

Abstract
Chimeric Antigen Receptor T-cell [CAR T] therapy has changed the treatment landscape of relapsed/refractory lymphoid malignancies. With an expanding pool of post CAR T-cell therapy survivors, prevention and management of late toxicities is emerging as an important component of survivorship care. This review summarizes the current state of evidence on late toxicities after CAR T-cell therapy in lymphoid malignancies. Late effects that are well described in clinical trials and observational studies include hypogammaglobulinemia, prolonged cytopenias, late infections, neurologic and neuropsychiatric effects, immune-related late effects, and subsequent malignancies. Hypogammaglobulinemia is the most common late effect in the setting of CD19-directed CAR T-cell therapy, which necessitates immunoglobulin replacement. Common determinants of late toxicities are age, underlying tumor type, prior therapy, CAR construct, and acute toxicities. Among currently approved indications, the incidence of hypogammaglobulinemia and prolonged cytopenia is higher in patients with acute lymphoblastic leukemia compared to aggressive non-Hodgkin lymphoma. Patient-reported physical and mental quality of life in long-term survivors is comparable to general population, albeit, with limited data thus far. This review provides an overview of the incidence, known risk-factors, and strategies for prevention and management of late toxicities in this population. Further research is needed to characterize the trajectory of late effects from population-based registries and long-term follow-up of ongoing clinical trials.
1.0. Introduction
Chimeric antigen receptor T-cell therapy [CAR T] has revolutionized the treatment of relapsed/refractory hematologic malignancies. The Food and Drug Administration [FDA] has approved CAR T-cell therapies targeting CD19 in relapsed/refractory B-cell acute lymphoblastic leukemia [B-ALL], aggressive B-cell non-Hodgkin lymphoma [NHL], and mantle cell lymphoma, based on impressive response rate and response durability in this difficult-to-treat patient population1–4. Currently, CAR T-cell therapies are being tested in other hematologic malignancies including multiple myeloma [MM], chronic lymphocytic leukemia [CLL], and acute myeloid leukemia as well as in metastatic solid tumors. Long-term follow-up of the pivotal ZUMA-1 trial testing axicabtagene ciloleucel [axi-cel] in relapsed/refractory B-cell NHL showed approximately 40% of patients to be in a durable complete remission [CR] at 2 years, with a plateau in the survival curve2. With a growing pool of CAR T-cell therapy survivors, a subset of whom may potentially be cured of their primary malignancy, research on the incidence, risk-factors, and timeline of late effects will be critical for optimal survivorship care. Although early toxicities of CAR T-cell therapy, including cytokine release syndrome [CRS] and CAR T-related encephalopathy syndrome or Immune effector Cell Associated Neurotoxicity Syndrome [CRES/ICANS], has been well-described in literature5, there is lack of a comprehensive review synthesizing the current evidence on observed and potential late-effects of CAR T-cell therapy.
The aim of this review is to summarize the current evidence on late-effects of CAR T-cell therapy and provide practical insights on surveillance and management. For the purpose of this review, we focus on toxicities that occur beyond 1–3 months after CAR T-cell infusion. Although there is a lack of consensus on the timeline of late toxicities after CAR T-cell therapy, most acute toxicities such as CRS or CRES/ICANS resolve and patients may be managed in the community beyond that time-period. We also acknowledge that late effects should be interpreted in the context of several factors including primary malignancy, prior therapies, antigen target [e.g. CD19], CAR construct, and patient-related factors such as age and frailty. Our review summarizes issues relevant to CD19 and BCMA directed constructs for lymphoid malignancies since current data are primarily available for this patient population. However, the same principles for long-term follow-up and preventive care apply to cellular therapies and disease groups that are presently under investigation and may transition to routine clinical care in the future.
2.0. Hematopoietic Cell Transplantation as Framework for CAR T-cell Late Effects
Although data on late complications and long-term follow-up after CAR T-cell therapy are limited, lessons can be translated from extensive research that has been conducted in the arena of hematopoietic cell transplantation [HCT] survivorship and late effects6,7. Conceptually, there are several similarities between the two procedures, such as pre-treatment exposures, issues around immune reconstitution and infections, and post-treatment recovery. In the setting of HCT, data on late effects have been collected individually by transplant centers as well as by the Center for International Blood and Marrow Transplant Research [CIBMTR] and other international registries. Furthermore, many transplant centers have dedicated survivorship clinics, which has been associated with improved survival outcome8. Systematically collected long-term follow up data has informed the field about the pathogenesis of late toxicities, and the importance of exposures and risk-factors for specific late-effects, their incidence and manifestations, and opportunities to prevent or mitigate them9. Quality of life issues and post-transplant rehabilitation have been well characterized10. Implementation of survivorship care plans led to reduced distress and improved mental quality of life11. Extensive research has also been conducted on a spectrum of care models for delivering care to HCT survivors12.
The HCT framework can serve as a foundation for research on late toxicities in CAR T-cell therapy recipients. The majority of care for these patients is being provided at transplant centers. As such, centers that administer cellular therapy are required to have accreditation by the Foundation for the Accreditation of Cellular Therapy [FACT], whose quality standards for immune effector cell therapy require a mechanism to monitor and care for late effects of cellular therapy. Furthermore, long-term follow up data of commercially available CAR T-cell therapies is being captured by registries such as the CIBMTR, which will enable us to better characterize late effects and develop evidence-based survivorship care guidelines in future.
3.0. Late Effects after CAR T-cell Therapy
Incidence and management of commonly observed late-effects are summarized in Table I. The timeline of common late effects is illustrated in Figure I.
Table I.
Incidence and Management of Late toxicities with CAR T-cell Therapy
| Late Effects | Incidence | Management |
|---|---|---|
| Hematological | ||
| Prolonged Cytopenias | Anemia: 17–53% Thrombocytopenia: 16–41% Neutropenia: 3–53% ALL>NHL1–3,13,25,26 |
|
| Hypogammaglobulinemia | 23–100%1–3,13,15 |
|
| Neurologic | ||
| Neurologic and Psychiatric Events | 4–9%W3,31 |
|
| Immune-related Adverse Events | 8%13 |
|
| Second Cancers | 1–15%2,13 Hematologic Cancers: 1–6% Solid Cancers: 0–9% |
|
| Late Infections | 8–61%1,3,13,31 |
|
| Cardiac Toxicities | Troponin elevation: 54% Decreased LVEF: 28% CV deaths: 12%52 |
|
Abbreviations: ALL: Acute Lymphoblastic Leukemia. NHL: Non-Hodgkin Lymphoma. IVIG: Intravenous Immunoglobulin. CR: Complete Response.
