

Abstract
Background
Accurate rapid diagnostic tests for SARS‐CoV‐2 infection could contribute to clinical and public health strategies to manage the COVID‐19 pandemic. Point‐of‐care antigen and molecular tests to detect current infection could increase access to testing and early confirmation of cases, and expediate clinical and public health management decisions that may reduce transmission.
Objectives
To assess the diagnostic accuracy of point‐of‐care antigen and molecular‐based tests for diagnosis of SARS‐CoV‐2 infection. We consider accuracy separately in symptomatic and asymptomatic population groups.
Search methods
Electronic searches of the Cochrane COVID‐19 Study Register and the COVID‐19 Living Evidence Database from the University of Bern (which includes daily updates from PubMed and Embase and preprints from medRxiv and bioRxiv) were undertaken on 30 Sept 2020. We checked repositories of COVID‐19 publications and included independent evaluations from national reference laboratories, the Foundation for Innovative New Diagnostics and the Diagnostics Global Health website to 16 Nov 2020. We did not apply language restrictions.
Selection criteria
We included studies of people with either suspected SARS‐CoV‐2 infection, known SARS‐CoV‐2 infection or known absence of infection, or those who were being screened for infection. We included test accuracy studies of any design that evaluated commercially produced, rapid antigen or molecular tests suitable for a point‐of‐care setting (minimal equipment, sample preparation, and biosafety requirements, with results within two hours of sample collection). We included all reference standards that define the presence or absence of SARS‐CoV‐2 (including reverse transcription polymerase chain reaction (RT‐PCR) tests and established diagnostic criteria).
Data collection and analysis
Studies were screened independently in duplicate with disagreements resolved by discussion with a third author. Study characteristics were extracted by one author and checked by a second; extraction of study results and assessments of risk of bias and applicability (made using the QUADAS‐2 tool) were undertaken independently in duplicate. We present sensitivity and specificity with 95% confidence intervals (CIs) for each test and pooled data using the bivariate model separately for antigen and molecular‐based tests. We tabulated results by test manufacturer and compliance with manufacturer instructions for use and according to symptom status.
Main results
Seventy‐eight study cohorts were included (described in 64 study reports, including 20 pre‐prints), reporting results for 24,087 samples (7,415 with confirmed SARS‐CoV‐2). Studies were mainly from Europe (n = 39) or North America (n = 20), and evaluated 16 antigen and five molecular assays.
We considered risk of bias to be high in 29 (37%) studies because of participant selection; in 66 (85%) because of weaknesses in the reference standard for absence of infection; and in 29 (37%) for participant flow and timing. Studies of antigen tests were of a higher methodological quality compared to studies of molecular tests, particularly regarding the risk of bias for participant selection and the index test. Characteristics of participants in 35 (45%) studies differed from those in whom the test was intended to be used and the delivery of the index test in 39 (50%) studies differed from the way in which the test was intended to be used. Nearly all studies (97%) defined the presence or absence of SARS‐CoV‐2 based on a single RT‐PCR result, and none included participants meeting case definitions for probable COVID‐19.
Antigen tests
Forty‐eight studies reported 58 evaluations of antigen tests. Estimates of sensitivity varied considerably between studies. There were differences between symptomatic (72.0%, 95% CI 63.7% to 79.0%; 37 evaluations; 15530 samples, 4410 cases) and asymptomatic participants (58.1%, 95% CI 40.2% to 74.1%; 12 evaluations; 1581 samples, 295 cases). Average sensitivity was higher in the first week after symptom onset (78.3%, 95% CI 71.1% to 84.1%; 26 evaluations; 5769 samples, 2320 cases) than in the second week of symptoms (51.0%, 95% CI 40.8% to 61.0%; 22 evaluations; 935 samples, 692 cases). Sensitivity was high in those with cycle threshold (Ct) values on PCR ≤25 (94.5%, 95% CI 91.0% to 96.7%; 36 evaluations; 2613 cases) compared to those with Ct values >25 (40.7%, 95% CI 31.8% to 50.3%; 36 evaluations; 2632 cases). Sensitivity varied between brands. Using data from instructions for use (IFU) compliant evaluations in symptomatic participants, summary sensitivities ranged from 34.1% (95% CI 29.7% to 38.8%; Coris Bioconcept) to 88.1% (95% CI 84.2% to 91.1%; SD Biosensor STANDARD Q). Average specificities were high in symptomatic and asymptomatic participants, and for most brands (overall summary specificity 99.6%, 95% CI 99.0% to 99.8%).
At 5% prevalence using data for the most sensitive assays in symptomatic people (SD Biosensor STANDARD Q and Abbott Panbio), positive predictive values (PPVs) of 84% to 90% mean that between 1 in 10 and 1 in 6 positive results will be a false positive, and between 1 in 4 and 1 in 8 cases will be missed. At 0.5% prevalence applying the same tests in asymptomatic people would result in PPVs of 11% to 28% meaning that between 7 in 10 and 9 in 10 positive results will be false positives, and between 1 in 2 and 1 in 3 cases will be missed.
No studies assessed the accuracy of repeated lateral flow testing or self‐testing.
Rapid molecular assays
Thirty studies reported 33 evaluations of five different rapid molecular tests. Sensitivities varied according to test brand. Most of the data relate to the ID NOW and Xpert Xpress assays. Using data from evaluations following the manufacturer’s instructions for use, the average sensitivity of ID NOW was 73.0% (95% CI 66.8% to 78.4%) and average specificity 99.7% (95% CI 98.7% to 99.9%; 4 evaluations; 812 samples, 222 cases). For Xpert Xpress, the average sensitivity was 100% (95% CI 88.1% to 100%) and average specificity 97.2% (95% CI 89.4% to 99.3%; 2 evaluations; 100 samples, 29 cases). Insufficient data were available to investigate the effect of symptom status or time after symptom onset.
Authors' conclusions
Antigen tests vary in sensitivity. In people with signs and symptoms of COVID‐19, sensitivities are highest in the first week of illness when viral loads are higher. The assays shown to meet appropriate criteria, such as WHO's priority target product profiles for COVID‐19 diagnostics (‘acceptable’ sensitivity ≥ 80% and specificity ≥ 97%), can be considered as a replacement for laboratory‐based RT‐PCR when immediate decisions about patient care must be made, or where RT‐PCR cannot be delivered in a timely manner. Positive predictive values suggest that confirmatory testing of those with positive results may be considered in low prevalence settings. Due to the variable sensitivity of antigen tests, people who test negative may still be infected.
Evidence for testing in asymptomatic cohorts was limited. Test accuracy studies cannot adequately assess the ability of antigen tests to differentiate those who are infectious and require isolation from those who pose no risk, as there is no reference standard for infectiousness. A small number of molecular tests showed high accuracy and may be suitable alternatives to RT‐PCR. However, further evaluations of the tests in settings as they are intended to be used are required to fully establish performance in practice.
Several important studies in asymptomatic individuals have been reported since the close of our search and will be incorporated at the next update of this review. Comparative studies of antigen tests in their intended use settings and according to test operator (including self‐testing) are required.
Plain language summary
How accurate are rapid tests for diagnosing COVID‐19?
What are rapid point‐of‐care tests for COVID‐19?
Rapid point‐of‐care tests aim to confirm or rule out COVID‐19 infection in people with or without COVID‐19 symptoms. They:
‐ are portable, so they can be used wherever the patient is (at the point of care);
‐ are easy to perform, with a minimum amount of extra equipment or complicated preparation steps;
‐ are less expensive than standard laboratory tests;
‐ do not require a specialist operator or setting; and
‐ provide results ‘while you wait’.
We were interested in two types of commercially available, rapid point‐of‐care tests: antigen and molecular tests. Antigen tests identify proteins on the virus; they come in disposable plastic cassettes, similar to pregnancy tests. Rapid molecular tests detect the virus’s genetic material in a similar way to laboratory methods, but using smaller devices that are easy to transport or to set up outside of a specialist laboratory. Both test nose or throat samples.
Why is this question important?
People with suspected COVID‐19 need to know quickly whether they are infected, so that they can self‐isolate, receive treatment, and inform close contacts. Currently, COVID‐19 infection is confirmed by a laboratory test called RT‐PCR, which uses specialist equipment and often takes at least 24 hours to produce a result.
Rapid point‐of‐care tests could open access to testing for many more people, with and without symptoms, potentially in locations other than healthcare settings. If they are accurate, faster diagnosis could allow people to take appropriate action more quickly, with the potential to reduce the spread of COVID‐19.
What did we want to find out?
We wanted to know whether commercially available, rapid point‐of‐care antigen and molecular tests are accurate enough to diagnose COVID‐19 infection reliably, and to find out if accuracy differs in people with and without symptoms.
What did we do?
We looked for studies that measured the accuracy of any commercially produced, rapid antigen or molecular point‐of‐care test, in people tested for COVID‐19 using RT‐PCR. People could be tested in hospital or the community. Studies could test people with or without symptoms.
Tests had to use minimal equipment, be performed safely without risking infection from the sample, and have results available within two hours of the sample being collected.
What we found
We included 64 studies in the review. They investigated a total of 24,087 nose or throat samples; COVID‐19 was confirmed in 7415 of these samples. Studies investigated 16 different antigen tests and five different molecular tests. They took place mainly in Europe and North America.
Main results
Antigen tests
In people with confirmed COVID‐19, antigen tests correctly identified COVID‐19 infection in an average of 72% of people with symptoms, compared to 58% of people without symptoms. Tests were most accurate when used in the first week after symptoms first developed (an average of 78% of confirmed cases had positive antigen tests). This is likely to be because people have the most virus in their system in the first days after they are infected.
In people who did not have COVID‐19, antigen tests correctly ruled out infection in 99.5% of people with symptoms and 98.9% of people without symptoms.
Different brands of tests varied in accuracy. Pooled results for one test (SD Biosensor STANDARD Q) met World Health Organization (WHO) standards as ‘acceptable’ for confirming and ruling out COVID‐19 in people with signs and symptoms of COVID‐19. Two more tests met the WHO acceptable standards (Abbott Panbio and BIONOTE NowCheck) in at least one study.
Using summary results for SD Biosensor STANDARD Q, if 1000 people with symptoms had the antigen test, and 50 (5%) of them really had COVID‐19:
‐ 53 people would test positive for COVID‐19. Of these, 9 people (17%) would not have COVID‐19 (false positive result).
‐ 947 people would test negative for COVID‐19. Of these, 6 people (0.6%) would actually have COVID‐19 (false negative result).
In people with no symptoms of COVID‐19 the number of confirmed cases is expected to be much lower than in people with symptoms. Using summary results for SD Biosensor STANDARD Q in a bigger population of 10,000 people with no symptoms, where 50 (0.5%) of them really had COVID‐19:
‐ 125 people would test positive for COVID‐19. Of these, 90 people (72%) would not have COVID‐19 (false positive result).
‐ 9,875 people would test negative for COVID‐19. Of these, 15 people (0.2%) would actually have COVID‐19 (false negative result).
Molecular tests
Although overall results for diagnosing and ruling out COVID‐19 were good (95.1% of infections correctly diagnosed and 99% correctly ruled out), 69% of the studies used the tests in laboratories instead of at the point‐of‐care and few studies followed test manufacturer instructions. Most of the data relate to the ID NOW and Xpert Xpress tests. We noted a large difference in COVID‐19 detection between the two tests, but we cannot be certain about whether results will remain the same in a real world setting. We could not investigate differences in people with or without symptoms, nor time from when symptoms first showed because the studies did not provide enough information about their participants.
How reliable were the results of the studies?
In general, studies that assessed antigen tests used more rigorous methods than those that assessed molecular tests, particularly when selecting participants and performing the tests. Sometimes studies did not perform the test on the people for whom it was intended and did not follow the manufacturers’ instructions for using the test. Sometimes the tests were not carried out at the point‐of‐care. Nearly all the studies (97%) relied on a single negative RT‐PCR result as evidence of no COVID‐19 infection. Results from different test brands varied, and few studies directly compared one test brand with another. Finally, not all studies gave enough information about their participants for us to judge how long they had had symptoms, or even whether or not they had symptoms.
What does this mean?
Some antigen tests are accurate enough to replace RT‐PCR when used in people with symptoms. This would be most useful when quick decisions are needed about patient care, or if RT‐PCR is not available. Antigen tests may be most useful to identify outbreaks, or to select people with symptoms for further testing with PCR, allowing self‐isolation or contact tracing and reducing the burden on laboratory services. People who receive a negative antigen test result may still be infected.
Several point‐of‐care molecular tests show very high accuracy and potential for use, but more evidence of their performance when evaluated in real life settings is required.
We need more evidence on rapid testing in people without symptoms, on the accuracy of repeated testing, testing in non‐healthcare settings such as schools (including self‐testing), and direct comparisons of test brands, with testers following manufacturers’ instructions.
How up‐to‐date is this review?
This review updates our previous review and includes evidence published up to 30 September 2020.
Summary of findings
Summary of findings 1. Diagnostic accuracy of point‐of‐care antigen and molecular‐based tests for the diagnosis of SARS‐CoV‐2 infection.
| Question | What is the diagnostic accuracy of rapid point‐of‐care antigen and molecular‐based tests for the diagnosis of SARS‐CoV‐2 infection? | ||||||
| Population | Adults or children with suspected:
or populations undergoing screening for SARS‐CoV‐2 infection, including
|
||||||
| Index test | Any rapid antigen or molecular‐based test for diagnosis of SARS‐CoV‐2 meeting the following criteria:
|
||||||
| Target condition | Detection of current SARS‐CoV‐2 infection | ||||||
| Reference standard | For COVID‐19 cases: positive RT‐PCR alone or clinical diagnosis of COVID‐19 based on established guidelines or combinations of clinical features For non‐COVID‐19 cases: negative RT‐PCR or pre‐pandemic sources of samples |
||||||
| Action |
False negative results mean missed cases of COVID‐19 infection, with either delayed or no confirmed diagnosis and increased risk of community transmission due to false sense of security False positive results lead to unnecessary self‐isolation or quarantine, with the potential for new infection to be acquired |
||||||
| Quantity of evidence | Sample type | Number studies | Total samples | Samples from confirmed SARS‐CoV‐2 cases | |||
| Respiratory | 77 | 24,418 | 7484 | ||||
| Non‐respiratory | 1 | 79 | 29 | ||||
| Limitations in the evidence | |||||||
|
Risk of bias (based on 78 studies) |
Participants: high (29) or unclear (27) risk in 56 studies (72%) Index test (antigen tests): high (0) or unclear (19) risk in 19 studies (40% of 48 studies) Index test (molecular tests): high (3) or unclear (22) risk in 25 studies (83% of 30 studies) Reference standard: high (66) unclear (6) risk in 72 studies (92%) Flow and timing: high (29) or unclear (36) risk in 65 studies (83%) |
||||||
|
Concerns about applicability (based on 78 studies) |
Participants: high concerns in 35 studies (45%) Index test (antigen tests): high concerns in 23 studies (48% of 48 studies) Index test (molecular tests): high concerns in 16 studies (53% of 30 studies) Reference standard: high concerns in 76 studies (97%) |
||||||
| Findings: antigen tests | |||||||
| Evaluations (studies) | Samples (SARS‐CoV‐2 cases) |
Sensitivity (95% CI) [Range] |
Specificity (95% CI) [Range] |
||||
| Symptomatic | 37 (27) | 15,530 (4410) | 72.0 (63.7 to 79.0) [0% to 100%] |
99.5 (98.5 to 99.8) [8% to 100%] |
|||
| Symptomatic (up to 7 days from onset of symptoms)a | 26 (21) | 2320 (2320) | 78.3 (71.1 to 84.1) [15% to 95%] |
‐ | |||
| Asymptomatic | 12 (10) | 1581 (295) | 58.1 (40.2 to 74.1) [29% to 85%] |
98.9 (93.6 to 99.8) [14% to 100%] |
|||
| Examples of pooled results for individual antigen tests using data for evaluations compliant with manufacturer instructions for use according to symptom status | |||||||
| Tests | Evaluations | Samples |
SARS‐CoV‐2 cases |
Sensitivity (95% CI) | Specificity (95% CI) | ||
| Symptomatic participants | |||||||
| Coris Bioconcept ‐ COVID‐19 Ag Respi‐Strip | 3 | 780 | 414 | 34.1 (29.7 to 38.8) | 100 (99.0 to 100) | ||
| Abbott ‐ Panbio Covid‐19 Ag | 3 | 1094 | 252 | 75.1 (57.3 to 87.1) | 99.5 (98.7 to 99.8) | ||
| SD Biosensor ‐ STANDARD Q COVID‐19 Ag | 3 | 1947 | 336 | 88.1 (84.2 to 91.1) | 99.1 (97.8 to 99.6) | ||
| Asymptomatic participants | |||||||
| Coris Bioconcept ‐ COVID‐19 Ag Respi‐Strip | 2 | 45 | 14 | 28.6 (8.4 to 58.1) | 100 (88.8 to 100) | ||
| Abbott ‐ Panbio Covid‐19 Ag | 1 | 474 | 47 | 48.9 (35.1 to 62.9) | 98.1 (96.3 to 99.1) | ||
| SD Biosensor ‐ STANDARD Q COVID‐19 Ag | 1 | 127 | 13 | 69.2 (38.6 to 90.9) | 99.1 (95.2 to 100) | ||
| Symptomatic participants: average sensitivity and specificity (and 95% CIs) applied to a hypothetical cohort of 1000 patients where 50, 100 and 200 have COVID‐19 infection | |||||||
| Test | Prevalence | TP (95% CI) | FP (95% CI) | FN (95% CI) | TN (95% CI) | PPV | 1 – NPV |
| Coris Bioconcept | 5% | 17 (15 to 19) | 0 (0 to 10) | 33 (31 to 35) | 950 (941 to 950) | 100% | 3.4% |
| 10% | 34 (30 to 39) | 0 (0 to 9) | 66 (61 to 70) | 900 (891 to 900) | 100% | 6.8% | |
| 20% | 68 (59 to 78) | 0 (0 to 8) | 132 (122 to 141) | 800 (792 to 800) | 100% | 14.1% | |
| Abbott ‐ Panbio Covid‐19 Ag | 5% | 38 (29 to 44) | 5 (2 to 12) | 12 (6 to 21) | 945 (938 to 948) | 89% | 1.3% |
| 10% | 75 (57 to 87) | 5 (2 to 12) | 25 (13 to 43) | 896 (888 to 898) | 94% | 2.7% | |
| 20% | 150 (115 to 174) | 4 (2 to 10) | 50 (26 to 85) | 796 (790 to 798) | 97% | 5.9% | |
| SD Biosensor ‐ STANDARD Q COVID‐19 Ag | 5% | 44 (42 to 46) | 9 (4 to 21) | 6 (4 to 8) | 941 (929 to 946) | 84% | 0.6% |
| 10% | 88 (84 to 91) | 8 (4 to 20) | 12 (9 to 16) | 892 (880 to 896) | 92% | 1.3% | |
| 20% | 176 (168 to 182) | 7 (3 to 18) | 24 (18 to 32) | 793 (782 to 797) | 96% | 2.9% | |
| Asymptomatic participants: average sensitivity and specificity (and 95% CIs) applied to a hypothetical cohort of 10,000 patients where 50, 100 and 200 have COVID‐19 infection | |||||||
| Coris Bioconcept | 0.5% | 14 (4 to 29) | 0 (0 to 1114) | 36 (21 to 46) | 9950 (8836 to 9950) | 100% | 0.4% |
| 1% | 29 (8 to 58) | 0 (0 to 1109) | 71 (42 to 92) | 9900 (8791 to 9900) | 100% | 0.7% | |
| 2% | 57 (17 to 116) | 0 (0 to 1098) | 143 (84 to 183) | 9800 (8702 to 9800) | 100% | 1.4% | |
| Abbott ‐ Panbio Covid‐19 Ag | 0.5% | 24 (18 to 31) | 189 (90 to 368) | 26 (19 to 32) | 9761 (9582 to 9860) | 11% | 0.3% |
| 1% | 49 (35 to 63) | 188 (89 to 366) | 51 (37 to 65) | 9712 (9534 to 9811) | 21% | 0.5% | |
| 2% | 98 (70 to 126) | 186 (88 to 363) | 102 (74 to 130) | 9614 (9437 to 9712) | 34% | 1.0% | |
| SD Biosensor ‐ STANDARD Q COVID‐19 Ag | 0.5% | 35 (19 to 45) | 90 (0 to 478) | 15 (5 to 31) | 9860 (9472 to 9950) | 28% | 0.2% |
| 1% | 69 (39 to 91) | 89 (0 to 475) | 31 (9 to 61) | 9811 (9425 to 9900) | 44% | 0.3% | |
| 2% | 138 (77 to 182) | 88 (0 to 470) | 62 (18 to 123) | 9712 (9330 to 9800) | 61% | 0.6% | |
| Findings: rapid molecular tests | |||||||
| Evaluations (studies) | Samples | SARS‐CoV‐2 cases |
Average sensitivity (95% CI) [Range] |
Average specificity (95% CI) [Range] |
|||
| 29 (26) | 4351 | 1787 | 95.1 (90.5 to 97.6) [57% to 100%] |
98.8 (98.3 to 99.2) [ 92% to 100%] |
|||
| Pooled results for individual tests using data from compliant with manufacturer instructions for use | |||||||
| Tests | Evaluations | Samples |
SARS‐CoV‐2 cases |
Sensitivity (95% CI) | Specificity (95% CI) | ||
| Abbott ‐ ID NOW | 4 | 812 | 222 | 73.0 (66.8 to 78.4) | 99.7 (98.7 to 99.9) | ||
| Cepheid ‐ Xpert Xpress | 2 | 100 | 29 | 100 (88.1 to 100) | 97.2 (89.4 to 99.3) | ||
| DRW ‐ SAMBA II | 1 | 149 | 33 | 87.9 (71.8 to 96.6) | 97.4 (92.6 to 99.5) | ||
| DNANudge COVID Nudge | 1 | 386 | 71 | 94.4 (86.2 to 98.4) | 100 (98.8 to 100) | ||
| Average sensitivity and specificity (and 95% CIs) applied to a hypothetical cohort of 1000 patients where 50, 100 and 200 have COVID‐19 infection | |||||||
| Tests | Prevalence | TP (95% CI) | FP (95% CI) | FN (95% CI) | TN (95% CI) | PPVb | 1 – NPVc |
| ID NOW | 5% | 37 (33 to 39) | 3 (1 to 12) | 14 (11 to 17) | 947 (938 to 949) | 93% | 1.4% |
| 10% | 73 (67 to 78) | 3 (1 to 12) | 27 (22 to 33) | 897 (888 to 899) | 96% | 2.9% | |
| 20% | 146 (134 to 157) | 2 (1 to 10) | 54 (43 to 66) | 798 (790 to 799) | 98% | 6.3% | |
| Xpert Xpress | 5% | 50 (44 to 50) | 27 (7 to 101) | 0 (0 to 6) | 923 (849 to 943) | 65% | 0.0% |
| 10% | 100 (88 to 100) | 25 (6 to 95) | 0 (0 to 12) | 875 (805 to 894) | 80% | 0.0% | |
| 20% | 200 (176 to 200) | 22 (6 to 85) | 0 (0 to 24) | 778 (715 to 794) | 90% | 0.0% | |
| SAMBA II | 5% | 44 (36 to 48) | 25 (5 to 70) | 6 (2 to 14) | 925 (880 to 945) | 64% | 0.6% |
| 10% | 88 (72 to 97) | 23 (5 to 67) | 12 (3 to 28) | 877 (833 to 896) | 79% | 1.4% | |
| 20% | 176 (144 to 193) | 21 (4 to 59) | 24 (7 to 56) | 779 (741 to 796) | 89% | 3.0% | |
| COVID Nudge | 5% | 47 (43 to 49) | 0 (0 to 11) | 3 (1 to 7) | 950 (939 to 950) | 100% | 0.3% |
| 10% | 94 (86 to 98) | 0 (0 to 11) | 6 (2 to 14) | 900 (889 to 900) | 100% | 0.6% | |
| 20% | 189 (172 to 197) | 0 (0 to 10) | 11 (3 to 28) | 800 (790 to 800) | 100% | 1.4% | |
| 1 – NPV: 1 – negative predictive value (the percentage of people with negative results who are infected); Ag: antigen;CI: confidence interval; FN: false negative; FP: false positive;IFU: [manufacturers'] instructions for use; PPV: positive predictive value (the percentage of people with positive results who are infected); RT‐PCR: reverse transcription polymerase chain reaction; TN: true negative; TP: true positive | |||||||
aSpecificity only estimated in 8 of 26 evaluations by time after symptom onset. bPPV (positive predictive value) defined as the percentage of positive rapid test results that are truly positive according to the reference standard diagnosis. c1‐NPV (negative predictive value), where NPV is defined as the percentage of negative rapid test results that are truly negative according to the reference standard diagnosis.
Background
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) and the resulting COVID‐19 pandemic present important diagnostic evaluation challenges. These range from: understanding the value of signs and symptoms in predicting possible infection; assessing whether existing biochemical and imaging tests can identify infection or people needing critical care; and evaluating whether in vitro diagnostic tests can accurately identify and rule out current SARS‐CoV‐2 infection, and identify those with past infection, with or without immunity.
We are creating and maintaining a suite of living systematic reviews to cover the roles of tests and patient characteristics in the diagnosis of COVID‐19. This review is the first update of a review summarising evidence of the accuracy of rapid antigen and molecular tests that are suitable for use at the point of care. In some scenarios the tests could potentially be used as alternatives to standard laboratory‐based molecular assays, such as reverse transcription polymerase chain reaction (RT‐PCR) assays, that are relied on for identifying current infection, in others they may be used where no testing is currently done. If sufficiently accurate, point‐of‐care tests have the potential to greatly expand access and speed of testing, In turn, if accurate, they may have greater impact on public health than laboratory‐based molecular methods as they are less expensive, provide results more quickly and do not require the same technical expertise and laboratory capacity. These tests can be undertaken locally, avoiding the need for centralised testing facilities that rarely meet the needs of patients, caregivers, health workers and society as a whole, especially in low‐ and middle‐income countries. As these are rapid tests, their results can be returned within the same clinical encounter, facilitating timely decisions concerning the need for isolation and contract tracing activities.
Target condition being diagnosed
COVID‐19 is the disease caused by infection with the SARS‐CoV‐2 virus. The key target conditions for this suite of reviews are current SARS‐CoV‐2 infection, current COVID‐19 disease, and past SARS‐CoV‐2 infection. The tests included in this review concern the identification of current infection, as defined by reference standard methods of diagnosis, including molecular assays such as RT‐PCR, or internationally recognised clinical guidelines for diagnosis of SARS‐CoV‐2. In the context of test evaluation, and throughout this review, we use the term 'reference standard' to denote the best available method (test or tests) for diagnosing the target condition, as opposed to other uses of the term in diagnostic virology (such as reference methods or reference materials).
For current infection, the severity of the disease is of ultimate importance for patient outcomes. However, rapid testing does not establish severity of disease, and for this review we consider the role of point‐of‐care tests for detecting SARS‐CoV‐2 infection of any severity, distinguishing only between symptomatic and asymptomatic infection.
COVID‐19 public health interventions focus on reducing disease transmission, thus it is important to identify and isolate people who are infected before or whilst they are infectious. It is reasonably presumed that people with symptoms who meet national criteria for COVID‐19 testing, or who are identified through contact tracing, have a high enough risk of being infectious to ask them to isolate. However, assessing the risk of an individual being infectious in asymptomatic screening is more difficult, as there is no reference standard test for being ‘infectious’. Using RT‐PCR status as a reference standard (as is done for target condition of ‘infection’) will ensure that infectious people are not missed, but as RT‐PCR continues to detect viral RNA days and weeks after the onset of infection will wrongly classify some people as infectious. Alternative reference standards that have been proposed for infectiousness include assessing the viability of the virus using viral culture, or using a value of the cycle threshold (Ct value) from RT‐PCR results to group individuals above or below a particular value (as a proxy for viral load) as more or less likely to be infectious. Converting Ct values (also known as quantification cycle (Cq) or crossing point (Cp) values) into direct quantitative values of viral load (viral copies per cell) is possible but challenging, as the relationship between Ct values and viral load varies between machines and laboratories. Thus comparison at fixed Ct values is unlikely to be comparable across studies. Viral culture is unsuitable as a reference standard because it is technically complex and often unreliable, which leads to it being an insensitive test (the failure to culture virus potentially being a result of the culture technique and not an indicator of non‐infectiousness). The suitability of RT‐PCR is limited as the inverse relationship between viral load (Ct value) and risk of infection is a continuum of risk without there being a meaningful cut‐point (with virus being cultured from samples with Ct values as high as 35 (Singanayagam 2020)). Similarly, those with low viral loads at the onset of infection will be missed. A preferable alternative, of tracking contacts for evidence of secondary infections, requires longitudinal follow‐up and is better considered as a question about risk of transmission, which can be addressed using predictive modelling approaches (taking into account host, agent and environmental factors). This is in contrast to the diagnostic test accuracy paradigm which can only determine if individuals are infected at a single point in time.
For these reasons, this review only focuses on the target condition of 'infection' for both symptomatic and asymptomatic applications of tests. We do report results where they are presented split by an RT‐PCR Ct value to report on accuracy according to groups with higher and lower viral load, but advise caution on their interpretation considering the lack of standardisation of PCR Ct values. Given the current state of the scientific knowledge we do not consider it appropriate to consider these as groups which are defined as 'infectious' and 'not infectious'.
RT‐PCR carries a very small risk of false positive results for infection and a higher risk of false negative results. False positive results may result from failures in sampling or laboratory protocols (e.g. mislabelling), contamination during sampling or processing, or low‐level reactions during PCR (Healy 2020; Mayers 2020). At times when SARS‐CoV‐2 infections have been rare, population prevalence surveys using RT‐PCR have shown test positivity rates of 0.44% (95% credible interval: 0.22% to 0.76%) (August 2020; ONS 2020), and 0.077% (0.065%, 0.092%) (June to July 2020; Riley 2020 React‐1 study). These values can be used to place an upper bound on the possible false positive rate of RT‐PCR of less than 0.077% (as the total numbers testing positive will comprise both true positive and false positive RT‐PCR results). The World Health Organization (WHO) recently issued a notice of concern regarding interpretation of specimens at or near the limit for PCR positivity (i.e. those with high cycle threshold (Ct) values), citing potential difficulties in distinguishing the presence of the target virus from these types of background ‘noise’ (WHO 2020a). False negative rates have been estimated by looking at individuals with symptoms who initially test negative, but positive on a subsequent test. These rates have been estimated to be as high as 20% to 30% in the first week of symptom onset; Arevalo‐Rodriguez 2020; Yang 2020a; Zhao 2020; Kucirka 2020). Including probable COVID‐19 cases within the target condition, as defined by internationally recognised clinical guidelines for diagnosis of SARS‐CoV‐2 will partially mitigate these missed cases.
Index test(s)
The primary consideration for the eligibility of tests for inclusion in this review is that they should detect current infection and should have the capacity to be performed at the ‘point of care’ or in a ‘near‐patient’ testing role. There is an ongoing debate around the specific use and definitions of these terms, therefore for the purposes of this review, we consider ‘point‐of‐care’ and ‘near patient’ to be synonymous, but for consistency and avoidance of confusion, we use the term ‘point‐of‐care’ throughout.
We have adapted a definition of point‐of‐care testing, namely that it “refers to decentralized testing that is performed by a minimally trained healthcare professional near a patient and outside of central laboratory testing” (WHO 2018), with the additional caveat that test results must be available within a single clinical encounter (Pai 2012). Our criteria for defining a point‐of‐care test are therefore:
the equipment for running and or reading the assay must be portable or easily transported, although mains power may be required;
minimal sample preparation requirements, for example, single‐step mixing, with no requirement for additional equipment or precise sample volume transfer unless a disposable automatic fill or graduated transfer device is used;
minimal biosafety requirements, for example, personal protective equipment (PPE) for sample collector and test operator, good ventilation and a biohazard bag for waste disposal;
no requirement for a temperature‐controlled environment; and
test results available within two hours of sample collection.
Tests for detection of current infection that are currently suitable for use at the point of care include antigen tests and molecular‐based tests. Both types of test use the same respiratory‐tract samples acquired by swabbing, washing or aspiration as for laboratory‐based RT‐PCR. Rapid antigen tests use lateral flow immunoassays, which are disposable devices, usually in the form of plastic cassettes akin to a pregnancy test. Viral antigen is captured by dedicated antibodies that are either colloidal gold‐ or fluorescent‐labelled. Antigen detection is indicated by visible lines appearing on the test strip (colloidal gold‐based immunoassays, or CGIA), or through fluorescence, which can be detected using an immunofluorescence analyser (fluorescence immunoassays or FIA). Molecular‐based tests to detect viral ribonucleic acid (RNA) have historically been laboratory‐based assays using RT‐PCR technology (see Alternative test(s)). In recent years, automated, single‐step RT‐PCR methods have been developed, as well as other nucleic acid amplification methods, such as isothermal amplification, that do not require the sophisticated thermo cycling involved in RT‐PCR (Green 2020). These technological advances have allowed molecular technologies to be developed that are suitable for use in a point‐of‐care context (Kozel 2017), however they still require small portable machines and many take longer to produce results than antigen tests.
Following the emergence of COVID‐19 there has been prolific industry activity to develop accurate tests. The Foundation for Innovative Diagnostics (FIND) and Johns Hopkins Centre for Health Security have maintained online lists of available tests for SARS‐CoV‐2 (FIND 2020). At the time of writing (5 January 2021), FIND listed 129 rapid antigen tests, 118 of which are described as "commercialised" and 92 have been identified as having regulatory approval. These numbers are a substantial increase on the 48 listed, 32 commercialised and 21 with regulatory approval at the time of our original review (19 July 2020). A total of 142 molecular tests were described as automated, including both laboratory‐based assays and assays suitable for use outside of a laboratory setting (i.e. near or at the point of care). Further information from FIND indicates that 53 of the 142 assays were categorised as point‐of‐care or near point‐of‐care tests, including 43 with regulatory approval. This classification was based on the information provided to FIND by the test manufacturers and does not necessarily mean that these tests meet the criteria for point‐of‐care tests that we have specified for this review. The numbers of tests of these types will continue to increase over time.
Given the urgent need to identify the evidence base for tests that are available for purchase, the focus of this first update of the review is on tests that are commercially produced. All commercially produced assays are supplied with a specific product code, product inserts or instructions for use (IFU) sheets that document the intended use of the test; sample storage and preparation and testing procedures; who should deliver the test and in whom; and any restrictions around the type of samples that can be used.
There are many proposals for serial testing with lateral flow tests to detect infection, rather than a single use. In this case it would be appropriate to evaluate the accuracy of the strategy rather than a single test.
Clinical pathway
Patients may be tested for SARS‐CoV‐2 when they present with symptoms, have had known exposure to a confirmed case, or in a screening context, with no known exposure to SARS‐CoV‐2. The standard approach to diagnosis of SARS‐CoV‐2 infection is through laboratory‐based testing of swab samples taken from the upper respiratory (e.g. nasopharynx, oropharynx) or lower respiratory tract (e.g. bronchoalveolar lavage or sputum) with RT‐PCR. RT‐PCR is the primary method for detecting infection during the acute phase of the illness while the virus is still present. Both the WHO and the China CDC (National Health Commission of the People's Republic of China), have produced case definitions for COVID‐19 that include the presence of convincing clinical evidence (some including positive serology tests) when RT‐PCR is negative (Appendix 1).
Prior test(s)
Signs and symptoms are used in the initial diagnosis of suspected SARS‐CoV‐2 infection and to help identify those requiring tests. A number of key symptoms have been suggested as indicators of mild to moderate COVID‐19, including: cough, fever greater than 37.8 °C, headache, breathlessness, muscle pain, fatigue, and loss of sense of smell and taste (Struyf 2021). However, the recently published review of signs and symptoms found good evidence for the accuracy for these symptoms alone or in combination to be lacking (Struyf 2021).
Where people are asymptomatic but are being tested as part of screening (e.g. universal testing of students as part of a risk‐reduction effort) or on the basis of epidemiological risk factors, such as exposure to someone with confirmed SARS‐CoV‐2 or following travel to more highly endemic countries, no prior tests will have been conducted.
Role of index test(s)
For most settings in which testing for acute SARS‐CoV‐2 infection in symptomatic individuals takes place, results of molecular laboratory‐based RT‐PCR tests are unlikely to be available within a single clinical encounter. Point‐of‐care tests potentially have a role either as a replacement for RT‐PCR (if sufficiently accurate), or as a means of triaging and rapid management (quarantine or treatment, or both), with confirmatory RT‐PCR testing for those with negative rapid test results (CDC 2020; WHO 2020b). Obtaining quick results within a healthcare visit will allow faster decisions about isolation and healthcare interventions for those with positive test results, and allow contact tracing to begin in a more timely manner. Modelling studies suggest contact tracing is most effective if it starts within 24 hours of case detection, with delays in testing (e.g. due to laboratory turnaround time for reporting PCR results) leading to reductions in the proportion of onward transmissions per index case that can be prevented by track and trace (Kretzschmar 2020).
If sufficiently accurate, negative rapid test results in symptomatic patients could allow faster return to work or school, therefore conferring important economic and educational implications. Negative results also allow immediate consideration of other causes of symptoms, which may be time‐sensitive, for example bacterial pneumonia or thrombo‐embolism.
For asymptomatic individuals, if accurate, rapid tests may also be considered for screening at‐risk (exposed) populations, for example in hospital workers or in local outbreaks.
Rapid tests, particularly antigen tests which can be more easily delivered at scale, could also be used for mass screening purposes as recently piloted in Slovakia and in Liverpool UK (University of Liverpool 2020), or used in a more targeted fashion such as single test application at airports or for border entry, to allow entry to large public gatherings, or screening students as a risk‐reduction strategy (Ferguson 2020). Preliminary data on the rollout of such a policy in the UK has highlighted the many challenges in such an approach (Deeks 2020a; Nabavi 2021), and the requirement for full and proper field trial evaluations. Frequent repeated use of antigen tests in asymptomatic individuals with no known exposure to identify COVID‐19 cases has also been proposed (Larremore 2020), but field trial evaluations would be required to determine whether promising results from modelling studies can be borne out in practical settings (Crozier 2021).
Alternative test(s)
This review is one of seven that cover the range of tests and clinical characteristics being considered in the management of COVID‐19 (Deeks 2020b; McInnes 2020), five of which have already been published (Deeks 2020c; Salameh 2020; Stegeman 2020; Struyf 2021), including the first iteration of this review (Dinnes 2020). Full details of the alternative tests and evidence of their accuracy is summarised in these reviews. The SARS‐CoV‐2‐specific biomarker tests that might be considered as alternatives to point‐of‐care tests are considered here.
Laboratory‐based molecular tests
RT‐PCR tests for SARS‐CoV‐2 identify viral ribonucleic acid (RNA). Reagents for RT‐PCR were rapidly produced once the viral RNA sequence was published (Corman 2020). Testing is undertaken in central laboratories and can be very labour‐intensive, with several points along the path of performing a single test where errors may occur, although some automation of parts of the process is possible. The amplification process requires thermal cycling equipment to allow multiple temperature changes within a cycle, with cycles repeated up to 40 times until viral DNA is detected (Carter 2020). Although the amplification process for RT‐PCR can be completed in a relatively short timeframe, the stages of extraction, sample processing and data management (including reporting) mean that test results are typically only available in 24 to 48 hours. Where testing is undertaken in a centralised laboratory, transport times increase this further. The time to result for fully automated RT‐PCR assays is shorter than for manual RT‐PCR, however most assays still require sample preparation steps that make them unsuitable for use at the point of care. Other nucleic acid amplification methods, including loop‐mediated isothermal amplification (LAMP), or CRISPR‐based nucleic acid detection methods, that allow amplification at a constant temperature are now commercially available (Chen 2020). These methods have the potential to reduce the time to produce test results after extraction and sample processing to minutes, but the time for the whole process may still be significant. Laboratory‐based molecular tests are most often applied to upper and lower respiratory samples although they are also being used on faecal and urine samples.
Antibody tests
Serology tests to measure antibodies to SARS‐CoV‐2 have been evaluated in people with active infection and in convalescent cases (Deeks 2020c). Antibodies are formed by the body's immune system in response to infections, and can be detected in whole blood, plasma or serum. Antibody tests are available for laboratory use including enzyme‐linked immunosorbent assay (ELISA) methods, or more advanced chemiluminescence immunoassays (CLIA). There are also rapid lateral flow assays (LFA)s for antibody testing that use a minimal amount of whole blood, plasma or serum on a testing strip as opposed to the respiratory specimens that are used for rapid antigen tests; all assays for antibody detection are considered in Deeks 2020c.
Rationale
It is essential to understand the clinical accuracy of tests and clinical features to identify the best way they can be used in different settings to develop effective diagnostic and management pathways for SARS‐CoV‐2 infection and disease. The suite of Cochrane living systematic reviews summarises evidence on the clinical accuracy of different tests and diagnostic features. Estimates of accuracy from these reviews will help inform diagnosis, screening, isolation, and patient‐management decisions.
Summary of the previous version of the review
The first iteration of this review (Dinnes 2020), included 22 publications reporting on a total of 18 study cohorts with 3198 unique samples, 1775 of which had confirmed SARS‐CoV‐2 infection. We identified data for eight commercial tests (four antigen and four molecular) and one in‐house antigen test.
We did not find any studies at low risk of bias and had concerns about applicability of results across all studies. We judged patient selection to be at high risk of bias in 50% of the studies because of deliberate oversampling of samples with confirmed SARS‐CoV‐2 infection (sample enrichment) and unclear in 38% (7/18) because of poor reporting. Sixteen (89%) studies used only a single, negative RT‐PCR to confirm the absence of SARS‐CoV‐2 infection, risking missing infection. There was a lack of information on blinding of index test (n = 11), and about participant exclusions from analyses (n = 10). We did not observe differences in methodological quality between antigen and molecular test evaluations.
The eight evaluations of antigen tests reported considerable variation in sensitivity across studies (from 0% to 94%) with less variation in specificities (from 90% to 100%). The average sensitivity was 56.2% (95% CI 29.5 to 79.8%) and average specificity was 99.5% (95% CI 98.1% to 99.9%) (based on 943 samples, 596 with confirmed SARS‐CoV‐2). Data for individual antigen tests were limited with no more than two studies for any test.
We observed less variation in sensitivities across 13 evaluations of rapid molecular assays (range 68% to 100%) with similar variation in specificities (range 92% to 100%). Average sensitivity was 95.2% (95% CI 86.7% to 98.3%) and specificity 98.9% (95% CI 97.3% to 99.5%) based on a total of 2255 samples.
We were able to calculate pooled results for only two molecular tests: ID NOW (Abbott Laboratories; 5 evaluations) and Xpert Xpress (Cepheid Inc; 6 evaluations). Summary sensitivity for the Xpert Xpress assay (99.4%, 95% CI 98.0% to 99.8%) was 22.6 (95% CI 18.8 to 26.3) percentage points higher than that of ID NOW (76.8%, (95% CI 72.9% to 80.3%), whilst the specificity of Xpert Xpress (96.8%, 95% CI 90.6% to 99.0%) was marginally lower than ID NOW (99.6%, 95% CI 98.4% to 99.9%; a difference of −2.8 percentage points (95% CI from 6.4 percentage points lower to 0.8 higher).
Changes in the evidence base since the previous version
There has been a considerable increase in the number of evaluations available of antigen tests, and a lesser rise in the number of evaluations of molecular tests. More studies report key population features such as setting, and symptom status, and there has been an increase in direct swab testing as would occur in a point‐of‐care setting. However, due to the nature of sampling and the use of direct swab testing, few comparative studies are available. This review considers the available evidence in relevant population groups and settings according to test brand and compliance with manufacturer IFUs. We used the WHO's priority target product profiles for COVID‐19 diagnostics (i.e. acceptable performance criterion of sensitivity ≥ 80% and specificity ≥ 97%, or desirable criterion of ≥ 80% sensitivity and ≥ 99% specificity; WHO 2020c) as a benchmark against which to consider test performance.
We will update this review as often as is feasible to ensure that it provides current evidence about the accuracy of point‐of‐care tests.
This review follows a generic protocol that covers six of the seven Cochrane COVID‐19 diagnostic test accuracy reviews (Deeks 2020b). The Background and Methods sections of this review therefore use some text that was originally published in the protocol (Deeks 2020b), and text that overlaps some of our other reviews (Deeks 2020c; Struyf 2021).
Objectives
To assess the diagnostic accuracy of rapid point‐of‐care antigen and molecular‐based tests to determine if a person presenting in the community or in primary or secondary care has current SARS‐CoV‐2 infection, and to consider accuracy separately in symptomatic and asymptomatic population groups.
We estimated accuracy overall and separately according to symptom status (symptomatic and asymptomatic). Although we might expect to see differences in accuracy for testing of asymptomatic individuals with an epidemiological exposure to SARS‐CoV‐2 (targeted screening) compared to testing of asymptomatic individuals in a population screening setting, we did not anticipate finding sufficient numbers of studies for each testing application to allow any such difference to be explored. We will revisit this decision in subsequent iterations of this review.
Secondary objectives
Where data are available, we will investigate potential sources of heterogeneity that may influence diagnostic accuracy (either by stratified analysis or meta‐regression) according to test method and index test, participant or sample characteristics (duration of symptoms and viral load), study setting, study design and reference standard used.
We investigated adherence to manufacturers' IFUs in sensitivity analyses.
Methods
Criteria for considering studies for this review
Types of studies
We applied broad eligibility criteria to include all patient groups (that is, if patient population was unclear, we included the study) and all variations of a test.
We included studies of all designs that produce estimates of test accuracy or provide data from which we can compute estimates, including the following.
Studies restricted to participants confirmed to either have (or to have had) the target condition (to estimate sensitivity) or confirmed not to have (or have had) the target condition (to estimate specificity). These types of studies may be excluded in future review updates.
Single‐group studies, which recruit participants before disease status has been ascertained
Multi‐group studies, where people with and without the target condition are recruited separately (often referred to as two‐gate or diagnostic case‐control studies)
Studies based on either patients or samples
We excluded studies from which we could not extract data to compute either sensitivity or specificity.
We carefully considered the limitations of different study designs in the quality assessment and analyses.
We included studies reported in published journal papers, as preprints, and publicly available reports from independent bodies.
Participants
We included studies recruiting people presenting with suspicion of current SARS‐CoV‐2 infection or those recruiting populations where tests were used to screen for disease (for example, contact tracing or community screening).
We also included studies that recruited people known to have SARS‐CoV‐2 infection and known not to have SARS‐CoV‐2 infection (i.e. cases only or multi‐group studies).
We excluded small studies with fewer than 10 samples or participants. Although the size threshold of 10 is arbitrary, such small studies are likely to give unreliable estimates of sensitivity or specificity and may be biased.
Index tests
We included studies evaluating any rapid antigen or molecular‐based test for diagnosis of SARS‐CoV‐2, if it met the criteria outlined in the Background, that is:
requiring minimal equipment;
minimal sample preparation and biosafety considerations;
results available within two hours of sample collection; and
should be commercially produced (with test name and manufacturer or distributor documented).
All sample types (respiratory or non‐respiratory) were eligible. Strategies based on multiple applications of a test were also eligible for inclusion.
Target conditions
The target condition was current SARS‐CoV‐2 infection (either symptomatic or asymptomatic). We also refer to SARS‐CoV‐2 infection as ‘COVID‐19 infection’, particularly in the Plain Language Summary and Table 1.
Reference standards
We anticipated that studies would use a range of reference standards to define both the presence and absence of SARS‐CoV‐2 infection. For the QUADAS‐2 (Quality Assessment tool for Diagnostic Accuracy Studies; Whiting 2011), assessment we categorised each method of defining the presence of SARS‐CoV‐2 according to the risk of bias (the chances that it would misclassify the presence or absence of infection) and whether it defined COVID‐19 in an appropriate way that reflected cases encountered in practice. Likewise, we considered the risk of bias in definitions of the absence of SARS‐CoV‐2, and whether the definition captured all those who might be tested in practice.
Evaluations of molecular tests generally consider agreement between molecular assays, for example, agreement of a new rapid test against a more standard RT‐PCR test. For the purposes of this review, we considered RT‐PCR to be the ‘reference standard’ for SARS‐CoV‐2 infection, and present results as ‘sensitivity’ and ’specificity’ as opposed to percentage agreement. The result of further RT‐PCR analysis of discrepant cells (samples with results disagreeing on the rapid test and the RT‐PCR) were also considered in sensitivity analyses. As discrepant analysis involves retesting only a sub‐sample of patients selected according to index and reference standard results, it can introduce bias (Hadgu 1999). Retesting of all samples with a second test in a composite reference standard would be preferable when there are concerns over the accuracy of the first reference test.
Search methods for identification of studies
Electronic searches
We used two main sources for our electronic searches through 30 September 2020, which were devised with the help of an experienced Cochrane Information Specialist with diagnostic test accuracy review expertise (RSp). These searches aimed to identify all articles related to COVID‐19 and SARS‐CoV‐2 and were not restricted to those evaluating a particular type of test. Thus, the searches used no terms that specifically focused on an index test, diagnostic accuracy or study methodology.
Cochrane COVID‐19 Study Register searches
We used the Cochrane COVID‐19 Study Register (covid-19.cochrane.org/), for searches conducted from inception of the Register to 28 March 2020. At that time, the register was populated by searches of PubMed, as well as trials registers at US National Institutes of Health Ongoing Trials Register ClinicalTrials.gov (clinicaltrials.gov) and the WHO International Clinical Trials Registry Platform (apps.who.int/trialsearch).
Search strategies were designed for maximum sensitivity, to retrieve all human studies on COVID‐19 and with no language limits. See Appendix 2.
COVID‐19 Living Evidence Database from the University of Bern
From 28 March 2020, we used the COVID‐19 Living Evidence database from the Institute of Social and Preventive Medicine (ISPM) at the University of Bern (www.ispm.unibe.ch), as the primary source of records for the Cochrane COVID‐19 diagnostic test accuracy reviews. This search includes PubMed, Embase, and preprints indexed in bioRxiv and medRxiv databases. The strategies as described on the ISPM website are described here (ispmbern.github.io/covid-19/). See Appendix 3. To ensure comprehensive coverage we also downloaded records from the ‘Bern feed’ from 1 January to 28 March 2020 and de‐duplicated them against those obtained via the Cochrane COVID‐19 Study Register.
Due to the increased volume of published and preprint articles, from 25 May 2020 onwards we used artificial intelligence text analysis to conduct an initial classification of documents, based on their title and abstract information, for relevant and irrelevant documents (Appendix 4).
The decision to focus primarily on the Bern feed was because of the exceptionally large numbers of COVID‐19 studies available only as preprints. We are continuing to monitor the coverage of the Cochrane COVID‐19 Study Register and may move back to it as the primary source of records for subsequent review updates.
Other electronic sources
Prior to 28 March 2020 (when we began using the ‘Bern feed’), we identified Embase records through the Centers for Disease Control and Prevention (CDC), Stephen B Thacker CDC Library, COVID‐19 Research Articles Downloadable Database (cdc.gov/library/researchguides/2019novelcoronavirus/researcharticles.html), and de‐duplicated them against results from the Cochrane COVID‐19 Study Register. See Appendix 5.
We also checked our search results against two additional repositories of COVID‐19 publications up to 30 September 2020:
the Evidence for Policy and Practice Information and Co‐ordinating Centre (EPPI‐Centre) 'COVID‐19: Living map of the evidence' (eppi.ioe.ac.uk/COVID19_MAP/covid_map_v4.html);
the Norwegian Institute of Public Health 'NIPH systematic and living map on COVID‐19 evidence' (www.nornesk.no/forskningskart/NIPH_diagnosisMap.html)
Both repositories allow their contents to be filtered according to studies potentially relating to diagnosis, and both have agreed to provide us with updates of new diagnosis studies added.
Searching other resources
We have also contacted or accessed the websites of independent research groups undertaking test evaluations (for example, UK Public Health England (PHE), the Société Française Microbiologie (SFM), the Dutch National Institute for Public Health and the Environment (RIVM)) and studies co‐ordinated by FIND (finddx.org/covid-19/sarscov2-eval) and accessed the Diagnostics Global Health listing of manufacturer independent evaluations of antigen detecting rapid diagnostic tests (Ag‐RDTs) for SARS‐CoV‐2 (diagnosticsglobalhealth.org). We last accessed these additional resources on 16 November 2020.
We appeal to researchers to supply details of additional published or unpublished studies at the following email address, which we will consider for inclusion in future updates (coviddta@contacts.bham.ac.uk).
Data collection and analysis
Selection of studies
A team of experienced systematic review authors from the University of Birmingham screened the titles and abstracts of all records retrieved from the literature searches following the application of artificial intelligence text analysis (described in Electronic searches). Two review authors independently screened studies in Covidence. A third, senior review author resolved any disagreements. We tagged all records selected as potentially eligible according to the Cochrane COVID‐19 diagnostic test accuracy review(s) for which they might be eligible and we then exported them to separate Covidence reviews for each review title.
We obtained the full texts for all studies flagged as potentially eligible. Two review authors independently screened the full texts for one of the COVID‐19 biomarker reviews (molecular, antigen or antibody tests). We resolved any disagreements on study inclusion through discussion with a third review author.
Data extraction and management
One review author extracted the characteristics of each study, which a second review author checked. Items that we extracted are listed in Appendix 6.
Both review authors independently performed data extraction of 2x2 contingency tables of the number of true positives, false positives, false negatives and true negatives. They resolved disagreements by discussion. Where possible, we separately extracted data according to symptom status (symptomatic, asymptomatic, mixed symptom status or not reported), viral load (high or low, according to Ct cut‐offs defined within each study), and time post‐symptom onset (week one versus week two) and for molecular assays, before and after re‐analysis of samples in discrepant cells. For categorisation by symptom status, we classed studies reporting at least 75% of participants as symptomatic as ‘mainly symptomatic', we considered studies with less than 75% symptomatic participants to report ‘mixed’ groups along with those that reported recruiting both symptomatic and asymptomatic participants but did not provide the percentages in each group. We considered studies that provided no information as to the symptom status of included participants ‘not reported’. We also coded evaluations according to compliance with manufacturer IFUs. We based coding on three aspects of testing:
sample type (use of any sample not explicitly mentioned on the IFU scored 'No', otherwise scored 'Yes'),
provision of instructions for samples in viral transport medium ((VTM); only scored for evaluations using samples in VTM and only scored 'Yes' if specific instructions provided; scored 'Unclear' if VTM used and instructions for use of samples in VTM not documented in IFU); and
timing between sample collection and testing (scored 'Yes' only if all tests were carried out within specified time period, e.g. immediate on‐site testing, or for testing in laboratories if all tests reported to have been carried out within specified time period; scored 'Unclear' if time frame for testing was not reported and 'No' if any testing was carried out beyond the maximum stipulated timeframe).
We encourage study authors to contact us regarding missing details on the included studies (coviddta@contacts.bham.ac.uk).
Assessment of methodological quality
Two review authors independently assessed risk of bias and applicability concerns using the QUADAS‐2 checklist tailored to this review (Appendix 7; Whiting 2011). The two review authors resolved any disagreements by discussion.
Ideally, studies examining the use of tests in symptomatic people should prospectively recruit a representative sample of participants presenting with signs and symptoms of COVID‐19, either in community or primary care settings or to a hospital setting, and they should clearly record the time of testing after the onset of symptoms. Studies in asymptomatic people at risk of infection should document time from exposure. Studies applying tests in a screening setting should document eligibility criteria for screening, particularly if a targeted approach is used and should take care to record any previous confirmed or suspected SARS‐CoV‐2 infection or any relevant epidemiological exposures. Studies should perform tests in their intended use setting, using appropriate samples with or without viral transport medium and within the time period following specimen collection as indicated in the IFU document. Tests should be performed by relevant personnel (e.g. healthcare workers), and should be interpreted blinded to the final diagnosis (presence or absence of SARS‐CoV‐2). The reference standard diagnosis should be blinded to the result of the rapid test, and should not incorporate the result of the rapid test. If the reference standard includes clinical diagnosis of COVID‐19 for RT‐PCR‐negative patients, then established criteria should be used. Studies including samples from participants known not to have COVID‐19 should use pre‐pandemic sources or if contemporaneous samples then at least two RT‐PCR‐negative tests were required to confirm the absence of infection. Data should be reported for all study participants, including those where the result of the rapid test was inconclusive, or participants in whom the final diagnosis of COVID‐19 was uncertain. Studies should report whether results relate to participants (one sample per participant), or samples (multiple samples per participant).
Statistical analysis and data synthesis
We analysed rapid antigen and molecular tests separately. Studies often referred to ‘samples’ rather than ‘patients’, especially for the rapid molecular tests, however for many studies we do not suspect that inclusion of multiple samples per study participant was a significant issue. For consistency of terminology throughout the review, we refer to results on a per‐sample basis. If studies evaluated multiple tests in the same samples, we included them multiple times. We present estimates of sensitivity and specificity per study for each test brand using paired forest plots, and summarise results using average sensitivity and specificity in tables as appropriate. As heterogeneity is apparent in many analyses, these point estimates must be interpreted as the average of a distribution of values.
We did not make any formal comparisons between antigen assay brands because of the large number of different assays and small study numbers for many of them. We did however carry out a formal comparison (based on between‐study comparisons) for studies using two brands of molecular tests (ID NOW (Abbott Laboratories) and Xpert Xpress (Cepheid Inc)).
We estimated summary sensitivities and specificities with 95% confidence intervals (CI) using the bivariate model (Reitsma 2005), via the meqrlogit command of Stata/SE 16.0. When few studies were available, we simplified models by first assuming no correlation between sensitivity and specificity estimates and secondly by setting near‐zero variance estimates of the random effects to zero (Takwoingi 2017). In cases where there was only one study per test, we reported individual sensitivities and specificities with 95% CI constructed using the binomial exact method.
Where studies presented only estimates of sensitivity or of specificity, we fitted univariate, random‐effects, logistic regression models. In a number of instances where there was 100% sensitivity or specificity for all evaluations, we computed estimates and 95% CIs by summing the counts of TP, FP, FN and TN across 2x2 tables. These analyses are clearly marked in the tables. We present all estimates with 95% confidence intervals.
Investigations of heterogeneity
We examined heterogeneity between studies by visually inspecting the forest plots of sensitivity and specificity. Where adequate data were available, we investigated heterogeneity related to symptom status, time post‐symptom onset, viral load, test brand, and test method by including indicator variables in the random‐effects logistic regression models. Absolute differences between the sensitivity or specificity and the P values were reported from the model. In instances where only one study was available per test or when tests were being directly compared following summing of counts of the 2x2 tables, we performed test comparison using the two‐sample test of proportions. Few studies reported specificity estimates by time after symptom onset, therefore for this variable and for analyses by viral load, we considered only effects on sensitivity.
Sensitivity analyses
We performed four sensitivity analyses.
We estimated summary sensitivities and specificities according to test brand and symptom status using only studies that were compliant to the IFU.
We estimated sensitivity with and without studies that only evaluated samples with RT‐PCR‐confirmed SARS‐CoV‐2 (and thus did not estimate specificity).
We performed the same analysis for specificity in studies that only evaluated RT‐PCR‐negative control samples.
We made comparisons between analyses using the primary reference standard and analyses using results adjusted after retesting of samples with discrepant results with a second RT‐PCR test (discrepant analysis).
Assessment of reporting bias
We made no formal assessment of reporting bias but have indicated where we were aware that study results were available but unpublished.
Summary of findings
We summarised key findings in a 'Summary of findings' table indicating the strength of evidence for each test and findings, and highlighted important gaps in the evidence.
Updating
We are aware of additional studies published since the electronic searches were conducted on 30 September 2020 and plan to update this review. We have already conducted the next search to 1 January 2021.
Results
Results of the search
We screened 34,742 unique records (published or preprints) for inclusion in the complete suite of reviews to assist in the diagnosis of COVID‐19 (Deeks 2020b; McInnes 2020). Of 1749 records selected for further assessment for inclusion in any of the four molecular, antigen or antibody test reviews, we identified 199 full‐text reports requiring assessment for inclusion in this review; 90 for the first iteration of the review and 109 for this review update. See Figure 1 for the PRISMA flow diagram of search and eligibility results (McInnes 2018; Moher 2009).
1.
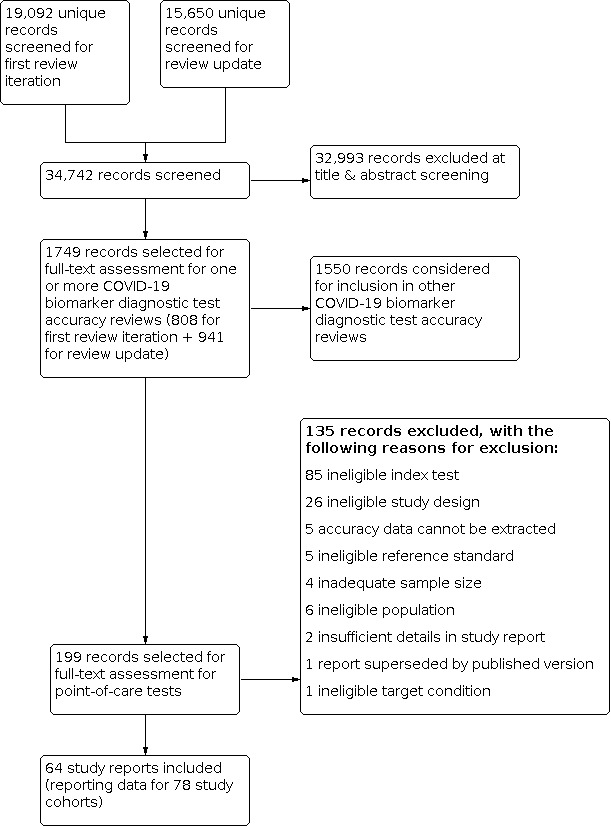
Study flow diagram
We included 64 reports in this review, and we excluded 135 publications that did not meet our inclusion criteria. Exclusions were mainly based on index test (n = 85) or ineligible study designs (n = 26), for example, designs that did not allow estimation of test accuracy. The reasons for exclusion of all 135 publications are provided in Characteristics of excluded studies. Appendix 8 provides a list of studies evaluating eligible tests but excluded for other reasons (n = 5), and studies evaluating technologies not yet suitable for use at the point of care (n = 41).
Of the 64 study reports, 18 were available only as preprints, 38 were published papers and eight were publicly available reports either from independent reference laboratories (one from Public Health England and two identified via the SMF) or were independent evaluations co‐ordinated by FIND (n = 5).
We contacted the authors of 10 study reports for further information (Blairon 2020; Courtellemont 2020; Diao 2020; Gibani 2020; Gremmels 2020(a); Linares 2020; Nash 2020; Porte 2020a; Schildgen 2020 [A]; Weitzel 2020 [A]), and received replies and the requested information with one exception (Linares 2020). We also contacted the evaluation teams at FIND and Public Health England and received additional information about study methods from FIND and some additional data from Public Health England.
The 64 included study reports relate to 78 separate studies. Please note when naming studies, we use the letters [A], [B], [C] etc. in square brackets to indicate data on different tests evaluated in the same study and (a), (b), (c) to indicate data from different participant cohorts from the same study report. For example, the five included reports from FIND correspond to eight ‘studies’ because three reports separately provided data from more than one evaluation centre.
Of the 78 studies, 77 reported data for respiratory samples and one (Szymczak 2020), reported data for faecal samples. The main results, Tables and Figures focus on the respiratory samples, with Szymczak 2020 reported separately.
Description of included studies
The 77 studies using respiratory samples included a total of 24,418 unique samples, with 7484 samples with RT‐PCR‐confirmed SARS‐CoV‐2 (some samples were analysed by more than one index test). Forty‐eight studies evaluated antigen tests (Albert 2020; Alemany 2020; Billaud 2020; Blairon 2020; Cerutti 2020; Courtellemont 2020; Diao 2020; Fenollar 2020(a); Fenollar 2020(b); FIND 2020a; FIND 2020b; FIND 2020c (BR); FIND 2020c (CH); FIND 2020d (BR); FIND 2020d (DE); FIND 2020e (BR); FIND 2020e (DE); Fourati 2020 [A]; Gremmels 2020(a); Gremmels 2020(b); Gupta 2020; Kruger 2020(a); Kruger 2020(b); Kruger 2020(c); Lambert‐Niclot 2020; Linares 2020; Liotti 2020; Mak 2020; Mertens 2020; Nagura‐Ikeda 2020; Nash 2020; PHE 2020(a); PHE 2020(b); PHE 2020(c) [non‐HCW tested]; PHE 2020(d) [HCW tested]; PHE 2020(d) [Lab tested]; PHE 2020(e); Porte 2020a; Porte 2020b [A]; Schildgen 2020 [A]; Scohy 2020; Shrestha 2020; Takeda 2020; Van der Moeren 2020(a); Van der Moeren 2020(b); Veyrenche 2020; Weitzel 2020 [A]; Young 2020) and 29 studies evaluated molecular tests (Assennato 2020; Broder 2020; Chen 2020a; Collier 2020; Cradic 2020(a); Cradic 2020(b); Dust 2020; Ghofrani 2020; Gibani 2020; Goldenberger 2020; Harrington 2020; Hogan 2020; Hou 2020; Jin 2020; Jokela 2020; Lephart 2020 [A]; Lieberman 2020; Loeffelholz 2020; Mitchell 2020; Moore 2020; Moran 2020; Rhoads 2020; Smithgall 2020 [A]; SoRelle 2020; Stevens 2020; Thwe 2020; Wolters 2020; Wong 2020; Zhen 2020 [A]). Summary study characteristics are presented in Table 2 with further details of study design and index test details in Appendix 9 and Appendix 10 for antigen assays and Appendix 11 and Appendix 12 for molecular assays. Full details are provided in the Characteristics of included studies table.
1. Description of studies.
| No. of studies (%) | |||
| Participants | Antigen tests | Rapid molecular | |
| Number of studies | 48 | 29 | |
| Sample size (by test type) | Median (IQR) | 291.5 (155 to 502.5) | 104 (75 to 172) |
| Range | 56 to 1676 | 19 to 524 | |
| Number of COVID‐19 cases (by test type) | Median (IQR) | 99.5 (45.5 to 128.5) | 50 (20 to 88) |
| Range | 0, 951 | 6, 220 | |
| Setting | COVID‐19 test centre | 22 (46) | 0 (0) |
| Contacts | 4 (8) | 0 (0) | |
| Hospital A&E | 3 (6) | 3 (10) | |
| Hospital inpatient | 2 (4) | 2 (7) | |
| Laboratory‐based | 11 (23) | 20 (69) | |
| Mixed | 4 (8) | 4 (14) | |
| Unclear | 2 (4) | 0 (0) | |
| Symptom status | Asymptomatic | 3 (6) | 0 (0) |
| Symptomatic | 16 (33) | 12 (41) | |
| Mainly symptomatica | 11 (23) | 0 (0) | |
| Mixed | 8 (17) | 3 (10) | |
| Not reported | 10 (21) | 14 (48) | |
| Study design | |||
| Recruitment structure | Single group – sensitivity and specificity | 29 (60) | 17 (59) |
| Two or more groups ‐ sensitivity and specificity | 10 (21) | 7 (24) | |
| Unclear | 2 (4) | 2 (7) | |
| Single group – sensitivity only | 6 (13) | 3 (10) | |
| Single group – specificity only | 1 (2) | 0 (0) | |
| Reference standard for COVID‐19 cases | All RT‐PCR‐positive | 47 (98) | 29 (100) |
| No. of studies = 42 | No. of studies = 26 | ||
| Reference standard for non‐COVID‐19 | COVID suspects (single RT‐PCR‐negative) | 39 (93) | 24 (92) |
| COVID suspects (double+ RT‐PCR‐negative) | 1 (2) | 1 (4) | |
| Current other disease (RT‐PCR‐negative) | 0 (0) | 1 (4) | |
| Pre‐pandemic (not described) | 1 (2) | 0 (0) | |
| Pre‐pandemic other disease | 1 (2) | 0 (0) | |
| Tests | No. of evaluations (%) | ||
| Total number of test evaluations | 58 | 32 | |
| Number of tests per study | 1 | 44 (92) | 26 (90) |
| 2 | 1 (2) | 3 (10) | |
| 3 | 1 (2) | 0 (0) | |
| 4 | 1 (2) | 0 (0) | |
| 5 | 1 (2) | 0 (0) | |
| Test method | CGIA | 41 (71) | 0 (0) |
| FIA | 9 (16) | 0 (0) | |
| LFA (alkaline phosphatase labelled) | 2 (3) | 0 (0) | |
| LFA (not otherwise specified) | 6 (10) | 0 (0) | |
| Automated RT‐PCR | 0 (0) | 18 (56) | |
| Isothermal amplification | 0 (0) | 13 (41) | |
| Other molecular (PCR + LFA) | 0 (0) | 1 (3) | |
| Sample type | NP alone | 30 (52) | 16 (50) |
| NP + OP combined | 12 (21) | 2 (6) | |
| Nasal alone | 2 (3) | 2 (6) | |
| OP alone | 1 (2) | 1 (3) | |
| Two or more of NP, or nasal or OP | 8 14) | 8 (25) | |
| Saliva | 1 (2) | 1 (3) | |
| Other | 3 (5) | 0 (0) | |
| Mixed (including lower respiratory) | 4 (7) | 1 (3) | |
| Not specified | 0 (0) | 1 (3) | |
| Sample storage | Direct | 28 (48) | 7 (22) |
| VTM | 20 (35) | 12 (38) | |
| Saline | 1 (2) | 0 (0) | |
| Direct or VTM | 0 (0) | 1 (3) | |
| VTM or PBS | 1 (2) | 0 (0) | |
| VTM or other | 0 (0) | 6 (19) | |
| Not specified | 8 (14) | 6 (19) | |
| Sample collection | HCW | 15 (26) | 2 (6) |
| Trained non‐HCW | 3 (5) | 0 (0) | |
| Self‐collected | 6 (10) | 0 (0) | |
| HCW or self‐collection | 0 | 1 (3) | |
| Not specified | 34 (59) | 29 (91) | |
| Sample testing | HCW (on‐site) | 13 (22) | 0 |
| Trained non‐HCW (on‐site) | 3 (5) | 0 | |
| HCW or on‐site laboratory personnel | 0 (0) | 1 (3) | |
| Not specified (on‐site testing) | 5 (9) | 1 (3) | |
| Laboratory staff | 12 (21) | 4 (13) | |
| Not stated (laboratory setting) | 15 (26) | 16 (50) | |
| IFU compliance | No | 16 (28) | 16 (50) |
| Yes | 29 (50) | 9 (28) | |
| Unclear | 13 (22) | 7 (22) | |
| A&E: accident and emergency department; CGIA: colloidal gold immunoassay; CI: confidence intervals; DRW: Diagnostics for the Real World; FIA: fluorescent immunoassay; HCW: healthcare worker; IFU: instructions for use; IQR: inter‐quartile range; LFA: lateral flow assay; NP: nasopharyngeal; OP: oropharyngeal; PBS: phosphatase‐buffered saline; RT‐PCR: reverse transcription polymerase chain reaction; VTM: viral transport medium | |||
a‘mainly’ symptomatic indicates ≥ 75% of included participants reported as symptomatic.
The median sample size of the included studies is 182 (interquartile range (IQR) 104 to 400) and median number of SARS‐CoV‐2 confirmed samples included is 63 (IQR 38 to 119). Sample sizes for antigen test evaluations were larger than those for molecular test evaluations (median 291.5 (IQR 155 to 502.5) compared to 104 (IQR 75 to 172)). Half of the studies (39/77, 51%) were conducted in Europe, 20 in North America, seven in South America, seven in Asia, one study included samples from more than one country and in one, the country of sample origin was unclear.
Participant characteristics
Antigen tests
Over half of the antigen test studies included samples from participants presenting in the community for COVID‐19 testing at: community test centres (22/48, 46%); emergency departments (3, 6%); or as part of contact tracing or outbreak investigations (4, 8%) (Table 2). Eleven antigen test studies (23%) selected samples from those submitted to laboratories for routine RT‐PCR testing with limited detail of the participants providing the samples (‘laboratory‐based’ studies), or included multiple (8%) or unclear (2%) settings. Over half of antigen test studies were conducted in symptomatic (16, 33%) or mainly symptomatic (11, 23%) populations, with only three (6%) exclusively in asymptomatic populations (two in asymptomatic contacts of confirmed cases (Fenollar 2020(b); Shrestha 2020), and one involved staff screening, all of whom were RT‐PCR‐negative (PHE 2020(e)). The remaining antigen studies included samples from populations with mixed symptom status (8, 17%) or provided no information regarding symptom status (10, 21%). Of the 10 that provided no information, seven were laboratory‐based studies providing no details of the settings from which the tested samples had been obtained, one included samples from a COVID‐19 test centre, one was an outbreak investigation and in one the study setting could not be derived. There were no studies evaluating strategies of multiple tests.
A total of 13 studies provided accuracy data for people with no symptoms at the time of testing (3 studies exclusively in asymptomatic populations, and 10 studies providing subgroup data for people with no reported symptoms); one study provided only specificity data. Of the 12 datasets reporting both sensitivity and specificity, one (Alemany 2020), purportedly described preventive screening of the general population (although the reported prevalence of 24% is very high for such a scenario), one (Cerutti 2020), described targeted traveller screening, four (Billaud 2020; Fenollar 2020(b); Gupta 2020; Shrestha 2020), tested contacts of confirmed cases (one as part of an outbreak investigation) and the remaining six datasets were subgroups of samples from people presenting for routine testing. We identified one additional asymptomatic dataset in a report of several substudies but we did not include it as participants underwent antigen testing up to five days after a positive PCR test and it was not possible to determine the time point at which symptom status was recorded; it was also not possible to determine which 'substudy’ the data related to (PHE 2020(d) [HCW tested]; PHE 2020(d) [Lab tested]).
Thirty‐one of the 48 studies evaluating antigen tests reported results for SARS‐CoV‐2‐confirmed samples above and below a Ct value from the reference standard RT‐PCR. The median proportion of participants with 'high' viral load was 52% (IQR 35% to 60%). The most commonly used threshold was 24 or 25 Ct or less (n = 29 studies (or 36/58 test evaluations); 11 studies (15/58 test evaluations) reported results with at a threshold of between 31 and 33 Ct or less ; and 13 studies (13 evaluations) reported other thresholds including less than: 28 Ct (n = 3), 30 Ct (n = 5), 31 Ct (n = 3), or 35 Ct (n = 2)
Molecular tests
In contrast, studies evaluating molecular tests were mainly laboratory‐based (20, 69%), with three (10%) including samples from participants presenting to emergency department or urgent care settings, two in hospital inpatients (7%), and four (14%) including samples from participants presenting in multiple settings. Twelve of the 29 studies (41%) reported included only samples from symptomatic patients, four reported mixed symptom status (10%) and 14 (48%) provided no information regarding symptom status. Of the 14 that provided no information, one was based in a hospital Accident and Emergency department, and the remaining 13 were laboratory‐based studies, only three of which gave any details of the settings from which the tested samples had been obtained (three reported inclusion of samples from either inpatients and outpatients (n = 1), inpatients and ambulatory patients (n = 1) or inpatients and emergency department patients (n = 1) but did not provide the number of samples from each source). There were no studies evaluating strategies of multiple tests.
Five studies evaluating molecular assays, reported proportions with high viral load ranging from 33% to 80%, median 46%. All five studies reported results above and below a Ct value of 30.
Study design and reference standards
Table 2 shows a similar distribution of study designs between those evaluating antigen and molecular tests. Overall, 60% of studies (n = 46) used a ‘single group’ design to estimate both sensitivity and specificity and 22% (n = 17) used a ‘two group’ design with separate selection of RT‐PCR‐positive and RT‐PCR‐negative samples. In four studies (5%), the design could not be fully determined but probably deliberate separate sampling of RT‐PCR‐positive and RT‐PCR‐negative samples had been used.
Nine studies included only samples with confirmed SARS‐CoV‐2, thus only allowing estimation of sensitivity (six antigen and three molecular assay studies), and one study included only SARS‐CoV‐2‐negative samples allowing estimation of specificity only. All studies defined the presence or absence of SARS‐CoV‐2 infection based on RT‐PCR. Of the 68 studies that included SARS‐CoV‐2‐negative samples, 63 (93%) required a single, negative PCR to confirm absence of infection and two (3%) required two negative PCR results. The remaining three studies used pre‐pandemic samples (n = 2) or contemporaneous samples with other respiratory infections.
Thirty‐three studies (43%), obtained paired swabs for index and reference standard, 39 (51%) used the same swab for point‐of‐care and RT‐PCR (18 antigen and 21 molecular studies) and five studies used a mix of paired and same swabs (n = 1) or it was not possible to determine this information from the study report.
Index tests
Fifteen studies evaluated only one test, seven compared two or more tests in the same participants (four with two tests each, one with three tests and one each with four or five tests). In total the 77 studies that used respiratory samples reported on a total of 90 test evaluations. Appendix 13 provides details extracted from the manufacturer’s instructions for use documents for all included tests.
Antigen tests
Forty‐eight studies reported 58 evaluations of antigen tests; 41 of CGIAs, nine FIA, two alternative type of LFA using alkaline phosphatase‐labelled antibodies, and six where assay type could not be determined. Studies evaluated 16 different commercially produced assays, as documented, with full assay identification details, in Appendix 13. One study reported the development of the Shenzhen Bioeasy assay (Diao 2020), but it is not clear whether the commercially available assay is identical to the one reported in the study or whether it has undergone further refinement. One study reported evaluating a Roche SARS‐CoV‐2 assay, which appears to be the SD Biosensor STANDARD Q (Schildgen 2020 [A]). Only 12 studies provided product codes for the tests evaluated (FIND 2020a; FIND 2020b; FIND 2020c (BR); FIND 2020c (CH); FIND 2020d (BR); FIND 2020d (DE); FIND 2020e (BR); FIND 2020e (DE); Gremmels 2020(a); Gremmels 2020(b); Porte 2020a; Weitzel 2020 [A]). The study reports or manufacturer IFUs for 11 assays reported targeting the nucleocapsid protein; this information was not reported for the Beijing Savant, Bionote, Biosynex, Liming Bio‐Products, or RapiGEN Inc assays (Appendix 13). We were unable to identify any information for Beijing Savant, E25Bio or Liming Bio‐Products assays online.
Multiple combinations of sample types and use of direct swab testing or swabs in viral transport medium or saline were reported across the studies (Table 2). Forty‐one of 58 evaluations used nasopharyngeal (n = 30), oropharyngeal (n = 1) or nasal (n = 2) samples (type of nasal sample was not reported), or combinations of nasopharyngeal, nasal or oropharyngeal samples (n = 8; nasopharyngeal or nasal mid‐turbinate in one, nasopharyngeal or combined naso‐ and oropharyngeal in two, naso‐ or oropharyngeal in two, and naso‐ or oropharyngeal or combined naso‐ and oropharyngeal samples in three. Thirteen evaluations used combined naso‐ and oropharyngeal samples for all participants, one used saliva samples and three evaluations (from one study) used bronchoalveolar lavage or throat wash samples. Of the six studies using nasal samples either alone (n = 2) or for at least some participants (n = 4), one reported that these were nares swabs, and the remaining five did not specify the type of nasal sample. Almost half of studies used direct swab testing (n = 28, 48%), 22 (38%) tested samples in viral transport medium, saline or other medium, and in 8 (14%) this information was not provided.
IFUs for five assays explicitly recommend against using any transport medium for swab testing (assays from Becton Dickinson, Bionote, Quidel and SD Biosensor; Appendix 13), one (Coris BioConcept) states that viral transport medium may be used, and the other nine do not mention use of transport medium, although two of the nine (from AAZ and Biosynex) imply that viral transport medium should not be used (using statements such as "use within one hour, stored in clean unused plastic tube"). We considered 29 of 58 antigen evaluations (50%) to be compliant with manufacturer IFUs in terms of sample type, use of viral transport medium and time interval between collection and testing. Sixteen evaluations were not compliant with IFUs; nine used viral transport medium, four used freezing, four tested samples not listed on the IFUs, and in two testing was not always conducted within the one‐hour time period specified in the IFU. For the remaining 13 evaluations either no IFU was available (n = 4), viral transport medium or saline was used but the IFU did not specifically address whether viral transport medium was recommended or not (n = 7), or insufficient detail was provided in the study.
Samples were collected by healthcare workers in 15 (26%) evaluations, by trained non‐healthcare workers, such as firefighters or Ministry of Health employees in three (5%) evaluations, self‐collected in six (10%) and the collection was not described in 34 evaluations (59%). Sample testing was conducted ‘on‐site’ immediately or within one hour of collection in 21 (36%) evaluations by the same healthcare workers (n = 13), trained non‐healthcare workers (n = 3) who collected the samples, or this information was not provided (n = 5). In the remaining 27 evaluations (47%), testing was conducted by laboratory staff (n = 12) or was inferred to be by laboratory staff (n = 15). For the latter group, the time interval between sample collection and testing was on receipt at the laboratory, some reporting delays of up to six hours.
Molecular tests
Twenty‐nine studies reported 32 evaluations of five different commercially available rapid molecular tests: 13 evaluating ID NOW (Abbott Laboratories), 15 evaluating Xpert Xpress (Cepheid Inc), two of SAMBA II (Diagnostics for the Real World), and one evaluation each of Accula (Mesa Biotech Inc.) and COVID Nudge (DNANudge). None of the studies reported product codes for the tests evaluated. One study of Xpert Xpress used the 'research use only' (RUO) version of the test but reported that the RUO version contains the same reagents as the 'emergency use authorisation' (EUA) version. The RUO test allows the user to view the amplification curves for the RdRp gene as well as for the E‐gene and N2 targets whereas the EUA version restricts the amplification curves to E and N2 only. ID NOW and SAMBA‐II use isothermal techniques, Xpert Xpress and COVID Nudge are based on RT‐PCR, and Accula is described as a PCR plus lateral flow assay.
Multiple combinations of sample types and use of direct swab testing or swabs in viral transport medium or saline were reported across the studies (Table 2). The sample types used included combined naso‐ and oropharyngeal samples (n = 2), nasopharyngeal samples alone (n = 16), nasal alone (n = 2), oropharyngeal samples alone (n = 1), or a combination of two or more of either nasopharyngeal or nasal or oropharyngeal samples (n = 8). One evaluation used throat saliva or lower respiratory tract specimens, one used saliva samples alone and one did not specify the sample type used. Of the six studies using nasal samples either alone (n = 2) or for at least some participants (n = 4), one reported using nares swabs, and the remaining five did not specify the type of nasal sample used.
Eight evaluations (25%) reported direct swab testing in some (n = 1) or all (n = 7) samples, 18 (59%) used swabs in viral transport medium only (n = 12) or in viral transport medium or some other transport medium (n = 6), and six did not report whether they used any transport medium.
Sample collection was described in only three evaluations (9%) (Gibani 2020; Harrington 2020; Rhoads 2020; Table 2); the remaining studies did not describe sample collection but it is likely that samples were collected as part of routine care by healthcare workers. Sample testing was clearly described as conducted on‐site by medical personnel or by laboratory personnel at local laboratories in one of the studies reporting sample collection (Harrington 2020), while a second implied testing as soon as possible after collection, possibly by the same healthcare worker (Gibani 2020). Four (12.5%) evaluations stated that laboratory staff carried out the tests. In 16 of the remaining 26 studies, testing by laboratory staff was inferred, based on delays between collection and testing of 18 hours to seven days (n = 10), or reported use of archived or frozen samples (n = 6). The remaining eight evaluations provided no useful information regarding who carried out the test (Assennato 2020; Dust 2020; Ghofrani 2020; Jin 2020; Jokela 2020; Moran 2020; Rhoads 2020; SoRelle 2020).
Two of the five manufacturers document IFU for samples stored in transport medium (Xpert Xpress and SAMBA II assays); two explicitly recommend against the use of viral transport medium (ID NOW and Accula), although at the time of the test evaluations some viral transport media were documented as acceptable for ID NOW; and one IFU does not mention the use of viral transport medium (COVID Nudge). Although immediate sample testing is preferred, all manufacturers document an acceptable period of refrigerated storage of between eight hours (COVID Nudge), and seven days with refrigeration (Xpert Xpress). See Appendix 13.
We considered only nine of 32 (28%) evaluations to be compliant with manufacturer IFUs in regard to sample type, use of viral transport medium and time interval between collection and testing. Sixteen evaluations were not compliant with IFUs; eight used viral transport medium, six used frozen samples, and two tested samples not listed on the IFUs. For the remaining seven evaluations, either the testing interval from sample collection was unclear (n = 5) or saline was used but the IFU did not specifically address whether this was recommended or not (n = 2).
Methodological quality of included studies
We report the overall methodological quality assessed using the QUADAS‐2 tool for all included studies (n = 78) in Figure 2 (Whiting 2011). See Appendix 14 for separate summary plots by test method and for a plot of study‐level ratings by quality domain. We explain how we reached these judgements in the Characteristics of included studies table.
2.
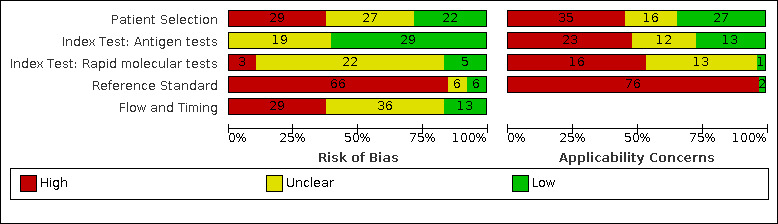
Risk of bias and applicability concerns graph: review authors' judgements about each domain presented as percentages across included studies. Numbers in the bars indicate the number of studies
We considered whether the findings of individual studies were at risk of bias, and whether there were concerns that results might not apply to standard use of the tests. We did not judge any study at low risk of bias, although in 11 of 78 studies the only concern was that a single negative RT‐PCR was used to confirm absence of COVID infection rather than the preferred two negative tests. All studies raised concerns regarding the applicability of their results, but in 13 of 78 studies the only concern was the reliance on only PCR to identify SARS‐CoV‐2 cases (and nine of these 13 are in common with the 11 using a single negative RT‐PCR).
Participant selection
We judged 22 studies (28%) to be at low risk of bias, and 29 (37%) at high risk of bias because of deliberate sampling of participants based on the reference standard result (n = 25; 16 two‐group studies and nine that only included samples with confirmed SARS‐CoV‐2 infection or absence of infection) or use of convenience sampling (n = 4). In 27 studies (35%) the risk of bias was unclear because of poor reporting of recruitment procedures or inclusion criteria (Figure 2).
A third (27/78) of studies were likely to have selected an appropriate patient group, recruiting participants from COVID‐19 test centres, urgent care or emergency departments or identified through contact tracing. We had high concerns about the applicability of the selected participants in almost half of studies (35/78). Recruited participants were unlikely to be similar to those in whom the test would be used in clinical practice because of deliberate sampling (n = 25) or sample inclusion based on the availability of residual and sometimes frozen samples, or both (n = 22).
Index tests
Poor reporting meant we could not clearly assess whether there was a risk of bias through performance of the index test in 41 (53%) studies. In general, antigen test studies were of a higher methodological standard for the index test domain compared to studies of molecular tests (Figure 2).
For antigen tests, we observed low risk of bias in 60% of studies (29/48). Risk of bias was unclear in the remaining studies because we could not judge whether interpretation of the index test was undertaken with knowledge of the reference standard result. For molecular tests, risk of bias was low in only 17% of studies (5/30). We observed high risk of bias in three studies (Moran 2020; Smithgall 2020 [A]; Wolters 2020) because they did not follow the manufacturer’s prespecified threshold for the Xpert Xpress test (re‐testing of samples with presumptive positive results). Risk of bias was unclear in 73% (22/30) of studies because they did not report blinding to the reference standard (n = 22), six of these studies also did not report how they handled presumptive positive results on Xpert Xpress.
Fourteen studies (18%), including 13 antigen and one molecular test study, conducted testing as would be expected in practice (low concern regarding applicability). We had high concerns about applicability in half of all studies (39/78); 48% (23/48) of antigen and 57% (16/30) of molecular studies. Twenty‐seven (11 antigen and 16 molecular) did not comply with manufacturers’ IFU and a further 10 (all antigen studies), did not carry out tests as would occur in practice (i.e. trained, centralised laboratory staff carried out testing). In another two antigen studies concerns for applicability were high because tests were not available for purchase (Diao 2020; Nash 2020). Of the remaining 25 studies (12 antigen and 13 molecular) 16 conducted the test within the manufacturer IFU but none clearly described the setting for testing or personnel conducting the test.
Reference standards
Six studies were at low risk of bias for the reference standard. Although 12 used an appropriate reference standard, half (6/12) did not clearly implement blinding of the reference standard to the index test. High risk of bias (66/78) was present because studies did not use an adequate reference standard (Figure 2); they used either a single negative RT‐PCR to define absence of SARS‐CoV‐2 infection (n = 64) or the index test formed part of a composite reference standard (n = 2).
A total of 36 studies reported blinded RT‐PCR interpretation, two (with composite reference standard) did not implement blinding, and 40 (51%) provided insufficient information about blinding of the reference standard to the index test to judge risk of bias.
We judged 76 of the 78 studies to raise concerns about applicability (97%) because of defining the presence of SARS‐CoV‐2 infection based on a single RT‐PCR‐positive result. These studies will have excluded individuals who are RT‐PCR‐negative but have exposure and clinical features that meet the case definitions for COVID‐19.
Flow and timing
Only 13 (17%) studies (all of antigen tests) were at low risk of bias for participant flow and timing (Figure 2). Twenty‐nine (37%) were at high risk of bias (19 antigen and 10 molecular) because of exclusion of samples following invalid index test results (n = 23); delays between ‘paired’ swabs of up to three days (n = 4), different reference standards used (n = 3), or because they provided results on a per sample instead of per patient basis (n = 2). These categories are not mutually exclusive.
We judged risk of bias unclear for 36 (46%) studies, primarily because of lack of clarity about participant inclusion and exclusion from analyses (n = 34), with no missing data or indeterminate test results reported and no Standards for Reporting Diagnostic Accuracy Studies (STARD)‐style participant flow diagram and checklist (Bossuyt 2015), to fully report outcomes for all samples.
Conflicts of interest
In 27 studies all authors declared no conflicts of interest, although one study that reported the validation of a new test included a co‐author affiliated to the test manufacturing company. Of these 27 studies, 19 were independent evaluations published by FIND or were from national reference laboratories. Twenty studies did not provide a conflict of interest statement, including 13 published studies and one study that reported affiliations to the test manufacturer. In the 12 remaining studies at least one author declared potential conflicts of interest in relation to the test.
Twenty‐six studies provided no funding statement, 12 reported no funding sources to declare, and the remainder (n = 40) reported one or more funding sources.
Findings
Of the 78 included studies, eight reported evaluations of more than one test using the same samples and one reported evaluations of three tests using different samples (Table 2). To include all results from all tests in these analyses we have treated results from different tests of the same samples within a study as separate data points, such that data are available on 91 test evaluations (58 evaluations of antigen tests in 48 studies and 33 evaluations of rapid molecular tests in 30 studies).
As previously stated, 77 of the 78 studies reported data for respiratory samples and one (Szymczak 2020), reported data for non‐respiratory (faecal) samples. The main results, Tables and Figures focus on the respiratory samples, with Szymczak 2020 reported separately.
The results tables identify where estimates are based on multiple assessments of the same samples by including both the number of test evaluations and the number of studies. Nine datasets are from ‘cases only’ studies reporting only sensitivity estimates (six for antigen tests and three for molecular assays), and one antigen test evaluation is for ‘non‐COVID‐19’ cases reporting only specificity. Summary results are presented for studies providing both sensitivity and specificity data and then adding in the data from sensitivity‐ or specificity‐only evaluations. The numbers of true positives, false positives, and total samples with and without confirmed SARS‐CoV‐2 infection are based on test result counts.
We present results for antigen tests overall and by subgroup in Table 3. Table 4 and Table 5 present results by test brand overall and by symptom status, and give results of sensitivity analyses restricting by compliance with manufacturer IFU. Forest plots of study data for the primary analysis are in Figure 3 and for subgroup analyses by symptom status and time after symptom onset are in Figure 4 and Figure 5. Appendix 15 provides forest plots for study data according to Ct value and study design. Individual plots by test brand are provided in Figure 6 for test brands with three or more evaluations and Figure 7 for test brands with one or two evaluations. Figure 8 shows data from studies comparing the accuracy of two or more antigen assays. Full identification details for studies of antigen‐based assays are provided in Appendix 9 and Appendix 10.
2. Antigen tests: summary of sensitivity and specificity analyses.
| Subgroup | Test | Evaluations | Samples | Cases | Average sensitivity, % (95% CI) | Average specificity, % (95% CI) |
| Overall analysis | ||||||
| Evaluations reporting both sensitivity and specificity | 51 | 21,614 | 6136 | 68.9 (61.8 to 75.1) | 99.6 (99.0 to 99.8) | |
| Evaluations reporting sensitivity dataa | 57 | 22,605 | 7127 | 67.7 (60.8 to 74.0) | N/A | |
| Evaluations reporting specificity dataa | 52 | 22,152 | 6136 | N/A | 99.5 (99.0 to 99.8) | |
| Subgroup analyses (with sensitivity analyses restricting to direct comparisons) | ||||||
| Symptom status (all) | Symptomatic | 37 | 15,530 | 4410 | 72.0 (63.7 to 79.0) | 99.5 (98.5 to 99.8) |
| Asymptomatic | 12 | 1581 | 295 | 58.1 (40.2 to 74.1) | 98.9 (93.6 to 99.8) | |
| Difference | −13.8 (−33.1 to 5.4) P = 0.159 |
−0.6 (−2.6 to 1.4) P = 0.551 |
||||
| Symptomatic: direct comparison | 9 | 2437 | 890 | 68.0 (51.4 to 81.1) | 99.2 (83.9 to 100) | |
| Asymptomatic: direct comparison | 9 | 1182 | 213 | 53.6 (35.0 to 71.3) | 99.2 (85.5 to 100) | |
| Difference | −14.4 (−38.8 to 10.0) P = 0.246 |
−0.01 (−3.2 to 3.2), P = 0.995 |
||||
| Mixed symptoms or not reported | 19 | 6220 | 2392 | 63.0 (52.2 to 72.6) | 98.4 (98.0 to 98.8) | |
|
Time post‐symptom onset (sensitivity only) |
Week 1 | 26 | 5769 | 2320 | 78.3 (71.1 to 84.1)a | N/A |
| Week 2 | 22 | 935 | 692 | 51.0 (40.8 to 61.0)a | N/A | |
| Difference | −27.3 (−32.8 to −21.9) P < 0.0001 |
|||||
| Week 1: direct comparison | 22 | 4978 | 2164 | 76.6 (68.2 to 83.4)a | N/A | |
| Week 2: direct comparison | 22 | 935 | 692 | 48.8 (37.9 to 59.8)a | N/A | |
| Difference | −27.9 (−33.3 to −22.5) P < 0.0001 |
|||||
|
Ct value (sensitivity only) |
Higher viral load (< or ≤ 25 Ct threshold)b | 36 | 2613 | 2613 | 94.5 (91.0 to 96.7)a | N/A |
| Lower viral load (> or >= 25 Ct threshold)b | 36 | 2632 | 2632 | 40.7 (31.8 to 50.3)a | N/A | |
| Difference | −53.8 (−63.6 to −44.1) P < 0.0001 |
|||||
| Higher viral load (≤ 32 or33 Ct threshold)c | 15 | 2127 | 2127 | 82.5 (74.0 to 88.6)a | N/A | |
| Lower viral load (> 32 or 33 Ct threshold)c | 15 | 346 | 346 | 8.9 (3.3 to 21.7)a | N/A | |
| Difference | −73.5 (−84.7 to −62.4) P < 0.0001 |
|||||
| Study design | Single group: sensitivity and specificity | 29 | 15,336 | 3536 | 72.1 (64.8 to 78.3) | 99.6 (99.1 to 99.8) |
| Two or more groups: sensitivity and specificity | 20 | 5729 | 2396 | 64.1 (48.5 to 77.2) | 97.3 (96.7 to 97.8) | |
| −8.0 (−24.2 to 8.2) P = 0.334 |
−2.3 (−2.9 to −1.6) P < 0.0001 |
|||||
| Unclear | 2 | 549 | 204 | 65.2 (39.6 to 84.3) | 96.3 (88.0 to 98.9) | |
| Test method | CGIA | 36 | 17,448 | 5085 | 64.0 (55.7 to 71.6) | 99.0 (98.8 to 99.2) |
| FIA | 9 | 2820 | 712 | 79.6 (67.5 to 88.0) | 97.7 (95.3 to 98.8) | |
| Difference |
15.6 (2.6 to 28.5) P = 0.019 |
−1.3 (−3.0 to 0.3) P = 0.113 |
||||
| LFA (not otherwise specified) | 5 | 1184 | 277 | 78.0 (46.0 to 93.7) | 96.0 (94.5 to 97.1) | |
| LFA (ALP) | 1 | 162 | 62 | 80.6 (68.6 to 89.6) | 100 (96.4 to 100) | |
| ALP: alkaline phosphatase labelled; CGIA: colloidal gold immunoassay; CI: confidence intervals; Ct: cycle threshold; FIA: fluorescent immunoassay; LFA: lateral flow assay; N/A: not applicable | ||||||
aSeparate pooling of sensitivity or specificity, or both.
b threshold for 'higher' viral load was < 25 Ct in 18 evaluations and ≤ 25 Ct in 18 evaluations
c threshold for 'higher' viral load ≤ 33 Ct in 13 evaluations and < 32 in 2 evaluations
3. Antigen tests: summary data by test brand and compliance with manufacturers' instructions for use.
| Test | All | IFU‐compliant | ||||
| Number of evaluations; samples (cases) | Average sensitivity, % (95% CI) | Average specificity, % (95% CI) | Number of evaluations; samples (cases) | Average sensitivity, % (95% CI) | Average specificity, % (95% CI) | |
|
AAZ ‐ COVID‐VIRO (2 studies not pooled) |
1; 632 (295) | 61.7 (55.9 to 67.3) | 100 (98.9 to 100) | |||
| 1; 248 (101) | 96.0 (90.2 to 98.9) | 86.4 (79.8 to 91.5) | 1; 248 (101) | 96.0 (90.2 to 98.9) | 86.4 (79.8 to 91.5) | |
| Abbott ‐ Panbio Covid‐19 Ag | 10; 5509 (1849) | 72.0 (60.6 to 81.1) | 99.3 (99.0 to 99.6) | 5; 1776 (362) | 72.0 (56.5 to 83.5) | 99.2 (98.5 to 99.5) |
| including sensitivity‐only cohort | 11; 2031 (2031) | 72.8 (62.6 to 81.0)a | 6; 544 (544) | 73.5 (61.1 to 83.0)a | ||
| Becton Dickinson ‐ BD Veritor | 2; 602 (55) | 82.3 (62.1 to 93.0) | 99.5 (98.3 to 99.8) | |||
| including sensitivity‐only cohort | 3; 180 (180) | 79.4 (72.9 to 84.7)a | ||||
| BIONOTE ‐ NowCheck COVID‐19 Ag | 1; 400 (102) | 89.2 (81.5 to 94.5) | 97.3 (94.8 to 98.8) | 1; 400 (102) | 89.2 (81.5 to 94.5) | 97.3 (94.8 to 98.8) |
| Biosynex ‐ Biosynex COVID‐19 Ag BSS | 1; 634 (297) | 59.6 (53.8 to 65.2) | 100 (98.9 to 100) | |||
| Coris Bioconcept ‐ COVID‐19 Ag Respi‐Strip | 7; 1781 (707) | 39.7 (31.3 to 48.7) | 98.3 (97.4 to 98.9) | 7; 1781 (707) | 39.7 (31.3 to 48.7) | 98.3 (97.4 to 98.9) |
| E25Bio ‐ DART (N‐based) | 1; 190 (100) | 80.0 (70.8 to 87.3) | 91.1 (83.2 to 96.1) | |||
|
Fujirebio ‐ ESPLINE SARS‐CoV‐2 (2 studies not pooled) |
1; 162 (62) | 80.6 (68.6 to 89.6) | 100 (96.4 to 100) | |||
| 1; 103 (103) | 11.6 (6.2 to 19.5) | |||||
| Innova Medical Group ‐ Innova SARS‐CoV‐2 Ag | 3; 2945 (596) | 47.9 (34.3 to 61.8) | 99.8 (99.5 to 99.9) | 1; 1676 (372) | 57.5 (52.3 to 62.6) | 99.6 (99.1 to 99.9) |
| including sensitivity‐only cohorts | 5; 1017 | 59.0 (43.4to 73.0)a | 3; 793 | 69.1 (58.3to 78.2)a | ||
| including specificity‐only cohort | 4; 2887 | 99.8 (99.5to 99.9)a | 2; 1842 | 99.7 (99.3to 99.9)a | ||
| Liming Bio‐Products ‐ StrongStep® COVID‐19 Ag | 1; 19 (9) | 0 (0 to 33.6) | 90.0 (55.5 to 99.7) | |||
| Quidel Corporation ‐ SOFIA SARS Ag | 1; 64 (32) | 93.8 (79.2 to 99.2) | 96.9 (83.8 to 99.9) | |||
| RapiGEN ‐ BIOCREDIT COVID‐19 Ag | 5; 2010 (310) | 63.3 (45.7 to 78.0) | 99.5 (99.1 to 99.8) | 3; 1828 (189) | 73.0 (57.4 to 84.4) | 99.8 (99.4 to 99.9) |
| including sensitivity‐only cohort | 6; 470 (470) | 57.7 (39.8to 73.8)a | ||||
| Roche ‐ SARS‐CoV‐2 | 1; 73 (42) | 88.1 (74.4 to 96.0) | 19.4 (7.5 to 37.5) | |||
| Savant Biotech ‐ Huaketai SARS‐CoV‐2 N Protein | 1; 109 (78) | 16.7 (9.2 to 26.8) | 100 (88.8 to 100) | |||
| SD Biosensor ‐ STANDARD F COVID‐19 Ag | 4; 1552 (295) | 72.6 (54.0 to 85.7) | 97.5 (96.4 to 98.2) | 2; 1129 (159) | 75.5 (68.2 to 81.5) | 97.2 (96.0 to 98.1) |
| SD Biosensor ‐ STANDARD Q COVID‐19 Ag | 6; 3480 (821) | 79.3 (69.6 to 86.6) | 98.5 (97.9 to 98.9) | 4; 2522 (421) | 85.8 (80.5 to 89.8) | 99.2 (98.2 to 99.6) |
|
Shenzhen Bioeasy Biotech ‐ 2019‐nCoV Ag development‐phase publication |
3; 965 (177) | 86.2 (72.4 to 93.7) | 93.8 (91.9 to 95.3) | 1; 727 (15) | 66.7 (38.4 to 88.2) | 93.1 (91.0 to 94.9) |
| 1; 239 (208) | 67.8 (61.0 to 74.1) | 100 (88.8 to 100) | ||||
| Ag: antigen; CI: confidence interval; IFU: [manufacturers'] instructions for use; N: nucleoprotein | ||||||
aSeparate pooling of sensitivity or specificity. b2x2 tables combined prior to calculating estimates.
4. Antigen tests: summary data by symptom status, test brand and compliance with manufacturers' instructions for use.
| All | IFU‐compliant | |||||
| Number of evaluations; samples (cases) | Average sensitivity, % (95% CI) | Average specificity, % (95% CI) | Number of evaluations; samples (cases) | Average sensitivity, % (95% CI) | Average specificity, % (95% CI) | |
| SYMPTOMATIC participants by test | ||||||
|
AAZ ‐ COVID‐VIRO (2 studies not pooled) |
1; 632 (295) | 61.7 (55.9 to 67.3) | 100 (98.9 to 100) | |||
| 1; 248 (101) | 96.0 (90.2 to 98.9) | 86.4 (79.8 to 91.5) | 1; 248 (101) | 96.0 (90.2 to 98.9) | 86.4 (79.8 to 91.5) | |
| Abbott ‐ Panbio Covid‐19 Ag | 8; 3699 (1162) | 74.1 (60.8 to 84.0) | 99.8 (99.5 to 99.9) | 3; 1094 (252) | 75.1 (57.3 to 87.1) | 99.5 (98.7 to 99.8) |
| including sensitivity‐only cohort | 9; 1344 (1344) | 74.8 (63.4 to 83.6)a | 4; 434 (434) | 76.2 (63.6to 85.4)a | ||
| Becton Dickinson ‐ BD Veritor | 2; 602 (55) | 82.3 (62.1 to 93.0) | 99.5 (98.3 to 99.8) | |||
| including sensitivity‐only cohort | 3; 180 (180) | 79.4 (72.9to 84.7)a | ||||
| BIONOTE ‐ NowCheck COVID‐19 Ag | 1; 400 (102) | 89.2 (81.5 to 94.5) | 97.3 (94.8 to 98.8) | 1; 400 (102) | 89.2 (81.5 to 94.5) | 97.3 (94.8 to 98.8) |
| Biosynex ‐ Biosynex COVID‐19 Ag BSS | 1; 634 (297) | 59.6 (53.8 to 65.2) | 100 (98.9 to 100) | |||
| Coris Bioconcept ‐ COVID‐19 Ag Respi‐Strip | 3; 780 (414) | 34.1 (29.7 to 38.8)a | 100 (99.0 to 100)a,b | 3; 780 (414) | 34.1 (29.7 to 38.8)a | 100 (99.0 to 100)a,b |
| Fujirebio ‐ ESPLINE SARS‐CoV‐2 | 1; 88 (88) | 11.4 (5.6 to 19.9) | ||||
| Innova Medical Group ‐ Innova SARS‐CoV‐2 Ag | 2; 2794 (550) | 56.2 (52.0 to 60.3) | 99.8 (99.5 to 99.9) | 1; 1676 (372) | 57.5 (52.3 to 62.6) | 99.6 (99.1 to 99.9) |
| including sensitivity‐only cohorts | 4; 971 (971) | 65.5 (54.8to 74.9)† | 3; 793 (793) | 69.1 (58.3to 78.2)† | ||
| Liming Bio‐Products ‐ StrongStep® COVID‐19 Ag | 1; 19 (9) | 0 (0 to 33.6) | 90.0 (55.5 to 99.7) | |||
