of P-glycoprotein in multidrug-resistant cancer. Ecancermedicalscience. 2017;11:768. doi: 10.3332/ecancer.2017.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu D, van Dam EM, Brymora A, Duggin IG, Robinson PJ, Roufogalis BD. The small GTPases Rab5 and RalA regulate intracellular traffic of P-glycoprotein. Biochim Biophys Acta. 2007;1773:1062–1072. doi: 10.1016/j.bbamcr.2007.03.023. [DOI] [PubMed] [Google Scholar]
- 33.Xie Y, Burcu M, Linn DE, Qiu Y, Baer MR. Pim-1 kinase protects P-glycoprotein from degradation and enables its glycosylation and cell surface expression. Mol Pharmacol. 2010;78:310–318. doi: 10.1124/mol.109.061713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xie Y, Xu K, Linn DE, Yang X, Guo Z, Shimelis H, Nakanishi T, Ross DD, Chen H, Fazli L, et al. The 44-kDa Pim-1 kinase phosphorylates BCRP/ABCG2 and thereby promotes its multimerization and drug-resistant activity in human prostate cancer cells. J Biol Chem. 2008;283:3349–3356. doi: 10.1074/jbc.M707773200. [DOI] [PubMed] [Google Scholar]
- 35.Perego P, Gatti L, Beretta GL. The ABC of glycosylation. Nat Rev Cancer. 2010;10:523. doi: 10.1038/nrc2789-c1. [DOI] [PubMed] [Google Scholar]
- 36.Hawkins BT, Rigor RR, Miller DS. Rapid loss of blood-brain barrier P-glycoprotein activity through transporter internalization demonstrated using a novel in situ proteolysis protection assay. J Cereb Blood Flow Metab. 2010;30:1593–1597. doi: 10.1038/jcbfm.2010.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Storch CH, Ehehalt R, Haefeli WE, Weiss J. Localization of the human breast cancer resistance protein (BCRP/ABCG2) in lipid rafts/caveolae and modulation of its activity by cholesterol in vitro. J Pharmacol Exp Ther. 2007;323:257–264. doi: 10.1124/jpet.107.122994. [DOI] [PubMed] [Google Scholar]
- 38.Hoshi Y, Uchida Y, Tachikawa M, Ohtsuki S, Terasaki T. Actin filament-associated protein 1 (AFAP-1) is a key mediator in inflammatory signaling-induced rapid attenuation of intrinsic P-gp function in human brain capillary endothelial cells. J Neurochem. 2017;141:247–262. doi: 10.1111/jnc.13960. [DOI] [PubMed] [Google Scholar]
- 39.Erdei Z, Sarkadi B, Brozik A, Szebenyi K, Varady G, Mako V, Pentek A, Orban TI, Apati A. Dynamic ABCG2 expression in human embryonic stem cells provides the basis for stress response. Eur Biophys J. 2013;42:169–179. doi: 10.1007/s00249-012-0838-0. [DOI] [PubMed] [Google Scholar]
- 40.Komori H, Yamada K, Tamai I. Hyperuricemia enhances intracellular urate accumulation via down-regulation of cell-surface BCRP/ABCG2 expression in vascular endothelial cells. Biochim Biophys Acta Biomembr. 2018;1860:973–980. doi: 10.1016/j.bbamem.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 41.Tohidpour A, Morgun AV, Boitsova EB, Malinovskaya NA, Martynova GP, Khilazheva ED, Kopylevich NV, Gertsog GE, Salmina AB. Neuroinflammation and infection: molecular mechanisms associated with dysfunction of neurovascular unit. Front Cell Infect Microbiol. 2017;7:276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Poller B, Drewe J, Krahenbuhl S, Huwyler J, Gutmann H. Regulation of BCRP (ABCG2) and P-glycoprotein (ABCB1) by cytokines in a model of the human blood-brain barrier. Cell Mol Neurobiol. 2010;30:63–70. doi: 10.1007/s10571-009-9431-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nem de Oliveira Souza I, Frost PS, França JV, Nascimento-Viana JB, Neris RLS, Freitas L, Pinheiro DJLL, Nogueira CO, Neves G, Chimelli L, et al. Acute and chronic neurological consequences of early-life Zika virus infection in mice. Sci Transl Med. 2018;10:eaar2749. doi: 10.1126/scitranslmed.aar2749. [DOI] [PubMed] [Google Scholar]
- 44.von Wedel-parlow M, Wolte P, Galla HJ. Regulation of major efflux transporters under inflammatory conditions at the blood-brain barrier in vitro. J Neurochem. 2009;111:111–118. doi: 10.1111/j.1471-4159.2009.06305.x. [DOI] [PubMed] [Google Scholar]