Note: Management recommendations in this table are based on current state of evidence and expert opinion.
Figure 1.
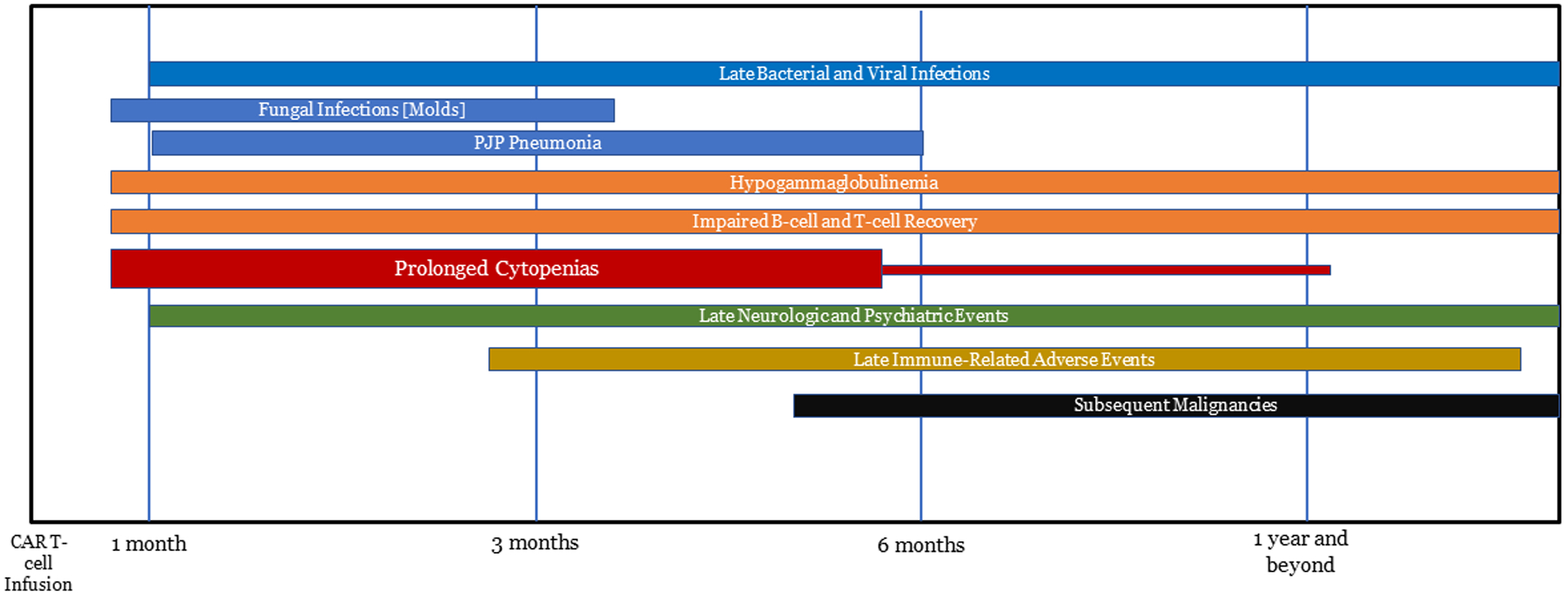
Time-line of Late Toxicities with CAR T-cell Therapy.
3.1. Hypogammaglobulinemia
Hypogammaglobulinemia, defined as immunoglobulin G [IgG] level <400 mg/dl or need for immunoglobulin replacement due to recurrent infections, is the most common late adverse effect of CTL01913. It is an “on-target off-tumor” toxicity in this context. Furthermore, approximately 35% of patients have baseline hypogammaglobulinemia likely secondary to underlying disease and prior therapies14. In a long-term follow-up study of CD-19 directed CAR T in R/R B-cell NHL or CLL, 67% of patients had hypogammaglobulinemia beyond 90 days13. Notably, approximately half of these patients were in ongoing complete remission [CR]. Updated results of ZUMA-1 trial, which tested axi-cel in R/R aggressive B-cell NHL, showed that 31% of patients received intravenous immunoglobulin [IVIG], among which, only 8% had received IVIG prior to their first hospital discharge after axi-cel infusion2. Among the 39 patients with ongoing responses at a median follow-up of 27 months in this study, 44% needed IVIG therapy, indicating prolonged B-cell aplasia in responders2. In the JULIET trial testing tisagenlecleucel in R/R B-cell NHL, 30% of patients received IVIG after cell infusion3. Notably, only 16% of patients with ongoing CR in this study had peripheral CD19+ B-cell count return to normal range. Interestingly, a post-hoc analysis from JULIET study showed that IgG levels were comparable between patients receiving or not receiving IVIG14.
The incidence of hypogammaglobulinemia is higher in patients with B-cell ALL after CAR T-cell therapy. In the ELIANA trial, which tested tisagenlecleucel in children and young adults with R/R B-cell ALL, the probability of maintenance of B-cell aplasia at 6 months after infusion was 83%, with the median time to B-cell recovery not reached at a median follow-up of 13 months1. More than 90% of responding patients required immunoglobulin replacement in this study. In ZUMA-2 trial testing KTE-X19 in relapsed/refractory mantle cell lymphoma, 32% of patients have required immunoglobulin replacement at a median follow-up of 12 months4. Data from initial patients treated with CD-19 directed CAR T for CLL show that those with ongoing responses beyond four years have persistent B-cell aplasia and hypogammaglobulinemia15.
In MM, ablation of malignant and normal plasma cells by BCMA-directed CARs as well as prior plasma cell-directed will likely induce prolonged hypogammaglobulinemia and increased infection risk. In a study on dual CAR against CD19 and BCMA in MM, at a median follow-up of 6 months, IgM had recovered but IgG and IgA had not16 Long-term follow up data from currently ongoing myeloma CAR trials will highlight the kinetics of immunoglobulin reconstitution and need for immunoglobulin replacement in these patients.