| Quidel Corporation ‐ SOFIA SARS Ag | 1; 64 (32) | 93.8 (79.2 to 99.2) | 96.9 (83.8 to 99.9) | |||
| RapiGEN ‐ BIOCREDIT COVID‐19 Ag | 3; 608 (206) | 58.4 (36.3 to 77.5) | 96.4 (82.8 to 99.3) | 1; 476 (117) | 74.4 (65.5 to 82.0) | 98.9 (97.2 to 99.7) |
| Roche ‐ SARS‐CoV‐2 | 1; 23 (10) | 100 (69.2 to 100) | 7.7 (0.2 to 36.0) | |||
| Savant Biotech ‐ Huaketai SARS‐CoV‐2 N Protein | 1; 109 (78) | 16.7 (9.2 to 26.8) | 100 (88.8 to 100) | |||
| SD Biosensor ‐ STANDARD F COVID‐19 Ag | 3; 1193 (191) | 78.0 (71.6 to 83.3) | 97.2 (96.0 to 98.1) | 2; 1129 (159) | 75.5 (68.2 to 81.5) | 97.2 (96.0 to 98.1) |
| SD Biosensor ‐ STANDARD Q COVID‐19 Ag | 5; 2760 (731) | 80.1 (68.5 to 88.1) | 98.1 (97.4 to 98.6) | 3; 1947 (336) | 88.1 (84.2 to 91.1) | 99.1 (97.8 to 99.6) |
| Shenzhen Bioeasy Biotech ‐ 2019‐nCoV Ag | 3; 965 (177) | 86.2 (72.5 to 93.7) | 93.8 (91.9 to 95.3) | 1; 727 (15) | 66.7 (38.4 to 88.2) | 93.1 (91.0 to 94.9) |
| ASYMPTOMATIC participants by test | ||||||
| Abbott ‐ Panbio Covid‐19 Ag | 6; 1097 (190) | 58.1 (41.7 to 72.9) | 98.4 (92.2 to 99.7) | 2; 474 (47) | 48.9 (35.1 to 62.9) | 98.1 (96.3 to 99.1) |
| Coris Bioconcept ‐ COVID‐19 Ag Respi‐Strip | 1; 45 (14) | 28.6 (8.4 to 58.1) | 100 (88.8 to 100) | 1; 45 (14) | 28.6 (8.4 to 58.1) | 100 (88.8 to 100) |
| Fujirebio ‐ ESPLINE SARS‐CoV‐2 | 1; 15 (15) | 13.3 (1.7 to 40.5) | N/A | |||
| RapiGEN ‐ BIOCREDIT COVID‐19 Ag | 2; 140 (60) | 63.2 (21.7 to 91.4) | 98.9 (82.9 to 99.9) | 1; 113 (47) | 85.1 (71.7 to 93.8) | 100 (94.6 to 100) |
| Roche ‐ SARS‐CoV‐2 | 1; 27 (13) | 84.6 (54.6 to 98.1) | 14.3 (1.8 to 42.8) | |||
| SD Biosensor ‐ STANDARD Q COVID‐19 Ag | 2; 272 (18) | 61.1 (37.9 to 80.2) | 99.6 (97.3 to 99.9) | 1; 127 (13) | 69.2 (38.6 to 90.9) | 99.1 (95.2 to 100) |
| Ag: antigen; CI: confidence interval; N: nucleoprotein; N/A: not applicable | ||||||
aseparate pooling of sensitivity or specificity. b2x2 tables combined prior to calculating estimates.
3.
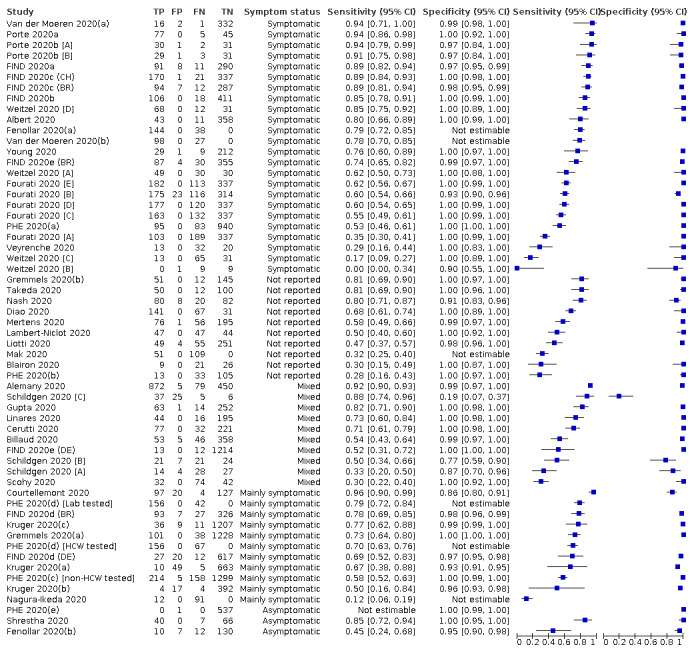
Forest plot of studies evaluating antigen tests. BR: Brazil; CH: Switzerland; DE: Germany; HCW: healthcare worker; Lab: laboratory
4.
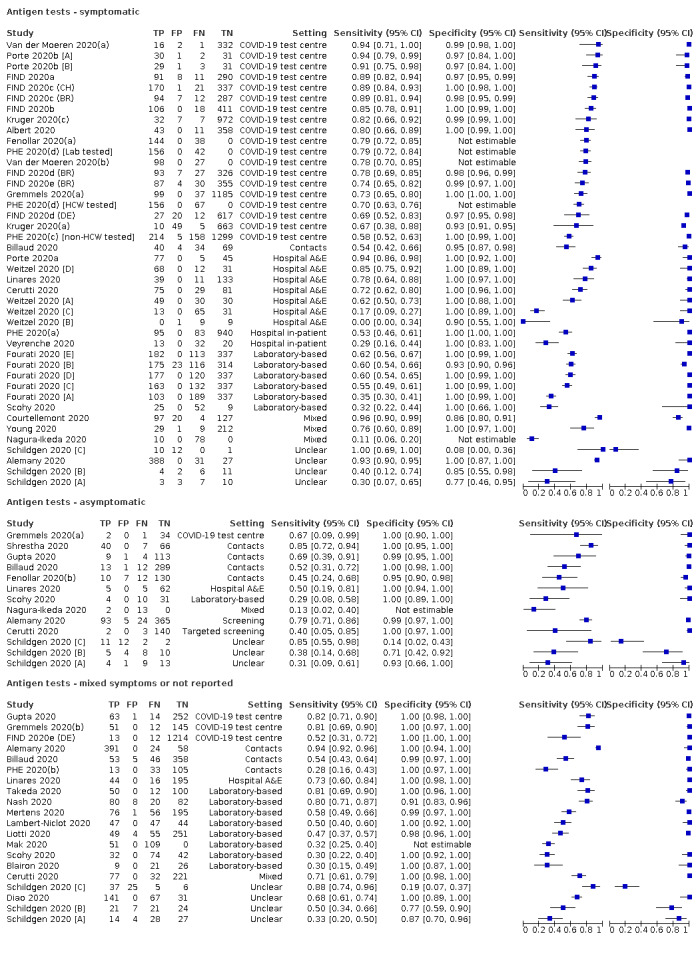
Forest plot of data for antigen tests according to symptom status. A&E: accident and emergency; BR: Brazil; CH: Switzerland; DE: Germany; HCW: healthcare worker; Lab: laboratory
5.
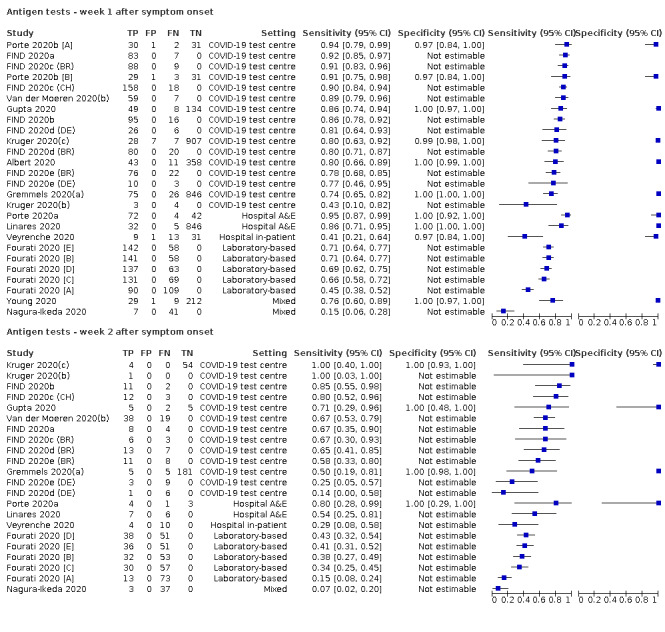
Forest plot of antigen test evaluations by week post symptom onset (pso). A&E: accident and emergency; Ag: antigen; BR: Brazil; CH: Switzerland; DE: Germany
6.
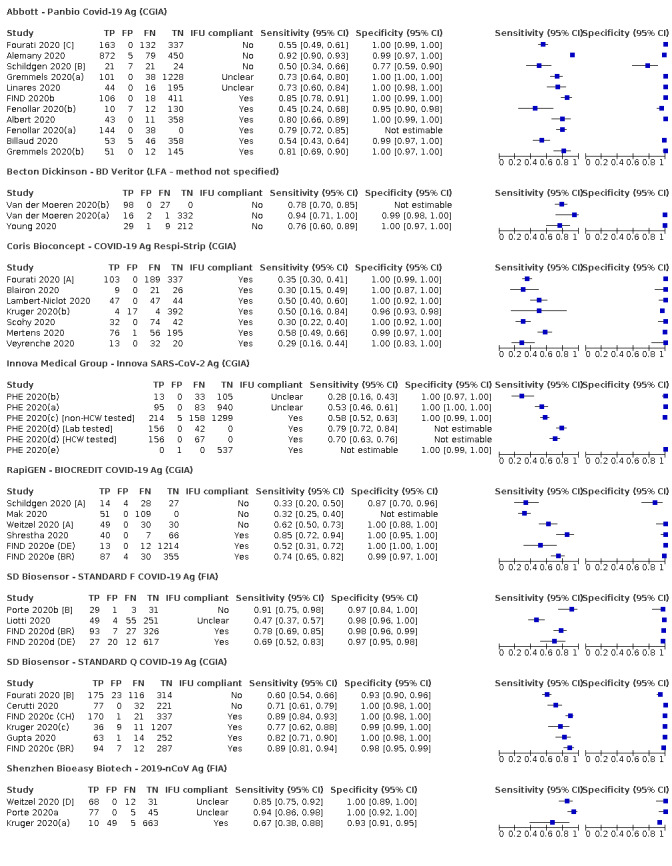
Forest plot by test brand for assays with ≥ 3 evaluations. BR: Brazil; CGIA: colloidal‐gold immunoassay; CH: Switzerland; DE: Germany; FIA: fluorescent immunoassay; HCW: healthcare worker; IFU: instructions for use; Lab: laboratory; LFA: lateral flow assay
7.
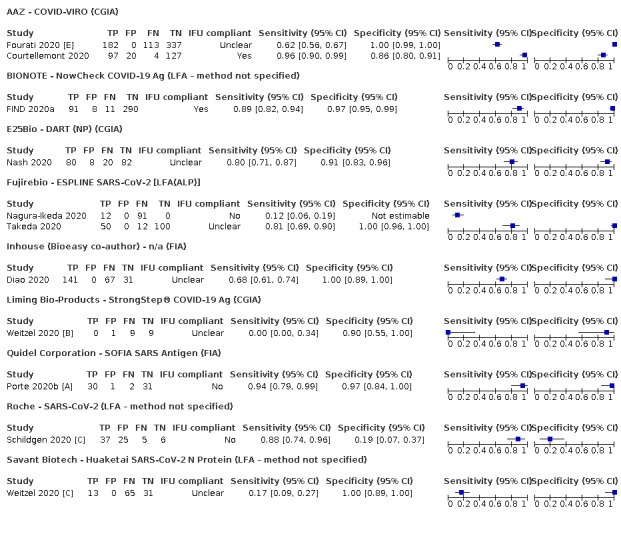
Forest plot by test brand for assays with < 3 evaluations; CGIA: colloidal‐gold immunoassay; FIA: fluorescent immunoassay; IFU: instructions for use; LFA: lateral flow assay
8.
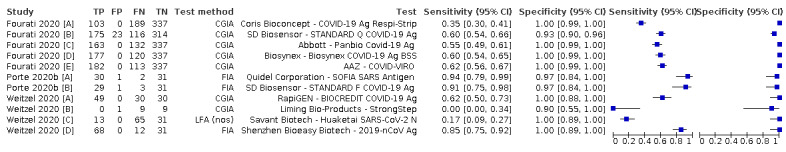
Forest plot of studies reporting comparative data. CGIA: colloidal‐gold immunoassay; FIA: fluorescent immunoassay; LFA: lateral flow assay; nos: not otherwise specified
Results for molecular tests overall and by subgroup are reported in Table 6. Forest plots of study data for the primary analysis is in Figure 9 and for subgroup analyses by Ct value, study design and sensitivity analyses by pre‐ and post‐discrepant analysis in Appendix 16. Individual plots by test brand are provided in Figure 10. Full identification details for studies of molecular‐based assays are provided in Appendix 11 and Appendix 12. Appendix 17 provides forest plots for study data according to Ct value and discrepant analysis.
5. Molecular tests: summary of sensitivity and specificity analyses.
| Test or subgroup | Evaluations | Samples | Cases | Average sensitivity, % (95% CI) | Average specificity, % (95% CI) | |
| Overall analysis | ||||||
| Evaluations reporting both sensitivity and specificity | 29 | 4351 | 1787 | 95.1 (90.5 to 97.6) | 98.8 (98.3 to 99.2) | |
| Evaluations reporting sensitivity dataa | 32 | 4537 | 1973 | 95.5 (91.5 to 97.7) | N/A | |
| Subgroup analyses (with sensitivity analyses restricting to direct comparisons) | ||||||
|
Viral load (sensitivity only) |
High viral load (≤ 30 Ct) | 6 | 204 | 204 | 100 (98.2 to 100)a,b | N/A |
| Low viral load (> 30 Ct) | 6 | 149 | 149 | 95.6 (55.7 to 99.7) | N/A | |
| By study design | Single group – sensitivity and specificity | 18 | 2899 | 976 | 93.2 (85.5 to 97.0) | 99.4 (98.4 to 99.8) |
| Two or more groups ‐ sensitivity and specificity | 9 | 1265 | 718 | 97.2 (90.7 to 99.2) | 99.3 (96.5 to 99.8) | |
| Difference |
4.0 (‐2.2to 10.1) P = 0.211 |
‐0.2 (‐1.3to 1.0) P = 0.771 |
||||
| Unclear designs | 2 | 187 | 93 | 93.2 (71.0 to 98.7)a | 100 (96.2 to 100)a,b | |
| Test brand | Abbott – ID NOW | 12 | 1853 | 634 | 78.6 (73.7 to 82.8) | 99.8 (99.2 to 99.9) |
| Cepheid – Xpert Xpress | 13 | 1691 | 911 | 99.1 (97.7 to 99.7) | 97.9 (94.6 to 99.2) | |
| Difference |
19.8 (14.9to 24.7) P < 0.0001 |
‐1.9 (‐3.8to ‐0.1) P = 0.036 |
||||
| Abbott – ID NOW (including sensitivity only cohort) | 13 | 1949 | 730 | 81.5 (75.2 to 86.5)a | N/A | |
| Cepheid – Xpert Xpress (including sensitivity only cohorts) | 15 | 1781 | 1001 | 99.1 (97.8 to 99.6)a | N/A | |
| DNANudge – COVID Nudge | 1 | 386 | 71 | 94.4 (86.2 to 98.4) | 100 (98.8 to 100) | |
| Diagnostics for the Real World – SAMBA II | 2 | 321 | 121 | 96.0 (81.1 to 99.3) | 97.0 (93.5 to 98.6) | |
| Mesa Biotech – Accula | 1 | 100 | 50 | 68.0 (53.3 to 80.5) | 100 (92.9 to 100) | |
|
Test brand (restricted to IFU‐compliant) |
Abbott – ID NOW | 4 | 812 | 222 | 73.0 (66.8 to 78.4) | 99.7 (98.7 to 99.9) |
| Cepheid – Xpert Xpress | 2 | 100 | 29 | 100 (88.1 to 100)a | 97.2 (89.4 to 99.3)a | |
| DRW – SAMBA II | 1 | 149 | 33 | 87.9 (71.8 to 96.6) | 97.4 (92.6 to 99.5) | |
| DNANudge – COVID Nudge | 1 | 386 | 71 | 94.4 (86.2 to 98.4) | 100 (98.8 to 100) | |
| Discrepant analysis | Before discrepant analysis | 6 | 1533 | 623 | 97.9 (88.1 to 99.7) | 97.8 (96.6 to 98.6) |
| After discrepant analysis | 6 | 1533 | 632 | 99.2 (93.6 to 99.9) | 99.6 (98.8 to 99.8) | |
| Difference |
1.3 (‐2.8to 5.4) P = 0.528 |
1.8 (0.7to 2.8) P = 0.001 |
||||
| CI: confidence interval; Ct: cycle threshold; IFU: [manufacturers'] instructions for use; N/A: not applicable | ||||||
aSeparate pooling of sensitivity or specificity. b2x2 tables combined prior to calculating estimates.
9.
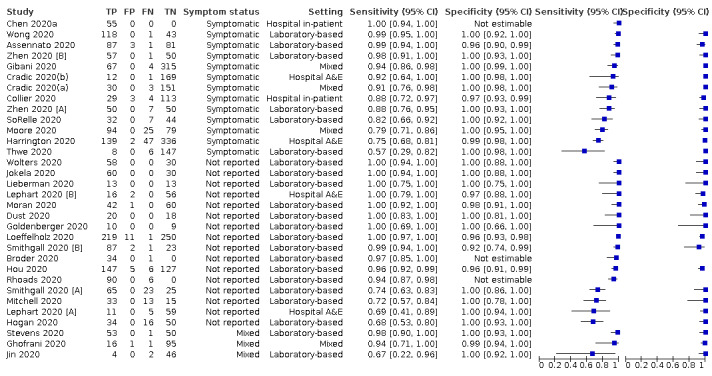
Forest plot of studies evaluating rapid molecular tests. A&E: accident and emergency
10.
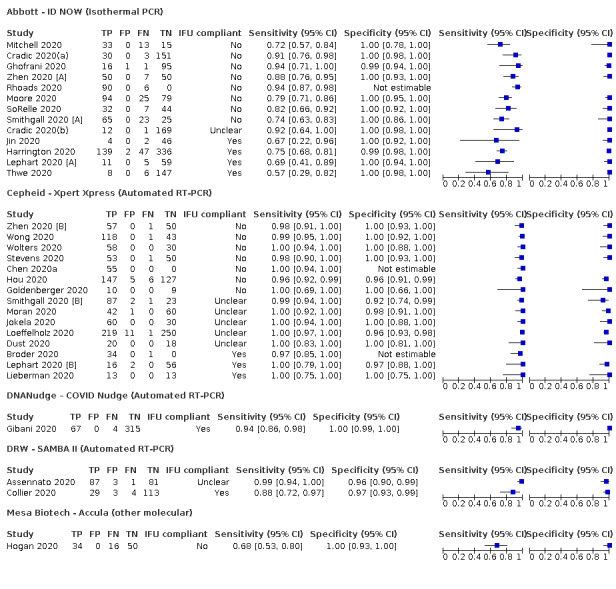
Forest plot by test brand for molecular assays. A&E: accident and emergency; IFU: instructions for use
Accuracy of antigen tests overall and by subgroup
Results showed high levels of heterogeneity in sensitivity. Average sensitivity was 68.9% (95% CI 61.8% to 75.1%) and average specificity was 99.6% (95% CI 99.0% to 99.8%) across the 51 evaluations of antigen tests reporting both sensitivity and specificity (based on 21,614 samples, including 6136 samples with confirmed SARS‐CoV‐2; Table 3; Figure 3). Adding the six ‘sensitivity only’ datasets and single ‘specificity only’ datasets had a negligible impact on results (Table 3). In the sections below we show that there are substantial differences between subgroups of studies according to symptom status, timing, test method and brand, therefore this average value is unlikely to accurately predict the performance of the test in a given setting and should not be used for this purpose.
Subgroup analysis by symptom status
Subgroup analysis by symptom status suggests that average test sensitivity to detect infection is 13.8 percentage points lower in asymptomatic (58.1%, 95% CI 40.2% to 74.1%; based on 12 evaluations, 1581 samples and 295 cases) compared to symptomatic (72.0%, 95% CI 63.7% to 79.0%; based on 37 evaluations, 15,530 samples and 4410 cases) participants (95% CI for the difference in sensitivity: 33.1 percentage points lower to 5.4 percentage points higher; Table 3; Figure 4). Restricting the comparison by symptom status to the nine evaluations reporting data for both symptomatic and asymptomatic subgroups (thus ensuring the comparison is made between the same tests used in the same way) showed a similar difference in sensitivity (14.4 percentage points lower in asymptomatic participants, 95% CI 38.8 lower to 10.0 percentage points higher; Table 3). Average results for the 19 evaluations in participants with mixed symptom status (n = 10) or symptom status not reported (n = 9) were between those observed for the symptomatic and asymptomatic subgroups: sensitivity 63.0% (95% CI 52.2% to 72.6%) and specificity 98.4% (95% CI 98.0% to 98.8%) (6220 samples; 2392 cases).
We did not observe any important differences in specificity according to symptom status (Table 3).
Subgroup analysis by time from symptom onset
We pooled data by time from symptom onset separately for sensitivity and specificity because the majority of evaluations did not report these data for people without SARS‐CoV‐2 (Table 3; Figure 5). Sensitivity was 78.3% (95% CI 71.1% to 84.1%) (26 evaluations; 5769 samples, 2320 cases) in the first seven days after symptom onset compared to 51.0% (40.8% to 61.0%) (22 evaluations; 935 samples, 692 cases) in the second week of symptoms (a decrease of 27.3 percentage points, 95% CI −32.8 to −21.9 percentage points decrease). This difference remained on restriction to the 22 evaluations reporting data for people in both week one and week two of symptoms (removing other between‐study differences; Table 3).
We did not observe any differences in specificity according to time after symptom onset (Table 3).
Subgroup analysis by Ct value
A total of 36 evaluations reported sensitivity according to Ct value using a threshold of 24 (n = 18) or 25 (n = 18) Ct or less to define higher viral load (Table 3; Appendix 15). Summary sensitivity in those with higher viral load was 94.5% (95% CI 91.0% to 96.7%) (based on 2613 cases), compared to 40.7% in those with lower viral load (95% CI 31.8% to 50.3%) (based on 2632 cases) (i.e. sensitivity was 53.8 percentage points lower for those with lower viral load; 95% CI 63.6 to 44.1 percentage points lower)). Applying a Ct threshold of ≤ 33 (n = 13) or < 32 (n = 2) led to a bigger difference in sensitivity although the number of samples in the lower viral load subgroup was considerably smaller: sensitivity associated with higher viral load was 82.5% (95% CI 74.0% to 88.6%) (based on 2127 samples) and for lower viral load was 8.9% (3.3% to 21.7%) (based on 346 samples), a difference of 73.5 percentage points (95% CI 84.7 to 62.4 percentage points lower).
Subgroup analysis by study design
We did not observe any clear differences in average sensitivity or specificity when studies were grouped by study design (15,336 samples and 3536 cases in 29 single group studies and 5729 samples and 2396 cases in 20 two‐group studies; Table 3; Appendix 15). Average sensitivity was lower in two‐group studies (64.1%, 95% CI 48.5% to 77.2%) compared to single‐group studies (72.1%, 95% CI 64.8% to 78.3%), however confidence intervals overlapped and the difference was within that which may be expected by chance (8.0 percentage points lower, 95% CI from 24.2 percentage points lower to 8.2 higher). Average specificities were 2.3 percentage points lower in the two‐group studies (95% CI from 2.9 to 1.6 percentage points lower), at 97.3% (95% CI 96.7% to 97.8%) compared to 99.6% (95% CI 99.1% to 99.8%) in single‐group studies.
Subgroup analysis by test method
We observed differences in accuracy according to test method (Table 3). The majority of evaluations (n = 36; 17,448 samples, 5085 cases) reported using a CGIA, average sensitivity was lower (64.0%, 95% CI 55.7% to 71.6%) than for FIAs (79.6%, 95% CI 67.5% to 88.0%; n = 9; 2820 samples, 712 cases; absolute difference of 15.6 percentage points, 95% CI 2.6 to 28.5 percentage points). We also observed marginal differences in specificity, with estimates of 99.0% (95% CI 98.8% to 99.2%) for CGIA and 97.7% (95% CI 95.3% to 98.8%) for FIA, a difference of 1.3 percentage points (95% from 3.0 percentage points lower to 0.3 higher). Results for lateral flow assays where the method could not be determined (n = 5) and for the single evaluation of an alkaline phosphatase (ALP)‐labelled assay were heterogeneous but largely in the realms of those observed for the other assay types (Table 3).
Results by test brand according to symptom status and IFU compliance
Results by test brand overall and sensitivity analyses by IFU compliance (based on sample type, use of viral transport medium, and time period between sample collection and test procedure) are reported in Table 4. Results by test brand for symptomatic and asymptomatic subgroups overall and by IFU compliance are in Table 5. Given the mixed settings in which asymptomatic individuals were tested (Results of the search), the data for asymptomatic subgroups cannot be considered applicable to any particular scenario for asymptomatic testing. Only three studies reported direct comparisons of tests, two using nasopharyngeal or oropharyngeal samples (Fourati 2020 [A]; Weitzel 2020 [A]).
We observed considerable heterogeneity in sensitivities for all assays.
AAZ – COVID‐VIRO
Two evaluations of the COVID‐VIRO assay included 880 samples and 396 SARS‐CoV2‐positive samples (Figure 7). We did not pool the studies due to the heterogeneity in both sensitivity and specificity, although both were conducted in symptomatic or mainly symptomatic participants using nasopharyngeal samples.
In one study that compared antigen assays using nasopharyngeal samples in viral transport medium, sensitivity was 61.7% (95% CI 55.9% to 67.3%) and specificity (in pre‐pandemic samples) 100% (95% CI 98.9% to 100%; 632 samples, 295 cases;'Fourati 2020 [E]).
The second study used direct swab testing in compliance with the manufacturer’s IFU. Twenty participants in the study who previously tested positive on PCR retested negative with PCR at the time of the antigen test. All twenty samples showed weak lines on antigen testing. We considered these as false positives in the review (based on the negative result of the concurrent PCR test) whereas the study authors considered them to be true positives. With our re‐calculation, the test demonstrated sensitivity of 96.0% (95% CI 90.2% to 98.9%) and specificity of 86.4% (95% CI 79.8% to 91.5%; Courtellemont 2020). Sensitivity in this study may have been inflated by the inclusion of hospitalised, confirmed SARS‐CoV‐2‐positive participants.
Abbott – Panbio Covid‐19 Ag
We identified 11 evaluations of the Panbio assay, including 5691 unique samples, with 2031 SARS‐CoV‐2‐positive cases (Figure 6). One of the 11 evaluations included only SARS‐CoV‐2‐positive cases (n = 182 samples). Studies were conducted in community COVID‐19 test centres or emergency departments (n = 6), in contacts of confirmed cases (n = 2), and laboratory‐based evaluations (n = 2). The setting was not clear in one study. Participants were reportedly symptomatic (n = 5), asymptomatic (n = 1), with mixed symptom status (n = 4), or symptom status was not reported (n = 1). Nine evaluations used nasopharyngeal samples (Albert 2020; Billaud 2020; Fenollar 2020(b); FIND 2020b; Fourati 2020 [C]; Gremmels 2020(a); Gremmels 2020(b); Linares 2020), one (Alemany 2020), tested nasopharyngeal or nasal samples and one (Schildgen 2020 [A]), used bronchoalveolar lavage or throat wash samples. Only three of the 11 evaluations reported product codes for the assays used, one of which was for the assay for use with nasopharyngeal swabs (41FK10) and two (from the same study report) were for the assay for use with nasal swabs (41FK11), although the study reports using nasopharyngeal samples (Gremmels 2020(a); Gremmels 2020(b)).
Five of the 11 evaluations complied with manufacturer IFU for the test. Reasons for non‐compliance included use of viral transport medium, frozen storage, type of swab tested, or lack of clear reporting of test procedures used.
The average sensitivity and specificity of the Panbio assay were:
72.0% (95% CI 60.6% to 81.1%) and 99.3% (95% CI 99.0% to 99.6%) overall (n = 10; 5509 samples; 1849 cases; Table 4);
74.1% (95% CI 60.8% to 84.0%) and 99.8% (95% CI 99.5% to 99.9%) in symptomatic people (n = 8; 3699 samples, 1162 cases); and
58.1% (95% CI 41.7% to 72.9%) and 98.4% (95% CI 92.2% to 99.7%) in asymptomatic people (n = 6; 1097 samples, 190 cases; Table 5).
Restricting to IFU‐compliant evaluations, average sensitivities and specificities were:
72.0% (95% CI 56.5% to 83.5%) and 99.2% (95% CI 98.5% to 99.5%) overall (n = 5; 1776 samples, 362 cases; Table 4);
75.1% (95% CI 57.3% to 87.1%) and 99.5% (95% CI 98.7% to 99.8%) in symptomatic people (n = 3; 1094 samples, 252 cases); and
48.9% (95% CI 35.1% to 62.9%) and 98.1% (95% CI 96.3% to 99.1%) in asymptomatic people (n = 2; 474 samples, 47 cases; Table 5).
The addition of one evaluation that reported sensitivity only in symptomatic participants led to only marginal differences in average sensitivity (Fenollar 2020(a); Table 5).
Becton Dickinson ‐ BD Veritor
We identified three evaluations of the BD Veritor assay, including 727 unique samples, with 180 SARS‐CoV‐2‐positive cases (Figure 6). One of the three evaluations included only SARS‐CoV‐2‐positive cases (n = 125 samples). Studies were conducted in community COVID‐19 test centres (n = 2), or in multiple settings (n = 1). All participants were symptomatic. Two evaluations used combined naso‐ and oropharyngeal samples and one tested nasal samples.
None of the evaluations complied with manufacturer IFU for the test because the interval between sample collection and testing was greater than the maximum of one hour.
Average sensitivity and specificity of the BD Veritor assay were:
82.3% (95% CI 62.1% to 93.0%) and 99.5% (95% CI 98.3%, 99.8%) in symptomatic people (n = 2; 602 samples, 55 cases; Van der Moeren 2020(a); Young 2020; Table 4; Table 5).
Adding the ‘cases only’ evaluation reduced average sensitivity to 79.4% (95% CI 72.9% to 84.7%) (n = 3; 180 cases; Van der Moeren 2020(b)).
The BD Veritor assay requires interpretation using a Veritor analyzer device, but Van der Moeren 2020(a) found that visual inspection of the test device resulted in the same sensitivity as with the Analyzer device, and similar specificity (100% compared to 99% using the Analyzer device).
BIONOTE ‐ NowCheck COVID‐19 Ag
We identified a single IFU‐compliant evaluation of the NowCheck assay in symptomatic participants (FIND 2020a;Figure 7). The study included 400 samples with 102 SARS‐CoV‐2‐positive cases, from participants presenting at a community‐based COVID‐19 test centre.
The sensitivity and specificity in this study were 89.2% (95% CI 81.5% to 94.5%) and 97.3% (95% CI 94.8% to 98.8%; Table 4; Table 5).
Biosynex ‐ Biosynex COVID‐19 Ag BSS
We identified a single evaluation of the Biosynex assay in symptomatic participants (Fourati 2020 [D]), including 634 samples with 297 with confirmed SARS‐CoV‐2 (Figure 7). The evaluation was not in compliance with the manufacturer’s IFU because samples were stored in viral transport medium and frozen prior to testing. The setting in which participants presented for testing was not reported.
Observed sensitivity was 59.6% (95% CI 53.8% to 65.2%) and specificity 100% (95% CI 98.9% to 100%; Table 4; Table 5).
Coris Bioconcept ‐ COVID‐19 Ag Respi‐Strip
The seven evaluations of the Coris Bioconcept assay included 1781 samples, with 707 SARS‐CoV‐2‐positive cases (Blairon 2020; Fourati 2020 [A]; Kruger 2020(b); Lambert‐Niclot 2020; Mertens 2020; Scohy 2020; Veyrenche 2020; Figure 6). Five of the seven were laboratory‐based evaluations with limited detail regarding study participants. One study recruited from community‐based COVID‐19 test centres and one included samples from hospital inpatients. Three studies included only or mainly symptomatic participants, one was in a mixed group and three did not report symptom status.
All evaluations tested naso‐ or oropharyngeal swabs and were compliant with the manufacturer IFU, however, it may be worth noting that the IFU for this assay permits the use of viral transport medium and freezing of samples, although immediate testing is recommended.
The average sensitivity and specificity of the COVID‐19 Ag Respi‐Strip were:
39.7% (95% CI 31.3% to 48.7%) and 98.3% (95% CI 97.4% to 98.9%) overall (n = 7; 1781 samples, 707 cases; Table 4);
34.1% (95% CI 29.7% to 38.8%) and 100% (95% CI 99.0% to 100%) in symptomatic people (n = 3; 780 samples, 414 cases); and
28.6% (95% CI 8.4% to 58.1%) and 100% (95% CI 88.8% to 100%) in asymptomatic people (n = 1; 45 samples, 14 cases; Scohy 2020;Table 5).
E25Bio ‐ DART (nasopharyngeal)
We identified a single evaluation of the E25Bio DART assay that included 190 samples, 100 with SARS‐CoV‐2 (Nash 2020; Figure 7). The symptom status of included participants was not reported and the manufacturer IFU is not yet available as the assay has been submitted for Emergency Use Authorisation (EUA) approval with the US Food and Drug Administration (FDA).
Sensitivity was 80.0% (95% CI 70.8% to 87.3%) and specificity 91.1% (95% CI 83.2% to 96.1%; Table 4).
Fujirebio ‐ ESPLINE SARS‐CoV‐2
We included two eligible evaluations were included, with a total of 265 samples, 165 were SARS‐COV‐2‐positive (Nagura‐Ikeda 2020; Takeda 2020; Figure 7). One study reported only sensitivity data (Nagura‐Ikeda 2020).
Takeda 2020 reported sensitivity of 80.6% (95% CI 68.6% to 89.6%) and specificity of 100% (95% CI 96.4% to 100%) in nasopharyngeal samples (162 samples, 62 cases; Table 4). They did not report symptom status of participants and provided insufficient detail to allow us to judge IFU compliance.
Nagura‐Ikeda 2020 evaluated the assay using saliva samples in symptomatic participants (not within IFU specifications), the ESPLINE assay correctly identified 12 of 103 PCR‐positive samples (sensitivity 11.6%, 95% CI 6.2% to 19.5%; Table 4; Table 5).
Innova Medical Group ‐ Innova SARS‐CoV‐2 Ag
We included one report that evaluated the Innova study as six separate substudies; three reported both sensitivity and specificity (PHE 2020(a); PHE 2020(b); PHE 2020(c) [non‐HCW tested]), two reported sensitivity alone (PHE 2020(d) [HCW tested]; PHE 2020(d) [Lab tested]), and one reported specificity alone (PHE 2020(e); Figure 6). The studies reported a total of 3904 participants, including 1017 SARS‐CoV‐2‐positive cases. Detail regarding symptom status, was limited, however the study populations were coded as: symptomatic (samples from hospital inpatients in PHE 2020(a)), mainly symptomatic for samples from COVID‐19 testing centres (PHE 2020(c) [non‐HCW tested]; PHE 2020(d) [HCW tested]; PHE 2020(d) [Lab tested]), although data on symptom status were reported for only two of these studies (PHE 2020(d) [HCW tested]; PHE 2020(d) [Lab tested]), not reported for the outbreak investigation in PHE 2020(b) and asymptomatic staff screening for PHE 2020(e). The study authors for the outbreak evaluation study did not report the sensitivity value of 28.3% (95% CI 16.0% to 43.5%) in the publications but provided it to us on request.
All evaluations used naso‐ or oropharyngeal samples, two in viral transport medium (PHE 2020(a); PHE 2020(b)), and four using direct swab testing in compliance with manufacturer IFU (PHE 2020(c) [non‐HCW tested]; PHE 2020(d) [HCW tested]; PHE 2020(d) [Lab tested]; PHE 2020(e)).
For studies reporting both sensitivity and specificity, average sensitivity and specificity were:
47.9% (95% CI 34.3% to 61.8%) and 99.8% (95% CI 99.5% to 99.9%) overall (n = 3; 2945 samples, 596 cases; Table 4); and
56.2% (95% CI 52.0% to 60.3%) and 99.8% (95% CI 99.5% to 99.9%) in symptomatic people (n = 2; 2794 samples, 550 cases; Table 5).
Only one of the three studies that reported both sensitivity and specificity was compliant with manufacturer IFU, the sensitivity and specificity were:
57.5% (95% CI 52.3% to 62.6%) and 99.6 (95% CI 99.1%, 99.9%) overall (n = 1; 1676 samples, 372 cases).
Summary results from the four IFU‐compliant evaluations were calculated as follows:
average sensitivity across three evaluations of mainly symptomatic participants 69.1% (95% CI 58.3% to 78.2%; n = 3; 793 cases; Table 4; Table 5);
average specificity from two evaluations of 99.7% (95% CI 99.3% to 99.9%; n = 2; 1842 samples with no SARS‐CoV‐2; Table 4).
Adding data from single‐group evaluations in either RT‐PCR‐positive or RT‐PCR‐negative participants:
average sensitivity was 59.0% (43.4%, 73.0%) (n = 5; 1015 cases)
average specificity was 99.8% (99.5%, 99.9%) (n = 4; 2887 RT‐PCR negative samples) (Table 5).
Results for each of the three IFU‐compliant evaluations by test operator were (Figure 6):
sensitivity of 57.5% (95% CI 52.3% to 62.6%) and specificity 99.6% (95% CI 99.1% to 99.9%), when the test was used by self‐trained, non‐healthcare workers (n = 1; 1676 samples, 372 cases; PHE 2020(c) [non‐HCW tested]);
sensitivity of 70.0% (95% CI 63.5% to 75.9%) when the test was used by healthcare workers (n = 1; 223 cases; PHE 2020(d) [HCW tested]);
sensitivity of 78.8% (95% CI 72.4% to 84.3%) when the test was used by laboratory scientists (n = 1; 198 cases; PHE 2020(d) [Lab tested]).
Liming Bio‐Products ‐ StrongStep® COVID‐19 Ag
We identified a single evaluation of the StrongStep assay in 19 symptomatic participants with nine SARS‐CoV‐2 positive samples ((Weitzel 2020 [B]; Figure 7). We could not identify the manufacturer’s IFU for this assay. The study authors terminated the evaluation early following poor early results for this assay.
Sensitivity was 0% (95% CI 0% to 33.6%) and specificity 90.0% (95% CI 55.5% to 99.7%; 19 samples, 9 cases; Table 4; Table 5).
Quidel Corporation ‐ SOFIA SARS Antigen
We identified a single evaluation of the SOFIA assay in symptomatic participants, including 64 samples with 32 SARS‐CoV‐2‐positive cases (Porte 2020b [A]; Figure 7). The study used combined naso‐ and oropharyngeal swab samples in viral transport medium, therefore the evaluation was not compliant with the manufacturer IFU.
Sensitivity was 93.8% (95% CI 79.2% to 99.2%) and specificity was 96.9% (95% CI 83.8% to 99.9%; Table 4; Table 5).
RapiGEN ‐ BIOCREDIT COVID‐19 Ag
We identified six evaluations of the RapiGen BIOCREDIT assay; these reported data for 2170 samples, with 470 confirmed SARS‐COV‐2‐positive cases (FIND 2020e (BR); FIND 2020e (DE); Mak 2020;Schildgen 2020 [A]; Shrestha 2020; Weitzel 2020 [A]; Figure 6). One laboratory‐based study included cases only (n = 160). The other evaluations included participants from community‐based COVID‐19 test centres (n = 2), emergency departments (n = 1), contact tracing (n = 1) or did not clearly report the setting (n = 1). Two studies included only symptomatic participants, two reported including both symptomatic and asymptomatic participants (mixed group) and one did not report symptom status. All evaluations apart from one (Schildgen 2020 [A]), tested nasopharyngeal or combined naso‐ or oropharyngeal samples.
Only three of the six evaluations complied with manufacturer IFU, with non‐compliance because of the use of viral transport medium, or the type of swab tested.
The average sensitivity and specificity of the BIOCREDIT assay were:
63.3% (95% CI 45.7% to 78.0%) and 99.5% (95% CI 99.1 to 99.8) overall (n = 5; 2010 samples, 310 cases; Table 4);
58.4% (95% CI 36.3% to 77.5%) and 96.4% (95% CI 82.8% to 99.3%) in symptomatic people (n = 3; 608 samples, 206 cases);
63.2% (95% CI 21.7% to 91.4%) and 98.9% (95% CI 82.9% to 99.9%) in asymptomatic people (n = 2; 140 samples, 60 cases) (Table 5).
Restricting to IFU‐compliant evaluations, average sensitivities and specificities were:
73.0% (95% CI 57.4% to 84.4%) and 99.8% (95% CI 99.4% to 99.9%) overall (n = 3; 1828 samples, 189 cases; Table 4);
74.4% (95% CI 65.5% to 82.0%) and 98.9% (95% CI 97.2% to 99.7%) in symptomatic people (n = 1; 476 samples, 117 cases);
85.1% (95% CI 71.7% to 93.8%) and 100% (95% CI 94.6% to 100%) in asymptomatic people (n = 1; 113 samples, 47 cases; Shrestha 2020;Table 5).
The addition of one evaluation that reported sensitivity only led to a decrease in overall average sensitivity of 5.6 percentage points (Mak 2020; Table 5).
Roche ‐ SARS‐CoV‐2
According to the manufacturer IFU, the Roche SARS‐CoV‐2 assay is available under a partnership with SD Biosensor.
There was a single evaluation of the Roche assay using 73 bronchoalveolar lavage or throat wash samples (not covered by the IFU) in participants with mixed symptom status (Figure 7); 42 of the 73 samples were RT‐PCR‐positive (Schildgen 2020 [A]).
Overall, using bronchoalveolar lavage or throat wash samples, the sensitivity and specificity were 88.1% (95% CI 74.4% to 96.0%) and 19.4% (95% CI 7.5% to 37.5%) (73 samples, 42 cases; Table 4). Only the results for the subgroup of 50 throat wash samples could be separated by symptom status:
in symptomatic participants, sensitivity was 100% (95% CI 69.2% to 100%) and specificity was 7.7% (95% CI 0.2% to 36.0%) with 23 throat wash samples and 10 cases;
in asymptomatic participants, sensitivity was 84.6% (95% CI 54.6% to 98.1%) and specificity was 14.3% (95% CI 1.8% to 42.8%), with 27 throat wash samples, 13 cases; Table 5).
Savant Biotech ‐ Huaketai SARS‐CoV‐2 N Protein
We identified a single evaluation of the Huaketai assay in 109 symptomatic participants, using combined naso‐ or oropharyngeal swabs in viral transport medium (Weitzel 2020 [C]; Figure 7). We could not obtain the manufacturer IFU.
Sensitivity was 16.7% (95% CI 9.2% to 26.8%) and specificity was 100% (95% CI 88.8% to 100%; 109 samples, 78 cases; Table 4; Table 5).
SD Biosensor ‐ STANDARD F COVID‐19 Ag
We identified four evaluations of the STANDARD F assay; these reported data for 1552 samples, with 295 confirmed SARS‐COV‐2‐positive cases (FIND 2020d (BR); FIND 2020d (DE); Liotti 2020; Porte 2020b [B]; Figure 6). Three evaluations included all or mainly symptomatic participants from community‐based COVID‐19 test centres and one was a laboratory‐based study that did not provide details regarding symptom status.
All evaluations tested nasopharyngeal or combined naso‐ or oropharyngeal samples, however only two complied with manufacturer IFU. Reasons for non‐compliance were the use of viral transport medium, or lack of information concerning viral transport medium.
The average sensitivity and specificity of the STANDARD F COVID‐19 Ag assay were:
72.6% (95% CI 54.0% to 85.7%) and 97.5% (95% CI 96.4% to 98.2%) overall (n = 4; 1552 samples, 295 cases; Table 4);
78.0% (95% CI 71.6% to 83.3%) and 97.2% (95% CI 96.0% to 98.1%) in symptomatic people (n = 3; 1193 samples, 191 cases; Table 5).
No data for asymptomatic people were available.
Restricting to IFU‐compliant evaluations, average sensitivity and specificity were:
75.5% (95% CI 68.2% to 81.5%) and 97.2% (95% CI 96.0 to 98.1%), both studies in symptomatic people (n = 2; 1129 samples, 159 cases; Table 5).
SD Biosensor ‐ STANDARD Q COVID‐19 Ag
We identified six evaluations of the STANDARD Q assay; these reported data for 3480 samples, with 821 confirmed SARS‐CoV‐2‐positive cases (Figure 6). Four evaluations included participants from community‐based COVID‐19 test centres, one was a laboratory‐based study, and one included multiple settings. Four evaluations included symptomatic or mainly symptomatic participants, and two included mixed symptomatic and asymptomatic participants.
All evaluations tested nasopharyngeal or combined naso‐ or oropharyngeal samples, four of which were compliant with manufacturer’s IFUs, the other two used samples in viral transport medium.
The average sensitivity and specificity of the STANDARD Q COVID‐19 Ag assay were:
79.3% (95% CI 69.6% to 86.6%) and 98.5% (95% CI 97.9% to 98.9%) overall (n = 6; 3480 samples, 821 cases; Table 4);
80.1% (95% CI 68.5% to 88.1%) and 98.1% (95% CI 97.4% to 98.6%) in symptomatic people (n = 5; 2760 samples, 731 cases); and
61.1% (95% CI 37.9% to 80.2%) and 99.6% (95% CI 97.3% to 99.9%) in asymptomatic people (n = 2; 272 samples, 18 cases; Table 5).
Restricting to IFU‐compliant evaluations, average sensitivities and specificities were:
85.8% (95% CI 80.5% to 89.8%) and 99.2% (95% CI 98.2% to 99.6%) overall (n = 4; 2522 samples, 421 cases; Table 4);
88.1% (95% CI 84.2% to 91.1%) and 99.1% (95% CI 97.8% to 99.6%) in symptomatic people (n = 3; 1947 samples, 336 cases); and
69.2% (95% CI 38.6% to 90.9%) and 99.1% (95% CI 95.2% to 100%) in asymptomatic people (n = 1; 127 samples, 13 cases; Table 5).
Shenzhen Bioeasy Biotech ‐ 2019‐nCoV Ag
We included three evaluations of the Bioeasy FIA; these included 965 samples with 177 SARS‐CoV‐2‐positive cases ((Kruger 2020(a); Porte 2020a; Weitzel 2020 [D]; Figure 6). Studies were conducted in hospital emergency departments (n = 2) or a community COVID‐19 test centre (n = 1). Participants in studies were all symptomatic or mainly symptomatic.
Two evaluations used combined naso‐ or oropharyngeal swabs and one tested either nasopharyngeal or oropharyngeal swabs. Two evaluations used swabs in viral transport medium, which was not documented as suitable for use on the manufacturer IFU.
The average sensitivity and specificity of the Shenzhen Bioeasy assay were :
86.2% (95% CI 72.4% to 93.7%) and 93.8 (95% CI 91.9% to 95.3%) overall (all symptomatic; n = 3; 965 samples, 177 cases; Table 4; Table 5).
The single IFU‐compliant evaluation Kruger 2020(a) reported sensitivity of 66.7% (95% CI 38.4% to 88.2%) and specificity of 93.1% (95% CI 91.0% to 94.9%; 727 samples, 15 cases).
We also included an additional study that reported the development of this assay but we did not pool data with the other evaluations as it was a development and not a validation study (Diao 2020; Figure 7). Sensitivity was 67.8% (95% CI 61.0% to 74.1%) and specificity was 100% (95% CI 88.8% to 100%; 239 samples, 208 cases).
Direct test comparisons
Three studies reported direct comparisons of different antigen assays in naso‐ or oropharyngeal samples; however none of the studies had any assay comparisons in common. All three studies utilised swabs in viral transport medium and all were conducted in symptomatic participants. We cannot derive any clear conclusions about comparative performance of tests from these studies.
Figure 8 shows variable diagnostic performance between and to some extent within studies. Four of the five assays in Fourati 2020 [A] demonstrated sensitivities in the range of 55% to 62% (SD Biosensor STANDARD Q, Abbott Panbio Covid‐19 Ag, Biosynex COVID‐19 Ag, AAZ – COVID‐VIRO), with one outlier (Coris Bioconcept – Covid‐19 Ag) at 35% (maximum of 297 cases). Specificity was 100% for all assays apart from SD Biosensor SDQ (specificity 93%; 337 pre‐pandemic samples).
In Porte 2020b [A] (32 cases) both assays had sensitivities over 90% (SD Biosensor STANDARD F and Quidel Sofia SARS Antigen), with specificities 97% (32 non‐COVID‐19 samples)
Weitzel 2020 [A] observed a range in assay sensitivities from 0% for the Liming Bio‐Products assay (based on only nine cases), to 17% (for Savant Biotech – Huaketai SARS‐CoV‐2 N), 62% (RapiGEN – BIOCREDIT COVID‐19 Ag) and 85% for Shenzhen Bioeasy Biotech – 2019 nCov Ag (78 to 80 cases for the latter three assays). Specificities were 100% for all assays (based on 30 to 31 samples) apart from the one from Liming Bio‐Products (specificity 90% based on 10 samples).
Accuracy of rapid molecular tests overall and by subgroup
Average sensitivity and specificity for the 29 rapid molecular test evaluations that included samples with and without SARS‐CoV‐2, were 95.1% (95% CI 90.5% to 97.6%) and 98.8% (95% CI 98.3% to 99.2%; 4351 samples, 1781 with confirmed SARS‐CoV‐2; Table 6). Adding the three 'cases only' studies made little difference to the average sensitivity (95.5%, 95% CI 91.5% to 97.7%; 1973 cases).
Figure 9 demonstrates heterogeneity in sensitivity estimates (ranging from 57% to 100%), with consistently high specificities (92% to 100%, but with upper limits of 95% CIs of 99% or 100% in every study).
Subgroup analyses by viral load
We extracted sensitivity data according to viral load from 10 evaluations of molecular tests, six of which reported data at a Ct threshold for higher viral load of 30 or less (Jokela 2020; Lieberman 2020; Mitchell 2020; Smithgall 2020 [A]; Smithgall 2020 [B]; Wolters 2020), four using Xpert Xpress and two using ID NOW. (Appendix 16)
All sensitivity estimates for the higher viral load subgroups were 100% (based on 204 samples with confirmed SARS‐CoV‐2), with a 95% CI for the average of 98.2% to 100%. For the lower viral load group, average sensitivity was 95.6% (95% CI 55.7% to 99.7%) (149 samples with confirmed SARS‐CoV‐2; Table 6).
We observed a similar pattern for the studies using alternative Ct thresholds to define higher and lower viral load (Appendix 17).
Subgroup analysis by study design
We did not observe any clear differences in average sensitivity or specificity when studies were separated by study design (2899 samples and 976 cases in 18 single‐group studies and 1265 samples and 718 cases in nine two‐group studies; Table 6; Appendix 17). Average sensitivity was higher in two‐group studies (97.2%, 95% CI 90.7% to 99.2%) compared to single‐group studies (93.2%, 95% CI 85.5% to 97.0%); a difference of 4.0 percentage points (95% CI from 2.2 percentage points lower to 10.1 higher). Average specificities had almost identical point estimates at 99.4% (95% CI 98.4 to 99.8%) and 99.3% (95% CI 96.5% to 99.8%) respectively (Table 6).
Abbott – ID NOW
Thirteen studies evaluated the ID NOW assay, with 1949 samples and 730 confirmed SARS‐CoV‐2 cases; one study included only SARS‐CoV‐2‐positive cases (n = 36; Figure 10). Seven evaluations were laboratory‐based, three recruited participants from emergency department settings and three were conducted in multiple settings. Seven studies included only symptomatic participants, two included both symptomatic and asymptomatic people, and four did not report symptom status.
Eleven evaluations used nasopharyngeal or nasal swab samples, one was conducted using saliva samples and one did not specify the sample type. Only four evaluations were compliant with manufacturer IFUs; lack of compliance was based on the use of viral transport medium, sample type, and interval between sample collection and testing.
Pooled analyses demonstrated average sensitivity and specificity of:
78.6% (95% CI 73.7% to 82.8%) and 99.8% (95% CI 99.2% to 99.9%) overall (n = 12; 1853 samples, 634 cases); and
73.0% (95% CI 66.8% to 78.4%) and 99.7% (95% CI 98.7% to 99.9%), restricted to evaluations that were compliant with the manufacturer’s IFU (n = 4; 812 samples, 222 cases; Table 6).
Average sensitivity increased to 81.5% (95% CI 75.2% to 86.5%), with the addition of the cases only study (730 cases; Rhoads 2020).
Cepheid Inc – Xpert Xpress
The Xpert Xpress assay was evaluated in 15 studies using respiratory specimens, with 1781 samples and 1001 confirmed SARS‐CoV‐2 cases; two of the studies included only SARS‐CoV‐2‐positive cases (n = 90; Figure 10). Thirteen evaluations were laboratory‐based, one recruited participants from emergency department settings and one included samples from hospital inpatients. Three studies included only symptomatic participants, one included both symptomatic and asymptomatic people (mixed symptom status), and 11 did not report symptom status.
Fourteen evaluations used nasopharyngeal, oropharyngeal or nasal swab samples, and one was conducted using throat saliva or lower respiratory samples. Only three evaluations were compliant with manufacturer IFUs. Lack of compliance with the IFU was because of the use of frozen samples (n = 8), or sample type (n = 1) or concerns about the timing between sample collection and testing (n = 3).
Pooled analyses demonstrated average sensitivity and specificity of:
99.1% (95% CI 97.7% to 99.7%) and 97.9% (95% CI 94.6 % to 99.2%) overall (n = 13; 1691 samples, 911 with confirmed SARS‐CoV‐2);
100% (95% CI 88.1% to 100%) and 97.2% (95% CI 89.4%, 99.3%), restricted to evaluations that were compliant with the manufacturer’s IFU (n = 2; 100 samples, 29 cases; Table 6)
Average sensitivity did not change with addition of two cases‐only studies (99.1%, 95% CI 97.8% to 99.6%; n = 15; 730 cases; Broder 2020; Chen 2020a).
One additional study considered accuracy in non‐respiratory samples using Xpert Xpress (Szymczak 2020). Sensitivity in stool samples obtained up to 33 days after symptom onset was 93.1% (95% CI 77.2% to 99.1%) and specificity was 96.0% (95% CI 86.3% to 99.5%; 79 samples, 29 cases).
Comparison of ID NOW with Xpert Xpress
Comparing the overall pooled results between ID NOW and Xpert Xpress, the average sensitivity of Xpert Xpress was 19.8 (95% CI 14.9 to 24.7) percentage points higher than that of ID NOW (P < 0.0001; Table 6).
The average specificity of Xpert Xpress was marginally lower than that of ID NOW, a difference of −1.9 percentage points (95% CI −3.8 to −0.1).
DNAnudge – COVID Nudge
We included one evaluation of COVID Nudge with a total of 386 participants and 71 SARS‐CoV‐2‐positive cases (Gibani 2020; Figure 10). Participants were recruited from multiple settings including hospital inpatients (n = 88), accident and emergency (n = 15) and healthcare workers and their families (n = 280). All participants were symptomatic and direct testing of nasopharyngeal samples was used (within manufacturer IFU).
The sensitivity of the COVID Nudge assay was 94.4% (95% CI 86.2 to 98.4%) and specificity was 100% (95% CI 98.8% to 100%; 386 samples and 71 cases; Table 6).
Diagnostics for the Real World (DRW) – SAMBA II
We included two evaluations of SAMBA II with 321 samples (121 with confirmed SARS‐CoV‐2; Figure 10). All participants were symptomatic. One study conducted direct testing of combined naso‐ or oropharyngeal samples from hospital inpatients and the other obtained combined naso‐ or oropharyngeal samples in viral transport medium from Public Health England. It was not reported whether the PHE samples were stored or frozen prior to testing so we could not determine whether they complied with the IFU for the assay.
The average sensitivity and specificity of SAMBA‐II were 96.0% (95% CI 81.1% to 99.3%) and 97.0% (95% CI 93.5% to 98.6%; 2 studies; 321 samples, 121 with confirmed SARS‐CoV‐2; Table 6).
In the IFU‐compliant evaluation, sensitivity was 87.9% (95% CI 71.8% to 96.6%) and specificity was 97.4% (95% CI 92.6% to 99.5%; 149 samples, 33 cases; Collier 2020; Table 6).
Mesa Biotech – Accula
We included one evaluation of the Accula assay with a total of 100 samples (50 SARS‐CoV‐2 positive; Hogan 2020; Figure 10). The study was laboratory‐based and symptom status was not reported.
The study used nasopharyngeal samples in viral transport medium or saline, therefore the evaluation was not compliant with IFU requirements.
The sensitivity and specificity of the Accula test were 68.0% (95% CI 53.3% to 80.5%) and 100% (95% CI 92.9% to 100%; 100 samples, 50 cases; Table 6).
Sensitivity analysis of the impact of discrepant analysis
Six evaluations of molecular tests (in 1533 samples) reported results before and after discrepant analysis where selected samples were re‐tested with either the same (Collier 2020; Harrington 2020; Moran 2020; Stevens 2020), or an alternative RT‐PCR assay (Assennato 2020; Loeffelholz 2020). Four studies also reported re‐testing of samples with the index test (Assennato 2020; Collier 2020; Harrington 2020; Moran 2020; Appendix 16; Appendix 17).
Discrepant analysis reduces the number of samples deemed to be false negative or false positive errors. Discrepant analysis reduced the false negative proportion (1‐sensitivity) from 2.1% to 0.8% and the false positive rate (1‐specificity) from 2.2% to 0.4%. Three of the five studies reporting initially false positive results reported zero false positives after sample re‐testing and one reported a drop in false positives from 11 to 3 (Loeffelholz 2020; Appendix 16). Three of the four studies that reported re‐testing of initially false negative results reported reclassification as true negative on re‐testing, and in the other the single false negative remained as a false negative. Given the bias inherent in choosing the reference test dependent on the observed results, we caution against these findings.
An additional study tested all samples with two different RT‐PCR assays, and hence used a more accurate reference standard in all samples, not just samples with discrepant results (Moore 2020). Six initial true negatives were reclassified as false negatives after the second RT‐PCR. Had discrepant analysis been undertaken these misclassifications would have been missed, further underlining the methodological flaws inherent to discrepant analysis.
Other sources of heterogeneity
We also planned to evaluate the effect of sample type and reference standard.
For sample type, the use of variable combinations of sample types with or without viral transport media created numerous sparse subgroups by sample type (Appendix 18). Instead we considered study compliance with manufacturer IFU requirements which is a more pragmatic classification.
All studies used RT‐PCR alone as the reference standard for diagnosing SARS‐CoV‐2 infection.
Publication bias
We did not formally test for publication bias evident in the pattern of results, but did note that the identity of tests not meeting the PHE assessment criteria were not reported due to confidentiality agreements (PHE 2020(a)).
Discussion
This is the second iteration of a Cochrane living review summarising the accuracy of point‐of‐care antigen and molecular tests for detecting current SARS‐CoV‐2 infection. This version of the review is based on published journal articles or studies available as preprints from 1 January 2020 up until 30 September 2020. In addition, we also included evaluations of antigen assays that were available as independent national reference laboratory publications or that were co‐ordinated and published by FIND, and journal articles that were listed on the Diagnostics Global Health website to 16 November 2020.
Summary of main results
We included data from 77 studies using respiratory specimens, including 24,418 samples (7484 samples with confirmed SARS‐CoV‐2), and one study of faecal specimens (79 samples, 29 with confirmed SARS‐CoV‐2). Forty‐eight studies (reporting 58 test evaluations) considered antigen tests; 30 studies (reporting 33 test evaluations) considered rapid molecular tests, including the single study (evaluation) in faecal samples. Key findings are presented in the Table 1.
We summarise six key findings from this review:
1. Despite a considerable increase in the number of studies evaluating point‐of‐care tests, particularly antigen tests, there are still no published or preprint reports of accuracy for a significant number of commercially produced point‐of‐care tests. This review located evaluations for 16 antigen tests (three of which we could not identify as available for purchase) and five molecular assays. These represent a small proportion of assays currently on the market (118 commercialised antigen tests and 53 molecular assays).
2. The new studies have more robust and appropriate study designs compared to those in the first version of this review. Particularly for antigen tests where there are now studies recruiting participants from community‐based COVID‐19 testing clinics. Reporting of key details, such as settings and symptom status have improved, and studies are now evaluating direct swab testing as would occur in a point‐of‐care setting. However, concerns about risk of bias and applicability of results remain, and further improvements in study methods and reporting are needed before strong conclusions can be drawn about the accuracy of many antigen and molecular tests reviewed here. As it is not known whether these limitations will lead to over‐ or underestimates of test accuracy, estimates should be cautiously interpreted in context of their methodological limitations and the settings in which they were conducted. More direct comparisons of test brands are needed, with evaluations undertaken in the intended use settings for these tests.
Particular methodological concerns include the use of deliberate sampling according to known presence or absence of SARS‐CoV‐2 infection; use of anonymised samples submitted to laboratories for routine RT‐PCR testing (with no setting or participant details); and no information on symptoms or time from symptom onset. Differences in case‐mix related to symptomatic status, time post‐symptom onset and distribution of viral load are likely to have contributed to the observed variation in accuracy.
RT‐PCR was the reference standard in all studies ‐ no study defined the presence of COVID‐19 using clinical or radiological features in the absence of a negative RT‐PCR result.
3. Studies frequently did not follow the manufacturer’s instructions or did not use the test at the point of care. Fewer than half conducted the tests according to the manufacturers' IFU (41% (37/91); 29/58 antigen test evaluations and 8/33 molecular test evaluations). Reasons for non‐compliance included use of frozen samples, use of viral transport media, or lengthy intervals between sample collection and testing. Almost a third of studies (23/78) undertook on‐site, direct swab testing immediately or within an hour of sample collection; trained laboratory staff conducted tests in 16 (21%) studies, and 31 (40%) studies did not clearly describe the test operator and setting for the test procedure but we inferred that tests were carried out in a centralised laboratory setting, for example based on reported delays between collection and testing or reported use of archived or frozen samples.
4. For antigen test evaluations in symptomatic participants, we observed considerable heterogeneity in sensitivities (and to a lesser extent the specificities). Whilst the average sensitivity was 72.0% (95% CI 63.7% to 79.0%) and specificity was 99.5% (95% CI 98.5% to 99.8%), average sensitivity decreased with time since onset of symptoms, being higher in the first week (78.3%, 95% CI 71.1% to 84.1%) than when done later (51.0 95% CI 40.8% to 61.0%). Sensitivity was high in those with higher viral loads defined by Ct values ≤ 25 (94.5% 95% CI 91.0% to 96.7%) compared to those with lower viral loads (40.7%, 95% CI 31.8% to 50.3%). Focusing on studies that used the test in accordance with the manufacturer’s instructions, sensitivities for different brands varied from 34% to 96% (either based on pooled results or single studies). WHO have set a minimum 'acceptable' sensitivity requirement of 80%, and acceptable and ideal (or 'desirable') specificity requirements of 97% and 99% respectively (WHO 2020c). Only one assay (SD Biosensor STANDARD Q) met the WHO acceptable criterion for sensitivity based on pooled results of several studies. One further test (BIONOTE NowCheck) also met the acceptable sensitivity criterion, but only one study evaluated it. Abbott Panbio met the sensitivity criterion in individual studies but not overall. The acceptable performance criterion of 97% specificity was also met for all three tests, and two tests met the desirable criterion of more than 99% specificity (Abbott Panbio and SD Biosensor STANDARD Q).
Considerable heterogeneity in sensitivities remained after restricting analyses by test brand and symptom status, suggesting an effect not only from participant characteristics but from setting, sample type and collection method, sample storage and preparation, and testing procedures that cannot be easily unpicked. The PHE studies included in this review allow some consideration of the effect of test operator experience on the accuracy of the Innova test although different samples were tested by each test operator such that only an indirect comparison of sensitivity can be made. Sensitivity increased from 57.5% (95% CI 52.3%, 62.6%; 372 samples) when testing was conducted on‐site by trained non‐healthcare workers (PHE 2020(c) [non‐HCW tested]), to 70.0% (95% CI 63.5% to 75.9%; 223 samples) in samples tested on‐site by healthcare workers ((PHE 2020(d) [HCW tested]), to 78.8% (95% CI 72.4% to 84.3%; 198 samples) for those tested by laboratory scientists (PHE 2020(d) [Lab tested]). The effect of test operator on accuracy has been observed for rapid diagnostic tests for other infectious diseases such as malaria (Boyce 2018; Landier 2018), and is worthy of further investigation for diagnosis of SARS‐CoV‐2.
5. Twelve studies evaluated the accuracy of antigen tests in asymptomatic people for detection of SARS‐CoV‐2 infection defined by PCR status. As discussed, this does not address the issue of whether the test is identifying those who are infectious (as there is no reference standard that can be used). The average sensitivity for detecting infection in asymptomatic participants was 58.1% (95% CI 40.2% to 74.1%) with specificity of 98.9% (95% CI 93.6% to 99.8%), both lower than in symptomatic people. Only half of studies reported clearly defined asymptomatic cohorts (e.g. preventive screening in the general population (n = 1), in returning travellers (n = 1), or in contacts of confirmed cases (n = 4)), the other six reported asymptomatic subgroups from mixed symptom cohorts. Only one of the 12 studies provided data by viral load (Fenollar 2020(b)); 5% (1/22) of RT‐PCR‐positive samples had a Ct value of 25 or less, but 50% (11/22) had Ct values of 30 or less. No information on time after exposure to infection was reported.
6. For rapid molecular assays there were differences between test brands. Most data were for ID NOW and Xpert Xpress assays; average sensitivity for ID NOW was 78.6% (95% CI 73.7% to 82.8%) and Xpert Xpress 99.1% (95% CI 97.7% to 99.7%). Specificity for ID NOW was 99.8% (95% CI 99.23%, 99.9%) and Xpert Xpress 97.9% (95% CI 94.6% to 99.2%). These differences are beyond those expected by chance (P < 0.0001).
We were not able to investigate the effects of symptomatic status, or time from symptom onset: 12/29 were from symptomatic populations, three from ‘mixed’ symptomatic and asymptomatic populations (percentage from each group not reported), and the remaining 14 evaluations provided no information on symptom status (2/14 recruited from A&E and 12 were laboratory‐based). These and other methodological limitations in the studies mean that we do not know how the assays would perform in any specific clinical setting when used in people suspected of having SARS‐CoV‐2 infection on the basis of symptoms, or of exposure to a confirmed case in the absence of symptoms. It is likely however that some difference in sensitivity between ID NOW and Xpert Xpress would be maintained in the absence of bias. The difference in specificity between the tests is small (ID NOW being 1.9% more specific compared to Xpert Xpress), but potentially important especially if used in a low‐prevalence setting. However, this difference in specificity would not be an issue should test‐positives be confirmed by a laboratory‐based RT‐PCR assay.
7. There are proposals for repeated use of antigen tests in different asymptomatic groups, such as school children and staff, hospital and care home workers, and even the general public, with a variety of different testing strategies. We found no data or studies evaluating the accuracy of any of these serial screening strategies.
We did not formally compare antigen with molecular assays because there were no head‐to‐head comparisons of the two test types. Instead, we illustrate predicted numbers of true positives, false positives, false negatives and true negatives, applying summary estimates of test accuracy to a hypothetical cohort of people suspected of SARS‐CoV‐2 infection across a range in prevalence of SARS‐CoV‐2 infection (Table 1). For both antigen and molecular assays, we only use summary data from evaluations conducted in accordance with manufacturers’ IFUs, and for antigen tests we used separate results from symptomatic and asymptomatic participants.
Illustration of predicted effect of antigen testing by symptom status
For antigen test evaluations in symptomatic people, we selected three assays representing the range in observed average sensitivities: Coris Bioconcept COVID‐19 Ag Respi‐Strip (34.1% to 95% CI 29.7% to 38.8%), Abbott ‐ Panbio Covid‐19 Ag (75.1% to 95% CI 57.3% to 87.1%); and SD Biosensor ‐ STANDARD Q COVID‐19 Ag (88.1% to 95% CI 84.2% to 91.1%). Average specificities for the same three assays were 100% (95% CI 99.0% to 100%) to 99.5% (95% CI 98.7% to 99.8%) and 99.1% (95% CI 97.8% to 99.6%) respectively. Applied to a cohort of 1000 people with signs and symptoms of COVID‐19, in whom 50 people had confirmed infection (prevalence of 5%), for the three assays above we predicted that:
17, 43 or 53 people would have a positive test result, of which 0, 5 and 9 would be false positives (positive predictive values (PPV) 100%, 88.4% and 83.0%, respectively), and
33, 12 and 6 people with negative test results would be falsely negative (negative predictive values (NPV) 96.6%, 98.7%, and 99.4%).
Increasing the prevalence to 10% or 20%, increases PPV and decreases NPV. As there is considerable heterogeneity in the estimates of sensitivity, the values observed in practice could vary considerably from these figures as shown by the estimates derived from the confidence intervals (Table 1).
For antigen test evaluations in asymptomatic participants there was considerably less available data from IFU‐compliant evaluations. We selected the same three exemplars, average sensitivities for identification of any infection (whether infectious or not) were lower than for symptomatic populations: 28.6% (95% CI 8.4% to 58.1%) for the Coris Bioconcept assay; 48.9% (95% CI 35.1% to 62.9%) for the Abbott assay; and 69.2% (95% CI 38.6% to 90.9%) for the SD Biosensor assay. Average specificities for the same three assays were: 100% (95% CI 88.8% to 100%), 98.1% (95% CI 96.3% to 99.1%), and 99.1% (95% CI 95.2% to 100%).
Applying the average values to a larger cohort of 10,000 people asymptomatic for COVID‐19 and with a lower prevalence of 0.5% in whom 50 people had confirmed infection (infectious or not):
14, 213 or 125 individuals would have a positive test result of which 0, 189 and 90 would be false positives (PPVs of 100%, 11% and 28%, respectively), and
36, 26 and 15 people with negative test results would be falsely negative (NPVs 99.6%, 99.7%, and 99.8%).
We derived the summary estimates used in these calculations from asymptomatic participants identified for testing in a number of scenarios and they cannot be directly translated to a particular setting, such as mass screening, for example. The confidence intervals for the average estimates used in these calculations are also extremely wide for both sensitivities and specificities, such that the numbers of false positives and false negatives observed in practice could differ substantially from these figures. Increasing the prevalence of confirmed SARS‐CoV‐2 infection to 1% or 2% makes little difference to the absolute number of false positive results for these assays, but has a large relative effect when considered in relation to the number of positive test results (PPVs for the Abbott and SD Biosensor assays increasing to 40% and 61% at 2% prevalence).
Illustration of predicted effect of rapid molecular tests for symptomatic testing
For molecular assays, data from IFU‐compliant evaluations were available for four of the five assays: ID NOW (Abbott Laboratories), Xpert Xpress (Cepheid Inc), SAMBA II (Diagnostics for the Real World) and COVID Nudge (DNAnudge). Average sensitivities were derived as 73.0% (95% CI 66.8% to 78.4%), 100% (95% CI 88.1% to 100%), 87.9% (95% CI 71.8% to 96.6%) and 94.4% (95% CI 86.2% to 98.4%). Average specificities were 99.7% (95% CI 98.7% to 99.9%), 97.2% (95% CI 89.4% to 99.3%), 97.4% (95% CI 92.6% to 99.5%) and 100% (95% CI 98.8% to 100%), respectively (Table 1).
Data by symptom status for these assays were very limited, therefore we assumed that the intended use is most likely to be for diagnosis of acute infection in symptomatic individuals and have applied the average estimates of accuracy to a hypothetical cohort of 1000 people, at prevalences of 5%, 10% and 20% (Table 1). If 50 of 1000 people had confirmed infection (5% prevalence):
40, 77, 69 and 47 individuals would have a positive test result of which 3, 27, 25 or 0 would be false positive (PPVs of 93.0%, 64.9%, 63.8%, and 100% respectively).
14, 0, 6 and 3 people with negative test results would be falsely negative (NPVs 98.6%, 100%, 99.4% and 99.7%).
Increasing the prevalence of confirmed SARS‐CoV‐2 infection to 10% or 20% has a large relative effect when considered in relation to the number of positive test results for both Xpert Xpress and SAMBA II (PPVs were 64.9% and 63.8% at 5% prevalence compared to 90.1% and 89.3% at 20% prevalence). Less variation in PPV was observed for ID NOW and COVID‐Nudge because of the higher observed specificities. The NPV for the molecular assays is not affected to the same degree by these prevalence changes because of their relatively high sensitivities and the relatively low‐prevalence scenarios being considered.
Across all exemplar assays in the Table 1, we observed the widest variation in NPV for the Coris Bioconcept antigen assay in symptomatic participants (86% to 97%), demonstrating that even in a low‐prevalence setting, tests with poor sensitivity can have a considerable impact on the level of confidence that can be had in a negative test result.
Strengths and weaknesses of the review
Our review used a broad search screening all articles concerning COVID‐19 or SARS‐CoV‐2. We undertook all screening and eligibility assessments, QUADAS‐2 assessments (Whiting 2011), and data extraction of study findings independently and in duplicate. Although it is possible that the use of artificial intelligence text analysis to identify studies most relevant to diagnostic questions may have led to some eligible studies being missed, we believe that the multi‐stranded search strategy used will have identified most if not all relevant literature. Whilst we have reasonable confidence in the completeness and accuracy of the findings up until the search date, should errors be noted please inform us at coviddta@contacts.bham.ac.uk so that we can verify and correct in our next update.
We undertook a careful assessment of sample preparation and biosafety requirements as well as time to test result, to ensure that included tests were suitable for use at the point of care. The application of these index test criteria led to the exclusion of 39 of the 85 studies that we excluded on the basis of the index tests evaluated. Evaluations of alternative laboratory‐based molecular technologies are under consideration for inclusion in another review in our series of Cochrane COVID‐19 diagnostic test accuracy reviews. Furthermore, for this iteration of the review, we explicitly considered whether the test evaluations were conducted in accordance with the manufacturer IFU, regarding the sample types used, the use of viral transport medium and the permitted time between sample collection and testing.
We did not consider any manufacturer statements on the intended use of the tests by population, but we are aware that some IFUs recommend testing only in symptomatic people and within certain time frames after symptom onset (e.g. the Innova assay). Where possible, however, we did provide data separately for symptomatic and asymptomatic participants and identified clear trends towards lower sensitivities in asymptomatic individuals for detection of infection. We were unable to assess the accuracy of antigen tests for identification of infectious individuals, as there is no established reference standard for infectiousness (and it seems unlikely that one will ever be established). We have presented results by Ct value where it has been reported by the individual studies. We recognise the limitations from this approach, and given the extent to which RT‐PCR Ct values vary between assays (Vogels 2020), and between laboratories, we strongly caution against the direct application of our results in high and low Ct value subgroups to any particular clinical context. There is no 'step change' in 'infectiousness' according to any fixed Ct value; increasing numbers of studies demonstrate successful viral culture in individuals considered to have 'low' viral load (Jaafar 2020; Singanayagam 2020), and, more importantly, that transmission of infection does occur from index cases with low RT‐PCR Ct values (Lee 2021; Marks 2021). Ultimately, viral load on its own is only one factor influencing an individual's ability to transmit infection, 'infectiousness' being modified by host factors such as the health of an individual’s immune system or presence of comorbidities, and environmental risk factors including closeness and length of contact with others.
Weaknesses of the review primarily reflect the weaknesses in the primary studies and their reporting. Although study quality improved in comparison to the first iteration of this review, many studies continue to omit descriptions of participants, and key aspects of study design and execution. In order to include data for all tests in pooled analyses we had to include some samples multiple times. We have been explicit about these issues where they arose. It is possible that eligible studies have been missed by our search strategy however we believe the risk to be very low considering our broad approach to identification of literature. Despite our best efforts to be as comprehensive as possible, new evaluations are continuously becoming available and it is impossible for any published and peer‐reviewed systematic review to be fully up to date.
Around a quarter (18/78) of the studies we have included are currently only available as preprints, and as yet, have not undergone peer review. As published versions of these studies are identified in the future, we will double‐check study descriptions, methods and findings, and update the review as required.
Applicability of findings to the review question
There are an increasing number of roles and testing strategies for which antigen and rapid molecular assays are considered, and it is likely that the performance of these tests needs to be considered separately for each of the use cases.
Our review shows that antigen tests do not appear to perform as well in asymptomatic populations compared to symptomatic populations for detecting infection. The amount of available data for asymptomatic populations is less than that from symptomatic populations and is also based on asymptomatic individuals tested in a range of scenarios, from preventive or targeted screening, to contact tracing or testing at dedicated COVID‐19 test centres, which may explain some of the observed variability. It is also not clear whether individuals in these studies were truly cases of asymptomatic infection as opposed to pre‐ or post‐symptomatic, or were even mildly symptomatic and mislabelled as asymptomatic. Incomplete symptom assessment and lack of adequate follow‐up to identify subsequent development of symptoms or previous history of symptoms can all contribute to inappropriate classification of individuals as asymptomatic infection (Meyerowitz 2020). As the studies in our review did not systematically attempt to identify pre‐ or post‐symptomatic individuals, it may be more appropriate to consider the estimates for test accuracy for asymptomatic populations as primarily representing accuracy in those without clearly defined symptoms at the time of testing.
We are aware that several important studies in asymptomatic individuals have been reported since the close of our search. In mass screening in Liverpool, Innova was positive in 28 of 70 PCR‐detected cases (sensitivity for infection 40.0%, 95% CI 28.5% to 52.4%) and 26 of 39 with Ct values less than 25 (sensitivity 66.7%, 95% CI 49.8% to 80.9%). Screening University of Birmingham students found 2 of 7185 students positive with Innova, and estimated sensitivity of 3.2% (95% CI 0.6% to 15.6%) for detecting any infection, 9.1% (95% CI 1.0% to 49.1%) for Ct values less than 30 and 100% (95% CI 15.8% to 100%) for Ct less than 25 (Ferguson 2020). BinaxNOW (which uses the same test strip as PanBio) has been tested in asymptomatic groups: in San Francisco the test detected 7 of 11 PCR‐positive cases (sensitivity 63.6%, 95% CI 30.8% to 89.1%), and 6 of 6 with Ct values less than 30 (100%, 95% CI 54.1% to 100%; Pilarowski 2021); in a drive‐through centre in Massachusetts it detected the virus in 70 of 107 in adults (sensitivity 65.4%, 95% CI 55.6 to 74.4) and 40 of 57 in children (70.2%, 95% CI 56.6% to 81.6%)); no breakdown by viral load is available (Pollock 2020). The specificity of the tests in all studies has remained high (above 99%). This selection of results is not based on a systematic search (this will occur in the next update) but these results suggest that emerging evidence is illustrating a range of sensitivity values for the ability of the tests to detect infection, with high detection rates only in groups with very high viral loads.
Given the superior test performance characteristics for symptomatic populations in the first week of symptoms and in those with higher viral loads, the observed poorer performance in those without symptoms is perhaps not surprising. Evidence suggests that higher viral loads are observed in the first week of illness, beginning two days prior to the development of symptoms (Cevik 2021). Viral load patterns in asymptomatic people are less clear but similarly high titers of SARS‐CoV‐2 have been observed at the onset of infection with a suggestion of faster clearance (Cevik 2021). However, variation in viral trajectories means that even if an asymptomatic person can identify a clear contact with a confirmed case of SARS‐CoV‐2 infection, it is not possible to pinpoint when (or even if) that individual will have a sufficient viral load to be detected on antigen testing. A serial testing policy would be likely to identify at least some infected asymptomatic contacts, but comes at the cost of increased numbers of false positives, especially in low‐prevalence settings. There were no evaluations of serial testing in any of the studies.
For molecular tests, we observed a lack of studies undertaken in intended use settings, with most data being from laboratory testing. Although more evidence is available for accuracy in symptomatic people, applicability issues regarding the way in which the tests are carried out and in how cases of SARS‐CoV‐2 infection are defined remain, and it is not yet possible to determine how tests will perform in practice.
We recommend caution in applying the results outside of the individual study (or closely related) contexts and use case scenarios.
Authors' conclusions
Implications for practice.
We consider the implications for practice for this review separately for symptomatic and for asymptomatic testing.
In the Role of index test(s) section, we suggested that for symptomatic individuals, and if sufficiently accurate, point‐of‐care testing could be used either to replace laboratory‐based RT‐PCR or as a triage to RT‐PCR. As point‐of‐care tests are more accessible and provide a result more quickly than RT‐PCR, theoretically their use may increase detection and speed up isolation and contact‐tracing, leading to reduction in disease spread and reduce the burden on laboratory services.
The evidence included to date suggests that:
1. For diagnosis in symptomatic individuals in the first few days of symptoms, the most accurate rapid antigen tests are a useful alternative to laboratory‐based RT‐PCR where immediate results are required for timely patient management or where there are significant logistical or financial challenges in delivering RT‐PCR in a timely manner. Rapid antigen tests are only sufficiently sensitive in the first week since onset of symptoms.
Antigen tests vary in sensitivity, and only those shown to meet appropriate criteria, such as WHO's priority target product profiles for COVID‐19 diagnostics (i.e. sensitivity ≥ 80% and specificity ≥ 97%; WHO 2020c), could be considered as a rational substitute for RT‐PCR.
Tests had high specificity, thus in symptomatic populations (where prevalence is likely to be high) the risk of false positives is low. At 80% sensitivity compared to RT‐PCR, the probability that infected individuals are missed is 20% higher than for RT‐PCR. Thus the possibility of false negative results should be considered in those with a high clinical suspicion of COVID‐19, particularly if tested several days after onset of symptoms when viral load levels may have fallen.
2. Rapid antigen tests may be used simultaneously in combination with RT‐PCR for symptomatic people, particularly where RT‐PCR turn‐around times are slow, to exploit the benefits of earlier results and consequent contact‐tracing and isolation. Given the risk of false‐negative results, isolation may be required until RT‐PCR‐negative results are obtained. Similarly, for investigation of local outbreaks, rapid antigen testing in a clearly defined population may establish cases and contacts that require isolation whilst awaiting results from RT‐PCR.
In other circumstances rapid antigen tests may be used to triage to follow‐on RT‐PCR tests (rather than all receiving PCR tests) dependent on prevalence and the consideration of the consequences of false positive and false negative results.
Where prevalence is low, positive rapid test results require confirmatory testing to avoid unnecessary quarantine measures (PPVs around 85% to 90% for antigen assays mean that between 1 in 10 and 1 in 7 positive results will be falsely positive). If unverified, negative rapid test results should be delivered with appropriate advice on self‐isolation procedures for the duration of symptoms in order to minimise the effect on transmission of infection from missed cases. RT‐PCR tests should still be considered for people with a high clinical suspicion of COVID‐19 and negative rapid test..
Where prevalence is higher (i.e. 20% or higher), false positives are less of a concern (PPVs are 96% to 100%) but the impact from false negative results becomes increasingly important and all test negatives may be considered for verification. At 20% prevalence, and using data for the more sensitive of our three exemplar assays, between 3% and 6% of those with negative rapid test results are missed cases of SARS‐CoV‐2 (24 to 50 cases missed out of a total of 200 cases). The lower the NPV the greater the potential effect on transmission of infection from missed cases and greater the impact from delays in commencement of contact tracing. For scenarios in which positive results do not have confirmatory testing, it is important that assays with high specificities (in the range of 99% to 100%) are selected in order to minimise the impact from false positive results at higher prevalences of disease.
3. We identified virtually no evidence for mass screening of asymptomatic individuals using rapid antigen tests in people with no known exposure. A small study screening travellers returning from high‐risk countries (Cerutti 2020), identified only five SARS‐CoV‐2 infections (prevalence of 3%) with a reported sensitivity of antigen testing for detecting infection of 40%. However, important larger studies have been published since the end of our search, as mentioned above.
The key focus in mass screening is identification of individuals who are or will become infectious. PCR‐positives define those who had detectable viral particles on their swab, which will include most of those who are or will become infectious, but also include individuals post‐infection with residual viral particles. Without a reference standard for infectiousness, test accuracy studies cannot assess the ability of the test to detect the infectious subgroup of infections, and cannot provide evidence as to how well rapid antigen tests differentiate between individuals requiring isolation and those who provide no risk. The effectiveness of mass screening using these tests will only be established though outcome studies, such as cluster‐randomised community trials.
Given the low false positive rate of rapid tests, when used in a period of outbreak, those found testing positive will have a high chance of being true positives, and thus the test can be used to identify cases requiring isolation. Consideration should be made as to whether test positives should be confirmed with PCR to identify false positives. With a 1% prevalence, a test with 40% sensitivity and 99.6% specificity would yield as many false positives as true positives.
However, the low and variable sensitivity, and lack of evidence that those who test negative are not, or will not become, infectious indicates that those who are rapid antigen test‐negative cannot be considered free of risk of being, or of becoming, infectious. In any screening or mass testing programme people testing negative may still have a non‐negligible risk of infection.
4. We did not find any evidence of test accuracy in at‐risk asymptomatic groups, such as contacts of confirmed cases, hospital workers, or during local outbreaks at schools, workplaces, or care homes. The impact of low‐sensitivity tests in these settings is greater than in mass screening, as there will be higher numbers of false negatives, which could either create new outbreaks or will increase the severity of existing outbreaks. Positive cases will be more likely to be true positives than in mass screening settings.
5. We did not find any evidence evaluating the repeated use of tests. Although serial testing (over a number of days), or combinations of different rapid tests (e.g. an antigen test followed by a rapid molecular test) on the same sample are proposed to overcome the limitations of low test sensitivity, they all require validation. Use of multiple tests may increase false positive results, and there are likely to be many individuals with repeated false negative results reducing the expected benefit of subsequent tests. It is unlikely that models will be able to predict how well repeated tests and test combinations would work.
6. Some rapid molecular tests showed promising accuracy levels approximating those of laboratory‐based RT‐PCR and thus may have a role in small‐capacity settings where obtaining test results within two hours will enable appropriate decision making. Results for Xpert Xpress, COVID Nudge and SAMBA II all showed high sensitivity and specificity. However, we identified methodological concerns with many of the evaluations such that we cannot be certain as to how the tests will perform when used in a point‐of‐care setting. Any application in practice should be accompanied with a proper evaluation to ascertain performance in real‐world settings. Rapid molecular tests do not have all the logistical advantage of rapid antigen tests and the resource implications of their use at scale are potentially high, but they may be well suited for some testing scenarios. There is no evidence for use of rapid molecular tests in asymptomatic populations.
Our conclusions are in line with those in the first version of this review despite the increase in the evidence base. Ultimately, decisions around rapid testing will be driven not only by diagnostic accuracy but by acceptable levels of test complexity, time to result, access and acceptability to those being tested, and how test results influence individual behaviour, all of which might vary according to the setting in which the tests are to be used.
Implications for research.
There is now a considerable volume of research for point‐of‐care tests for SARS‐CoV‐2 infection. However further well designed prospective and comparative evaluations of individual tests and test strategies in clinically relevant settings are urgently needed. Studies should recruit consecutive series of eligible participants and should clearly describe the clinical status, document time from symptom onset or time since exposure. Point‐of‐care tests must be conducted in accordance with manufacturer instructions for use, and across the spectrum of point‐of care settings and test operators.
There needs to be evaluations of both individual tests and strategies of use of repeated tests. For molecular assays field trials are needed, not only to demonstrate test accuracy in these groups but acceptability and ease of use outside of centralised laboratories.
We observed a number of studies of molecular assays employing discrepant analysis to confirm the disease status of samples with false positive results in particular. There is a considerable risk of this type of selective re‐testing leading to distorted results. If there is sufficient concern about the reliability of a single RT‐PCR test then all samples should be tested with two RT‐PCR assays. Finally, any future research study needs to be clear about eligibility and exclusion decisions throughout the whole diagnostic pathway, and should conform to the updated Standards for Reporting of Diagnostic Accuracy (STARD) guideline (Bossuyt 2015).
Consideration needs to be made of the best method for evaluating mass screening programmes. Whilst test accuracy studies help indicate which tests are likely to detect the greatest numbers of cases with the fewest false positives, assessing whether detecting asymptomatic cases leads to worthwhile reductions in disease spread will only be properly answered by studies of impact not accuracy.
What's new
| Date | Event | Description |
|---|---|---|
| 19 July 2022 | Amended | Amended to a previous version following an unintentional amendment. An update is pending. |
History
Review first published: Issue 8, 2020
| Date | Event | Description |
|---|---|---|
| 15 April 2021 | Amended | Clarification in Appendices that isothermal amplification is not a RT‐PCR test. |
| 24 March 2021 | Amended | Correction of typo in abstract |
| 24 March 2021 | Amended | Amendment to PLS title |
| 9 March 2021 | New citation required and conclusions have changed | This review has been updated and the conclusions have changed |
| 30 September 2020 | New search has been performed | We have updated our review and now include 64 study reports in 78 study cohorts, evaluating 16 antigen and 5 molecular assays |
Acknowledgements
Members of the Cochrane COVID‐19 Diagnostic Test Accuracy Review Group include:
the project team (Deeks JJ, Dinnes J, Takwoingi Y, Davenport C, Leeflang MMG, Spijker R, Hooft L, Van den Bruel A, McInnes MDF, Emperador D, Dittrich S, Cunningham J);
-
the systematic review teams for each review:
Molecular, antigen, and antibody tests (Adriano A, Arevalo‐Rodriguez I, Beese S, Buitrago DC, Ciapponi A, Domen J, Dretzke J, Ferrante di Ruffano L, Harris I, Mateos M, Price M, Taylor M, Taylor‐Phillips S)
Signs and symptoms (Stuyf T, Domen J, Horn S)
Routine laboratory markers (Yang B, Langendam M, Ochodo E, Guleid F, Holtman G, Verbakel J, Wang J, Stegeman I)
Imaging tests (Salameh JP, McGrath TA, van der Pol CB, Frank RA, Prager R, Hare SS, Dennie C, Jenniskens K, Korevaar DA, Cohen JF, van de Wijgert J, Damen JAAG, Wang J);
the wider team of systematic reviewers from the University of Birmingham, UK who assisted with title and abstract screening across the entire suite of reviews for the diagnosis of COVID‐19 (Agarwal R, Baldwin S, Berhane S, Herd C, Kristunas C, Quinn L, Scholefield B).
The editorial process for this review was managed by Cochrane's Editorial and Methods Department Central Editorial Service in collaboration with Cochrane Infectious Diseases. We thank Helen Wakeford, Anne‐Marie Stephani and Deirdre Walshe for their comments and editorial management. We thank Liz Bickerdike for comments on the Abstract. We thank Robin Featherstone and Douglas M Salzwedel for comments on the search and Mike Brown and Paul Garner for sign‐off comments. We thank Denise Mitchell for her efforts in copy‐editing this review.
Thank you also to peer referees Kristien Verdonck, David Sinclair and Jim Hugget, consumer referees Brian Duncan and Ceri Dare, methodological referees Mia Schmidt‐Hansen and Jo Leonardi‐Bee, for their insights.
The editorial base of Cochrane Infectious Diseases is funded by UK aid from the UK Government for the benefit of low‐ and middle‐income countries (project number 300342‐104). The views expressed do not necessarily reflect the UK Government’s official policies.
The authors thank Dr Mia Schmidt‐Hansen who was the Cochrane Diagnostic Test Accuracy (DTA) Contact Editor for this review; the clinical and methodological referees; the Cochrane DTA Editorial Team; and Anne Lawson who copy‐edited the protocol. We would also like to thank all corresponding authors who provided additional information regarding their studies, and colleagues at both the Norwegian Insititute of Public Health and the EPPI‐Centre who provided updates from their COVID‐19 living evidence maps.
Jonathan Deeks is a UK National Institute for Health Research (NIHR) Senior Investigator Emeritus. Yemisi Takwoingi is supported by a NIHR Postdoctoral Fellowship. Jonathan Deeks, Jacqueline Dinnes, Yemisi Takwoingi, Clare Davenport and Malcolm Price are supported by the NIHR Birmingham Biomedical Research Centre. Sian Taylor‐Phillips is supported by an NIHR Career Development Fellowship. This paper presents independent research supported by the NIHR Birmingham Biomedical Research Centre at the University Hospitals Birmingham NHS Foundation Trust and the University of Birmingham. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care.
Appendices
Appendix 1. Summary of World Health Organization and Chinese National Health Commission Guidelines for the diagnosis of SARS‐CoV‐2
Table A: World Health Organization guidelines for the diagnosis of SARS‐CoV‐2a
Includes laboratory testing guidelines and global surveillance guidelines
| Date range (2020) | Definition of confirmed case | Definition of confirmed non‐case | Definition of suspect case | Definition of probable case | Role of serology in testing |
| 10‐30 January 2020 | 10‐30 January: no documentation to define at this time (before first date of global guidelines)
31 January onwards: a confirmed case is a person with laboratory confirmation of COVID‐19 infection, irrespective of clinical signs and symptoms. No prescribed test in laboratory guidelines, suggested tests from 10 January include broad coronavirus RT‐PCR (with sequencing of precise virus in test positives), whole genome sequencing, broad coronavirus serology on paired samples, microscopy, culture (Lab 10 January). Four suggested tests from 17 January: broad coronavirus RT‐PCR (with sequencing of precise virus in test positives), NAAT for SARS‐CoV‐2 when it becomes available, whole genome sequencing, and broad coronavirus serology on paired samples. States that once specific NAAT assays are developed and validated, confirmation will be based on specific detection of unique sequences of viral nucleic acid by RT‐PCR. |
None stated | No definition of 'suspect case' at this time, but case definitions for surveillance are defined as a combination of symptoms and exposure, with more severe symptoms requiring less evidence for exposure | No definition at this time | Serological testing may be useful to confirm immunologic response to a pathogen from a specific viral group, e.g. coronavirus. Best results from serologic testing requires the collection of paired serum samples (in the acute and convalescent phase) from cases under investigation. |
| 31 January‐26 February 2020 | None stated | Suspect case defined as combination of symptoms and exposure, with more severe symptoms requiring less evidence for exposure | A suspect case with inconclusive laboratory results or is test‐positive using a pan‐coronavirus assay without laboratory evidence of other respiratory pathogens (global 31 January) | ||
| 27 February‐1 March 2020 | None stated | Suspect case defined as combination of symptoms and exposure, with more severe symptoms requiring less evidence for exposure, OR defined by symptoms requiring hospitalisation and an absence of alternative explanation | A suspected case with inconclusive laboratory results (global 27 February) | ||
| 2 March‐19 March 2020 | A person with laboratory confirmation of COVID‐19 infection, irrespective of clinical signs and symptoms (global 31 January, 27 February, 20 March)
Laboratory confirmation of cases by NAAT specific to SARS‐CoV‐2 such as real‐time RT‐PCR with confirmation by nucleic acid sequencing when necessary. The viral genes targeted so far include the N, E, S and RdRP genes. In areas with no known COVID‐19 virus circulation confirmation requires:
Discordant results should be resampled. In areas where COVID‐19 virus is widely spread a simpler algorithm might be adopted (e.g. RT‐PCR of a single discriminatory target) |
One or more negative result does not rule out the possibility of COVID‐19 virus infection | In cases where NAAT assays are negative and there is a strong epidemiological link to COVID‐19 infection, paired serum samples (in the acute and convalescent phase) could support diagnosis once validated serology tests are available. Serological assays will play an important role in research and surveillance but are not currently recommended for case detection. |
||
| 19 March 2020‐current (12‐03‐21) | Probable case A suspect case for whom testing for the COVID‐19 virus is inconclusive OR A suspect case for whom testing could not be performed for any reason. | ||||
| NAAT: nucleic acids amplification test; RT‐PCR: reverse transcription polymerase chain reaction | |||||
aSource data from Laboratory testing of 2019 novel coronavirus (2019‐nCoV) in suspected human cases: interim guidance, World Health Organization. 10 January, 17 January, 2 March, 19 March, 21 March 2020 (WHO 2020d), and Global surveillance for COVID‐19 caused by human infection with COVID‐19 virus, interim guidance, 31 January, 27 February, and 20 March 2020 (WHO 2020e).
Table B: Summary of Chinese National Health Commission guidelines for diagnosis and treatment for novel coronavirus pneumonia (trial versions 1‐7)
| Dates in effect | Definition of confirmed case | Definition of confirmed non‐case | Definition of suspect case | Role of serology in testing |
| 16‐17 January 2020 (version 1) | Cases (not confirmed cases) defined as virus genome highly homologous to coronaviruses | Not defined | Observation cases: defined as combination of exposure in Wuhan and symptoms focused on pneumonia, leukopenia and lack of improvement. | No role |
| 18 January‐2 March 2020 (versions 2, 3, 4, 5, 5 revised, and 6) | Suspect cases with either
|
Suspect cases can be ruled out after 2 consecutive negative respiratory tract nucleic acid tests taken at least 24 hours apart. | Suspect cases: combination of exposure (such as residence in/travel to Wuhan or exposure to a confirmed case within 14 days of onset) AND clinical features (such as symptoms: fever, respiratory symptoms, and tests: chest imaging, white blood cell and lymphocyte count). Exact definition varies slightly with version | No role |
| 3 March 2020‐current (12‐03‐21 (version 7) | Suspect cases with either
|
Suspect cases can be ruled out after 2 negative NAATs, taken at least 24 hours apart, and the NCP virus‐specific IgM and IgG are negative after 7 days from onset. | Suspect cases: combination of exposure (such as residence in/travel to Wuhan or exposure to a confirmed case within 14 days of onset) AND clinical features (such as symptoms: fever, respiratory symptoms, and tests: chest imaging, white blood cell and lymphocyte count). | Part of definition of cases and confirmed non‐cases |
| NAAT: nucleic acids amplification test; NCP: novel coronavirus pneumonia; RT‐PCR: reverse transcription polymerase chain reaction; Source: Table from Cheng 2020 | ||||
Appendix 2. Cochrane COVID‐19 Study Register searches
| Source | Strategy |
| Clinical Trials.gov | COVID‐19 OR 2019‐nCoV OR SARS‐CoV‐2 OR 2019 novel coronavirus OR severe acute respiratory syndrome coronavirus 2 OR Wuhan coronavirus OR coronavirus |
| WHO International Clinical Trials Registry Platform | Screen the entire COVID‐19.csv file available from who.int/emergencies/diseases/novel-coronavirus-2019 |
| PubMed | (2019 nCoV[tiab] OR 2019nCoV[tiab] OR corona virus[tiab] OR corona viruses[tiab] OR coronavirus[tiab] OR coronaviruses[tiab] OR COVID[tiab] OR COVID19[tiab] OR nCov 2019[tiab] OR SARS‐CoV2[tiab] OR SARS CoV‐2[tiab] OR SARSCoV2[tiab] OR SARSCoV‐2[tiab] OR "Coronavirus"[Mesh:NoExp] OR "COVID‐19"[nm] OR "COVID‐19 drug treatment"[nm] OR "COVID‐19 diagnostic testing"[nm] OR "COVID‐19 serotherapy"[nm] OR "COVID‐19 vaccine"[nm] OR "LAMP assay"[nm] OR "severe acute respiratory syndrome coronavirus 2"[nm] OR "spike protein, SARS‐CoV‐2"[nm]) NOT ("animals"[mh] NOT "humans"[mh]) NOT (editorial[pt] OR newspaper article[pt]) |
Appendix 3. Living search from the University of Bern
The following information is taken from the university of Bern website (see: ispmbern.github.io/covid-19/living-review/collectingdata.html).
The register is updated daily and CSV file downloads are made available.
1 April 2020
From 1 April 2020, we will retrieve the curated BioRxiv/MedRxiv dataset (connect.medrxiv.org/relate/content/181).
26 to 31 March 2020
MEDLINE: (\"Wuhan coronavirus\" [Supplementary Concept] OR \"COVID‐19\" OR \"2019 ncov\"[tiab] OR ((\"novel coronavirus\"[tiab] OR \"new coronavirus\"[tiab]) AND (wuhan[tiab] OR 2019[tiab])) OR 2019‐nCoV[All Fields] OR (wuhan[tiab] AND coronavirus[tiab])))))
Embase: (nCoV or 2019‐nCoV or ((new or novel or wuhan) adj3 coronavirus) or covid19 or covid‐19 or SARS‐CoV‐2).mp.
BioRxiv/MedRxiv: ncov or corona or wuhan or COVID or SARS‐CoV‐2
With the kind support of the Public Health & Primary Care Library PHC (www.unibe.ch/university/services/university_library/faculty_libraries/medicine/public_health_amp_primary_care_library_phc/index_eng.html), and following guidance of the Medical Library Association (www.mlanet.org/p/cm/ld/fid=1713).
1 January 2020 to 25 March 2020
MEDLINE: ("Wuhan coronavirus" [Supplementary Concept] OR "COVID‐19" OR "2019 ncov"[tiab] OR (("novel coronavirus"[tiab] OR "new coronavirus"[tiab]) AND (wuhan[tiab] OR 2019[tiab])) OR 2019‐nCoV[All Fields] OR (wuhan[tiab] AND coronavirus[tiab])))))
Embase: ncov OR (wuhan AND corona) OR COVID
BioRxiv/MedRxiv: ncov or corona or wuhan or COVID
Appendix 4. Search classification model
We needed a more efficient approach to keep up with the rapidly increasing volume of COVID‐19 literature. A classification model for COVID‐19 diagnostic studies was built with the model building function within Eppi Reviewer, which uses the standard SGCClassifier in Scikit‐learn on word trigrams. As outputs, new documents receive a percentage (from the predict_proba function) where scores close to 100 indicate a high probability of belonging to the class ‘relevant document’ and scores close to 0 indicate a low probability of belonging to the class ‘relevant document’. We used three iterations of manual screening (title and abstract screening, followed by full‐text review) to build and test classifiers. The final included studies were used as relevant documents, while the remainder of the COVID‐19 studies were used as irrelevant documents. The classifier was trained on the first round of selected articles, and tested and retrained on the second round of selected articles. Testing on the second round of selected articles revealed poor positive predictive value but 100% sensitivity at a cut‐off of 10. The poor positive predictive value is mainly due to the broad scope of our topic (all diagnostic studies in COVID‐19), poor reporting in abstracts, and a small set of included documents. The model was retrained using the articles selected of the second and third rounds of screening, which added a considerable number of additional documents. This led to a large increase in positive predictive value, at the cost of a lower sensitivity, which led us to reduce the cut‐off to 5. The largest proportion of documents had a score between 0‐5. This set did not contain any of the relevant documents. This version of the classifier with a cut‐off 5 was used in subsequent rounds and accounted for approximately 80% of the screening burden.
Appendix 5. CDC Library, COVID‐19 Research Articles Downloadable Database
Embase records from the Stephen B. Thacker CDC Library, COVID‐19 Research articles Downloadable database
Records were obtained by the CDC library by searching Embase through Ovid using the following search strategy.
| Source | Strategy |
| Embase | coronavir* OR corona virus* OR betacoronavir* OR covid19 OR covid 19 OR nCoV OR novel CoV OR CoV 2 OR CoV2 OR sarscov2 OR 2019nCoV OR wuhan virus*).mp. OR ((wuhan OR hubei OR huanan) AND (severe acute respiratory OR pneumonia*) AND outbreak*).mp. OR Coronavirus infection/ OR coronavirinae/ OR exp betacoronavirus/ Limits: 2020‐ OR (novel coronavir* OR novel corona virus* OR covid19 OR covid 19 OR nCoV OR novel CoV OR CoV 2 OR CoV2 OR sarscov2 OR 2019nCoV OR wuhan virus*).mp. OR ((wuhan OR hubei OR huanan) AND (severe acute respiratory OR pneumonia*) AND outbreak*).mp. OR ((wuhan OR hubei OR huanan) AND (coronavir* OR betacoronavir*)).mp. Limits: 2019‐ |
Appendix 6. Data extraction items
| Patient sampling items | Patient characteristics and setting items | Index test items | Reference standard items | Flow and timing items | Notes items |
| A1 Purpose | B1 Setting | D1.1 Test name (please include product code if reported) | E1 Reference standard for cases including threshold | F1 What was the time interval between index and reference tests? | G1 Funding |
| A2 Design (and description of groups labelled [1] [2] …) | B2 Location (include name of institution if available) | D1.2 Manufacturer | E1.1 RT‐PCR genetic targets | F2 Did all patients receive the same reference standard? | G2 Publication status |
| A3 Recruitment | B3 Country | D1.3 Antigen or genetic target | E2 Samples used | F3 Missing data | G3 Source (preprint or journal name) |
| A4 Were cases recruited prospectively or retrospectively? | B4 Dates | D1.4 Antibodies used | E3 Timing of reference standard | F4 Uninterpretable results | G4 Study author CoI (including any manufacturer affiliations) |
| A5 Sample size (virus/COVID cases) | B5 Symptoms and severity | D1.5 POC or laboratory | E4 Was it blind to index test? | F5 Indeterminate results (index) | G5 Comment |
| A6 Inclusion and exclusion criteria | B6 Demographics | D1.6 Test method | E5 Did it incorporate index test? | F5.1 Indeterminate results (reference) | |
| A7 Comment | B7 Exposure history | D1.7 When were samples taken? | E6 Reference standard for non‐cases | F6 Samples or patients | |
| B8 Comment | D1.8 Samples used (include who collected by) | E7 Samples used | F7 Comment | ||
| Non‐COVID patients (if additional groups) | D1.8.1 Transport media (volume and manufacturer detail) | E8 Timing of reference standard | |||
| C1.1 Group name | D1.8.2 Sample storage and timing of test | E9 Was it blind to index test? | |||
| C1.2 Source and time | D1.9 Who applied the test (include reported training/e)? | E10 Did it incorporate index test? | |||
| C1.3 Characteristics | D1.10 How was positive defined? | E11 Comment | |||
| C2.1 Group name | D1.11 Blinded to reference standard | ||||
| C2.2 Source and time | D1.12 Threshold predefined | ||||
| C2.3 Characteristics | D1.13 Comment | ||||
| CoI: conflict of interest; POC: point of care; RT‐PCR: reverse transcription polymerase chain reaction | |||||
Appendix 7. Criteria for assessment of study quality (QUADAS‐2)
| DOMAIN: Participant selection | |
| Was a consecutive or random sample of patients enrolled? | This will be similar for all index tests, target conditions, and populations. Yes: if a study explicitly stated that all participants within a certain time frame were included; that this was done consecutively; or that a random selection was done. No: if it was clear that a different selection procedure was employed; for example, selection based on clinician's preference, or based on institutions, or based on result of RT‐PCR Unclear: if the selection procedure was not clear or not reported |
| Was a case‐control design avoided? | This will be similar for all index tests, target conditions, and populations. Yes: if a study explicitly stated that all participants came from the same group of (suspected) patients. No: if it was clear that a different selection procedure was employed for the participants depending on their COVID‐19 status or SARS‐CoV‐2 infection status; or if only participants with SARS‐CoV‐2 infection were included Unclear: if the selection procedure was not clear or not reported. |
| Did the study avoid inappropriate exclusions? | Studies may have excluded patients, or selected patients in such a way that they avoided including those who were difficult to diagnose or likely to be borderline. Although the inclusion and exclusion criteria will be different for the different index tests, inappropriate exclusions and inclusions will be similar for all index tests: for example, only elderly patients excluded, or children (as sampling may be more difficult). This needs to be addressed on a case‐by‐case basis. Yes: if a high proportion of eligible patients was included without clear selection. No: if a high proportion of eligible patients was excluded without providing a reason; if, in a retrospective study, participants without index test or reference standard results were excluded. Unclear: if the exclusion criteria were not reported. |
| Did the study avoid inappropriate inclusions? | Some laboratory studies may have intentionally included groups of patients in whom the accuracy was likely to differ, such as those with particularly low or high viral loads, or who had other diseases, such that the sample over‐represented these groups. This needs to be addressed on a case‐by‐case basis. Yes: if samples included were likely to be representative of the spectrum of disease. No: if the study oversampled patients with particular characteristics likely to affect estimates of accuracy. Unclear: if the exclusion criteria were not reported. |
| Could the selection of patients have introduced bias? | High: if one or more signalling questions were answered with no, as any deviation from the selection process may lead to bias. Low: if all signalling questions were answered with yes. Unclear: all other instances |
| Is there concern that the included participants do not match the review question? | High: for two‐group studies that included healthy or other disease controls, whether pre‐pandemic or contemporaneous; studies that only included people with COVID‐19 (whether RT‐PCR‐confirmed only, participants meeting official guideline criteria); Low: for single‐group studies recruiting participants with signs and symptoms of COVID‐19; or for two‐group studies where control groups suspected of COVID‐19 were separately recruited. Unclear: if a description about the participants was lacking. |
| DOMAIN: Index tests | |
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes: if blinding was explicitly stated or index test was recorded before the results from the reference standard were available. No: if it was explicitly stated that the index test results were interpreted with knowledge of the results of the reference standard. Unclear: if blinding was unclearly reported. |
| If a threshold was used, was it prespecified? | Yes: if the test was dichotomous by nature, or if the threshold was stated in the methods section, or if study authors stated that the threshold as recommended by the manufacturer was used. No: if a receiver operating characteristic curve was drawn or multiple threshold reported in the results section; and the final result was based on one of these thresholds. Unclear: if threshold selection was not clearly reported. |
| Could the conduct or interpretation of the index test have introduced bias? | High: if one or more signalling questions were answered with no, as even in a laboratory situation knowledge of the reference standard may lead to bias. Low: if all signalling questions were answered with yes. Unclear: all other instances |
| Is there concern that the index test, its conduct, or interpretation differ from the review question? | For all test types, if index test is 'in‐house' or not commercially available, then state 'High'. If any test procedures used in the study diverged from IFU ((use of VTM, or testing outwith stated time limit), also state High If testing carried out in centralised laboratory and not near patient then state High. Evaluations that withheld the name of the test, or that used mixed sample types or did not report the evaluation setting, state Unclear If samples used and any sample processing steps are in accordance with test IFU, or if study describes conducting the test according to the manufacturer's protocol, state Low |
| DOMAIN: Reference standard | |
| Is the reference standard likely to correctly classify the target condition? | We will define acceptable reference standards using a consensus process once the list of reference standards that have been used has been obtained from the eligible studies. For COVID‐19 cases Yes: RT‐PCR; confirmed or suspected case using official criteria (WHO, CDC) or a clearly set out combination of signs/symptoms/exposure No: RT‐PCR not used, or if inadequate combination of clinical characteristics used in PCR‐negatives, e.g. computed tomography alone Unclear: if definition of COVID‐19 was not reported For absence of COVID‐19 Yes: if at least 2 negative RT‐PCR results reported if suspected COVID‐19 based on signs/symptoms; single negative RT‐PCR test for asymptomatic contacts or contemporaneous controls with no clinical suspicion of COVID‐19; only pre‐pandemic sources of control samples used. No: single RT‐PCR or number of negative RT‐PCRs not reported for COVID‐19 suspects; no RT‐PCR reported (untested) for asymptomatic contacts or contemporaneous controls Unclear: if timing of control samples (pre‐pandemic or contemporaneous) was not reported |
| Were the reference standard results interpreted without knowledge of the results of the index test? | Yes: if it was explicitly stated that the reference standard results were interpreted without knowledge of the results of the index test, or if the result of the index test was obtained after the reference standard. No: if it was explicitly stated that the reference standard results were interpreted with knowledge of the results of the index test or if the index test was used to make the final diagnosis. Unclear: if blinding was unclearly reported. |
| Did the definition of the reference standard incorporate results from the index test(s)? | Yes: if results from the index test were a component of the reference standard definition. No: if the reference standard did not incorporate the index standard test. Unclear: if it was unclear whether the results of the index test formed part of the reference standard. |
| Could the conduct or interpretation of the reference standard have introduced bias? | High: if one or more signalling questions were answered with no. Low: if all signalling questions were answered with yes. Unclear: all other instances |
| Is there concern that the target condition as defined by the reference standard does not match the review question? | Applicability was judged primarily on the definition of disease‐positive. High: if RT‐PCR alone used to define cases Low: if clinical criteria, including RT‐PCR, were used to define cases, regardless of whether official criteria were used, as long as the criteria were explicitly described. Unclear: if definition of COVID‐19 cases was not provided, including if some clinically diagnosed cases were included but the clinical criteria used were not described. |
| DOMAIN: Flow and timing | |
| Was there an appropriate interval between index test and reference standard? | Yes: if same swab used, or swabs obtained at same time regardless of freezing (which is covered under index applicability) No: if different samples used with more than 24 hours between collection times Unclear: if can't tell |
| Did all participants receive the same reference standard? | Yes: if all participants received the same reference standard (clearly no differential verification). No: if (part of) the index test‐positives or index test‐negatives received a different reference standard. Unclear: if it was not reported |
| Were all participants included in the analysis? | Yes: if it is clear that all eligible participants were included in the analyses. No: if after the inclusion/exclusion process, participants were removed from the analyses for different reasons: no reference standard done, no index test done, intermediate results of both index test or reference standard, indeterminate results of both index test or reference standard, samples unusable. Unclear: if it is not possible to determine whether all participants were included (e.g. from a STARD‐style participant flow diagram) |
| Did all participants receive a reference standard? | Yes: if all participants received a reference standard (clearly no partial verification). No: if only (part of) the index test positives or index test negatives received the complete reference standard. Unclear: if it was not reported |
| Were results presented per participant? | Yes: if either only one sample per participant (regardless of disaggregation of results over time), or if multiple samples per participant but results are disaggregated by time period (at least week by week) No: if multiple samples per participant and results are not disaggregated by time period Unclear: if it is not possible to tell whether results presented are per participant or per sample |
| Could the participantflow have introduced bias? | High: if one or more signalling questions were answered with no. Low: if all signalling questions were answered with yes. Unclear: all other instances |
| CDC: Centers for Disease Control; ICU: intensive care unit; IFU: instructions for use; RT‐PCR: real‐time polymerase chain reaction; SARS‐CoV‐2: severe acute respiratory syndrome coronavirus 2; VTM: viral transport medium; WHO: World Health Organization | |
Appendix 8. Excluded studies
| Study | Exclusion reason | Notes | Other review inclusion |
| Studies 'almost' included | |||
| Basu 2020 | Ineligible reference standard | Assesses agreement between two POC tests | No; excluded |