- 45.Chaudhary V, Yuen KS, Chan JF, Chan CP, Wang PH, Cai JP, Zhang S, Liang M, Kok KH, Yuen KY, et al. Selective activation of type II interferon signaling by Zika Virus NS5 protein. J Virol. 2017;91. doi: 10.1128/JVI.00163-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Minagar A, Long A, Ma T, Jackson TH, Kelley RE, Ostanin DV, Sasaki M, Warren AC, Jawahar A, Cappell B, et al. Interferon (IFN)-beta 1a and IFN-beta 1b block IFN-gamma-induced disintegration of endothelial junction integrity and barrier. Endothelium. 2003;10:299–307. doi: 10.1080/10623320390272299. [DOI] [PubMed] [Google Scholar]
- 47.Li F, Wang Y, Yu L, Cao S, Wang K, Yuan J, Wang C, Wang K, Cui M, Fu ZF. Viral infection of the central nervous system and neuroinflammation precede blood-brain barrier disruption during japanese encephalitis virus infection. J Virol. 2015;89:5602–5614. doi: 10.1128/JVI.00143-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liebner S, Fischmann A, Rascher G, Duffner F, Grote EH, Kalbacher H, Wolburg H. Claudin-1 and claudin-5 expression and tight junction morphology are altered in blood vessels of human glioblastoma multiforme. Acta Neuropathol. 2000;100:323–331. doi: 10.1007/s004010000180. [DOI] [PubMed] [Google Scholar]
- 49.Liebner S, Kniesel U, Kalbacher H, Wolburg H. Correlation of tight junction morphology with the expression of tight junction proteins in blood-brain barrier endothelial cells. Eur J Cell Biol. 2000;79:707–717. doi: 10.1078/0171-9335-00101. [DOI] [PubMed] [Google Scholar]
- 50.Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M, Tsukita S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161:653–660. doi: 10.1083/jcb.200302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10:1366–1373. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- 52.Yang CM, Lin CC, Lee IT, Lin YH, Yang CM, Chen WJ, Jou MJ, Hsiao LD. Japanese encephalitis virus induces matrix metalloproteinase-9 expression via a ROS/c-Src/PDGFR/PI3K/Akt/MAPKs-dependent AP-1 pathway in rat brain astrocytes. J Neuroinflammation. 2012;9:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Daniels BP, Holman DW, Cruz-Orengo L, Jujjavarapu H, Durrant DM, Klein RS, Griffin DE. Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals. mBio. 2014;5:e01476–01414. doi: 10.1128/mBio.01476-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chai Q, He WQ, Zhou M, Lu H, Fu ZF. Enhancement of blood-brain barrier permeability and reduction of tight junction protein expression are modulated by chemokines/cytokines induced by rabies virus infection. J Virol. 2014;88:4698–4710. doi: 10.1128/JVI.03149-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lazear HM, Daniels BP, Pinto AK, Huang AC, Vick SC, Doyle SE, Gale M Jr., Klein RS, Diamond MS. Interferon-lambda restricts West Nile virus neuroinvasion by tightening the blood-brain barrier. Sci Transl Med. 2015;7:284ra259. doi: 10.1126/scitranslmed.aaa4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Andras IE, Toborek M. HIV-1-induced alterations of claudin-5 expression at the blood-brain barrier level. Methods Mol Biol. 2011;762:355–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bhargavan B, Kanmogne GD. Toll-like receptor-3 mediates HIV-1-induced interleukin-6 expression in the human brain endothelium via TAK1 and JNK pathways: implications for viral neuropathogenesis. Mol Neurobiol. 2018;55:5976–5992. doi: 10.1007/s12035-017-0816-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Persidsky Y, Heilman D, Haorah J, Zelivyanskaya M, Persidsky R, Weber GA, Shimokawa H, Kaibuchi K, Ikezu T. Rho-mediated regulation of tight junctions during monocyte migration across the blood-brain barrier in HIV-1 encephalitis (HIVE). Blood. 2006;107:4770–4780. doi: 10.1182/blood-2005-11-4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meng W, Takeichi M. Adherens junction: molecular architecture and regulation. Cold Spring Harb Perspect Biol. 2009;1:a002899. doi: 10.1101/cshperspect.a002899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nagyoszi P, Wilhelm I, Farkas AE, Fazakas C, Dung NT, Hasko J, Krizbai IA. Expression and regulation of toll-like receptors in cerebral endothelial cells. Neurochem Int. 2010;57:556–564. doi: 10.1016/j.neuint.