Management
With an increasing pool of patients surviving post CD-19 or BCMA-directed CAR T, management of hypogammaglobulinemia leading to secondary immunodeficiency will be critical to mitigate the risk of infections. Data from primary immunodeficiency disorders as well as rituximab-treated patients suggest that a persistent IgG level of <400 mg/dl is associated with a higher risk of infections17,18. Hence, immunoglobulin replacement [IVIG or subcutaneous formulation] is frequently offered in these patients with a goal IgG trough level of ≥400 mg/dl, particularly if there are recurrent or severe infections19. In patients with recurrent infections despite IgG level of greater than 400 mg/dl, checking IgG subclass levels and administering IVIG if low can be considered. Notably, the half-life of IgG is 3–4 weeks which requires monthly dosing. Other than infusion reaction, rare but serious adverse effects such as thromboembolic events, autoimmune hemolytic anemia, and acute kidney injury has been reported with IVIG20. Recovery of B-cells in peripheral blood is not always associated with rise in IgG production post-CAR T-cell therapy21. Hence, close monitoring of IgG level and discontinuing IVIG once trough levels are persistently ≥400 mg/dl without replacement is a rational strategy. T-cell recovery may also remain impaired after CAR T-cell therapy and checking for CD4/CD8 T-cell subsets in peripheral blood can also assist clinicians in assessing immune recovery. With preclinical development of a cellular antidote to CAR19 and expression of inducible caspase in CAR constructs22,23, future studies should investigate whether these strategies can be applied to patients in a durable CR with persistent B-cell aplasia or hypogammaglobulinemia. Development of suicide gene-modified CAR T-cell therapy can also help overcome late and long-term toxicities such as hypogammaglobulinemia and will have to be tested for safety and efficacy24.
Prolonged Cytopenia
Prolonged cytopenias involving all three cell lines have been described after CAR T-cell therapy, both in clinical trials and observational studies. The precise mechanism behind prolonged cytopenias remains unclear, with bone marrow biopsy in most cases demonstrating hypocellularity. In the context of NHL, factors associated with grade 3 or worse cytopenias at 30 days after CAR T-cell infusion include ECOG performance status of 1 or greater and more than three prior lines of therapy25.
In ZUMA-1 trial, 18 out of 108 patients [17%] had grade 3 or worse cytopenia beyond 3 months, with neutropenia in 11%, thrombocytopenia in 7%, and anemia in 3%2. However, at a median follow-up of 27 months, only two patients had grade 3 cytopenia [one anemia and one thrombocytopenia], indicating that most cytopenias resolve on long-term follow-up. In the JULIET trial, unresolved grade 3 or 4 neutropenia and thrombocytopenia was present at day 28 in 24% and 41% of patients respectively3. Although all cases of neutropenia resolved by 3 months, 38% had unresolved thrombocytopenia at the 3-month landmark. The burden of grade 3 or worse cytopenias beyond one month is greater in R/R B-cell ALL trials, with the incidence being 41% for platelets and 53% for neutrophils in the ELIANA trial1. At a median follow-up of 13 months, 29% and 20% of patients with grade 3 or 4 thrombocytopenia and neutropenia respectively did not have resolution to grade 2 or lower in this study. However, direct comparison of the incidence of prolonged cytopenias across trials should be performed with caution given different CAR constructs, patient demographics, and prior therapies. In the phase 1 study on bb-2121 in MM, six out of 17 patients [35%] with thrombocytopenia and one out of 32 patients with neutropenia did not recover by four weeks after CAR T-cell infusion26. Long-term follow-up of another phase I/II trial on CD19-directed CAR T showed three out of 19 evaluable patients [16%] to have prolonged cytopenia beyond 90 days, including one patient with ALL and two with NHL13. Two patients in this study had pancytopenia needing transfusions and growth factor support lasting 15 and 22 months before resolution whereas one patient had unresolved pancytopenia at latest follow-up [18 months]. In an observational study from MSKCC, the following factors were associated with a lower likelihood of complete count recovery at 1 month: >3 prior lines of therapy, baseline cytopenia, CAR construct [incidence of complete hematopoietic recovery being higher with 4–1BB costimulatory domain (70%)compared to that with CD28 or 19–28ᶻ costimulatory domain (17–19%), grade ≥3 CRS/ICANS, higher peak CRP, and higher peak ferritin level27. Another study from Israel showed late hematologic toxicity in 27 out of 29 [93%] responding patients with ALL or NHL after administration of CTL019 with CD28 co-stimulatory domain and fludarabine-cyclophosphamide lymphodepletion28. Notably, late neutropenia, thrombocytopenia, and anemia [requiring PRBC transfusion] occurred in 76%, 76%, and 17% respectively. Factors associated with late cytopenias were prior HSCT and higher CRS grade. Approximately one-half and one-third of patients with neutropenia and thrombocytopenia respectively had intermediate count recovery between immediate and late cytopenias resembling a biphasic pattern.
Management
Management of prolonged cytopenias entail supportive care measures such as blood product transfusion, prophylactic antibiotics in neutropenic patients, and growth factor support. Other causes of cytopenia such as nutritional deficiency, viral infections, and subsequent myeloid neoplasm should be excluded. The risk of prolonged cytopenia post-CAR T-cell therapy is higher in B-cell ALL compared to NHL, based on clinical trials and observational studies29. Thrombopoietin receptor agonists like eltrombopag has been anecdotally used for prolonged thrombocytopenia13, however, prospective data on efficacy are lacking. A case of successful infusion of cryopreserved autologous hematopoietic progenitor cells in a MM patient with prolonged pancytopenia after BCMA CAR T-cell therapy has been reported30, and further prospective studies are needed to assess safety and efficacy of this approach.
Late Infections
Both hypogammaglobulinemia and prolonged cytopenias can lead to increased risk of late infections in CAR T-cell therapy survivors. In the updated analysis of ZUMA-1 trial, there were nine grade 3 or higher infections in eight patients [8%] at a median of 7–19 months after CAR T-cell infusion31. The most common site of infection was respiratory tract. No infection-related mortality has been reported from ZUMA-1 to our knowledge. Another study on CD-19 directed CAR T in CLL reported one infection-related death from pseudomonas wound infection in a MRD-negative patient at 21 months after cell infusion15. The incidence of all-grade infection beyond 8 weeks after cell infusion was 39% in the JULIET trial testing tisagenlecleucel in R/R B-cell NHL, with 18% being grade 3 or 43. Late infections [beyond 90 days] with CD-19 directed CAR T in B-cell NHL and CLL has been systematically evaluated in a study from the Fred Hutchinson Cancer Research Center13. Notably, 61% of patients had at least one infection, with a total of 153 infection events and infection density of 2.08 per patient-year. Similar to ZUMA-1 trial, the most common site was respiratory tract. A vast majority of patients [80%] were treated in the outpatient setting, with only 5% requiring admission to the intensive care unit. Among patients with an identified causative organism, 60% were bacterial, 31% were viral, and 9% were fungal.