| FIND 2020f | Superseded by Kruger 2020(a) | Coris Bioconcept data | No; excluded |
| McDonald 2020 | Ineligible reference standard | Only antigen negatives get RT‐PCR | No; excluded |
| McCormick‐Baw 2020 | Ineligible reference standard | RT‐PCR (including Xpert XPress) using alternative sample types | Sampling methods comparison |
| Mlcochova 2020 | superseded by Collier 2020 | SAMBA‐II data (33 COVID cases; same recruitment dates | No; excluded |
| Studies excluded on index test technology | |||
| Anahtar 2020 | Ineligible index test | in‐house RT‐LAMP; direct testing | Technology comparison |
| Ar Gouilh 2020 | Ineligible index test | in‐house RT‐LAMP | Technology comparison |
| Arumugam 2020 | Ineligible index test | in‐house RT‐PCR; direct testing | Technology comparison |
| Azzi 2020 | Ineligible index test | in‐house RT‐LAMP | Technology comparison |
| Baek 2020 | Ineligible index test | in‐house RT‐LAMP | Technology comparison |
| Bokelmann 2020 | Ineligible index test | Cap‐iLAMP (capture and improved loop‐mediated isothermal amplification). | Technology comparison |
| Bordi 2020 | Ineligible index test | One step RT‐PCR; not suited to POC | Technology comparison |
| Broughton 2020 | Ineligible index test | in‐house CRISPR‐Cas12 based assay | Technology comparison |
| Chen 2020a | Ineligible index test | CRISPR/Cas12a | Technology comparison |
| Chow 2020 | Ineligible index test | in‐house RT‐LAMP | Technology comparison |
| Ding 2020a | Ineligible index test | in‐house RT‐LAMP | Technology comparison |
| Dong 2020 | Ineligible index test | One‐step RT‐dPCR | Technology comparison |
| Fowler 2020 | Ineligible index test | (direct) RT‐LAMP | Technology comparison |
| Freire‐Paspuel 2020b | Ineligible index test | compares two RT‐PCR kits | No; excluded |
| Hirotsu 2020 | Ineligible index test | Automated RT‐PCR; not suited to POC | No; excluded |
| Hu 2020 | Ineligible index test | in‐house RT‐LAMP | Technology comparison |
| Huang 2020 | Ineligible index test | in‐house Rt‐LAMP | Technology comparison |
| James 2020 | Ineligible index test | in‐house RT‐LAMP | Technology comparison |
| Jiang 2020 | Ineligible index test | in‐house RT‐LAMP | Technology comparison |
| Joung 2020 | Ineligible index test | SHERLOCK testing in one Pot | Technology comparison |
| Joung 2020a | Ineligible index test | in‐house RT‐LAMP | Technology comparison |
| Lee 2020 | Ineligible index test | in‐house RT‐LAMP | Technology comparison |
| Lu 2020a | Ineligible index test | in‐house RT‐LAMP | Technology comparison |
| Mohon 2020 | Ineligible index test | in‐house RT‐LAMP | Technology comparison |
| Newman 2020 | Ineligible index test | in‐house RT‐LAMP used in mobile setting; sample prep includes centrifuge | Technology comparison |
| Osterdahl 2020 | Ineligible index test | in‐house RT‐LAMP | Technology comparison |
| Peto 2020 | Ineligible index test | loop‐mediated isothermal amplification and nanopore sequencing | Technology comparison |
| Pollock 2020a | Ineligible index test | Laboratory‐based Ag assay | Technology comparison |
| Qian 2020 | Ineligible index test | Fast isothermal Nucleid acid detection (FIND) assay (RT‐RPA) | Technology comparison |
| Rauch 2020 | Ineligible index test | CREST ‐ CRISPr‐Cas13a | Technology comparison |
| Shirato 2020 | Ineligible index test | Intended for direct testing but used with extracted RNA | Technology comparison |
| Singh 2020b | Ineligible index test | targeted‐mass spectrometry | Technology comparison |
| Wang 2020a | Ineligible index test | RT‐RAA assay | Technology comparison |
| Wang 2020a | Ineligible index test | CRISPR/Cas12a‐based assay with a naked eye readout, CRISPR/Cas12a‐NER | Technology comparison |
| Yan 2020 | Ineligible index test | in‐house RT‐LAMP | Technology comparison |
| Yang 2020b | Ineligible index test | in‐house RT‐LAMP | Technology comparison |
| Yu 2020a | Ineligible index test | in‐house RT‐LAMP | Technology comparison |
| Yu 2020b | Ineligible index test | in‐house RT‐LAMP (iLACO) | Technology comparison |
| Yu 2020c | Ineligible index test | LFA technology but sample requires PCR amplification step first | Technology comparison |
| Zhang 2020 | Ineligible index test | (RT‐LAMP) coupled with nanoparticles‐based biosensor (NBS) assay (RT‐LAMP‐NBS) | Technology comparison |
| Zhu 2020 | Ineligible index test | RT‐LAMP with extracted RNA | Technology comparison |
| LFA: lateral flow assay; PCR: polymerase chain reaction; POC: point‐of‐care; RT‐LAMP: reverse transcription loop‐mediated isothermal amplification; RT‐PCR: reverse transcription polymerase chain reaction | |||
Appendix 9. Antigen tests: summary study characteristics
| Study | Study design; inclusion criteria | Setting; country (recruitment dates) | Participant characteristics |
Reference standard Reference samples and timing |
Missing data or indeterminate results |
|
Albert 2020 Preprint 412 (54) |
Single group (prospective); clinical suspicion of COVID‐19 (compatible signs or symptoms appearing within the prior week) | COVID‐19 test centre (primary care); Spain (2 September to 7 October 2020) |
Symptomatic: all < 7 days pso median age, 31 years (range, 1‐91); 42% male |
RT‐PCR (single assay) Target: ORF1ab, N and S genes NP in VTM Timing: as for index; tested within 24 h Interval: simultaneous; paired |
None reported Index: none reported Reference: none reported |
|
Alemany 2020 Preprint Total N 1406 (951 cases) [1] 446 (419) [2] 473 (415) [3] 487 (117) |
Single group (not stated); Samples from [1] symptomatic individuals in routine practice [2] contacts exposed to confirmed case [3] preventive screening of unexposed asymptomatic individuals |
Laboratory‐based); Spain (Not stated) |
Mixed: No details; 15 (1.1%) hospitalised Mean age 40.4 years (SD 24.5), 453 (32.2% male) |
RT‐PCR (single assay) Target: not stated; as per CDC protocol NP or nasal mid‐turbinate; as per index test Timing: fresh samples stored at 2–8 ºC for up to 72 h prior to RT‐PCR Interval: simultaneous (same swab) |
None reported Index: none reported Reference: none reported |
|
Billaud 2020 Published 462 (99); 47 missing, presumably with no paired data |
Single group (prospective); cluster investigation at higher education institute | Contacts (screening); France (September 16 and 17) |
Mixed: 166/509, 32.6% symptomatic Mean, median age Students 21.6 years, 21 years (18‐37 years) Teachers 47.2 years, 49 years (26‐64 years) |
RT‐PCR (single assay) Target: not stated NP (paired) Timing: as for index Interval: simultaneous |
47 missing, including 11 uninterpretable on Ag test Index: none reported Reference: none reported |
|
Blairon 2020 Published 56 (30) |
Single group (prospective) Samples sent for laboratory diagnosis |
Laboratory‐based (swabs obtained at hospital site; no further detail); Belgium (5 April‐4 May 2020) |
None reported | RT‐PCR (single assay) Target: E gene NP swabs (same as for Ag test) Timing: not stated Interval: not stated but infer short interval |
None reported; main cohort excluded None reported; 1 'invalid' sample excluded from main cohort Index: none reported; reference: none reported |
|
Cerutti 2020 Published 330 (109) |
Single group (not stated); (1) symptomatic patients at 1 of 2 EDs (n = 185) (2) asymptomatic travellers returning from high‐risk countries |
Mixed ((1) ED
(2) Possible contacts); Italy ((1) 3 Mar‐1 May (2) August 2020) |
Mixed: not stated; cohort (2) were asymptomatic (1) mean age 44.6, 95 % CI: 40.7–48.6 (2) mean age 35.9, 95 % CI: 32.7–39.1 |
RT‐PCR (single assay) Target: not stated Not stated Timing: not stated Interval: simultaneous; not clear if same sample used or paired swabs obtained |
None reported Index: none reported Reference: none reported |
|
Courtellemont 2020 Preprint 248 (121) |
Unclear; two group (Unclear) (1) Symptomatic or asymptomatic people voluntarily accessing the COVID‐19 Screening Department (2) hospitalised SARS‐CoV‐2‐positive patients |
Mixed (COVID testing unit and inpatient); France (12 Oct‐19 Oct) |
Mainly symptomatic (99/121 cases) median age 38 years, mean age 43 years (range: 18‐96) 117 male |
RT‐PCR (single assay) Target: ORF1ab, S and N genes NP in VTM; paired Timing: as for index Interval: simultaneous; paired |
None reported None reported Index: none reported Reference: none reported |
|
Diao 2020 Preprint (not peer reviewed) 239 (208) for nasopharyngeal swab; 20 (19) for urine |
Single group (retrospective) Samples from cases of suspected SARS‐CoV‐2 infection |
Unclear (not stated); China (not stated) |
Not reported | RT‐PCR (single assay);
Threshold ≤ 40 Ct Target: ORF1ab and N gene As for index test; NP swab Timing: not stated Interval: done in parallel |
Not reported Index: nR Reference: none reported |
|
Fenollar 2020(a) Accepted manuscript 182 (182) |
Single group (cases) (unclear) [1] symptomatic, all PCR+ Second cohort reported in Fenollar 2020(b) |
COVID‐19 test centre (unclear; no details); France (21 September‐2 October 2020) |
Symptomatic No other details | RT‐PCR (single assay ‐ VitaPCR, Credo) Target: not stated n/a NP (paired, from opposite nostril) Timing: not stated Interval: Paired swabs |
None reported Index: none reported Reference: none reported |
|
Fenollar 2020(b) Accepted manuscript 159 (22) |
Single group (unclear) [2] asymptomatic contacts of confirmed cases Second cohort reported in Fenollar 2020a |
Contacts (unclear); France (Sep 21‐Oct 2 2020) |
Asymptomatic: No other details |
RT‐PCR (single assay ‐ VitaPCR, Credo) Target: not stated NP (paired, from opposite nostril) Timing: not stated Interval: paired swabs |
None reported Index: none reported Reference: none reported |
|
FIND 2020a published 400 (102) |
Single group (prospective) Symptoms consistent with COVID‐19 (meeting national definition for testing) |
COVID‐19 test centre (community); Brazil (30 July‐21 August 2020) |
Symptomatic; no further details mean age 40 years (range 4‐84) (n = 396) 181 (45%) male |
RT‐PCR (single assay);
Threshold ≤ 37 Ct Target: N1, N2 NP swabs Timing: same as for index test Interval: as per PCR turnaround time |
Reports 0 invalid results None reported Index: none reported Reference: none reported |
|
FIND 2020b published 535 (124) |
Single group (prospective) Presenting either with symptoms compatible with SARS‐CoV2, or known positive contact or asymptomatic HCW |
COVID‐19 test centre (community); Switzerland (9‐16 Oct 2020) |
Symptomatic: 534/535 (99%) symptomatic Mean age 38.5y (16‐85y) 247, 46% male |
RT‐PCR (single assay);
Threshold <40 Ct (from Figure) Target: not stated NP swab (paired, from contralateral nostril) Timing: author contact advises only paired swabs. Interval: as per PCR turnaround time |
None reported Index: none reported Reference: none reported |
|
FIND 2020c (BR) published 400 (106) |
Single group (prospective) Ambulatory patients meeting national suspect definition for COVID‐19 testing |
COVID‐19 test centre (community); Brazil (13‐30 Jul 2020) |
Symptomatic: 392/397 (99%); no further details mean age 37y (2‐94) (397 participants); 229/398 male (57%) |
RT‐PCR (single assay);
Ct threshold not stated; author contact advises Ct thresholds as per assay IFUs Target: N1 and N2 NP swabs Timing: author contact advises only paired swabs used. Interval: as per PCR turnaround time |
Reports 0 missing data None reported Index: none reported Reference: none reported |
|
FIND 2020c(cH) published 529 (191) |
Single group (prospective) Patients seeking COVID‐19 either with symptoms compatible with a SARS‐CoV2 infection, or with a known positive contact or asymptomatic HCWs |
COVID‐19 test centre (community); Switzerland (9‐23 October 2020) |
Symptomatic: Not stated; time pso recorded for 183/191, 96% 141/183 COVID‐positive cases had symptoms for 0‐4 days (77%) Not stated |
RT‐PCR (single assay);
Threshold < 40 Ct (from Figure) Target: not stated NP swab (paired, from contralateral nostril) Timing: author contact advises only paired swabs used. Interval: as per PCR turnaround time |
None reported Index: none reported Reference: none reported |
|
FIND 2020d (BR) published 453 (120) |
Single group (prospective) Adults in community meeting national suspect definition for COVID‐19 testing |
COVID‐19 test centre (community clinic or tertiary hospital); Brazil ([1] 17 Aug‐9 September [2] 11 Jul‐8 Aug) |
Mainly symptomatic: 421/450 (94%); no further details mean age 39 years (0‐95 years) (451 participants); 185 male (41%) |
RT‐PCR (multiple assays);
Author contact advises Ct thresholds as per assay IFUs Target: 1. N1 and N2; 2. E and RdRp NP swabs Timing: author contact advises only paired swabs used. Interval: as per PCR turnaround time |
Reports 0 missing data None reported Index: none reported Reference: none reported |
|
FIND 2020d (DE) published 676 (39) |
Single group (prospective) Adults in community meeting national suspect definition for COVID‐19 testing presenting at [1] a drive‐in testing centre or [2] ambulatory testing clinic |
COVID‐19 test centre (community); Germany ([1] Heidelberg: 15 June‐18 July 2020 [2] Berlin: 6 July–23 September 2020) |
Mainly symptomatic: 517/669 (77%); no further details mean age 38 years (18‐85 years) (676 participants); 307 male (46%) |
RT‐PCR (multiple assays);
Author contact advises Ct thresholds as per assay IFUs Target: not stated apart from 3. E gene NP (n = 305), NOP (n = 342) and/or OP swabs (n = 32) Timing: author contact advises only paired swabs used. Interval: as per PCR turnaround time |
Reports 0 missing data None reported Index: none reported Reference: none reported |
|
FIND 2020e (BR) published 476 (117) |
Single group (prospective); adults in community meeting national suspect definition for COVID‐19 testing |
COVID‐19 test centre (community); Brazil (27 Jul‐16 Sep) |
Symptomatic: 470/476 (99%) symptomatic; no further details mean age 45 years (0‐106 years) (473 participants); 252 male (53%) |
RT‐PCR (single assay);
Ct threshold not stated Target: N1 and N2 NP swabs Timing: author contact advises only paired swabs used. Interval: as per PCR turnaround time |
Reports 0 missing data None reported Index: none reported Reference: none reported |
|
FIND 2020e (DE) published 1239 (25) |
Single group (prospective) Adults in community meeting national suspect definition for COVID‐19 testing |
COVID‐19 test centre (community); Germany ([1] Heidelberg: 4 May ‐ 3 September [2] Berlin: 4 May ‐ 18 Aug) |
Mixed: 733/1223 (59.9%) symptomatic; no further details mean age 39.5 years (17,59.2 years) (1239 participants); 607 male (50%) |
RT‐PCR (multiple assays)
Author contact advises Ct thresholds as per assay IFUs Target: not stated NP swabs Timing: author contact advises only paired swabs used. Interval: as per PCR turnaround time |
Reports 0 missing data None reported Index: none reported Reference: none reported |
|
Fourati 2020 [A] Published 634 (297); number of cases tested varied per assay |
Two group (retrospective) (1) residual samples from participants with positive SARS‐CoV‐2 PCR tested when they presented symptoms (2) pre‐pandemic samples |
Laboratory‐based (unclear; "consulted or were admitted"); France (9 March‐9 April 2020) |
Symptomatic No further details Not stated |
RT‐PCR (single assay) Target: not stated NP; same as for index Timing: as for index Interval: same swab; simultaneous |
Number of cases missing per assay varied; reasons for missing data not reported (presumably invalid assay results)
[A] 5, 1.7%
[B] 6, 2.0%
[C] 2, 0.7%
[D] 0
[E] 2, 0.7%
[F] 0 Not stated Index: not stated Reference: not stated |
|
Gremmels 2020(a) Preprint [1] 1369 (139) |
Single group (prospective) [1] community‐dwelling mildly symptomatic participants in a medium endemic area Second cohort reported in Gremmels 2020(b) |
COVID‐19 test centre (community) Netherlands [1] 22 September‐6 October |
Mainly symptomatic Cohort [1] only. Data on symptoms were missing from nine participants Asymptomatic 37, 2.7%, sore throat 907, 66.3%; coryza 943, 69%; cough 780, 57.1%; headache 601, 44.0%; tiredness 565, 41.3%; general malaise 365, 26.7% (further 19 documented) median age 36.4 years (IQR 27.0, 49.6 years); 523, 38.3% male |
RT‐PCR (single assay) Target: E‐, N‐, and RdRP‐gene NOP (paired) Timing: NOP swab obtained first for RT‐PCR Interval: paired |
2 patients excluded ('inappropriate application of NP swab and lab mislabelling'), dis status not reported. None reported Index: none; no bands were classified as unclear Reference: patients |
|
Gremmels 2020(b) Preprint [2] 208 (63) |
Single group (prospective) [2] Community‐dwelling mildly symptomatic participants in a high endemic area Second cohort reported in Gremmels 2020(a) |
COVID‐19 test centre (community); Netherlands [2] 23 September‐9 October |
Not reported Not stated; 'mildly symptomatic', presume mixed as per Gremmels 2020(a) Not stated |
RT‐PCR (single assay) Target: E‐, N‐, and RdRP‐gene NOP (paired) Timing: NOP swab obtained first for RT‐PCR Interval: paired |
None reported Index: none; no bands were classified as unclear by the independent observers Reference: none |
|
Gupta 2020 Published 330 (77) |
Single group (not stated; appears prospective); meeting Indian Council of Medical Research (ICMR) strategy for COVID‐19 testing (symptomatic or asymptomatic contacts between 5 and 10 days of exposure) |
COVID‐19 test centre (outpatient; tertiary care hospital) India (31 May‐24 July 2020.) |
Mixed 204 (62%) symptomatic; 126 (38%) asymptomatic. Median symptom duration: 1 day (range: 1‐10). Symptoms included: fever (31.5%), cough (25.4%), fatigue/malaise (11.8%), headache (3.3%), runny nose (3.3%) Median age 34.1 ± 12.6 years; 231 (70%) male |
RT‐PCR (single assay) Target: ORF1 ab nasal and throat swabs (NOP) in VTM Timing: as for index test; sequence for specimen collection was random for both the samples Interval: paired swabs |
None reported Index: none reported Reference: none reported |
|
Kruger 2020(a) Kruger 2020(b) Kruger 2020(c) Preprint Overall: 2407 (70) By assay: [A] 729 (15) [B] 425 (8) [C] 1263 (47) SD Biosensor data (assay [C]) can also be extracted by site (1) 334 (7) (2) 907 (39) (3) 19 (0) |
Single group (prospective) Participants at risk for SARS‐CoV‐2 infection based on exposure to a confirmed case, suggestive symptoms, or travel to a high‐risk area, presenting at 1 of 3 sites: (1) drive‐in testing station (n = 1213) (2) a clinical ambulatory testing facility (n = 1308) (3) secondary care facility (n = 53) |
COVID‐19 test centre or secondary care (in‐patient?) (1), (2) Germany (3) UK (17 April and 25 August 2020; dates varied by assay and site) |
Mainly symptomatic Symptomatic on testing day (n = 2355) Overall: 1901, 80.7% [A] 564, 81.2% [B] 283, 68.9% [C] 1054, 84.4% Prior negative test result (n = 1928) Overall: 236, 12.2% [A] 73, 11.7% [B] 38, 12.6% [C] 125, 12.5% Detailed symptoms are reported by site and test in supplementary materials Mean age (SD) (n = 2405) Overall 40.4 years (14.3) [A] 42.7 (14.9) [B] 44.9 (15.4) [C] 37.6 (12.7) Male (%) (n = 2361) Overall: 1115, 47.2% [A] 47.2% [B] 39.7% [C] 49.8% |
RT‐PCR (multiple assays) Target: not stated Paired swabs; as per index test (RT‐PCR swab obtained first) Drive‐in centre: NP or OP Other centres: combined NOP (OP conducted first) Timing: as per index test Interval: paired; simultaneous |
154 excluded following enrolment
[116 2nd swab refused
3 nose bleed after 1st swab
3 insufficient time for both swabs
31 other reasons
1 no reason available] Antigen tests: [A] 2 invalid (PCR‐negative) [B] 8 invalid (PCR‐negative) [C] 0 invalid reported PCR: 3 excluded as invalid (n = 2) or not available (n = 1) Index: none reported Reference: none reported |
|
Lambert‐Niclot 2020 Accepted manuscript 138 (94) |
Single group (unclear; testing conducted prospectively); Samples submitted for RT‐PCR testing |
Laboratory‐based (3 university hospital virology laboratories); France (1 April‐15 April 2020) |
Not reported | RT‐PCR (multiple assays) Target: E gene As for index test; NP swab Timing: within a few hours after collection Interval: same sample, both tests conducted within a few hours |
4 samples collected in COBAS VTM gave invalid results and all samples in COBAS medium were excluded Index: control lines reported as 'barely visible' for 9 positive and 8 negative tests Reference: none reported |
|
Linares 2020 Preprint 255 (60); NB 257 reported in sample collection |
Single group (unclear; appears to be prospective) 2 locations: [1] symptomatic patients admitted to ED with clinical suspicion of COVID‐19 (n = 135) or asymptomatic patients with history of contact with another COVID‐19 patient (n = 17) [2] symptomatic patients (n = 50) or asymptomatic (n = 55) patients attending one of two primary healthcare centres |
Hospital A&E (n = 135) or primary care (n = 50); Spain (10‐15 September) |
Mixed: 185, 72% symptomatic ED (n = 135): fever 40, dyspnoea 42, cough 22, headache 14 Prim care (n = 50): fever 14, dyspnoea 1, cough 18, headache 17 Mean(?) age (range): ED 51.5 years (37.0‐71.8 years); primary care 39.0 years (25.0‐56.0 years) Male: ED 77 (51%), primary care 49 (47%) |
RT‐PCR (single assay);
Threshold not stated Target: not stated NP (paired) Timing: not stated Interval: paired |
None reported however 257 reported in Methods and 255 in Results None reported Index: none reported Reference: none reported |
|
Liotti 2020 Published letter 329 (104) |
Unclear; two group (retrospective) Residual samples selected from one of two virology laboratories at two COVID‐19 reference hospitals |
Laboratory‐based (not reported) Italy (not stated) |
Not reported Not stated Of SARS‐CoV‐2‐positive samples, 21, 20% high viral load (< 25 Ct), 83, 80% low viral load (≥ 25) [28, 27% with Ct ≥ 35] Not stated |
RT‐PCR (multiple assays) Target: not stated NP (same as index) Timing: not stated Interval: simultaneous (same swab) |
None reported Index: none reported FP results were re‐tested with Ag assay, 3 of 4 remained positive (all blood contaminated) Reference: none reported |
|
Mak 2020 Published 160 samples from 152 patients (160) |
Single group (cases) (retrospective) RT‐PCR‐positive samples selected from Hong Kong's COVID‐19 reference laboratory |
Laboratory‐based (not stated) Hong Kong (1 February‐21 April 2020) |
Not reported Not stated High viral load (< 18.57 Ct) ‐ 64, 40% 'Normal' viral load > 18.57 ‐ 96, 60% Not stated |
RT‐PCR (single assay);
Threshold ≤ 40 Ct Target: RdRp NPA & TS, NPS & TS, sputum and throat saliva, as for index test Timing: not stated Interval: simultaneous; same samples |
None reported Index: none reported Reference: none reported |
|
Mertens 2020 Preprint (not peer‐reviewed) n = 328 samples (99 at LHUB‐ULB, 132 at CHU Liège, 97 at UZ Leuven); 132 COVID‐19 cases |
Single group (retrospectively) Samples from cases of suspected SARS‐CoV‐2 infection |
Laboratory‐based (university laboratory; discussion states no outpatients) Belgium (19‐30 March 2020) |
Not reported Not reported |
RT‐PCR (multiple assays)
Threshold ≤ 40 Ct Target: multiple As for index test Timing: analysed at time of collection Interval: same samples used; discussion report 'some delay' between PCR and antigen testing |
No None reported Index: weak T lines considered positive Reference: none reported |
|
Nagura‐Ikeda 2020 Accepted manuscript 103 (103) |
Single group (cases) (NR; samples appear to be collected prospectively) Patients with laboratory‐confirmed COVID‐19 referred for isolation and treatment, including symptomatic and asymptomatic |
Mixed (inpatient and asymptomatic (admitted or quarantined)) Japan (11 February‐13 May 2020) |
Mainly symptomatic 88 (85%) symptomatic, including 16 (15%) severe (showing clinical symptoms of pneumonia ‐ dyspnea, tachypnoea, saturation of percutaneous oxygen [SpO2] < 93%, and the need for oxygen therapy); 15 (15%) asymptomatic IPD provided Median age 46, range 18‐87; 66 (64%) male |
RT‐PCR (no details) Target: not reported NP or OP; appears to be same day as saliva collection Timing: specific timing in regard to symptom onset NR Interval: unclear; saliva collected on day of admission to quarantine/hospital but NP/OP conducted prior |
Not stated None reported Index: none reported Reference: none reported |
|
Nash 2020 Preprint 190 (100) |
Unclear; two group (retrospective) Samples from suspected patients submitted to 'PATH' (ww.path.org) for routine COVID diagnosis |
Laboratory‐based Not reported (not reported) |
Not reported Not reported |
RT‐PCR (single assay) Target: N, S, and ORF1ab genes Nasal (same swab) Timing: not stated Interval: simultaneous (same swab) |
None reported Index: none reported Reference: none reported |
|
PHE 2020(a) Published 1118 (178) |
Two group (retrospective) Residual swabs from [1] PCR+ in‐patients (n = 200, all frozen) [2] PCR‐ inpatient (n = 1000, all fresh samples) Swabs were sent to Porton Down following routine testing |
Inpatient UK (March 2020 (PCR+)) |
Symptomatic | RT‐PCR (may be Roche assay) Target: not stated Appears to be same sample as for Ag test Timing: as for index test Interval: same swab |
See below, plus 1 void PCR Failure rates reported as: [1] 12/212, 6% [2] 50/1040, 5.1% NB remaining samples per group (200 and 990) does not match with final numbers reported (178 and 940), no explanation given in report Index: unclear Reference: unclear |
|
PHE 2020(b) Published 157 (46) |
Single group (retrospective) Samples obtained during a COVID‐19 outbreak at a navy barracks (n = 157) |
Contacts (outbreak); UK (Not stated) |
Not reported | RT‐PCR (unclear, may be Roche Cobas assay) Target: unclear Appears to be same sample as for Ag test Timing: as for index test Interval: same swab |
None reported Failure rate reported as 6/157, 3.8% NB resulting number samples (n = 151) does not match with final number reported (n = 152) Index: unclear Reference: unclear |
|
PHE 2020(c) [non‐HCW tested] Published 1946 (372) |
Single group (not stated) Individuals presenting at a regional COVID‐19 testing centre |
COVID‐19 test centre UK (Not stated) |
Not reported; presumably symptomatic and meeting testing criteria | RT‐PCR (appears to be Roche assay) Target: not stated Not stated; paired swabs obtained Timing: as for index test Interval: paired swabs; simultaneous |
Initial sample of 1946 reported, 27 failed and PCR with void PCR. Data reported for only 1686 Failure rate reported as 27/1946 failed, 1.4% Index: unclear Reference: unclear |
|
PHE 2020(d) [HCW tested] PHE 2020(d) [Lab tested] Published 479 (479) |
Single group (cases) (not stated) Individuals presenting at one of 14 drive‐through regional COVID‐19 NHS test and trace centres. Those with a PCR +ve result returned for a re‐test within 5 days of the original result. It appears that only those with PCR +ve results at the second sampling were included. [A] HCW tested [B] Lab scientist tested |
COVID‐19 test centre (14 NHS test and trace centres; no further details); UK (not stated) |
Mainly symptomatic; 40/421 (9.5%) asymptomatic, 59 (14%) with no data, 322/421 with ≥ 1 symptom recorded. Unclear if symptoms were present at the time of the 1st swab or at the time of the 2nd sampling; data for asymptomatic group therefore not included in analyses NB: text reports data for 41 asymptomatic and 344 symptomatic from the Phase 3b study (total n = 385) |
RT‐PCR (may be Roche Cobas assay) Target: not stated Not stated; combined NOP swabs in VTM Timing: as for index test Interval: unclear, may have been paired |
HCW tested: 267 reported, 27 failed, leaving 240 for inclusion however data for only 223 HCW tested samples are provided lab scientist tested: Initial sample of 212 reported, 9 failed, leaving 203 for inclusion however data for only 198 lab scientist tested samples are provided. Index: unclear Reference: unclear |
|
PHE 2020(e) Published 538 (0) |
Single group (not stated) PHE and hospital staff volunteering for testing |
Screening UK (not stated) |
Asymptomatic | RT‐PCR (may be Roche assay) Target: not stated Not stated; paired swabs obtained Timing: as for index test Interval: paired swabs; simultaneous |
Initial sample of 570 reported, 36 failed, leaving 534 for inclusion. Data for 538 included Failure rate reported as 17/358, 4.7% Index: unclear Reference: unclear |
|
Porte 2020a Preprint (not peer reviewed) 127 samples; 82 PCR positive |
Single group (retrospectively) Patients with respiratory symptoms and/or fever and an epidemiological risk factor for SARS‐CoV‐2 infection (travel or contact with case) |
Hospital A&E (private hospital emergency room); Chile (16‐21 March 2020) |
Symptomatic Cough 94 (74.6%) Fever 77 (61.1%) Median duration of symptoms of 2 days (IQR 1–4) (range 0‐12) Duration of symptoms Day 0‐3 91 (72.2%) Day 4‐7 27 (22.4%) Day ≥ 8 8 (6.3%) 68 male (53.5%) median age 38 years (IQR 29.5–44) (range 1–91) |
RT‐PCR (single assay);
Threshold ≤ 40 Ct Target: not stated As for index test; same OP and NP swabs used Timing: median 2 d pso (IQR 1‐4, range 0‐12) Interval: same sample used; within 48 h |
No Not reported Index: not reported Reference: patients |
|
Porte 2020b [A] Accepted manuscript 64 (32) |
Multi‐group (retrospective) (1) COVID‐19 patients presenting within 5 days of symptom onset (n = 32) (2) symptomatic patients with negative PCR (n = 20) (3) asymptomatic patients screened prior to surgery (n = 12) |
COVID‐19 test centre (private clinic) Chile (Not stated) |
Symptomatic Not reported; 12 asymptomatic Total sample Median age 39 years (IQR 36.7‐57); 33, 52% male |
RT‐PCR (single assay)
Threshold ≤ 40 Ct Target: not stated NOP; as for index test Timing: not stated Interval: simultaneous; same sample |
None reported Index: none reported Reference: none reported |
|
Schildgen 2020 [A] preprint 73 (42) |
Two group (not stated; presume retrospective) [1] RT‐PCR positive BAL or throat wash samples [2] RT‐PCR‐negative samples |
Unclear (not stated) Germany (Not stated) |
Mixed Not stated for BAL samples, throat wash from 23 symptomatic and 27 asymptomatic people Not stated |
RT‐PCR (single assay) Target: not stated BAL or throat wash; as per index test Timing: not stated Interval: same swab |
8 PCR invalid samples also tested; 2/8 invalid in one AG assay each, 3/8 negative in all 3 Ag assays None reported Index: none reported Reference: none reported |
|
Scohy 2020 Published 148 (106) |
Single group (not stated) NP swabs submitted for testing at a large tertiary hospital |
Laboratory‐based (unclear) Belgium (6‐21 April 2020) |
Mixed 86 (58%) symptomatic, 45 (30%) asymptomatic, 17 (11%) symptom status not reported Median age 57.5 (0, 94 years); 64 (43%) male |
RT‐PCR (single assay)
Threshold ≤ 40 Ct Target: RdRp NP; same as for index Timing: not stated Interval: same sample |
None reported Index: none reported Reference: none reported |
|
Shrestha 2020 Published 113 (47) |
Single group (not stated; appears prospective) Close contacts of confirmed cases identified through contact tracing, and residing in quarantine centre |
Contacts (contact tracing) Nepal (August‐September 2020) |
Asymptomatic All asymptomatic; tested on day 5 Range 13‐74; 89, 79% male |
RT‐PCR (no details) Target: not stated NP in 3 mL VTM Timing: as for index test Interval: simultaneous, paired samples |
None reported Index: tests were repeated for samples with indistinct outcomes. Reference: none reported |
|
Takeda 2020 Preprint 162 (62) |
Two group (retrospective) [1] RT‐PCR‐confirmed COVID‐19 samples [2] Random sample of RT‐PCR‐negative samples |
Laboratory‐based (multiple clinical institutions) Japan ("Early April'' also later states 4 day period) |
Not reported Not stated |
RT‐PCR (single assay) Target: N2 NP, as for index test Timing: not stated Interval: simultaneous, same samples |
16 positive samples omitted; possibly because not initial samples but unclearly reported None reported Index: none reported Reference: none reported |
|
Van der Moeren 2020(a) Preprint 354 (17) |
Single group (prospective) [1] Adults presenting at a single community test centre for COVID‐19 testing Second cohort reported in Van der Moeren 2020(b) |
COVID‐19 test centre (community) Netherlands (28‐30 September) |
Symptomatic No details Day < 7 12, 70.6%, Day > 7 1, 5.9%, not reported 4, 23.5% |
RT‐PCR (multiple assays) Target: E‐ and RDRP‐gene (Cobas) or E‐gene and N‐gene (Abbott) NOP; specimen from the throat and nasal cavity up to the nasal bridge Timing: as for index test Interval: paired |
2 samples excluded due to RT‐PCR coding error 1 invalid on Ag test Index: none reported Reference: none reported |
|
Van der Moeren 2020(b) Preprint 132 (132) |
Single group (cases) (prospective) [2] Patients with a positive PCR test result at one of 2 community testing facilities who were retested at home within 72 h of initial positive result Second cohort reported in Van der Moeren 2020(a) |
COVID‐19 test centre (community) Netherlands (28 September‐6 October) |
Symptomatic At time of home visit: asymptomatic 3, 2% (2/3 still PCR +ve) Symptomatic 129 (123 still PCR +ve) Day < 7 66, 50% Day > 7 57, 43% Not stated |
RT‐PCR (multiple assays) Target: E‐ and RDRP‐gene or E and N‐gene NOP; specimen from the throat and nasal cavity up to the nasal bridge Timing: as for index test Interval: paired |
Review team excluded 7 no longer PCR+ at time of home visit (1 asymptomatic (antigen test positive), 6 symptomatic (antigen test result not given)) None reported Index: none reported Reference: none reported |
|
Veyrenche 2020 Preprint 65 (45) |
Two group (retrospective) [1] PCR+ hospital inpatients ( [2] Pre‐pandemic samples from 'patients' (not otherwise specified) |
Hospital in‐patient (no further detail); France (14 March‐11 April) |
Symptomatic: All hospitalised; 27/45, 60% cases 'severe' according to WHO guideline (similar numbers per Ct subgroup) Median age: Ct ≤ 25 66 years (IQR 48‐84) Ct 25‐35 63 years (50‐76) Ct ≥ 35 58 years (49‐67) Controls 64 (35‐93) 32/45, 71% male, all controls were male |
RT‐PCR (single assay) Target: RdRp, N, E NP; as for index Timing: as for index Interval: simultaneous; same swab |
None reported Index: none reported Reference: none reported |
|
Weitzel 2020 [A] Preprint 111 (80) |
Single group (retrospective) Patients with respiratory symptoms and/or fever |
Hospital A&E (emergency room at private hospital); Chile (16 March‐26 April 2020) |
Symptomatic Respiratory symptoms and/or fever; no further detail Median age 40 years; 50, 45% male (median age 38 years, 43% male for all samples tested during period) |
RT‐PCR (single assay)
Threshold ≤ 40 Ct Target: RdRp as for index; NOP swabs; Timing: as for index test; median 2 days (IQR 1‐5 days) Interval: same samples; index tests conducted after frozen storage |
2 invalid excluded Two tests invalid due to insufficient liquid migration Index: none reported Reference: none reported |
|
Young 2020 Preprint 251 (38); 9 excluded |
Single group (prospective) ≥ 1 symptoms of COVID‐19 (within ≤ 7 days post symptom onset) at 21 study sites Second cohort excluded as only discrepant results on the two Ag assays underwent RT‐PCR |
Mixed (drive‐through/tent (n = 42), outpatient clinic (n = 74), research clinic (n = 72), or skilled nursing facility (n = 66)) USA (5‐11 June 2020) |
Symptomatic 110 (43%) cough, 98 (39%) muscle pain, 95 (37%) headache, 90 (35%) sore throat, 78 (31%) fever. Of those at ≤ 6 days pso (n = 245): 94 (38%) with 1 symptom, 151 (62%) with ≥ 2 symptoms median age 43 (range 18‐90); 91 (36%) male |
RT‐PCR (single assay) Target: not stated NP (n = 217) or OP (n = 34); clinician collected Timing: swabs taken prior to any study swabs (potential for contamination of nasal cavity) Interval: simultaneous (paired) |
9 excluded; 6 did not meet eligibility criteria and 3 had invalid specimens/results (2 on RT‐PCR and 1 labelling error) 3 invalid on at least one assay Index: none reported Reference: none reported. Re‐test of 9 'FN' results with BD MAX RT‐PCR resulted in 2 confirmed FN (BD MAX +ve and sero +ve), 6 were BD Max ‐ve (incl 1 sero +ve) and 1 invalid (no result) |
| A&E: accident and emergency; BAL: bronchoalveolar lavage; CDC: National Health Commission of the People's Republic of China; Ct: cycle threshold; ED: emergency department; FP: false positive; HCW: healthcare worker; IFU: [manufacturers'] instructions for use; IPD: individual patient data; IQR: interquartile range; NHS: National Health Service (UK); NOP: naso‐oropharyngeal; NP: nasopharyngeal; OP: oropharyngeal; pso: post‐symptom onset; RT‐PCR: reverse transcription polymerase chain reaction; SD: standard deviation; VTM: viral transport medium | |||||
Appendix 10. Antigen tests: summary index test details
| Study | Index test (manufacturer) | Test method Target | Sample details | Test operator Test threshold |
| Albert 2020 | Panbio COVID‐19 AG Rapid Test Device (no product code reported) (Abbott Diagnostic GmbH, Jena, Germany) | CGIA (from IFU) Nucleoprotein |
Samples tested: NP; collected by trained nurses using flocked swabs (Direct) Timing of sampling: day < 7 pso Timing of test: immediate testing Storage: none |
Not stated Threshold: visible line within 15 min; as per manufacturer |
| Alemany 2020 | Panbio COVID‐19 Ag Test (no product codes) (Abbott Laboratories) [Selected from comparison of 4 assays using 40 NP samples] |
CGIA Not stated (SARS‐CoV‐2 antigen) |
Samples tested: varied by site [1] and [2] NP, [3] nasal mid‐turbinate (VTM); collection not reported
Timing of sampling: not stated (SARS‐CoV‐2 antigen) Timing of test: not stated; frozen samples Storage: stored at 2‐8 °C prior to PCR then frozen (‐80 °C) prior to Ag testing |
2 laboratory technicians Threshold: visible line; as per manufacturer |
| Billaud 2020 | ABBOTT SARS‐COV2 Antigenic Test (Abbott) (no product code reported) | CGIA (from IFU) Not stated |
Samples tested: NP; collected by firefighters (direct) Timing of sampling: not stated, includes people > 7 days pso Timing of test: immediate testing Storage: none |
Not stated Threshold: visual line; as per manufacturer |
| Blairon 2020 | COVID‐19 Ag Respi‐Strip (no product code reported) (Coris Biocencept (Gembloux, Belgium)) | LFA Not stated |
Samples tested: NP swabs; collection not reported (VTM) Timing of sampling: not stated; appears to be on presentation (repeat tests ordered at clinician's discretion were excluded) Timing of test: infer that Ag test conducted immediately on receipt of sample at on‐site laboratory Storage: no storage described |
Not stated; infer laboratory staff Threshold: as per manufacturer |
| Cerutti 2020 | STANDARD Q COVID‐19 Ag (SD‐Biosensor, RELAB, I) (no product code reported) | CGIA (from IFU) NP |
Samples tested: NP; collection not stated (VTM) Timing of sampling: not stated Timing of test: not stated Storage: primarily run in parallel with standard of care RT‐PCR; 13 were frozen residual samples |
Not stated; laboratory staff presumed Threshold: visual line after 15‐30 min; as per manufacturer |
| Courtellemont 2020 | COVID‐VIRO® (AAZ, Boulogne Billancourt, France) (no product code reported) | CGIA Nucleocapsid |
Samples tested: NP; collected by trained personnel (nurse,
doctors, or biologist)
Subgroup had OP or saliva collected (direct) Timing of sampling: median 5 days pso, mean 5.3 days, range 1‐20 d Timing of test: immediate testing Storage: none |
Not stated Threshold: visible line; as per manufacturer |
| Diao 2020 | Not stated (in‐house) | FIA Nucleocapsid protein (N‐antigen) |
Samples tested: NP (all), urine (subgroup) (saline) Timing of sampling: not stated Timing of test: not reported Storage: not reported |
Not stated; presume lab staff Threshold: mean value of the fluorescence signal plus 5 SD |
| Fenollar 2020(a) | PANBIO COVID‐19 Ag (Abbott) (no product code reported) | CGIA (from IFU) NP |
Samples tested: NP (direct) Timing of sampling: not stated Timing of test: tested within 1 h Storage: none |
Not stated; presume on‐site testing Threshold: visual line; as per manufacturer |
| Fenollar 2020(b) | PANBIO COVID‐19 Ag (Abbott) (no product code reported) | CGIA (from IFU) NP |
Samples tested: NP (direct) Timing of sampling: not stated Timing of test: tested within 1 h Storage: none |
Not stated; presume on‐site testing Threshold: visual line; as per manufacturer |
| FIND 2020a | NowCheck COVID‐19 Ag test (RG1901DG) (Bionote Inc) | LFA (nos) SARS‐CoV‐2 nucleocapsid antigen |
Samples tested: proprietary NP swab collected by HCW (direct) Timing of sampling: median 4 days pso (IQR 3, 6 days); day < 0‐3 152, 39% day 4‐7 180, 46% day ≥ 8 58, 15% Timing of test: not specified; as soon as possible after collection and within IFU recommendations Storage: room temperature for 1 h or 2‐8 °C for 4 h |
HCW Threshold: presence of visible control and test lines |
| FIND 2020b | Panbio COVID‐19 Ag Rapid Test (41FK10) (Abbott) (no product code reported) | CGIA (from IFU) Not reported |
Samples tested: NP (direct) Timing of sampling: time pso recorded for 115/124, 92% Day 0‐3 89, 78% Day 4‐7 23, 20% Day 8+ 3, 3% Timing of test: not specified; as soon as possible after collection and within IFU recommendations Storage: author contact advises tested as soon as possible and within the time limit specified in the IFU |
HCW Threshold: presence of visible control and test lines |
| FIND 2020c (BR) | STANDARD Q COVID‐19 Ag (09COV30D) (SD Biosensor Inc) | CGIA (from IFU) Not reported |
Samples tested: NP; collected by HCW (direct) Timing of sampling: median 5 days pso (IQR 4, 6 days) (for 397 patients); day < 0‐3 85, 21% day 4‐7 273, 69% day ≥ 8 39, 10% Timing of test: tested as soon as possible and within the time limit specified in the IFU Storage: none |
HCW Threshold: presence of visible control and test lines |
| FIND 2020c (CH) | STANDARD Q COVID‐19 Ag (09COV30D) (SD Biosensor Inc) | CGIA (from IFU) Not reported |
Samples tested: NP (direct) Timing of sampling: median not reported (range 0‐15); day < 0‐3 ‐ 122, 67%% day 4‐7 ‐ 54, 29% Day 8+ ‐ 7, 34% Timing of test: tested as soon as possible and within the time limit specified in the IFU Storage: none |
HCW Threshold: presence of visible control and test lines |
| FIND 2020d (BR) | STANDARD F COVID‐19 Ag FIA (F‐NCOV‐01G, 10COV30D) (SD Biosensor Inc) | FIA Not reported |
Samples tested: NP; collected by HCW (Direct) Timing of sampling: median 4 days pso (IQR 3, 6 days) (for 421 patients); day <0‐3 ‐ 131, 31% day 4‐7 ‐ 248, 59% day >=8 ‐ 42, 10% Timing of test: tested as soon as possible and within the time limit specified in the IFU Storage: none |
HCW Threshold: as per STANDARD F Analyzer; cut‐off index ≥ 1.0 (as per IFU) |
| FIND 2020d (DE) | STANDARD F COVID‐19 Ag FIA (F‐NCOV‐01G, 10COV30D) (SD Biosensor Inc) | FIA Not reported |
Samples tested: [1] NP; [2] Combined NOP swabs; collected by HCW (direct) Timing of sampling: median 3 days pso (IQR 2,5 days) (for 505 patients); day < 0‐3 ‐ 257, 51% day 4‐7 ‐ 202, 47% day ≥ 8 ‐ 46, 9% Timing of test: tested as soon as possible and within the time limit specified in the IFU Storage: none |
HCW Threshold: as per STANDARD F Analyzer; cut‐off index ≥ 1.0 (as per IFU) |
| FIND 2020e (BR) | BIOCREDIT COVID‐19 Ag (G61RHA20) (RapiGEN Inc) | CGIA (from IFU) Not reported |
Samples tested: NP; collected by HCW (direct) Timing of sampling: median 5 days pso (IQR 4, 7 days) (for 470 patients); day < 0‐3 ‐ 95, 20% day 4‐7 ‐ 296, 63% day ≥ 8 ‐ 79, 17% Timing of test: tested as soon as possible and within the time limit specified in the IFU Storage: none |
HCW Threshold: visual appearance of test and control lines |
| FIND 2020e (DE) | BIOCREDIT COVID‐19 Ag (G61RHA20) (RapiGEN Inc) | CGIA (from IFU) Not reported |
Samples tested: [1] NP; [2] NOP; collected by HCW (direct) Timing of sampling: median 3 days pso (IQR 2, 4 days) (for 701 patients); day < 0‐3 ‐ 472, 67% day 4‐7 ‐ 161, 23% day ≥ 8 ‐ 68, 10% Timing of test: tested as soon as possible and within the time limit specified in the IFU Storage: none |
HCW Threshold: visual appearance of test and control lines |
|
Fourati 2020 [A] Fourati 2020 [B] Fourati 2020 [C] Fourati 2020 [D] Fourati 2020 [E] |
[A] SARS‐CoV‐2 COVID‐19 Respi‐Strip [B] Standard Q COVID‐19 Ag [C] PanBio COVID‐19 Antigen Rapid Test [D] Biosynex COVID‐19 Ag BSS [E] COVID‐VIRO Antigen Rapid Test [F] NG Test SARS‐CoV‐2 Ag (assay excluded) (no product codes reported) ([A] Coris BioConcept, Gembloux, Belgium [B] SD BIOSENSOR, Inc., Korea [C] Abbott, Chicago, Illinois, USA [D] Biosynex, Strasbourg, France [E] AAZ, Boulogne‐Billancourt, France [F] NG Biotech, Guipry, France | [A] CGIA (from IFU)
[B] LFA (nos)
[C] CGIA (from IFU)
[D] CGIA (from IFU)
[E] CGIA (from IFU) Not stated |
Samples tested: NP; collection not reported (VTM) Timing of sampling: pso (reported for 289 samples): 0‐3 days 97, 34% 4‐7 days 103, 36% 8‐11 days 63, 22% ≥ 12 days 26, 9% No. samples reported at > 7 days varied per test, maximum was 289 Timing of test: not stated Storage: Frozen at ‐80 °C until use |
Laboratory staff Threshold: visual, as per manufacturer |
| Gremmels 2020(a) | Panbio COVID‐19 Ag Rapid Test (lot 41ADF011A) (Abbott (Lake Country, IL, U.S.A)) | CGIA (from IFU) NP |
Samples tested: NP; obtained after NOP swab for RT‐PCR; implies collected by HCW (unclear) Timing of sampling: cohort [1] (data on duration of symptoms reportedly missing for 201 participants; total reported here is 1138 but denominator for %s is 1166) day 1‐3 pso 387, 33.2% day 4‐7 560, 48.0% day > 7 191, 16.4% Timing of test: within 2 h of collection Storage: none described |
2 independent observers Threshold: visual line within 15 min; as per manufacturer |
| Gremmels 2020(b) | Panbio COVID‐19 Ag Rapid Test (lot 41ADF011A) (Abbott (Lake Country, IL, U.S.A)) | CGIA (from IFU) NP |
Samples tested: NP; obtained after NOP swab for RT‐PCR; implies collected by HCW (direct) Timing of sampling: not stated; on presentation Timing of test: within 2 h Storage: appears to be room temperature |
2 independent observers Threshold: visual line within 15 min; as per manufacturer |
| Gupta 2020 | Standard Q rapid antigen detection test (SD Biosensor, Inc., Gurugram) (no product code reported) | CGIA (from IFU) Not stated |
Samples tested: NP; collection method detailed but personnel not described; presume HCW. Sequence for specimen collection was random for both the samples (Ag and RT‐PCR) (direct) Timing of sampling: symptomatic: 192 (95%) ≤ 5 days pso (incl 57 cases) Timing of test: immediate testing Storage: none |
Same person who obtained swab; HCW Threshold: visual; test and control lines |
|
Kruger 2020(a) Kruger 2020(b) Kruger 2020(c) |
[A] Bioeasy 2019‐nCoV Ag Fluorescence Rapid Test Kit (Time‐Resolved Fluorescence)
[B] COVID‐19 Ag Respi‐Strip
[C] STANDARD Q COVID‐19 Ag Test ([A] Shenzhen Bioeasy Biotechnology Co. Ltd., Guangdong Province, China [B] Coris Bioconcept, Gembloux, Belgium [C] SD Biosensor, Inc. Gyeonggi‐do, Korea) (no product codes reported) |
[A] FIA
[B] and [C] CGIA Not stated |
Samples tested: drive‐in centre: NP or OP
Other centres: combined NOP (OP conducted first)
RT‐PCR swab obtained first, then same technique repeated for Ag test (direct) Timing of sampling: overall: mean 5 days pso (SD 9.6). [A] 7.0 (SD 12.2); [B] 6.2 (SD 14.0); [C] 3.7 (SD 5.6) Timing of test: not stated but no delay reported (on‐site testing) for drive‐in and ambulatory testing; secondary care samples transported to lab Storage: as above RT‐PCR swab obtained first, then same technique repeated for Ag test |
Drive‐in and ambulatory clinic: POC evaluation
Secondary care: laboratory staff Threshold: [A] as per Analyzer; [B] and [C] visual appearance were interpreted by 2 operators, each blinded to the result of the other. In case of discrepant results, both operators re‐read the result and agreed on a final result. Invalid results were repeated once using the remaining buffer according to the respective IFUs. Readouts were done within the recommended time for each Ag‐RDT (10 minutes for Bioeasy, 15 minutes for Coris and 15‐30 minutes for SD Biosensor). |
| Lambert‐Niclot 2020 | COVID‐19 Ag Respi‐Strip CORIS (no product code) (BioConcept®, Gembloux, Belgium) | CGIA SARS‐CoV‐2 NP |
Samples tested: NP swabs in VTM (collection process not described) (VTM) Timing of sampling: not stated Timing of test: not stated (soon after collection) Storage: none; no cooling or freezing step used |
Not stated; presume lab staff Threshold: as per manufacturer |
| Linares 2020 | PanBio COVID‐19 Ag Rapid Test Device (no product code) (Abbott Rapid Diagnostic Jena GmbH, Jena, Germany) | CGIA (from IFU) Nucleocapsid |
Samples tested: NP; HCW obtained (direct) Timing of sampling: ED: 2 days pso (IQR? 1‐5) PC: 4 days pso (IQR? 2‐8) Table 3 reports range of 0‐27 days pso or post COVID‐19 contact, and range of 0‐16 days for days pso for symptomatic cases only Timing of test: not stated; presume immediate on‐site testing Storage: not stated |
Not stated Threshold: not stated; as per manufacturer |
| Liotti 2020 | STANDARD F COVID‐19 Ag FIA (no product codes reported) (SD Biosensor (Suwon, South Korea)) | FIA NP |
Samples tested: NP; collection not reported (not specified) Timing of sampling: not reported Timing of test: within 24 h after collection Storage: samples kept at 4 °C until testing |
Not stated; lab staff Threshold: as per manufacturer |
| Mak 2020 | BIOCREDIT COVID‐19 Ag (no product code reported) (RapiGEN Inc) | CGIA Not stated |
Samples tested: throat saliva (TS, n = 45), nasopharyngeal swab and throat swab (NPS & TS, n = 103), nasopharyngeal aspirate and throat swab (NPA & TS, n = 81), sputum (n = 45); no details of collection methods (VTM or PBS) Timing of sampling: not stated Timing of test: not stated; frozen samples Storage: stored at −70 °C until used for study purposes |
Not stated; laboratory staff presumed Threshold: not stated |
| Mertens 2020 | COVID‐19 Ag Respi‐Strip (Coris BioConcept (Belgium)) (no product code reported) | CGIA SARS‐CoV and SARS‐CoV‐2 highly conserved nucleoprotein |
Samples tested: mixed (322 NP swabs, 4 NPA and 2 BAL) (VTM) Timing of sampling: not stated Timing of test: not described Storage: not reported |
Laboratory technician Threshold: visible reddish‐purple band appearing at the Test line position (T) |
| Nagura‐Ikeda 2020 | ESPLINE® SARS‐CoV‐2 (no product code reported) [Five other tests performed including RT‐PCR and RT‐LAMP, but not eligible for this review] (Fuji Rebio Inc) | LFA (no reader device required) NP |
Samples tested: saliva (self collected) (direct) Timing of sampling: saliva collected on admission to hospital; IPD reports this was median 7 days pso (1‐14) Timing of test: not stated; frozen samples Storage: stored at ‐80 °C until sample preparation |
Not stated; implies laboratory staff Threshold: not stated; appearance of test line implied |
| Nash 2020 | Direct antigen rapid test (DARTTM); NP‐based (E25Bio Inc (Cambridge MA)) | Immunochromatographic paper‐based (CGIA) NP |
Samples tested: nasal; collection not described (not specified) Timing of sampling: not stated Timing of test: not stated Storage: banked frozen prior to testing |
Not stated; presume lab staff Threshold: visual line |
| PHE 2020(a) | Innova SARS‐CoV‐2 Antigen Rapid Qualitative Test (Innova Medical Group) (no product code reported) | CGIA (from IFU) Not stated |
Samples tested: combined NP and OP swabs; inpatients so presumed HCW collected (VTM) Timing of sampling: not stated Timing of test: not stated Storage: frozen (PCR+); fresh (PCR‐) |
Laboratory staff Threshold: visual line; as per manufacturer |
| PHE 2020(b) | Innova SARS‐CoV‐2 Antigen Rapid Qualitative Test (Innova Medical Group) (no product code reported) | CGIA (from IFU) Not stated |
Samples tested: OP swabs; self‐collected (VTM) Timing of sampling: 1 week after outbreak; no further details Timing of test: not stated Storage: transported at 4 °C to Porton Down for testing |
Laboratory staff Threshold: visual line; as per manufacturer |
| PHE 2020(c) [non‐HCW tested] | Innova SARS‐CoV‐2 Antigen Rapid Qualitative Test (Innova Medical Group) (no product code reported) | CGIA (from IFU) Not stated |
Samples tested: anterior nasal and combined OP samples. Self‐collected (direct) Timing of sampling: not stated Timing of test: immediate testing Storage: none |
Self‐trained non‐HCW Threshold: visual line; as per manufacturer |
|
PHE 2020(d) [HCW tested] PHE 2020(d) [Lab tested] |
Innova SARS‐CoV‐2 Antigen Rapid Qualitative Test (Innova Medical Group) (no product code reported) | CGIA (from IFU) Not stated |
Samples tested: combined anterior nasal and OP swabs; self‐collected (direct) Timing of sampling: not stated Timing of test: immediate testing Storage: none |
[A] HCW on‐site
[B] laboratory scientist at PHE Threshold: visual line; as per manufacturer |
| PHE 2020(e) | Innova SARS‐CoV‐2 Antigen Rapid Qualitative Test (Innova Medical Group) (no product code reported) | CGIA (from IFU) Not stated |
Samples tested: OP swab for PHE staff; NP swab for hospital staff. All self‐collected (direct) Timing of sampling: not stated Timing of test: immediate testing Storage: none |
Lab scientist Threshold: visual line; as per manufacturer |
| Porte 2020a | Diagnostic Kit for 2019‐Novel Coronavirus (2019‐nCoV) Ag Test (Cat. N° YRLF04401025, lot N° 2002N408) (Bioeasy Biotechnology Co., Shenzhen, China ) | CGIA SARS‐CoV‐2 nucleocapsid protein |
Samples tested: mixed (322 NP swabs, 4 NPA and 2 BAL) (VTM) Timing of sampling: not stated Timing of test: not described Storage: not reported |
Laboratory technician Threshold: as per manufacturer |
|
Porte 2020b [A] Porte 2020b [B] |
[A] SOFIA SARS Antigen FIA
[B] STANDARD® F COVID‐19 Ag FIA
(no product codes reported) ([A] Quidel Corporation, San Diego, CA, USA [B] SD Biosensor Inc., Gyeonggi‐do, Republic of Korea) |
Both FIA NP |
Samples tested: naso‐oropharyngeal flocked swabs; obtained by trained personnel (VTM) Timing of sampling: all < 5 days pso; median PCR+: 2 days (IQR 1‐3) PCR‐: 1 day (IQR 0.75‐4) Timing of test: not stated; frozen samples Storage: stored at ‐80 °C following RT‐PCR |
Laboratory staff Threshold: as per manufacturer; both using analyzer device |
|
Schildgen 2020 [A] Schildgen 2020 [B] Schildgen 2020 [C] |
[A] BIOCREDIT [B] Panbio [C] SARS‐CoV‐2 Rapid Antigen test ([A] RapiGEN, [B] Abbott, [C] Roche) (no product code reported) | All CGIA Not stated |
Samples tested: BAL (n = 13); throat wash (n = 50, including 27 from asymptomatic) (not specified) Timing of sampling: not stated Timing of test: not stated Storage: not stated |
Not stated; presume lab staff Threshold: as per manufacturer |
| Scohy 2020 | COVID‐19 Ag Respi‐Strip (product code not reported) (Coris Bioconcept) | CGIA NP |
Samples tested: NP (not specified) Timing of sampling: not reported Timing of test: not stated; immediate or after period of storage Storage: none or stored at 4 °C until the test |
Not stated Threshold: visual appearance of T line; also states that "Two versions of the test were evaluated. On the second version, conjugate was coupled on a different way and the control line was optimized." |
| Shrestha 2020 | BIOCREDIT (RapiGen) (no product code reported) | Not stated Not stated |
Samples tested: NP (Direct) Timing of sampling: day 5 of quarantine Timing of test: not stated Storage: none reported; other sample from the same individual was processed for the results as instructed by the manufacturing company of antigen kit |
Lab technician (trained) Threshold: visual line; as per manufacturer |
| Takeda 2020 | ESPLINE SARS‐CoV‐2 (no product code reported) (Fujirebio Inc) | LFA using alkaline phosphatase (ALP)‐labelled antibodies SARS‐CoV‐2 antigen (from IFU) |
Samples tested: NP; collection not reported (not specified) Timing of sampling: not stated but all cases presumed by study authors to be from patients suspected of SARS‐CoV‐2 for the first time Timing of test: not stated Storage: swabs mixed with sample treatment solution; no storage reported |
Not stated; laboratory staff presumed Threshold: visual line, as per manufacturer |
| Van der Moeren 2020(a) | BD Veritor System for Rapid Detection of SARS‐CoV‐2 (Becton Dickinson) (no product code reported) | CGIA (from IFU) NP |
Samples tested: NOP? "specimen from the throat and the superficial nasal cavities (bilateral, 2.5 cm proximal from the nostril)"; collected by GGD employee (direct) Timing of sampling: time pso only provided for PCR+ cases: 12 < 7 d; 1 ≥ 7 d; 4 = no pso data Timing of test: within 6 h (at lab) Storage: stored dry in sterile test tubes and stored and transported on dry ice until processing at the laboratory; tested within 6 h after collection |
Trained laboratory technicians Threshold: [A] using Analyzer [B] visual inspection |
| Van der Moeren 2020(b) | BD Veritor System for Rapid Detection of SARS‐CoV‐2 (Becton Dickinson) (no product code reported) | CGIA (from IFU) NP |
Samples tested: NOP? "specimen from the throat and the superficial nasal cavities (bilateral, 2.5 cm proximal from the nostril)"; collected by GGD employee (direct) Timing of sampling: not reported; on presentation Timing of test: within 6 h (at lab) Storage: stored dry in sterile test tubes and stored and transported on dry ice until processing at the laboratory; tested within 6 h after collection |
Trained laboratory technicians Threshold: [A] using Analyzer [B] visual inspection |
| Veyrenche 2020 | Coris COVID‐19 Ag Respi‐Strip (BioConcept®, Gembloux, Belgium) (no product code reported) | CGIA NP |
Samples tested: NP; collection not described (VTM) Timing of sampling: day 1‐20 pso, median Ct ≤ 25 ‐ 7 (4, 10; presume this is IQR but could be range ‐ is described as SD in paper) Ct 25‐35 ‐ 8 (4, 12) Ct ≥ 35 ‐ 11 (7, 15) Timing of test: not stated Storage: not stated; RT‐PCR conducted prospectively within a few hours but not reported for Ag testing |
Not stated; presume lab staff Threshold: visual, as per manufacturer |
|
Weitzel 2020 [A] Weitzel 2020 [B] Weitzel 2020 [C] Weitzel 2020 [D] |
[A] Biocredit COVID‐19 Ag One Step SARS‐CoV‐2 Antigen Test (RapiGEN Inc., Anyang‐si, Gyeonggi‐do, Republic of Korea) [B] COVID‐19 Antigen Rapid Test Device StrongStep® COVID‐19 Antigen Test (Liming Bio‐Products Co., Jiangsu, China) [C] Huaketai New Coronavirus (SARS‐CoV‐2) N Protein Detection Kit (Fluorescence immunochromatography) (Savant Biotechnology Co., Beijing, China), [D] Diagnostic Kit for 2019‐Novel Coronavirus (2019‐nCoV) Ag Test (Fluorescence Immunochromatographic Assay) (Bioeasy Biotechnology Co., Shenzhen, China) | CGIA Not reported in study |
Samples tested: mixed (322 NP swabs, 4 NPA and 2 BAL) (VTM) Timing of sampling: not stated Timing of test: not described Storage: not reported |
Single trained laboratory technician under BSL2 cabinet; visual outputs read by 2 independent observers with referral to 3rd if needed Threshold: as per manufacturer; Savant test required use of manufacturer supplied UV torch due to unavailability of reader device in Chile |
| Young 2020 | BD Veritor SARS‐CoV‐2 antigen test (no product codes) (Becton, Dickinson and Company, BD Life Sciences—Integrated Diagnostic Solutions, San Diego, CA) | LFA (nos) NP |
Samples tested: nasal; clinician collected from both nostrils (same swab) (Direct) Timing of sampling: all ≤ 7 days pso; median 3.0 d, mean 3.2 d 38 (15%) 1 day pso, 57 (23%) 2 days, 54 (22%) 3 days, 40 (16%) 4 days, 37 (15%) 5 days 19 (8%) 6 days, 6 (2%) 7 days Timing of test: not stated; frozen samples Storage: swabs were shipped for testing on dry ice (−70 °C); |
Not stated; Veritor testing was performed internally at BD (San Diego, CA, USA) Threshold: as per manufacturer |
| Ag: antigen; BAL: bronchoalveolar lavage; CGIA: colloidal‐gold immunoassay; Ct: cycle threshold; FIA: fluorescent immunoassay; HCW: healthcare worker; IFU: [manufacturers'] instructions for use; IPD: individual patient data; IQR: interquartile range; LFA: lateral flow assay; NOP: naso‐oropharyngeal; NP: nasopharyngeal; NPA: nasopharyngeal aspirate; OP: oropharyngeal; pso: post‐symptom onset; PBS: phosphate‐buffered saline; PCR+: polymerase chain reaction‐positive; PCR‐: polymerase chain reaction‐negative; PHE: Public Health England; POC: point‐of‐care; RT‐PCR: reverse transcription polymerase chain reaction; SD: standard deviation; TS: throat swab; UV: ultraviolet; VTM: viral transport medium | ||||
Appendix 11. Molecular tests: summary study details
| Study | Study design; inclusion criteria | Setting; country (recruitment dates) | Participant characteristics |
Reference standard Samples and timing |
Missing data or uninterpretable results |
|
Assennato 2020 Preprint 172 (88; 91 after retesting with RT‐PCR) |
Single group; symptomatic individuals with suspected COVID‐19 sent for routine laboratory diagnosis; supplied via PHE Recruitment: not stated |
Laboratory‐based (no further details); UK (Not stated) |
‘symptomatic'; no further details Not stated |
RT‐PCR (single assay);
Threshold ≤ 36 Ct Target: (1) RdRp, E gene (2) RdRp 'different region' As for index; combined nose and throat swab in VTM Timing: not stated Interval: not stated; seems likely reference was carried out for routine diagnostic testing |
None reported Index: 3 FP and 1 FN result retested using SAMBA‐II; same results obtained on repeat Reference: 3 FP and 1 FN result were re‐tested* ‐ all 3 FPs found to be borderline +ve for ≥ 1 target gene on either Colindale or Cambridge (Wuhan) test (reclassified as TP) ‐ the FN result remained +ve on both RT‐PCR assays |
|
Broder 2020 Accepted manuscript 35 (35) |
Single group (cases); Samples +ve on RT‐PCR (Roche Cobas 6800) with lower range of viral load (E target Ct ≥ 30) Recruitment: not stated; deliberate sampling according to viral load |
Laboratory‐based (not stated); USA (Not stated) |
Not stated; lower viral load Not stated |
RT‐PCR (single assay) Target: E gene (unclear if other genetic targets as well) N/A As for index test; NP swab Timing: not stated; presume on presentation Interval: same samples; index within 3 days of reference |
None reported Index: none reported Reference: samples +ve on reference were tested by in‐house assay using modified CDC protocol |
|
Chen 2020a Published 58 (58); can only include data for 55 +ve on NP swabs |
Single group (cases); archived paired samples from COVID‐19 inpatients Recruitment: not stated |
Hospital in‐patient (no further detail); People’s Republic of China (Not stated) |
Not stated Median age 38 years; 28, 48% male |
RT‐PCR (single assay) Target: RdRp N/A only cases included Not stated; infer single ‐ve Same as index test Timing: not stated; prior to index test Interval: simultaneous; same samples |
None reported, however 3 samples +ve only on saliva excluded by review team Not stated Index: not stated Reference: none reported |
|
Collier 2020 Preprint and published version (25‐8‐20) 149 (32) |
Single group; patients admitted with a possible diagnosis of COVID‐19 Recruitment: consecutive |
Hospital inpatient (no further detail); UK (6 April‐2 May 2020) |
Not stated Mean age 62.7 years, 70, 47% male |
RT‐PCR (single assay) Target: not stated Not stated; separate swab used as participants were excluded if > 18‐h interval between swab collections Timing: not stated Interval: < 18 h |
Yes; 5 discarded VTM, 1 timing of PHE swab not reported, 1 inadequate SAMBA swab, 2 swab interval > 24 h Index: not described "Indeterminate … tests were repeated … until a valid result was obtained." Discrepant results re‐tested on original samples Reference: "indeterminate … tests were repeated on a replicate … swab until a valid result was obtained." Discrepant results re‐tested on original samples |
|
Cradic 2020(a) published 184 (33) |
Single group; symptomatic patients meeting criteria for testing Recruitment: not stated |
Mixed (ED or inpatients); USA (Not stated ) |
All symptomatic, no further details. Not stated |
Composite; result obtained from at least 2 of 3 commercial assays (includes 2 RT‐PCRs and Abbot ID NOW) Target: RdRp, S or ORF1ab gene (either present), ORF1ab or E gene (both present for +ve, either present for presumptive +ve) Same as index test Timing: not stated Interval: simultaneous ‐ same swab |
None reported Index: none reported Reference: none reported |
|
Cradic 2020(b) published 182 (13) |
Single group; presenting to ED with signs/symptoms of COVID‐19 submitted for routine laboratory testing (n = 182) Recruitment: not stated |
Hospital A&E (ED); USA (Not stated ) |
All symptomatic, no further details. Not stated |
RT‐PCR (single assay) Target: S or ORF1ab gene (either present) NP swab in UTM, same as index Timing: not stated Interval: simultaneous; paired swabs |
None reported Index: none reported Reference: none reported |
|
Dust 2020 Published 38 (20) |
Two‐group; [1] SARS‐CoV‐2 +ve samples submitted for routine viral diagnostic testing [2] samples +ve for other respiratory infection Convenience sampling Recruitment: retrospective |
Laboratory‐based (unclear; submitted to laboratory); Canada (Not stated) |
Not reported | RT‐PCR (single assay);
Ct threshold not stated Target: E, N1 NP (as for index) Timing: not stated Interval: simultaneous (same swab) |
None reported Index: none reported Reference: none reported |
|
Ghofrani 2020 Published 113 (17) |
Single group Patients with both RT‐PCR and POC test results available (n = 113), including: [1] symptomatic patients with a PCR swab test close to presentation and a re‐swab for POC testing [2] patients with +ve RT‐PCR results and remnant NP swabs available for POC test, [3] asymptomatic patients with +ve POC result on admission who were re‐swabbed for RT‐PCR confirmation. N per group was not reported Recruitment: convenience |
Mixed (hospital and community); USA (6 April‐21 April 2020) |
'Majority' symptomatic, no further details Not stated |
RT‐PCR (no details) Target: not stated Mixed; either paired swabs (within 3 days of each other) or same samples used Timing: not stated Interval: some same sample; paired samples could be up to 3 days apart |
None reported Index: none reported Reference: none reported |
|
Gibani 2020 Published 418 () |
Three sources of participants:
[1] self‐referred, HCWs or their family members with suspected COVID‐19 who were not admitted to hospital (n = 280)
[2] ED patients with suspected COVID‐19 (n = 15)
[3] hospital inpatient admissions with or without suspected COVID‐19 (n = 91)
Total N 418 paired samples; 32 excluded as invalid (patient group not reported), 24 invalid on DnaNudge and 8 on RT‐PCR) [1] and [2] not reported [3] consecutive Recruitment: prospective |
Mixed (community, A&E, inpatient); London or Oxford, UK ([1] 10 April‐12 May [2] 2‐24 April [3] 12‐18 May) |
Only group [3] were inpatient median age 46 years (IQR 31–66); 124, 32% male |
RT‐PCR (multiple assays) Target: see above NOP (paired) Timing: not stated Interval: simultaneous (paired) |
Additional 47 samples not 'paired'; not collected on same date 32 samples excluded; 24 invalid on DNANudge (failed to amplify RNaseP; 22/24 with associated RT‐PCR result were ‐ve) and 8 on RT‐PCR (all 8 from 1 site) Index: none reported Reference: none reported |
|
Goldenberger 2020 Published 19 (10) |
Two‐group; [1] SARS‐CoV‐2 +ve samples selected to reflect a broad range of Ct values [2] SARS‐CoV‐2 ‐ve samples (n = 9) Sampled from patients suspected of COVID‐19 undergoing routine diagnostics within a 1‐week period Convenience Recruitment: unclear |
Laboratory‐based (unclear); Switzerland (1 week during 2020 pandemic) |
Not reported | RT‐PCR (single assay);
Threshold NR but all PCR+ < 33 Ct Target: E, ORF1 NP (same as index) Timing: not stated Interval: simultaneous (same swab) |
None reported Index: none reported Reference: none reported |
|
Harrington 2020 Accepted manuscript 524 (186) |
Single group; Symptomatic patients meeting diagnostic criteria for COVID‐19 Recruitment: consecutive |
Hospital A&E (EDs (n = 3) or urgent care centres (n = 2)); USA (Not reported) |
Not stated | RT‐PCR (single assay) Target: not stated Not specifically stated; presume yes as central lab used NP swabs (paired) Timing: VTM (no detail) Interval: simultaneous swab collection (different swabs for index and reference) |
None reported Index: none reported Reference: 2 initial FPs had repeat sampling: ‐ 1 retested on RT‐PCR only and was +ve (designated as TP) ‐ 1 retested on RT‐PCR and ID Now and was ‐ve on both (designated as TN) |
|
Hogan 2020 Preprint 100 (50) |
Single group; adult patients from one hospital and paediatric and adult samples from surrounding hospitals Recruitment: unclear; equal numbers of +ve and ‐ve RT‐PCR samples |
Laboratory‐based (clinical virology laboratory); USA (7‐13 April 2020) |
Not stated | RT‐PCR (single assay) Target: E gene As for index test; NP swab Timing: not stated Interval: not stated implies tests undertaken soon after collection |
None reported 3 invalid results were re‐tested; 1 +ve and 2 ‐ve Index: 1 known RT‐PCR+ sample with faint +ve test line re‐tested (same result; considered +ve) Reference: none reported |
|
Hou 2020 Accepted manuscript 285 (153) |
Single group; remnant OP swabs submitted for SARS‐CoV‐2 testing Recruitment: not stated |
Laboratory‐based (mixed inpatient and outpatient); China (February‐April 2020) |
178 (62.5%) inpatient; 107 (37.5%) outpatients. Site 2 were all inpatients 220 (77.2%) aged ≤ 65 years; 159 (55.8%) male |
RT‐PCR (multiple assays) Target: not stated OP (same as for rapid test) Timing: not stated Interval: simultaneous (same swab); time period of frozen storage was not reported |
None reported Index: none reported Reference: none reported |
|
Jin 2020 Published 52 (6) |
Single group; paired dry swabs and NP or OP swabs in UTM (includes pre‐admission screening for surgical patients) Recruitment: unclear |
Laboratory‐based (unclear); USA (23‐26 April 2020) |
Not stated | RT‐PCR (single assay) Target: ORF1/a, E gene Not stated for paired samples, but for full cohort NP and OP swabs in VTM used (400 uL) Timing: not stated Interval: simultaneous (paired swabs) |
None reported Index: none reported Reference: none reported |
|
Jokela 2020 Preprint 107 (61); only 90 tested with Xpert Xpress |
Two‐group: NP or OP swab samples sent to university laboratory: [1] for SARS‐CoV‐2 testing (n = 97), [2] pre‐pandemic samples sent for testing due to suspicion of other respiratory virus infection (n = 10) Recruitment: not stated |
Laboratory‐based (not reported); Finland (March‐May 2020) |
Not stated | RT‐PCR (multiple assays) Target: 1) N gene, 2) orf1ab and E, 3) orf1ab and N NP or OP, as for index Timing: not stated Interval: simultaneous (same samples) |
107 samples tested with Novodiag but only 90 for Xpert None reported Index: none reported Reference: none reported |
|
Lephart 2020 [A] Preprint [1] 75 (16) |
Single group; ‐ patients presenting to ED (75) Recruitment: not stated Second cohort of 13 cases excluded |
Hospital A&E ([1] ED; [2] in‐patient); USA (22 April‐5 May 2020) |
Not reported | Composite: result from ≥ 2 of 4 commercial assays (includes ID NOW and 3 RT‐PCR assays (incl Xpert Xpress)) Target: not stated Three ‐ves (on different assays) required for absence of infection (same as for Xpert Xpress) Timing: within 24 h of sample collection (on presentation at ED); no further detail Interval: same swab [B], or paired collection [A] |
None reported Index: [A] no invalid results [B] 1 'invalid' result; not reported if this was a 'presumptive +ve' (E gene only) on Xpert Xpress or no result Reference: none reported |
|
Lieberman 2020 Accepted manuscript 169 (87) |
Single group; Samples submitted for clinical diagnostic testing Recruitment: not stated |
Laboratory‐based (not reported); USA (Not stated) |
Not stated | RT‐PCR (single assay);
Threshold not stated Target: NI, N2 As for index test; NP swab Timing: not stated Interval: all testing conducted within 72 h |
None reported; review team excluded data for 28 specimens comparing Panther Fusion with DiaSorin Simplexa Not stated Index: not stated Reference: inconclusive' (ie one genetic target detected) considered +ve |
|
Loeffelholz 2020 Accepted manuscript 486 (220) |
Two‐group; patients referred for COVID‐19 testing at according to the local criteria Recruitment: convenience; deliberate sampling to enrich for +ve specimens |
Laboratory‐based (not stated); USA, UK, France, Italy (1 March‐2 April 2020) |
Not stated Adults at all sites except New York City Dept. Health and Mental Hygiene and Niguarda Hospital where all age groups were tested (ages not stated) |
RT‐PCR (multiple assays) Target: different targets depending on RT‐PCR test used (see cut‐off index) Same as for index test Timing: as for index test Interval: same samples but majority of index tests performed after frozen storage for undefined period |
4 Xpert Xpress test results lost permanently (single instrument computer malfunction); + 1 invalid excluded 1 Xpert Xpress test invalid due to cartridge error Index: presumptive +ve results not reanalysed by Xpert Xpress; all discrepant results reanalysed by a 3rd RT‐PCR method Reference: inconclusive results analyzed by a 3rd RT‐PCR method |
|
Mitchell 2020 Accepted manuscript 61 (46) |
Single group; Samples +ve and ‐ve on one of two SARS‐CoV‐2 RT‐PCR assays Recruitment: not stated; possible deliberate sampling of +ve cases |
Laboratory‐based (2 independent laboratories); USA (Not stated) |
Not stated | RT‐PCR (one of two assays) Target: not stated As for index test Timing: as for index test Interval: same samples but used at different times (samples used for index test stored at −80 ℃) |
None reported Index: none reported Reference: none reported |
|
Moore 2020 Preprint 200 (125) |
Two‐group; symptomatic (fever or cough or shortness of breath) adult and pediatric outpatients, ED patients, and inpatients Recruitment: consecutive (first 94 participants), then deliberate sampling used |
Mixed (outpatients, ED patients and inpatients); USA (27 March‐9 April 2020) |
79 (39.5%) hospitalised including 29 in ICU, 76 (38%) ambulatory care including 55 seen in a designated COVID‐19 screening clinic), and 45 (23%) seen at ED. Mean age 50 years (SD 17 years), 92 (46%) men |
RT‐PCR (multiple assays);
Threshold ≤ 40 Ct or presence of amplification curve Target: a. N1, N2 b. N, RdRp As for index test; NP swab Timing: not stated Interval: all 3 tests conducted within 72 h of sample collection |
2 invalid excluded 2 results were invalid on ID Now and were not retested (excluded) Index: none reported Reference: discordant results on RT‐PCR had record review to determine presence/absence COVID‐19 infection |
|
Moran 2020 Accepted manuscript 103 (42) |
Single group; inpatients and ambulatory patients Recruitment: not stated |
Laboratory‐based (inpatient and ambulatory; samples selected from central laboratory); USA (Not stated) |
Not stated | RT‐PCR (single assay) Target: ORF1, E As for index; nasal or NP swabs Timing: not stated Interval: not stated; same sample |
None reported Index: single FP (‐ve on E gene and low +ve on N gene) retested with Xpert Xpress and considered ‐ve on both targets Reference: single FP was retested on RT‐PCR and found to be repeatedly ‐ve |
|
Rhoads 2020 Accpeted manuscript 96 (96) |
Single group (cases); Samples +ve using standard of care testing Recruitment: convenience |
Laboratory‐based (includes self‐collected and provided‐collected samples); USA (Not stated) |
Not stated | RT‐PCR (multiple assays) Target: N1 and N2 As for index test Timing: as for index test Interval: same samples used |
None reported Index: none reported Reference: RT‐PCR detected only one of two targets for two samples (both considered +ve (diagnosed as +ve on original sample testing); both were ‐ve on index test) |
|
Smithgall 2020 [A] Published 113 (88) |
Two‐group Routine clinical testing by RT‐PCR Recruitment: unclear; describes deliberate sampling of samples with high, medium and low Ct values on RT‐PCR |
Laboratory‐based (inpatient and ED); USA (8 April‐13 April) |
Not stated 111 adult (range 23‐101 years; average 65 years for RT‐PCR+ and 43 years for RT‐PCR‐); 2 paediatric (age 1 day and 5 days) 61, 54% male |
RT‐PCR (single assay)
Threshold ≤ 37 Ct on both target genes Target: ORF1 a/b, E‐gene Not stated As for index test Timing: as for index test Interval: simultaneous; same samples used |
None reported Index: Xpert: 1 sample was a presumptive +ve based on detection of E‐gene target but not the N2 target Reference: none reported |
|
SoRelle 2020 Published letter 83 (39) |
Unclear design; participants symptomatic for COVID‐19 Sampling: not stated Recruitment: not stated |
Laboratory‐based (unclear); USA (Not reported) |
All symptomatic Not reported |
RT‐PCR (multiple assays) Target: not stated NP in VTM (paired) Timing: not stated Interval: paired |
None reported Index: none reported Reference: none reported; presumptive +ves not mentioned |
|
Stevens 2020 Accepted manuscript 104 (54) |
Unclear design; Residual samples from symptomatic and asymptomatic individuals undergoing routine testing; selected to represent the full range of Ct values Sampling: convenience Recruitment: retrospective |
Laboratory‐based (serving adult and pediatric tertiary care hospitals); USA (31 March‐7 April) |
Unclear; 'symptomatic and asymptomatic';
Of 54 cases, 10 (19%) were low viral low (Ct > 35) Not reported |
RT‐PCR (single assay) Target: 2 regions of ORF1ab NP in VTM; as for index test Timing: not stated Interval: same sample |
6 samples excluded due to insufficient sample volume 1 RT‐PCR+ sample re‐tested on Xpert Xpress due to initial interpretation of no results (invalid); Xpert +ve on re‐test Index: no presumptive +ves were observed Reference: 1 RT‐PCR+ sample that was ‐ve on both targets for Xpert Xpress (FN) was re‐tested on Panther Fusion and found to be ‐ve (TN) |
|
Szymczak 2020 Published 79 (29 +ve on stool; 48 previously +ve on NP/OP swab) |
Single group; remnant samples from patients with symptomatic diarrhoea submitted for routine diagnostic testing Recruitment: convenience |
Laboratory‐based (unclear); USA (21 April‐15 May 2020) |
All symptomatic for diarrhoea Not stated |
RT‐PCR (single assay) Target: two ORF1a regions Stool, as for index Timing: some samples frozen at −80 °C prior to testing with Hologic Panther Fusion Interval: simultaneous; same swabs |
None reported Index: discrepant results re‐tested with both index and reference test Reference: as above |
|
Thwe 2020 Published 161 (14) |
Single group; symptomatic patients with paired samples Sampling: not stated Recruitment: retrospective |
Laboratory‐based (inpatient and ED); USA (April‐May 2020 ("4 weeks data")) |
All symptomatic Not stated |
RT‐PCR (single assay) Target: not stated NP in VTM (paired) Timing: not stated Interval: paired |
None reported (review team excluded 21 samples with Xpert Xpress as reference standard) None reported Index: none reported Reference: none reported; no discrepant analysis |
|
Wolters 2020 Accepted manuscript 88 (58) |
Two‐group; Samples selected from laboratories on the basis of presence/absence of 2 genetic targets on RT‐PCR Recruitment: not stated; deliberate sampling according to target gene |
Laboratory‐based (not stated; 3 laboratories); The Netherlands (January‐March 2020) |
Not stated | RT‐PCR (multiple assays) Target: mixed As for index test Timing: as for index test Interval: same samples used; index text seems to have been conducted after frozen storage |
None reported Index: samples +ve on only 1 target were both re‐tested on RT‐PCR only Reference: as above |
|
Wong 2020 Published 162 (119) |
Single group; samples submitted for routine testing from patients with suspected COVID‐19 infection Sampling: not stated Recruitment: both retrospective (n = 74) and prospective (n = 88) |
Laboratory‐based (A&E, inpatient and outpatient); China (Not stated) |
Not stated Median age 46 (IQR: 35 (28‐63); male = 69 (44%) |
RT‐PCR (single assay) Target: not stated deep throat saliva or lower respiratory tract; as per index test Timing: not stated Interval: simultaneous (same samples) |
None reported Index: none reported Reference: none reported |
|
Zhen 2020 [A] Accepted manuscript 108 (58) |
Two ‐group; Samples from symptomatic patients of all ages and gender Recruitment: not stated; deliberate sampling to represent the TP rate at authors' institution (50%‐60%), and to span low and high viral loads |
Laboratory‐based; USA (March‐April 2020) |
"Symptomatic"; no further details Not stated (all ages and gender) |
RT‐PCR (single assay) Target: 2 regions of ORF1ab; either +ve single RT‐PCR As for index; NP swabs Timing: not stated Interval: not stated in exact terms |
1 specimen with invalid result on ID Now was excluded from that dataset Index: none reported; no re‐testing conducted Reference: none reported; no re‐testing conducted |
| A&E: Accident and Emergency [Department]; CDC: National Health Commission of the People's Republic of China; Ct: cycle threshold; ED: Emergency Department; FN: false negative; FP: false positive; HCW: healthcare worker; ICU: intensive care unit; IQR: interquartile range; N/A: not applicable; NOP: naso‐oropharyngeal; NP: nasopharyngeal; NR: not reported; OP: oropharyngeal; PHE: Public Health England; RT‐PCR: reverse transcription polymerase chain reaction; TN: true negative; TP: true positive; UTM: universal transport media; VTM: viral transport medium | |||||
Appendix 12. Molecular tests: summary index test details
| Study | Index test (manufacturer) | Test method Target | Sample details | Test operator Test threshold |
| Assennato 2020 | SAMBA II SARS‐CoV‐2 Test (Diagnostics for the Real World) | Automated RT‐PCR ORF1ab, N2 |
Samples tested: combined nose and throat swab samples, provided as VTM Timing of sampling: not stated Timing of test: not stated Sample storage: not stated |
Not stated; presume laboratory staff Threshold: as per manufacturer; either target present |
| Broder 2020 | GeneXpert Xpress SARS‐CoV‐2 assay (no product code reported) (Cepheid) | Automated RT‐PCR Not stated E gene |
Samples tested: NP swabs (not specified) Timing of sampling: not stated Timing of test: within 3 days of RT‐PCR Sample storage: not stated |
Not stated; presume lab staff Threshold: as per manufacturer |
| Chen 2020a | Xpert Xpress SARS‐CoV‐2 assay (no product codes reported) (Cepheid, Sunnyvale, CA, USA) | Automated RT‐PCR E and N2 gene |
Samples tested: NP, saliva (posterior OP, self‐collected by clearing the throat and spitting c1 mL saliva directly into a sterile bottle in the early morning before mouth rinsing and breakfast) (VTM) Timing of sampling: not stated Timing of test: not stated; archived samples Sample storage: not stated; archived |
Not stated; infer laboratory staff Threshold: not stated |
| Collier 2020 | SAMBA II SARS‐CoV‐2 test (no product code reported) (Diagnostics for the Real World (DRW), University of Cambridge, Cambridge) | Automated RT‐PCR Orf1 and the E genes |
Samples tested: combined nasal/throat swab (NOP) on dry sterile swab. Collection not reported (direct) Timing of sampling: not stated; appears to be on presentation/admission but no further details Timing of test: test performed within 18 h of reference test Sample storage: not stated |
Not stated; infer laboratory staff Threshold: as per manufacturer |
| Cradic 2020(a) | [A] ID NOW COVID‐19 EUA; Study also evaluates [B] Diasorin Simplexa and [C] Roche cobas 6800 SARS‐CoV‐2; not eligible for this review (Abbott Laboratories) | Isothermal amplification RdRp |
Samples tested: NP swabs in UTM; collected on flocked swab, no other details, (VTM) Timing of sampling: unclear, infer upon presentation Timing of test: immediate or within 72 h Sample storage: asap, or stored for up to 72 h at 2 °C‐8 °C. Following routine testing, samples were stored frozen (≤ −80°C) until comparator testing with the Roche cobas assay could be completed |
Not stated; infer laboratory staff. Threshold: as per manufacturer |
| Cradic 2020(b) | [A] ID NOW COVID‐19 EUA; Study also evaluates [B] Diasorin Simplexa and [C] Roche cobas 6800 SARS‐CoV‐2; not eligible for this review (Abbott Laboratories) | Isothermal amplification RdRp |
Samples tested: NP swabs in UTM (collected as part of standard of care), plus direct testing of OP swabs and of nasal swabs (collected according to CDC instructions) (Direct) Timing of sampling: unclear, infer upon presentation Timing of test: not stated; presume as for Cradic 2020(a) (immediate or within 72 h) Sample storage: not stated; presume as for Cradic 2020(a) (asap, or stored for up to 72 h at 2 °C‐8 °C) |
Not stated; infer laboratory staff. Threshold: as per manufacturer |
| Dust 2020 | Xpert Xpress (no product code) [Also evaluates cobas SARS‐CoV‐2 RT‐PCR (Roche) and 3 in‐house RT‐PCR assays; not eligible for this review] (Cepheid Inc) | Automated RT‐PCR E, N2 |
Samples tested: NP swabs in VTM; collection not reported (VTM) Timing of sampling: not stated Timing of test: not stated Sample storage: not stated |
Not stated Threshold: not stated; presume as per manufacturer (presumptive positives not mentioned) |
| Ghofrani 2020 | ID NOW COVID‐19 assay (no product code reported) (Abbott Laboratories) | Isothermal amplification RdRp region |
Samples tested: nasal 58 (51.3%), NP 33 (29.2%), not stated 22 (19.5%)
Direct testing 58 (51.3%), UTM 26 (23.0%); not stated 29 (25.7%). (direct or VTM) Timing of sampling: not stated; implies mostly close to presentation Timing of test: not stated Sample storage: not stated |
Not stated; infer laboratory staff Threshold: not stated; presume as per manufacturer |
| Gibani 2020 | COVIDNudge (no product code) (DnaNudge, UK) | Automated RT‐PCR; Described as "integrated lab‐on‐chip device that enables sample‐to‐result (RT‐)PCR" rdrp1, rdrp2, e‐gene, n‐gene, n1, n2, and n3 |
Samples tested: NP; HCW obtained swabs using paediatric swab (Direct) Timing of sampling: on presentation; timing not reported Timing of test: not stated; appears to be as soon as possible after collection Sample storage: none described |
Unclear; possibly HCW Threshold: at least 2 replicates of at least one viral gene target amplified |
| Goldenberger 2020 | Xpert Xpress (no product code) (Cepheid Inc) | Automated RT‐PCR E, N2 |
Samples tested: NP (VTM) Timing of sampling: not stated Timing of test: not stated Sample storage: frozen at −80 °C until batch‐wise sample processing with the Xpert |
Laboratory technician Threshold: not stated; both targets reported |
| Harrington 2020 | ID Now COVID‐19 assay (no product code provided) (Abbott) | Automated RT‐PCR Not stated |
Samples tested: nasal swabs (provider collected) (direct) Timing of sampling: not stated Timing of test: not stated (soon after collection) Sample storage: none |
On‐site medical personnel (urgent care centres); laboratory personnel at each separate location (EDs)
Operators at 2 sites were reportedly experienced users of ID Now (one ED and 1 urgent care centre) and 3 sites received training) Threshold: as per manufacturer |
| Hogan 2020 | Accula SARS‐CoV‐2 PocT (no product code reported) (Mesa Biotech, Inc., San Diego, CA) | Automated RT‐PCR N gene |
Samples tested: NP swabs in VTM (n = 37) or saline (n = 63, including 37 positive on RT‐PCR) (VTM or other) Timing of sampling: not stated Timing of test: not stated (? soon after collection) Sample storage: not stated |
Not stated; performed at the SHC Clinical Virology Laboratory Threshold: as per manufacturer |
| Hou 2020 | Xpert Xpress (no product code reported) (Cepheid Inc) | Automated RT‐PCR E, N2 |
Samples tested: OP (not specified) Timing of sampling: not stated Timing of test: not stated; frozen samples Sample storage: stored at ‐80 ℃ within 24 h of collection |
Not stated Threshold: not stated; presumably as per manufacturer |
| Jin 2020 | ID NOW (product code not reported) (Abbott Laboratories) | Isothermal amplification RdRp |
Samples tested: dry swabs as per manufacturer EUA protocol (direct) Timing of sampling: not stated Timing of test: NR; appears immediate Sample storage: none |
Not stated; laboratory staff presumed Threshold: as per manufacturer |
| Jokela 2020 | Xpert Xpress (no product code reported) (Cepheid Inc) | Automated RT‐PCR E, N2 |
Samples tested: NP or OP; no details on collection (not specified) Timing of sampling: not stated Timing of test: not stated Sample storage: not stated |
Not stated Threshold: not stated |
|
Lephart 2020 [A] Lephart 2020 [B] |
[A] ID NOW
[B] Xpert Xpress
(No product codes reported)
([A] Abbott Molecular
[B] Cepheid) 2 additional RT‐PCR tests evaluated but not extracted |
[A] isothermal amplification
[B] Automated RT‐PCR Not reported in paper |
Samples tested: [A] nasal
[B] NP
Presume collected by HCW but not reported (direct) Timing of sampling: on presentation; timing pso not reported Timing of test: [A] within 24 h [B] stored at 4 ℃ and tested within 24 h Sample storage: [A] appears to be room temperature [B] stored at 4 ℃ |
Not stated; presume lab staff Threshold: each assay was performed according to manufacturer’s EUA instructions |
| Lieberman 2020 | [A] Xpert Xpress
[A] Cepheid Also evaluates 4 other assays not eligible for review [B] Panther Fusion RUO, Hologic [C] Panther Fusion EUA, Hologic [D] Simplexa, Diasorin [E] Cobas 6800, Roche |
Automated RT‐PCR [A] E, N2 [B] and [C] Orf1ab, 2ab [D] S, ORF1ab [E] ORF1ab, E |
Samples tested: NP swabs (collection not described) (VTM) Timing of sampling: not stated Timing of test: < 72 h Sample storage: 4 ℃ with no freeze‐thaws |
Not stated; presume lab staff Threshold: any one of two targets detected was considered positive for all assays; Xpert Xpress data extracted as per IFU definition (positive = both targets or N gene positive) |
| Loeffelholz 2020 | Cepheid Xpert Xpress SARS‐CoV‐2 (RUO version, no product code reported) (Cepheid Europe) | Automated RT‐PCR Nucleocapsid gene (N2) and the envelope gene (E) (also detects RdRp but this does not contribute to positivity) |
Samples tested: mixed [NP + saliva (NPS) (n = 339), OP + saliva (OPS) (n = 15), combined NPS/OPS in the same transport vial (n = 97)], and TA (n = 30):
a. Baltimore ‐ 61 NPS
b. Los Angeles ‐ 88 NPS
c. Manchester ‐ 54 NPS/OPS, 11 NPS
d. Paris ‐ 68 NPS
e. New York City ‐ NPS 11, OPS 15, TA 30, NPS/OPS 43
f. Milan ‐ 79 NPS
g. Newark ‐ 21 NPS (VTM or other) Timing of sampling: not stated Timing of test: not stated; except one site < 2 h (n = 21) Sample storage: stored at ‐80 °C; except 1 site tested in real time (n = 21) |
Not stated; presume lab staff Threshold: as per manufacturer: if both targets are detected, or if only N2 is detected, the test reports a positive result. If only the E target is detected the test reports a presumptive positive result |
| Mitchell 2020 | ID NOW COVID‐19 (product code not reported) (Abbott, Chicago, USA) | Automated RT‐PCR Not stated |
Samples tested: NP in VTM Timing of sampling: not stated Timing of test: not stated Sample storage: stored at −80 ℃ |
Certified laboratory personnel Threshold: as per manufacturer |
| Moore 2020 | ID NOW (no product code) (Abbott) | Automated RT‐PCR RdRp |
Samples tested: NP swabs in 3 mL VTM (collection not reported) (VTM) Timing of sampling: not stated Timing of test: < 72 h from collection Sample storage: none, or else stored at 4 °C (if testing could not be completed on the same day) |
Not stated; presume lab staff Threshold: as per manufacturer |
| Moran 2020 | Xpert Xpress SARS‐CoV‐2 assay (no product code) (Cepheid, Sunnyvale, CA) | Automated RT‐PCR E, N (N2 region) |
Samples tested: 8 nasal and 95 NP swabs (not specified) Timing of sampling: not stated Timing of test: not stated Sample storage: not stated |
Not stated; presume lab staff Threshold: as per manufacturer |
| Rhoads 2020 | [A] ID Now
([A] Abbott; Chicago, USA)
(product codes not reported) Also evaluates Simplexa, Diasorin (Saluggia, Italy) |
Automated RT‐PCR Not stated |
Samples tested: nasal swabs (self‐collected) and NP swabs (provider collected) (VTM or other) Timing of sampling: not stated Timing of test: not stated Sample storage: not stated |
Not stated; presume lab staff Threshold: as per manufacturer |
|
Smithgall 2020 [A] Smithgall 2020 [B] |
[A] ID Now [B] Xpert Xpress (product codes not reported) ([A] Abbott [B] Cepheid) | Automated RT‐PCR [A] RdRp gene [B] N2, E genes |
Samples tested: NP swabs (collection not described) (VTM or other) Timing of sampling: not stated Timing of test: within 48 h collection Sample storage: stored at 4°C |
Not stated; presume lab staff Threshold: as per manufacturer |
| SoRelle 2020 | ID NOW (no product codes) (Abbott Diagnostics) | Isothermal amplification Not stated |
Samples tested: saliva; collection not described (not specified) Timing of sampling: not stated; chart review of patients with FN results against either RT‐PCR (NP) Xpert Xpress (saliva) (n = 9) showed 6/9 tested > 2 weeks after symptom onset Timing of test: not stated Sample storage: not stated |
Not stated; presume lab staff Threshold: as per manufacturer |
| Stevens 2020 | Xpert Xpress (no product code) (Cepheid Inc) | Automated RT‐PCR E, N2 |
Samples tested: NP in VTM (VTM) Timing of sampling: not stated Timing of test: not stated Sample storage: Frozen at −80 °C |
Not stated; presume lab staff Threshold: presence of N2 +/‐ E gene; E gene only considered presumptive positive |
| Szymczak 2020 | Xpert Xpress (no product code reported) (Cepheid Inc) | Automated RT‐PCR N2 and E |
Samples tested: stool, collection not reported (saline) Timing of sampling: PCR+ stool samples collected 0‐33 days from initial respiratory PCR; 8/27 collected at ≥ 14 days and 6/27 collected at ≥ 21 days Timing of test: up to 7 days Sample storage: stored at 2‐8 ℃ |
Not stated Threshold: not stated |
| Thwe 2020 | ID NOW (no product code) (Abbott) | Isothermal amplification Not stated |
Samples tested: dry NP swabs (direct) Timing of sampling: not stated Timing of test: within 2 h Sample storage: appears to be room temperature |
Not stated Threshold: as per manufacturer |
| Wolters 2020 | Cepheid Xpert Xpress SARS‐CoV‐2 (product code not reported) (Cepheid Europe) | Automated RT‐PCR E‐gene (sarbeco specific) and N2‐gene (SARS‐CoV‐2 specific) |
Samples tested: NP or mid‐turbinate, and OP swabs (VTM or other) Timing of sampling: not stated Timing of test: not stated Sample storage: stored at −80 ℃ |
Not stated; presume lab staff Threshold: as per manufacturer: E‐gene only positive specimens considered ‘SARS‐CoV‐2 presumptive positive’ and require retesting, N2 only positives deemed positive |
| Wong 2020 | Xpert Xpress (Cepheid Inc) | Automated RT‐PCR E and N2 |
Samples tested: deep throat saliva (DTS) (n = 120), or lower respiratory tract (LRT) (n = 42; 35 sputum, 6 tracheal aspirate 1 BAL) (not specified) Timing of sampling: not stated Timing of test: transported on the same day and tested promptly Sample storage: not stated; transported to laboratory |
Lab staff Threshold: as per manufacturer; presumptive positives mentioned only in Introduction section |
|
Zhen 2020 [A] Zhen 2020 [B] |
[A] Xpert® Xpress SARS‐CoV‐2
[B] ID NOW COVID‐19
(no product codes reported) [A] Cepheid [B] Abbott [Also evaluates [C] ePlex SARS‐CoV‐2 Test, GenMark] |
Automated RT‐PCR [A] N2, E [B] RdRp |
Samples tested: NP swabs (VTM) Timing of sampling: not stated Timing of test: for routine testing up to 72 h; 20 samples tested prospectively after collection on all systems Sample storage: for routine testing (ePlex) stored at 2‐8 ⁰C; then stored at −80 ⁰C (ID Now, Xpert Xpress and Hologic RT‐PCR); 20 samples tested prospectively after collection on all systems |
Not stated; presume lab staff Threshold: as per manufacturer |
| BAL: bronchoalveolar lavage; ED: Emergency Department; FN: false negative; HCW: healthcare worker; NOP: naso‐oropharyngeal; NP: nasopharyngeal; NR: not reported; OP: oropharyngeal; PCR: polymerase chain reaction; pso: post‐symptom onset; RT‐PCR: reverse transcription polymerase chain reaction; TA: tracheal aspirate; VTM: viral transport medium | ||||
Appendix 13. Index test details from manufacturer instructions for use documents
| Index testa | Type of assay Throughput Time to result | Equipment Kit storage | Sample types | Transport medium | Sample storage | Test interpretation |
| Antigen tests | ||||||
| AAZ; COVID‐VIRO COVID‐19 Ag Rapid Test IFU: TR‐COV‐006 |
CGIA Single test 15 min |
Provides: test device, buffer, NP swabs, extraction tubes, nozzles and filters; 2‐30 °C |
NP | Not stated | Test ASAP after collection; can be stored in clean, unused sealed plastic tube at room temperature (15‐30 °C) for up to 1 h prior to testing. If > 1 h delay occurs, dispose of sample | Visual: negative if control line only; positive if both test and control lines appear no matter how faint; invalid if no control line visible |
| Abbott Rapid Diagnostics; PanbioTM COVID‐19 Ag Rapid Test Device IFU: 41FK10 |
CGIA Single test 15 min |
Provides: buffer, extraction tubes and caps, positive and negative control swabs, NP swabs for collection, tube rack 2‐30 °C |
NP | Not mentioned; implies not recommended | Test direct swab specimens immediately after collection. If not possible, swab specimen can be kept in an extraction tube filled with extraction buffer (300 μL) at room temperature (15‐30 °C) for up to 2 h prior to testing | Visual: negative if control line only; positive if both test and control lines appear no matter how faint; invalid if no control line visible |
| Becton Dickinson; BD Veritor™ System for Rapid Detection of SARS‐CoV‐2 IFU: 256082 |
Not stated; LFA Single test 15 min (up to 60 min if 'walk‐away' mode enabled) |
Provides: test device, extraction reagent, specimen sampling swabs, positive and negative control swabs. Also requires: BD Veritor™ Plus Analyzer (Cat. No. 256066) 2–30 °C |
Nasal | Not recommended; "NOT INTENDED for testing liquid samples such as wash or aspirate samples or swabs in transport media as results can be compromised by over dilution" | Test ASAP after collection, and no later than 1 h after specimen collection | Automated: 'CoV2: +' indicates positive result; 'CoV2: ‐' for presumptive negative; 'CONTROL INVALID' for invalid result |
| Beijing Savant; SARS‐Cov‐2 Antigen Fluorescence Rapid Detection Kit IFU: not obtained; no mention of any COVID tests on website |
IFU not obtained | IFU not obtained | IFU not obtained | IFU not obtained | IFU not obtained | IFU not obtained |
| Bionote; NOWCHECK COVID‐19 Ag test IFU: not stated |
Not stated; LFA Single test 15 min |
Provides: test device, extraction buffer tube and nozzle cap, sterile swab, paper stand 2‐30 °C / 36‐86 °F |
NP | Do not use transport media | Use the collected specimen immediately. Specimens may be stored at room temperature for up to 1 h or at 2‐8 °C/ 36‐46 °F for up to 4 h prior to testing | Visual: negative if control line only; positive if both test and control lines appear no matter how faint; invalid if no control line visible |
| Biosynex; NowCheck COVID‐19 Ag test IFU: SW4000605 |
CGIA Single test 15 min |
Provides: test cassettes, extraction buffer, sterile swabs (CE 0197), extraction tubes, end caps 2‐30 °C |
NP | Not stated | Test ASAP after collection; can be stored in clean, unused sealed plastic tube at room temperature (15‐30 °C) for up to 1 h prior to testing. If > 1 h delay occurs, dispose of sample | Visual: negative if control line only; positive if both test and control lines appear no matter how faint; invalid if no control line visible |
| Coris BioConcept; COVID‐19 Ag Respi‐Strip IFU: 5723/TB/V03 |
CGIA (paper strip method) Single test 15 min |
Paper strips in a bottle with desiccant; LY‐S dilution buffer (3.5 mL or 15 mL; tubes and stoppers) 4‐30 °C |
NPs or culture extracted solution; samples must be liquid | A gel or a sponge matrix can be used | ASAP, any delay may result in a low signal intensity. If not, store frozen at −20 °C | Visual; read through collection tube Control line only (negative), T line (with or without control (positive), no control line (invalid) |
| e25bio; DART (Direct antigen rapid test) IFU: n/a |
CGIA | IFU not obtained | IFU not obtained | IFU not obtained | IFU not obtained | IFU not obtained |
| Fujirebio Inc; ESPLINE SARS‐CoV‐2 IFU: FRI46955 (K4B01TE) |
LFA (alkaline phosphatase‐labelled) Single test 30 min |
Reaction cassette, sample extraction solution (squeeze tube), applicator tip 1‐30 °C |
NP fluid | Not stated; recommends samples are prepared immediately after collection (placing swab in provided sample extraction solution), however documented clinical validation results were from swabs immersed in VTM prior to use | Samples must be prepared immediately after specimen collection | Visual; positive if blue test line (T) and reference line (r) positions, negative if blue r line only, invalid if no blue r line appears or if red r line still present. If the r and T lines appear before 30 min, the sample must be considered "positive"; samples that only turn “positive” after 30 min, must be considered “negative” |
| Innova Medical Group; Innova SARS‐CoV‐2 Antigen Rapid Qualitative Test IFU: A/02 |
CGIA Single test 20‐30 min |
Provides: test cartridge, extraction tube, extraction solution, QC card 2‐30 °C |
Nasal or OP; intended for use within the first 5 days of the onset of symptoms | Not mentioned | Test ASAP after collection. Based on data generated with influenza virus, throat swabs are stable for up to 24 h at room temperature or 2°‐8 °C | Visual: negative if control line only; positive if both test and control lines appear no matter how faint; invalid if no control line visible |
| Liming Bio‐Products Co., Ltd; COVID‐19 Antigen Rapid Test Device (StrongStep®) IFU: obtained via Weitzel 2020 [A]; REF 500200 v1 |
CGIA Single test 15 min |
Test device, extraction buffer vial, extraction tubes, workstation for holding tubes 2‐30 °C |
NP or OP | Not mentioned in IFU | ASAP; can be held in clean, dry plastic tube or sleeve up to 72 h at 15‐30 °C, or 2‐8 °C before processing | Visual; 2 coloured bands for positive; control band only for negative; test line only is invalid |
| Quidel; Sofia SARS Antigen FIA IFU: 1439000EN00 (04/20) |
FIA Single test 15 min |
Provided: test cassette, reagent tubes, reagent solution, nasal swabs, 120 μL fixed volume pipette, SARS positive control swab, negative control swab
Required: Sofia or Sofia 2 reader device, Calibration Cassette 15 °C‐30 °C |
Nasal or NP | Updated IFU: directly test patient specimens without transport media. Original IFU: if transport of samples with VTM is required, minimal dilution of the sample is recommended, e.g. ≤ 1 mL | Test ASAP after collection. Based on data generated with influenza virus, nasal or NP swabs are stable for up to 24 h at room temperature or 2‐8 °C, and nasal or NP swabs in VTM are stable for up to 72 h at 2‐8 °C | The Sofia screen will display results for the procedural control as being 'tick' or 'cross', and will individually provide a '+' or '‐' result for SARS. If the procedural control is 'X' retest with a new patient sample and a new test cassette. Results must not be interpreted past 30 minutes after inoculation |
| RapiGEN Inc; BIOCREDIT COVID‐19 Ag IFU: I‐H0734‐E00(2020.04.03) |
CGIA Single test 5‐8 min |
Test device, assay diluent tube and filter cap, swab for NP collection; 1‐40 °C |
NP swab | Not mentioned in IFU | Test ASAP after collection; if storage required then 2‐8 °C for up to 12 h, or −20 °C for up to 24 h | Visual; control line only (negative), control and test lines (positive), no control line (invalid) |
| SD Biosensor Inc; Standard Q COVID‐19 Ag IFU: Q‐NCOV‐01G |
LFA (conjugated with colour particles) Single test 30 min |
Provides: test device, extraction buffer tube, filter cap, sterile swab; room temperature, 2‐30 °C/36‐86 °F |
NP | Not recommended; "Do not use transport media" | Test ASAP after collection; may be stored at room temperature for up to 1 h or at 2‐8 °C/36‐46 °F for up to 4 h prior to testing | Visual; the presence of 'control' and 'test' lines, no matter how faint the result is considered positive; negative if control line only; invalid if test line only |
| SD Biosensor Inc; Standard F COVID‐19 Ag FIA IFU: F‐NCOV‐01G |
FIA Single test 30 min |
Provides: test device, extraction buffer tube, filter cap, sterile swab. Standard F Analyzer also required (F100 or F200) room temperature, 2‐30 °C /36‐86 °F |
NP | Not recommended; "Do not use transport media" | Test ASAP after collection; may be stored at room temperature for up to 24 h or at 2‐8 °C/36‐46 °F for up to 48 h prior to testing | Automatic; the analyzer will automatically display the test result in 30 min. Cut‐off index value ≥ 1.0 is positive, < 1.0 is negative, cut‐off index not displayed is invalid result |
| Shenzhen Bioeasy Biotechnology Co, Ltd; BIOEASY 2019‐nCoV Ag Fluorescence Rapid Test Kit (Time‐Resolved Fluorescence) IFU: TS‐IU‐F027‐A2 (YRLF04401025/ YRLF04401050/ YRLF04401100) |
FIA Single test 10 min |
Test card, extraction solution, extraction tube, dripper, swab and ID chip. Test runs on immunofluorescence analyser (supplied separately), transfer pipette also required | Nasal swabs, throat swabs and deep sputum samples | Not mentioned in IFU | ASAP after collection, or store at 2‐8 °C for ≤ 24 h; or store at −70 °C for longer periods. Avoid repeated freezing and thawing (no more than 3 times). | Automatic; positive if both detection line and control line detect a fluorescent signal, and the detection line detection value is ≥ 0.005 ng/mL; negative if fluorescent signal on control line only; invalid if no fluorescent signal, or signal only on test line |
| Rapid molecular testsa | ||||||
| Abbott Diagnostics Scarborough Inc; ID NOW COVID‐19 IFU: IN190000 v1 |
Isothermal nucleic acid amplification 1 cartridge per run 5‐13 min |
Sample receiver (with elution/lysis buffer), test base (with 2 sealed reaction tubes, each containing a lyophilised pellet), transfer cartridge for transfer of the eluted sample to the test base, positive and negative control swabs; requires ID NOW Instrument | Throat, nasal, NP and OP swabs (direct testing or in listed VTM) | Early versions of IFU documents multiple options but now not recommended (ID NOW COVID‐19 Product Insert, IN190000 Rev.3 2020/04:6‐8) | ASAP after collection, otherwise hold in original package at room temperature (15‐30 °C) for up to 2 h. If longer then store at 2‐8 °C for up to 24 h from collection. No mention of frozen storage | Automatic; results displayed on the instrument screen as positive, negative or presence or absence of COVID‐19 Viral RNAs cannot be determined |
| Cepheid Inc.; Xpert Xpress SARS‐CoV‐2 test IFU: XPRSARS‐COV2‐10 |
Automated RT‐PCR 1‐80 cartridges according to GeneExpert system used 45 min |
Single‐use disposable cartridges that hold the RT‐PCR reagents and host the RT‐PCR process, transfer pipette; run on GeneExpert System | NP swab in VTM | Swab stored in viral transport tube containing 3 mL transport medium | Store at room temperature (15–30 °C) for up to 8 h or refrigerate (2–8 °C) up to 7 days until testing performed | Automatic; displayed positive (N2+ and E+, or N2+ only), presumptive positive (E+ only), negative (both negative), no result (repeat test), instrument error |
| Diagnostics for the Real World Ltd; SAMBA II COVID‐19 Test IFU: REF 8500‐12 |
Isothermal amplification Single test per run 1.5 h |
Each test set contains 4 cartridges for extraction, amplification and detection of the amplification products, 2 mL SCoV buffer, fixed‐volume pipette, 300 μL + pipette tips or transfer pipettes 300 μL, sample collection tube and sample card; SAMBA II Assay Module and Tablet module both required to run the test 2‐37 °C |
Combined nose and throat swabs, NP/OP swabs | Direct testing or UTM/VTM can be used; no limitations on type of VTM recorded in IFU | Store at 2‐30 °C for up to 18 h prior to testing. Freezing of samples should be avoided |
Automatic; presented and stored on the connected tablet Tablet module result: negative, positive, invalid, halted, read failure or no results Visual reading of test strip: internal control line only (negative), ≥ 1 of 2 test lines (ORF and or N lines) with or without internal control line (positive), no lines (invalid); other combinations possible in rare cases |
| dnanudge; CovidNudge IFU: 9501001 10‐2020 (v 4.1) |
RT‐PCR 1.5h |
Includes: DnaNudge COVID Nudge Cartridge, NP sample kit, or sputum sample kit
Requires: DnaNudge NudgeBox, Oragene OG‐500 sample collection tube (for sputum) ≤ 25°C |
NP or sputum | Not mentioned | Swab should be immediately inserted into the DnaCartridge and sealed. DnaCartridges containing swab specimens can be stored at room temperature (15–30 °C) for up to 8 h | Positive: if ≥ 3 viral gene replicates amplify in any of the assays Indeterminate: if 1 or 2 of the viral gene replicates amplify in any of the assays Negative: if none of the assays except the control assay amplifies Invalid: if ≤ 2 replicates of the control assay amplifies Error: in the event of any technical error during the sample preparation phase of the test, the NudgeBox will indicate with flashing red LEDs |
| Mesa Biotech Inc.; Accula SARS‐Cov‐2 Test IFU: LBL‐60058 Rev A (COV4100) |
RT‐PCR + LFA 1 cartridge per run 30 min |
Each test kit contains: test cassette, SARS‐CoV‐2 buffer (5.0 mL), single‐use fixed‐volume pipette, positive + negative control swabs; Accula or Silaris dock required to run test | Throat swab and nasal swab per test; direct testing only * check this ‐ Hogan 2020 reports use of NP swabs only | Not recommended and will invalidate the test | Prepared sample (in buffer vial) may be stored at room temperature for up to 24 h or refrigerated (2‐8 °C) and tested within 72 h of sample collection. Sample may be stored for up to 1 week at −20 °C | Visually interpretation (shown as blue test and control lines on exterior of test cassette): positive (any test line at T position, with or without control line C, but with no negative control line), negative (control line only with no negative control line), invalid (appearance of negative control line or all lines absent) |
| ASAP: as soon as possible; CGIA: colloidal gold immunoassay; FIA: fluorescent immunoassay; IFU: instructions for use; LFA: lateral flow assay; NP: nasopharyngeal; NPS: nasopharyngeal swab; OP: oropharyngeal; RNA: ribonucleic acid; RT‐PCR: reverse transcription polymerase chain reaction; UTM: universal transfer medium; VTM: viral transport medium | ||||||
| aThe reported product codes are as reported in the instructions for use documents and may diverge from those evaluated in the included studies (product codes were reported in only two of 18 studies). | ||||||
Appendix 14. Study quality by test group and at study‐level
11.
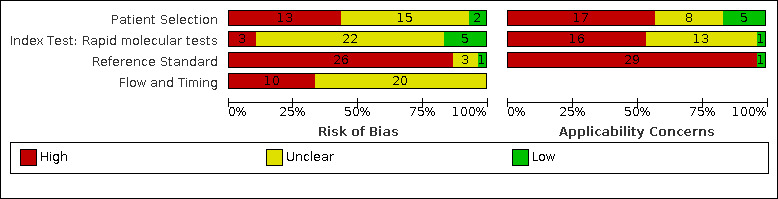
Risk of bias and applicability concerns graph: review authors' judgements about each domain presented as percentages across included studies
12.
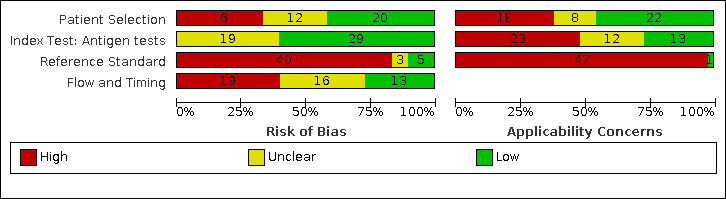
Risk of bias and applicability concerns graph: review authors' judgements about each domain presented as percentages across included studies
13.
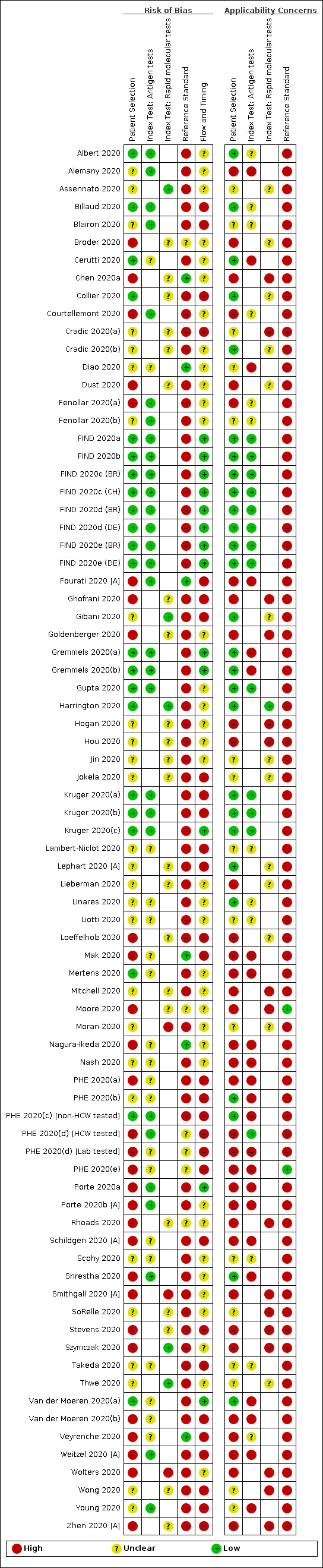
Risk of bias and applicability concerns summary: review authors' judgements about each domain for each included study
Appendix 15. Antigen tests: additional figures for subgroup analyses
14.