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 61.Camire RB, Beaulac HJ, Willis CL. Transitory loss of glia and the subsequent modulation in inflammatory cytokines/chemokines regulate paracellular claudin-5 expression in endothelial cells. J Neuroimmunol. 2015;284:57–66. doi: 10.1016/j.jneuroim.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hollo Z, Homolya L, Davis CW, Sarkadi B. Calcein accumulation as a fluorometric functional assay of the multidrug transporter. Biochim Biophys Acta. 1994;1191:384–388. doi: 10.1016/0005-2736(94)90190-2. [DOI] [PubMed] [Google Scholar]
- 63.Feng B, Mills JB, Davidson RE, Mireles RJ, Janiszewski JS, Troutman MD, de Morais SM. In vitro P-glycoprotein assays to predict the in vivo interactions of P-glycoprotein with drugs in the central nervous system. Drug Metab Dispos. 2008;36:268–275. doi: 10.1124/dmd.107.017434. [DOI] [PubMed] [Google Scholar]
- 64.Hu Z, Zhou Z, Hu Y, Wu J, Li Y, Huang W, Lebedeva IV. HZ08 reverse P-glycoprotein mediated multidrug resistance in vitro and in vivo. PLoS One. 2015;10:e0116886. doi: 10.1371/journal.pone.0116886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sukhaphirom N, Vardhanabhuti N, Chirdchupunseree H, Pramyothin P, Jianmongkol S. Phyllanthin and hypophyllanthin inhibit function of P-gp but not MRP2 in Caco-2 cells. J Pharm Pharmacol. 2013;65:292–299. doi: 10.1111/j.2042-7158.2012.01593.x. [DOI] [PubMed] [Google Scholar]
- 66.Tsai AC, Pai HC, Wang CY, Liou JP, Teng CM, Wang JC, Pan SL. In vitro and in vivo anti-tumour effects of MPT0B014, a novel derivative aroylquinoline, and in combination with erlotinib in human non-small-cell lung cancer cells. Br J Pharmacol. 2014;171:122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Knops N, van den Heuvel LP, Masereeuw R, Bongaers I, de Loor H, Levtchenko E, Kuypers D. The functional implications of common genetic variation in CYP3A5 and ABCB1 in human proximal tubule cells. Mol Pharm. 2015;12:758–768. doi: 10.1021/mp500590s. [DOI] [PubMed] [Google Scholar]
- 68.Polli JW, Wring SA, Humphreys JE, Huang L, Morgan JB, Webster LO, Serabjit-Singh CS. Rational use of in vitro P-glycoprotein assays in drug discovery. J Pharmacol Exp Ther. 2001;299:620–628. [PubMed] [Google Scholar]
- 69.Bubik M, Ott M, Mahringer A, Fricker G. Rapid assessment of p-glycoprotein-drug interactions at the blood-brain barrier. Anal Biochem. 2006;358:51–58. doi: 10.1016/j.ab.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 70.Hollo Z, Homolya L, Hegedus T, Sarkadi B. Transport properties of the multidrug resistance-associated protein (MRP) in human tumour cells. FEBS Lett. 1996;383:99–104. doi: 10.1016/0014-5793(96)00237-2. [DOI] [PubMed] [Google Scholar]
- 71.Evers R, Kool M, Smith AJ, van Deemter L, de Haas M, Borst P. Inhibitory effect of the reversal agents V-104, GF120918 and Pluronic L61 on MDR1 Pgp-, MRP1- and MRP2-mediated transport. Br J Cancer. 2000;83:366–374. doi: 10.1054/bjoc.2000.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Essodaigui M, Broxterman HJ, Garnier-Suillerot A. Kinetic analysis of calcein and calcein-acetoxymethylester efflux mediated by the multidrug resistance protein and P-glycoprotein. Biochemistry. 1998;37:2243–2250. doi: 10.1021/bi9718043. [DOI] [PubMed] [Google Scholar]
- 73.Bastos MS, Lessa N, Naveca FG, Monte RL, Braga WS, Figueiredo LT, Ramasawmy R, Mourão MP. Detection of herpesvirus, enterovirus, and arbovirus infection in patients with suspected central nervous system viral infection in the Western Brazilian Amazon. J Med Virol. 2014;86:1522–1527. doi: 10.1002/jmv.23953. [DOI] [PubMed] [Google Scholar]
- 74.Simonsen KA, Anderson-Berry AL, Delair SF, Davies HD. Early-onset neonatal sepsis. Clin Microbiol Rev. 2014;27:21–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Doran KS, Fulde M, Gratz N, Kim BJ, Nau R, Prasadarao N, Schubert-Unkmeir A, Tuomanen EI, Valentin-Weigand P. Host-pathogen interactions in bacterial meningitis. Acta Neuropathol. 2016;131:185–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.