In the ELIANA trial, 18 out of 40 B-ALL patients with prolonged neutropenia beyond day 28 developed grade 3 or 4 infection1. Some atypical infections were noted, including human herpes virus-6 encephalitis and systemic mycosis. Notably, three out of 17 late deaths [18%] beyond 30 days were infection-related in this study. In patients with B-ALL, grade 3 or higher CRS is a predictor of subsequent infection within 6 months after CD-19 directed CAR T infusion32. Increased steroid use in patients developing CRS or ICANS could be a contributor to higher infection risk in such patients. The phase 1 study from Sloan Kettering in B-ALL patients also showed a 6% infection-related mortality, indicating that the risk of fatal infection may be higher in B-ALL compared to other lymphoid malignancies. In the BCMA CAR T-cell trial in MM, 26% of patients had upper respiratory tract infections from week 8 through month 6 after infusion, 7% being grade 326.
Management
One of the key aspects of infection management beyond 30 days after CAR T-cell therapy is prevention and early detection. As mentioned above, using immunoglobulin replacement therapy for primary prophylaxis in hypogammaglobulinemic patients and G-CSF [Granulocyte-Colony Stimulating Factor] support for neutropenic patients should be considered to mitigate the risk of infection. However, the impact of G-CSF has not been prospectively studied in this context and further studies are necessary to delineate safety and efficacy. Furthermore, clinicians should have a strong index of suspicion for infection prior to CAR T-cell therapy, as many patients have received prior autologous or allogeneic HCT and may be severely immunocompromised33. At our institution, we use antiviral prophylaxis with acyclovir beginning with lymphodepleting chemotherapy and through at least 90 days post-infusion. Continuing antiviral prophylaxis till 6 months post-infusion is also a reasonable approach. Ciprofloxacin [or equivalent for patients who are intolerant or allergic to quinolones] is used for antibacterial prophylaxis, beginning on day 0 and continuing until recovery of absolute neutrophil count. Antifungal prophylaxis with fluconazole should be initiated during the neutropenic period and switching to a mold-active agent such as posaconazole should be considered if patient requires prolonged high-dose corticosteroids or has prolonged neutropenia [>3 weeks]33. Prophylaxis for Pneumocystis jiroveci pneumonia [PJP] with trimethoprim-sulfamethoxazole is continued at our institution until CD4+ T-cell count is greater than 200 cells/μL. Some experts recommend continuing PJP prophylaxis until at least 6 months34. There is a lack of robust evidence on the need and optimal duration of PJP prophylaxis in this setting. Currently, there are no specific guidelines on vaccination after CAR T-cell therapy. Due to prolonged B-cell aplasia after CD-19 directed CAR T-cell therapy, the immunogenicity of vaccinations may be reduced. Experts recommend administering killed or inactivated vaccine beyond 6 months and live vaccines beyond 1 year after CAR T-cell therapy in patients who are in remission and do not require any further cancer-directed therapy34.
Late Neurologic Events
Acute neurotoxicity [CRES/ICANS] is a well-known adverse event early after CAR T-cell therapy, with onset of symptoms by 5 days after infusion5. Clinical manifestations include confusion, aphasia, agraphia, ataxia, memory loss, hallucinations, and apraxia. Three distinct patterns of neurologic symptoms have been reported after CAR T-cell therapy, one that is concurrent with CRS, one that happens shortly after CRS subsides, and a third form with a delayed onset to 3 or more weeks after CAR T-cell infusion35. It is unclear whether clinical predictors of CRES/ICANS, e.g., high disease burden or high peak CAR T-cell count, will apply to late neurologic events as well. A phase I/II trial of CD-19 directed CAR T has systematically evaluated late neurologic and psychiatric events at a median follow-up of 28 months [range, 13–63 months] from infusion13. Notably, new neurologic and psychiatric events beyond day 90 were noted in 9 [10%] and 8 [9%] patients respectively. Specific neurologic events of interest were three cerebrovascular accidents [CVA] [6–35 months], one transient ischemic attack [4 months], one Alzheimer’s dementia [14 months], and one peripheral neuropathy [17 months]. Psychiatric events of interest were newly diagnosed or exacerbation of mood disorders [onset at 1.5–33 months]13. In the ZUMA-1 trial, one patient had grade 1 ongoing memory impairment at a median-follow up of 9 months, which subsequently resolved. Furthermore, four patients had unresolved neurologic events, who eventually died of progressive disease [n=2] or non-neurologic adverse events [n=2]31. In the ELIANA trial, 25% of patients with R/R B-cell ALL had unresolved grade 3 neurologic events at day 18 post-infusion1. Notably, three out of 10 patients with grade 3 neurologic events did not experience resolution at study discontinuation, among which, one had no response and two died of leukemia progression and encephalitis. In summary, 4–5% of clinical trial patients have unresolved neurotoxicity at study discontinuation1,31. In the BCMA CAR T-cell study for MM [bb2121], there were no new neurologic events reported from week 8 through month 6 after infusion26. However, the upper limit of range of neurotoxicity duration was 251 days, indicating that some patients had prolonged symptoms beyond 1 month.
Management
There is lack of data on management of late neurologic events, including utility of corticosteroids or anti-IL-6 monoclonal antibody in this context. Furthermore, attribution of causality to CAR T-cell therapy for late neurologic events like CVA or peripheral neuropathy is difficult in uncontrolled trials or observational studies. Prospective studies to define the nature and trajectory of late neurologic events to identify clinical and biological correlates are urgently needed to better manage these patients in future. Furthermore, patients with CNS involvement by their underlying cancer have been traditionally excluded in CAR T-cell therapy trials. However, since these patients are receiving CAR T in clinical practice, they should be carefully followed for development of late neurologic events. Randomized controlled trials on CAR T-cell therapies will provide further comparative data on late neurotoxicity.