Forest plot of antigen test evaluations by study design. BR: Brazil; CH: Switzerland; DE: Germany; HCW: healthcare worker
15.
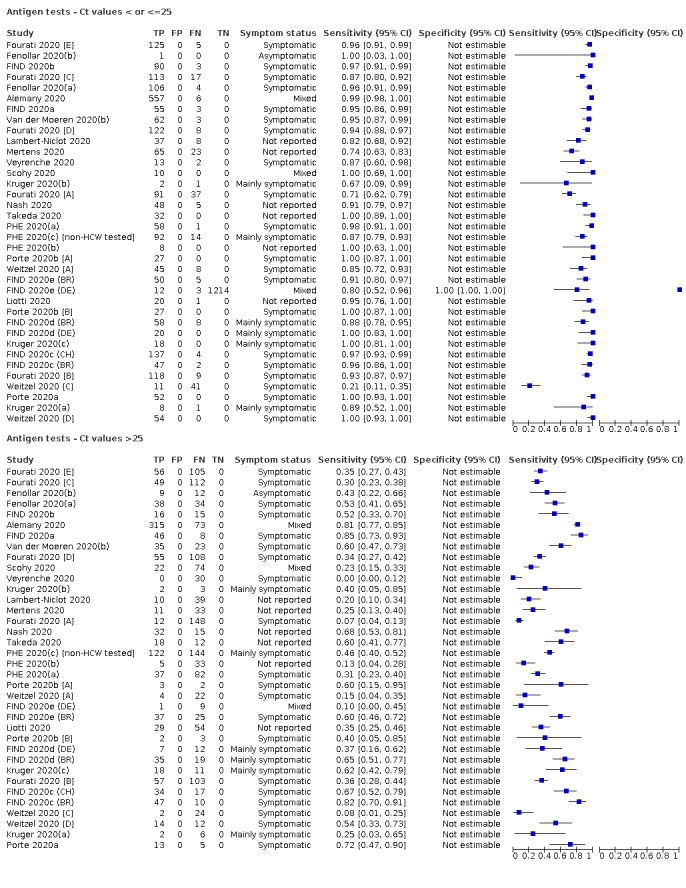
Forest plot of studies evaluating antigen tests: higher versus lower viral load (< or > 25 Ct). BR: Brazil; CH: Switzerland; Ct: cycle threshold; DE: Germany; HCW: healthcare worker
16.
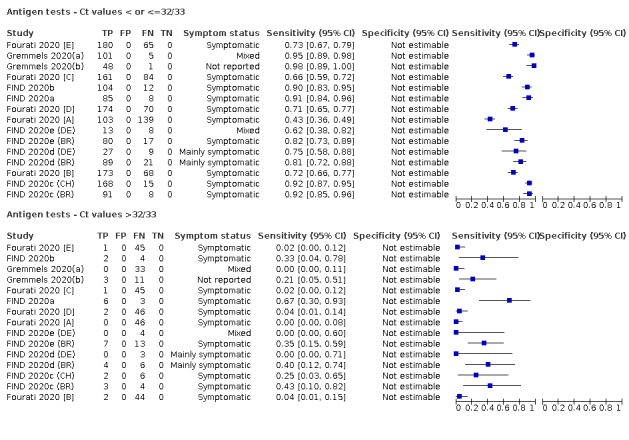
Forest plot of studies evaluating antigen tests: higher versus lower viral load (< or > 32/33 Ct threshold). BR: Brazil; CH: Switzerland; ; Ct: cycle threshold; DE: Germany
17.
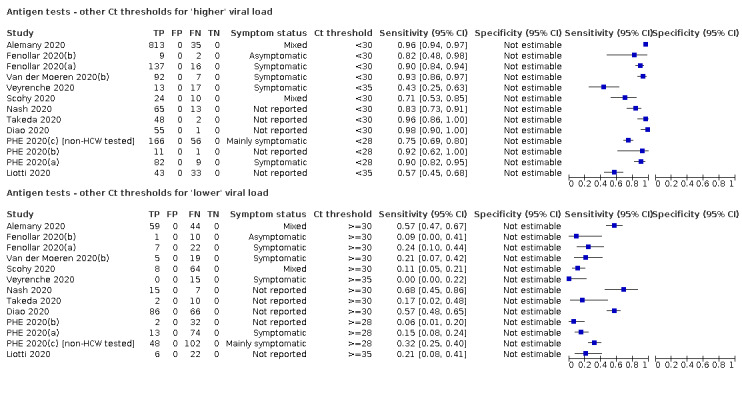
Forest plot of studies evaluating antigen tests: higher versus lower viral load (other Ct thresholds). Ct: cycle threshold; HCW: healthcare worker
Appendix 16. Effect of sample re‐testing and discrepant analysis
| Study | Index test (target genes) | First RT‐PCR | Target gene | Second RT‐PCR | Target gene | False positives | False negatives | Index test re‐test | Reference standard re‐test |
| Discrepant analysis | |||||||||
| Assennato 2020 | SAMBA II (ORF1ab, N2) | PHE Cambridge (Wuhan) assay | RdRp, E gene | PHE Colindale RT‐PCR assay | RdRp 'different region' | 3 → 0 | 1 → 1 | Yes; same results obtained | Yes 3 FPs (reclassified as TP), all borderline positive for ≥ 1 target gene on either RT‐PCR test 1 FN (remained FN), positive on both RT‐PCR assays |
| Collier 2020 | SAMBA II (ORF1ab, N2) |
In‐house PHE assay | Not stated | Appears to be same assay | Not stated | 3 → 1 | 4→ 1 | Yes; same results obtained | Yes 2 FPs (recalsssified as TP) positive by PHE on retest and had high clinical suspicion on notes review 3 FNs (reclassified as TN) were negative by PHE on retest and were considered negative after clinical notes review and therefore were true negatives |
| Harrington 2020 | ID NOW (RdRp) | Abbott RealTime | Not stated | Same RT‐PCR | Same | 2 → 0 | 47 no‐retest | 1 FP reclassified as TN with repeat sampling 1 FP not re‐tested | 1 FP reclassified as TP 1 FP reclassified as TN (both with repeat sampling) |
| Loeffelholz 2020 | Xpert Xpress (RUO) (E, N2) | Multiple RT‐PCR assays according to site | Varied by assay | Different RT‐PCR | Varied by assay | 11 → 3 | 1 → 0 | None reported | 1 FN re‐classified as TN (inconclusive positive on Quest assay; negative on CDC assay) 3 FP remained as FP (2 negative on NY assay, 1 negative on Charité Virologie assay; all confirmed negative with Hologic Panther Fusion) 8 FP re‐classified as TP (all negative on Charité Virologie assay; positive on re‐test with Roche Tib Molbiol assay) |
| Moran 2020 | Xpert Xpress (E, N2) | Roche cobas 6800 | ORF1, E | Same RT‐PCR | Same | 1 → 0 | 0 | 1 FP reclassified as TN (was initially E gene negative and low positive for N2; negative for both targets on re‐test) | 1 FP 'repeatedly negative' on RT‐PCR re‐test (re‐classified as TN based on index re‐test) |
| Stevens 2020 | Xpert Xpress (E, N2) | Panther Fusion SARS‐CoV‐2 Assay (Hologic, Inc., San Diego, CA) | ORF1ab | Same RT‐PCR | Same | 0 | 1 → 0 | No | 1 FN (reclassified as TN) was negative on both targets for Xpert Xpress (FN), negative on re‐test with Panther Fusion |
| Additional studies reporting sample re‐testing (not discrepant analysis) | |||||||||
| Broder 2020 | Xpert Xpress (E, N2) | Roche cobas 6800 | ORF1a, E | modified CDC protocol | NR | 0 | 1 | None reported No presumptive positive results reported | Yes 1 FN (became TN) |
| Hogan 2020 | Accula (N) | In‐house assay | E gene | N/A | N/A | 0 | 16 | Yes 1 TP remained as TP; faint positive Accula test line was repeated on re‐test | None reported |
| Lieberman 2020 | Xpert Xpress (E, N2) | CDC EUA‐based in‐house test (positive if 1 of 2 targets detected) | NI, N2 | N/A | N/A | 0 | 0 | Yes 1 presumptive positive (E‐gene only positive) became positive (N‐gene only positive) on re‐test | None reported |
| Moore 2020 | ID NOW (RdRp) | Modified CDC RT‐PCR | N1, N2 | Abbott RealTime | N, RdRp | 0 → 0 | 25 → 31 | None reported | All samples tested with both RT‐PCR assays 25 FN remained as FN (2 were inconclusive but considered positive on CDC assay, confirmed positive with Abbott RealTime assay) 6 TN reclassified as FN (negative on CDC assay, confirmed positive with Abbott RealTime assay) All 8 discordant results between the two RT‐PCR's were confirmed SARS‐CoV‐2‐positive based on record review |
| Wolters 2020 | Xpert Xpress (E, N2) | In‐house assays per site | Varied by site | Same RT‐PCR per laboratory | Same | 0 → 2 | 0 | None reported 1 presumptive positive considered TP by review team | 2 TP samples (both positive on only one target; 1 presumptive positive (E positive) and 1 positive (N2 positive)) re‐classified as FP; both considered SARS‐CoV‐2 negative on RT‐PCR re‐test *authors note that viral loads were at the limit of detection for Xpert Xpress and that multiple freeze‐thaw steps of samples could have had a significant impact on detection. |
| CDC: center for disease control; EUA: emergency use authorisation; FN: false negative; FP: false positive; PHE: Public Health England; RT‐PCR: reverse transcriptase polymerase chain reaction; RUO: research use only; TN: true negative; TP: true positive | |||||||||
Appendix 17. Molecular tests ‐ Additional figures for subgroup analyses
18.
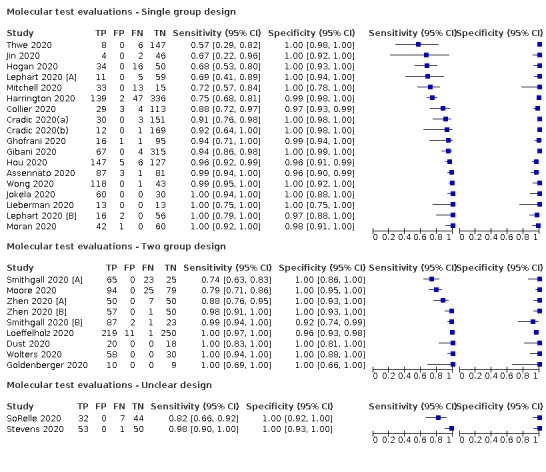
Forest plot of molecular test evaluations by study design
19.
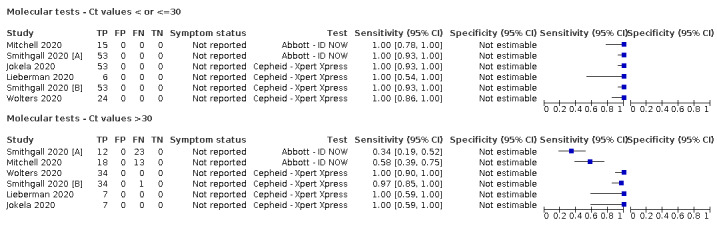
Forest plot of studies evaluating rapid molecular tests: high versus low viral load (30 Ct threshold). Ct: cycle threshold
20.
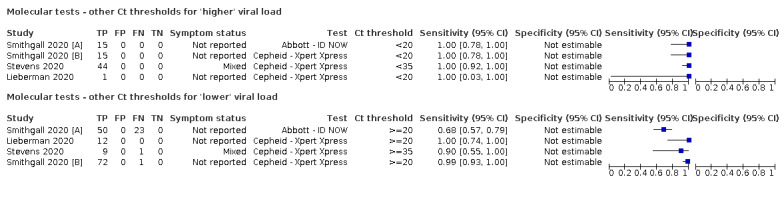
Forest plot of studies evaluating rapid molecular tests: high versus low viral load (other Ct thresholds). Ct: cycle threshold
21.
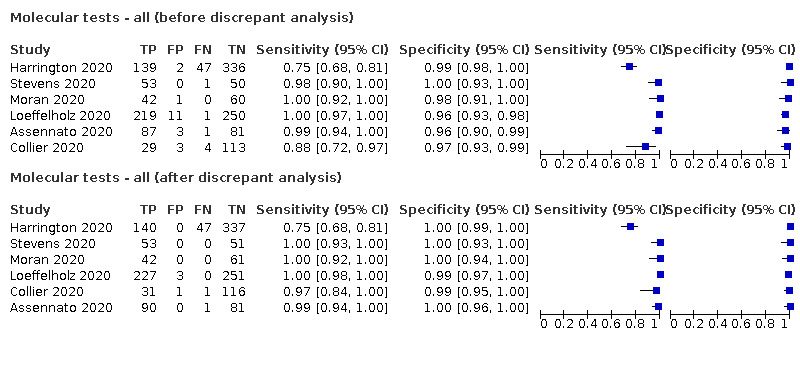
Rapid molecular assays before and after discrepant analysis
Appendix 18. Planned heterogeneity investigations
| Test subgroups | Number of studies | |||
| Sample type | Overall | Direct testing | Using VTM | Other or mixed |
| Antigen tests | n = 48 | |||
| NP only | 32 | 19 | 8 | 5 |
| Nasal | 2 | 1 | 1 | |
| Saliva | 1 | 1 | ‐ | ‐ |
| NP+OP | 5 | 2 | 3 | ‐ |
| NP or OP or combined NP + OP or nasal (≥ 2 evaluated) | 7 | 5 | 1 | 1 |
| BAL or throat wash | 1 | 0 | 0 | 1 |
| Rapid molecular tests | n = 30 | |||
| NP only | 14 | 3 | 9 | 2 |
| OP only | 1 | ‐ | ‐ | 1 |
| Nasal | 2 | 2 | ||
| Saliva | 1 | ‐ | ‐ | 1 |
| NP+OP | 2 | 1 | 1 | 0 |
| NP or OP or NOP or nasal (≥ 2) | 7 | ‐ | ‐ | 7 |
| Throat saliva or LRT | 1 | ‐ | ‐ | 1 |
| Stool | 1 | ‐ | ‐ | 1 |
| Not stated | 1 | 1 | ‐ | ‐ |
| BAL: bronchoalveolar lavage; LRT: lower respiratory tract; NP: nasopharyngeal; OP: oropharyngeal, VTM: viral transport medium | ||||
Data
Presented below are all the data for all of the tests entered into the review.
Tests. Data tables by test.
1. Test.
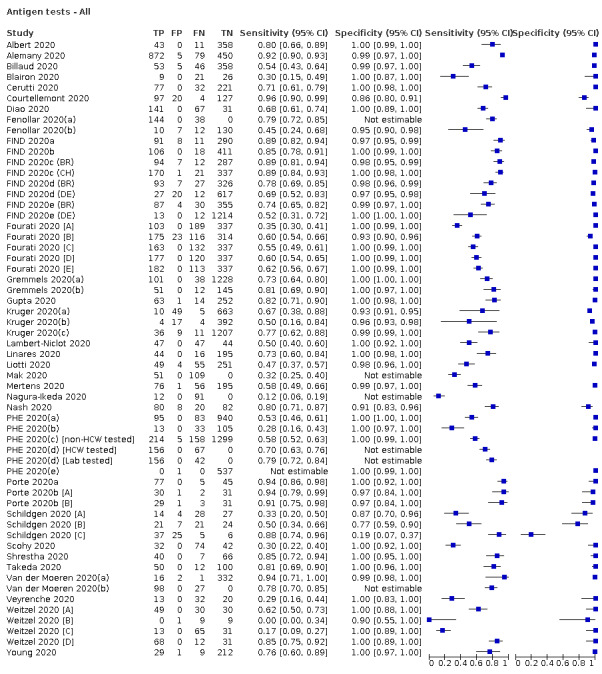
Antigen tests ‐ All
2. Test.
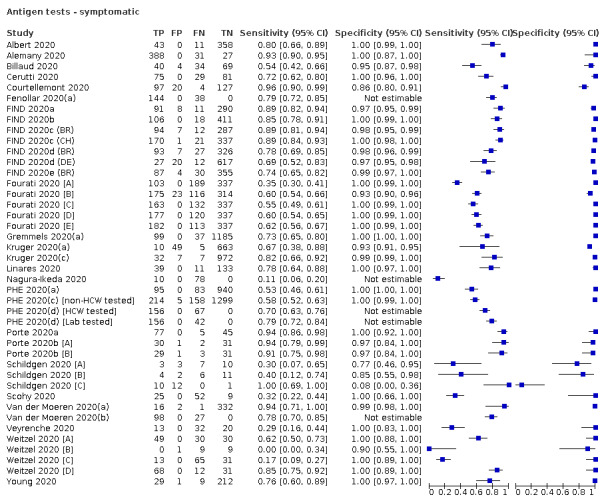
Antigen tests ‐ symptomatic
3. Test.
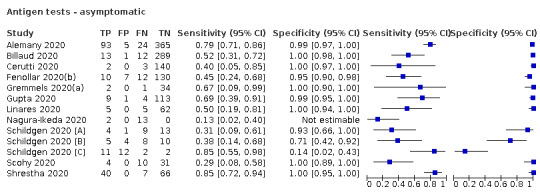
Antigen tests ‐ asymptomatic
4. Test.
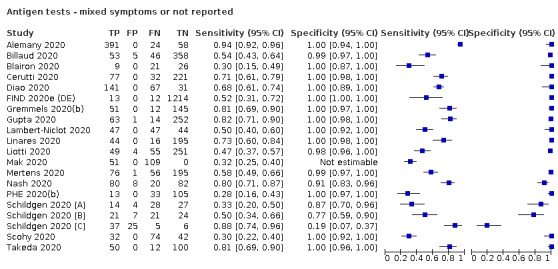
Antigen tests ‐ mixed symptoms or not reported
5. Test.
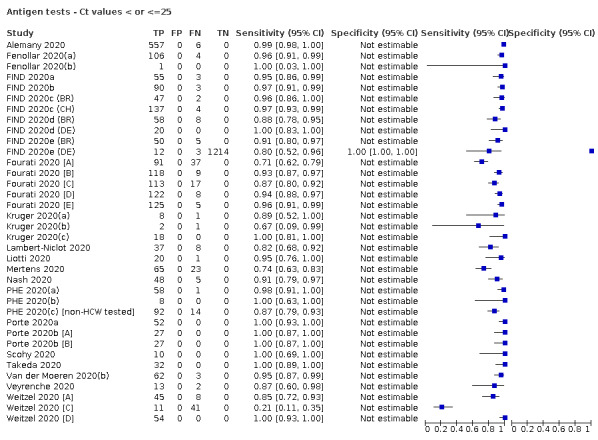
Antigen tests ‐ Ct values < or <=25
6. Test.
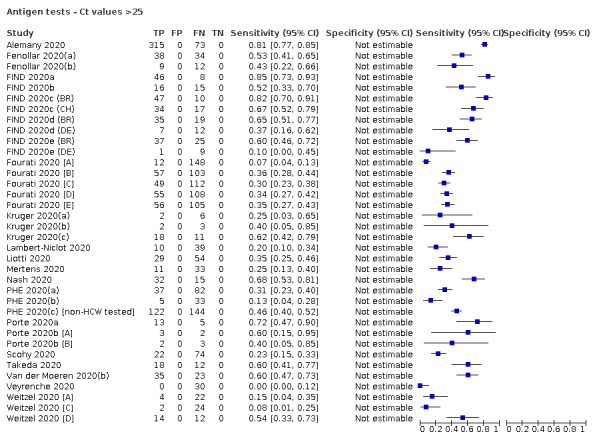
Antigen tests ‐ Ct values >25
7. Test.
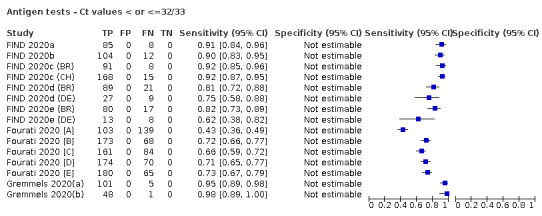
Antigen tests ‐ Ct values < or <=32/33
8. Test.
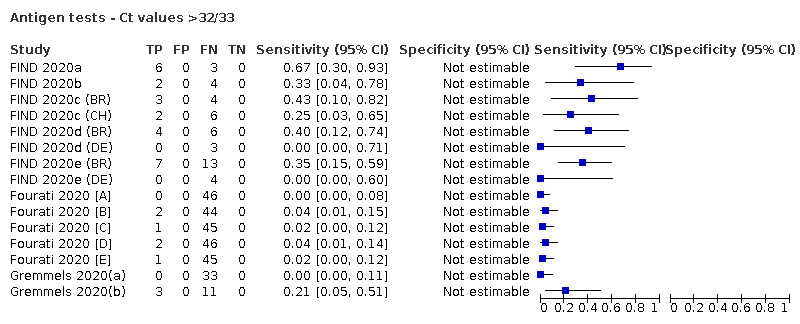
Antigen tests ‐ Ct values >32/33
9. Test.
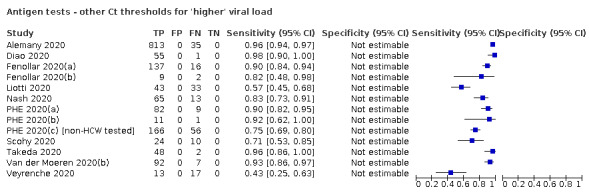
Antigen tests ‐ other Ct thresholds for 'higher' viral load
10. Test.
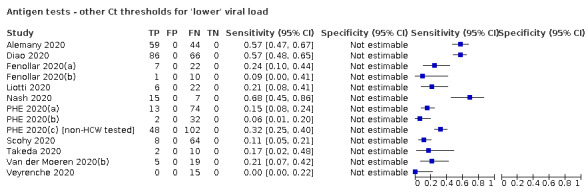
Antigen tests ‐ other Ct thresholds for 'lower' viral load
11. Test.
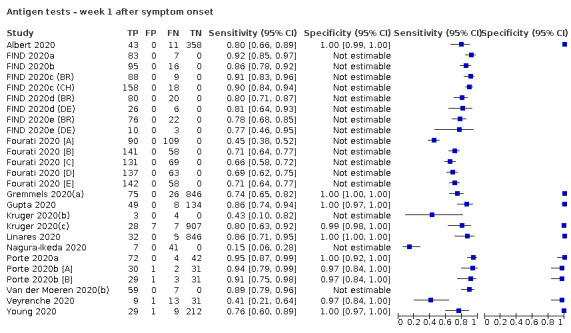
Antigen tests ‐ week 1 after symptom onset
12. Test.
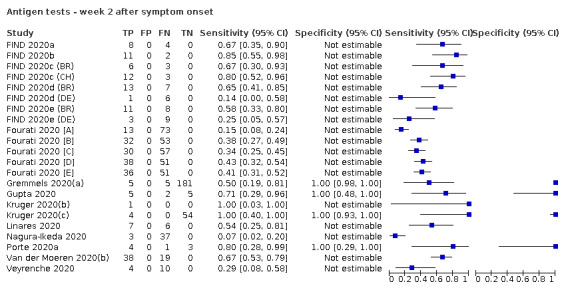
Antigen tests ‐ week 2 after symptom onset
13. Test.
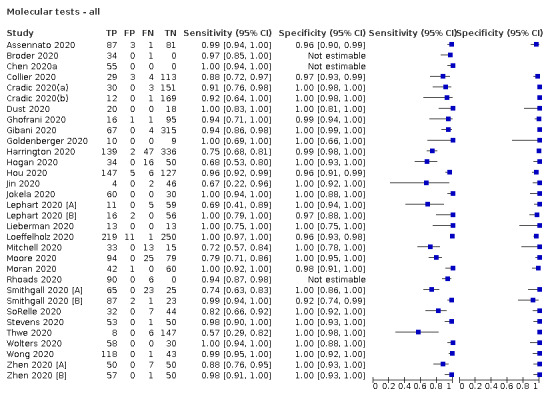
Molecular tests ‐ all
14. Test.
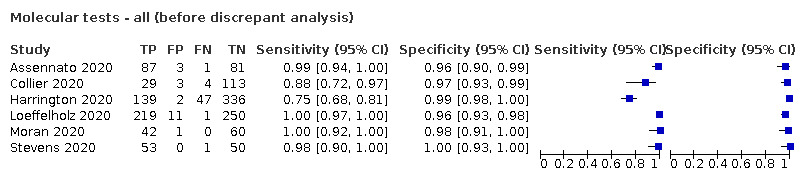
Molecular tests ‐ all (before discrepant analysis)
15. Test.
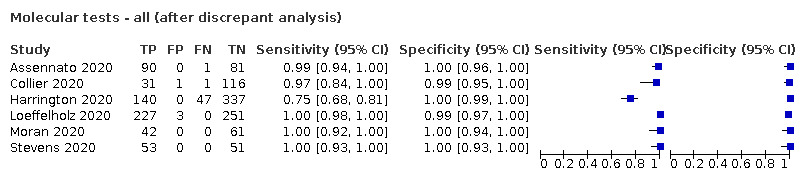
Molecular tests ‐ all (after discrepant analysis)
16. Test.