Immune-related Late Effects
Autoimmune reactions, with development of new or exacerbation of pre-existing autoimmune disease, is a potential concern after CAR T-cell therapy, similar to that seen with immune checkpoint inhibitors [ICI]. Among the clinical trials which led to FDA approvals thus far, late autoimmune reactions have not been reported at a median follow-up of 13–27 months from CAR T-cell infusion1–3. However, in the Fred Hutchinson Cancer Research Center study of CD-19 directed CAR T in lymphoid malignancies, possible late immune-related adverse events were seen in 7 [8%] patients at a median of 234 days after infusion [range, 67–1099 days]13. The specific events were lymphocytic alveolitis, persistent skin rash, eosinophilic pneumonia, pneumonitis not otherwise specified [NOS], granulomatous disease NOS, persistent flu-like syndrome, and collagenous colitis. Notably, CAR T-cells were noted on skin biopsy in the patient with a persistent skin rash. However, correlative studies showed no evidence of CAR T-cell re-expansion in the peripheral blood at the time of immune-related events13. Another group reported three patients with R/R aggressive lymphoma who were treated sequentially with CD-19 directed CAR T and pembrolizumab, and subsequently developed autoimmune thyroiditis, pneumonitis, and T-cell mediated autoimmune skin rash 1–6 months after CTL019 infusion36. However, definitive attribution of autoimmune toxicities to CAR T-cell therapy is difficult due to uncontrolled nature of these studies. In patients with prior allogeneic hematopoietic cell transplantation, the incidence of developing graft-versus-host-disease [GVHD] after CTL019 therapy is 20%, at a latency of 2–3 months from cell infusion13.
Management
There is a lack of consensus guidelines on how to best manage late immune-related adverse events after CAR T-cell therapy. Based on available data for the treatment of immune-related adverse events of ICI37, inducing transient immunosuppression by administration of glucocorticoids is a rational strategy. In the Fred Hutchinson Cancer Research Center study, most immune-related events were treated with corticosteroids. The persistent skin rash did not respond to steroids and was subsequently treated with PUVA [photochemotherapy] since biopsy showed spongiosis and psoriaform dermatitis, leading to a partial response13. Future studies should explore the role of additional immunosuppressive agents such as infliximab [monoclonal antibody against tumor necrosis factor-α] or vedolizumab [monoclonal antibody against integrin α4ᵦ7] in this context, especially in patients non-responsive to corticosteroids, given their activity in immune-related adverse events of ICIs. Patients receiving CAR T-cell therapy for relapse post-allogeneic HCT should be closely monitored for development of GVHD. Similarly, patient receiving off-the-shelf allogeneic CARs, many of which are in clinical trials currently, should also be closely monitored for GVHD.
Subsequent Malignancies
In the long-term follow-up of ZUMA-1 trial, one patient developed myelodysplastic syndrome [MDS] at 19 months, which was attributed to prior cytotoxic therapy2. Other than that, to the best of our knowledge, no secondary malignancies have been reported thus far from clinical trials that led to FDA approval of CD-19 directed CAR T in B-cell ALL and B-cell NHL, although follow-up is short. In a phase I/II study from Fred Hutchinson Cancer Research Center, subsequent hematologic cancers were seen in 5 patients [6%], including four cases of MDS and one MM13. Notably, the median time from CAR T-cell infusion to diagnosis of MDS was 6 months [range, 4–17 months]. Furthermore, two out of four patients with MDS had pre-existing cytogenetic abnormalities prior to CAR T-cell therapy and one patient with MM had pre-existing monoclonal gammopathy of undetermined significance. Eight patients [9%] developed solid tumors, including six with non-melanoma skin cancer, one with melanoma, and one non-invasive bladder cancer. Given these patients had received extensive prior cytotoxic therapies, it is unclear whether CAR T-cells can be attributed to development of subsequent cancers. Notably, a case of unintentional introduction of CAR gene into a leukemic B-cell has been reported, leading to relapse of CD19-negative leukemia 9 months after infusion of CD-19 directed CAR T38. Insertional oncogenesis due to insertion of a viral vector near an oncogene in the engineered T-cells is a possibility, however, no such cases have been reported till date to our knowledge39. Malignant transformation of engineered T-cells leading to T-cell leukemia has not been reported to date. Studies conducted by CIBMTR and other registries will inform incidence and risk-factors for second cancers in CAR T-cell therapy recipients.
Management
Since secondary malignancies are rare events with a potentially long latency, long-term follow up of ongoing clinical trials and epidemiologic data are needed to accurately estimate the incidence of second cancers. FDA has mandated at least 15-year follow up for axi-cel and tisagenlecleucel, and these prospective post-marketing registry studies are being conducted by the CIBMTR in which incidence of subsequent malignancies is a key endpoint. An anti-CAR19 idiotype CAR [αCAR19] has been developed by the Penn group, which can potentially target and kill CAR19+ cells, and can be clinically applied as an antidote for unintentional transduction of leukemic B-cells by CAR gene or development of T-cell leukemia23. However, data on clinical application of this approach is not available till date to our knowledge. Until such further studies provide an evidence base to suggest appropriate second cancer screening, CAR T-cell therapy recipients can follow recommendations for cancer screening similar to what has been recommended for HCT survivors40–42.
Patient-Reported Outcomes
Since CAR T-cell therapies are associated with several symptomatic adverse events and can potentially impact health-related quality of life [HRQoL], measurement of patient-reported outcomes [PROs] is crucial for assessing tolerability and comparative effectiveness43. The ELIANA trial on tisagenlecleucel in R/R B-ALL recently reported data on PROs measured up until 12 months after CAR T-cell infusion44. PROs were a secondary outcome of the trial, measured by standardized generic instruments for assessing quality of life [QoL], including the Pediatric Quality of Life Inventory [PedsQL] and the European Quality of Life-5 Dimensions [EQ-5D]. The PRO-specific objective was change in summary scores from baseline to follow-up time-points [day 28, and months 3, 6, 9, and 12]. Notably, there was a clinically meaningful improvement in mean total PedsQL score at each time-point from 3–12 months after infusion compared to baseline. Furthermore, the magnitude of improvement was greatest for physical functioning and smallest for social functioning. Clinically meaningful improvement was also observed in psychosocial health summary score and emotional functioning score beyond 3 months. These findings empirically demonstrated that deep and durable hematologic responses from CD-19 directed CAR T translate into an improved global QoL, including physical, psychosocial, and emotional functioning. Similarly, in ZUMA-2 trial on KTE-X19 in relapsed/refractory mantle cell lymphoma, PRO was a secondary endpoint, with the PRO-specific objective being change in score from baseline to month 6 in the five-level version of EQ-5D [European Quality of Life-5 Dimensions] questionnaire45. After a transient decrease in patient reported HRQoL on week 4, overall health returned to baseline of better in most patients at month 6. In the JULIET trial testing tisagenlecleucel in relapsed/refractory B-cell NHL, sustained improvement in all HRQoL domains was observed among patients achieving a CR or partial response to therapy at all follow-up time points46. Another cross-sectional study investigated patient-reported long-term neuropsychiatric outcomes in 40 CAR T-cell therapy survivors with B-cell NHL, CLL or ALL as their primary disease47. Notably, 63% of patients had acute neurotoxicity after CD-19 directed CAR T infusion in this study. The investigators used Patient Reported Outcomes Measurement Information System [PROMIS] measures, with the median time to PRO questionnaire completion being 3 years [range, 1–5]. Notably, the mean global physical health, global mental health, social function, anxiety, fatigue, pain, and sleep disturbance scores for the study cohort did not differ significantly from that of general population. However, 15 patients [38%] reported some cognitive difficulty, with pre-CAR T depression and acute neurotoxicity being associated with subsequent cognitive impairment. Furthermore, presence of cognitive difficulties was associated with significantly worse global physical and mental health scores. Younger age was associated with worse global mental health, anxiety, and depression. This study highlights the importance of assessing neuropsychiatric outcomes like anxiety, depression, and cognitive impairment in CAR T survivors so they can be appropriately referred to mental health or cognitive rehabilitation services.
Integration of PROs in clinical trials is important to assess “tolerability” of anti-cancer therapies48. With different highly effective CAR T-cell products and non-cellular therapies competing for similar treatment spaces in hematologic malignancies, comparative safety and tolerability will play a major role in treatment decision making and regulatory assessments. One of the major challenges in interpretation of PRO data is lack of consistency regarding the use of PRO instruments, time-points of PRO assessment, and statistical design to handle missing data. With several recently published guidelines for inclusion of PROs in clinical trials49,50, the quality of PRO-specific trial design and reporting will likely further improve in future. In patients receiving CAR T-cell therapies as standard-of-care, a centralized PRO collection system should be established using the infrastructure already available at CIBMTR for HSCT recipients51.
Other Potential Late Effects
Potential organ-specific late effects of CAR T-cell therapy include cardiac and renal toxicities. Cardiac toxicities have been described after CAR T-cell therapy, mostly in the context of CD-19 directed CAR T for lymphoid malignancies52. Among patients with paired pre- and post-CAR T troponin measurement, which is a marker for cardiomyocyte injury, 54% had troponin elevation after CAR T-cell infusion, with the median time to troponin increase being 16 days [range, 6–31 days]. Notably, the incidence of troponin elevation was significantly higher in patients with grade 2 or higher CRS compared to those with grade 1 or no CRS. Furthermore, approximately one-third of patients with available data had a clinically significant reduction in left ventricular ejection fraction [LVEF] after CAR T-cell infusion. At a median follow-up of 10 months, there were 17 cardiovascular [CV] events [12%], including six CV deaths, six decompensated congestive heart failure [CHF], and five new onset arrhythmias. Notably, the CV event rate was significantly higher in patients with positive troponin [55%] compared to those with negative troponin [4%]. On multivariable analysis for risk factors, higher time-lag between CRS onset and tocilizumab administration was associated with a higher odds of subsequent CV events. This study highlighted the role of serum troponin as a biomarker for CV toxicity after CAR T-cell therapy and the potential cardio-protective role of tocilizumab, especially in the context of CRS. Further prospective studies should evaluate the role of brain natriuretic peptide and global longitudinal strain in predicting CV toxicities53. As CAR T survivor pool continues to increase, late cardiac events in specific populations like older patients or those with prior exposure to cardiotoxic chemotherapy like anthracycline or carfilzomib should be investigated for early intervention. To our knowledge, immune-related cardiac toxicities such as myocarditis, which are seen with ICIs, have not yet been reported with CAR T-cell therapy.
Acute kidney injury [AKI] after CAR T-cell therapy happens in around 20% of children and young adults receiving CD-19 directed CAR T for R/R B-ALL54. Notably, patients with grade 3/4 CRS have a five-fold higher risk of developing all-grade AKI and 10-fold higher risk of developing severe AKI compared to those with grade 1/2 CRS. Approximately 90% of patients recover their kidney function by 30 days. In adult population, the incidence of all-grade AKI by day 100 is higher at 30%, with 91% recovering their renal function to baseline by 30 days from AKI onset55. Risk factors in this population includes prior autologous or allogeneic HCT, requiring intensive care unit admission, and grade 3–4 CRS. Long-term follow up is required to investigate whether a subset of these patients is at risk of developing chronic kidney disease.
Table II.
Recommended Screening and Preventative Care for Specific Late Effects
| Late Effects | Screening and Preventative Care at Specific Time-points |
|---|---|
| Prolonged Cytopenia | CBC with differential at least every 30 days after the acute phase of CAR T cell infusion until normalization of blood counts |
| Hypogammaglobulinemia | Obtain serum IgG level monthly beyond day 30 after CAR T-cell infusion, until IgG>400 mg/dl. Consider obtaining IgG subclass level if active infections despite total IgG>400 mg/dl |
| Neuropsychiatric Late effects | Clinical evaluation for signs and symptoms of neuropsychiatric dysfunction monthly after day 30, with diagnostic tests [e.g. objective neuropsychological testing, MRI] in patients with signs or symptoms |
| Immune-related Adverse Events | Clinical evaluation for signs or symptoms of immune-relate adverse events such as pneumonitis or colitis at least every month until one year and every six months thereafter and targeted diagnostic tests in those with clinical suspicion |
| Second Cancer | Age and sex-appropriate screening for solid cancers Periodic monitoring of blood counts to screen for therapy-related myeloid neoplasms and low threshold to perform bone marrow examination in patients with unexplained or worsening cytopenia |
| Late Infections | CDC panel for CD4+ T-cell count monthly beyond day 30 until CD4 count is greater than 200 cells/μL |
Note: Management recommendations in this table are based on current state of evidence and expert opinion.