Molecular tests ‐ Ct values < or <=30
17. Test.

Molecular tests ‐ Ct values >30
18. Test.

Molecular tests ‐ other Ct thresholds for 'higher' viral load
19. Test.

Molecular tests ‐ other Ct thresholds for 'lower' viral load
20. Test.

Molecular tests ‐ other sites
21. Test.
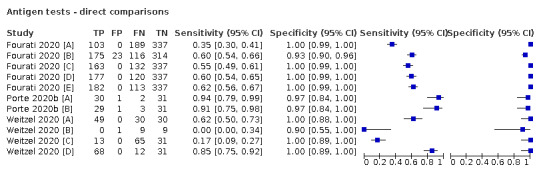
Antigen tests ‐ direct comparisons
22. Test.
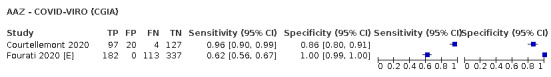
AAZ ‐ COVID‐VIRO (CGIA)
23. Test.
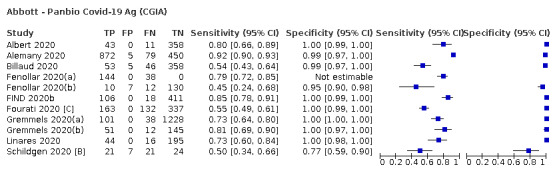
Abbott ‐ Panbio Covid‐19 Ag (CGIA)
24. Test.
Becton Dickinson ‐ BD Veritor (LFA – method not specified)
25. Test.

BIONOTE ‐ NowCheck COVID‐19 Ag (LFA – method not specified)
26. Test.

Biosynex ‐ Biosynex COVID‐19 Ag BSS (CGIA)
27. Test.
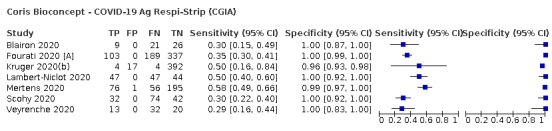
Coris Bioconcept ‐ COVID‐19 Ag Respi‐Strip (CGIA)
28. Test.

E25Bio ‐ DART (NP) (CGIA)
29. Test.

Fujirebio ‐ ESPLINE SARS‐CoV‐2 [LFA(ALP)]
30. Test.

Inhouse (Bioeasy co‐author) ‐ n/a (FIA)
31. Test.
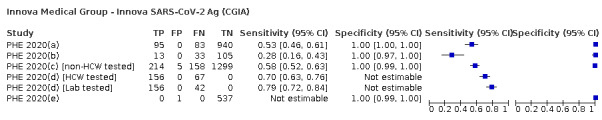
Innova Medical Group ‐ Innova SARS‐CoV‐2 Ag (CGIA)
32. Test.

Liming Bio‐Products ‐ StrongStep® COVID‐19 Ag (CGIA)
33. Test.

Quidel Corporation ‐ SOFIA SARS Antigen (FIA)
34. Test.
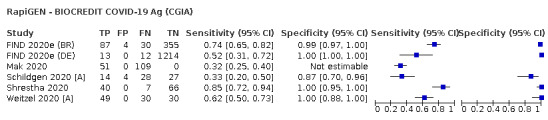
RapiGEN ‐ BIOCREDIT COVID‐19 Ag (CGIA)
35. Test.

Roche ‐ SARS‐CoV‐2 (LFA – method not specified)
36. Test.

Savant Biotech ‐ Huaketai SARS‐CoV‐2 N Protein (LFA – method not specified)
37. Test.
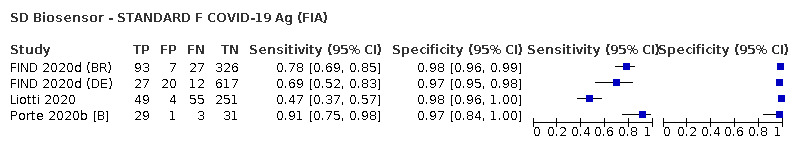
SD Biosensor ‐ STANDARD F COVID‐19 Ag (FIA)
38. Test.
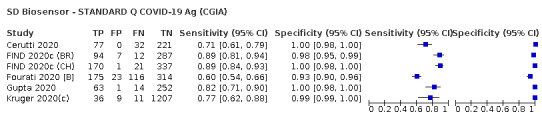
SD Biosensor ‐ STANDARD Q COVID‐19 Ag (CGIA)
39. Test.

Shenzhen Bioeasy Biotech ‐ 2019‐nCoV Ag (FIA)
40. Test.
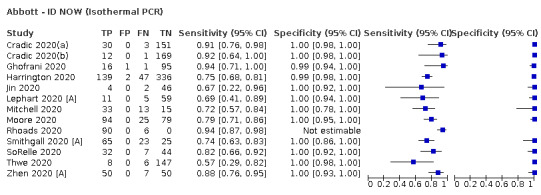
Abbott ‐ ID NOW (Isothermal PCR)
41. Test.
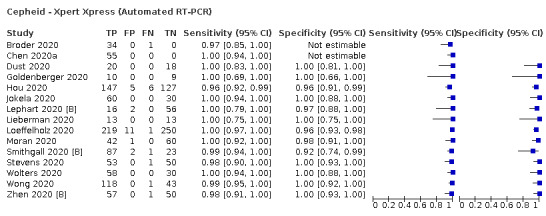
Cepheid ‐ Xpert Xpress (Automated RT‐PCR)
42. Test.

DNANudge – COVID Nudge (Automated RT‐PCR)
43. Test.

DRW ‐ SAMBA II (Automated RT‐PCR)
44. Test.

Mesa Biotech ‐ Accula (other molecular)
45. Test.
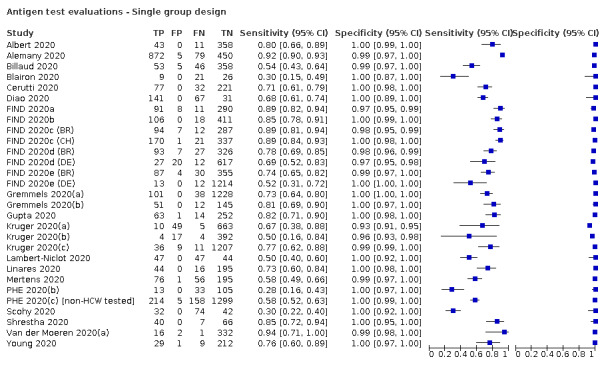
Antigen test evaluations ‐ Single group design
46. Test.
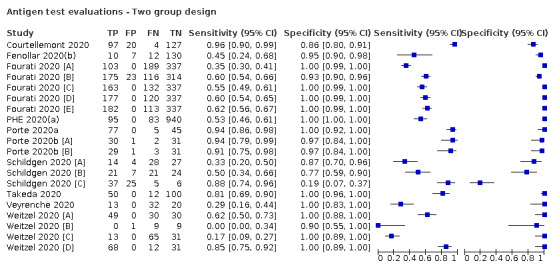
Antigen test evaluations ‐ Two group design
47. Test.

Antigen test evaluations ‐ Unclear design
48. Test.
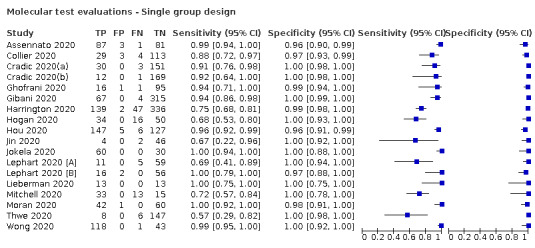
Molecular test evaluations ‐ Single group design
49. Test.
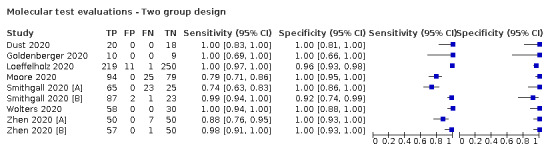
Molecular test evaluations ‐ Two group design
50. Test.

Molecular test evaluations ‐ Unclear design
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Albert 2020.
| Study characteristics | |||
| Patient Sampling | Single group study estimating sensitivity and specificity:
Patients with clinical suspicion of COVID‐19 (compatible signs or symptoms appearing within the prior week) attending one of 8 primary care centres (n=412) Recruitment: Not stated; likely consecutive Prospective or retrospective: Prospective |
||
| Patient characteristics and setting | Setting: Primary care Location: 8 primary care centres of the Health Department Clínico‐Malvarrosa in Valencia. Country: Spain Dates: Sep 2nd to Oct 7 2020 Symptoms and severity: All symptomatic (<7 days p.s.o) Demographics: median age, 31 y (range, 1‐91); 42% male 327 adults; median, 36 y (17‐91y) 85 children; median, 11 y (1‐16y) Exposure history: Not stated |
||
| Index tests | Test name: Panbio™ COVID‐19 AG Rapid Test Device (no product code reported) Manufacturer: Abbott Diagnostic GmbH, Jena, Germany Antibody: Nucleoprotein Antigen target: Not stated Test method: Not stated Samples used: NP; collected by trained nurses using flocked swabs Transport media: None for Ag testing Sample storage: None Test operator: Not stated; immediate testing Definition of test positivity: Visible line within 15 mins; As per manufacturer Blinding reported: Yes Timing of samples: Day <7 pso |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; TaqPath COVID‐19 Combo Kit (Thermo Fisher Scientific, Massachusetts, USA) Definition of non‐COVID cases: As for cases; single negative Genetic target(s): ORF1ab, N and S genes Samples used: NP in UTM Timing of reference standard: As for index; tested within 24h Blinded to index test: Not stated; presume Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous; paired All patients received same reference standard: Yes Missing data: None reported; no participant flow diagram reported Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: This work received no public or private funds. Abbott Diagnostics provided Panbio™ COVID‐19 AG Rapid Test Device kits. Publication status: Pre‐print Source: medRxiv Author COI: The authors declare no conflicts of interest |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Alemany 2020.
| Study characteristics | |||
| Patient Sampling | Single group study including particpants from three settings:
[1] symptomatic individuals with suspected COVID‐19 seen in routine practice (n=446)
[2] contacts exposed to positive PCR confirmed COVID‐19 cases (n=473)
[3] preventive screening of unexposed asymptomatic individuals in the general population (n=487) Recruitment: Retrospective (frozen swabs) Prospective or retrospective: Not stated |
||
| Patient characteristics and setting | Setting: Mixed/Unclear (laboratory‐based) Location: Not reported; multiple author institutions reported Country: Spain Dates: Not stated Symptoms and severity: Not stated; 15/1406 (1.1%) reportedly hospitalised (all PCR+) Viral load of cases: Ct <20: 258 (18.3%); Ct 20‐24 305 (21.7%); Ct 25‐29 285 (30.3%); Ct >30 103 (7.3%) Demographics: All samples: mean age 40.4y (SD 24.5), 453 (32.2% male) Exposure history: 473/1406 (33.6%) identified through contact tracing; |
||
| Index tests | Test name: PanbioTM COVID‐19 Ag Test (no product codes) [Selected following validation exercise using 40 NP samples to compare PanBio with Coris Bioconcept COVID‐19 Ag RespiStrip, SD Biosensor Standard F COVID‐19 Ag FIA and Standard Q COVID‐19 Ag Test] Manufacturer: Abbott Laboratories, Illinois, USA Antibody: Not stated Antigen target: SARS‐CoV‐2 Test method: CGIA Samples used: [1] and [2] NP, [3] nasal mid‐turbinate; collection not reported Transport media: VTM (DeltaSwab Virus) Sample storage: stored at 2‐8C prior to PCR then frozen (‐80C) prior to Ag testing; "Internal validation showed no significant change in the test performance using Abbot test Kit buffer or a mix of the Kit buffer and transport media at 1:3 dilution; likewise, the use of frozen specimens showed no significant differences compared with fresh ones" Test operator: two laboratory technicians Definition of test positivity: Visible line; as per manufacturer Blinding reported: Yes Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; in‐house following CDC protocol Definition of non‐COVID cases: As per cases; single negative PCR for absence of infection Genetic target(s): Not stated; as per CDC protocol Samples used: NP or nasal mid‐turbinate; as per index test Timing of reference standard: fresh samples stored at 2 – 8 ºC for up to 72 hours prior to RT‐PCR Blinded to index test: Yes; conducted first Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous (same swab) All patients received same reference standard: Yes Missing data: None reported; no participant flow diagram reported Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: The test kits were purchased to Abbott Rapid Diagnostics Healthcare SL (Spain). The funders of the study had no role in the study conception, design, conduct, data analysis, or writing of the report. Publication status: Pre‐print Source: medRxiv Author COI: Authors declare no conflicts of interest |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Assennato 2020.
| Study characteristics | |||
| Patient Sampling | Single‐group study to estimate sensitivity and specificity:
‐ samples from symptomatic individuals with suspected COVID‐19 sent for routine laboratory diagnosis; supplied via PHE (n = 172) Recruitment: not stated Prospective or retrospective: retrospective Number of samples (samples with confirmed SARS‐CoV‐2): 172 (88) |
||
| Patient characteristics and setting | Setting: not stated; supplied by PHE Location: PHE, Cambridge Laboratory (samples from East of England) Country: UK Dates: not stated Symptoms and severity: symptomatic; no further details Demographics: not stated Exposure history: not stated |
||
| Index tests | Test name: SAMBA II SARS‐CoV‐2 Test Manufacturer: Diagnostics for the Real World Antigen target: ORF1ab, N2 Antibody: N/A Test method: rapid PCR Samples used: combined nose and throat swab samples, provided as VTM Transport media: samples diluted 1:2 with SAMBA SCoV buffer Sample storage: not stated Test operator: not stated; presume laboratory staff Definition of test positivity: as per manufacturer; either target present Blinding reported: yes; states that samples were rendered anonymous and provided blinded for the purpose of test validation Timing of samples: not stated |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; (1) Cambridge RdRp gene (Wuhan) assay on the Rotor gene Q real‐time PCR assay routinely used by PHE; Ct ≤ 36 considered positive. (2) Samples also tested with the PHE Colindale (Reference Laboratory) assay Definition of non‐COVID cases: Single RT‐PCR negative Genetic target(s): (1) RdRp, E gene, (2) RdRp 'different region' Samples used: combined nose and throat swab in VTM; same as for index test Timing of reference standard: not stated; Cambridge assay seems to have been part of routine testing near to time of sample collection; not clear if Colindale assay was at a later date after a period of storage Blinded to index test: not stated but seems yes for Cambridge assay Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: not stated; seems likely reference was carried out for routine diagnostic testing All participants received same reference standard: yes (all samples underwent both RT‐PCR tests) Missing data: none reported, no participant flow diagram reported Uninterpretable results: none reported Indeterminate results (index test): 3 FP and 1 FN result retested using SAMBA‐II; same results obtained on repeat Indeterminate results (reference standard): 3 FP and 1 FN result were re‐tested ‐ all 3 FPS found to be borderline positive for ≥ 1 target gene on either Colindale or Cambridge (Wuhan) test (reclassified as TP) ‐ the FN result remained positive on both RT‐PCR assays Unit of analysis: refers to participants rather than samples |
||
| Comparative | |||
| Notes | Funding: RKG is funded by Wellcome Senior Fellowship In Clinical Science award no WT108082AIA Publication status: preprint Source: medRxiv Author COI: no COI statement reported; 3 co‐authors are affiliated to test manufacturer |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Billaud 2020.
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity: ‐ teachers (n=90) and students (n=419) screened for COVID‐19 as part of a cluster investigation (n=509) Recruitment: Not stated; appears to be open to all Prospective or retrospective: Prospective |
||
| Patient characteristics and setting | Setting: Screening Location: College, Lyon Country: France Dates: September 16 and 17 Symptoms and severity: 166/509, 32.6% symptomatic including 152/419 (36%) students Demographics: Mean, median age Students 21.6y, 21y (18 to 37y) Teachers 47.2y, 49y (26 to 64y) Exposure history: Outbreak investigation |
||
| Index tests | Test name: Described as "ABBOTT SARS‐COV2 Antigenic Test"; presumed to be Panbio COVID‐19 Ag Test Manufacturer: Abbott Antibody: Not stated Antigen target: Not stated Test method: Not stated Samples used: NP; collected by firefighters Transport media: None used Sample storage: n/a; tested immediately on site Test operator: Not stated Definition of test positivity: Visual line; as per manufacturer Blinding reported: Yes, performed first Timing of samples: Not stated but includes people >7 days pso |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; SARS‐COV‐2 (Thermofisher) Definition of non‐COVID cases: As for cases; single negative Genetic target(s): Not stated Samples used: NP (paired) Timing of reference standard: As for index Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous All patients received same reference standard: Yes Missing data: 47 missing, including 11 uninterpretable Uninterpretable results: 11 uninterpretable on Ag test Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: Not stated, public funding Publication status: Published Source: Report accessed via SFM Microbiologie website Author COI: None |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Blairon 2020.
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity: sampled from cohort of suspected COVID‐19 patient samples sent for laboratory diagnosis (n=56)
[Excluded data for full cohort, as only those with negative antigen test underwent confirmatory RT‐PCR; of 912 submitted samples during time period, 776 remained after removing repeat tests and were reported in main study] Recruitment: Selection of 56 for verification analysis was not reported. Prospective or retrospective: prospectively |
||
| Patient characteristics and setting | Setting: Unclear; swabs obtained at hospital site (no further detail) Location: Not stated; author institution Iris Hospitals South, Brussels Country: Belgium Dates: April 5 ‐ May 4 2020 Symptoms and severity: Not stated Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: COVID‐19 Ag Respi‐Strip (no product code reported) Manufacturer: Coris Bioconcept (Gembloux, Belgium) Antibody: Not stated Antigen target: Not stated Test method: LFA Samples used: NP swabs; collection not reported Transport media: Samples for antigen testing taken from UTM‐RT swabs (Copan spa, Brescia, IT) Sample storage: No storage described; infer that antigen test was conducted immediately on receipt of sample at on‐site laboratory 'after antigenic testing was performed, the molecular assessment of SARS‐CoV‐2 was outsourced to a university centre' Test operator: Not stated; infer laboratory staff Definition of test positivity: As per manufacturer Blinding reported: Not stated; infer yes as conducted prior to PCR confirmation Timing of samples: Not stated; appears to be on presentation (repeat tests ordered at clinician's discretion were excluded) |
||
| Target condition and reference standard(s) | Reference standard: qRT‐PCR Definition of non‐COVID cases: As above, single PCR negative to confirm absence of disease Genetic target(s): E gene Samples used: NP swabs (same as for Ag test) Timing of reference standard: Not stated Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Not stated but infer short interval; samples sent to university centre laboratory for PCR confirmation All patients received same reference standard: Yes (only if author confirms Ag+ also got PCR) Missing data: None reported; review team excluded main cohort data as no reference standard for antigen test positive samples Uninterpretable results: None reported; 1 'invalid' sample excluded from main cohort Indeterminate results (index test): None reported; 1 'non‐conform' sample excluded from main cohort Indeterminate results (reference standard): None reported Unit of analysis: Unclear; main cohort includes unique patient samples but not reported for separate group of 56 |
||
| Comparative | |||
| Notes | Funding: None to declare Publication status: Published Source: Journal of Clinical Virology Author COI: None to declare |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Broder 2020.
| Study characteristics | |||
| Patient Sampling | Single‐group study to estimate sensitivity:
‐ samples positive on Roche cobas 6800 assay in lower range of viral load (E target Ct ≥ 30) (n = 35) Recruitment: not stated; deliberate sampling according to viral load Prospective or retrospective: unclear Number of samples (samples with confirmed SARS‐CoV‐2): 35 (35) |
||
| Patient characteristics and setting | Setting: not stated Location: not stated; author institution Emory University School of Medicine, Atlanta Country: USA Dates: not stated Symptoms and severity: not stated; lower viral load Demographics: not stated Exposure history: not stated |
||
| Index tests | Test name: GeneXpert Xpress SARS‐CoV‐2 assay (no product code reported) Manufacturer: Cepheid Antigen target: not stated E gene Antibody: N/A Test method: rapid PCR Samples used: NP swabs in VTM Transport media: not stated Sample storage: within 3 days of initial testing (with RT‐PCR) Test operator: not stated; presume laboratory staff Definition of test positivity: “all specimens were tested using the manufacturer’s protocol”, no mention of presumptive positives Blinding reported: not stated Timing of samples: not stated |
||
| Target condition and reference standard(s) | Reference standard: Roche cobas 6800 SARS‐CoV‐2 assay Definition of non‐COVID cases: N/A Genetic target(s): E gene (unclear if other genetic targets as well) Samples used: NP swabs (as for index test) Timing of reference standard: not stated; presume on presentation Blinded to index test: not stated; presume yes Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: same samples; index within 3 days of reference All participants received same reference standard: yes Missing data: none reported Uninterpretable results: none reported, no participant flow diagram reported Indeterminate results (index test): none reported Indeterminate results (reference standard): discrepancies resolved using modified CDC RT‐PCR; 1 FN confirmed as disease negative (i.e. a TN) Unit of analysis: not stated; refers only to samples |
||
| Comparative | |||
| Notes | Funding: no funding described Publication status: accepted manuscript Source: Journal of Clinical Microbiology Author COI: Dr. Kraft participated on a Roche advisory board regarding COVID serology. All other study authors have no conflicts |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Cerutti 2020.
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity in two cohorts:
(1) symptomatic patients attending one of two Emergency departments (n=185)
(2) asymptomatic travellers returning home from European high risk countries (Croatia, Spain, Malta) (n=145) Recruitment: (1) Random; (2) Not stated, presume consecutive Prospective or retrospective: Not stated |
||
| Patient characteristics and setting | Setting: Mixed; (1) Emergency department; (2) Possible contacts Location: (1) two Infectious Disease reference centres in North‐Italy (ASL Citt`a di Torino, Turin and San Martino University Hospital, Genoa); (2) Not stated; samples sent to Microbiology and Virology Laboratory, Amedeo di Savoia Hospital, Torino Country: Italy Dates: (1) Mar 3 to May 1; (2) August 2020 Symptoms and severity: Not stated; cohort (2) were asymptomatic Demographics: (1) mean age 44.6, 95 %CI: 40.7–48.6; (2) mean age 35.9, 95 % CI: 32.7–39.1 Exposure history: (1) Not stated; (2) High risk country visit |
||
| Index tests | Test name: STANDARD Q COVID‐19 Ag Manufacturer: SD‐Biosensor, RELAB, I Antibody: NP Antigen target: Not stated Test method: Not stated Samples used: NP; collection not stated Transport media: UTM (Copan, I) Sample storage: Primarily run in parallel with standard of care RT‐PCR; 13 were frozen residual samples Test operator: Not stated; laboratory staff presumed Definition of test positivity: Visual line after 15‐30 mins; as per manufacturer. Blinding reported: Not stated Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; Seegene Allplex® 2019 n‐CoV Assay (N = 159), DiaSorin Simplexa® (n = 28), and Cobas 6800 Roche® (N = 118). Definition of non‐COVID cases: Single negative Genetic target(s): Not stated Samples used: Not stated Timing of reference standard: Not stated Blinded to index test: Unclear Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous; not clear if same sample used or paired swabs obtained All patients received same reference standard: Yes; different assays Missing data: None reported; no participant flow diagram reported Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: Authors thank RELAb for the donation of the STANDARD Q COVID‐19 SD‐Biosensor kits to pursue the study. No other specific grant from public funding agencies was received. Publication status: Published Source: J Clin Virol Author COI: The authors report no declarations of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Chen 2020a.
| Study characteristics | |||
| Patient Sampling | Single group study using:
‐ archived paired samples from COVID‐19 inpatients (n=58). Aim is to compare diagnostic yield between saliva and NP swabs but can also extract sensitivity for each using rapid test. Recruitment: Not stated Prospective or retrospective: Retrospective |
||
| Patient characteristics and setting | Setting: In‐patients Location: Queen Mary Hospital, Pokfulam, Hong Kong Country: People’s Republic of China Dates: Not stated Symptoms and severity: Not stated Demographics: Median age 38 y; 28, 48% male Exposure history: Not stated |
||
| Index tests | Test name: Xpert Xpress SARS‐CoV‐2 assay (no product codes reported) Manufacturer: Cepheid, Sunnyvale, CA, USA Target gene(s): E and N2 gene Antigen target: n/a Test method: Automated RT‐PCR Samples used: NP, saliva (posterior oropharyngeal, self‐collected by clearing the throat and spitting c1 mL saliva directly into a sterile bottle in the early morning before mouth rinsing and breakfast) Transport media: Both sample types immersed in 2ml of viral transport solution Sample storage: Not stated; archived Test operator: Not stated; infer laboratory staff Definition of test positivity: Not stated; tested 'according to manufacturer’s instruction' ‐ no mention of presumptive positives Blinding reported: Not stated; Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Reference standard: in‐house SARS‐CoV‐2 RNA dependent RNA polymerase/ Helicase (RdRp/Hel) real‐time
RT–PCR assay Definition of non‐COVID cases: n/a only cases included Genetic target(s): RdRp Samples used: same as index test Timing of reference standard: Not stated; prior to index test Blinded to index test: Not stated; infer yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous; same samples All patients received same reference standard: Yes Missing data: None reported, no participant flow diagram reported. THree samples positive only on saliva excluded by review team Uninterpretable results: Not stated Indeterminate results (index test): Not stated Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: This study was partly supported by Consultancy Services for Enhancing Laboratory Surveillance of Emerging Infectious Diseases and Research Capability on Antimicrobial Resistance, and the Theme‐Based Research Scheme (T11/707/15) of the Research Grants Council, the donations of Richard Yu and Carol Yu, the Shaw Foundation Hong Kong Michael Seak‐Kan Tong, May Tam Mak Mei Yin Respiratory Viral Research Foundation Limited, Hui Ming, Hui Hoy, and Chow Sin Lan Charity Fund Limited, Chan Yin Chuen Memorial Charitable Foundation, Marina Man‐Wai Lee, the Jessie & George Ho Charitable Foundation, Perfect Shape Medical Limited, and Kai Chong Tong. Publication status: Published Source: Emerging microbes and infections Author COI: No potential conflict of interest was reported by the author(s); Xpert Xpress cartridges provided by the test manufacturer via an Investigator‐Initiated Study agreement (Cepheid‐IIS‐2020‐0009). |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Collier 2020.
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity: suspected COVID‐19 patients admitted with a possible diagnosis of COVID‐19 (n=149) Recruitment: Consecutive Prospective or retrospective: prospectively |
||
| Patient characteristics and setting | Setting: In‐patients Location: Cambridge University Hospitals NHS Foundation Trust Country: UK Dates: April 6 ‐ May 2 2020 Symptoms and severity: Not stated Demographics: Mean age 62.7 y, 70, 47% male Exposure history: Not stated |
||
| Index tests | Test name: SAMBA II SARS‐CoV‐2 test (no product code reported) Manufacturer: Diagnostics for the Real World (DRW), University of Cambridge, Cambridge Target gene(s): Orf1 and the E genes Antigen target: n/a Test method: RT‐PCR Samples used: combined nasal/throat swab (NOP) on dry sterile swab. Collection not reported Transport media: None used; samples inactivated in SCov buffer prior to testing Sample storage: Not stated. Test performed within 18 hours of reference test Test operator: Not stated; infer laboratory staff Definition of test positivity: As per manufacturer Blinding reported: Unclear; yes if always conducted before reference test but not explicitly described, i.e. 'SAMBA swab must be taken within 18 hours of the standard laboratory swab' Timing of samples: Not stated; appears to be on presentation/admission but no further details |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; in‐house PHE assay Definition of non‐COVID cases: As above, single PCR negative to confirm absence of disease Genetic target(s): Not stated Samples used: Not stated; separate swab used as participants were excluded if >18h interval between swab collections Timing of reference standard: Not stated Blinded to index test: Yes; 'The results of the SAMBA II SARS‐CoV‐2 was not known to the assessors of the standard lab RT‐PCR prior.' Not stated. Possibly if done prior to index test. Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: <18 hours All patients received same reference standard: Yes Missing data: Yes; 5 discarded VTM, 1 timing of PHE swab not reported, 1 inadequate SAMBA swab, 2 interval between swabs >24h Uninterpretable results: None reported Indeterminate results (index test): Not stated 'Indeterminate SAMBA II SARS CoV‐2 tests were repeated with a 1:2 dilution of sample to inactivation buffer according to manufacturer standard operating procedures until a valid result was obtained.' Discrepant results between index and reference were also re‐tested using SAMBA‐II on original samples Indeterminate results (reference standard): 1 false negative Indeterminate standard lab RT PCR tests were repeated on a replicate nose/throat swab until a valid result was obtained. Discrepant results between index and reference were re‐tested using RT‐PCR on original samples, with reference to clinical notes to determine clinical suspicion. Remaining discrepant results were re‐tested using alternative sample, i.e. sample in SCov buffer tested on RT‐PCR and sample in VTM tested on SAMBA‐II Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: The Wellcome Trust (Senior Research Fellowship to RKG WT108082AIA and PhD Research Fellowship to DAC; Principal Research Fellowship 210688/Z/18/Z to PJL), Addenbrooke’s Charitable Trust to PJL, National Institute of Health Research (NIHR) Cambridge BRC Publication status: Pre‐print and published version (25‐8‐20) Source: Pre‐print; Cell Reports Medicine Author COI: Pre‐print ‐ Dr. Besser reports personal fees from STAGO, personal fees from Novartis, personal fees from Cosmopharma, personal fees from Werfen, personal fees from Agios, grants from Mitsubishi Pharma, outside the submitted work; RKG reports fees from ad hoc consulting from ViiV, Gilead and UMOVIS. Published version ‐ The authors declare no competing interests (Three co‐authors affiliated to test manufacturer) |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | No | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Courtellemont 2020.
| Study characteristics | |||
| Patient Sampling | Unclear design estimating sensitivity and specificity (coded as two group because of deliberate sampling of PCR positive cases):
(1) Symptomatic (headache, fatigue, fever, or respiratory signs) or asymptomatic people voluntarily accessing the COVID‐19 Screening Department (n=231)
(2) hospitalized SARS‐CoV‐2 positive patients (n=17) [review team excluded 20 cases with a previous positive RT‐qPCR within 5 days but a negative RTqPCR at the time of study sampling] Recruitment: Unclear Prospective or retrospective: Unclear |
||
| Patient characteristics and setting | Setting: Mixed Location: COVID‐19 Screening Department and SARS CoV‐2 positive patients hospitalized in the Infectious Diseases Department of the Centre Hospitalier Régional (CHR) of Orléans, France, or the Department of Infectious and Tropical Diseases of the Centre Hospitalier Universitaire (CHU) Tenon, Paris Country: France Dates: Oct 12 to Oct 19 Symptoms and severity: 99/121, 82% cases were symptomatic; 22 asymptomatic Demographics: median age 38y, mean age 43y (range: 18‐96), 117, 47% male Exposure history: Not stated |
||
| Index tests | Test name: COVID‐VIRO® Manufacturer: AAZ, Boulogne Billancourt, France Antibody: Nucleocapsid Antigen target: monoclonal Test method: CGIA Samples used: NP; collected by trained personnel (nurse, doctors, or biologist); subgroup also had OP or saliva collected Transport media: Direct testing for Ag test Sample storage: None Test operator: Not stated Definition of test positivity: Visible line; As per manufacturer Blinding reported: Yes Timing of samples: median 5 days pso, mean 5.3 days, range 1 to 20d |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; TaqPath Covid‐19 Multiplex RT‐PCR, Thermofisher Definition of non‐COVID cases: single negative PCR Genetic target(s): ORF1ab, S and N genes Samples used: NP in VTM; paired Timing of reference standard: As for index Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous; paired All patients received same reference standard: Yes Missing data: None reported, no participant flow diagram reported; review team excluded 20 cases with a previous positive RT‐qPCR within 5 days but a negative RTqPCR at the time of study sampling Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: No funding statement reported Publication status: Preprint Source: medRxiv Author COI: No COI statement reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Cradic 2020(a).
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity:
‐ symptomatic patients suspected of COVID‐19 that met criteria for testing, either presenting to ED or as inpatients at single hospital (n=184) Recruitment: Not stated Prospective or retrospective: Prospective [Second cohort of paired samples from patients presenting to ED with signs/symptoms of COVID‐19 submitted for routine laboratory testing (n=182), extracted as Cradic 2020(b)] |
||
| Patient characteristics and setting | Setting: Mixed (ED/inpatients) Location: OhioHealth Riverside Methodist Hospital, Columbus Country: USA Dates: Not stated Symptoms and severity: All symptomatic, no further details. Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: [A] ID NOW COVID‐19 EUA [Study also evaluates [B] Diasorin Simplexa and [C] Roche cobas 6800 SARS‐CoV‐2; not eligible for this review] Manufacturer: Abbott Laboratories Target gene(s): RdRp Antigen target: n/a Test method: Isothermal PCR Samples used: NP swabs in UTM; collected on flocked swab, no other details, Transport media: 3 mL of sterile UVT (Becton Dickinson) Sample storage: asap, or stored for up to 72 hours at 2°C to 8°C. Following routine testing, samples were stored frozen (≤–80°C) until comparator testing with the Roche cobas assay could be completed Test operator: Not stated; infer laboratory staff. Definition of test positivity: as per manufacturer Blinding reported: Not stated Timing of samples: Unclear, infer upon presentation |
||
| Target condition and reference standard(s) | Reference standard: Composite reference standard, defined as the result obtained from at least 2 of the 3 assays conducted (Abbot ID NOW, Diasorin Simplexa or Roche cobas 6800 SARS‐CoV‐2) Definition of non‐COVID cases: Same as index test; single negative for absence disease Genetic target(s): RdRp, S or ORF1ab gene (either present), ORF1ab or E gene (both present for +ve, either present for presumptive +ve) Samples used: Same as index test Timing of reference standard: Not stated Blinded to index test: No (>=2 +ve) Incorporated index test: Yes |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous ‐ same swab All patients received same reference standard: Yes Missing data: None reported, no participant flow diagram reported Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: No funding statement reported Publication status: published Source: American Journal of Clinical Pathology Author COI: No COI statement reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | No | ||
| Reference standard does not incorporate result of index test? | No | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Cradic 2020(b).
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity: paired samples from patients presenting to ED with signs/symptoms of COVID‐19 submitted for routine laboratory testing (n=182) Recruitment: Not stated Prospective or retrospective: Prospective [Second cohort of symptomatic patients suspected of COVID‐19 that met criteria for testing, either presenting to ED or as inpatients at single hospital (n=184), extracted as Cradic 2020(a)] |
||
| Patient characteristics and setting | Setting: Emergency department Location: OhioHealth Laboratory Services, Columbus (presume ED at OhioHealth Riverside Methodist Hospital) Country: USA Dates: Not stated Symptoms and severity: All symptomatic, no further details. Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: [A] ID NOW COVID‐19 EUA [Study also evaluates [B] Diasorin Simplexa and [C] Roche cobas 6800 SARS‐CoV‐2; not eligible for this review] Manufacturer: Abbott Laboratories Target gene(s): RdRp Antigen target: n/a Test method: Isothermal PCR Samples used: NP swabs in UTM (collected as part of standard of care), plus direct testing of OP swabs and of nasal swabs (collected according to CDC instructions) Transport media: presume as above for NP in UTM Sample storage: not stated Test operator: Not stated; infer laboratory staff. Definition of test positivity: as per manufacturer Blinding reported: Not stated Timing of samples: Unclear, infer upon presentation |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; Diasorin Simplexa Definition of non‐COVID cases: Same as index test; single negative for absence disease Genetic target(s): S or ORF1ab gene (either present) Samples used: NP swab in UTM Timing of reference standard: Not stated Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous; paired swabs All patients received same reference standard: Yes Missing data: None reported, no participant flow diagram reported Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: No funding statement reported Publication status: published Source: American Journal of Clinical Pathology Author COI: No COI statement reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Diao 2020.
| Study characteristics | |||
| Patient Sampling | Single group estimating sensitivity and specificity for detecting active disease
‐ samples from cases of suspected SARS‐CoV‐2 infection (n = 239) Recruitment: not stated if participants were consecutive Prospective or retrospective: retrospective Number of samples (samples with confirmed SARS‐CoV‐2): 239 (208) |
||
| Patient characteristics and setting | Setting: hospital (inpatients) Location: 7 centres, including General Hospital of Central Theatre Command, Wuhan No.7 People’s Hospital, Wuhan Pulmonary Hospital, Hubei Maternal and Child Hospital, Taikang Hospital, Hanyang Hospital and Wuguo Hospital. Urine study done in Southwest Hospital in Chongqing Country: China Dates: not stated Symptoms and severity: not stated Demographics: not stated Exposure history: not stated |
||
| Index tests | Test name: not stated Manufacturer: in house (but study authors affiliated to Bioeasy Technology) Antibody: monoclonal antibody Antigen target: nucleocapsid protein (N‐antigen) Test method: FIA (fluorescence immunochromatographic); requires immunofluorescence analyser Samples used: NP (all), urine (subgroup) Transport media: samples diluted and mixed in 500 μL saline solution; 100 μL transferred to the sample well of the test card Sample storage: not reported Test operator: not stated; presume laboratory staff Definition of test positivity: cut‐off value was determined by testing 100 nasal swab samples of healthy people and calculated as the mean value of the fluorescence signal plus 5 SD. Blinding reported: done in parallel; blinded Timing of samples: not stated |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR (Daan Gene kit); performed on ABI Prism 7500 and Light Cycler 480 real‐time PCR system. Threshold < 40 Ct; threshold < 30 Ct also investigated
Definition of non‐COVID cases: all participants underwent 3 nucleic acid tests, and the results of each nucleic acid test were verified by 2 COVID‐19 nucleic acid test kits. Genetic target(s): ORF1ab and N gene Samples used: NP swab, same as for index test Timing of reference standard: not stated Blinded to index test: done in parallel; blinded Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: done in parallel All participants received same reference standard: yes Missing data: not reported, no participant flow diagram reported Uninterpretable results: not reported Indeterminate results (index test): none reported Indeterminate results (reference standard): none described Unit of analysis: participants |
||
| Comparative | |||
| Notes | Funding: this research was supported by grants from National Key R&D Program of China (2016YFA0502204); Chongqing Health Commission COVID‐19 Project (2020ZX01). Publication status: preprint (not peer‐reviewed) Source: medRxiv preprint Author COI: study authors declare no COI present; 1 affiliated to Shenzhen Bioeasy Biotechnology Co. Ltd. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Dust 2020.
| Study characteristics | |||
| Patient Sampling | Design unclear; coded as two group study:
[1] SARS‐CoV‐2 positive samples submitted for routine viral diagnostic testing (n=20 evaluated with Xpert Xpress)
[2] samples positive for other respiratory infection from those submitted for routine viral diagnostic testing (n=18)
(Sampled from total n of 177; 65 SARS‐CoV‐2 positive, 112 SARS‐CoV‐2 negative, including 57 positive for other respiratory viruses)
[Study also reports results for reference panel of simulated specimens; not extracted for this review) Recruitment: Convenience Prospective or retrospective: Retrospective |
||
| Patient characteristics and setting | Setting: Unclear; submitted to laboratory Location: Cadham Provincial Laboratory (CPL), Manitoba Country: Canada Dates: Not stated Symptoms and severity: Not reported Demographics: Not reported Exposure history: Not reported |
||
| Index tests | Test name: Xpert Xpress (no product code)
[also evaluates cobas SARS‐CoV‐2 RT‐PCR (Roche) and three in‐house RT‐PCR assays; not eligible for this review] Manufacturer: Cepheid Inc Antibody: E, N2 Antigen target: n/a Test method: automated RT‐PCR Samples used: NP swabs in VTM; collection not reported Transport media: VTM; no further detail Sample storage: Not stated, could be archived samples Test operator: Not stated Definition of test positivity: Not stated; presume as per manufacturer (presumptive positives not mentioned) Blinding reported: Not stated Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Reference standard: In‐house RT‐PCR (extraction with MagMAX™ reagents on a KingFisher™ instrument (Thermo Scientific™) and RT‐PCR performed on a Bio‐Rad CFX96 real time PCR detection system); Ct threshold NR Definition of non‐COVID cases: As for cases; single negative Genetic target(s): E, N1 Samples used: NP (as for index) Timing of reference standard: Not stated Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous (same swab) All patients received same reference standard: Yes Missing data: None reported, no participant flow diagram reported Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Not stated |
||
| Comparative | |||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | No | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Fenollar 2020(a).
| Study characteristics | |||
| Patient Sampling | Two cohorts of patients presenting for COVID‐19 testing at the same institution. This extraction relates to:
[1] Single group study to estimate sensitivity alone: symptomatic patients, all PCR positive (n=182)
Fenollar 2020(b) reports data for [2] Single group study to estimate both sensitivity and specificity: asymptomatic contacts of confirmed cases (n=159) Recruitment: Prospective Prospective or retrospective: Unclear |
||
| Patient characteristics and setting | Setting: Unclear; COVID‐19 testing Location: Institut Hospitalo‐universitaire Méditerranée Infection, Marseille, Country: France Dates: Sep 21 to Oct 2 2020 Symptoms and severity: Not stated; all symptomatic Ct values for 154 pts: Ct <=20: 58, 38%; Ct 21‐25: 49, 32%; Ct 26‐30: 39, 25%; Ct 31‐34: 8, 5% Demographics: Not reported Exposure history: [1] Not stated |
||
| Index tests | Test name: Panbio COVID‐19 Ag Manufacturer: Abbott Antibody: NP Antigen target: Not stated Test method: Not stated Samples used: NP Transport media: Not stated; appears to be direct testing Sample storage: Tested within 1 hour Test operator: Not stated Definition of test positivity: Visual line; as per manufacturer Blinding reported: Not stated, but presume yes as conducted within 1h of collection Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Reference standard: Automated RT‐PCR; VitaPCR (Credo diagnostics, Singapore) Definition of non‐COVID cases: n/a Genetic target(s): Not stated Samples used: NP (paired, from opposite nostril) Timing of reference standard: Not stated Blinded to index test: Unclear Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous; paired swabs All patients received same reference standard: Yes Missing data: None reported Uninterpretable results: None reported, no participant flow diagram reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: Supported by the Méditerranée‐Infection Foundation and the French Agence Nationale de la Recherche under reference Investissements d’Avenir Méditerranée Infection 10‐IAHU‐03 and Région Provence‐Alpes‐Côte d’Azur and European funding FEDER IHUBIOTK. Source: Accepted manuscript Author COI: Pr Raoult and Pr Drancourt are co‐founders of the Pocrame startup that develops diagnostic devices for infectious diseases |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Fenollar 2020(b).
| Study characteristics | |||
| Patient Sampling | Two cohorts of patients presenting for COVID‐19 testing at the same institution. This extraction relates to:
[2] Single group study to estimate both sensitivity and specificity: asymptomatic contacts of confirmed cases (n=159)
See Fenollar 2020(a) for extraction of additional cohort:
[1] Single group study to estimate sensitivity alone: symptomatic patients, all PCR positive (n=182) Recruitment: Prospective Prospective or retrospective: Unclear |
||
| Patient characteristics and setting | Setting: Unclear Location: Institut Hospitalo‐universitaire Méditerranée Infection, Marseille, Country: France Dates: Sep 21 to Oct 2 2020 Symptoms and severity: All asymptomatic; 21/22 cases had Ct >25 Demographics: Not reported Exposure history: [2] All described as contacts |
||
| Index tests | Test name: PANBIO COVID‐19 Ag Manufacturer: Abbott Antibody: NP Antigen target: Not stated Test method: Not stated Samples used: NP Transport media: Not stated; appears to be direct testing Sample storage: Tested within 1 hour Test operator: Not stated Definition of test positivity: Visual line; as per manufacturer Blinding reported: Not stated, conducted first Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Reference standard: Automated RT‐PCR; VitaPCR (Credo diagnostics, Singapore) Definition of non‐COVID cases: As for cases; single negative Genetic target(s): Not stated Samples used: NP (paired, from opposite nostril) Timing of reference standard: Not stated Blinded to index test: Unclear Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous; paired swabs All patients received same reference standard: Yes Missing data: None reported, no participant flow diagram reported Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: Supported by the Méditerranée‐Infection Foundation and the French Agence Nationale de la Recherche under reference Investissements d’Avenir Méditerranée Infection 10‐IAHU‐03 and Région Provence‐Alpes‐Côte d’Azur and European funding FEDER IHUBIOTK. Source: Accepted manuscript Author COI: Pr Raoult and Pr Drancourt are co‐founders of the Pocrame startup that develops diagnostic devices for infectious diseases |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
FIND 2020a.
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity:
‐ patients with symptoms consistent with COVID‐19 (meeting national definition for testing) presenting at a community testing clinic Recruitment: Consecutive recruitment Prospective or retrospective: Prospective |
||
| Patient characteristics and setting | Setting: Community (COVID‐19 testing clinic) Location: Institution not described; Marica, Rio de Janeiro Country: Brazil Dates: 30 Jul to 21 Aug 2020 Symptoms and severity: All symptomatic; no further details Demographics: mean age 40y (range 4 to 84); reported for 396 participants 181 (45%) male Exposure history: Not stated |
||
| Index tests | Test name: NowCheck COVID‐19 Ag test (RG1901DG) Manufacturer: Bionote Inc Antibody: SARS‐CoV‐2 nucleocapsid antigen Antigen target: Mouse monoclonal SARS‐CoV‐2 antibodies Test method: Rapid chromatographic immunoassay in lateral flow format Samples used: Proprietary NP swab collected by HCW Transport media: No transport media. Sample is immediately transferred to proprietary tube containing extraction buffer. Sample storage: Test should be performed as soon as possible after collection. Specimens may be stored at RT for 1h or 2‐8°C for 4h. Test operator: HCW Definition of test positivity: Presence of visible control and test lines Blinding reported: Yes Timing of samples: median 4 days p.s.o (IQR 3, 6 days); day <0 to 3 152, 39% day 4 to 7 180, 46% day >=8 58, 15% |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR (in‐house assay based on the US CDC protocol); Ct threshold of 37 Definition of non‐COVID cases: Same as for cases. Single negative PCR required for absence of infection Genetic target(s): N1, N2 Samples used: NP swabs Timing of reference standard: Same timing as per NP swabs for index test Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: 0 to several days based on PCR turnaround times at the lab All patients received same reference standard: Yes Missing data: Reports 0 invalid results Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: FIND Publication status: published Source: FIND website/IFU index test Author COI: None stated (these are independent evaluations) |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Low risk | ||
FIND 2020b.
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity at single site:
‐ patients seeking COVID‐19 testing at main testing centre; described as presenting either with symptoms compatible with a SARS‐CoV‐2 infection, or with a known positive contact or asymptomatic HCWs (n=535) Recruitment: Consecutive recruitment Prospective or retrospective: Prospective |
||
| Patient characteristics and setting | Setting: Community (main testing centre) Location: Hopitaux Universitaires de Geneve (HUG), Geneva Country: Switzerland Dates: 9‐16 Oct 2020 Symptoms and severity: 534/535 symptomatic (99%) Demographics: Mean age 38.5y (16 to 85y) 247, 46% male Exposure history: Not stated |
||
| Index tests | Test name: PanbioTM Covid‐19 Ag Rapid Test (41FK10) Manufacturer: Abbott Antibody: Not reported Antigen target: Not reported Test method: CGIA (from product insert) Samples used: NP Transport media: No transport media; assay buffer used Sample storage: Author contact advises tested as soon as possible and within the time limit specified in the IFU Test operator: HCW Definition of test positivity: Presence of visible control and test lines Blinding reported: Yes Timing of samples: time pso recorded for 115/124, 92%. Day 0‐3 89, 78%; Day 4‐7 23, 20%; Day 8+ 3, 3% |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR Roche Cobas; Ct threshold <40 (from Figure) Definition of non‐COVID cases: Same as for cases. Single negative PCR required for absence of infection Genetic target(s): Not stated Samples used: NP swab (paired, from contralateral nostril) Timing of reference standard: Not stated; author contact advises only paired swabs used. Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Paired swabs; 0 to several days based on PCR turnaround times at the lab All patients received same reference standard: Yes Missing data: Reports 0 invalid. Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: FIND Publication status: published Source: FIND/HUG website/IFU index test Author COI: None stated (these are independent evaluations) |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Low risk | ||
FIND 2020c (BR).
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity at three sites; this extraction is for data from Brazil (see FIND 2020c (CH) and Kruger 2020(c) for extraction of data from other sites):
‐ ambulatory patients meeting national suspect definition for COVID‐19 testing presenting at a community testing clinic in Brazil Recruitment: Consecutive recruitment Prospective or retrospective: Prospective |
||
| Patient characteristics and setting | Setting: Community testing clinic Location: Macae, state of Rio de Janeiro Country: Brazil Dates: 13‐30 Jul 2020 Symptoms and severity: 392/397 (99%) symptomatic; no further details Demographics: mean age 37y (2‐94) (397 participants); 229/398 male (57%) Exposure history: Not stated |
||
| Index tests | Test name: STANDARD Q COVID‐19 Ag (09COV30D) Manufacturer: SD Biosensor Inc Antibody: Not reported Antigen target: Not reported Test method: Rapid chromatographic immunoassay in lateral flow format Samples used: NP; collected by HCW Transport media: Proprietary swab/media provided by SD Biosensor Sample storage: Author contact advises tested as soon as possible and within the time limit specified in the IFU Test operator: HCW Definition of test positivity: Presence of visible control and test lines Blinding reported: Yes Timing of samples: median 5 days p.s.o (IQR 4, 6 days) (for 397 patients); day <0 to 3 85, 21%; day 4 to 7 273, 69%; day >=8 39, 10% |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR (In‐house; Lab‐developed assay based on the US CDC protocol; Ct threshold not stated; author contact advises Ct thresholds as per assay IFUs Definition of non‐COVID cases: Same as for cases. Single negative PCR required for absence of infection Genetic target(s): N1 and N2 Samples used: NP swabs Timing of reference standard: Not stated; author contact advises only paired swabs used. Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Paired swabs; 0 to several days based on PCR turnaround times at the lab All patients received same reference standard: Yes Missing data: Reports 0 missing data Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: FIND Publication status: published Source: FIND website/IFU index test Author COI: None stated (these are independent evaluations) |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Low risk | ||
FIND 2020c (CH).
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity at single site; this extraction is for data from Switzerland (see FIND 2020c (BR) and Kruger 2020(c) for extraction of data from other sites):
‐ patients seeking COVID‐19 testing at main testing centre; described as presenting either with symptoms compatible with a SARS‐CoV2 infection, or with a known positive contact or asymptomatic HCWs (n=529; from total cohort of 1064 volunteers) Recruitment: Consecutive recruitment Prospective or retrospective: Prospective |
||
| Patient characteristics and setting | Setting: Community (main testing centre) Location: Hopitaux Universitaires de Geneve (HUG), Geneva Country: Switzerland Dates: 9‐23 Oct 2020 Symptoms and severity: Not stated; time pso recorded for 183/191, 96% 141/183 COVID positive cases had symptoms for 0‐4days (77%) Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: STANDARD Q COVID‐19 Ag (09COV30D) Manufacturer: SD Biosensor Inc Antibody: Not reported Antigen target: Not reported Test method: Rapid chromatographic immunoassay in lateral flow format Samples used: NP Transport media: Proprietary swab/media provided by SD Biosensor Sample storage: Author contact advises tested as soon as possible and within the time limit specified in the IFU Test operator: HCW Definition of test positivity: Presence of visible control and test lines Blinding reported: Yes Timing of samples: median not reported (range 0 to 15); day <0 to 3 ‐ 122, 67%; day 4‐7 ‐ 54, 29%; Day 8+ ‐ 7, 34% |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR Roche Cobas; Ct threshold <40 (from Figure) Definition of non‐COVID cases: Same as for cases. Single negative PCR required for absence of infection Genetic target(s): Not stated Samples used: NP swab (paired, from contralateral nostril) Timing of reference standard: Not stated; author contact advises only paired swabs used. Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Paired swabs; 0 to several days based on PCR turnaround times at the lab All patients received same reference standard: Yes Missing data: Reports 0 missing data Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: FIND Publication status: published Source: FIND & HUG websites/IFU index test Author COI: None stated (these are independent evaluations) |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Low risk | ||
FIND 2020d (BR).
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity at two sites; this extraction is for data from Brazil (see FIND 2020d (DE) for extraction of data from other site):
‐ adults in community meeting national suspect definition for COVID‐19 testing presenting at [1] a community testing clinic or
[2] a tertiary level hospital Recruitment: Consecutive recruitment Prospective or retrospective: Prospective |
||
| Patient characteristics and setting | Setting: Mixed; community testing clinic and tertiary hospital Location: [1] Macae, state of Rio de Janeiro, [2] Universidade Federal do Rio de Janeiro (UFRJ) Country: Brazil Dates: [1] 17 Aug to 9 Sept, [2] 11 Jul to 8 Aug Symptoms and severity: 421/450 (94%) symptomatic; no further details Demographics: mean age 39 y (0‐95y) (451 participants); 185 male (41%) Exposure history: Not stated |
||
| Index tests | Test name: STANDARD F COVID‐19 Ag FIA (F‐NCOV‐01G, 10COV30D) Manufacturer: SD Biosensor Inc Antibody: Not reported Antigen target: Not reported Test method: FIA Samples used: NP; collected by HCW Transport media: Proprietary swab/media provided by SD Biosensor Sample storage: Author contact advises tested as soon as possible and within the time limit specified in the IFU Test operator: HCW Definition of test positivity: As per STANDARD F Analyzer; cut‐off index (COI) ≥ 1.0 (as per IFU) Blinding reported: Yes Timing of samples: median 4 days p.s.o (IQR 3, 6 days) (for 421 patients). Day <0 to 3 ‐ 131, 31%; day 4 to 7 ‐ 248, 59%; day >=8 ‐ 42, 10% |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; one of two in‐house assays:
1. Lab‐developed assay based on the US CDC protocol;
2. Lab‐developed assay based on the Charité Universitätsmedizin Berlin protocol.
Ct thresholds not stated; author contact advises Ct thresholds as per assay IFUs Definition of non‐COVID cases: Same as for cases. Single negative PCR required for absence of infection Genetic target(s): 1. N1 and N2; 2. E and RdRp Samples used: NP swabs Timing of reference standard: Not stated; author contact advises only paired swabs used. Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Paired swabs; 0 to several days based on PCR turnaround times at the lab All patients received same reference standard: Yes Missing data: Reports 0 missing data Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: FIND Publication status: published Source: FIND website/IFU for index test Author COI: None stated (these are independent evaluations) |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Low risk | ||
FIND 2020d (DE).
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity at two sites; this extraction is for data from Germany (see FIND 2020d (BR) for extraction of data from other site):
‐ adults in community meeting national suspect definition for COVID‐19 testing presenting at
[1] a drive‐in testing centre or
[2] ambulatory testing clinic Recruitment: Consecutive recruitment Prospective or retrospective: Prospective |
||
| Patient characteristics and setting | Setting: Community Location: [1] Heidelberg drive in testing, [2] Berlin: Ambulatory testing clinic of Charité – University Hospital Country: Germany Dates: [1] Heidelberg: 15 June‐18July 2020, [2] Berlin: 6 July – 23 Sept 2020 Symptoms and severity: 517/669 (77%) symptomatic; no further details Demographics: mean age 38 y (18‐85y) (676 participants); 307 male (46%) Exposure history: Not stated |
||
| Index tests | Test name: STANDARD F COVID‐19 Ag FIA (F‐NCOV‐01G, 10COV30D) Manufacturer: SD Biosensor Inc Antibody: Not reported Antigen target: Not reported Test method: FIA Samples used: [1] NP; [2] Combined NOP swabs; collected by HCW Transport media: Proprietary swab/media provided by SD Biosensor Sample storage: Author contact advises tested as soon as possible and within the time limit specified in the IFU Test operator: HCW Definition of test positivity: As per STANDARD F Analyzer; cut‐off index (COI) ≥ 1.0 (as per IFU) Blinding reported: Yes Timing of samples: median 3 days p.s.o (IQR 2,5 days) (for 505 patients). Day <0 to 3 ‐ 257, 51%; day 4 to 7 ‐ 202, 47%; day >=8 ‐ 46, 9% |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; one of 5 assays:
1. Cobas SARS‐CoV‐2 (Roche Diagnostics Inc); N = 342
2. Abbott RealTime SARS‐CoV‐2 (Abbott Molecular, Inc) N = 1
3. Allplex 2019‐nCov Assay (Seegene Inc); N = 20
4. LightMix® Modular SARS‐CoV (COVID19) E‐gene (Tib Molbiol); N = 233
5. Cobas (Roche) or Thermofisher (Multiplex TaqPath COVID‐19 CE‐IVD RT‐PCR Kit); N = 80
Ct thresholds not stated; author contact advises Ct thresholds as per assay IFUs Definition of non‐COVID cases: Same as for cases. Single negative PCR required for absence of infection Genetic target(s): Not stated apart from 3. E gene Samples used: NP (n=305), NOP (n=342) and/or OP swabs (n=32) Timing of reference standard: Not stated; author contact advises only paired swabs used. Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Paired swabs; 0 to several days based on PCR turnaround times at the lab All patients received same reference standard: Yes Missing data: Reports 0 missing data Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: FIND Publication status: published Source: FIND website/IFU for index test Author COI: None stated (these are independent evaluations) |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Low risk | ||
FIND 2020e (BR).
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity; this extraction is for data from Brazil (see FIND 2020e (DE) for extraction of data from other site):
‐ adults in community meeting national suspect definition for COVID‐19 testing presenting at a community testing clinic (n=476) Recruitment: Consecutive recruitment Prospective or retrospective: Prospective |
||
| Patient characteristics and setting | Setting: Community testing clinic Location: Marica, state of Rio de Janeiro Country: Brazil Dates: 27 Jul to 16 Sep Symptoms and severity: 470/476 (99%) symptomatic; no further details Demographics: mean age 45 y (0‐106 y) (473 participants); 252 male (53%) Exposure history: Not stated |
||
| Index tests | Test name: BIOCREDIT COVID‐19 Ag (G61RHA20) Manufacturer: RapiGEN Inc Antibody: Not reported Antigen target: Not reported Test method: LFA (CGIA, from IFU) Samples used: NP; collected by HCW Transport media: Assay diluent provided by manufacturer Sample storage: Author contact advises tested as soon as possible and within the time limit specified in the IFU Test operator: HCW Definition of test positivity: Visual appearance of test and control lines Blinding reported: Yes Timing of samples: median 5 days p.s.o (IQR 4, 7 days) (for 470 patients). Day <0 to 3 ‐ 95, 20%; day 4 to 7 ‐ 296, 63%; day >=8 ‐ 79, 17% |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; Lab‐developed assay based on the US CDC protocol.
Ct threshold not stated; author contact advises Ct thresholds as per assay IFUs Definition of non‐COVID cases: Same as for cases. Single negative PCR required for absence of infection Genetic target(s): N1 and N2 Samples used: NP swabs Timing of reference standard: Not stated; author contact advises only paired swabs used. Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Paired swabs; 0 to several days based on PCR turnaround times at the lab All patients received same reference standard: Yes Missing data: Reports 0 missing data Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: FIND Publication status: published Source: FIND website/IFU for index test Author COI: None stated (these are independent evaluations) |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Low risk | ||
FIND 2020e (DE).
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity at two sites; this extraction is for data from Germany (see FIND 2020e (BR) for extraction of data from other site):
‐ adults in community meeting national suspect definition for COVID‐19 testing presenting at
[1] a drive‐in testing centre or
[2] ambulatory testing clinic Recruitment: Consecutive recruitment Prospective or retrospective: Prospective |
||
| Patient characteristics and setting | Setting: Community Location: [1] Heidelberg drive in testing; [2] Berlin: Ambulatory testing clinic of Charité – University Hospital Country: Germany Dates: [1] Heidelberg: 4 May ‐ 3 Sept; [2] Berlin: 4 May ‐ 18 Aug Symptoms and severity: 733/1223 symptomatic; no further details Demographics: mean age 39.5 y (17,59.2 y) (1239 participants); 607 male (50%) Exposure history: Not stated |
||
| Index tests | Test name: BIOCREDIT COVID‐19 Ag (G61RHA20) Manufacturer: RapiGEN Inc Antibody: Not reported Antigen target: Not reported Test method: LFA (CGIA, from IFU) Samples used: [1] NP; [2] NOP; collected by HCW Transport media: Assay diluent provided by manufacturer Sample storage: Author contact advises tested as soon as possible and within the time limit specified in the IFU Test operator: HCW Definition of test positivity: Visual appearance of test and control lines Blinding reported: Yes Timing of samples: median 3 days p.s.o (IQR 2,4days) (for 701 patients). Day <0 to 3 ‐ 472, 67%; day 4 to 7 ‐ 161, 23%; day >=8 ‐ 68, 10% |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; one of 5 assays:
1. Cobas SARS‐CoV‐2 (Roche Diagnostics Inc); N = 344
2. Abbott RealTime SARS‐CoV‐2 (Abbott Molecular, Inc) N = 114
3. Allplex 2019‐nCov Assay (Seegene Inc); N = 571
4. LightMix® Modular SARS‐CoV (COVID19) E‐gene (Tib Molbiol); N = 132
5. RealStar® SARS‐CoV‐2 RT‐PCR Kit (Altona Diagnostics); N = 80
Ct thresholds not stated; author contact advises Ct thresholds as per assay IFUs Definition of non‐COVID cases: Same as for cases. Single negative PCR required for absence of infection Genetic target(s): Not stated Samples used: NP swabs Timing of reference standard: Not stated; author contact advises only paired swabs used. Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Paired swabs; 0 to several days based on PCR turnaround times at the lab All patients received same reference standard: Yes Missing data: Reports 0 missing data Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: FIND Publication status: published Source: FIND website/IFU for index test Author COI: None stated (these are independent evaluations) |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Low risk | ||
Fourati 2020 [A].
| Study characteristics | |||
| Patient Sampling | Two group study to estimate sensitivity and specificity:
(1) residual samples from subjects with positive SARS‐CoV‐2 PCR tested when they presented symptoms at the time of the first epidemic wave (n=297)
(2) pre‐pandemic samples (n=337) Recruitment: Random (stratified by Ct and time pso) Prospective or retrospective: Retrospective |
||
| Patient characteristics and setting | Setting: Mixed; likely outpatient and in‐patient "consulted or were admitted" Location: Henri Hospital Mondor de Créteil Country: France Dates: March 9 to April 9, 2020. Symptoms and severity: Not stated; all apparently symptomatic Data by viral load reported for 293/297 cases: <=20 Ct ‐ 39, 13%; 20 to 25 Ct ‐ 88, 30%; 25 to 30 Ct ‐ 72, 25%; >30 Ct ‐ 88, 30% Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Comparative study of six Ag tests (no product codes reported); Fourati 2020 [A] data relate to test [A], see additional entries for tests [B] to [E] [A] SARS‐CoV‐2 COVID‐19 Respi‐Strip [B] Standard Q COVID‐19 Ag [C] PanBio COVID‐19 Antigen Rapid Test [D] Biosynex COVID‐19 Ag BSS [E] COVID‐VIRO Antigen Rapid Test [F] NG Test SARS‐CoV‐2 Ag (assay excluded from review due to Vortex requirement as stated in IFU) (no product codes reported) Manufacturer: [A] Coris BioConcept, Gembloux, Belgium [B] SD BIOSENSOR, Inc., Korea [C] Abbott, Chicago, Illinois, USA [D] Biosynex, Strasbourg, France [E] AAZ, Boulogne‐Billancourt, France [F] NG Biotech, Guipry, France Antibody: Not stated Antigen target: Not stated Test method: Not stated Samples used: NP; collection not reported Transport media: VTM (Cepheid® or Deltalab®); 100 μL used for testing Sample storage: frozen at ‐80 °C until use Test operator: Laboratory staff Definition of test positivity: Visual, as per manufacturer. Blinding reported: Yes; each test was interpreted independently by two different laboratory technicians. A third reading was carried out in the event of discrepancy Timing of samples: post‐symptom onset (reported for 289 samples): 0‐3 days 97, 34%; 4‐7 days 103, 36%; 8=11 days 63, 22%; >=12 days 26, 9% No. samples reported at >7 days varied per test, maximum was 289 |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; in‐house assay developed by CNR (Institut Paster) or RealStar SARS‐CoV‐2 (Altona Diagnostics, Germany) Definition of non‐COVID cases: Pre‐pandemic Genetic target(s): Not stated Samples used: NP; same as for index Timing of reference standard: As for index Blinded to index test: Yes, seems to be at time of sampling Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Same swab; simultaneous All patients received same reference standard: Yes Missing data: Number of cases missing per assay varied; reasons for missing data not reported (presumably invalid assay results) [A] 5, 1.7% [B] 6, 2.0% [C] 2, 0.7% [D] 0 [E] 2, 0.7% [F] 0 Uninterpretable results: Not stated Indeterminate results (index test): Not stated Indeterminate results (reference standard): Not stated Unit of analysis: Presume patients |
||
| Comparative | |||
| Notes | Funding: Evaluation of [A] and [B] conducted in collaboration with Médecins sans Frontières and Epicenter Publication status: Published Source: Laboratory report obtained via SFM Microbiologie website Author COI: No COI present |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Fourati 2020 [B].
| Study characteristics | |||
| Patient Sampling | Comparative study of six Ag tests (no product codes reported); Fourati 2020 [A] reports full study characteristics and QUADAS | ||
| Patient characteristics and setting | |||
| Index tests | Comparative study of six Ag tests (no product codes reported); Fourati 2020 [B] relates to test [B] in the list below; see Fourati 2020 [A] for full study characteristics and QUADAS entries [A] SARS‐CoV‐2 COVID‐19 Respi‐Strip [B] Standard Q COVID‐19 Ag [C] PanBio COVID‐19 Antigen Rapid Test [D] Biosynex COVID‐19 Ag BSS [E] COVID‐VIRO Antigen Rapid Test [F] NG Test SARS‐CoV‐2 Ag (assay excluded from review due to Vortex requirement as stated in IFU) (no product codes reported) Manufacturer: [A] Coris BioConcept, Gembloux, Belgium [B] SD BIOSENSOR, Inc., Korea [C] Abbott, Chicago, Illinois, USA [D] Biosynex, Strasbourg, France [E] AAZ, Boulogne‐Billancourt, France [F] NG Biotech, Guipry, France Antibody: Not stated Antigen target: Not stated Test method: Not stated Samples used: NP; collection not reported Transport media: VTM (Cepheid® or Deltalab®); 100 μL used for testing Sample storage: frozen at ‐80 °C until use Test operator: Laboratory staff Definition of test positivity: Visual, as per manufacturer. Blinding reported: Yes; each test was interpreted independently by two different laboratory technicians. A third reading was carried out in the event of discrepancy Timing of samples: post‐symptom onset (reported for 289 samples): 0‐3 days 97, 34%; 4‐7 days 103, 36%; 8=11 days 63, 22%; >=12 days 26, 9% No. samples reported at >7 days varied per test, maximum was 289 |
||
| Target condition and reference standard(s) | Comparative study of six Ag tests (no product codes reported); Fourati 2020 [A] reports full study characteristics and QUADAS | ||
| Flow and timing | Comparative study of six Ag tests (no product codes reported); Fourati 2020 [A] reports full study characteristics and QUADAS | ||
| Comparative | |||
| Notes | |||
Fourati 2020 [C].
| Study characteristics | |||
| Patient Sampling | Comparative study of six Ag tests (no product codes reported); Fourati 2020 [A] reports full study characteristics and QUADAS | ||
| Patient characteristics and setting | Comparative study of six Ag tests (no product codes reported); Fourati 2020 [A] reports full study characteristics and QUADAS | ||
| Index tests | Comparative study of six Ag tests (no product codes reported); Fourati 2020 [C] relates to test [C] in the list below; see Fourati 2020 [A] for full study characteristics and QUADAS entries [A] SARS‐CoV‐2 COVID‐19 Respi‐Strip [B] Standard Q COVID‐19 Ag [C] PanBio COVID‐19 Antigen Rapid Test [D] Biosynex COVID‐19 Ag BSS [E] COVID‐VIRO Antigen Rapid Test [F] NG Test SARS‐CoV‐2 Ag (assay excluded from review due to Vortex requirement as stated in IFU) Manufacturer: [A] Coris BioConcept, Gembloux, Belgium [B] SD BIOSENSOR, Inc., Korea [C] Abbott, Chicago, Illinois, USA [D] Biosynex, Strasbourg, France [E] AAZ, Boulogne‐Billancourt, France [F] NG Biotech, Guipry, France Antibody: Not stated Antigen target: Not stated Test method: Not stated Samples used: NP; collection not reported Transport media: VTM (Cepheid® or Deltalab®); 100 μL used for testing Sample storage: frozen at ‐80 °C until use Test operator: Laboratory staff Definition of test positivity: Visual, as per manufacturer. Blinding reported: Yes; each test was interpreted independently by two different laboratory technicians. A third reading was carried out in the event of discrepancy Timing of samples: post‐symptom onset (reported for 289 samples): 0‐3 days 97, 34%; 4‐7 days 103, 36%; 8=11 days 63, 22%; >=12 days 26, 9% No. samples reported at >7 days varied per test, maximum was 289 |
||
| Target condition and reference standard(s) | Comparative study of six Ag tests (no product codes reported); Fourati 2020 [A] reports full study characteristics and QUADAS | ||
| Flow and timing | Comparative study of six Ag tests (no product codes reported); Fourati 2020 [A] reports full study characteristics and QUADAS | ||
| Comparative | |||
| Notes | |||
Fourati 2020 [D].
| Study characteristics | |||
| Patient Sampling | Comparative study of six Ag tests (no product codes reported); Fourati 2020 [A] reports full study characteristics and QUADAS | ||
| Patient characteristics and setting | Comparative study of six Ag tests (no product codes reported); Fourati 2020 [A] reports full study characteristics and QUADAS | ||
| Index tests | Comparative study of six Ag tests (no product codes reported); Fourati 2020 [D] relates to test [D] in the list below; see Fourati 2020 [A] for full study characteristics and QUADAS entries [A] SARS‐CoV‐2 COVID‐19 Respi‐Strip [B] Standard Q COVID‐19 Ag [C] PanBio COVID‐19 Antigen Rapid Test [D] Biosynex COVID‐19 Ag BSS [E] COVID‐VIRO Antigen Rapid Test [F] NG Test SARS‐CoV‐2 Ag (assay excluded from review due to Vortex requirement as stated in IFU) Manufacturer: [A] Coris BioConcept, Gembloux, Belgium [B] SD BIOSENSOR, Inc., Korea [C] Abbott, Chicago, Illinois, USA [D] Biosynex, Strasbourg, France [E] AAZ, Boulogne‐Billancourt, France [F] NG Biotech, Guipry, France Antibody: Not stated Antigen target: Not stated Test method: Not stated Samples used: NP; collection not reported Transport media: VTM (Cepheid® or Deltalab®); 100 μL used for testing Sample storage: frozen at ‐80 °C until use Test operator: Laboratory staff Definition of test positivity: Visual, as per manufacturer. Blinding reported: Yes; each test was interpreted independently by two different laboratory technicians. A third reading was carried out in the event of discrepancy Timing of samples: post‐symptom onset (reported for 289 samples): 0‐3 days 97, 34%; 4‐7 days 103, 36%; 8=11 days 63, 22%; >=12 days 26, 9% No. samples reported at >7 days varied per test, maximum was 289 |
||
| Target condition and reference standard(s) | Comparative study of six Ag tests (no product codes reported); Fourati 2020 [A] reports full study characteristics and QUADAS | ||
| Flow and timing | Comparative study of six Ag tests (no product codes reported); Fourati 2020 [A] reports full study characteristics and QUADAS | ||
| Comparative | |||
| Notes | |||
Fourati 2020 [E].
| Study characteristics | |||
| Patient Sampling | Comparative study of six Ag tests (no product codes reported); Fourati 2020 [A] reports full study characteristics and QUADAS | ||
| Patient characteristics and setting | Comparative study of six Ag tests (no product codes reported); Fourati 2020 [A] reports full study characteristics and QUADAS | ||
| Index tests | Comparative study of six Ag tests (no product codes reported); Fourati 2020 [E] relates to test [E] in the list below; see Fourati 2020 [A] for full study characteristics and QUADAS entries [A] SARS‐CoV‐2 COVID‐19 Respi‐Strip [B] Standard Q COVID‐19 Ag [C] PanBio COVID‐19 Antigen Rapid Test [D] Biosynex COVID‐19 Ag BSS [E] COVID‐VIRO Antigen Rapid Test [F] NG Test SARS‐CoV‐2 Ag (assay excluded from review due to Vortex requirement as stated in IFU) Manufacturer: [A] Coris BioConcept, Gembloux, Belgium [B] SD BIOSENSOR, Inc., Korea [C] Abbott, Chicago, Illinois, USA [D] Biosynex, Strasbourg, France [E] AAZ, Boulogne‐Billancourt, France [F] NG Biotech, Guipry, France Antibody: Not stated Antigen target: Not stated Test method: Not stated Samples used: NP; collection not reported Transport media: VTM (Cepheid® or Deltalab®); 100 μL used for testing Sample storage: frozen at ‐80 °C until use Test operator: Laboratory staff Definition of test positivity: Visual, as per manufacturer. Blinding reported: Yes; each test was interpreted independently by two different laboratory technicians. A third reading was carried out in the event of discrepancy Timing of samples: post‐symptom onset (reported for 289 samples): 0‐3 days 97, 34%; 4‐7 days 103, 36%; 8=11 days 63, 22%; >=12 days 26, 9% No. samples reported at >7 days varied per test, maximum was 289 |
||
| Target condition and reference standard(s) | Comparative study of six Ag tests (no product codes reported); Fourati 2020 [A] reports full study characteristics and QUADAS | ||
| Flow and timing | Comparative study of six Ag tests (no product codes reported); Fourati 2020 [A] reports full study characteristics and QUADAS | ||
| Comparative | |||
| Notes | |||
Ghofrani 2020.
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity in patients with both RT‐PCR and POCT results available (n=113), including:
[1] symptomatic patients with a PCR swab test close to presentation and a re‐swab for POC testing,
[2] patients with positive RT‐PCR results and remnant NP swabs available for POC test,
[3] asymptomatic patients with positive POC result on admission who were re‐swabbed for RT‐PCR confirmation.
N per group was not reported Recruitment: Convenience Prospective or retrospective: Retrospective |
||
| Patient characteristics and setting | Setting: Unclear; primarily in‐patients? Location: PeaceHealth Medical Group (10 hospitals and numerous clinics serving suburban and rural communities in three states) Country: USA Dates: April 6‐ April 21 2020 Symptoms and severity: Majority' symptomatic, no further details. Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: ID NOW COVID‐19 assay (no product code reported) Manufacturer: Abbott Laboratories Target gene(s): RdRp region Antigen target: n/a Test method: Isothermal PCR Samples used: Nasal 58 (51.3%), NP 33 (29.2%), not stated 22 (19.5%). Direct testing 58 (51.3%), UTM 26 (23.0%); not stated 29 (25.7%). Transport media: None or UTM; no further details Sample storage: Not stated Test operator: Not stated; infer laboratory staff. Definition of test positivity: Not stated; presume as per manufacturer Blinding reported: Not stated Timing of samples: Not stated; implies mostly close to presentation |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; not described (conducted at one of two commercial laboratories, one of two State Public Health laboratories, an academic medical center, or tested in‐house) Definition of non‐COVID cases: Same as index test; infer single negative Genetic target(s): not stated Samples used: Mixed; either paired swabs (within 3 days of each other) or same samples used Timing of reference standard: Not stated Blinded to index test: unclear; probably mixed depending on where RT‐PCR was conducted Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Some same sample; paired samples could be up to 3 days apart All patients received same reference standard: Yes Missing data: None reported Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: No funding received Publication status: Published Source: Unclear Author COI: none reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | Unclear | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | No | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | No | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Gibani 2020.
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity with three sources of participants:
[1] self‐referred, health‐care workers or their family members with suspected COVID‐19 who were not admitted to hospital (n=280)
[2] emergency department patients with suspected COVID‐19 (n=15)
[3] hospital inpatient admissions with or without suspected COVID‐19 (n=91)
Total N was 418 paired samples; 32 excluded as invalid (patient group not reported), 24 invalid on DnaNudge and 8 on RT‐PCR) Recruitment: [1] and [2] Not reported; [3] consecutive Prospective or retrospective: Prospective |
||
| Patient characteristics and setting | Setting: Mixed ([1] community, [2] A&E, [3] Inpatient) Location: [1] St Mary’s Hospital and the John Radcliffe Hospital, [2] St Mary’s Hospital, [3] Chelsea & Westminster Hospital Country: London or Oxford, UK Dates: [1] April 10 to May 12, [2] April 2 to 24, [3] May 12 to 18 Symptoms and severity: Only group [3] were inpatient Demographics: median age 46 y (IQR 31–66); 124, 32% male Exposure history: Not reported |
||
| Index tests | Test name: CovidNudge (no product code) Manufacturer: DnaNudge, UK Antibody: rdrp1, rdrp2, e‐gene, n‐gene, n1, n2, and n3 Antigen target: n/a Test method: Automated RT‐PCR; Described as "integrated lab‐on‐chip device that enables sample‐to‐result (RT‐)PCR" Samples used: NP; HCW obtained swabs using pediatric swab Transport media: None Sample storage: No delay reported Test operator: Unclear; possibly HCW Definition of test positivity: at least two replicates of at least one viral gene target amplified Blinding reported: Yes; results from CovidNudge testing reported before laboratory results were available Timing of samples: On presentation; timing not reported |
||
| Target condition and reference standard(s) | Reference standard: SARS‐CoV‐2 RT‐PCR; assay varied by site.
A. AusDiagnostics MT‐PCR (Orf1ab, Orf8); n=74
b. Roche RT‐PCR (Orf1ab, E); N=81
c. Abbott RT‐PCR (RdRp, N); n=66
d. ThermoFisher (orf1ab, the spike (S) gene and the nucleocapsid (N) gene); n=21
e. PHE in‐house RT‐PCR (RdRp); n=120
f. Imperial Molecular Diagnostics Unit (E); n=24 Definition of non‐COVID cases: As above (single negative) Genetic target(s): See above Samples used: NOP (paired) Timing of reference standard: Not stated Blinded to index test: Yes; centralised laboratory testing and point‐of‐care testing were done by separate staff members. Staff doing the centralised laboratory testing were masked to the point of‐ care test results and vice‐versa Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous (paired) All patients received same reference standard: Yes (different assays) Missing data: Additional 47 samples not 'paired'; not collected on same date Uninterpretable results: 32 samples excluded; 24 invalid on DNANudge (failed to amplify RNaseP; 22/24 with associated RT‐PCR result were negative) and 8 on RT‐PCR (all 8 from one site) Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: Supported by the National Institute for Health Research (NIHR) Imperial NHS Trust Biomedical Research Centre (London, UK). Part of this work was supported by the NIHR Health Protection Research Unit in Healthcare Associated Infections and Antimicrobial Resistance at Oxford University (Oxford, UK) in partnership with Public Health England (grant HPRU‐2012‐10041). DnaNudge supplied the test cartridges and NudgeBox processing units. Publication status: Published Source: Lancet Microbe Author COI: CT, RS, MS, MK, T‐KH, SDM, K‐YFL, JB, and AO are employees of DnaNudge. CT is the co‐inventor of the DnaNudge CovidNudge system and is named on the patent for the method and apparatus for analysing biological specimens on the DnaNudge platform (US Patent No: US 10 093 965.B2).16 LSPM has consulted for bioMerieux (2013–20), DNAelectronics (2015), Dairy Crest (2017–18), Pfizer (2018–20), and Umovis Lab (2020), received speaker fees from Profile Pharma (2018), received research grants from the UK National Institute for Health Research (NIHR; 2013–2019), Leo Pharma (2016), and CW+ Charity (2018–19), and received educational support from Eumedica (2016–17). NM has received speaker fees from Beyer (2016) and Pfizer (2019), and received educational support from Eumedica (2016) and Baxter (2017). MMG and GC are partly supported by the NIHR Imperial Biomedical Research Centre. GC is an NIHR research professor and investigator within the NIHR London in‐vitro diagnostic co‐operative. All other authors declare no competing interests. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Goldenberger 2020.
| Study characteristics | |||
| Patient Sampling | Design unclear but appears to be a two group study to estimate sensitivity and specificity:
[1] SARS‐CoV‐2 positive samples selected to reflect a broad range of Ct values (n=10)
[2] SARS‐CoV‐2 negative samples (n=9)
Groups [1] and [2] from patients suspected of COVID‐19 undergoing routine diagnostics within a one week period
[third cohort of pre‐pandemic samples positive for other coronaviruses reported but not included in review (n=8)] Recruitment: Convenience Prospective or retrospective: Unclear |
||
| Patient characteristics and setting | Setting: Unclear Location: University Hospital Basel Country: Switzerland Dates: One week during 2020 pandemic Symptoms and severity: Not reported Demographics: Not reported Exposure history: Not reported |
||
| Index tests | Test name: Xpert Xpress (no product code) Manufacturer: Cepheid Inc Antibody: E, N2 Antigen target: n/a Test method: Automated RT‐PCR Samples used: NP Transport media: UTM or eSwab media (Copan) Sample storage: frozen at −80 °C until batch‐wise sample processing with the Xpert Test operator: laboratory technician Definition of test positivity: Not stated; both targets reported in all samples Blinding reported: Unclear Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Reference standard: Roche cobas RT‐PCR; threshold not reported but all positive samples <33 Ct Definition of non‐COVID cases: [2] COVID‐19 suspects; as for cases (single negative PCR) Genetic target(s): E, ORF1 Samples used: NP (same as index) Timing of reference standard: Not stated Blinded to index test: Yes, conducted first Incorporated index test: Not stated |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous (same swab) All patients received same reference standard: Yes Missing data: None reported, no participant flow diagram reported Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Unclear |
||
| Comparative | |||
| Notes | Funding: None reported Publication status: Published Source: Journal of Virological Methods Author COI: None reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Gremmels 2020(a).
| Study characteristics | |||
| Patient Sampling | Report of two cohorts of patients presenting for COVID‐19 testing. Gremmels 2020(a) entry relates to:
[1] community‐dwelling mildly symptomatic subjects in a medium endemic area (n=1369) Gremmels 2020(b) entry reports data for second cohort in a high endemic area Recruitment: Yes; all individuals invited to participate Prospective or retrospective: Prospective |
||
| Patient characteristics and setting | Setting: Community testing centre Location: [1] University Medical Center Utrecht (UMCU) Country: Netherlands Dates: [1] Sep 22 to Oct 6 Symptoms and severity: Cohort [1] only. Data on symptoms were missing from nine subjects Asymptomatic 37, 2.7%, Sore throat 907, 66.3%; Coryza 943, 69%; Cough 780, 57.1%; Headache 601, 44.0%; Tiredness 565, 41.3%; General malaise 365, 26.7% (further 19 documented) Demographics: median age 36.4y (IQR 27.0, 49.6y); 523, 38.3% male Exposure history: 233, 17% contact with confirmed case |
||
| Index tests | Test name: Panbio™ COVID‐19 Ag Rapid Test (lot 41ADF011A) Manufacturer: Abbott (Lake Country, IL, U.S.A) Antibody: NP Antigen target: Not stated Test method: Not stated Samples used: NP; obtained after NOP swab for RT‐PCR; implies collected by HCW Transport media: Unclear; states transferred to 3 ml UTM after collection until further processing but also describes collected swabs transferred into dedicated sample collection tubes containing a sampling buffer for Ag test Sample storage: Not stated; within 2 hours of collection Test operator: Two independent observers Definition of test positivity: Visual line within 15 mins; as per manufacturer Blinding reported: Yes; observers (blinded to each other and to the PCR results) Timing of samples: Cohort [1] (data on duration of symptoms reportedly missing for 201 subjects; total reported here is 1138 but denominator for %s is 1166) day 1‐3 pso 387, 33.2%; day 4‐7 560, 48.0%; day >7 191, 16.4% |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; Seegene Allplex
positive result on amplification of any of the three SARS‐CoV‐2 genes Definition of non‐COVID cases: As for cases; single negative result Genetic target(s): E‐, N‐, and RdRP‐gene Samples used: NOP (paired) Timing of reference standard: NOP swab obtained first for RT‐PCR Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Paired All patients received same reference standard: Yes Missing data: 2 patients excluded ('inappropriate application of NP swab and lab mislabelling'), disease status not reported. [Considered overall low risk of bias due to small numbers] Uninterpretable results: None reported Indeterminate results (index test): None; no bands were classified as unclear by the independent observers Indeterminate results (reference standard): Patients Unit of analysis: |
||
| Comparative | |||
| Notes | Funding: This study was investigator initiated. No external funding was received Publication status: Pre‐print Source: medRxiv Author COI: No COI statement reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Low risk | ||
Gremmels 2020(b).
| Study characteristics | |||
| Patient Sampling | Report of two cohorts of patients presenting for COVID‐19 testing. Gremmels 2020(b) entry relates to:
[2] community‐dwelling mildly symptomatic subjects in a high endemic area (n=208) Gremmels 2020(a) entry reports data for second cohort in a medium endemic area Recruitment: Yes; all individuals invited to participate Prospective or retrospective: Prospective |
||
| Patient characteristics and setting | Setting: Community testing centre Location: [2] Horacio Oduber Hospital on Aruba Country: Netherlands Dates: [2] Sep 23 to Oct 9 Symptoms and severity: Not stated; 'mildly symptomatic', presume mixed as per Gremmels 2020a Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: Panbio™ COVID‐19 Ag Rapid Test (lot 41ADF011A) Manufacturer: Abbott (Lake Country, IL, U.S.A) Antibody: NP Antigen target: Not stated Test method: Not stated Samples used: NP; obtained after NOP swab for RT‐PCR; implies collected by HCW Transport media: No UTM used for Ag samples; collected swabs transferred into dedicated sample collection tubes containing a sampling buffer Sample storage: Not stated; within 2 hours of collection Test operator: Two independent observers Definition of test positivity: Visual line within 15 mins; as per manufacturer Blinding reported: Yes; observers (blinded to each other and to the PCR results) Timing of samples: Not stated; on presentation |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; Seegene Allplex
positive result = amplification of any of the three SARS‐CoV‐2 genes Definition of non‐COVID cases: As for cases; single negative result Genetic target(s): E‐, N‐, and RdRP‐gene Samples used: NOP (paired) Timing of reference standard: NOP swab obtained first for RT‐PCR Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Paired All patients received same reference standard: Yes Missing data: None reported for Aruba site Uninterpretable results: None reported Indeterminate results (index test): None; no bands were classified as unclear by the independent observers Indeterminate results (reference standard): none Unit of analysis: patients |
||
| Comparative | |||
| Notes | Funding: This study was investigator initiated. No external funding was received Publication status: Pre‐print Source: medRxiv Author COI: No COI statement reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Low risk | ||
Gupta 2020.
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity:
‐ symptomatic patients with suspected COVID‐19 and asymptomatic contacts of laboratory‐confirmed cases between 5 and 10 days of exposure, meeting Indian Council of Medical Research (ICMR) strategy for COVID‐19 testing Recruitment: Consecutive Prospective or retrospective: Not stated; appears prospective |
||
| Patient characteristics and setting | Setting: Outpatient (tertiary care hospital) Location: All India Institute of Medical Sciences (AIIMS), New Delhi Country: India Dates: May 31 to July 24, 2020. Symptoms and severity: 204 (62%) symptomatic; 126 (38%) asymptomatic. median symptom duration: 1 day (range: 1‐10). Symptoms included: fever (31.5%), cough (25.4%), fatigue/malaise (11.8%), headache (3.3%), runny nose (3.3%) Demographics: median age 34.1±12.6 yr; 231 (70%) male Exposure history: 127 asymptomatic were in contact with confirmed case |
||
| Index tests | Test name: Standard Q rapid antigen detection test Manufacturer: SD Biosensor, Inc., Gurugram Antibody: Not stated Antigen target: Not stated Test method: Not stated Samples used: NP; collection method detailed but personnel not described; presume HCW. Sequence for specimen collection was random for both the samples (Ag and RT‐PCR) Transport media: None Sample storage: None Test operator: Same person who obtained swab; HCW Definition of test positivity: Visual; test and control lines Blinding reported: Yes; conducted first Timing of samples: Symptomatic: 192 (95%) <=5 days pso (incl 57 cases) |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; commercial assay (BGI Genomics Co. Ltd., China). Psoitive defined as per manufacturer IFU Definition of non‐COVID cases: As for cases; single negative Genetic target(s): ORF1 ab Samples used: nasal and throat swabs (NOP) in VTM Timing of reference standard: As for index test; states the sequence for specimen collection was random for both the samples Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous; paired swabs All patients received same reference standard: Yes Missing data: None reported, no participant flow diagram reported Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: Study was financially supported by the Indian Council of Medical Research, New Delhi (for the Regional Virus Research and Diagnostic Laboratory at the All India Institute of Medical Sciences, New Delhi). Publication status: Published Source: Indian J Med Res Author COI: Author report no COI present |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Harrington 2020.
| Study characteristics | |||
| Patient Sampling | Single‐group study to estimate sensitivity and specificity:
‐ symptomatic patients meeting diagnostic criteria for COVID‐19 (n = 524) Recruitment: consecutive Prospective or retrospective: unclear; presume prospective Number of samples (samples with confirmed SARS‐CoV‐2): 524 (186) |
||
| Patient characteristics and setting | Setting: ED (n = 3) or urgent (immediate) care centres (n = 2) Location: not stated; author institutions Loyola University Medical Centre, Cedars‐Sinai Medical Centre Country: USA Dates: not reported Symptoms and severity: not stated Demographics: not stated Exposure history: not stated |
||
| Index tests | Test name: ID NOW COVID‐19 assay (no product code provided) Manufacturer: Abbott Antigen target: not stated Antibody: N/A Test method: not stated; isothermal PCR Samples used: nasal swabs (provider collected) Transport media: none; direct testing after heat inactivation Sample storage: ED swabs transported in sterile transport containers (using cups or conical tubes) Test operator: on‐site medical personnel (urgent care centres); laboratory personnel at each separate location (EDs) ‐ 2 sites reportedly experienced users of ID NOW (one ED and one urgent care centre) and 3 sites received training) Definition of test positivity: as per manufacturer Blinding reported: yes (RT‐PCR performed at separate central lab) Timing of samples: not stated; on presentation |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR (Abbott RealTime SARS‐CoV‐2 (ACOV) assay performed on the Abbott m2000 system (Abbott Molecular Inc. Des Plaines, IL); threshold not stated Definition of non‐COVID cases: not specifically stated; presume yes as central lab used Genetic target(s): not stated Samples used: NP swabs Timing of reference standard: VTM (no detail) Blinded to index test: not stated, transferred to central clinical laboratory; samples heat inactivated for 30 min at 60 °C prior to testing Incorporated index test: no (paired collection with swabs for index test) |
||
| Flow and timing | Time interval between index and reference tests: simultaneous swab collection (different swabs for index and reference) All participants received same reference standard: yes Missing data: none reported, no participant flow diagram reported Uninterpretable results: none reported Indeterminate results (index test): none reported Indeterminate results (reference standard): 2 initial FPs had repeat sampling: ‐ 1 retested on RT‐PCR only and was positive (designated as TP) ‐ 1 retested on RT‐PCR and ID NOW and was negative on both (designated as FP based on original sampling) Unit of analysis: participants |
||
| Comparative | |||
| Notes | Funding: study authors received "received no specific grant from any funding agency in the public, commercial, or not‐for‐profit sectors" Publication status: accepted manuscript Source: Journal of Clinical Microbiology Author COI: COI not mentioned |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Hogan 2020.
| Study characteristics | |||
| Patient Sampling | Single‐group design to estimate sensitivity and specificity
‐ samples from adult patients from 1 hospital and paediatric and adult samples from surrounding hospitals Recruitment: unclear; equal numbers of positive and negative RT‐PCR samples (suspect deliberate sampling by PCR result) Prospective or retrospective: not stated Number of samples (samples with confirmed SARS‐CoV‐2): 100 (50) |
||
| Patient characteristics and setting | Setting: hospital; not stated if inpatient or outpatient (samples selected from clinical virology laboratory) Location: Stanford Health Care (hospital), and surrounding hospitals (not named) Country: USA Dates: 7‐13 April 2020 Symptoms and severity: not stated Demographics: not stated Exposure history: not stated |
||
| Index tests | Test name: Accula SARS‐CoV‐2 POCT (no product code reported) Manufacturer: Mesa Biotech, Inc., San Diego, CA Antigen target: N gene Antibody: N/A Test method: rapid PCR Samples used: NP swabs in VTM (n = 37) or saline (n = 63, including 37 positive on RT‐PCR) Transport media: not stated; 10 μL of VTM or saline was transferred to 60 μL of SARS‐CoV‐2 buffer within a biosafety cabinet (not covered by manufacturer IFU) Sample storage: not stated; testing appears to have been conducted soon after sample collection Test operator: not stated; presume laboratory staff Definition of test positivity: as per manufacturer Blinding reported: not stated Timing of samples: not stated |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; in‐house SHC assay (cites Hogan 2020 10.1016/j.jcv.2020.104383:104383) Definition of non‐COVID cases: single RT‐PCR negative Genetic target(s): E gene Samples used: NP swabs, same as for index test Timing of reference standard: not stated Blinded to index test: not stated Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: not stated but implies that both tests undertaken in laboratory soon after sample collection All participants received same reference standard: yes Missing data: none reported Uninterpretable results: 3 invalid results were re‐tested; 1 positive and 2 negative Indeterminate results (index test): 1 known RT‐PCR‐positive sample that showed a faint positive test line was re‐tested and again showed the same faint test line (considered positive) Indeterminate results (reference standard): none reported Unit of analysis: refers to participants |
||
| Comparative | |||
| Notes | Funding: study authors report no specific funding Publication status: preprint Source: medRxiv Author COI: authors declare no COI present |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Unclear | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Hou 2020.
| Study characteristics | |||
| Patient Sampling | Single group study using remnant OP swabs submitted for SARS‐CoV‐2 testing at three medical centers (n = 285) Recruitment: Not stated Prospective or retrospective: Retrospective |
||
| Patient characteristics and setting | Setting: Mixed inpatient and outpatient Location: Three sites in Wuhan: Wuhan Tongji hospital (n=99), Wuhan Pulmonary hospital (n=96); Wuhan No. 1 hospital (n=90) Country: China Dates: Feb to Apr 2020 Symptoms and severity: 178 (62.5%) inpatient; 107 (37.5%) outpatients. Site 2 were all inpatients Demographics: 220 (77.2%) aged ≤65 years; 159 (55.8%) male Exposure history: No details; all Wuhan |
||
| Index tests | Test name: Xpert Xpress (no product code reported) Manufacturer: Cepheid Inc Target gene(s): E, N2 Antigen target: N/A Test method: Automated RT‐PCR Samples used: OP Transport media: Not stated; 'aliquot made' Sample storage: stored at ‐80°C within 24 h of collection Test operator: Not stated Definition of test positivity: Not stated; presume as per manufacturer (company funded study) ‐ no mention of presumptive positive results Blinding reported: Not stated Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR assays approved by Chinese National Medical Products Administration (NMPA) for the detection of SARS‐CoV‐2 Definition of non‐COVID cases: As for cases; single negative RT‐PCR Genetic target(s): Not stated Samples used: OP (same as for rapid test) Timing of reference standard: Not stated; conducted at time of sample collection Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous (same swab); time period of frozen storage was not reported All patients received same reference standard: Yes, although could be different RT‐PCR assays at different sites Missing data: None reported, no participant flow diagram reported Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients; states 'samples from unique patients' |
||
| Comparative | |||
| Notes | Funding: funded in part by the National Mega Project on Major Infectious Disease Prevention (2017ZX10103005‐007) and by the Cepheid Investigator‐Initiated Study award (Cepheid‐IIS‐2020‐005). Publication status: Accepted manuscript Source: J Clin Microbiol Author COI: YWT is an employee of Cepheid, the commercial manufacturer of the Xpert Xpress SARS‐CoV‐2 test. The other authors declare no competing interests. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Jin 2020.
| Study characteristics | |||
| Patient Sampling | Laboratory‐based study presenting data on a total of 8043 specimens for different RT‐PCR tests (n=7251) and ID NOW (n=792). States that a significant proportion of specimens tested by ID NOW were pre‐admission screening specimens for surgical patients but does not report percentage.
Eligible data refer to
[1] single group study to estimate sensitivity and specificity in paired dry swabs and NP or OP swabs in UTM (n=52)
[Additional cases only set: [2] 124 RT‐PCR positive NP/OP samples in UTM samples included 117 'retested with ID NOW' and 7 samples diluted in UTM from 4 positive specimens (the diluted samples cannot be distinguished from the set of 117 and data have been excluded from review) Recruitment: Unclear Prospective or retrospective: Retrospective |
||
| Patient characteristics and setting | Setting: Unclear; may be predominantly screening of surgical patients Location: Molecular & Genomic Pathology Laboratory, Thomas Jefferson University Hospital, Philadelphia Country: USA Dates: April 23 to 26, 2020 Symptoms and severity: Not stated Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: ID NOW (product code not reported) Manufacturer: Abbott Laboratories Target gene(s): RdRp Antigen target: n/a Test method: Isothermal PCR Samples used: 'dry swabs' as per manufacturer EUA protocol Transport media: None Sample storage: No storage reported (appears to be immediate testing) Test operator: Not stated; laboratory staff presumed Definition of test positivity: As per manufacturer Blinding reported: Not stated 'tested in parallel' Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; cobas SARS‐CoV‐2 Test (Roche Molecular Systems, Inc., Pleasanton, CA) using a cobas 6800 analyzer (Roche Molecular Systems, Inc). Either target present considered positive Definition of non‐COVID cases: As above; single PCR negative required Genetic target(s): ORF1/a, E gene Samples used: Not specifically described for subset of paired samples, but for full cohort NP and OP swabs in VTM used (400 uL) Timing of reference standard: Not stated Blinded to index test: Not stated; tested in parallel Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous (paired swabs) All patients received same reference standard: Yes Missing data: None reported Uninterpretable results: None reported, no participant flow diagram reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Not stated; described as 'paired patient specimens' |
||
| Comparative | |||
| Notes | Funding: No funding statement reported Publication status: Published Source: Arch Path Lab Med Author COI: No COI statement reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Jokela 2020.
| Study characteristics | |||
| Patient Sampling | Two group study to estimate sensitivity and specificity including NP or OP swab samples sent to university laboratory:
[1] for SARS‐CoV‐2 testing (n=97),
[2] pre‐pandemic samples sent for testing due to suspicion of other respiratory virus infection (n=10)
Recruitment: Not stated Prospective or retrospective: Not stated; presume retrospective [Also reports results for third cohort of samples from participants attending tertiary care EDs (n=362), however index test is ineligible for this review (Novodiag))] |
||
| Patient characteristics and setting | Setting: Not reported Location: Helsinki University Hospital Laboratory (HUSLAB), Helsinki Country: Finland. Dates: Mar to May 2020 Symptoms and severity: Not stated Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: Xpert Xpress (no product code reported) Manufacturer: Cepheid Inc Target gene(s): E, N2 Antigen target: n/a Test method: Automated RT‐PCR Samples used: NP or OP; no details on collection Transport media: Not stated Sample storage: Not stated Test operator: Not stated Definition of test positivity: Not stated; presume as per manufacturer ‐ no mention of presumptive positive results Blinding reported: Not stated Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR, one of three assays including 1) in‐house LDT, 2) cobas SARS‐CoV‐2 test kit (Roche), or 3) Amplidiag COVID‐19 test on the Amplidiag Easy platform (Mobidiag) Definition of non‐COVID cases: As above for COVID‐19 suspects (single PCR negative); for pre‐pandemic either Allplex Respiratory Panel 1/2/3 (Seegene, Seoul, Republic of Korea) and two by xTAG RVP Fast (Luminex Diagnostics, Toronto, Canada). Genetic target(s): 1) N gene, 2) orf1ab and E, 3) orf1ab and N Samples used: NP or OP, as for index Timing of reference standard: Not stated Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous (same samples) All patients received same reference standard: Yes (different assays) Missing data: 107 samples tested with Novodiag but only 90 for Xpert Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Not reported |
||
| Comparative | |||
| Notes | Funding: No funding statement reported Publication status: Preprint Source: medRxiv Author COI: No COI statement reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Kruger 2020(a).
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity of three assays (each tested on a separate cohort of individuals, and extracted as three entries Kruger 2020(a), Kruger 2020(b), Kruger 2020(c).
Participants at risk for SARS‐CoV‐2 infection based on exposure to a confirmed case, suggestive symptoms, or travel to a high risk area, presenting at one of three sites:
(1) drive‐in testing station (n=1213)
(2) a clinical ambulatory testing facility (n=1308)
(3) secondary care facility (n=53) This entry (Kruger 2020(a)) relates to the 727 participants tested with assay (a) from Shenzhen Bioeasy Biotechnology; it is unclear whether some particpants may have receieved more than one assay *This study was also reported as three independent FIND evaluations; author contact advised including data from the Kruger et al pre‐print Recruitment: Not stated; recorded as consecutive, as per FIND evaluation protocol Prospective or retrospective: Prospective |
||
| Patient characteristics and setting | Setting: Mixed; (1), (2) Community (drive‐in or clinical ambulatory testing); (3) secondary care Location: Three sites: (1) Heidelberg, Germany; (2) Berlin, Germany and (3) Liverpool University Hospital Foundation Trust, Liverpool Country: (1), (2) Germany, (3) UK Dates: April 17th and August 25th, 2020; dates varied by assay and site Whole sample: Symptomatic on testing day (n=1901/2355, 80.7%) N with prior negative test result (n=236/1928, 12.2%) Mean age (SD) (n=2405: 40.4y (14.3)) Male (%) (n=1115/2361, 47.2%) Participants undergoing assay (a) (denominator back‐calculated from n and %) Symptomatic on testing day: 564/694, 81.2% N with prior negative test result: 73/624, 11.7% Mean age (SD): 42.7y (14.9y) Male (%): 47.2% |
||
| Index tests | Study reports data for three Ag assays, each tested on a separate cohort of individuals. This entry (Kruger 2020(a)) relates to assay [A]. See Kruger 2020(b) and Kruger 2020(c) for assays (b) and (c) Test name: Bioeasy 2019‐nCoV Ag Fluorescence Rapid Test Kit (Time‐Resolved Fluorescence) Manufacturer: Shenzhen Bioeasy Biotechnology Co. Ltd., Guangdong Province, China Antibody: Not stated Antigen target: Not stated Test method: FIA Samples used: Drive‐in centre: NP or OP; Other centres: combined NOP (OP conducted first) RT‐PCR swab obtained first, then same technique repeated for Ag test. Transport media: None; used manufacturer supplied buffer solution as per IFU (for the Bioeasy assay, "the developer requested for pipettes to be used to transfer adequate quantities of liquid; in the IFU no pipette is needed and a nozzle is provided"). Sample storage: Drive‐in centre and ambulatory testing: tested on site (presume short time frame) Secondary care: transported on ice to a category 3 facility for testing RT‐PCR swab obtained first, then same technique repeated for Ag test. Test operator: Drive‐in and ambulatory clinic: POC evaluation Secondary care: laboratory staff Definition of test positivity: as per Analyzer Invalid results were repeated once using the remaining buffer according to the respective IFUs. Readouts were done within the recommended time for each Ag‐RDT (10 minutes for Bioeasy, 15 minutes for Coris and 15 to 30 minutes for SD Biosensor). Blinding reported: Yes; "Staff performing the Ag‐RDTs were blinded to results of RT‐PCR tests and vice versa" Timing of samples: Overall: mean 5 days pso (SD 9.6); for this assay 7.0 days (SD 12.2); |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; varied by site
Drive‐in samples (Heidelberg): TibMolbiol (Berlin, Germany); the Allplex SARS‐CoV‐2 Assay from Seegene (Seoul, South Korea); or the Abbott (Illinois, US) RealTime 2019‐nCoV assay
Ambulatory testing (Berlin): Roche Cobas SARS CoV‐2 assay (Pleasanton, CA United States) on the Cobas® 6800 or 8800 system; SARS CoV‐2 assay from TibMolbiol (Berlin, Germany)
Secondary care (UK): Genesig® Real‐Time Coronavirus COVID‐19 PCR assay (Genesig, UK)
Samples that showed a signal above the threshold in the relevant RT‐PCR target regions for each assay were considered to be positive Definition of non‐COVID cases: As per cases; single negative result Genetic target(s): Not stated Samples used: Paired swabs; as per index test (RT‐PCR swab obtained first,) Drive‐in centre: NP or OP Other centres: combined NOP (OP conducted first) Timing of reference standard: As per index test Blinded to index test: Yes; "Staff performing the Ag‐RDTs were blinded to results of RT‐PCR tests and vice versa" Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Paired; simultaneous All patients received same reference standard: Yes (different assays) Missing data: 154 excluded following enrolment [116 2nd swab refused, 3 nose bleed after 1st swab, 3 insufficient time for both swabs, 31 other reasons, 1 no reason available] Uninterpretable results: 2 invalid (PCR negative); PCR: 3 excluded as invalid (n=2) or not available (n=1) Indeterminate results (index test): None reported; Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Study reports an ease of use assessment; for this assay:
Funding: Study was supported by FIND, Heidelberg University Hospital and Charité – University Hospital internal funds. Pfizer funded the clinical team in Liverpool, UK. Publication status: Pre‐print Source: medRxiv Author COI: No COI statement reported; "external funders of the study had no role in study design, data collection, or data analysis" |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Kruger 2020(b).
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity of three assays (each tested on a separate cohort of individuals, and extracted as three entries Kruger 2020(a), Kruger 2020(b), Kruger 2020(c).
Participants at risk for SARS‐CoV‐2 infection based on exposure to a confirmed case, suggestive symptoms, or travel to a high risk area, presenting at one of three sites:
(1) drive‐in testing station (n=1213)
(2) a clinical ambulatory testing facility (n=1308)
(3) secondary care facility (n=53) This entry (Kruger 2020(c)) relates to the 425 participants tested with assay (b) from Coris Bioconcept; it is unclear whether some particpants may have receieved more than one assay *This study was also reported as three independent FIND evaluations; author contact advised including data from the Kruger et al pre‐print Recruitment: Not stated; recorded as consecutive, as per FIND evaluation protocol Prospective or retrospective: Prospective |
||
| Patient characteristics and setting | Setting: Mixed; (1), (2) Community (drive‐in or clinical ambulatory testing); (3) secondary care Location: Three sites: (1) Heidelberg, Germany; (2) Berlin, Germany and (3) Liverpool University Hospital Foundation Trust, Liverpool Country: (1), (2) Germany, (3) UK Dates: April 17th and August 25th, 2020; dates varied by assay and site Whole sample: Symptomatic on testing day (n=1901/2355, 80.7%) N with prior negative test result (n=236/1928, 12.2%) Mean age (SD) (n=2405: 40.4y (14.3)) Male (%) (n=1115/2361, 47.2%) Participants undergoing assay (b) (denominator back‐calculated from n and %) Symptomatic on testing day: 283/411, 68.9% N with prior negative test result: 38/301, 12.6% Mean age (SD): 44.9y (15.4y) Male (%): 39.7% |
||
| Index tests | Study reports data for three Ag assays, each tested on a separate cohort of individuals. See Kruger 2020(a) and Kruger 2020(c) for details of the other assays Test name: COVID‐19 Ag Respi‐Strip Manufacturer: Coris Bioconcept, Gembloux, Belgium Antibody: Not stated Antigen target: Not stated Test method: CGIA Samples used: Drive‐in centre: NP or OP Other centres: combined NOP (OP conducted first) RT‐PCR swab obtained first, then same technique repeated for Ag test. Transport media: None; used manufacturer supplied buffer solution as per IFU Sample storage: Drive‐in centre and ambulatory testing: tested on site (presume short time frame) Secondary care: transported on ice to a category 3 facility for testing RT‐PCR swab obtained first, then same technique repeated for Ag test. Test operator: Drive‐in and ambulatory clinic: POC evaluation Secondary care: laboratory staff Definition of test positivity: Visual appearance were interpreted by two operators, each blinded to the result of the other. In case of discrepant results, both operators re‐read the result and agreed on a final result. Invalid results were repeated once using the remaining buffer according to the respective IFUs. Readouts were done within the recommended time for each Ag‐RDT (15 minutes for Coris). Blinding reported: Yes; "Staff performing the Ag‐RDTs were blinded to results of RT‐PCR tests and vice versa" Timing of samples: Overall: mean 5 days pso (SD 9.6); this assay 6.2 days (SD 14.0) |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; varied by site
Drive‐in samples (Heidelberg): TibMolbiol (Berlin, Germany); the Allplex SARS‐CoV‐2 Assay from Seegene (Seoul, South Korea); or the Abbott (Illinois, US) RealTime 2019‐nCoV assay
Ambulatory testing (Berlin): Roche Cobas SARS CoV‐2 assay (Pleasanton, CA United States) on the Cobas® 6800 or 8800 system; SARS CoV‐2 assay from TibMolbiol (Berlin, Germany)
Secondary care (UK): Genesig® Real‐Time Coronavirus COVID‐19 PCR assay (Genesig, UK)
Samples that showed a signal above the threshold in the relevant RT‐PCR target regions for each assay were considered to be positive Definition of non‐COVID cases: As per cases; single negative result Genetic target(s): Not stated Samples used: Paired swabs; as per index test (RT‐PCR swab obtained first,) Drive‐in centre: NP or OP Other centres: combined NOP (OP conducted first) Timing of reference standard: As per index test Blinded to index test: Yes; "Staff performing the Ag‐RDTs were blinded to results of RT‐PCR tests and vice versa" Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Paired; simultaneous All patients received same reference standard: Yes (different assays) Missing data: 154 excluded following enrolment [116 2nd swab refused, 3 nose bleed after 1st swab, 3 insufficient time for both swabs, 31 other reasons, 1 no reason available] Uninterpretable results: 8 invalid (PCR negative) PCR: 3 excluded as invalid (n=2) or not available (n=1) Indeterminate results (index test): None reported; Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Study reports an ease of use assessment; for this assay:
Funding: Study was supported by FIND, Heidelberg University Hospital and Charité – University Hospital internal funds. Pfizer funded the clinical team in Liverpool, UK. Publication status: Pre‐print Source: medRxiv Author COI: No COI statement reported; "external funders of the study had no role in study design, data collection, or data analysis" |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Kruger 2020(c).
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity of three assays (each tested on a separate cohort of individuals, and extracted as three entries Kruger 2020(a), Kruger 2020(b), Kruger 2020(c).
Participants at risk for SARS‐CoV‐2 infection based on exposure to a confirmed case, suggestive symptoms, or travel to a high risk area, presenting at one of three sites:
(1) drive‐in testing station (n=1213)
(2) a clinical ambulatory testing facility (n=1308)
(3) secondary care facility (n=53) This entry (Kruger 2020(c)) relates to the 1263 participants tested with assay (c) from SD Biosensor; it is unclear whether some particpants may have receieved more than one assay *This study was also reported as three independent FIND evaluations; author contact advised including data from the Kruger et al pre‐print Recruitment: Not stated; recorded as consecutive, as per FIND evaluation protocol Prospective or retrospective: Prospective |
||
| Patient characteristics and setting | Setting: Mixed; (1), (2) Community (drive‐in or clinical ambulatory testing); (3) secondary care Location: Three sites: (1) Heidelberg, Germany; (2) Berlin, Germany and (3) Liverpool University Hospital Foundation Trust, Liverpool Country: (1), (2) Germany, (3) UK Dates: April 17th and August 25th, 2020; dates varied by assay and site Whole sample: Symptomatic on testing day (n=1901/2355, 80.7%) N with prior negative test result (n=236/1928, 12.2%) Mean age (SD) (n=2405: 40.4y (14.3)) Male (%) (n=1115/2361, 47.2%) Participants undergoing assay (b) (denominator back‐calculated from n and %) Symptomatic on testing day: 1054/1249, 84.4% N with prior negative test result: 125/1000, 12.5% Mean age (SD): 37.6 (12.7) Male (%): 49.8% Exposure history: Not stated |
||
| Index tests | Study reports data for three Ag assays, each tested on a separate cohort of individuals. See Kruger 2020(a) and Kruger 2020(b) for details of the other assays Test name:STANDARD Q COVID‐19 Ag Test Manufacturer: SD Biosensor, Inc. Gyeonggi‐do, Korea Antibody: Not stated Antigen target: Not stated Test method: CGIA Samples used: Drive‐in centre: NP or OP Other centres: combined NOP (OP conducted first) RT‐PCR swab obtained first, then same technique repeated for Ag test. Transport media: None; used manufacturer supplied buffer solution as per IFU Sample storage: Drive‐in centre and ambulatory testing: tested on site (presume short time frame) Secondary care: transported on ice to a category 3 facility for testing RT‐PCR swab obtained first, then same technique repeated for Ag test. Test operator: Drive‐in and ambulatory clinic: POC evaluation Secondary care: laboratory staff Definition of test positivity: Visual appearance were interpreted by two operators, each blinded to the result of the other. In case of discrepant results, both operators re‐read the result and agreed on a final result. Invalid results were repeated once using the remaining buffer according to the respective IFUs. Readouts were done within the recommended time for each Ag‐RDT (10 minutes for Bioeasy, 15 minutes for Coris and 15 to 30 minutes for SD Biosensor). Blinding reported: Yes; "Staff performing the Ag‐RDTs were blinded to results of RT‐PCR tests and vice versa" Timing of samples: Overall: mean 5 days pso (SD 9.6); this assay 3.7 days (SD 5.6) |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; varied by site
Drive‐in samples (Heidelberg): TibMolbiol (Berlin, Germany); the Allplex SARS‐CoV‐2 Assay from Seegene (Seoul, South Korea); or the Abbott (Illinois, US) RealTime 2019‐nCoV assay
Ambulatory testing (Berlin): Roche Cobas SARS CoV‐2 assay (Pleasanton, CA United States) on the Cobas® 6800 or 8800 system; SARS CoV‐2 assay from TibMolbiol (Berlin, Germany)
Secondary care (UK): Genesig® Real‐Time Coronavirus COVID‐19 PCR assay (Genesig, UK)
Samples that showed a signal above the threshold in the relevant RT‐PCR target regions for each assay were considered to be positive Definition of non‐COVID cases: As per cases; single negative result Genetic target(s): Not stated Samples used: Paired swabs; as per index test (RT‐PCR swab obtained first,) Drive‐in centre: NP or OP Other centres: combined NOP (OP conducted first) Timing of reference standard: As per index test Blinded to index test: Yes; "Staff performing the Ag‐RDTs were blinded to results of RT‐PCR tests and vice versa" Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Paired; simultaneous All patients received same reference standard: Yes (different assays) Missing data: 154 excluded following enrolment [116 2nd swab refused, 3 nose bleed after 1st swab, 3 insufficient time for both swabs, 31 other reasons, 1 no reason available] Uninterpretable results: 2 invalid (PCR negative); [B] 8 invalid (PCR negative); [C] 0 invalid reported PCR: 3 excluded as invalid (n=2) or not available (n=1) Indeterminate results (index test): None reported; Ease of use assessment reported: [A] a high number of test execution steps (including precision pipetting) … challenges when performing multiple tests at the same time possibly hindering the test’s wide‐spread use [B] challenges due to inconsistent test result interpretation (often only very faint lines visible) and deficiencies in both the test kit quality and design [C] no dissatisfactory scores identified Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Study reports an ease of use assessment; for this assay:
Funding: Study was supported by FIND, Heidelberg University Hospital and Charité – University Hospital internal funds. Pfizer funded the clinical team in Liverpool, UK. Publication status: Pre‐print Source: medRxiv Author COI: No COI statement reported; "external funders of the study had no role in study design, data collection, or data analysis" |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Low risk | ||
Lambert‐Niclot 2020.
| Study characteristics | |||
| Patient Sampling | Single‐group study to estimate sensitivity and specificity:
‐ samples submitted for RT‐PCR testing (n = 138) Recruitment: not stated Prospective or retrospective: unclear; testing conducted prospectively Number of samples (samples with confirmed SARS‐CoV‐2): 138 (94) |
||
| Patient characteristics and setting | Setting: not stated Location: samples collected from virology laboratories of 3 university hospital groups from Assistance‐Publique‐Hôpitaux de Paris (APHP), (Saint‐Antoine‐Tenon‐Trousseau, Saint‐Louis‐Lariboisière and Kremlin Bicêtre‐Paul Brousse) Country: France Dates: 1‐15 April 2020 Symptoms and severity: not stated Demographics: not stated Exposure history: not stated |
||
| Index tests | Test name: COVID‐19 Ag Respi‐Strip CORIS (no product code) Manufacturer: BioConcept, Gembloux, Belgium Antigen target: SARS‐CoV‐2 NP Antibody: monoclonal antibodies Test method: CGIA Samples used: NP swabs in VTM (collection process not described) Transport media: either of: COPAN UTM 3 mL, Virocult 1 mL, Eswab Amies 1 mL, 4MRT 3 mL, 0.9% NaCl buffer and cobas ROCHE Sample storage: no cooling or freezing step used Test operator: not stated; presume laboratory staff Definition of test positivity: not stated; as per manufacturer Blinding reported: not stated Timing of samples: not stated; presume on presentation |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR (different kits used including RealStar Altona®, Anatolia®, cobas 6800 Roche®, Allplex™ 2019‐nCoV Assay Seegene®) Definition of non‐COVID cases: single negative PCR Genetic target(s): E gene Samples used: NP swabs (same as for index) Timing of reference standard: within a few hours after collection; time post onset of symptoms not reported Blinded to index test: unclear Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: same sample, both tests conducted within a few hours All participants received same reference standard: yes (different kits) Missing data: none reported Uninterpretable results: 4 samples collected in cobas VTM gave invalid results and all samples in cobas medium were excluded Indeterminate results (index test): control lines reported as "barely visible" for 9 positive and 8 negative tests Indeterminate results (reference standard): none reported Unit of analysis: not reported, but samples tested on day of collection so considered to be 1 per participant |
||
| Comparative | |||
| Notes | Funding: no funding sources reported Publication status: accepted manuscript Source: Journal of Clinical Microbioloby Author COI: no conflict of interest statement reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Lephart 2020 [A].
| Study characteristics | |||
| Patient Sampling | Single group study including samples from:
[1] patients presenting to emergency department (n=75), or Recruitment: Not stated Prospective or retrospective: Not reported [Study also reports results for second group of recovering inpatients with previously laboratory‐confirmed COVID‐19 (n=13); for purposes of this review only those in group [1] were included] |
||
| Patient characteristics and setting | Setting: [1] ED Location: Not stated; pathology lab at University of Michigan Medical School Country: USA Dates: 22 Apr to 5 May 2020 Symptoms and severity: Not reported Demographics: Not reported Exposure history: Not reported |
||
| Index tests |
Test name: [A] ID NOW (second index test [B] Xpert Xpress, extracted as Lephart 2020 [B]; two additional RT‐PCR tests evaluated in study but not included in this review). No product codes reported Manufacturer: [A] Abbott Molecular Target gene: Not reported in paper Test method: [A] isothermal PCR Samples used: [A] Nasal; Presume collected by HCP but not reported Transport media: [A] None ‐ transported dry swabs in sealed sterile collection bags Sample storage: [A] within 24h Test operator: Not stated; presume lab staff Definition of test positivity: Each assay was performed according to manufacturer’s EUA instructions. Blinding reported: Not stated; unlikely Timing of samples: On presentation; timing pso not reported |
||
| Target condition and reference standard(s) | Reference standard: Composite: positive on >=2 of 4 NATs tested considered D+, including [A] ID NOW, [B] Xpert Xpress, [C] Simplexa COVID‐19 Direct (Diasorin) (this was the standard of care assay), [D] RealTime m2000 SARS‐CoV‐2 Assay (Abbott Molecular) Definition of non‐COVID cases: Three negatives (on different assays) required for D‐ Genetic target(s): Not stated Samples used: NP swabs (Same as for Xpert Xpress) Timing of reference standard: Within 24h of sample collection (on presentation at ED); no further detail Blinded to index test: Not stated; seems unlikely Incorporated index test: Yes |
||
| Flow and timing | Time interval between index and reference tests: Same swab [B], or paired collection [A] All patients received same reference standard: Yes, all had all 4 assays Missing data: None reported, no participant flow diagram reported Uninterpretable results: None reported Indeterminate results (index test): [A] no invalid results, [B] 1 'invalid' result; not reported if this was a 'presumptive positive' (E gene only) on Xpert Xpress or no result Indeterminate results (reference standard): None reported Unit of analysis: Unclear; text refers to 'patients' so presumed patient‐based |
||
| Comparative | |||
| Notes | Funding: No funding statement reported Publication status: Pre‐print Source: bioRxiv Author COI: No COI statement provided |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | No | ||
| Reference standard does not incorporate result of index test? | No | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Lephart 2020 [B].
| Study characteristics | |||
| Patient Sampling | See Lephart 2020 [A] for full study details and QUADAS entries | ||
| Patient characteristics and setting | |||
| Index tests | Test name: [B] Xpert Xpress (second index test [A] ID NOW, extracted as Lephart 2020 [A], also see see Lephart 2020 [A] for full study details and QUADAS entries; two additional RT‐PCR tests evaluated in study but not included in this review). No product codes reported Manufacturer: [B] Cepheid Target gene: Not reported in paper Test method: [B] Automated RT‐PCR Samples used: [B] NP; presume collected by HCP but not reported Transport media: [B] M4‐RT VTM (Thermo Fisher) Sample storage: [B] stored at 4°C and tested within 24h Test operator: Not stated; presume lab staff Definition of test positivity: each assay was performed according to manufacturer’s EUA instructions (presumptive positives not described) Blinding reported: Not stated; unlikely Timing of samples: On presentation; timing pso not reported |
||
| Target condition and reference standard(s) | See Lephart 2020 [A] for full study details and QUADAS entries | ||
| Flow and timing | See Lephart 2020 [A] for full study details and QUADAS entries | ||
| Comparative | |||
| Notes | |||
Lieberman 2020.
| Study characteristics | |||
| Patient Sampling | Single‐group study to estimate sensitivity and specificity:
‐ samples submitted for clinical diagnostic testing (n = 169; not all samples analysed for all tests) Recruitment: not stated Prospective or retrospective: retrospective (residual samples) Number of samples (samples with confirmed SARS‐CoV‐2): 169 (87) |
||
| Patient characteristics and setting | Setting: not stated; sampled from laboratory Location: Washington State Public Health Laboratory Country: USA Dates: not stated Symptoms and severity: not stated Demographics: not stated Exposure history: not stated |
||
| Index tests | Test name: Xpert Xpress Manufacturer: Cepheid Antigen target: E, N2 Antibody: N/A Test method: rapid PCR Samples used: NP swabs (collection not described) Transport media: 300 μL of VTM sample Sample storage: all same‐sample comparisons were performed on specimens stored at 4 °C for < 72 h with no freeze‐thaws Test operator: not stated; presume laboratory staff Common panel of 26 specimens tested at UW by the UW CDC EUA‐based LDT or at LabCorp Seattle Definition of test positivity: 1 of 2 targets detected was considered positive for all assays; Xpert Xpress data extracted as per IFU definition (positive = both targets or N gene positive; E‐gene‐positive requires retest) Blinding reported: not stated Timing of samples: not stated Also evaluates: [B] Hologic Panther Fusion RUO, [C] Hologic Panther Fusion EUA, [D] Diasorin Simplexa, [E] Roche cobas 6800 in same 26 samples and in additional residual specimens (n = 115) at UW (different N per test) |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; UW CDC EUA‐based in‐house test (positive if 1 of 2 targets detected ‐ presume at < 40 Ct) Definition of non‐COVID cases: single negative PCR Genetic target(s): NI, N2 Samples used: NP swabs, as for index test Timing of reference standard: not stated Blinded to index test: not stated Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: all testing conducted within 72 h All participants received same reference standard: yes Missing data: none reported, no participant flow diagram reported; review team excluded data for 28 specimens comparing Panther Fusion with DiaSorin Simplexa Uninterpretable results: not stated Indeterminate results (index test): ‘Inconclusive' results (i.e. 1 genetic target detected) were considered positive due to the high specificity of all assays and limited cross‐reactivity seen for SARS‐CoV‐2 primer sets. For Xpert Xpress only 12/13 were positive according to IFU specifications on first test (both targets present, or N gene positive); on retesting the presumptive positive became positive (detection of E‐gene but not N‐gene) Indeterminate results (reference standard): as for index test Unit of analysis: not stated, only refers to samples |
||
| Comparative | |||
| Notes | Funding: no funding statement reported Publication status: accepted manuscript Source: Journal of Clinical Microbioloby Author COI: no COI statement reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Linares 2020.
| Study characteristics | |||
| Patient Sampling | Single group study estimating sensitivity and specificity, recruiting at two locations:
[1] symptomatic patients admitted to ED with clinical suspicion of COVID‐19 (n=135) or asymptomatic patients with history of contact with another COVID‐19 patient (n=17)
[2] symptomatic patients (n=50) or asymptomatic (n=55) patients attending one of two primary healthcare centres Recruitment: Not stated Prospective or retrospective: Unclear; appears to be prospective |
||
| Patient characteristics and setting | Setting: Mixed; A&E or primary care Location: Hospital Universitario Príncipe de Asturias, Madrid Country: Spain Dates: Sep 10 to Sep 15 Symptoms and severity: 185, 72% symptomatic ED (n=135): fever 40, dyspnoea 42, cough 22, headache 14 Prim care (n=50): fever 14, dyspnoea 1, cough 18, headache 17 Demographics: Mean(?) age (range): ED 51.5y (37.0 to 71.8y); primary care 39.0y (25.0 to 56.0y) Male: ED 77 (51%), primary care 49 (47%) Exposure history: Not stated |
||
| Index tests | Test name: PanBio COVID‐19 Ag Rapid Test Device (no product code) Manufacturer: Abbott Rapid Diagnostic Jena GmbH, Jena, Germany Antibody: Nucleocapsid Antigen target: Not stated Test method: Not stated; qualitative membrane‐based immunoassay (immunochromatography) Samples used: NP; HCW obtained Transport media: None reported Sample storage: Not stated Test operator: Not stated Definition of test positivity: Not stated; as per manufacturer Blinding reported: Not stated Timing of samples: ED: 2 days pso (IQR? 1‐5) PC: 4 days pso (IQR? 2‐8) Table 3 reports range of 0 to 27 days post symptom onset or post COVID‐19 contact, and range of 0 to 16 days for days post symptoms onset for symptomatic cases only |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; Allplex SARS‐CoV‐2 assay (Seegene, Seoul, South Korea); appears to be <40 Ct threshold Definition of non‐COVID cases: As for cases (single ‐ve) Genetic target(s): Not stated Samples used: NP (paired) Timing of reference standard: Not stated Blinded to index test: Unclear Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Paired All patients received same reference standard: Yes Missing data: None reported however 257 reported in Methods and 255 in Results, no participant flow diagram reported Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: No funding statement provided Publication status: Pre‐print Source: medRxiv Author COI: No COI statement provided |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Liotti 2020.
| Study characteristics | |||
| Patient Sampling | Unclear design estimating sensitivity and specificity; residual samples selected from one of two virology laboratories at two Covid‐19 reference hospitals:
[1] RT‐PCR positive for SARS‐CoV‐2 (n=104)
[2] RT‐PCR negative for SARS‐CoV‐2 (n=255) Recruitment: Not stated Prospective or retrospective: Retrospective |
||
| Patient characteristics and setting | Setting: Unclear; laboratory samples Location: From authors' institutions: Fondazione Policlinico Universitario A. Gemelli IRCCS, and Istituto Nazionale per le Malattie Infettive (INMI) Lazzaro Spallanzani IRCCS, Rome Country: Italy Dates: Not stated Symptoms and severity: Not stated; Of SARS‐CoV‐2 positive samples, 21, 20% high viral load (<25 Ct), 83, 80%low viral load (>=25) [28, 27% with Ct >=35] Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: STANDARD F COVID‐19 Ag FIA (no product codes reported) Manufacturer: SD Biosensor (Suwon, South Korea) Antibody: NP Antigen target: monoclonal anti‐SARS‐CoV‐2 antibody Test method: FIA Samples used: NP; collection not reported Transport media: Not stated Sample storage: performed within 24 hr after collection on samples kept at 4 C until testing Test operator: Not stated; presume laboratory staff Definition of test positivity: As per manufacturer Blinding reported: Not stated Timing of samples: Not reported |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR (one of 4 assays); Altona Diagnostics RealStar® SARS‐CoV‐2 RT‐PCR, the Seegene Allplex™ 2019‐nCoV, the DiaSorin Simplexa™COVID‐19 Direct or the Roche Diagnostics Cobas® SARS‐CoV‐2 test Definition of non‐COVID cases: As for cases (single negative) Genetic target(s): Not stated Samples used: NP (same as index) Timing of reference standard: Not stated Blinded to index test: Yes (performed first) Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous (same swab) All patients received same reference standard: Yes Missing data: None reported, no participant flow diagram reported Uninterpretable results: None reported Indeterminate results (index test): None reported; FP results were re‐tested with Ag assay, 3 of 4 remained positive (all blood contaminated) Indeterminate results (reference standard): None reported Unit of analysis: Not stated |
||
| Comparative | |||
| Notes | Funding: Study supported by funds to the Istituto Nazionale per le Malattie Infettive (INMI) Lazzaro Spallanzani IRCCS, Rome, Italy, from the Ministero della Salute (Ricerca Corrente, linea 1; COVID‐ 2020‐12371817), the European Commission e Horizon 2020 (EU project 101003544 e CoNVat; EU project 101003551 e EXSCALATE4CoV; EU project 12371675 e EXCALATE4CoV; EU project 101005075 e KRONO) and the European Virus Archive e GLOBAL (grants no. 653316 and no. 871029). Publication status: Published letter Source: Clin Microbiol Infect Author COI: All authors report no relevant conflicts of interest |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Unclear | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Loeffelholz 2020.
| Study characteristics | |||
| Patient Sampling | Two‐group study to estimate sensitivity and specificity for diagnosis of active disease
‐ suspected patients referred for COVID‐19 testing at 7 sites according to the local criteria (n = 486); sampled to enrich for RT‐PCR‐positive specimens (not further described) Recruitment: convenience (in addition, 1 site (LAC+USC) tested specimens from a 4‐day point prevalence survey of patients presenting with COVID‐19 symptoms) Prospective or retrospective: retrospective Number of samples (samples with confirmed SARS‐CoV‐2): 486 (220) |
||
| Patient characteristics and setting | Setting: not stated Location: 7 sites: Johns Hopkins University, Baltimore; LAC+USC Medical Centre, University of Southern California, Los Angeles; Manchester University NHS Foundation Trust Manchester; Mondor Hospital, Paris; New York City Dept. Health and Mental Hygiene, NYC; Niguarda Hospital, Milan; University Hospital, Newark. Country: USA, UK, France, Italy Dates: 1 March‐2 April 2020 Symptoms and severity: not stated Demographics: adults at all sites except New York City Dept. Health and Mental Hygiene and Niguarda Hospital where all age groups were tested (ages not stated) Exposure history: not stated |
||
| Index tests | Test name: Cepheid Xpert Xpress SARS‐CoV‐2 (RUO version, no product code reported) Manufacturer: Cepheid Europe Antigen target: nucleocapsid gene (N2) and the envelope gene (E) (RUO version also detects RdRp gene but this does not contribute to definition of positive) Antibody: N/A Test method: automated point‐of‐care PCR Samples used: swabs (NP (n = 339), OP (n = 15), combined NP/OP in the same transport vial (n = 97)), and TA (n = 30):
Transport media: VTM (swabs), diluted in saline (TA). 1 site (Manchester) pretreated specimens with an equal volume (≥ 30‐< 50% (w/w)) of a guanidine hydrochloride buffer and heated at 80 °C Sample storage: stored at −80 °C prior to index test, except at 1 site (University Hospital, Newark) where specimens were tested in real time, within 2 h by the Xpert test (n = 21). Test operator: not stated; presume laboratory staff Definition of test positivity: as per manufacturer: if both targets are detected, or if only N2 is detected, the test reports a positive result. If only the E target is detected the test reports a presumptive positive result "because this target is shared among some members of the sarbecovirus subgenus of coronaviruses". The RUO version of the test shows the amplification curves and PCR cycle threshold for all 3 genetic targets. The study reports that "The EUA test version cartridge contains the same reagents as the RUO cartridge. The only difference between the tests is the software which in the EUA version allows the user to see amplification curves and results for the N2 and E targets only". Blinding reported: not stated Timing of samples: not stated, presume on presentation |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR (sites using each kit not reported, added by review team based on number of samples per site and per RT‐PCR kit)
Definition of non‐COVID cases: yes (performed prior to index test) Genetic target(s): different targets depending on RT‐PCR test used:
Tie‐breaker methods (for discrepant results), included: Hologic Panther Fusion (San Diego, USA), Tib‐Molbiol LightMix Modular Wuhan Coronavirus E‐gene RT‐PCR (Roche, Basel, Switzerland); and the CDC assay (IDT primers and probes) Samples used: as for index test Timing of reference standard: as for index test Blinded to index test: no storage; tested in real time Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: same samples but index performed after frozen storage for undefined period of time except at University Hospital, Newark where specimens were tested in real time, within 2 h by the Xpert test All participants received same reference standard: no Missing data: 4 Xpert Xpress test results were lost permanently due to a single instrument computer malfunction Uninterpretable results: 1 Xpert Xpress test was invalid due to a cartridge error (inadequate sample volume) Indeterminate results (index test) presumptive positive results on Xpert Xpress were not reanalysed by Xpert Xpress, but all discrepant results were reanalysed by a third RT‐PCR method Indeterminate results (reference standard): specimens with inconclusive results by a test, and those with discrepant results between Xpert and the RT‐PCR tests were analysed by a third RT‐PCR method 1 FN result was inconclusive on Quest SARS‐CoV‐2, and negative on CDC RT‐PCR; re‐considered as TN Of 11 FPs (including 1 presumptive positive on Xpert Xpress), 2 were negative on both New York SARS‐CoV‐2 and Panther Fusion (remained as FPs), and 9 were negative on in‐house RT‐PCR but positive on Roche RT‐PCR (reclassified as TP) In addition, 12 specimens (8 NP, 4 NP/OP) were inconclusive by the NY (RT)‐ PCR Diagnostic Panel and considered positive for data analysis purposes in the study. Of these, 11 were positive by the Xpert test and 1 was presumptive positive (EUA version of Xpert test). In 4 of these only the N1 target was detected and in 8 only the N2 target was detected by the New York EUA method, all with Ct values > 36 One NP specimen was inconclusive by the Quest SARS‐CoV‐2 rRT‐PCR test and negative by the Xpert test. The Quest test reports inconclusive if only a single target (N1 or N3) is detected. They were unable to determine which target was detected by the Quest test. This specimen was negative by a tie‐breaker NAAT. Unit of analysis: not stated; only samples reported |
||
| Comparative | |||
| Notes | Funding: not stated; presume funded by test manufacturer (see COI statement) Publication status: accepted manuscript Source: Journal of Clinical Microbiolobyogy Author COI: the study was designed and supervised by the sponsor, Cepheid. Data were collected by investigators at each study site, and statistical analyses were performed by a Cepheid author. Cepheid authors wrote the first draft of the manuscript. All study authors vouch for the accuracy and completeness of the data reported. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Mak 2020.
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity alone:
[1] RT‐PCR positive samples selected from Hong Kong's COVID‐19 reference laboratory (n=160 samples from 152 patients) Recruitment: Convenience; deliberate sampling of specific numbers of different respiratory sample types (selected from cohort of all available positive samples with sufficient quantity) Prospective or retrospective: Retrospective |
||
| Patient characteristics and setting | Setting: Not stated Location: Public Health Laboratory Services Branch, Hong Kong Country: Hong Kong Dates: Feb 1 to Apr 21 2020 Symptoms and severity: Not stated; High viral load (<18.57 Ct) ‐ 64, 40% 'Normal' viral load >18.57 ‐ 96, 60% Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: BIOCREDIT COVID‐19 Ag (no product code reported) Manufacturer: RapiGEN Inc Antibody: Not stated Antigen target: Not stated Test method: CGIA Samples used: throat saliva (TS, n = 45), nasopharyngeal swab and throat swab (NPS & TS, n=103), nasopharyngeal aspirate and throat swab (NPA & TS, n=81), sputum (n=45); no details of collection methods Transport media: Samples were placed in viral transport media (VTM) or Phosphate‐Buffered Saline (PBS). 100 μL sample volume was used; less viscous samples were added directly to sample well of the device, for more viscous samples the swab provided with the kit was used to collect the samples and was immersed in the provided assay diluent tube. The subsequent procedures were carried out according to the manufacturer’s instructions. Sample storage: stored at −70 °C until used for study purposes Test operator: Not stated; laboratory staff presumed Definition of test positivity: Not stated Blinding reported: Not stated but all positive samples Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Reference standard: In‐house RT‐PCR; <=40Ct Definition of non‐COVID cases: n/a Genetic target(s): RdRp Samples used: NPA & TS, NPS & TS, sputum and throat saliva, as for index test Timing of reference standard: Not stated Blinded to index test: Yes, prior to index test Incorporated index test: Not stated |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous; same samples All patients received same reference standard: Yes Missing data: None reported, no participant flow diagram reported Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Samples (160 from 152 patients) |
||
| Comparative | |||
| Notes | Funding: No funding statement reported Publication status: Published Source: J Clin Virol Author COI: Authors report no COI |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Mertens 2020.
| Study characteristics | |||
| Patient Sampling | Single‐group study to estimate sensitivity and specificity for diagnosis of active disease:
‐ samples from patients suspected of SARS‐COV‐2 infections (n = 328) Recruitment: random sampling of samples submitted to 3 laboratories 322/328 NP samples (NP swabs) were randomly selected Prospective or retrospective: retrospectively Number of samples (samples with confirmed SARS‐CoV‐2): 328 (132) |
||
| Patient characteristics and setting | Setting: unclear; samples from university laboratories (discussion states that no outpatient population has been sampled, therefore assume inpatients and HCW samples) Location: laboratories at Université Libre de Bruxelles (LHUB‐ULB), UZ Leuven and Centre Hospitalier Universitaire Sart‐Tilman (CHU) Liège Country: Belgium Dates: 19‐30 March 2020 Symptoms and severity: not reported Demographics: not reported Exposure history: unclear; 53/328 samples were from HCW |
||
| Index tests | Test name: COVID‐19 Ag Respi‐Strip Manufacturer: Coris BioConcept (Belgium) Antigen target: SARS‐CoV and SARS‐CoV‐2 highly conserved nucleoprotein Antibody: monoclonal antibodies directed against SARS‐CoV and SARS‐CoV‐2 highly conserved nucleoprotein antigen Test method: immunochromatographic assay using colloidal gold (CGIA) Samples used: remnant respiratory specimens (322 NP swabs, 4 NP aspirate and 2 BAL) Transport media: NP: flocked swab + UTM 3 mL (or 1 mL of Amies) (Copan, Brescia, Italy); NPA: 3 mL VTM (veal infusion broth (Difco, Becton Dickinson, Sparks, MD, USA) supplemented with bovine albumin (Sigma Aldrich, St Louis, MO, USA)) BAL: N/A Sample storage: not described Test operator: laboratory technician Definition of test positivity: visible reddish‐purple band appearing at the Test line position (T) Blinding reported: not stated Timing of samples: not clear |
||
| Target condition and reference standard(s) | Reference standard: qRT‐PCR: RealStar SARS‐CoV‐2 RT‐PCR Kit from Altona‐diagnostics with a cut‐off set at 40 Ct (LHUB‐ULB); Roche LC480 thermocycler using Taqman Fast Virus 1‐Step Master Mix (Thermo Fisher) (Liege); QuantStudio Dx (Thermo Fisher Scientific) or Panther Fusion (PF, Hologic, San Diego, USA) (UZ Leuven) Definition of non‐COVID cases:
Samples used: as for index test (respiratory specimens (322 NP swabs, 4 NP aspirate and 2 BAL) Timing of reference standard: not stated; same samples as for index test but analysed at time of collection Blinded to index test: yes (undertaken for diagnostic purposes at time of collection) Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: same samples used; discussion report 'some delay' between PCR and antigen testing All participants received same reference standard: yes but different RT‐PCR kits Missing data: none reported, no participant flow diagram reported Uninterpretable results: none reported; discussion reports some difficulties in visualising the strip through the closed tube requiring the lab technician to open the test tube in the laminar air flow cabinet and pull out the strip with forceps Indeterminate results (index test): weak T lines considered positive Indeterminate results (reference standard): none reported Unit of analysis: refers to participants |
||
| Comparative | |||
| Notes | Funding: not stated Publication status: preprint (not peer‐reviewed) Sourcepreprint server (medRxiv) Author COI: the IVD medical device has been developed by the investigator Pascal Mertens, Henri Magein, and Justine Bouzet working for Coris BioConcept (potential conflict of interest declared even though they don’t have any share in this company); Thierry Leclipteux was involved in the development of this test and is the CEO of Coris Bioconcept (potential conflict of interest declared). All scientific investigators that are external to Coris BioConcept declare having no conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Mitchell 2020.
| Study characteristics | |||
| Patient Sampling | Single‐group study to estimate sensitivity and specificity for diagnosis of active disease:
‐ samples positive and negative on 1 of 2 SARS‐CoV‐2 RT‐PCR assays Recruitment: not stated; suggests possible deliberate sampling of positive cases Prospective or retrospective: retrospective (residual samples) Number of samples (samples with confirmed SARS‐CoV‐2): 61 (46) |
||
| Patient characteristics and setting | Setting: not stated; 2 independent laboratories (Class II biosafety cabinet (BSC)) Location: not stated; author institutions University of Pittsburgh School of Medicine, Pittsburgh and Laboratory of Viral Diseases, Wadsworth Centre, New York State Department of Health, Albany, NY Country: USA Dates: not stated Symptoms and severity: not stated Demographics: not stated Exposure history: not stated |
||
| Index tests | Test name: ID NOW COVID‐19 (product code not reported) Manufacturer: Abbott, Chicago, USA Antigen target: not stated Antibody: N/A Test method: not stated (should be isothermal PCR) Samples used: NP samples (residual samples) Transport media: VTM; no further detail (no longer covered on IFU) Sample storage: stored at −80 ℃ prior to testing Test operator: certified laboratory personnel Definition of test positivity: not stated; as per manufacturer Blinding reported: not stated Timing of samples: not stated |
||
| Target condition and reference standard(s) | Reference standard: CDC EUA or the New York EUA RT‐PCR assays Definition of non‐COVID cases: single RT‐PCR negative Genetic target(s): not stated Samples used: as for index test Timing of reference standard: as for index test Blinded to index test: not stated; samples analysed at or near time of collection Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: same samples but used at different times (samples used for index test stored at −80 ℃) All participants received same reference standard: no, either the CDC EUA or the New York EUA assays Missing data: none reported, no participant flow diagram reported Uninterpretable results: none reported Indeterminate results (index test): none reported Indeterminate results (reference standard): none reported Unit of analysis: not stated; only samples reported |
||
| Comparative | |||
| Notes | Funding: not stated Publication status: accepted manuscript Source: Journal of Clinical Virology Author COI: COI not mentioned by study authors |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Moore 2020.
| Study characteristics | |||
| Patient Sampling | 2‐group study to estimate sensitivity and specificity:
‐ samples from symptomatic (fever or cough or shortness of breath) adult and paediatric outpatients, ED patients, and inpatients Recruitment: consecutive (first 94 participants), then all PCR‐positive samples plus the next PCR‐negative sample after each positive sample, to a total of 200 samples Prospective or retrospective: retrospective (participant and sample details extracted from the electronic medical record) Number of samples (samples with confirmed SARS‐CoV‐2): 200 (125) |
||
| Patient characteristics and setting | Setting: mixed (outpatients, ED patients and inpatients) Location: Rush University Medical Centre (RUMC) or Rush Oak Park Hospital (ROPH), Chicago Country: USA Dates: 27 March‐9 April 2020 Symptoms and severity: 79 (39.5%) hospitalised including 29 in ICU, 76 (38%) ambulatory care including 55 seen in a designated COVID‐19 screening clinic), and 45 (23%) seen at ED Demographics: mean age 50 years (SD 17 years), 92 (46%) men Exposure history: not stated |
||
| Index tests | Test name: ID NOW (no product code) Manufacturer: Abbott Antigen target: RdRp Antibody: N/A Test method: isothermal amplification test Samples used: NP swabs in 3 mL VTM (collection not reported) Transport media: M4‐RT VTM (Remel, Lenexa, KS) Sample storage: stored at 4 °C if all testing could not be completed on the same day; all tests completed within 72 h of collection Test operator: not stated; presume laboratory staff Definition of test positivity: as per manufacturer Blinding reported: not stated Timing of samples: not stated; presumably on presentation but no information on symptom status |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; 2 methods used in the study
Record review used to verify status of 8 samples positive on RealTime assay and negative (6) or inconclusive (2) on CDC assay (all considered disease‐positive) Definition of non‐COVID cases: single RT‐PCR negative Genetic target(s):
Samples used: NP swabs in VTM, as for index test Timing of reference standard: not stated Blinded to index test: not stated Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: all 3 tests conducted within 72 h of sample collection All participants received same reference standard: no? (all received both RT‐PCR tests, only discordant results on RT‐PCR had record review) Missing data: none reported, no participant flow diagram reported Uninterpretable results: 2 results were invalid on ID NOW and were not retested (excluded) Indeterminate results (index test): none reported Indeterminate results (reference standard): discordant results between 2 RT‐PCR assays had record review to determine presence/absence COVID‐19 infection Unit of analysis: participants (specimens from 200 unique participants) |
||
| Comparative | |||
| Notes | Funding: none reported (some reagents supplied from NIH) Publication status: preprint Source: medRxiv Author COI: no COI statement was reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Moran 2020.
| Study characteristics | |||
| Patient Sampling | Single‐group study to estimate sensitivity and specificity:
‐ specimens collected from inpatients and ambulatory patients at the University of Chicago Recruitment: not stated Prospective or retrospective: not stated Number of samples (samples with confirmed SARS‐CoV‐2): 103 (42) |
||
| Patient characteristics and setting | Setting: inpatient and ambulatory; samples selected from central laboratory Location: Clinical Microbiology Laboratory, University of Chicago Country: USA Dates: not stated Symptoms and severity: not stated Demographics: not stated Exposure history: not stated |
||
| Index tests | Test name: Xpert Xpress SARS‐CoV‐2 assay (no product code) Manufacturer: Cepheid, Sunnyvale, CA Antigen target: E, N (N2 region) Antibody: N/A Test method: rapid PCR Samples used: 8 nasal and 95 NP swabs Transport media: none described Sample storage: not stated Test operator: not stated; presume laboratory staff Definition of test positivity: not stated; re‐testing using Xpert Xpress was undertaken for an N‐gene positive result due discrepancy with RT‐PCR (not in line with IFU recommendation) Blinding reported: not stated Timing of samples: not stated |
||
| Target condition and reference standard(s) | Reference standard: Roche cobas SARS‐CoV‐2 assay on the cobas 6800 system (Roche Molecular Systems, Branchburg, NJ) Definition of non‐COVID cases: single RT‐PCR negative Genetic target(s): ORF1, E Samples used: nasal and NP swabs; same as for index test Timing of reference standard: not stated Blinded to index test: not stated Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: not stated; same sample and appear to have both been conducted soon after sample collection All participants received same reference standard: yes Missing data: none reported, no participant flow diagram reported Uninterpretable results: none reported Indeterminate results (index test): single FP (negative on E gene and low positive on N gene) was retested with Xpert Xpress and considered negative on both targets Indeterminate results (reference standard): single FP was retested on RT‐PCR and found to be repeatedly negative Unit of analysis: refers to participants |
||
| Comparative | |||
| Notes | Funding: none described Publication status: accepted manuscript Source: Journal of Clinical Microbioloby Author COI: no COI statement was reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | No | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Nagura‐Ikeda 2020.
| Study characteristics | |||
| Patient Sampling | Single group study of patients with laboratory confirmed COVID‐19 referred for isolation and treatment (n=103); participants had undergone qRT‐PCR tests using NP or OP swabs collected at public health institutes or hospitals (presumably symptomatic), asymptomatic patients were tested as a result of mass‐screening due to an outbreak or family cluster Recruitment: Not stated Prospective or retrospective: NR; samples appear to be collected prospectively but states that patient information was retrospectively collected from the hospital electronic medical records. |
||
| Patient characteristics and setting | Setting: Inpatient and asymptomatic (admitted or quarantined) Location: Self‐Defense Forces Central Hospital, Tokyo Country: Japan Dates: Feb 11 to May 13, 2020 Symptoms and severity: 88 (85%) symptomatic, including 16 (15%) severe (showing clinical symptoms of pneumonia ‐ dyspnea, tachypnea, saturation of percutaneous oxygen [SpO2] < 93%, and the need for oxygen therapy); 15 (15%) asymptomatic (including 4 pre‐symptomatic) Demographics: IPD provided ‐ median age 46, range 18‐87; 66 (64%) male Exposure history: Not reported |
||
| Index tests | Test name: ESPLINE® SARS‐CoV‐2 (no product code reported)
[Five other tests performed including RT‐PCR and RT‐LAMP, but not eligible for this review] Manufacturer: Fuji Rebio Inc Antibody: NP Antigen target: Not stated Test method: LFA (no reader device required) Samples used: Saliva (self‐collected) Transport media: None; around 500 μL saliva collected Sample storage: Stored at ‐80C until sample preparation Test operator: Not stated; implies laboratory staff Definition of test positivity: Not stated; appearance of test line implied Blinding reported: Not stated Timing of samples: saliva collected on admission to hospital; IPD reports this was median 7 days p.s.o (1‐14) |
||
| Target condition and reference standard(s) | Reference standard: RT‐qPCR on initial presentation (RT‐PCR was conducted on saliva samples as part of the study but this did not form part of the reference standard diagnosis) Definition of non‐COVID cases: Single RT‐PCR negative Genetic target(s): Not reported Samples used: NP or OP Timing of reference standard: On presentation or as part of mass screening; specific timing in regard to symptom onset was not reported for the original RT‐PCR and unclear if same day as saliva collection Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Unclear; saliva collected on day of admission to quarantine/hospital but NP/OP conducted at some point prior to that All patients received same reference standard: Yes Missing data: Not stated, no participant flow diagram reported Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: work was supported by the Health, Labour and Welfare Policy Research Grants, Research on Emerging and Re‐emerging Infectious Diseases and Immunization [grant number 20HA2002]. Publication status: Accepted manuscript Source: J Clin Microbiol Author COI: The authors declare that they have no conflicts of interests |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Unclear | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Nash 2020.
| Study characteristics | |||
| Patient Sampling | Unclear design to estimate sensitivity and specificity:
‐ samples from suspected patients submitted to 'PATH' (ww.path.org) for routine COVID diagnosis
[Second cohort of samples also tested using Spike‐based assay; excluded as assay requires use of centrifuge) Recruitment: Not stated Prospective or retrospective: Retrospective |
||
| Patient characteristics and setting | Setting: Unclear; samples provided to study authors by PATH (non‐profit organisation), protocol number 00004244 Location: Not reported Country: Not reported Dates: Not reported Symptoms and severity: Not reported Demographics: Not reported Exposure history: Not reported |
||
| Index tests | Test name: Direct antigen rapid test (DARTTM); NP‐based Manufacturer: E25Bio Inc (Cambridge MA); not yet available Antibody: NP Antigen target: anti‐N mouse monoclonal antibodies Test method: immunochromatographic paper‐based (CGIA) Samples used: Nasal; collection not described Transport media: Not stated Sample storage: banked frozen prior to testing Test operator: Not stated; presume lab staff Definition of test positivity: Visual line Blinding reported: Not stated Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Reference standard: qRT PCR; ThermoFisher/ AppliedBiosystems TaqPATH COVID‐19 Combo Kit (ThermoFisher, Waltham, MA USA) Definition of non‐COVID cases: As for cases; single negative PCR required Genetic target(s): N, S, and ORF1ab genes Samples used: Nasal (same swab) Timing of reference standard: Not stated Blinded to index test: Yes, conducted first Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous (Same swab) All patients received same reference standard: Yes Missing data: None reported Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Not stated |
||
| Comparative | |||
| Notes | Funding: The study is funded, in part, by a Bill and Melinda Gates Foundation Award (INV‐017872) to E25Bio, Inc. EN is funded by Tufts University DISC Seed Grant. MLN is supported by a FAPESP grant (#2020/04836‐0) and is a CNPq Research Fellow. AFV is supported by a FAPESP Fellow grant (#18/17647‐0). GRFC is supported by a FAPESP Fellow grant (#20/07419‐0). BHGAM 798 is supported by a FAPESP Scholarship (#19/06572‐2). Publication status: pre‐print Source: medRxiv Author COI: BN, AB, AR, MB, NS, AG, IB, and BBH are employed by or affiliated with E25Bio Inc. (www.e25bio.com), a company that develops diagnostics for epidemic viruses. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Unclear | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
PHE 2020(a).
| Study characteristics | |||
| Patient Sampling | Set of studies conducted by PHE and University of Oxford. This extraction relates to a two group study estimating sensitivity and specificity:
[1] residual frozen swabs from PCR+ in‐patients (n=200)
[2] residual fresh swab samples from PCR‐ patients (n=1000)
Swabs were sent to PHE Porton Down aafter routine testing
See other PHE 2020 extractions for other sub‐studies of Innova assay Recruitment: Unclear Prospective or retrospective: Retrospective |
||
| Patient characteristics and setting | Setting: Unclear; appears to be in‐patients (samples obtained from secondary healthcare setting; cases decsribed as from patients admitted to hsopital) Location: John Radcliffe Hospital, Oxford (Ag testing at PHE Porton Down) Country: UK Dates: March‐June 2020 (PCR+); August 2020 (PCR‐) Symptoms and severity: Not stated Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: Innova SARS‐CoV‐2 Antigen Rapid Qualitative Test Manufacturer: Innova Medical Group Antibody: Not stated Antigen target: Not stated Test method: Not stated Samples used: Naso‐ and oropharyngeal swabs Transport media: VTM (1ml) Sample storage: Frozen (PCR+); fresh (PCR‐) Test operator: Laboratory staff Definition of test positivity: Visual line; as per manufacturer Blinding reported: Not stated Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; not described. The pre‐print supplementary materials describes using the 'Roche platform' under the Phase 3b heading, and also provides the following text under the Phase 2 evaluation heading "Unless otherwise stated, all RT‐PCR testing was undertaken on the Roche Cobas® 6800 or 8800 system using their proprietary SARS‐CoV‐2 assay as per manufacturer’s instructions (with off‐board lysis using AVL buffer (Qiagen) and 5% Triton‐X100 (Sigma Aldrich)). This assay detects ORF‐1a/b as a SARS‐CoV‐2 specific target, and the E‐gene as a pan‐sarbecovirus target." Definition of non‐COVID cases: single negative PCR Genetic target(s): Not stated Samples used: Appears to be same sample as for Ag test Timing of reference standard: As for index test Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Same swab All patients received same reference standard: Yes Missing data: See below, plus 1 void PCR Uninterpretable results: Failure rates reported as: [1] 12/212, 6%; [2] 50/1040, 5.1% NB remaining samples per group (200 and 990) does not match with final numbers reported (178 and 940), however no explanation given in report. Indeterminate results (index test): Unclear Indeterminate results (reference standard): Unclear Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: PHE evaluation Publication status: Published Source: Online PHE report Author COI: None reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
PHE 2020(b).
| Study characteristics | |||
| Patient Sampling | Set of studies conducted by PHE and University of Oxford. This extraction relates to a single group study estimating sensitivity and specificity:
‐ samples obtained during a COVID‐19 outbreak at a Navy barracks (n=157 samples reported in pre‐print; 2x2 data provided by study investigators)
See other PHE extractions for other sub‐studies of Innova assay Recruitment: Unclear; presume consecutive Prospective or retrospective: Retrospective |
||
| Patient characteristics and setting | Setting: Outbreak investigation Location: Not stated Country: UK Dates: Not stated Symptoms and severity: Not stated Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: Innova SARS‐CoV‐2 Antigen Rapid Qualitative Test Manufacturer: Innova Medical Group Antibody: Not stated Antigen target: Not stated Test method: Not stated Samples used: OP swab used; self‐collected Transport media: VTM Sample storage: Transported at 4C to Porton Down for testing Test operator: Laboratory staff Definition of test positivity: Visual line; as per manufacturer Blinding reported: Not stated Timing of samples: One week after outbreak; no further details |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; not described. The pre‐print supplementary materials describes using the 'Roche platform' under the Phase 3b heading, and also provides the following text under the Phase 2 evaluation heading "Unless otherwise stated, all RT‐PCR testing was undertaken on the Roche Cobas® 6800 or 8800 system using their proprietary SARS‐CoV‐2 assay as per manufacturer’s instructions (with off‐board lysis using AVL buffer (Qiagen) and 5% Triton‐X100 (Sigma Aldrich)). This assay detects ORF‐1a/b as a SARS‐CoV‐2 specific target, and the E‐gene as a pan‐sarbecovirus target." Definition of non‐COVID cases: single negative PCR Genetic target(s): Not stated Samples used: Appears to be same sample as for Ag test Timing of reference standard: As for index test Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Same swab All patients received same reference standard: Yes Missing data: None reported Uninterpretable results: Failure rate reported as 6/157, 3.8% (Table 4 of pre‐print)NB resulting no. samples per group (n=151) does not quite match with final number reported (n=152) Indeterminate results (index test): Unclear Indeterminate results (reference standard): Unclear Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: PHE evaluation Publication status: Published and unpublished Source: Online PHE report, plus additional data provided by evaluation team Author COI: None reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
PHE 2020(c) [non‐HCW tested].
| Study characteristics | |||
| Patient Sampling | Set of studies conducted by PHE and University of Oxford. This extraction relates to a single group study estimating sensitivity and specificity:
‐ individuals presenting at a regional COVID‐19 testing centre as part of a Phase 4 community field service evaluation (n=1946; according to Table 3 of pre‐print)
See other PHE extractions for other sub‐studies of Innova assay Recruitment: Not stated; presume consecutive Prospective or retrospective: Not stated |
||
| Patient characteristics and setting | Setting: regional COVID‐19 testing centres as part of an NHS Test and Trace service evaluation involving the general public Location: Not stated Country: UK Dates: Not stated Symptoms and severity: Not stated, presumed 'mainly symptomatic' for purposes of review analyses Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: Innova SARS‐CoV‐2 Antigen Rapid Qualitative Test Manufacturer: Innova Medical Group Antibody: Not stated Antigen target: Not stated Test method: Not stated Samples used: Anterior nasal and combined oropharyngeal samples Transport media: Dry swab Sample storage: None; immediate testing Test operator: self‐trained non‐HCW ('Boots' member of staff); described in pre‐print as an “operator” or as 'self‐trained members of the public'. Definition of test positivity: Visual line; as per manufacturer Blinding reported: Yes; conducted on site Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; no details. The pre‐print supplementary materials describes using the 'Roche platform' under the Phase 3b heading, and also provides the following text under the Phase 2 evaluation heading "Unless otherwise stated, all RT‐PCR testing was undertaken on the Roche Cobas® 6800 or 8800 system using their proprietary SARS‐CoV‐2 assay as per manufacturer’s instructions (with off‐board lysis using AVL buffer (Qiagen) and 5% Triton‐X100 (Sigma Aldrich)). This assay detects ORF‐1a/b as a SARS‐CoV‐2 specific target, and the E‐gene as a pan‐sarbecovirus target." Definition of non‐COVID cases: Cases only study Genetic target(s): Not stated Samples used: Not stated; paired swabs obtained Timing of reference standard: As for index test Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Paired swabs; simultaneous All patients received same reference standard: Yes Missing data: Initial sample of 1946 reported, 27 failed, leaving 1919 for inclusion, however data for only 1686 samples are provided in the pre‐print (1314 PCR‐ in Table 3 and 372 PCR+ in text pg 7), a difference of 233 samples. Uninterpretable results: Failure rate reported as 27/1946 failed, 1.4% Indeterminate results (index test): Unclear Indeterminate results (reference standard): Unclear Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: PHE evaluation Publication status: Published Source: Online PHE report Author COI: none reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
PHE 2020(d) [HCW tested].
| Study characteristics | |||
| Patient Sampling | Set of studies conducted by PHE and University of Oxford. This extraction relates to a single group study estimating sensitivity alone:
‐ individuals presenting at one of 14 regional drive‐through COVID‐19 NHS test and trace centres as part of the FALCON C‐19 (Facilitating Accelerated Clinical validation Of Novel diagnostics for COVID‐19, 20/WA/0169, IRAS 284229) phase 3b study; those with a positive PCR result were asked to return for a re‐test within 5 days of the original test result. From the originally published report (Nov 2020) it appears that only participants with samples that were positive on PCR at the second sampling were included. PHE 2020(d) [HCW tested] is for health care worker tested samples, and PHE 2020(d) [Lab tested] is for laboratory scientist tested samples See other PHE extractions for other sub‐studies of Innova assay Recruitment: Not stated; presume consecutive Prospective or retrospective: Prospective Number of samples (cases): 479 (479) ; 267 tested by HCWs, 212 tested by laboratory scientists |
||
| Patient characteristics and setting | Setting: NHS drive through test and trace centres; no further details Location: 14 regional centres Country: UK Dates: 17 Sept to 23 Oct 2020 Symptoms and severity: Only described for all 421 included participants in PHE 2020(d) [HCW tested] and PHE 2020(d) [Lab tested] combined: Suppl Table 2 reports 40 (9.5%) asymptomatic, 59 (14%) with no data, leaving 322 with >=1 symptom recorded. It is not stated whether symptoms were present at the time of the original swab or at the time of the second sampling therefore data for the asymptomatic group have not been included in analyses . NB: text reports data for 41 asymptomatic and 344 symptomatic from the Phase 3b study (total n = 385) Demographics: For the 421 participants: median age 33 y, 168, 40% male Exposure history: Not stated |
||
| Index tests | Test name: Innova SARS‐CoV‐2 Antigen Rapid Qualitative Test Manufacturer: Innova Medical Group Antibody: Not stated Antigen target: Not stated Test method: Not stated Samples used: combined anterior nasal and oropharyngeal swabs (1 stored as a dry swab and 1 swab placed in VTM; swabs were self‐collected Transport media: Dry swab Sample storage: None; immediate testing (delay to testing at PHE for [B] is unclear) Test operator: PHE 2020(d) [HCW tested] HCW on‐site, PHE 2020(d) [Lab tested] Laboratory scientist at PHE Definition of test positivity: Visual line; as per manufacturer Blinding reported: Yes Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; no details. The pre‐print supplementary materials describes using the 'Roche platform' under the Phase 3b heading, and also provides the following text under the Phase 2 evaluation heading "Unless otherwise stated, all RT‐PCR testing was undertaken on the Roche Cobas® 6800 or 8800 system using their proprietary SARS‐CoV‐2 assay as per manufacturer’s instructions (with off‐board lysis using AVL buffer (Qiagen) and 5% Triton‐X100 (Sigma Aldrich)). This assay detects ORF‐1a/b as a SARS‐CoV‐2 specific target, and the E‐gene as a pan‐sarbecovirus target." Definition of non‐COVID cases: Genetic target(s): Not stated Samples used: Appears to be combined NOP swabs in VTM; obtained at same time as second sampling for Ag testing (5 days after 1st positive PCR) Timing of reference standard: As for index test Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: appears to be simultaneous (if 2nd PCR result was used). All patients received same reference standard: Yes Missing data: Initial sample of 267 reported, 27 failed, leaving 240 for inclusion however data for only 223 HCW tested samples are provided in the pre‐print (text pg 7). The original report (Nov 2020) documented 16 samples in this cohort that were either PCR‐ (n=15) or void (n=1) presumably at the time of the second sampling (as only PCR+ were invited for Ag testing. Although the numbers don't quite add up, it seems likely that this could explain the difference between the 240 and 223 samples. Uninterpretable results: Failure rates reported as: [A] 28/296, 10.4%; [B] 9/221, 4.2% Indeterminate results (index test): Unclear Indeterminate results (reference standard): Unclear Unit of analysis: Patients |
||
| Comparative | |||
| Notes | |||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Low concern | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | No | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
PHE 2020(d) [Lab tested].
| Study characteristics | |||
| Patient Sampling | Set of studies conducted by PHE and University of Oxford. This extraction relates to a single group study estimating sensitivity alone: ‐ individuals presenting at one of 14 regional drive‐through COVID‐19 NHS test and trace centres as part of the FALCON C‐19 (Facilitating Accelerated Clinical validation Of Novel diagnostics for COVID‐19, 20/WA/0169, IRAS 284229) phase 3b study; those with a positive PCR result were asked to return for a re‐test within 5 days of the original test result. From the originally published report (Nov 2020) it appears that only participants with samples that were positive on PCR at the second sampling were included. PHE 2020(d) [HCW tested] is for health care worker tested samples, and PHE 2020(d) [Lab tested] is for laboratory scientist tested samples See other PHE extractions for other sub‐studies of Innova assay Recruitment: Not stated; presume consecutive Prospective or retrospective: Prospective Number of samples (cases): 479 (479) ; 267 tested by HCWs, 212 tested by laboratory scientists |
||
| Patient characteristics and setting | Setting: NHS drive trhough test and trace centres; no further details Location: 14 regional centres Country: UK Dates: 17 Sept to 23 Oct 2020 Symptoms and severity: Only described for all 421 included participants in PHE 2020(d) [HCW tested] and PHE 2020(d) [Lab tested] combined: Suppl Table 2 reports 40 (9.5%) asymptomatic, 59 (14%) with no data, leaving 322 with >=1 symptom recorded. It is not stated whether symptoms were present at the time of the original swab or at the time of the second sampling therefore data for the asymptomatic group have not been included in analyses . NB: text reports data for 41 asymptomatic and 344 symptomatic from the Phase 3b study (total n = 385) Demographics: For the 421 participants: median age 33 y, 168, 40% male Exposure history: Not stated |
||
| Index tests | Test name: Innova SARS‐CoV‐2 Antigen Rapid Qualitative Test Manufacturer: Innova Medical Group Antibody: Not stated Antigen target: Not stated Test method: Not stated Samples used: combined anterior nasal and oropharyngeal swabs (1 stored as a dry swab and 1 swab placed in VTM; swabs were self‐collected Transport media: Dry swab Sample storage: None; immediate testing (delay to testing at PHE for [B] is unclear) Test operator: PHE 2020(d) [HCW tested] HCW on‐site, PHE 2020(d) [Lab tested] Laboratory scientist at PHE Definition of test positivity: Visual line; as per manufacturer Blinding reported: Yes for [A] unclear for [B] Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; no detailsThe pre‐print supplementary materials describes using the 'Roche platform' under the Phase 3b heading, and also provides the following text under the Phase 2 evaluation heading "Unless otherwise stated, all RT‐PCR testing was undertaken on the Roche Cobas® 6800 or 8800 system using their proprietary SARS‐CoV‐2 assay as per manufacturer’s instructions (with off‐board lysis using AVL buffer (Qiagen) and 5% Triton‐X100 (Sigma Aldrich)). This assay detects ORF‐1a/b as a SARS‐CoV‐2 specific target, and the E‐gene as a pan‐sarbecovirus target." Definition of non‐COVID cases: Genetic target(s): Not stated Samples used: Appears to be combined NOP swabs in VTM; obtained at same time as second sampling for Ag testing (5 days after 1st positive PCR) Timing of reference standard: As for index test Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: appears to be simultaneous (if 2nd PCR result was used). All patients received same reference standard: Yes Missing data: Initial sample of 212 reported, 9 failed, leaving 203 for inclusion however data for only 198 lab scientist tested samples are provided in the pre‐print (text pg 7). The original report (Nov 2020) documented 8 samples in this cohort that were PCR‐ presumably at the time of the second sampling (as only PCR+ were invited for Ag testing. Although the numbers don't quite add up, it seems likely that this could explain the difference between the 203 and 198 samples. Uninterpretable results: Failure rate reported as: 9/212, 4.2% Indeterminate results (index test): Unclear Indeterminate results (reference standard): Unclear Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: PHE evaluation Publication status: Published Source: Online PHE report Author COI: None reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | No | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
PHE 2020(e).
| Study characteristics | |||
| Patient Sampling | Set of studies conducted by PHE and University of Oxford. This extraction relates to a single group study estimating specificity alone:
‐ PHE and hospital staff volunteering for testing (n=538)
See other PHE extractions for other sub‐studies of Innova assay Recruitment: Not stated; presume consecutive Prospective or retrospective: Not stated |
||
| Patient characteristics and setting | Setting: Screening Location: PHE and John Radcliffe Hospital, Oxford Country: UK Dates: Not stated Symptoms and severity: Not stated; hospital staff described as asymptomatic Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: Innova SARS‐CoV‐2 Antigen Rapid Qualitative Test Manufacturer: Innova Medical Group Antibody: Not stated Antigen target: Not stated Test method: Not stated Samples used: N OP swab for PHE staff; NP swab for hospital staff. All self‐collected Transport media: Dry swab Sample storage: None; immediate testing Test operator: Not stated; presumably laboratory scientist at PHE Definition of test positivity: Visual line; as per manufacturer Blinding reported: Unclear Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; no details (single negative PCR ok for asymptomatic). The pre‐print supplementary materials describes using the 'Roche platform' under the Phase 3b heading, and also provides the following text under the Phase 2 evaluation heading "Unless otherwise stated, all RT‐PCR testing was undertaken on the Roche Cobas® 6800 or 8800 system using their proprietary SARS‐CoV‐2 assay as per manufacturer’s instructions (with off‐board lysis using AVL buffer (Qiagen) and 5% Triton‐X100 (Sigma Aldrich)). This assay detects ORF‐1a/b as a SARS‐CoV‐2 specific target, and the E‐gene as a pan‐sarbecovirus target." DGenetic target(s): Not stated Samples used: Not stated; presume same or paired swab Timing of reference standard: As for index test Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Unclear, may have been a few days All patients received same reference standard: Yes Missing data: Initial sample of 570 reported (358 hospital staff and 212 PHE staff), 36 failed (Table 4: 17 hospital staff and 19 PHE staff), leaving 534 for inclusion. Data for 538 included Uninterpretable results: Failure rate reported as 17/358, 4.7% (hospital) 19/212, 8.9% (PHE) Indeterminate results (index test): Unclear Indeterminate results (reference standard): Unclear Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: PHE evaluation Publication status: Published Source: Online PHE report Author COI: none reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | Low concern | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Porte 2020a.
| Study characteristics | |||
| Patient Sampling | Two‐group study to estimate sensitivity and specificity for diagnosis of active disease:
‐ samples from suspected COVID‐19 cases (n = 1453) with deliberate sampling of PCR‐positive and negative cases on a 2:1 basis (n = 127) Recruitment: convenience sampling Prospective or retrospective: retrospectively Number of samples (samples with confirmed SARS‐CoV‐2): 127 (82) |
||
| Patient characteristics and setting | Setting: outpatients attending ED at private medical centre (hospital) Location: Clínica Alemana, Santiago Country: Chile Dates: 16‐21 March 2020 Symptoms and severity: cough 94 (74.6%) Fever 77 (61.1%) Median duration of symptoms of 2 days (IQR 1–4; range 0‐12) Duration of symptoms: day 0‐3 91 (72.2%); day 4‐7 27 (22.4%); day ≥ 8 8 (6.3%) Demographics: 68 male (53.5%), median age 38 years (IQR 29.5–44; range 1–91) Exposure history: not stated |
||
| Index tests | Test name: diagnostic Kit for 2019‐Novel Coronavirus (2019‐nCoV) Ag Test (Cat. N° YRLF04401025, lot N° 2002N408) Manufacturer: Bioeasy Biotechnology Co., Shenzhen, China Antigen target: SARS‐CoV‐2 nucleocapsid protein Antibody: not stated Test method: FIA Samples used: remnant OP and NP swabs in 3 mL UTM Transport media: UTM‐RT System, Copan Diagnostics, Murrieta, CA, USA Sample storage: stored at 4 °C and tested within 48 h Test operator: laboratory technician Definition of test positivity: not stated; test "automatically delivers a positive or negative qualitative result" Positive or negative defined qualitatively Blinding reported: yes Timing of samples: on presentation Within 48 h of the PCR test but it doesn't say when PCR test was performed (median duration of symptoms reported in D9) |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR (COVID‐19 Genesig Real‐Time PCR assay (Primer Design Ltd., Chandler's Ford, UK)); Ct ≤ 40 considered positive Definition of non‐COVID cases: single RT‐PCR negative Genetic target(s): not stated Samples used: as for index test; same OP and NP swabs used Timing of reference standard: median 2 d post symptom onset (IQR 1‐4; range 0‐12) Blinded to index test: yes (index test done within 48 h of PCR test) Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: same sample used; within 48 h All participants received same reference standard: yes Missing data: None; partipant flow diagram reported Uninterpretable results: not reported Indeterminate results (index test): not reported Indeterminate results (reference standard): not reported Unit of analysis: participants |
||
| Comparative | |||
| Notes | Funding: this work did not receive funding Publication status: preprint (not peer‐reviewed) Source: SSRN Author COI: all study authors declare no competing interests |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Yes | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Low risk | ||
Porte 2020b [A].
| Study characteristics | |||
| Patient Sampling | Multi group study to estimate sensitivity and specificity:
(1) Covid‐19 patients presenting within 5 days of symptom onset (n=32)
(2) symptomatic patients with negative PCR (n=20)
(3) asymptomatic patients screened prior to surgery (n=12)
[27 PCR+ and 19 PCR‐ samples were used in Weitzel 2020 (different assays)] Recruitment: Not stated; appears to be convenience Prospective or retrospective: Retrospective |
||
| Patient characteristics and setting | Setting: Private clinic (classed as Emergence Dept) Location: Clínica Alemana, Santiago Country: Chile Dates: Not stated Symptoms and severity: Not reported; 12 asymptomatic Demographics: Total sample median age 39 y (IQR 36.7‐57); 33, 52% male Exposure history: Not reported |
||
| Index tests | Comparative study of two Ag tests (no product codes reported); Porte 2020b [A] data relate to test [A], see Porte 2020b [B] tests [B] data. [A] SOFIA SARS Antigen FIA [B] STANDARD® F COVID‐19 Ag FIA Manufacturer: [A] Quidel Corporation, San Diego, CA, USA [B] SD Biosensor Inc, Gyeonggi‐do, Republic of Korea Antibody: NP (both) Antigen target: Not stated Test method: Both FIA Samples used: naso‐oropharyngeal flocked swabs; obtained by trained personnel Transport media: UTM‐RT® System, Copan Diagnostics Sample storage: stored at ‐80 degrees C following RT‐PCR Test operator: Laboratory staff Definition of test positivity: As per manufacturer; both using analyzer device Blinding reported: Yes; blinded to RT‐PCR result Timing of samples: All <5 days p.s.o; median PCR+: 2 days (IQR 1‐3); PCR‐: 1 day (IQR 0.75‐4) |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; COVID‐19 Genesig®, Primerdesign Ltd., Chandler´s Ford, UK;
(Ct) values ≤40 were considered positive Definition of non‐COVID cases: As for cases Genetic target(s): Not stated Samples used: NOP; as for index test Timing of reference standard: Not stated Blinded to index test: Unclear Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous; same sample All patients received same reference standard: Yes Missing data: None reported, no participant flow diagram reported Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: This research did not receive any specific grant from funding agencies in the public, commercial, or not‐for‐profit sectors. Publication status: Published Source: Int J Infect Dis Author COI: All authors declare no competing interests |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Porte 2020b [B].
| Study characteristics | |||
| Patient Sampling | Comparative study of two Ag tests; Porte 2020b [A] reports full study characteristics and QUADAS | ||
| Patient characteristics and setting | |||
| Index tests | Comparative study of two Ag tests (no product codes reported); Porte 2020b [B] data relate to test [B], see Porte 2020b [A] for data relate to test [A] and QUADAS entries [A] SOFIA SARS Antigen FIA [B] STANDARD® F COVID‐19 Ag FIA Manufacturer: [A] Quidel Corporation, San Diego, CA, USA [B] SD Biosensor Inc, Gyeonggi‐do, Republic of Korea Antibody: NP (both) Antigen target: Not stated Test method: Both FIA Samples used: naso‐oropharyngeal flocked swabs; obtained by trained personnel Transport media: UTM‐RT® System, Copan Diagnostics Sample storage: stored at ‐80 degrees C following RT‐PCR Test operator: Laboratory staff Definition of test positivity: As per manufacturer; both using analyzer device Blinding reported: Yes; blinded to RT‐PCR result Timing of samples: All <5 days p.s.o; median PCR+: 2 days (IQR 1‐3); PCR‐: 1 day (IQR 0.75‐4) |
||
| Target condition and reference standard(s) | Comparative study of two Ag tests; Porte 2020b [A] reports full study characteristics and QUADAS | ||
| Flow and timing | Comparative study of two Ag tests; Porte 2020b [A] reports full study characteristics and QUADAS | ||
| Comparative | |||
| Notes | |||
Rhoads 2020.
| Study characteristics | |||
| Patient Sampling | Single‐group study to estimate sensitivity:
‐ samples positive using standard of care testing (n = 96)
(14 negative controls (UTM) included to control for carry‐over contamination only) Recruitment: convenience Prospective or retrospective: retrospective (remnant samples) Number of samples (samples with confirmed SARS‐CoV‐2): 96 (96) |
||
| Patient characteristics and setting | Setting: not stated; includes self‐collected and provided‐collected samples Location: not stated; author institutions University Hospitals Cleveland Medical Centre and Case Western Reserve University Country: USA Dates: not stated Symptoms and severity: not stated Demographics: not stated Exposure history: not stated |
||
| Index tests | Test name: ID NOW (product codes not reported) Manufacturer: Abbott; Chicago, USA Also reports evaluation of Diasorin Simplexa (not eligible for this review) Antigen target: not stated Antibody: N/A Test method: isothermal amplification test Samples used: nasal swabs (self‐collected) and NP swabs (provider collected); all remnant samples Transport media: nasal swabs (2 mL normal saline) and NP swabs (3 mL UTM) Sample storage: not stated Test operator: not stated; presume laboratory staff Definition of test positivity: not stated; as per manufacturer Blinding reported: not stated Timing of samples: not stated |
||
| Target condition and reference standard(s) | Reference standard: standard of care testing for original samples; remnant samples re‐tested with modified CDC RT‐PCR (using 7500 Fast instrument and using alternate RNA extraction method (Maxwell RSC 6 instrument with Viral TNA Kit (Cat# AS1330; Promega, Madison, USA)); samples with 1 positive target detected considered positive instead of "inconclusive" Definition of non‐COVID cases: as for index test Genetic target(s): N1 and N2 Samples used: as for index test Timing of reference standard: as for index test Blinded to index test: as for index test Incorporated index test: as for index test |
||
| Flow and timing | Time interval between index and reference tests: same samples used All participants received same reference standard: yes Missing data: none reported, no participant flow diagram reported Uninterpretable results: none reported Indeterminate results (index test): none reported Indeterminate results (reference standard): RT‐PCR detected only 1 of 2 targets for 2 samples (both considered positive (diagnosed as positive on original sample testing); both were negative on index test) Unit of analysis: not stated; only samples reported |
||
| Comparative | |||
| Notes | Funding: no outside funding used to support the investigation Publication status: accepted manuscript Source: Journal of Clinical Microbioloby Author COI: COI not mentioned by study authors |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Unclear risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Schildgen 2020 [A].
| Study characteristics | |||
| Patient Sampling | Unclear design; appears to be single cohort with deliberate sampling of PCR+/PCR‐:
[1] RT‐PCR positive BAL or throat wash samples (n=42)
[2] RT‐PCR negative samples (n=31)
Described as pilot sample panel Recruitment: Appears to be convenience Prospective or retrospective: Not stated; presume retrospective |
||
| Patient characteristics and setting | Setting: Not stated Location: Authors institution: Kliniken der Stadt Köln gGmbH (Koln city clinics) Country: Germany Dates: Not stated Symptoms and severity: Not stated for BAL samples, throat wash from 23 symptomatic and 27 asymptomatic people. Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Comparative study of three Ag tests (no product codes reported); Schildgen 2020 [A] data relate to test [A], see Schildgen 2020 [B] and Schildgen 2020 [C] for data relate to tests [B] and [C]. Test name: [A] BIOCREDIT [B] Panbio [C] SARS‐CoV‐2 Rapid Antigen test Manufacturer: [A] RapiGEN [B] Abbott [C] Roche Antibody: Not stated Antigen target: Not stated Test method: All LFA Samples used: BAL (n=13); throat wash (n=50, including 27 from asymptomatic) Transport media: Not stated Sample storage: Not stated Test operator: Not stated; presume lab staff Definition of test positivity: As per manufacturer Blinding reported: Not stated Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; RealStar® SARS‐CoV‐2 RT‐PCR Kit, Altona, Germany Definition of non‐COVID cases: As for cases Genetic target(s): Not stated Samples used: BAL or throat wash; As per index test Timing of reference standard: Not stated Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Same swab All patients received same reference standard: Yes Missing data: 8 PCR invalid samples also tested; 2/8 invalid in one AG assay each, 3/8 negative in all 3 Ag assays Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Unclear |
||
| Comparative | |||
| Notes | Funding: The study did not receive any external funding Publication status: preprint Source: medRxiv Author COI: The authors declare that they have no conflicts of interest |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Schildgen 2020 [B].
| Study characteristics | |||
| Patient Sampling | Comparative study of three Ag tests; Schildgen 2020 [A] reports full study characteristics and QUADAS | ||
| Patient characteristics and setting | Comparative study of three Ag tests; Schildgen 2020 [A] reports full study characteristics and QUADAS | ||
| Index tests | Comparative study of three Ag tests (no product codes reported); Schildgen 2020 [B] data relate to test [B], see Schildgen 2020 [A] and Schildgen 2020 [C] for data relate to tests [A] and [C], and for QUADAS entries. Test name: [A] BIOCREDIT [B] Panbio [C] SARS‐CoV‐2 Rapid Antigen test Manufacturer: [A] RapiGEN [B] Abbott [C] Roche Antibody: Not stated Antigen target: Not stated Test method: All LFA Samples used: BAL (n=13); throat wash (n=50, including 27 from asymptomatic) Transport media: Not stated Sample storage: Not stated Test operator: Not stated; presume lab staff Definition of test positivity: As per manufacturer Blinding reported: Not stated Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Comparative study of three Ag tests; Schildgen 2020 [A] reports full study characteristics and QUADAS | ||
| Flow and timing | Comparative study of three Ag tests; Schildgen 2020 [A] reports full study characteristics and QUADAS | ||
| Comparative | |||
| Notes | |||
Schildgen 2020 [C].
| Study characteristics | |||
| Patient Sampling | Comparative study of three Ag tests; Schildgen 2020 [A] reports full study characteristics and QUADAS | ||
| Patient characteristics and setting | Comparative study of three Ag tests; Schildgen 2020 [A] reports full study characteristics and QUADAS | ||
| Index tests | Comparative study of three Ag tests (no product codes reported); Schildgen 2020 [C] data relate to test [C], see Schildgen 2020 [A] and Schildgen 2020 [B] for data relate to tests [A] and [B], and for QUADAS entries. Test name: [A] BIOCREDIT [B] Panbio [C] SARS‐CoV‐2 Rapid Antigen test Manufacturer: [A] RapiGEN [B] Abbott [C] Roche Antibody: Not stated Antigen target: Not stated Test method: All LFA Samples used: BAL (n=13); throat wash (n=50, including 27 from asymptomatic) Transport media: Not stated Sample storage: Not stated Test operator: Not stated; presume lab staff Definition of test positivity: As per manufacturer Blinding reported: Not stated Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Comparative study of three Ag tests; Schildgen 2020 [A] reports full study characteristics and QUADAS | ||
| Flow and timing | Comparative study of three Ag tests; Schildgen 2020 [A] reports full study characteristics and QUADAS | ||
| Comparative | |||
| Notes | |||
Scohy 2020.
| Study characteristics | |||
| Patient Sampling | Single group study including NP swabs submitted to laboratory at a large tertiary hospital (n=148) Recruitment: Random sample Prospective or retrospective: Not stated |
||
| Patient characteristics and setting | Setting: Unclear; presume microbiology laboratory takes samples from number of sources Location: Cliniques universitaires Saint‐Luc Hospital, Brussels Country: Belgium Dates: Apr 6 to Apr 21, 2020 Symptoms and severity: 86 (58%) symptomatic, 45 (30%) asymptomatic, 17 (11%) symptom status not reported; Cases only: viral load <25 Ct 10 (9%), >=25 Ct 96 (91%) Demographics: median age 57.5 (0, 94y); 64 (43%) male Exposure history: Not reported |
||
| Index tests | Test name: COVID‐19 Ag Respi‐Strip (product code not reported) Manufacturer: Coris Bioconcept Antibody: NP Antigen target: monoclonal antibody Test method: CGIA Samples used: NP Transport media: Not stated Sample storage: "If the rapid antigen test was not performed immediately, samples were stored at 4 °C until the test" Test operator: Not stated Definition of test positivity: Visual appearance of T line; also states that "Two versions of the test were evaluated. On the second version, conjugate was coupled on a different way and the control line was optimized." Blinding reported: Unclear Timing of samples: Not reported |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR: genesig® Real‐Time PCR assay (Primerdesign Ltd, Chandler’s Ford, UK); <40 Ct Definition of non‐COVID cases: Single PCR negative Genetic target(s): RdRp Samples used: NP; same as for index Timing of reference standard: Not stated Blinded to index test: Yes Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Same sample All patients received same reference standard: Yes Missing data: None reported, no participant flow diagram reported Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: No funding statement reported; COVID‐19 Ag Respi‐Strip tests provided by Coris BioConcept. Publication status: Published Source: J Clin Virol Author COI: The authors declare no conflicts of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Shrestha 2020.
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity:
‐ subjects who were close contacts of confirmed cases identified through contact tracing, residing in quarantine centre (n=113) Recruitment: Convenience Prospective or retrospective: Not stated; appears prospective |
||
| Patient characteristics and setting | Setting: Contact tracing Location: Not applicable; author institutions include Shukraraaj Tropical and Infectious Disease Hospital, Kathmandu Country: Nepal Dates: Aug to Sep 2020 Symptoms and severity: All asymptomatic Demographics: Range 13 to 74; 89, 79% male Exposure history: All exposed to confirmed case |
||
| Index tests | Test name: BIOCREDIT Manufacturer: RapiGen Antibody: Not stated Antigen target: Not stated Test method: Not stated Samples used: NP Transport media: None used Sample storage: None reported; other sample from the same individual was processed for the results as instructed by the manufacturing company of antigen kit Test operator: Lab technician (trained) Definition of test positivity: Visual line; as per manufacturer. Blinding reported: Unclear; appears to be Yes Timing of samples: Day 5 of quarantine |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; not detailed, "followed the standard protocol regulated by WHO, instruction manual of company and as per NHTC training regarding sample collection and transport" Definition of non‐COVID cases: As for cases; single negative Genetic target(s): Not stated Samples used: NP in 3mL VTM Timing of reference standard: As for index test Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous, paired samples All patients received same reference standard: Yes Missing data: None reported Uninterpretable results: None reported Indeterminate results (index test): Tests were repeated for samples with indistinct outcomes. Indeterminate results (reference standard): Unit of analysis: Patient |
||
| Comparative | |||
| Notes | Funding: No funding statement provided Publication status: Published Source: KATHMANDU UNIVERSITY MEDICAL JOURNAL Author COI: No COI statement provided |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Smithgall 2020 [A].
| Study characteristics | |||
| Patient Sampling | Two‐group study to estimate sensitivity and specificity:
‐ patients undergoing routine clinical testing by RT‐PCR (n = 113) Recruitment: unclear; describes deliberate sampling of samples with high, medium and low Ct values on the reference standard RT‐PCR Prospective or retrospective: unclear; residual swabs used but testing undertaken within 48 h of sample collection Number of samples (samples with confirmed SARS‐CoV‐2): 113 (88) |
||
| Patient characteristics and setting | Setting: inpatient and ED (n from each not reported) Location: not stated; author institution is Columbia University Irving Medical Centre Country: USA Dates: 8‐13 April 2020 Symptoms and severity: not stated Demographics: 111 adult (range 23‐101 years; average 65 years for RT‐PCR‐positive and 43 years for RT‐PCR‐negative); 2 paediatric (age 1 day and 5 days) 61, 54% male Exposure history: not stated |
||
| Index tests | Test name: [A] ID NOW (see Smithgall 2020 [B] for details of comparator test) (product codes not reported) Manufacturer: [A] Abbott Antigen target: [A] RdRp gene Antibody: N/A Test method: [A] isothermal PCR Samples used: residual NP swabs (collection not described) Transport media: 3 mL VTM (M4RT VTM; ThermoFisher Scientific, Waltham, MA) or UTM (UTM; Becton Dickinson and Co., Franklin Lakes, NJ) Sample storage: stored at 4 °C; testing completed within 48 h of sample collection Test operator: not stated; presume laboratory staff Definition of test positivity: automated as per manufacturer Blinding reported: not stated Timing of samples: not stated; presume on admission or presentation at ED |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR with cobas SARS‐CoV‐2 assay on the 6800 platform (Roche Diagnostics, Indianapolis, IN); threshold not stated, all Ct values < 37 on both target genes Definition of non‐COVID cases: not stated; presume single RT‐PCR negative Genetic target(s): ORF1 a/b, E‐gene Samples used: as for index test Timing of reference standard: as for index test Blinded to index test: as for index test Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: simultaneous; same samples used All participants received same reference standard: yes Missing data: none reported Uninterpretable results: Indeterminate results (index test): Xpert: 1 sample was a presumptive positive based on detection of E‐gene target but not the N2 target Indeterminate results (reference standard): none reported Unit of analysis: participants |
||
| Comparative | |||
| Notes | Funding: none reported Publication status: published Source: Journal of Clinical Virology Author COI: study authors report no conflicts of interest present |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | No | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Smithgall 2020 [B].
| Study characteristics | |||
| Patient Sampling | See Smithgall 2020 [A] for full study details and QUADAS‐2 entries | ||
| Patient characteristics and setting | See Smithgall 2020 [A] for full study details and QUADAS‐2 entries | ||
| Index tests | Test name: [B] Xpert Xpress (product codes not reported) (see Smithgall 2020 [A] for details of comparator test) Manufacturer: [B] Cepheid Antigen target: [B] N2, E genes Antibody: N/A Test method: [B] automated RT‐PCR Samples used: residual NP swabs (collection not described) Transport media: 3 mL VTM (M4RT VTM; ThermoFisher Scientific, Waltham, MA) or UTM (UTM; Becton Dickinson and Co., Franklin Lakes, NJ) Sample storage: stored at 4 °C; testing completed within 48 h of sample collection. Test operator: not stated; presume laboratory staff Definition of test positivity: presumptive positive (only E gene present) considered positive (re‐testing recommended on IFU) Blinding reported: not stated Timing of samples: not stated; presume on admission or presentation at ED |
||
| Target condition and reference standard(s) | See Smithgall 2020 [A] for full study details and QUADAS‐2 entries | ||
| Flow and timing | See Smithgall 2020 [A] for full study details and QUADAS‐2 entries | ||
| Comparative | |||
| Notes | See Smithgall 2020 [A] for full study details and QUADAS‐2 entries | ||
SoRelle 2020.
| Study characteristics | |||
| Patient Sampling | Unclear design to estimate sensitivity and specificity:
paired saliva and NP samples from participants symptomatic for COVID‐19 (n=83)
[Additional saliva samples included for comparison of ID NOW with Xpert Xpress; not extracted for this review] Recruitment: Not stated Prospective or retrospective: Not stated |
||
| Patient characteristics and setting | Setting: Unclear Location: From authors institutions: University of Texas Southwestern Medical Center, Dallas Country: USA Dates: Not reported Symptoms and severity: Not reported Demographics: Not reported Exposure history: Not reported |
||
| Index tests | Test name: ID NOW (no product codes) Manufacturer: Abbott Diagnostics Antibody: Not stated Antigen target: n/a Test method: Isothermal PCR Samples used: Saliva; collection not described Transport media: Not stated Sample storage: Not stated Test operator: Not stated; presume lab staff Definition of test positivity: As per manufacturer Blinding reported: Not stated Timing of samples: Not stated; chart review of patients with FN results against either RT‐PCR (NP) Xpert Xpress (Saliva) (n=9) showed 6/9 tested >2 weeks after symptom onset |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; either Xpert® Xpress SARS‐CoV‐2 (Cepheid) or Abbott RealTime SARS‐CoV‐2 (Abbott Molecular) RT‐PCR assays; n per assay is not reported Definition of non‐COVID cases: As for cases (single negative) Genetic target(s): Not stated Samples used: NP in VTM (paired) Timing of reference standard: Not stated Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Paired All patients received same reference standard: Yes Missing data: None reported, no participant flow diagram reported Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported; presumptive positives not mentioned Unit of analysis: Patients? |
||
| Comparative | |||
| Notes | Funding: No funding statement reported Publication status: Published letter Source: Clin Chim Acta Author COI: No COI statement reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Unclear | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Stevens 2020.
| Study characteristics | |||
| Patient Sampling | Unclear design to estimate sensitivity and specificity:
‐ selected residual samples from symptomatic and asymptomatic individuals undergoing routine testing; selected to represent the full range of Ct values Recruitment: Convenience Prospective or retrospective: Retrospective |
||
| Patient characteristics and setting | Setting: Unclear; laboratory‐based, serving adult and pediatric tertiary care hospitals Location: Stanford Healthcare Virology Laboratory, Stanford Country: USA Dates: Mar 31 to Apr 7 Symptoms and severity: Unclear; 'symptomatic and asymptomatic'; Of 54 cases, 10 (19%) were low viral low (Ct>35) Demographics: Not reported Exposure history: Not reported |
||
| Index tests | Test name: Xpert Xpress (no product code) Manufacturer: Cepheid Inc Antibody: E, N2 Antigen target: n/a Test method: Automated RT‐PCR Samples used: NP in VTM Transport media: VTM (MicroTest M4RT, Remel Inc., San Diego, CA) Sample storage: All samples frozen at ‐80°C prior to testing on the Xpert system Test operator: Not stated; presume lab staff Definition of test positivity: Presence of N2 +/‐ E gene; E gene only considered presumptive positive Blinding reported: Not stated Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; Panther Fusion SARS‐CoV‐2 Assay (Hologic, Inc., San Diego, CA); interpreted based on the manufacturer’s cycle threshold cut‐off value Definition of non‐COVID cases: As for cases; single negative Genetic target(s): Two regions of ORF1ab Samples used: NP in VTM; as for index test Timing of reference standard: Not stated Blinded to index test: Yes, conducted first Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Same sample All patients received same reference standard: Yes Missing data: 6 samples excluded due to insufficient sample volume Uninterpretable results: 1 RT‐PCR positive sample re‐tested on Xpert Xpress due to initial interpretation of no results (invalid); Xpert +ve on re‐test Indeterminate results (index test): No presumptive positives were observed Indeterminate results (reference standard): 1 RT‐PCR positive sample that was negative on both targets for Xpert Xpress (FN) was re‐tested on Panther Fusion and found to be negative (TN) Unit of analysis: Unclear |
||
| Comparative | |||
| Notes | Funding: No funding statement reported Publication status: Accepted manuscript Source: J Appl Lab Med Author COI: No authors declared any potential conflicts of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | Unclear | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Szymczak 2020.
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity:
‐ remnant samples from patients with symptomatic diarrhea submitted for routine diagnostic testing (n=79 from 77 patients) Recruitment: Convenience Prospective or retrospective: Retrospective |
||
| Patient characteristics and setting | Setting: Unclear Location: Clinical Microbiology Laboratory at Montefiore Medical Center, New York Country: USA Dates: Apr 21 to May 15 2020 Symptoms and severity: All symptomatic for diarrhoea Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: Xpert Xpress (no product code reported) Manufacturer: Cepheid Inc Target gene(s): N2 and E Antigen target: n/a Test method: Automated RT‐PCR Samples used: Stool, collection not reported Transport media: Not stated; coated swabs transferred to 1 ml 0.85% saline for testing Sample storage: Stored at 2 to 8C for up to 7 days prior to testing Test operator: Not stated Definition of test positivity: Describes 'following the package insert instructions' ‐ presumptive positives not reported Blinding reported: Yes; conducted first Timing of samples: PCR +ve stool samples collected 0 to 33 days from initial respiratory PCR; 8/27 collected at >=14 days and 6/27 collected at >=21 days |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; Hologic Panther Fusion Definition of non‐COVID cases: As for cases (single PCR negative) Genetic target(s): two ORF1a regions Samples used: Stool, as for index Timing of reference standard: Some samples frozen at ‐80oC prior to testing with Hologic Panther Fusion Blinded to index test: Unclear Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous; same swabs All patients received same reference standard: Yes Missing data: None reported, no participant flow diagram reported Uninterpretable results: None reported Indeterminate results (index test): discrepant results re‐tested with both index and reference test using both a new aliquot and a shared aliquot tested on both instruments on the same day Indeterminate results (reference standard): discrepant results re‐tested with both index and reference test using both a new aliquot and a shared aliquot tested on both instruments on the same day Unit of analysis: Samples (79 from 77 patients) |
||
| Comparative | |||
| Notes | Funding: No funding statement reported Publication status: Published Source: J Clin Microbiol Author COI: No COI statement reported |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | Unclear | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Takeda 2020.
| Study characteristics | |||
| Patient Sampling | Two group study to estimate sensitivity and specificity, in:
[1] RT‐PCR confirmed COVID‐19 samples selected from a total of 88 positive samples during time period (n=62);
[2] Random sample of RT‐PCR negative samples selected from 1363 negative specimens tested during same time frame (n=100) Recruitment: Unclear for cases (may have been all 'initial' samples tested); random sample of non‐cases Prospective or retrospective: Unclear |
||
| Patient characteristics and setting | Setting: Not stated; multiple clinical institutions Location: SRL Inc, Tokyo Country: Japan Dates: early April'' also later states 4 day period Symptoms and severity: Not stated; High viral load (< 25 Ct) ‐ 32/60, 53% Low viral load (>=25 Ct) ‐ 28/60, 47% Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: ESPLINE SARS‐CoV‐2 (no product code reported) Manufacturer: Fujirebio Inc Antibody: SARS‐CoV‐2 antigen (from IFU) Antigen target: Anti‐SARS‐CoV‐2 monoclonal antibodies (mouse) (from IFU) Test method: LFA using alkaline phosphatase (ALP) labelled antibodies Samples used: NP; collection not reported Transport media: Not described Sample storage: Swabs mixed with sample treatment solution; no storage reported Test operator: Not stated; laboratory staff presumed Definition of test positivity: Visual line, as per manufacturer Blinding reported: Not stated Timing of samples: Not stated but all cases are first samples presumed by authors to be from patient suspected of SARS‐CoV‐2 for the first time; negative samples were 'probably … from … COVID‐19 patients for monitoring purposes and to check for negative conversion' |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; QuantiTect Probe RT‐PCR Kit (Qiagen). Definition of non‐COVID cases: As for cases; single negative required Genetic target(s): N2 Samples used: NP, as for index test Timing of reference standard: Not stated Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous, same samples All patients received same reference standard: Yes Missing data: 16 positive samples omitted; possibly because not initial samples but unclearly reported Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients (for cases), not clear for non‐cases |
||
| Comparative | |||
| Notes | Funding: None reported, however laboratory wholly owned by test manufacturer Publication status: Pre‐print Source: medRxiv Author COI: SRL Inc. is a subsidiary of Miraca Holdings Inc. Miraca Holdings Inc. holds all stock of Fujirebio Inc. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Unclear | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Thwe 2020.
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity:
symptomatic patients with paired samples tested with both ID NOW (dry NP swabs) and a real‐time RT‐PCR assay (NP swabs in VTM) (n=182)
[samples with RT‐PCR using Xpert Xpress (n=21) were excluded from this review] Recruitment: Not stated Prospective or retrospective: Retrospective |
||
| Patient characteristics and setting | Setting: Mixed (inpatient and ED); lab‐based study Location: University of Texas Medical Branch, Galveston Country: USA Dates: April to May 2020 ('4 weeks data') Symptoms and severity: Not stated Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: ID NOW (no product code) Manufacturer: Abbott Antibody: Not stated Antigen target: n/a Test method: Isothermal PCR Samples used: dry NP swabs Transport media: None Sample storage: in plain untreated sterile urine collection tubes Test operator: Not stated Definition of test positivity: As per manufacturer Blinding reported: Yes; conducted first Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Reference standard: One of 4 RT‐PCR assays;
1. Abbott RealTime SARS‐CoV‐2 (Abbott Park, IL, USA) (n=22)
2. Panther Fusion® SARS‐COV‐2 (San Diego, CA, USA) (n=129)
3. Cepheid Xpert® Xpress SARS‐CoV‐2 (Sunnyvale, CA, USA)) (n=21; excluded from this review)
4. a laboratory developed
test (LDT) (n=10) Definition of non‐COVID cases: As for cases (single negative) Genetic target(s): Not stated Samples used: NP in VTM (paired) Timing of reference standard: Not stated Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Paired All patients received same reference standard: Yes Missing data: None reported (review team excluded 21 samples tested with RT‐PCR) Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported; no discrepant analysis Unit of analysis: Patient |
||
| Comparative | |||
| Notes | Funding: This project did not receive any funding support from any agencies in the public, commercial, or not for‐profit sectors Publication status: Published Source: Diagnostic Microbiol Infect Dis Author COI: All authors have no conflict of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Van der Moeren 2020(a).
| Study characteristics | |||
| Patient Sampling | Study reports data for two cohorts. Van der Moeren 2020(a) relates to cohort [1] Single group study to estimate sensitivity and specificity: all adults presenting at a single community test centre for COVID‐19 testing (n=354)