Highlights:
Hypogammaglobulinemia is the most common late effect of CD-19 directed CAR T-cell therapy
The incidence of hypogammaglobulinemia and prolonged cytopenia is greater in ALL compared to NHL trials
Common determinants of late toxicity are age, prior therapies, tumor type, acute toxicities [CRS/ICANS], and CAR construct
Funding Source:
Navneet Majhail is partially supported by a grant from the National Cancer Institute (R01-CA215134). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations:
- ALL
Acute Lymphoblastic Leukemia
- NHL
Non-Hodgkin Lymphoma
- IVIG
Intravenous Immunoglobulin
- CR
Complete Response
Footnotes
Conflicts of Interest: No relevant financial conflicts of interest.
Ethics Committee Approval: Not Applicable
References
- 1.Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. New England Journal of Medicine. 2018;378(5):439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Locke FL, Ghobadi A, Jacobson CA, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. The Lancet Oncology. 2019;20(1):31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. The New England journal of medicine. 2019;380(1):45–56. [DOI] [PubMed] [Google Scholar]
- 4.Wang M, Munoz J, Goy A, et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. The New England journal of medicine. 2020;382(14):1331–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neelapu SS, Tummala S, Kebriaei P, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15(1):47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Majhail NS, Rizzo JD. Surviving the cure: long term followup of hematopoietic cell transplant recipients. Bone marrow transplantation. 2013;48(9):1145–1151. [DOI] [PubMed] [Google Scholar]
- 7.Majhail NS. Long-term complications after hematopoietic cell transplantation. Hematol Oncol Stem Cell Ther. 2017;10(4):220–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Majhail NS, Mau LW, Chitphakdithai P, et al. Transplant center characteristics and survival after allogeneic hematopoietic cell transplantation in adults. Bone marrow transplantation. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Majhail NS, Rizzo JD, Lee SJ, et al. Recommended screening and preventive practices for long-term survivors after hematopoietic cell transplantation. Bone marrow transplantation. 2012;47(3):337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pidala J, Anasetti C, Jim H. Quality of life after allogeneic hematopoietic cell transplantation. Blood. 2009;114(1):7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Majhail NS, Murphy E, Laud P, et al. Randomized controlled trial of individualized treatment summary and survivorship care plans for hematopoietic cell transplantation survivors. Haematologica. 2019;104(5):1084–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hashmi SK, Bredeson C, Duarte RF, et al. National Institutes of Health Blood and Marrow Transplant Late Effects Initiative: The Healthcare Delivery Working Group Report. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2017;23(5):717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cordeiro A, Bezerra ED, Hirayama AV, et al. Late Events after Treatment with CD19-Targeted Chimeric Antigen Receptor Modified T Cells. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2020;26(1):26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaeger U T C, Borchmann P, McGuirk J, Holte H, Waller E, Jaglowski S, Andreadis C, Foley SR, Fleury I, Westin J, Teshima T, Mielke S, Salles G, Ho PJ, Izutsu K, Schuster S, Bachanova V, Maziarz R, Van Besien K, Kersten MJ Wagner‐Johnston N Kato K Corradini P Tiwari R Forcina A Pacaud L Bishop M. INTRAVENOUS IMMUNOGLOBULIN THERAPY USE IN PATIENTS WITH RELAPSED/REFRACTORY DIFFUSE LARGE B‐CELL LYMPHOMA TREATED WITH TISAGENLECLEUCEL IN THE JULIET TRIAL. International Conference on Malignant Lymphoma Palazzo dei Congressi; 12 June 2019, 2019; Lugano, Switzerland. [Google Scholar]
- 15.Porter DL, Hwang WT, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7(303):303ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan Z, Cao J, Cheng H, et al. A combination of humanised anti-CD19 and anti-BCMA CAR T cells in patients with relapsed or refractory multiple myeloma: a single-arm, phase 2 trial. The Lancet Haematology. 2019;6(10):e521–e529. [DOI] [PubMed] [Google Scholar]
- 17.Quinti I, Soresina A, Guerra A, et al. Effectiveness of immunoglobulin replacement therapy on clinical outcome in patients with primary antibody deficiencies: results from a multicenter prospective cohort study. J Clin Immunol. 2011;31(3):315–322. [DOI] [PubMed] [Google Scholar]
- 18.Makatsori M, Kiani-Alikhan S, Manson AL, et al. Hypogammaglobulinaemia after rituximab treatment-incidence and outcomes. QJM. 2014;107(10):821–828. [DOI] [PubMed] [Google Scholar]
- 19.Ogba N, Arwood NM, Bartlett NL, et al. Chimeric Antigen Receptor T-Cell Therapy. 2018;16(9):1092. [DOI] [PubMed] [Google Scholar]
- 20.Ueda M, Berger M, Gale RP, Lazarus HM. Immunoglobulin therapy in hematologic neoplasms and after hematopoietic cell transplantation. Blood Rev. 2018;32(2):106–115. [DOI] [PubMed] [Google Scholar]
- 21.Doan A, Pulsipher MA. Hypogammaglobulinemia due to CAR T-cell therapy. Pediatr Blood Cancer. 2018;65(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Stasi A, Tey SK, Dotti G, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. The New England journal of medicine. 2011;365(18):1673–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruella M, Barrett DM, Shestova O, et al. A cellular antidote to specifically deplete anti-CD19 chimeric antigen receptor positive cells. Blood. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minagawa K, Al-Obaidi M, Di Stasi A. Generation of Suicide Gene-Modified Chimeric Antigen Receptor-Redirected T-Cells for Cancer Immunotherapy. Methods in molecular biology (Clifton, NJ). 2019;1895:57–73. [DOI] [PubMed] [Google Scholar]
- 25.Strati P, Adkins S, Nastoupil LJ, et al. Hematopoietic recovery and immune reconstitution after axi-cel CAR T-cell therapy in patients with relapsed/refractory large B-cell lymphoma. Journal of Clinical Oncology. 2019;37(15_suppl):7545–7545. [Google Scholar]
- 26.Raje NS, Berdeja JG, Lin Y, et al. bb2121 anti-BCMA CAR T-cell therapy in patients with relapsed/refractory multiple myeloma: Updated results from a multicenter phase I study. Journal of Clinical Oncology. 2018;36(15_suppl):8007–8007. [Google Scholar]
- 27.Jain T, Knezevic A, Pennisi M, et al. Hematopoietic recovery in patients receiving chimeric antigen receptor T-cell therapy for hematologic malignancies. Blood advances. 2020;4(15):3776–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fried S, Avigdor A, Bielorai B, et al. Early and late hematologic toxicity following CD19 CAR-T cells. Bone marrow transplantation. 2019;54(10):1643–1650. [DOI] [PubMed] [Google Scholar]
- 29.Jain T, Knezevic A, Pennisi M, et al. Hematopoietic Recovery Following Chimeric Antigen Receptor T Cell (CAR T) Therapy in Hematological Malignancies. Biology of Blood and Marrow Transplantation. 2020;26(3):S63–S64. [Google Scholar]
- 30.Yan L, Shang J, Shi X, et al. Successful treatment of marrow failure after CARTs for myeloma by the infusion of cryopreserved stem cells. Am J Hematol. 2020;95(1):E20–E23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. New England Journal of Medicine. 2017;377(26):2531–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park JH, Romero FA, Taur Y, et al. Cytokine Release Syndrome Grade as a Predictive Marker for Infections in Patients With Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia Treated With Chimeric Antigen Receptor T Cells. Clin Infect Dis. 2018;67(4):533–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hill JA, Seo S. How we prevent infections in patients receiving CD19-targeted chimeric antigen receptor T-cells for B-cell malignancies. Blood. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hill JA, Seo SK. How I prevent infections in patients receiving CD19-targeted chimeric antigen receptor T cells for B-cell malignancies. Blood. 2020;136(8):925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hunter BD, Jacobson CA. CAR T-cell associated neurotoxicity: Mechanisms, clinicopathologic correlates, and future directions. J Natl Cancer Inst. 2019. [DOI] [PubMed] [Google Scholar]
- 36.Kambhampati S, Gray L, Fakhri B, et al. Immune-related Adverse Events Associated With Checkpoint Inhibition in the Setting of CAR T Cell Therapy: A Case Series. Clin Lymphoma Myeloma Leuk. 2019. [DOI] [PubMed] [Google Scholar]
- 37.Postow MA, Sidlow R, Hellmann MD. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. The New England journal of medicine. 2018;378(2):158–168. [DOI] [PubMed] [Google Scholar]
- 38.Ruella M, Xu J, Barrett DM, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med. 2018;24(10):1499–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Molecular therapy oncolytics. 2016;3:16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Majhail NS, Rizzo JD, Lee SJ, et al. Recommended screening and preventive practices for long-term survivors after hematopoietic cell transplantation. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2012;18(3):348–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Inamoto Y, Shah NN, Savani BN, et al. Secondary solid cancer screening following hematopoietic cell transplantation. Bone marrow transplantation. 2015;50(8):1013–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morton LM, Saber W, Baker KS, et al. National Institutes of Health Hematopoietic Cell Transplantation Late Effects Initiative: The Subsequent Neoplasms Working Group Report. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2017;23(3):367–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chakraborty R, Sidana S, Shah GL, Scordo M, Hamilton BK, Majhail NS. Patient-Reported Outcomes with Chimeric Antigen Receptor T Cell Therapy: Challenges and Opportunities. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2019;25(5):e155–e162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laetsch TW, Myers GD, Baruchel A, et al. Patient-reported quality of life after tisagenlecleucel infusion in children and young adults with relapsed or refractory B-cell acute lymphoblastic leukaemia: a global, single-arm, phase 2 trial. The Lancet Oncology. 2019;20(12):1710–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang M, Munoz J, Goy A, et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. New England Journal of Medicine. 2020;382(14):1331–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maziarz RT, Waller EK, Jaeger U, et al. Patient-reported long-term quality of life after tisagenlecleucel in relapsed/refractory diffuse large B-cell lymphoma. Blood advances. 2020;4(4):629–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ruark J, Mullane E, Cleary N, et al. Patient-Reported Neuropsychiatric Outcomes of Long-Term Survivors after Chimeric Antigen Receptor T Cell Therapy. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2020;26(1):34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sacks CA, Miller PW, Longo DL. Talking about Toxicity - “What We’ve Got Here Is a Failure to Communicate”. The New England journal of medicine. 2019;381(15):1406–1408. [DOI] [PubMed] [Google Scholar]
- 49.Coens C, Pe M, Dueck AC, et al. International standards for the analysis of quality-of-life and patient-reported outcome endpoints in cancer randomised controlled trials: recommendations of the SISAQOL Consortium. The Lancet Oncology. 2020;21(2):e83–e96. [DOI] [PubMed] [Google Scholar]
- 50.Calvert M, Kyte D, Mercieca-Bebber R, et al. Guidelines for Inclusion of Patient-Reported Outcomes in Clinical Trial Protocols: The SPIRIT-PRO Extension. Jama. 2018;319(5):483–494. [DOI] [PubMed] [Google Scholar]
- 51.Shaw BE, Brazauskas R, Millard HR, et al. Centralized patient-reported outcome data collection in transplantation is feasible and clinically meaningful. Cancer. 2017;123(23):4687–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alvi RM, Frigault MJ, Fradley MG, et al. Cardiovascular Events Among Adults Treated With Chimeric Antigen Receptor T-Cells (CAR-T). J Am Coll Cardiol. 2019;74(25):3099–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lancellotti P, Moonen M, Galderisi M. Chimeric Antigen Receptor T-Cells and Cardiovascular Toxicity: Cause for Concern? J Am Coll Cardiol. 2019;74(25):3109–3111. [DOI] [PubMed] [Google Scholar]
- 54.Myers RM, Fitzgerald J, Elgarten CW, et al. Acute Kidney Injury after Chimeric Antigen Receptor T-Cell Therapy for Pediatric Acute Lymphoblastic Leukemia. Biology of Blood and Marrow Transplantation. 2019;25(3, Supplement):S168–S169. [Google Scholar]
- 55.Gutgarts V, Jain T, Zheng J, et al. Acute Kidney Injury after CAR-T Cell Therapy: Low Incidence and Rapid Recovery. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]