see Van der Moeren 2020(b) for cohort [2] data [2] Single group study to estimate sensitivity alone: patients with a positive PCR test result at one of 3 community testing facilities who were retested at home within 72h of initial positive result (n=132) Recruitment: Consecutive; 'all' adults invited to participate Prospective or retrospective: Prospective |
||
| Patient characteristics and setting | Setting: COVID‐19 test centre (community) Location: Municipal Health Service (GGD) regional test centre at Breda Country: Netherlands Dates: Sep 28 to Sep 30 Symptoms and severity: Not stated; symptomatic Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: BD Veritor System for Rapid Detection of SARS‐CoV‐2 Manufacturer: Becton Dickinson Antibody: NP Antigen target: Not stated Test method: LFA; no further detail Samples used: NOP; "specimen from the throat and the superficial nasal cavities (bilateral, 2.5 cm proximal from the nostril)"; collected by GGD employee Transport media: Direct testing Sample storage: stored dry in sterile test tubes and stored and transported on dry ice until processing at the laboratory; tested within 6 hours after collection Test operator: trained laboratory technicians Definition of test positivity: reported using Analyzer (included in main analysis for review), and by naked eye inspection alone Blinding reported: Not stated Timing of samples: Not reported; on presentation time pso only provided for PCR+ cases: 12 < 7d; 1 ≥ 7d; 4=no pso data |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; either Cobas 6800 (Roche) or the m2000 (Abbott). Definition of non‐COVID cases: As for cases; single negative Genetic target(s): E‐ and RDRP‐gene (Cobas) or E‐gene and N‐gene (Abbott) Samples used: NOP; specimen from the throat and nasal cavity up to the nasal bridge Timing of reference standard: As for index test Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Paired All patients received same reference standard: Yes; different assays Missing data: 2 samples excluded due to RT‐PCR coding error [Considered overall low risk of bias due to small numbers] Uninterpretable results: 1 invalid on Ag test Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: The VRD (antigen) tests for this study were provided by the Dutch Ministry of Health, Welfare and Sport (VWS). Publication status: Pre‐print Source: medRxiv Author COI: Jan Kluytmans is member of the National Outbreak Management Team of The Netherlands and of a committee which supports the implementation of the Corona‐reporting App. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Yes | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Low risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Low concern | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | Low risk | ||
Van der Moeren 2020(b).
| Study characteristics | |||
| Patient Sampling | Study reports data for two cohorts. Van der Moeren 2020(b) relates to cohort
[2] Single group study to estimate sensitivity alone: patients with a positive PCR test result at one of twp community testing facilities who were retested at home within 72h of initial positive result (n=132)
see Van der Moeren 2020(a) for data related to cohort [1] Single group study to estimate sensitivity and specificity: all adults presenting at a single community test centre for COVID‐19 testing (n=354) Recruitment: Unclear; implies 'all' those with positive PCR invited to participate Prospective or retrospective: Prospective |
||
| Patient characteristics and setting | Setting: Community Location: Municipal Health Service (GGD) regional test centres at Breda or Roosendaal Country: Netherlands Dates: Sep 28 to Oct 6 Symptoms and severity: At time of home visit: Asymptomatic 3, 2% (2/3 still PCR +ve) Symptomatic 129 (123 still PCR +ve) Day <7 66, 50% Day >7 57, 43% Demographics: Not stated Exposure history: Not stated |
||
| Index tests | Test name: BD Veritor System for Rapid Detection of SARS‐CoV‐2 Manufacturer: Becton Dickinson Antibody: NP Antigen target: Not stated Test method: LFA; no further detail Samples used: NOP? "specimen from the throat and the superficial nasal cavities (bilateral, 2.5 cm proximal from the nostril)"; collected by GGD employee Transport media: Direct testing Sample storage: stored dry in sterile test tubes and stored and transported on dry ice until processing at the laboratory; tested within 6 hours after collection Test operator: trained laboratory technicians Definition of test positivity: reported using Analyzer (included in main analysis for review), and by naked eye inspection alone Blinding reported: Not stated Timing of samples: Not reported; on presentation |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; either Cobas 6800 (Roche) or the m2000 (Abbott). Definition of non‐COVID cases: n/a Genetic target(s): E‐ and RDRP‐gene (Roche) or E‐gene and N‐gene (Abbott) Samples used: NOP; specimen from the throat and nasal cavity up to the nasal bridge Timing of reference standard: As for index test Blinded to index test: Not stated Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Paired All patients received same reference standard: Yes; different assays Missing data: Review team excluded 7 no longer PCR+ at time of home visit (1 asymptomatic, 6 symptomatic) ‐ VRD result for 1 asymptomatic PCR‐ is given (VRD‐) Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: The VRD (antigen) tests for this study were provided by the Dutch Ministry of Health, Welfare and Sport (VWS). Publication status: Pre‐print Source: medRxiv Author COI: Jan Kluytmans is member of the National Outbreak Management Team of The Netherlands and of a committee which supports the implementation of the Corona‐reporting App. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Veyrenche 2020.
| Study characteristics | |||
| Patient Sampling | Two group study estimating sensitivity and specificity:
[1] PCR+ hospital inpatients (n=45)
[2] pre‐pandemic samples from 'patients' (not otherwise specified) (n=20) Recruitment: Not stated; appears to be convenience as equal numbers per Ct value subgroup Prospective or retrospective: Retrospective |
||
| Patient characteristics and setting | Setting: Inpatient Location: Montpellier University hospitals (Centre Hospitalier Universitaire de Montpellier, Montpellier) Country: France Dates: 14 March to 11 April Symptoms and severity: 27/45, 60% cases 'severe' according to WHO guideline (similar numbers per Ct subgroup) Demographics: Median age: Ct<=25 ‐ 66 (IQR 48, 84) Ct 25‐35 ‐ 63 (50, 76) Ct>=35 ‐ 58 (49‐67) Controls 64 (35, 93); 32/45, 71% male, all controls were male Exposure history: Not stated |
||
| Index tests | Test name: Coris COVID‐19 Ag Respi‐Strip Manufacturer: BioConcept®, Gembloux, Belgium Antibody: NP Antigen target: monoclonal ab Test method: CGIA Samples used: NP; collection not described Transport media: Yes; "swabs were collected in various transport media (eSwab™ COPAN Amies 1 ml, Σ‐Transwab® liquid Amies, viral transport medium tube VTM‐M 2.0ml)." Sample storage: Unclear; RT‐PCR conducted prospectively within a few hours but not reported for Ag testing Test operator: Not stated; presume lab staff Definition of test positivity: Visual, as per manufacturer Blinding reported: Not stated Timing of samples: day 1 to 20 pso, median Ct<=25 ‐ 7 (4, 10; presume this is IQR but could be range ‐ is described as SD in paper) Ct 25‐35 ‐ 8 (4, 12) Ct>=35 ‐ 11 (7, 15) |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; Allplex™ 2019‐nCoV Assay (Seegene, Seoul, South Korea) Definition of non‐COVID cases: pre‐pandemic Genetic target(s): RdRp, N, E Samples used: NP; as for index Timing of reference standard: As for index Blinded to index test: Yes, conducted first Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous; same swab All patients received same reference standard: No Missing data: None reported, no participant flow diagram reported Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: supported by Grants from Montpellier University Hospital and Montpellier University (MUSE). Publication status: pre‐print Source: medRxiv Author COI: The authors declare that there are no conflicts of interest |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | Unclear | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | Yes | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | Low risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | No | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Weitzel 2020 [A].
| Study characteristics | |||
| Patient Sampling | Single‐group study to estimate sensitivity and specificity:
‐ samples from patients with respiratory symptoms and/or fever attending a private hospital ED Recruitment: convenience with deliberate sampling of positive cases to ensure a 2:1 distribution reported (5276 samples processed during study period) Prospective or retrospective: retrospective Number of samples (samples with confirmed SARS‐CoV‐2): 111 (80) *17 samples included in Porte 2020a |
||
| Patient characteristics and setting | Setting: ED (private hospital) Location: Clínica Alemana de Santiago Country: Chile Dates: 16 March‐26 April 2020 Symptoms and severity: respiratory symptoms and/or fever; no further detail Demographics: median age 40 years; 50, 45% male (median age 38 years, 43% male for all samples tested during period) Exposure history: none reported |
||
| Index tests |
Weitzel 2020 [A] entry is for test [A] in the list below Test name: [A] Biocredit COVID‐19 Ag One Step SARS‐CoV‐2 Antigen Test (RapiGEN Inc., Anyang‐si, Gyeonggi‐do, Republic of Korea) [B] COVID‐19 Antigen Rapid Test Device StrongStep COVID‐19 Antigen Test (Liming Bio‐Products Co., Jiangsu, China) [C] Huaketai New Coronavirus (SARS‐CoV‐2) N Protein Detection Kit (Fluorescence immunochromatography) (Savant Biotechnology Co., Beijing, China), [D] Diagnostic Kit for 2019‐Novel Coronavirus (2019‐nCoV) Ag Test (Fluorescence Immunochromatographic Assay) (Bioeasy Biotechnology Co., Shenzhen, China). Manufacturer: [A] RapiGEN Inc., Anyang‐si, Gyeonggi‐do, Republic of Korea [B] Liming Bio‐Products Co., Jiangsu, China [C] Savant Biotechnology Co., Beijing, China [D] Bioeasy Biotechnology Co., Shenzhen, China Antigen target: not reported in study Antibody: not reported in study Test method: [A] and [B] CGIA [C] and [D] FIA Samples used: NOP swabs in 3 mL UTM Transport media: UTM‐RT System (Copan Diagnostics, Murrieta, CA, USA) Sample storage: stored at −80 °C; index tests applied on 28 and 29 April 2020 Test operator: single, trained laboratory technician under BSL2 cabinet; visual outputs read by 2 independent observers with referral to third if needed Definition of test positivity: as per manufacturer; Beijing Savant test required use of manufacturer supplied UV torch due to unavailability of reader device in Chile Blinding reported: yes; blinding stated Timing of samples: median 2 days (IQR 1‐5 days); 88% (96/109) during the first week of symptoms |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; COVID‐19 Genesig Real‐Time PCR assay (Primerdesign Ltd., Chandler's Ford, UK). Ct ≤ 40 considered positive Definition of non‐COVID cases: single PCR negative Genetic target(s): RdRp Samples used: NOP swabs; as for index Timing of reference standard: as for index test; median 2 days (IQR 1‐5 days) Blinded to index test: yes; prior to index Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: same samples; index tests conducted after frozen storage All participants received same reference standard: yes Missing data: none reported; evaluation of Liming test was discontinued after initial poor performance (zero TP) Uninterpretable results: 2 tests had invalid results due to insufficient liquid migration (2 results excluded for each test) Indeterminate results (index test): visual interpretation of the Beijing Savant assay (using manufacturer supplied UV torch) was reportedly difficult under daylight conditions; manufacturer's fluorescence reader not available in Chile. Indeterminate results (reference standard): none reported Unit of analysis: participants |
||
| Comparative | |||
| Notes | Funding: study authors report that the work received no funding; Savant Biotechnology Co. provided test kits free of charge Publication status: preprint Source: medRxiv Author COI: all authors declare no competing interests |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Weitzel 2020 [B].
| Study characteristics | |||
| Patient Sampling | See Weitzel 2020 [A] for full study details and QUADAS entries | ||
| Patient characteristics and setting | |||
| Index tests |
Weitzel 2020 [B] entry is for test [B] in the list below; see Weitzel 2020 [A] for full study details and QUADAS entries Test name: [A] Biocredit COVID‐19 Ag One Step SARS‐CoV‐2 Antigen Test (RapiGEN Inc., Anyang‐si, Gyeonggi‐do, Republic of Korea) [B] COVID‐19 Antigen Rapid Test Device StrongStep COVID‐19 Antigen Test (Liming Bio‐Products Co., Jiangsu, China) [C] Huaketai New Coronavirus (SARS‐CoV‐2) N Protein Detection Kit (Fluorescence immunochromatography) (Savant Biotechnology Co., Beijing, China), [D] Diagnostic Kit for 2019‐Novel Coronavirus (2019‐nCoV) Ag Test (Fluorescence Immunochromatographic Assay) (Bioeasy Biotechnology Co., Shenzhen, China). Manufacturer: [A] RapiGEN Inc., Anyang‐si, Gyeonggi‐do, Republic of Korea [B] Liming Bio‐Products Co., Jiangsu, China [C] Savant Biotechnology Co., Beijing, China [D] Bioeasy Biotechnology Co., Shenzhen, China Antigen target: not reported in study Antibody: not reported in study Test method: [A] and [B] CGIA [C] and [D] FIA Samples used: NOP swabs in 3 mL UTM Transport media: UTM‐RT System (Copan Diagnostics, Murrieta, CA, USA) Sample storage: stored at −80 °C; index tests applied on 28 and 29 April 2020 Test operator: single, trained laboratory technician under BSL2 cabinet; visual outputs read by 2 independent observers with referral to third if needed Definition of test positivity: as per manufacturer; Savant test required use of manufacturer supplied UV torch due to unavailability of reader device in Chile Blinding reported: yes; blinding stated Timing of samples: median 2 days (IQR 1‐5 days); 88% (96/109) during the first week of symptoms |
||
| Target condition and reference standard(s) | See Weitzel 2020 [A] for full study details and QUADAS entries | ||
| Flow and timing | See Weitzel 2020 [A] for full study details and QUADAS entries | ||
| Comparative | |||
| Notes | |||
Weitzel 2020 [C].
| Study characteristics | |||
| Patient Sampling | See Weitzel 2020 [A] for full study details and QUADAS entries | ||
| Patient characteristics and setting | See Weitzel 2020 [A] for full study details and QUADAS entries | ||
| Index tests |
Weitzel 2020 [C] entry is for test [C] in the list below; see Weitzel 2020 [A] for full study details and QUADAS entries Test name: [A] Biocredit COVID‐19 Ag One Step SARS‐CoV‐2 Antigen Test (RapiGEN Inc., Anyang‐si, Gyeonggi‐do, Republic of Korea) [B] COVID‐19 Antigen Rapid Test Device StrongStep COVID‐19 Antigen Test (Liming Bio‐Products Co., Jiangsu, China) [C] Huaketai New Coronavirus (SARS‐CoV‐2) N Protein Detection Kit (Fluorescence immunochromatography) (Savant Biotechnology Co., Beijing, China), [D] Diagnostic Kit for 2019‐Novel Coronavirus (2019‐nCoV) Ag Test (Fluorescence Immunochromatographic Assay) (Bioeasy Biotechnology Co., Shenzhen, China). Manufacturer: [A] RapiGEN Inc., Anyang‐si, Gyeonggi‐do, Republic of Korea [B] Liming Bio‐Products Co., Jiangsu, China [C] Savant Biotechnology Co., Beijing, China [D] Bioeasy Biotechnology Co., Shenzhen, China Antigen target: not reported in study Antibody: not reported in study Test method: [A] and [B] CGIA [C] and [D] FIA Samples used: NOP swabs in 3 mL UTM Transport media: UTM‐RT System (Copan Diagnostics, Murrieta, CA, USA) Sample storage: stored at −80 °C; index tests applied on 28 and 29 April 2020 Test operator: single, trained laboratory technician under BSL2 cabinet; visual outputs read by 2 independent observers with referral to third if needed Definition of test positivity: as per manufacturer; Savant test required use of manufacturer supplied UV torch due to unavailability of reader device in Chile Blinding reported: yes; blinding stated Timing of samples: median 2 days (IQR 1‐5 days); 88% (96/109) during the first week of symptoms |
||
| Target condition and reference standard(s) | See Weitzel 2020 [A] for full study details and QUADAS entries | ||
| Flow and timing | See Weitzel 2020 [A] for full study details and QUADAS entries | ||
| Comparative | |||
| Notes | |||
Weitzel 2020 [D].
| Study characteristics | |||
| Patient Sampling | See Weitzel 2020 [A] for full study details and QUADAS entries | ||
| Patient characteristics and setting | See Weitzel 2020 [A] for full study details and QUADAS entries | ||
| Index tests |
Weitzel 2020 [D] entry is for test [D] in the list below; see Weitzel 2020 [A] for full study details and QUADAS entries Test name: [A] Biocredit COVID‐19 Ag One Step SARS‐CoV‐2 Antigen Test (RapiGEN Inc., Anyang‐si, Gyeonggi‐do, Republic of Korea) [B] COVID‐19 Antigen Rapid Test Device StrongStep® COVID‐19 Antigen Test (Liming Bio‐Products Co., Jiangsu, China) [C] Huaketai New Coronavirus (SARS‐CoV‐2) N Protein Detection Kit (Fluorescence immunochromatography) (Savant Biotechnology Co., Beijing, China), [D] Diagnostic Kit for 2019‐Novel Coronavirus (2019‐nCoV) Ag Test (Fluorescence Immunochromatographic Assay) (Bioeasy Biotechnology Co., Shenzhen, China). Manufacturer: [A] RapiGEN Inc., Anyang‐si, Gyeonggi‐do, Republic of Korea [B] Liming Bio‐Products Co., Jiangsu, China [C] Savant Biotechnology Co., Beijing, China [D] Bioeasy Biotechnology Co., Shenzhen, China Antigen target: not reported in study Antibody: not reported in study Test method: [A] and [B] CGIA [C] and [D] FIA Samples used: NOP swabs in 3 mL UTM Transport media: UTM‐RT System (Copan Diagnostics, Murrieta, CA, USA) Sample storage: stored at −80°C; index tests applied on 28 and 29 April 2020 Test operator: single, trained laboratory technician under BSL2 cabinet; visual outputs read by 2 independent observers with referral to third if needed Definition of test positivity: as per manufacturer; Savant test required use of manufacturer supplied UV torch due to unavailability of reader device in Chile Blinding reported: yes; blinding stated Timing of samples: median 2 days (IQR 1‐5 days); 88% (96/109) during the first week of symptoms |
||
| Target condition and reference standard(s) | See Weitzel 2020 [A] for full study details and QUADAS entries | ||
| Flow and timing | See Weitzel 2020 [A] for full study details and QUADAS entries | ||
| Comparative | |||
| Notes | |||
Wolters 2020.
| Study characteristics | |||
| Patient Sampling | 2‐group study to estimate sensitivity and specificity for diagnosis of active disease:
‐ samples selected from laboratories on the basis of presence/absence of 2 genetic targets on RT‐PCR: SARS‐CoV‐2 E‐gene +/RdRp gene + (n = 30); SARS‐CoV‐2 E‐gene +/RdRp gene – (n = 28); SARS‐CoV‐2 E‐gene ‐/RdRp gene (n = 30)
(A separate set of samples were tested in triplicate at all 3 laboratories to determine limits of detection and analytical specificity) Recruitment: not stated; deliberate sampling used Prospective or retrospective: retrospective Sample size (cases): 88 (58) |
||
| Patient characteristics and setting | Setting: not stated; 3 laboratories Location: Radboud UMC in Nijmegen, PAMM in Veldhoven and the RIVM in Bilthoven Country: The Netherlands Dates: January‐March 2020 Symptoms and severity: not stated Demographics: not stated Exposure history: not stated |
||
| Index tests | Test name: Cepheid Xpert Xpress SARS‐CoV‐2 (product code not reported) Manufacturer: Cepheid Europe Antigen target: E‐gene (sarbeco‐specific) and N2‐gene (SARS‐CoV‐2‐specific) Antibody: N/A Test method: not stated (it should be automated PCR) Samples used: NP or mid‐turbinate, and OP swabs Transport media: UTM or GLY medium; no further details Sample storage: stored at −80 ℃ Test operator: not stated; presume laboratory staff Definition of test positivity: as per manufacturer; reported E‐gene‐only positive specimens as presumptive positive but no re‐testing with Xpert Xpress was reported. N2‐only positives were considered positive (but re‐tested with RT‐PCR) Blinding reported: not stated (see comment section) Timing of samples: not stated |
||
| Target condition and reference standard(s) | Reference standard: in‐house RT‐PCR:
Radboud UMC Lab: MagNApure 96 (Roche) (isolation platform); MagNApure 96 DNA and Viral NA Small Volume (extraction kit); Roche LC480 II (PCR platform); Life Technologies Taqman FastVirus 1‐step mastermix (RT‐PCR mastermix)
PAMM Lab: Roche cobas 4800 (isolation platform); CT/NG extraction protocol (extraction kit); Roche LC480 II (PCR platform); Roche LightCycler Multiplex RNA Virus Master (RT‐PCR mastermix);
RIVM Lab: BioMérieux NucliSens (isolation platform); easyMAG EasyMAG extraction reagents (extraction kit); Thermo Fisher QuantStudio 6 (PCR platform); Life Technologies Taqman FastVirus 1‐step mastermix (RT‐PCR mastermix) Definition of non‐COVID cases: yes (performed prior to index test) Genetic target(s): Radboud UMC lab: E‐gene and RdRp‐gene PAMM Lab: started with E‐gene and RdRp‐gene and mid‐March moved on to E‐gene testing only RIVM Lab: started with E‐gene and RdRp‐gene and at the beginning of April moved on to E‐gene and CDC N1‐gene primer and probes Samples used: as for index test Timing of reference standard: as for index test Blinded to index test: storage prior to freezing was not reported; samples were analysed at or near time of collection ("processed … in the routine diagnostic procedure using the locally implemented RT‐PCR") Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: same samples used; index text seems to have been conducted after frozen storage Missing data: none reported, no participant flow diagram reported Uninterpretable results: none reported Indeterminate results (index test): 1 sample was positive only on N2 gene (positive according to IFU) and 1 was positive only on E gene (presumptive positive, requires re‐testing according to IFU). Both samples were re‐tested on RT‐PCR only Indeterminate results (reference standard): re‐testing of the two ‘FN’ samples (one TP and 1 presumptive positive according to IFU definition) with RT‐PCR found both samples to be disease‐negative (reclassed as 1 TN and 1 FP); study authors note that the viral loads of these samples are at the limit of detection for Xpert Xpress and that multiple freeze‐thaw steps of samples could have had a significant impact on detection. Unit of analysis: not stated; only samples reported |
||
| Comparative | |||
| Notes | Funding: not stated Publication status: accepted manuscript Source: Journal of Clinical Virology Author COI: the study authors declare no COI present |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | No | ||
| Could the conduct or interpretation of the index test have introduced bias? | High risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | Unclear risk | ||
Wong 2020.
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity:
‐ samples submitted for routine testing from patients with suspected COVID‐19 infection presenting at A&E (n=93), in‐patient (n=47) or outpatient n=18) (total n=158 providing 162 samples) Recruitment: Not stated Prospective or retrospective: Both retrospective (n=74) and prospective (n=88) |
||
| Patient characteristics and setting | Setting: Mixed; A&E, inpatient and outpatient Location: Prince of Wales Hospital, Hong Kong Country: China Dates: Not stated Symptoms and severity: Not stated Demographics: Median age 46 (IQR: 35(28‐63); males = 69 (44%) Exposure history: Not stated |
||
| Index tests | Test name: Xpert Xpress Manufacturer: Cepheid Inc Antibody: E and N2 Antigen target: n/.a Test method: Automated RT‐PCR Samples used: deep throat saliva (DTS) (n=120), or lower respiratory tract (LRT) (n=42; 35 sputum, 6 tracheal aspirate 1 BAL) Transport media: None; collected in plain sterile container. Prior to testing, PBS was added to was added into neat DTS specimens (ratio 1:1) and vortexed for homogenization and allowed to settle for 5‐ 10 min. 2mL of homogenized sample transferred to another vial for centrifugation at 2000 g for 5 min. 1mL of LRT specimens added to 3 mL of in‐house prepared Maintenance Medium (MM) (10X Minimum Essential Medium (MEM), 200 mM glutamine, 1 M HEPES, 7.5 % NaHCO3, 12 mg gentamicin, 0.5 mg amphotericin B, 10,000 units penicillin, 10 mg streptomycin, pH 7.1–7.4); mixture was emulsified by pipetting up and down, followed by centrifugation at 2000 g for 5 min. Supernatant was used for testing as per manufacturer’s instructions for both RT‐PCR and Xpert Xpress Sample storage: transported to laboratory on the same day and tested promptly Test operator: Lab staff Definition of test positivity: As per manufacturer; presumptive positives mentioned only in Introduction section Blinding reported: Not stated Timing of samples: Not stated |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; TIB‐Molbiol LightMix® SarbecoV E‐gene assay; all positive cases confirmed by reference laboratory of Hong Kong (Public Health Laboratory Service Branch, PHLSB). Definition of non‐COVID cases: As for cases (single negative) Genetic target(s): Not stated Samples used: DTS or LRT; as per index test Timing of reference standard: Not stated Blinded to index test: Yes; conducted first (upon receipt, all samples were screened with our standard‐of‐care assay) Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous (Same samples) All patients received same reference standard: Yes Missing data: None reported Uninterpretable results: None reported Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported Unit of analysis: Samples (162/158) |
||
| Comparative | |||
| Notes | Funding: This research did not receive any specific grant from funding agencies in the public, commercial, or not‐for‐profit sectors. Publication status: Published Source: J Clin Virol Author COI: The authors report no declarations of interest. |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Unclear | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Unclear | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | Unclear | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | No | ||
| Could the patient flow have introduced bias? | High risk | ||
Young 2020.
| Study characteristics | |||
| Patient Sampling | Single group study to estimate sensitivity and specificity:
‐ Patients with one or more symptoms of COVID‐19 (within <=7 days post symptom onset) at 21 study sites (n=260)
[Second cohort of 361 samples from COVID suspects <=5 days p.s.o. also evaluated to compare BD Veritor with Quidel Sofia® 2 SARS Antigen FIA but excluded from review as only discrepant results on the two Ag assays underwent RT‐PCR] Recruitment: Not stated Prospective or retrospective: Prospective |
||
| Patient characteristics and setting | Setting: Mixed; drive‐through/tent (n=42), outpatient clinic (n=74), research clinic (n=72), or skilled nursing facility (n=66) Location: Unclear; 21 geographically diverse study sites [Author institutions BD Life Sciences, Louisiana State University Health Sciences Center, Tricore Reference Laboratory) Country: USA Dates: June 5‐11, 2020 Symptoms and severity: 110 (43%) cough, 98 (39%) muscle pain, 95 (37%) headache, 90 (35%) sore throat, 90 (35%) sore throat, 78 (31%) fever. Of those at <=6 days p.s.o (n=245): 94 (38%) with one symptom, 151 (62%) with >= 2 symptoms Demographics: median age 43 (range 18 to 90); 91 (36%) male Exposure history: |
||
| Index tests | Test name: BD Veritor SARS‐CoV‐2 antigen test (no product codes) Manufacturer: Becton, Dickinson and Company, BD Life Sciences—Integrated Diagnostic Solutions, San Diego, CA Antibody: NP Antigen target: not stated Test method: Not stated; chromatographic immunoassay with analyser Samples used: Nasal; clinician collected from both nostrils (same swab) Transport media: dry nasal swabs Sample storage: Swabs were shipped for testing on dry ice (‐70°C); Test operator: Not stated; Veritor testing was performed internally at BD (San Diego, CA, USA) Definition of test positivity: As per manufacturer Blinding reported: Yes; all personnel blinded to all other test results Timing of samples: All <=7 days p.s.o; median 3.0 d, mean 3.2 d. 38 (15%) 1 day p.s.o, 57 (23%) 2 days, 54 (22%) 3 days, 40 (16%) 4 days, 37 (15%) 5 days, 19 (8%) 6 days, 6 (2%) 7 days |
||
| Target condition and reference standard(s) | Reference standard: Lyra® SARS‐CoV‐2 PCR Assay (Quidel Corporation. Athens, OH); BD MAX™ real time SARS‐CoV‐2 PCR assay used for discordant testing Definition of non‐COVID cases: As for cases (single negative) Genetic target(s): Not stated Samples used: NP (n= 217) or OP (n=34); clinician collected (if an NP swab was collected as part of SOC, the participant had the option of having an OP study swab taken in lieu of a second NP swab) Timing of reference standard: Swabs taken prior to any study swabs (potential for contamination of nasal cavity) Blinded to index test: Yes; performed at TriCore Reference Laboratories. "All testing was conducted with all personnel blinded to all other test results" Incorporated index test: No |
||
| Flow and timing | Time interval between index and reference tests: Simultaneous (paired) All patients received same reference standard: Yes Missing data: 9 excluded; 6 did not meet eligibility criteria and 3 had invalid specimens/results (2 on RT‐PCR and 1 labelling error) Uninterpretable results: 3 invalid on at least one assay Indeterminate results (index test): None reported Indeterminate results (reference standard): None reported. Re‐test of 9 'FN' results with BD MAX RT‐PCR resulted in 2 confirmed FN (BD MAX +ve and sero +ve), 6 were BD Max ‐ve (incl 1 sero +ve) and 1 invalid (no result) Unit of analysis: Patients |
||
| Comparative | |||
| Notes | Funding: Study was funded by Becton, Dickinson and Company; BD Life Sciences—Integrated Diagnostics Solutions. Non‐BD employee authors received research funds as part of this work Publication status: Pre‐print Source: medRxiv Author COI: CRD, CF, KE, JCA, HR, and CKC are employees of Becton, Dickinson and Company; SY, None; CC, None; AM, None; CGF, None; CB, None; JA, None; RA, CEO and PI of Comprehensive Clinical Research LLC |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | Unclear | ||
| Was a case‐control design avoided? | Yes | ||
| Did the study avoid inappropriate exclusions? | Yes | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | Unclear risk | ||
| Are there concerns that the included patients and setting do not match the review question? | Unclear | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Yes | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Low risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Yes | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Yes | ||
| Could the patient flow have introduced bias? | High risk | ||
Zhen 2020 [A].
| Study characteristics | |||
| Patient Sampling | 2‐group study to estimate sensitivity and specificity:
‐ samples from symptomatic patients of all ages and gender Recruitment: not stated; specimens selected to represent the true positivity rate at authors' institution (50% to 60%), and to span low and high viral loads Prospective or retrospective: mixed; included frozen samples (n = 88) and prospectively tested (n = 20) Number of samples (samples with confirmed SARS‐CoV‐2):108 (58) |
||
| Patient characteristics and setting | Setting: not stated; selected from laboratory Location: not stated; authors' institutions were Northwell Health Laboratories, and Dept Pathology and Laboratory Medicine, The Donald and Barbara Zucker School of Medicine Country: USA Dates: March‐April 2020 Symptoms and severity: "symptomatic"; no further details Demographics: not stated (all ages and genders) Exposure history: not stated |
||
| Index tests |
Zhen 2020 [A] is the entry for test [A] from the list below Test name: [A] Xpert® Xpress SARS‐CoV‐2 [B] ID NOW COVID‐19 (no product codes reported) Manufacturer: [A] Cepheid, [B] Abbott Antigen target: [A] N2, E; [B] RdRp Antibody: N/A Test method: rapid PCR Samples used: NP swabs Transport media: UTM (various manufacturers) Sample storage: on collection, stored at 2‐8 ⁰C for up to 72 h; after routine testing, stored at −80 ⁰C 88 samples tested using ePlex on collection, then frozen prior to testing with ID NOW, Xpert Xpress and Hologic RT‐PCR; 20 samples tested prospectively after collection on all systems Test operator: not stated; presume laboratory staff Definition of test positivity: not stated; states “testing was performed according to the manufacturer’s instructions” but no presumptive positives reported Blinding reported: not stated Timing of samples: not stated Study also evaluates [C] GenMar kePlex® SARS‐CoV‐2 Test (not eligible for this review) |
||
| Target condition and reference standard(s) | Reference standard: RT‐PCR; Hologic Panther Fusion SARS‐CoV‐2 assay, performed according to manufacturer's IFU Definition of non‐COVID cases: single RT‐PCR Genetic target(s): 2 regions of ORF1ab; either positive Samples used: NP swabs; same as for index test Timing of reference standard: not stated Blinded to index test: not stated Incorporated index test: no |
||
| Flow and timing | Time interval between index and reference tests: not stated in exact terms; delay between index and reference only for GenMark assay, as 88 samples tested at time of collection with ePlex then frozen before testing with all other assays. All participants received same reference standard: yes Missing data: none reported, no participant flow diagram reported Uninterpretable results: 1 specimen with invalid result on ID NOW was excluded from that dataset Indeterminate results (index test): none reported; no re‐testing conducted Indeterminate results (reference standard): none reported; no re‐testing conducted Unit of analysis: not stated only refers to samples |
||
| Comparative | |||
| Notes | Funding: none stated; study authors thank Cepheid for providing the reagents used Publication status: accepted manuscript Source: Journal of Clinical Microbioloby Author COI: Gregory Berry has previously given education seminars for Abbott, Cepheid, and Hologic, Inc. and has received Honorariums |
||
| Methodological quality | |||
| Item | Authors' judgement | Risk of bias | Applicability concerns |
| DOMAIN 1: Patient Selection | |||
| Was a consecutive or random sample of patients enrolled? | No | ||
| Was a case‐control design avoided? | No | ||
| Did the study avoid inappropriate exclusions? | Unclear | ||
| Did the study avoid inappropriate inclusions? | Yes | ||
| Could the selection of patients have introduced bias? | High risk | ||
| Are there concerns that the included patients and setting do not match the review question? | High | ||
| DOMAIN 2: Index Test (Antigen tests) | |||
| DOMAIN 2: Index Test (Rapid molecular tests) | |||
| Were the index test results interpreted without knowledge of the results of the reference standard? | Unclear | ||
| If a threshold was used, was it pre‐specified? | Yes | ||
| Could the conduct or interpretation of the index test have introduced bias? | Unclear risk | ||
| Are there concerns that the index test, its conduct, or interpretation differ from the review question? | High | ||
| DOMAIN 3: Reference Standard | |||
| Is the reference standards likely to correctly classify the target condition? | No | ||
| Were the reference standard results interpreted without knowledge of the results of the index tests? | Unclear | ||
| Reference standard does not incorporate result of index test? | Yes | ||
| Could the reference standard, its conduct, or its interpretation have introduced bias? | High risk | ||
| Are there concerns that the target condition as defined by the reference standard does not match the question? | High | ||
| DOMAIN 4: Flow and Timing | |||
| Was there an appropriate interval between index test and reference standard? | Yes | ||
| Did all patients receive the same reference standard? | Yes | ||
| Were all patients included in the analysis? | No | ||
| Did all participants receive a reference standard? | Yes | ||
| Were results presented per patient? | Unclear | ||
| Could the patient flow have introduced bias? | High risk | ||
Zhen 2020 [B].
| Study characteristics | |||
| Patient Sampling | See Zhen 2020 [A] for full study details and QUADAS entries | ||
| Patient characteristics and setting | See Zhen 2020 [A] for full study details and QUADAS entries | ||
| Index tests |
Zhen 2020 [B] is the entry for test [B] from the list below, see Zhen 2020 [A] for full study details and QUADAS entries Test name: [A] Xpert® Xpress SARS‐CoV‐2 [B] ID NOWCOVID‐19 (no product codes reported) Manufacturer: [A] Cepheid, [B] Abbott Antigen target: [A] N2, E; [B] RdRp Antibody: N/A Test method: isothermal amplification test Samples used: NP swabs Transport media: UTM (various manufacturers) Sample storage: on collection, stored at 2‐8 ⁰C for up to 72 h; after routine testing, stored at −80 ⁰C 88 samples tested using ePlex on collection, then frozen prior to testing with ID NOW, Xpert Xpress and Hologic RT‐PCR; 20 samples tested prospectively after collection on all systems Test operator: not stated; presume laboratory staff Definition of test positivity: not stated; states “testing was performed according to the manufacturer’s instructions” but no presumptive positives reported Blinding reported: not stated Timing of samples: not stated Study also evaluates [C] GenMar kePlex® SARS‐CoV‐2 Test (not eligible for this review) |
||
| Target condition and reference standard(s) | See Zhen 2020 [A] for full study details and QUADAS entries | ||
| Flow and timing | See Zhen 2020 [A] for full study details and QUADAS entries | ||
| Comparative | |||
| Notes | Funding: none stated; study authors thank Cepheid for providing the reagents used Publication status: accepted manuscript Source: Journal of Clinical Microbioloby Author COI: Gregory Berry has previously given education seminars for Abbott, Cepheid, and Hologic, Inc. and has received Honorariums |
||
BAL: bronchoalveolar lavage; CDC: Center for Disease Control; CGIA: colloidal gold immunoassay; COI: conflict of interest; Ct: cycle threshold; ED: Emergency Department; EUA: emergency use authorisation; FIA: fluorescence immunochromatographic; FN: false negative; FP: false positive; GLY: Glucose‐Lactalbumin‐Yeast; HCW: healthcare worker; ICU: intensive care unit; IFU: instructions for use; IQR: interquartile range; LDT: laboratory‐developed test; N/A: not applicable; NAAT: nucleic acids amplification test; NIH: National Institutes of Health; NOP: naso‐oropharyngeal; NP: nasopharyngeal; OP: oropharyngeal; PCR: polymerase chain reaction; PHE: Public Health England; qRT‐PCR: quantitative reverse transcription polymerase chain reaction; RNA: ribonucleic acid; RT‐PCR: reverse transcription polymerase chain reaction; SD: standard deviation; TA: tracheal aspirate; TN: true negative; TP: true positive; UTM: universal transport medium; UV: ultraviolet; UW: University of Washington; VTM: viral transport medium;
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Ai 2020 | Ineligible index test |
| Anahtar 2020 | Ineligible index test |
| Ar Gouilh 2020 | Ineligible index test |
| Arizti‐Sanz 2020 | Ineligible index test |
| Arumugam 2020 | Ineligible index test |
| Avetyan 2020 | Ineligible index test |
| Azhar 2020 | Ineligible index test |
| Azzi 2020 | Ineligible index test |
| Baek 2020 | Ineligible index test |
| Barra 2020 | Ineligible study design |
| Basu 2020 | Ineligible reference standard |
| Behrmann 2020 | Accuracy data cannot be extracted |
| Bokelmann 2020 | Ineligible index test |
| Bordi 2020 | Ineligible index test |
| Brandsma 2020 | Ineligible index test |
| Broughton 2020 | Ineligible index test |
| Bull 2020 | Ineligible index test |
| Bulterys 2020 | Ineligible index test |
| Callahan 2020a | Accuracy data cannot be extracted |
| Callahan 2020b | Ineligible index test |
| Chandler‐Brown 2020 | Ineligible study design |
| Chen 2020b | Ineligible index test |
| Chow 2020 | Ineligible index test |
| CNR 2020 | Insufficient details in study report |
| CNR 2020a | Insufficient details in study report |
| Colson 2020 | Inadequate sample size |
| Comar 2020 | Ineligible reference standard |
| Comer 2020 | Ineligible population |
| Crone 2020 | Ineligible index test |
| Curti 2020 | Ineligible study design |
| Davda 2020 | Ineligible index test |
| Ding 2020a | Ineligible study design |
| Ding 2020b | Ineligible index test |
| Dohla 2020 | Ineligible index test |
| Dong 2020 | Ineligible index test |
| El‐Tholoth 2020 | Ineligible study design |
| Farfan 2020 | Ineligible study design |
| FIND 2020f | Superseded by Kruger 2020(a) |
| Fowler 2020 | Ineligible index test |
| Francis 2020 | Ineligible study design |
| Freire‐Paspuel 2020a | Ineligible study design |
| Freire‐Paspuel 2020b | Ineligible index test |
| Ganguli 2020 | Ineligible population |
| Giamarellos‐Bourboulis 2020 | Ineligible study design |
| Gonzalez‐Gonzalez 2020a | Ineligible study design |
| Gonzalez‐Gonzalez 2020b | Ineligible population |
| Grant 2020 | Ineligible index test |
| Hass 2020 | Ineligible target condition |
| Herrera 2020 | Ineligible reference standard |
| Hirotsu 2020 | Ineligible index test |
| Hogan 2020a | Ineligible index test |
| Howson 2020 | Ineligible study design |
| Hu 2020 | Ineligible index test |
| Huang 2020 | Ineligible index test |
| Huang 2021 | Ineligible study design |
| James 2020 | Ineligible index test |
| Jiang 2020 | Ineligible index test |
| Joung 2020 | Ineligible index test |
| Joung 2020a | Ineligible index test |
| Kalikiri 2020 | Ineligible index test |
| Kashiwagi 2020 | Inadequate sample size |
| Kim 2019 | Ineligible study design |
| Kim 2020 | Ineligible index test |
| Konrad 2020 | Ineligible study design |
| Kurstjens 2020 | Ineligible index test |
| Kyosei 2020 | Ineligible study design |
| Lalli 2020 | Inadequate sample size |
| Lamb 2020 | Ineligible study design |
| Landry 2020 | Ineligible index test |
| Le Hingrat 2020 | Ineligible index test |
| Lee 2020 | Ineligible index test |
| Li 2020 | Ineligible index test |
| Lin 2020 | Ineligible population |
| Liotti 2020a | Ineligible index test |
| Lowe 2020 | Ineligible index test |
| Lu 2020 | Ineligible study design |
| Lu 2020a | Ineligible index test |
| Lubke 2020 | Ineligible index test |
| Mahari 2020 | Ineligible study design |
| Marais 2020 | Ineligible index test |
| Marzinotto 2020 | Accuracy data cannot be extracted |
| McCormick‐Baw 2020 | Ineligible reference standard |
| McDonald 2020 | Ineligible reference standard |
| McRae 2020 | Ineligible index test |
| Mei 2020 | Ineligible index test |
| Meyerson 2020 | Ineligible index test |
| Michel 2020 | Ineligible index test |
| Mlcochova 2020 | Ineligible index test |
| Mohon 2020 | Ineligible index test |
| Moses 2020 | Ineligible index test |
| Mostafa 2020 | Ineligible study design |
| Muraoka 2020 | Ineligible study design |
| Nachtigall 2020 | Ineligible index test |
| Newman 2020 | Ineligible index test |
| Noerz 2020 | Ineligible index test |
| Ogawa 2020 | Inadequate sample size |
| Osterdahl 2020 | Ineligible index test |
| Paden 2020 | Ineligible study design |
| Patchsung 2020 | Ineligible index test |
| Pellanda 2020 | Ineligible index test |
| Peto 2020 | Ineligible index test |
| Pfefferle 2020 | Ineligible study design |
| Pollock 2020a | Ineligible index test |
| Qian 2020 | Ineligible index test |
| Rabe 2020 | Ineligible population |
| Rauch 2020 | Ineligible index test |
| Rodel 2020 | Ineligible index test |
| Rodriguez‐Manzano 2020 | Ineligible index test |
| Seo 2020 | Accuracy data cannot be extracted |
| Shirato 2020 | Ineligible index test |
| Singh 2020a | Ineligible index test |
| Singh 2020b | Ineligible index test |
| Smyrlaki 2020 | Ineligible index test |
| St Hilaire 2020 | Ineligible index test |
| Tan 2020 | Ineligible study design |
| Tanida 2020 | Ineligible index test; also preselected on cycle threshold (only < 34 cycle threshold included) |
| Tibbetts 2020 | Ineligible index test |
| Tran 2020 | Ineligible population |
| Visseaux 2020 | Ineligible index test |
| Wang 2020a | Ineligible index test |
| Wang 2020b | Accuracy data cannot be extracted |
| Wang 2020c | Ineligible index test |
| Wee 2020 | Ineligible study design |
| Wu 2020 | Ineligible index test |
| Xue 2020 | Ineligible index test |
| Yan 2020 | Ineligible index test |
| Yang 2020b | Ineligible index test |
| Yu 2020a | Ineligible index test |
| Yu 2020b | Ineligible index test |
| Yu 2020c | Ineligible index test |
| Zamecnik 2020 | Ineligible index test |
| Zeng 2020 | Ineligible study design |
| Zhang 2020 | Ineligible index test |
| Zhao 2020 | Ineligible study design |
| Zhu 2020 | Ineligible index test |
Differences between protocol and review
We planned to check the following websites for eligible index tests, however these did not prove to be very accessible or easy to use and, after initial review, were not further considered:
National Institute for Health Research (NIHR) Innovation Observatory (www.io.nihr.ac.uk/)
We planned to check the following evidence repository for additional eligible studies however, the EPPI‐Centre and Norwegian Institute of Public Health resources proved to be more accessible therefore we decided to prioritise our other sources of evidence.
Meta‐evidence (meta-evidence.co.uk/the-role-of-evidence-synthesis-in-covid19/)
We intended for two authors to independently perform data extraction, however one review author extracted study characteristics, and a second author checked them. Contingency table data were extracted independently by two review authors as planned.
We planned to evaluate the effect of additional sources of heterogeneity, including reference standard and sample type. However, additional formal investigations using meta‐regression were not possible because of lack of variability across the studies in these features.
We planned to conduct a sensitivity analysis excluding studies that are solely published as preprints. We have inadequate study numbers to allow this at present but will reconsider for the next update.
Contributions of authors
JD was the contact person with the editorial base. JDI co‐ordinated contributions from the co‐authors and wrote the final draft of the review. JJD, JDi, YT, CD, STP, IH, AA, LFR, MP, MT, JDr, SB screened papers against eligibility criteria. RS conducted the literature searches. JDi, MT and AA appraised the quality of papers. JDi, MT and AA extracted data for the review and sought additional information about papers. JDi entered data into Review Manager 2020. JDi, JJD, YT and SB, analysed and interpreted data. JJD, JDi, YT, CD, STP, RS, ML, LH, AVB, DE, SD, JC worked on the methods sections and commented on the draft review. JJD and JDi responded to the comments of the referees. JJD is the guarantor of the update.
Sources of support
Internal sources
Liverpool School of Tropical Medicine, UK
University of Birmingham, UK
External sources
-
Department for International Development, UK
Project number: 300342‐104
National Institute for Health Research (NIHR), UK
NIHR Birmingham Biomedical Research Centre at the University Hospitals Birmingham NHS Foundation Trust and the University of Birmingham, UK
Declarations of interest
Jonathan J Deeks: JD has published or been quoted in opinion pieces in scientific publications, and in the mainstream and social media related to diagnostic testing. JD was the statistician on the Birmingham evaluation of the Innova test which is mentioned in the discussion of the paper. There was no funding for this evaluation of the Innova test. JD is a member of the Royal Statistical Society (RSS) COVID‐19 taskforce steering group, and co‐chair of the RSS Diagnostic Test Advisory Group. He is a consultant adviser to the WHO Essential Diagnostic List. JD receives payment from the BMJ as their Chief Statistical advisor.
Jacqueline Dinnes: none known
Yemisi Takwoingi: none known
Clare Davenport: none known
Mariska MG Leeflang: none known
René Spijker: none known
Lotty Hooft: none known
Ann Van den Bruel: none known
Devy Emperador: is employed by FIND with funding from DFID and KFW. FIND is a global non‐for profit product development partnership and WHO Diagnostic Collaboration Centre. It is FIND’s role to accelerate access to high‐quality diagnostic tools for low‐resource settings and this is achieved by supporting both R&D and access activities for a wide range of diseases, including COVID‐19. FIND has several clinical research projects to evaluate multiple new diagnostic tests against published Target Product Profiles that have been defined through consensus processes. These studies are for diagnostic products developed by private sector companies who provide access to know‐how, equipment/reagents, and contribute through unrestricted donations as per FIND policy and external SAC review.
Sabine Dittrich: is employed by FIND with funding from DFID and Australian Aid. FIND is a global non‐for profit product development partnership and WHO Diagnostic Collaboration Centre. It is FIND’s role to accelerate access to high‐quality diagnostic tools for low‐resource settings and this is achieved by supporting both R&D and access activities for a wide range of diseases, including COVID‐19. FIND has several clinical research projects to evaluate multiple new diagnostic tests against published Target Product Profiles that have been defined through consensus processes. These studies are for diagnostic products developed by private sector companies who provide access to know‐how, equipment/reagents, and contribute through unrestricted donations as per FIND policy and external SAC review.
Ada Adriano: none known
Sophie Beese: none known
Janine Dretzke: none known
Lavinia Ferrante di Ruffano: none known
Isobel Harris: none known
Malcolm Price: none known
Sian Taylor‐Phillips: none known
Sarah Berhane: none known
Jane Cunningham: none known
Edited (no change to conclusions)
References
References to studies included in this review
Albert 2020 {published data only}
- Albert E, Torres I, Bueno F, Huntley D, Molla E, Fernández-Fuentes MÁ, et al. Field evaluation of a rapid antigen test (Panbio™ COVID-19 Ag Rapid Test Device) for COVID-19 diagnosis in primary healthcare centers. Clinical Microbiology and Infection 2020 Nov 13 [Epub ahead of print]. [DOI: 10.1016/j.cmi.2020.11.004] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert E, Torres I, Bueno F, Huntley D, Molla E, Fernández-Fuentes MÁ, et al. Field evaluation of a rapid antigen test (Panbio™ COVID-19 Ag Rapid Test Device) for the diagnosis of COVID-19 in primary healthcare centers. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Alemany 2020 {published data only}
- Alemany A, Baro B, Ouchi D, Ubals M, Corbacho-Monné M, Vergara-Alert J, et al. Analytical and clinical performance of the Panbio COVID-19 Antigen-Detecting Rapid Diagnostic Test. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Assennato 2020 {published data only}
- Assennato SM, Ritchie AV, Nadala C, Goel N, Tie C, Nadala LM, et al. Performance evaluation of the SAMBA II SARS-CoV-2 test for point-of-care detection of SARS-CoV-2. Journal of Clinical Microbiology 2020;59:e01262-20. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assennato SM, Ritchie AV, Nadala C, Goel N, Zhang H, Datir R, et al. Performance evaluation of the point-of-care SAMBA II SARS-CoV-2 test for detection of SARS-CoV-2. medRxiv [Preprint] 24 May 2020. [DOI: 10.1101/2020.05.24.20100990] [DOI] [PMC free article] [PubMed] [Google Scholar]
Billaud 2020 {published data only}
- Billaud G, Gaymard A, Lina B, Laboratoire de Virologie des HCL CNR des virus des infections respiratoires. Evaluation du Test Antigénique ABBOTT SARS-COV2 ABBOT. Lyon, France: SFM (French Society of Microbiology), 2020. [Google Scholar]
Blairon 2020 {published data only}
- Blairon L, Wilmet A, Beukinga I, Tre-Hardy M. Implementation of rapid SARS-CoV-2 antigenic testing in a laboratory without access to molecular methods: experiences of a general hospital. Journal of Clinical Virology 2020;129:104472. [DOI: 10.1016/j.jcv.2020.104472] [DOI] [PMC free article] [PubMed] [Google Scholar]
Broder 2020 {published data only}
- Broder K, Babiker A, Myers C, White T, Jones H, Cardella J, et al. Test agreement between Roche cobas 6800 and Cepheid GeneXpert Xpress SARS-CoV-2 assays at high cycle threshold ranges. Journal of Clinical Microbiology 2020;58:e01187-20. [DOI: 10.1128/JCM.01187-20] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broder KJ, Babiker A, Myers C, White T, Jones H, Cardella J, et al. Test agreement between Roche cobas 6800 and Cepheid GeneXpert Xpress SARS-CoV-2 assays at high cycle threshold ranges. bioRxiv [Preprint] 5 May 2020:1-13. [DOI: 10.1101/2020.05.05.078501] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cerutti 2020 {published data only}
- Cerutti F, Burdino E, Milia MG, Allice T, Gregori G, Bruzzone B, et al. Urgent need of rapid tests for SARS CoV-2 antigen detection: evaluation of the SD-Biosensor antigen test for SARS-CoV-2. Journal of Clinical Virology 2020;132:104654. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chen 2020a {published data only}
- Chen JH, Yip CC, Poon RW, Chan KH, Cheng VC, Hung IF, et al. Evaluating the use of posterior oropharyngeal saliva in a point-of-care assay for the detection of SARS-CoV-2. Emerging Microbes and Infections 2020;9(1):1356-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Collier 2020 {published data only}
- Collier DA, Assennato SM, Sithole N, Sharrocks K, Ritchie A, Ravji P, et al. Rapid point of care nucleic acid testing for SARS-CoV-2 in hospitalised patients: a clinical trial and implementation study. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Courtellemont 2020 {published data only}
- Courtellemont L, Guinard J, Guillaume C, Giaché S, Rzepecki V, Seve A, et al. Real-life performance of a novel antigen detection test on nasopharyngeal specimens for SARS-CoV-2 infection diagnosis: a prospective study. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cradic 2020(a) {published data only}
- Cradic K, Lockhart M, Ozbolt P, Fatica L, Landon L, Lieber M, et al. Clinical evaluation and utilization of multiple molecular in vitro diagnostic assays for the detection of SARS-CoV-2. American Journal of Clinical Pathology 2020;154(2):201-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cradic 2020(b) {published data only}
- Cradic K, Lockhart M, Ozbolt P, Fatica L, Landon L, Lieber M, et al. Clinical evaluation and utilization of multiple molecular in vitro diagnostic assays for the detection of SARS-CoV-2. American Journal of Clinical Pathology 2020;154(2):201-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Diao 2020 {published data only}
- Diao B, Wen K, Chen J, Liu Y, Yuan Z, Han C, et al. Diagnosis of acute respiratory syndrome coronavirus 2 infection by detection of nucleocapsid protein. medRxiv [Preprint] 10 March 2020:1-13. [DOI: 10.1101/2020.03.07.20032524] [DOI] [Google Scholar]
- Diao B, Wen K, Zhang J, Chen J, Han C, Chen Y, et al. Accuracy of a nucleocapsid protein antigen rapid test in the diagnosis of SARS-CoV-2 infection. Clin Microbiol Infect 2020 Oct 5 [Epub ahead of print]. [DOI: 10.1016/j.cmi.2020.09.057] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dust 2020 {published data only}
- Dust K, Hedley A, Nichol K, Stein D, Adam H, Karlowsky JA, et al. Comparison of commercial assays and laboratory developed tests for detection of SARS-CoV-2. Journal of Virological Methods 2020;285:113970. [DOI: 10.1016/j.jviromet.2020.113970] [DOI] [PMC free article] [PubMed] [Google Scholar]
Fenollar 2020(a) {published data only}
- Fenollar F, Bouam A, Ballouche M, Fuster L, Prudent E, Colson P, et al. Evaluation of the Panbio Covid-19 rapid antigen detection test device for the screening of patients with COVID-19. Journal of Clinical Microbiology 2020. [DOI: 10.1128/JCM.02589-20] [DOI] [PMC free article] [PubMed] [Google Scholar]
Fenollar 2020(b) {published data only}
- Fenollar F, Bouam A, Ballouche M, Fuster L, Prudent E, Colson P, et al. Evaluation of the Panbio Covid-19 rapid antigen detection test device for the screening of patients with COVID-19. Journal of Clinical Microbiology 2020. [DOI: 10.1128/JCM.02589-20] [DOI] [PMC free article] [PubMed] [Google Scholar]
FIND 2020a {published data only}
- Foundation for Innovative New Diagnostics (FIND). Evaluation of Bionote, Inc. NowCheck COVID-19 Ag Test - External Report. Switzerland: FIND, 2020. [Google Scholar]
FIND 2020b {published data only}
- Foundation for Innovative New Diagnostics (FIND). Evaluation of Abbott Panbio COVID-19 Ag Rapid Test Device - External Report. Switzerland: FIND, 2020. [Google Scholar]
FIND 2020c (BR) {published data only}
- Foundation for Innovative New Diagnostics (FIND). Evaluation of SD Biosensor, Inc. STANDARD Q COVID-19 Ag Test - External Report. Switzerland: FIND, 2020. [Google Scholar]
FIND 2020c (CH) {published data only}
- Foundation for Innovative New Diagnostics (FIND). Evaluation of SD Biosensor, Inc. STANDARD Q COVID-19 Ag Test - External Report. Switzerland: FIND, 2020. [Google Scholar]
FIND 2020d (BR) {published data only}
- Foundation for Innovative New Diagnostics (FIND). Evaluation of SD Biosensor, Inc. STANDARD F COVID-19 Ag FIA - External Report. Switzerland: FIND, 2020. [Google Scholar]
FIND 2020d (DE) {published data only}
- Foundation for Innovative New Diagnostics (FIND). Evaluation of SD Biosensor, Inc. STANDARD F COVID-19 Ag FIA - External Report. Switzerland: FIND, 2020. [Google Scholar]
FIND 2020e (BR) {published data only}
- Foundation for Innovative New Diagnostics (FIND). Evaluation of RapiGEN Inc. BIOCREDIT COVID-19 Ag - External Report. Switzerland: FIND, 2020. [Google Scholar]
FIND 2020e (DE) {published data only}
- Foundation for Innovative New Diagnostics (FIND). Evaluation of RapiGEN Inc. BIOCREDIT COVID-19 Ag - External Report. Switzerland: FIND, 2020. [Google Scholar]
Fourati 2020 [A] {published data only}
- Fourati S, Audureau E, Chevaliez S, Pawlotsky JM. Évaluation de la performance diagnostique des tests rapides d’orientation diagnostique antigéniques COVID-19. France: AP-HP Hopitaux universitaires Henri-Mondor, 2020. [Google Scholar]
Fourati 2020 [B] {published data only}
- Fourati S, Audureau E, Chevaliez S, Pawlotsky JM. Évaluation de la performance diagnostique des tests rapides d’orientation diagnostique antigéniques COVID-19. France: AP-HP Hopitaux universitaires Henri-Mondor, 2020. [Google Scholar]
Fourati 2020 [C] {published data only}
- Fourati S, Audureau E, Chevaliez S, Pawlotsky JM. Évaluation de la performance diagnostique des tests rapides d’orientation diagnostique antigéniques COVID-19. France: AP-HP Hopitaux universitaires Henri-Mondor, 2020. [Google Scholar]
Fourati 2020 [D] {published data only}
- Fourati S, Audureau E, Chevaliez S, Pawlotsky JM. Évaluation de la performance diagnostique des tests rapides d’orientation diagnostique antigéniques COVID-19. France: AP-HP Hopitaux universitaires Henri-Mondor, 2020. [Google Scholar]
Fourati 2020 [E] {published data only}
- Fourati S, Audureau E, Chevaliez S, Pawlotsky JM. Évaluation de la performance diagnostique des tests rapides d’orientation diagnostique antigéniques COVID-19. France: AP-HP Hopitaux universitaires Henri-Mondor, 2020. [Google Scholar]
Ghofrani 2020 {published data only}
- Ghofrani M, Casas MT, Pelz RK, Kroll C, Blum N, Foster SD. Performance characteristics of the ID NOW COVID-19 assay: a regional health care system experience. medRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
Gibani 2020 {published data only}
- Gibani MM, Toumazou C, Sohbati M, Sahoo R, Karvela M, Hon TK, et al. Assessing a novel, lab-free, point-of-care test for SARS-CoV-2 (CovidNudge): a diagnostic accuracy study. Lancet Microbe 2020;1(7):E300-E307. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Goldenberger 2020 {published data only}
- Goldenberger D, Leuzinger K, Sogaard KK, Gosert R, Roloff T, Naegele K, et al. Brief validation of the novel GeneXpert Xpress SARS-CoV-2 PCR assay. Journal of Virologica. Methods 2020;284:113925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gremmels 2020(a) {published data only}
- Gremmels H, Winkel BM, Schuurman R, Rosingh A, Rigter NA, Rodriguez O, et al. Real-life validation of the Panbio COVID-19 Antigen Rapid Test (Abbott) in community-dwelling subjects with symptoms of potential SARS-CoV-2 infection. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremmels H, Winkel BM, Schuurman R, Rosingh A, Rigter NA, Rodriguez O, et al. Real-life validation of the PanbioTM COVID-19 antigen rapid test (Abbott) in community-dwelling subjects with symptoms of potential SARS-CoV-2 infection. EClinicalMedicine 2021;31:100677. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gremmels 2020(b) {published data only}
- Gremmels H, Winkel BM, Schuurman R, Rosingh A, Rigter NA, Rodriguez O, et al. Real-life validation of the PanbioTM COVID-19 antigen rapid test (Abbott) in community-dwelling subjects with symptoms of potential SARS-CoV-2 infection. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gupta 2020 {published data only}
- Gupta A, Khurana S, Das R, Srigyan D, Singh A, Mittal A, et al. Rapid chromatographic immunoassay-based evaluation of COVID-19: a cross-sectional, diagnostic test accuracy study & its implications for COVID-19 management in India. Indian Journal of Medical Research 2020 Oct 31 [Epub ahead of print]. [DOI: 10.4103/ijmr.IJMR_3305_20] [DOI] [PMC free article] [PubMed] [Google Scholar]
Harrington 2020 {published data only}
- Harrington A, Cox B, Snowdon J, Bakst J, Ley E, Grajales P, et al. Comparison of Abbott ID NOW and Abbott m2000 methods for the detection of SARS-CoV-2 from nasopharyngeal and nasal swabs from symptomatic patients. Journal of Clinical Microbiology 2020;58(8):e00798-20. [DOI: 10.1128/JCM.00798-20.] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hogan 2020 {published data only}
- Hogan CA, Garamani N, Lee AS, Tung JK, Sahoo MK, Huang C, et al. Comparison of the Accula SARS-CoV-2 test with a laboratory-developed assay for detection of SARS-CoV-2 RNA in clinical nasopharyngeal specimens. bioRxiv [Preprint] 2020. [DOI: 10.1101/2020.05.12.092379v1] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hou 2020 {published data only}
- Hou H, Chen J, Wang Y, Lu Y, Zhu Y, Zhang B, et al. Multicenter evaluation of the Cepheid Xpert Xpress SARS-CoV-2 Assay for the detection of SARS-CoV-2 in oropharyngeal swab specimens. Journal of Clinical Microbiology 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jin 2020 {published data only}
- Jin R, Pettengill MA, Hartnett NL, Auerbach HE, Peiper SC, Wang Z. Commercial severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) molecular assays: superior analytical sensitivity of cobas SARS-CoV-2 relative to NxTAG Cov Extended Panel and ID NOW COVID-19 test. Archives of Pathology and Laboratory Medicine 2020;144(11):1303-10. [DOI] [PubMed] [Google Scholar]
Jokela 2020 {published data only}
- Jokela P, Jääskeläinen AE, Jarva H, Holma T, Ahava M, Mannonen L, et al. SARS-CoV-2 sample-to-answer nucleic acid testing in a tertiary care emergency department: evaluation and utility. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokela P, Jääskeläinen AE, Jarva H, Holma T, Ahava MJ, Mannonen L, et al. SARS-CoV-2 sample-to-answer nucleic acid testing in a tertiary care emergency department: evaluation and utility. Journal of Clinical Virology 2020;131:104614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kruger 2020(a) {published data only}
- Krüger LJ, Gaeddert M, Köppel L, Brümmer LE, Gottschalk C, Miranda IB, et al. Evaluation of the accuracy, ease of use and limit of detection of novel, rapid, antigen-detecting point-of-care diagnostics for SARS-CoV-2. medRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
Kruger 2020(b) {published data only}
- Krüger LJ, Gaeddert M, Köppel L, Brümmer LE, Gottschalk C, Miranda IB, et al. Evaluation of the accuracy, ease of use and limit of detection of novel, rapid, antigen-detecting point-of-care diagnostics for SARS-CoV-2. medRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
Kruger 2020(c) {published data only}
- Krüger LJ, Gaeddert M, Köppel L, Brümmer LE, Gottschalk C, Miranda IB, et al. Evaluation of the accuracy, ease of use and limit of detection of novel, rapid, antigen-detecting point-of-care diagnostics for SARS-CoV-2. medRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
Lambert‐Niclot 2020 {published data only}
- Lambert-Niclot S, Cuffel A, Le Pape S, Vauloup-Fellous C, Morand-Joubert L, Roque-Afonso AM, et al. Evaluation of a rapid diagnostic assay for detection of SARS CoV-2 antigen in nasopharyngeal swab. Journal of Clinical Microbiology 2020;58(8):e00977-20. [DOI: 10.1128/JCM.00977-20] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lephart 2020 [A] {published data only}
- Lephart PR, Bachman M, LeBar W, McClellan S, Barron K, Schroeder L, et al. Comparative study of four SARS-CoV-2 nucleic acid amplification test (NAAT) platforms demonstrates that ID NOW performance is impaired substantially by patient and specimen type. bioRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lephart 2020 [B] {published data only}
- Lephart PR, Bachman M, LeBar W, McClellan S, Barron K, Schroeder L, et al. Comparative study of four SARS-CoV-2 nucleic acid amplification test (NAAT) platforms demonstrates that ID NOW performance is impaired substantially by patient and specimen type. bioRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lieberman 2020 {published data only}
- Lieberman JA, Pepper G, Naccache SN, Huang ML, Jerome KR, Greninger AL. Comparison of commercially available and laboratory developed assays for in vitro detection of SARS-CoV-2 in clinical laboratories. Journal of Clinical Microbiology 2020;58(8):e00821-20. [DOI: 10.1128/JCM.00821-20] [DOI] [PMC free article] [PubMed] [Google Scholar]
Linares 2020 {published data only}
- Linares M, Pérez TR, Romanyk J, Pérez García F, Gómez-Herruz P, Arroyo T, et al. Panbio antigen rapid test is reliable to diagnose SARS-CoV-2 infection in the first 7 days after the onset of symptoms. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linares M, Pérez-Tanoira R, Carrero A, Romanyk J, Pérez-García F, Gómez-Herruz P, et al. Panbio antigen rapid test is reliable to diagnose SARS-CoV-2 infection in the first 7 days after the onset of symptoms. Journal of Clinical Virology 2020;133:104659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Liotti 2020 {published data only}
- Liotti FM, Menchinelli G, Lalle E, Palucci I, Marchetti S, Colavita F, et al. Performance of a novel diagnostic assay for rapid SARS-CoV-2 antigen detection in nasopharynx samples. Clinical Microbiology and Infection 2020 Sep 23 [Epub ahead of print]. [DOI: 10.1016/j.cmi.2020.09.030] [DOI] [PMC free article] [PubMed] [Google Scholar]
Loeffelholz 2020 {published data only}
- Loeffelholz MJ, Alland D, Butler-Wu SM, Pandey U, Perno CF, Nava A, et al. Multicenter evaluation of the Cepheid Xpert Xpress SARS-CoV-2 test. Journal of Clinical Microbiology 2020;58(8):e00926-20. [DOI: 10.1128/JCM.00926-20] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mak 2020 {published data only}
- Mak GC, Cheng PK, Lau SS, Wong KK, Lau CS, Lam ET, et al. Evaluation of rapid antigen test for detection of SARS-CoV-2 virus. Journal of Clinical Virology 2020;129:104500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mertens 2020 {published data only}
- Mertens P, De Vos N, Martiny D, Jassoy C, Mirazimi A, Cuypers L, et al. Development and potential usefulness of the COVID-19 Ag Respi-Strip Diagnostic Assay in a pandemic context. Frontiers in Medicine (Lausanne) 2020;7:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens P, De Vos N, Martiny D, Jassoy C, Mirazimi A, Cuypers L, et al. Development and potential usefulness of the COVID-19 Ag Respi-Strip diagnostic assay in a pandemic context. medRxiv [Preprint] 24 April 2020:1-29. [DOI: 10.1101/2020.04.24.20077776] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mitchell 2020 {published data only}
- Mitchell SL, George KS. Evaluation of the COVID19 ID NOW EUA assay. Journal of Clinical Virology 2020;128:104429. [DOI: 10.1016/j.jcv.2020.104429] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2020 {published data only}
- Moore NM, Li H, Schejbal D, Lindsley J, Hayden M. Comparison of two commercial molecular tests and a laboratory-developed modification of the CDC 2019-nCOV RT-PCR assay for the qualitative detection of SARS-CoV-2 from upper respiratory tract specimens. medRxiv [Preprint] 2020:1-22. [DOI: 10.1101/2020.05.02.20088740] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore NM, Li H, Schejbal D, Lindsley J, Hayden MK. Comparison of two commercial molecular tests and a laboratory-developed modification of the CDC 2019-nCoV RT-PCR assay for the detection of SARS-CoV-2. Journal of Clinical Microbiology 2020;58:e00938-20. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moran 2020 {published data only}
- Moran A, Beavis KG, Matushek SM, Ciaglia C, Francois N, Tesic V, et al. The detection of SARS-CoV-2 using the Cepheid Xpert Xpress SARS-CoV-2 and Roche cobas SARS-CoV-2 assays. Journal of Clinical Microbiology 2020;58(8):e00772-20. [DOI: 10.1128/JCM.00772-20] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nagura‐Ikeda 2020 {published data only}
- Nagura-Ikeda M, Imai K, Tabata S, Miyoshi K, Murahara N, Mizuno T, et al. Clinical evaluation of self-collected saliva by quantitative reverse transcription-PCR (RT-qPCR), direct RT-qPCR, reverse transcription-loop-mediated isothermal amplification, and a rapid antigen test to diagnose COVID-19. Journal of Clinical Microbiology 2020;58(9):e01438-20. [DOI: 10.1128/JCM.01438-20] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nash 2020 {published data only}
- Nash B, Badea A, Reddy A, Bosch M, Salcedo N, Gomez AR, et al. The impact of high frequency rapid viral antigen screening on COVID-19 spread and outcomes: a validation and modeling study. medRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
PHE 2020(a) {published data only}
- Peto T. COVID-19: rapid antigen detection for SARS-CoV-2 by lateral flow assay: a national systematic evaluation for mass-testing. medRxiv [Preprint] 2021. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Public Health England (PHE). Preliminary report from the Joint PHE Porton Down & University of Oxford SARS-CoV-2 test development and validation cell: rapid evaluation of lateral flow viral antigen detection devices (LFDs) for mass community testing. Public Health England, 2020. [Google Scholar]
PHE 2020(b) {published data only}
- Public Health England (PHE). Preliminary report from the Joint PHE Porton Down & University of Oxford SARS-CoV-2 test development and validation cell: rapid evaluation of lateral flow viral antigen detection devices (LFDs) for mass community testing. Public Health England, 2020. [Google Scholar]
PHE 2020(c) [non‐HCW tested] {published data only}
- Public Health England (PHE). Preliminary report from the Joint PHE Porton Down & University of Oxford SARS-CoV-2 test development and validation cell: rapid evaluation of lateral flow viral antigen detection devices (LFDs) for mass community testing. Public Health England, 2020. [Google Scholar]
PHE 2020(d) [HCW tested] {published data only}
- Public Health England (PHE). Preliminary report from the Joint PHE Porton Down & University of Oxford SARS-CoV-2 test development and validation cell: rapid evaluation of lateral flow viral antigen detection devices (LFDs) for mass community testing. Public Health England, 2020. [Google Scholar]
PHE 2020(d) [Lab tested] {published data only}
- Public Health England (PHE). Preliminary report from the Joint PHE Porton Down & University of Oxford SARS-CoV-2 test development and validation cell: rapid evaluation of lateral flow viral antigen detection devices (LFDs) for mass community testing. Public Health England, 2020. [Google Scholar]
PHE 2020(e) {published data only}
- Public Health England (PHE). Preliminary report from the Joint PHE Porton Down & University of Oxford SARS-CoV-2 test development and validation cell: rapid evaluation of lateral flow viral antigen detection devices (LFDs) for mass community testing. Public Health England, 2020. [Google Scholar]
Porte 2020a {published data only}
- Porte L, Legarraga P, Vollrath V, Aguilera X, Munita JM, Araos R, et al. Evaluation of novel antigen-based rapid detection test for the diagnosis of SARS-CoV-2 in respiratory samples. International Journal of Infectious Diseases 2020;99:328-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porte L, Legarraga P, Vollrath V, Aguilera X, Munita JM, Araos R, et al. Evaluation of novel antigen-based rapid detection test for the diagnosis of SARS-CoV-2 in respiratory samples. papers.ssrn.com/abstract=3569871 [Preprint] 14 April 2020;(dx.doi.org/10.2139/ssrn.3569871):1-23. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Porte 2020b [A] {published data only}
- Porte L, Legarraga P, Iruretagoyena M, Vollrath V, Pizarro G, Munita JM, et al. Rapid SARS-CoV-2 antigen detection by immunofluorescence – a new tool to detect infectivity. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.10.04.20206466] [DOI] [Google Scholar]
Porte 2020b [B] {published data only}
- Porte L, Legarraga P, Iruretagoyena M, Vollrath V, Pizarro G, Munita JM, et al. Rapid SARS-CoV-2 antigen detection by immunofluorescence – a new tool to detect infectivity. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.10.04.20206466] [DOI] [Google Scholar]
Rhoads 2020 {published data only}
- Rhoads DD, Cherian SS, Roman K, Stempak LM, Schmotzer CL, Sadri N. Comparison of Abbott ID NOW, Diasorin Simplexa, and CDC FDA EUA methods for the detection of SARS-CoV-2 from nasopharyngeal and nasal swabs from individuals diagnosed with COVID-19. Journal of Clinical Microbiology 2020;58(8):e00760-20. [DOI: 10.1128/JCM.00760-20] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schildgen 2020 [A] {published data only}
- Schildgen V, Demuth S, Lüsebrink J, Schildgen O. Limits and opportunities of SARS-CoV-2 antigen rapid tests – an experience based perspective. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.09.22.20199372] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schildgen 2020 [B] {published data only}
- Schildgen V, Demuth S, Lüsebrink J, Schildgen O. Limits and opportunities of SARS-CoV-2 antigen rapid tests – an experience based perspective. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.09.22.20199372] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schildgen 2020 [C] {published data only}
- Schildgen V, Demuth S, Lüsebrink J, Schildgen O. Limits and opportunities of SARS-CoV-2 antigen rapid tests – an experience based perspective. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.09.22.20199372] [DOI] [PMC free article] [PubMed] [Google Scholar]
Scohy 2020 {published data only}
- Scohy A, Anantharajah A, Bodeus M, Kabamba-Mukadi B, Verroken A, Rodriguez-Villalobos H. Low performance of rapid antigen detection test as frontline testing for COVID-19 diagnosis. Journal of Clinical Virology 2020;129:104455. [DOI: 10.1016/j.jcv.2020.104455] [DOI] [PMC free article] [PubMed] [Google Scholar]
Shrestha 2020 {published data only}
- Shrestha B, Neupane AK, Pant S, Shrestha A, Bastola, A. Sensitivity and specificity of lateral flow antigen test kits for COVID-19 in asymptomatic population of quarantine centre of Province 3. Kathmandu University Medical Journal 2020;18(70):36-9. [PubMed] [Google Scholar]
Smithgall 2020 [A] {published data only}
- Smithgall MC, Scherberkova I, Whittier S, Green D. Comparison of Cepheid Xpert Xpress and Abbott ID Now to Roche cobas for the rapid detection of SARS-CoV-2. bioRxiv [Preprint] 25 April 2020:1-16. [DOI: 10.1101/2020.04.22.055327] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithgall MC, Scherberkova I, Whittier S, Green DA. Comparison of Cepheid Xpert Xpress and Abbott ID Now to Roche Cobas for the rapid detection of SARS-CoV-2. Journal of Clinical Virology 2020;128:104428. [DOI: 10.1016/j.jcv.2020.104428] [DOI] [PMC free article] [PubMed] [Google Scholar]
Smithgall 2020 [B] {published data only}
- Smithgall MC, Scherberkova I, Whittier S, Green DA. Comparison of Cepheid Xpert Xpress and Abbott ID Now to Roche Cobas for the rapid detection of SARS-CoV-2. Journal of Clinical Virology 2020;128:104428. [DOI: 10.1016/j.jcv.2020.104428] [DOI] [PMC free article] [PubMed] [Google Scholar]
SoRelle 2020 {published data only}
- SoRelle JA, Mahimainathan L, McCormick-Baw C, Cavuoti D, Lee F, Thomas A, et al. Saliva for use with a point of care assay for the rapid diagnosis of COVID-19. Clinica Chimica Acta 2020;510:685-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SoRelle Jeffrey, Mahimmainathan Lenin, McCormick-Baw Clare, Cavuoti Dominick, Lee Franceca, Bararia Anjali, et al. Evaluation of symptomatic patient saliva as a sample type for the Abbott ID NOW COVID-19 assay. medRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
Stevens 2020 {published data only}
- Stevens B, Hogan CA, Sahoo MK, Huang C, Garamani N, Zehnder J, et al. Comparison of a point-of-care assay and a high-complexity assay for detection of SARS-CoV-2 RNA. Journal of Applied Laboratory Medicine 2020;5(6):1307-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Szymczak 2020 {published data only}
- Szymczak WA, Goldstein DY, Orner EP, Fecher RA, Yokoda RT, Skalina KA, et al. Utility of stool PCR for the diagnosis of COVID-19: comparison of two commercial platforms. Journal of Clinical Microbiology 2020;58:e01369-20. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Takeda 2020 {published data only}
- Takeda Y, Mori M, Omi K. SARS-CoV-2 qRT-PCR Ct value distribution in Japan and possible utility of rapid antigen testing kit. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.06.16.20131243] [DOI] [Google Scholar]
Thwe 2020 {published data only}
- Thwe PM, Ren P. How many are we missing with ID NOW COVID-19 assay using direct nasopharyngeal swabs? Findings from a mid-sized academic hospital clinical microbiology laboratory. Diagnostic Microbiology and Infectious Disease 2020;98(2):115123. [DOI: 10.1016/j.diagmicrobio.2020.115123] [DOI] [PMC free article] [PubMed] [Google Scholar]
Van der Moeren 2020(a) {published data only}
- Van der Moeren N, Zwart VF, Lodder EB, Van den Bijllaardt W, Van Esch HR, Stohr JJ, et al. Performance evaluation of a SARS-CoV-2 rapid antigen test: test performance in the community in the Netherlands. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.10.19.20215202] [DOI] [Google Scholar]
Van der Moeren 2020(b) {published data only}
- Van der Moeren N, Zwart VF, Lodder EB, Van den Bijllaardt W, Van Esch HR, Stohr JJ, et al. Performance evaluation of a SARS-CoV-2 rapid antigen test: test performance in the community in the Netherlands. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.10.19.20215202] [DOI] [Google Scholar]
Veyrenche 2020 {published data only}
- Veyrenche N, Bollore K, Pisoni A, Bedin A-S, Mondain A-M, Ducos J, et al. Diagnosis value of SARS-CoV-2 antigen/antibody combined testing using rapid diagnostic tests at hospital admission. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.09.19.20197855] [DOI] [PMC free article] [PubMed] [Google Scholar]
Weitzel 2020 [A] {published data only}
- Weitzel T, Legarraga P, Iruretagoyena M, Pizarro G, Vollrath V, Araos R, et al. Head-to-head comparison of four antigen-based rapid detection tests for the diagnosis of SARS-CoV-2 in respiratory samples. bioRxiv [Preprint] 30 May 2020:1-21. [DOI: 10.1101/2020.05.27.119255] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzel T, Legarraga P, Iruretagoyena M, Pizarro G, Vollrath V, Porte L, et al. Comparative evaluation of four rapid SARS-CoV-2 antigen detection tests using universal transport medium. Travel Medicine and Infectious Diseases 2020 Dec 2 [Epub ahead of print]:101942. [DOI: 10.1016/j.tmaid.2020.101942] [DOI] [PMC free article] [PubMed] [Google Scholar]
Weitzel 2020 [B] {published data only}
- Weitzel T, Legarraga P, Iruretagoyena M, Pizarro G, Vollrath V, Araos R, et al. Head-to-head comparison of four antigen-based rapid detection tests for the diagnosis of SARS-CoV-2 in respiratory samples. bioRxiv [Preprint] 30 May 2020:1-21. [DOI: 10.1101/2020.05.27.119255] [DOI] [PMC free article] [PubMed] [Google Scholar]
Weitzel 2020 [C] {published data only}
- Weitzel T, Legarraga P, Iruretagoyena M, Pizarro G, Vollrath V, Araos R, et al. Head-to-head comparison of four antigen-based rapid detection tests for the diagnosis of SARS-CoV-2 in respiratory samples. bioRxiv [Preprint] 30 May 2020:1-21. [DOI: 10.1101/2020.05.27.119255] [DOI] [PMC free article] [PubMed] [Google Scholar]
Weitzel 2020 [D] {published data only}
- Weitzel T, Legarraga P, Iruretagoyena M, Pizarro G, Vollrath V, Araos R, et al. Head-to-head comparison of four antigen-based rapid detection tests for the diagnosis of SARS-CoV-2 in respiratory samples. bioRxiv [Preprint] 30 May 2020:1-21. [DOI: 10.1101/2020.05.27.119255] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wolters 2020 {published data only}
- Wolters F, Van de Bovenkamp J, Van den Bosch B, Van den Brink S, Broeders M, Chung NH, et al. Multi-center evaluation of Cepheid Xpert(R) Xpress SARS-CoV-2 point-of-care test during the SARS-CoV-2 pandemic. Journal of Clinical Virology 2020;128:104426. [DOI: 10.1016/j.jcv.2020.104426] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wong 2020 {published data only}
- Wong RC, Wong AH, Ho YI, Leung EC, Lai RW. Evaluation on testing of deep throat saliva and lower respiratory tract specimens with Xpert Xpress SARS-CoV-2 assay. Journal of Clinical Virology 2020;131:104593. [DOI: 10.1016/j.jcv.2020.104593 ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Young 2020 {published data only}
- Young S, Taylor S, Cammarata C, Roger-Dalbert C, Montano A, Griego-Fullbright C, et al. Clinical evaluation of BD Veritor SARS-CoV-2 point-of-care test performance compared to PCR-based testing and versus the Sofia 2 SARS Antigen point-of-care test. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.09.01.20185777] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young S, Taylor SN, Cammarata CL, Varnado KG, Roger-Dalbert C, Montano A, et al. Clinical evaluation of BD Veritor SARS-CoV-2 point-of-care test performance compared to PCR-based testing and versus the Sofia 2 SARS Antigen point-of-care test. Journal of Clinical Microbiology 2020. [DOI: 10.1128/JCM.02338-20] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhen 2020 [A] {published data only}
- Zhen W, Smith E, Manji R, Schron D, Berry GJ. Clinical evaluation of three sample-to-answer platforms for the detection of SARS-CoV-2. Journal of Clinical Microbiology 2020;58(8):e00783-20. [DOI: 10.1128/JCM.00783-20] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhen 2020 [B] {published data only}
- Zhen W, Smith E, Manji R, Schron D, Berry GJ. Clinical evaluation of three sample-to-answer platforms for the detection of SARS-CoV-2. Journal of Clinical Microbiology 2020;58(8):e00783-20. [DOI: 10.1128/JCM.00783-20] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Ai 2020 {published data only}
- Ai JW, Zhang HC, Xu T, Wu J, Zhu M, Yu YQ, et al. Optimizing diagnostic strategy for novel coronavirus pneumonia, a multi-center study in Eastern China. medRxiv [Preprint] 17 February 2020:1-18. [DOI: 10.1101/2020.02.13.20022673] [DOI] [Google Scholar]
Anahtar 2020 {published data only}
- Anahtar MN, McGrath GE, Rabe BA, Tanner NA, White BA, Lennerz JK, et al. Clinical assessment and validation of a rapid and sensitive SARS-CoV-2 test using reverse-transcription loop-mediated isothermal amplification. medRxiv [Preprint] 18 May 2020:1-22. [DOI: 10.1101/2020.05.12.20095638] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ar Gouilh 2020 {published data only}
- Ar Gouilh M, Cassier R, Maille E, Schanen C, Rocque L-M, Vabret Astrid. An easy, reliable and rapid SARS-CoV2 RT-LAMP based test for Point-of-Care and diagnostic lab. medRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
Arizti‐Sanz 2020 {published data only}
- Arizti-Sanz J, Freije CA, Stanton AC, Boehm CK, Petros BA, Siddiqui S, et al. Integrated sample inactivation, amplification, and Cas13-based detection of SARS-CoV-2. bioRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Arumugam 2020 {published data only}
- Arumugam A, Faron ML, Yu P, Markham C, Wong S. A rapid COVID-19 RT-PCR detection assay for low resource settings. bioRxiv [Preprint] 30 April 2020:1-13. [DOI: 10.1101/2020.04.29.069591] [DOI] [Google Scholar]
Avetyan 2020 {published data only}
- Avetyan D, Chavushyan A, Ghazaryan H, Melkonyan A, Stepanyan A, Zakharyan R, et al. SARS-CoV-2 detection by extraction-free qRT-PCR for massive and rapid COVID-19 diagnosis during a pandemic. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Azhar 2020 {published data only}
- Azhar M, Phutela R, Kumar M, Ansari AH, Rauthan R, Gulati S, et al. Rapid, accurate, nucleobase detection using FnCas9. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Azzi 2020 {published data only}
- Azzi L, Baj A, Alberio T, Lualdi M, Veronesi G, Carcano G, et al. Rapid salivary test suitable for a mass screening program to detect SARS-CoV-2: a diagnostic accuracy study. Journal of Infection 2020;81(3):e75-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Baek 2020 {published data only}
- Baek YH, Um J, Antigua KJ, Park JH, Kim Y, Oh S, et al. Development of a reverse transcription-loop-mediated isothermal amplification as a rapid early-detection method for novel SARS-CoV-2. Emerging Microbes & Infections 2020;9(1):998-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Barra 2020 {published data only}
- Barra GB, Ticiane Henriques SR, Goes MP, Henriques JR, Nery LF. Analytical sensibility and specificity of two RT-qPCR protocols for SARS-CoV-2 detection performed in an automated workflow. medRxiv [Preprint] 10 March 2020:1-5. [DOI: 10.1101/2020.03.07.20032326] [DOI] [PMC free article] [PubMed] [Google Scholar]
Basu 2020 {published data only}
- Basu A, Zinger T, Inglima K, Woo KM, Atie O, Yurasits L, et al. Performance of Abbott ID NOW COVID-19 rapid nucleic acid amplification test in nasopharyngeal swabs transported in viral media and dry nasal swabs, in a New York City academic institution. Journal of Clinical Microbiology 2020;58(8):e01136-20. [DOI: 10.1128/JCM.01136-20] [DOI] [PMC free article] [PubMed] [Google Scholar]
Behrmann 2020 {published data only}
- Behrmann O, Bachmann I, Spiegel M, Schramm M, El Wahed AA, Dobler G, et al. Rapid detection of SARS-CoV-2 by low volume real-time single tube reverse transcription recombinase polymerase amplification using an exo probe with an internally linked quencher (exo-IQ). Clinical Chemistry 8 May 2020 [Epub ahead of print]:hvaa116. [DOI: 10.1093/clinchem/hvaa116] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bokelmann 2020 {published data only}
- Bokelmann L, Nickel O, Maricic T, Paabo S, Meyer M, Borte S, et al. Rapid, reliable, and cheap point-of-care bulk testing for SARS-CoV-2 by combining hybridization capture with improved colorimetric LAMP (Cap-iLAMP). medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bordi 2020 {published data only}
- Bordi L, Piralla A, Lalle E, Giardina F, Colavita F, Tallarita M, et al. Rapid and sensitive detection of SARS-CoV-2 RNA using the Simplexa COVID-19 direct assay. Journal of Clinical Virology 2020;128:104416. [DOI] [PMC free article] [PubMed] [Google Scholar]
Brandsma 2020 {published data only}
- Brandsma E, Verhagen HJ, Van de Laar TJ, Claas EC, Cornelissen M, Van den Akker E. Rapid, sensitive and specific SARS coronavirus-2 detection: a multi-center comparison between standard qRT-PCR and CRISPR based DETECTR. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Broughton 2020 {published data only}
- Broughton JP, Deng X, Yu G, Fasching CL, Singh J, Streithorst J, et al. Rapid detection of 2019 novel coronavirus SARS-CoV-2 using a CRISPR-based DETECTR lateral flow assay. medRxiv [Preprint] 27 March 2020:1-28. [DOI: 10.1101/2020.03.06.20032334] [DOI] [Google Scholar]
Bull 2020 {published data only}
- Bull RA, Adikari TN, Ferguson JM, Hammond JM, Stevanovski I, Beukers AG, et al. Analytical validity of nanopore sequencing for rapid SARS-CoV-2 genome analysis. Nature Communications 2020;11(1):6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bulterys 2020 {published data only}
- Bulterys PL, Garamani N, Stevens B, Sahoo MK, Huang C, Hogan CA, et al. Comparison of a laboratory-developed test targeting the envelope gene with three nucleic acid amplification tests for detection of SARS-CoV-2. Journal of Clinical Virology 2020;129:104427. [DOI] [PMC free article] [PubMed] [Google Scholar]
Callahan 2020a {published data only}
- Callahan CJ, Lee R, Zulauf K, Tamburello L, Smith KP, Previtera J, et al. Open development and clinical validation of multiple 3D-printed sample-collection swabs: rapid resolution of a critical COVID-19 testing bottleneck. medRxiv [Preprint] 7 May 2020:1-16. [EMBASE: 10.1101/2020.04.14.20065094] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan CJ, Lee R, Zulauf KE, Tamburello L, Smith KP, Previtera J, et al. Open development and clinical validation of multiple 3D-printed nasopharyngeal collection swabs: rapid resolution of a critical COVID-19 testing bottleneck. Journal of Clinical Microbiology 2020;58(8):e00876-20. [DOI: 10.1128/JCM.00876-20] [DOI] [PMC free article] [PubMed] [Google Scholar]
Callahan 2020b {published data only}
- Callahan C, Lee R, Lee G, Zulauf K E, Kirby J E, Arnaout R. Nasal-swab testing misses patients with low SARS-CoV-2 viral loads. medRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
Chandler‐Brown 2020 {published data only}
- Chandler-Brown D, Bueno AM, Atay O, Tsao DS. A highly scalable and rapidly deployable RNA extraction-free COVID-19 assay by quantitative Sanger sequencing. medRxiv [Preprint] 10 April 2020:1-15. [DOI: 10.1101/2020.04.07.029199] [DOI] [Google Scholar]
Chen 2020b {published data only}
- Chen Y, Shi Y, Chen Y, Yang Z, Wu H, Zhou Z, et al. Contamination-free visual detection of SARS-CoV-2 with CRISPR/Cas12a: a promising method in the point-of-care detection. Biosens Bioelectron 2020;169:112642. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chow 2020 {published data only}
- Chow FW, Chan TT, Tam AR, Zhao S, Yao W, Fung J, et al. A rapid, simple, inexpensive, and mobile colorimetric assay COVID-19-LAMP for mass on-site screening of COVID-19. International Journal of Molecular Sciences 2020;21(15):5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
CNR 2020 {published data only}
- Centre National de Référence des virus des infections respiratoires. Evaluation des performances analytiques du test VitaPCR™ SARS-CoV-2 Assay, BIOSYNEX. Lyon, France: SFM (French Society of Microbiology), 2020. [Google Scholar]
CNR 2020a {published data only}
- Centre National de Référence des virus des infections respiratoires. Résultats d’évaluation de la performance en analytique pour la détection du SARS-CoV-2 dans le cadre de l’épidémie de COVID-19 comparaison avec la technique de référence du CNR IPP. Lyon, France: SFM (French Society of Microbiology), 2020. [Google Scholar]
Colson 2020 {published data only}
- Colson P, Lagier JC, Baudoin JP, Bou Khalil J, La Scola B, Raoult D. Ultrarapid diagnosis, microscope imaging, genome sequencing, and culture isolation of SARS-CoV-2. European Journal of Clinical Microbiology & Infectious Diseases 2020;39(8):1601-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Comar 2020 {published data only}
- Comar M, Brumat M, Concas MP, Argentini G, Bianco A, Bicego L, et al. COVID-19 experience: first Italian survey on healthcare staff members from a Mother-Child Research hospital using combined molecular and rapid immunoassays test. medRxiv [Preprint] 22 April 2020:1-12. [DOI: 10.1101/2020.04.19.20071563] [DOI] [Google Scholar]
Comer 2020 {published data only}
- Comer SW, Fisk D. An extended laboratory validation study and comparative performance evaluation of the Abbott ID NOW COVID-19 Assay in a Coastal California tertiary care medical center. medRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
Crone 2020 {published data only}
- Crone MA, Priestman M, Ciechonska M, Jensen K, Sharp DJ, Randell P, et al. A new role for biofoundries in rapid prototyping, development, and validation of automated clinical diagnostic tests for SARS-CoV-2. medRxiv [Preprint] 12 May 2020:1-31. [DOI: 10.1101/2020.05.02.20088344] [DOI] [Google Scholar]
Curti 2020 {published data only}
- Curti L, Pereyra-Bonnet F, Gimenez CA. An ultrasensitive, rapid, and portable coronavirus SARS-CoV-2 sequence detection method based on CRISPR-Cas12. bioRxiv [Preprint] 2 March 2020:1-10. [DOI: 10.1101/2020.02.29.971127] [DOI] [Google Scholar]
Davda 2020 {published data only}
- Davda JN, Frank K, Prakash S, Purohit G, Vijayashankar DP, Vedagiri D, et al. An inexpensive RT-PCR endpoint diagnostic assay for SARS-CoV-2 using nested PCR: direct assessment of detection efficiency of RT-qPCR tests and suitability for surveillance. bioRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
Ding 2020a {published data only}
- Ding X, Yin K, Li Z, Liu C. All-in-One Dual CRISPR-Cas12a (AIOD-CRISPR) assay: a case for rapid, ultrasensitive and visual detection of novel coronavirus SARS-CoV-2 and HIV virus. bioRxiv [Preprint] 21 March 2020:1-19. [DOI: 10.1101/2020.03.19.998724] [DOI] [Google Scholar]
Ding 2020b {published data only}
- Ding X, Yin K, Li Z, Lalla RV, Ballesteros E, Sfeir MM, et al. Ultrasensitive and visual detection of SARS-CoV-2 using all-in-one dual CRISPR-Cas12a assay. Nature Communications 2020;11(1):4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
Dohla 2020 {published data only}
- Dohla M, Boesecke C, Schulte B, Diegmann C, Sib E, Richter E, et al. Rapid point-of-care testing for SARS-CoV-2 in a community screening setting shows low sensitivity. Public Health 2020;182:170-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Dong 2020 {published data only}
- Dong Y, Wu X, Li S, Lu R, Wan Z, Qin J, et al. Comparative evaluation of 19 reverse transcription loop-mediated isothermal amplification assays for detection of SARS-CoV-2. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
El‐Tholoth 2020 {published data only}
- El-Tholoth M, Bau HH, Song J. A single and two-stage, closed-tube, molecular test for the 2019 novel coronavirus (COVID-19) at home, clinic, and points of entry. chemRxiv [Preprint] 2020;19:19. [DOI: 10.26434/chemrxiv.11860137] [DOI] [Google Scholar]
Farfan 2020 {published data only}
- Farfan MJ, Torres JP, Oryan M, Olivares M, Gallardo P, Salas C. Optimizing RT-PCR detection of SARS-CoV-2 for developing countries using pool testing. medRxiv [Preprint] 17 April 2020:1-10. [DOI: 10.1101/2020.04.15.20067199] [DOI] [PubMed] [Google Scholar]
FIND 2020f {published data only}
- FIND. FIND Evaluation of Coris BioConcept COVID-19 Ag Respi-Strip - External Report. Switzerland: FIND, 2020. [Google Scholar]
Fowler 2020 {published data only}
- Fowler VL, Armson B, Gonzales JL, Wise EL, Howson EL, Vincent-Mistiaen Z, et al. A reverse-transcription loop-mediated isothermal amplification (RT-LAMP) assay for the rapid detection of SARS-CoV-2 within nasopharyngeal and oropharyngeal swabs at Hampshire Hospitals NHS Foundation Trust. medRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
Francis 2020 {published data only}
- Francis R, Le Bideau M, Jardot P, Grimaldier C, Raoult D, Khalil JY, et al. High speed large scale automated isolation of SARS-CoV-2 from clinical samples using miniaturized co-culture coupled with high content screening. bioRxiv [Preprint] 19 May 2020:1-23. [DOI: 10.1101/2020.05.14.097295] [DOI] [PMC free article] [PubMed] [Google Scholar]
Freire‐Paspuel 2020a {published data only}
- Freire-Paspuel B, Vega-Marino P, Velez A, Cruz M, Bereguiain MA. High sensitivity CDC EUA SARS-CoV-2 kit-based End Point-PCR assay. medRxiv [Preprint] 18 May 2020:1-7. [DOI: 10.1101/2020.05.11.20098590] [DOI] [Google Scholar]
Freire‐Paspuel 2020b {published data only}
- Freire-Paspuel B, Vega-Marino P, Velez A, Castillo P, Cruz M, Garcia-Bereguiain MA. Evaluation of nCoV-QS (MiCo BioMed) for RT-qPCR detection of SARS-CoV-2 from nasopharyngeal samples using CDC FDA EUA qPCR kit as a gold standard: An example of the need of validation studies. Journal of Clinical Virology 2020;128:104454. [DOI: 10.1016/j.jcv.2020.104454] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ganguli 2020 {published data only}
- Ganguli A, Mostafa A, Berger J, Aydin M, Sun F, Valera E, et al. Rapid isothermal amplification and portable detection system for SARS-CoV-2. bioRxiv [Preprint] 21 May 2020:1-31. [DOI: 10.1101/2020.05.21.108381] [DOI] [PMC free article] [PubMed] [Google Scholar]
Giamarellos‐Bourboulis 2020 {published data only}
- Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host & Microbe 2020;27(6):992-1000 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gonzalez‐Gonzalez 2020a {published data only}
- Gonzalez-Gonzalez E, Lara-Mayorga IM, Rodriguez-Sanchez IP, Yee-de Leon F, Garcia-Rubio A, Garciamendez-Mijares CE, et al. Scaling diagnostics in times of COVID-19: rapid prototyping of 3D-printed water circulators for loop-mediated isothermal amplification (LAMP) and detection of SARS-CoV-2 virus. medRxiv [Preprint] 19 June 2020:1-39. [DOI: 10.1101/2020.04.09.20058651] [DOI] [Google Scholar]
Gonzalez‐Gonzalez 2020b {published data only}
- Gonzalez-Gonzalez E, Trujillo-de Santiago G, Lara-Mayorga IM, Martinez-Chapa SO, Alvarez MM. Portable and accurate diagnostics for COVID-19: combined use of the miniPCR thermocycler and a well-plate reader for SARS-CoV-2 virus detection. PLoS One 2020;15(8):e0237418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Grant 2020 {published data only}
- Grant PR, Turner MA, Shin GY, Nastouli E, Levett LJ. Extraction-free COVID-19 (SARS-CoV-2) diagnosis by RT-PCR to increase capacity for national testing programmes during a pandemic. bioRxiv [Preprint] 9 April 2020:1-6. [DOI: ] [Google Scholar]
Hass 2020 {published data only}
- Hass KN, Bao M, He Q, Park M, Qin P, Du K. Integrated Micropillar Polydimethylsiloxane Accurate CRISPR Detection (IMPACT) system for rapid viral DNA sensing. bioRxiv [Preprint] 20 March 2020:1-10. [DOI: 10.1101/2020.03.17.994137] [DOI] [PMC free article] [PubMed] [Google Scholar]
Herrera 2020 {published data only}
- Herrera V, Hsu V, Adewale A, Hendrix T, Johnson L, Kuhlman J, et al. Testing of healthcare workers exposed to COVID19 with rapid antigen detection. medRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
Hirotsu 2020 {published data only}
- Hirotsu Y, Maejima M, Shibusawa M, Nagakubo Y, Hosaka K, Amemiya K, et al. Comparison of automated SARS-CoV-2 antigen test for COVID-19 infection with quantitative RT-PCR using 313 nasopharyngeal swabs, including from seven serially followed patients. International Journal of Infectious Diseases 2020;99:397-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hogan 2020a {published data only}
- Hogan CA, Sahoo MK, Huang C, Garamani N, Stevens B, Zehnder J, et al. Comparison of the Panther Fusion and a laboratory-developed test targeting the envelope gene for detection of SARS-CoV-2. Journal of Clinical Virology 2020;127:104383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Howson 2020 {published data only}
- Howson E, Kidd S, Sawyer J, Cassar C, Cross D, Lewis T, et al. Preliminary optimisation of a simplified sample preparation method to permit direct detection of SARS-CoV-2 within saliva samples using reverse-transcription loop-mediated isothermal amplification (RT-LAMP). medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hu 2020 {published data only}
- Hu X, Deng Q, Li J, Chen J, Wang Z, Zhang X, et al. Development and clinical application of a rapid and sensitive loop-mediated isothermal amplification test for SARS-CoV-2 infection. medRxiv [Preprint] 29 May 2020:1-28. [DOI: 10.1101/2020.05.20.20108530] [DOI] [PMC free article] [PubMed] [Google Scholar]
Huang 2020 {published data only}
- Huang WE, Lim B, Hsu CC, Xiong D, Wu W, Yu Y, et al. RT-LAMP for rapid diagnosis of coronavirus SARS-CoV-2. Microbial Biotechnology 2020;13(4):950-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Huang 2021 {published data only}
- Huang L, Ding L, Zhou J, Chen S, Chen F, Zhao C, et al. One-step rapid quantification of SARS-CoV-2 virus particles via low-cost nanoplasmonic sensors in generic microplate reader and point-of-care device. Biosensors & Bioelectronics 2021;171:112685. [DOI] [PMC free article] [PubMed] [Google Scholar]
James 2020 {published data only}
- James P, Stoddart D, Harrington ED, Beaulaurier J, Ly L, Reid SW, et al. LamPORE: rapid, accurate and highly scalable molecular screening for SARS-CoV-2 infection, based on nanopore sequencing. medRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
Jiang 2020 {published data only}
- Jiang M, Pan W, Arastehfar A, Fang W, ling L, Fang H, et al. Development and validation of a rapid single-step reverse transcriptase loop-mediated isothermal amplification (RT-LAMP) system potentially to be used for reliable and high-throughput screening of COVID-19. medRxiv [Preprint] 27 March 2020:1-12. [DOI: 10.1101/2020.03.15.20036376] [DOI] [PMC free article] [PubMed] [Google Scholar]
Joung 2020 {published data only}
- Joung J, Ladha A, Saito M, Segel M, Bruneau R, Huang MW, et al. Point-of-care testing for COVID-19 using SHERLOCK diagnostics. medRxiv [Preprint] 8 May 2020:1-21. [DOI: 10.1101/2020.05.04.20091231] [DOI] [Google Scholar]
Joung 2020a {published data only}
- Joung J, Ladha A, Saito M, Kim NG, Woolley AE, Segel M, et al. Detection of SARS-CoV-2 with SHERLOCK One-Pot Testing. New England Journal of Medicine 2020;383(15):1492-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kalikiri 2020 {published data only}
- Kalikiri MK, Hasan M, Mirza F, Xaba T, Tang P, Lorenz S. High-throughput extraction of SARS-CoV-2 RNA from nasopharyngeal swabs using solid-phase reverse immobilization beads. medRxiv [Preprint] 11 April 2020:1-5. [DOI: 10.1101/2020.04.08.20055731] [DOI] [Google Scholar]
Kashiwagi 2020 {published data only}
- Kashiwagi K, Ishii Y, Aoki K, Yagi S, Maeda T, Miyazaki T, et al. Immunochromatographic test for the detection of SARS-CoV-2 in saliva. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kim 2019 {published data only}
- Kim JH, Kang M, Park E, Chung DR, Kim J, Hwang ES. A simple and multiplex Loop-Mediated isothermal Amplification (LAMP) assay for rapid detection of SARS-CoV. Biochip Journal 2019;13(4):341-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kim 2020 {published data only}
- Kim Y, Yaseen AB, Kishi JY, Hong F, Saka SK, Sheng K, et al. Single-strand RPA for rapid and sensitive detection of SARS-CoV-2 RNA. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.08.17.20177006] [Google Scholar]
Konrad 2020 {published data only}
- Konrad R, Eberle U, Dangel A, Treis B, Berger A, Bengs K, et al. Rapid establishment of laboratory diagnostics for the novel coronavirus SARS-CoV-2 in Bavaria, Germany, February 2020. Euro Surveillance 2020;25(9):2000173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kurstjens 2020 {published data only}
- Kurstjens S, Van der Horst A, Herpers R, Geerits MW, Kluiters-de Hingh YC, Göttgens E-L, et al. Rapid identification of SARS-CoV-2-infected patients at the emergency department using routine testing. bioRxiv [Preprint] 4 April 2020:1-21. [DOI: 10.1101/2020.04.20.20067512] [DOI] [PubMed] [Google Scholar]
Kyosei 2020 {published data only}
- Kyosei Y, Namba M, Yamura S, Takeuchi R, Aoki N, Nakaishi K, et al. Proposal of de novo antigen test for COVID-19: ultrasensitive detection of spike proteins of SARS-CoV-2. Diagnostics (Basel) 2020;10(8):594. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lalli 2020 {published data only}
- Lalli MA, Chen X, Langmade SJ, Fronick CC, Sawyer CS, Burcea LC, et al. Rapid and extraction-free detection of SARS-CoV-2 from saliva with colorimetric LAMP. medRxiv [Preprint] 11 May 2020:1-25. [DOI: 10.1101/2020.05.07.20093542] [DOI] [Google Scholar]
Lamb 2020 {published data only}
- Lamb LE, Bartolone SN, Ward E, Chancellor MB. Rapid detection of novel coronavirus (COVID-19) by reverse transcription-loop-mediated isothermal amplification. medRxiv [Preprint] 24 February 2020:1-17. [DOI: 10.1101/2020.02.19.20025155] [DOI] [PMC free article] [PubMed] [Google Scholar]
Landry 2020 {published data only}
- Landry ML, Criscuolo J, Peaper DR. Challenges in use of saliva for detection of SARS CoV-2 RNA in symptomatic outpatients. Journal of Clinical Virology 2020;130:104567. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lee 2020 {published data only}
- Lee JY, Best N, McAuley J, Porter JL, Seemann T, Schultz MB, et al. Validation of a single-step, single-tube reverse transcription loop-mediated isothermal amplification assay for rapid detection of SARS-CoV-2 RNA. Journal of Medical Microbiology 2020;69(9):1169-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Best N, McAuley J, Porter JL, Seemann T, Schultz MB, et al. Validation of a single-step, single-tube reverse transcription-loop-mediated isothermal amplification assay for rapid detection of SARS-CoV-2 RNA. bioRxiv [Preprint] 30 April 2020. [DOI: 10.1101/2020.04.28.067363] [DOI] [PMC free article] [PubMed] [Google Scholar]
Le Hingrat 2020 {published data only}
- Le Hingrat Q, Visseaux B, Laouenan C, Tubiana S, Bouadma L, Yazdanpanah Y, et al. SARS-CoV-2 N-antigenemia: a new alternative to nucleic acid amplification techniques. medRxiv [Preprint] 2020. [Google Scholar]
Li 2020 {published data only}
- Li M, Zhao Y, Li Y, Chen X, Luo D, Luo M, et al. Development and evaluation of a novel RT-PCR system for reliable and rapid SARS-CoV-2 screening of blood donations. Transfusion 2020;60(12):2952-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lin 2020 {published data only}
- Lin CY, Hwang D, Chiu NC, Weng LC, Liu HF, Mu JJ, et al. Increased detection of viruses in children with respiratory tract infection using PCR. International Journal of Environmental Research and Public Health 2020;17(2):564. [DOI] [PMC free article] [PubMed] [Google Scholar]
Liotti 2020a {published data only}
- Liotti FM, Menchinelli G, Marchetti S, Morandotti GA, Sanguinetti M, Posteraro B, et al. Evaluating the newly developed BioFire COVID-19 test for SARS-CoV-2 molecular detection. Clinical Microbiology and Infection 2020;26(12):1699-700. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lowe 2020 {published data only}
- Lowe CF, Matic N, Ritchie G, Lawson T, Stefanovic A, Champagne S, et al. Detection of low levels of SARS-CoV-2 RNA from nasopharyngeal swabs using three commercial molecular assays. Journal of Clinical Virology 2020;128:104387. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lu 2020 {published data only}
- Lu R, Wu X, Wan Z, Li Y, Zuo L, Qin J, et al. Development of a novel reverse transcription loop-mediated isothermal amplification method for rapid detection of SARS-CoV-2. Virologica Sinica 2020;35(3):344-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lu 2020a {published data only}
- Lu R, Wu X, Wan Z, Li Y, Jin X, Zhang C. A novel reverse transcription loop-mediated isothermal amplification method for rapid detection of SARS-CoV-2. International Journal of Molecular Sciences 2020;21(8):2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lubke 2020 {published data only}
- Lubke N, Senff T, Scherger S, Hauka S, Andree M, Adams O, et al. Extraction-free SARS-CoV-2 detection by rapid RT-qPCR universal for all primary respiratory materials. Journal of Clinical Virology 2020;130:104579. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mahari 2020 {published data only}
- Mahari S, Roberts A, Shahdeo D, Gandhi S. eCovSens-Ultrasensitive novel in-house built printed circuit board based electrochemical device for rapid detection of nCOVID-19 antigen, a spike protein domain 1 of SARS-CoV-2. bioRxiv [Preprint] 11 May 2020:1-20. [DOI: 10.1101/2020.04.24.059204] [DOI] [Google Scholar]
Marais 2020 {published data only}
- Marais G, Naidoo M, Hsiao NY, Valley-Omar Z, Smuts H, Hardie D. The implementation of a rapid sample preparation method for the detection of SARS-CoV-2 in a diagnostic laboratory in South Africa. PLoS One 2020;15(10):e0241029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Marzinotto 2020 {published data only}
- Marzinotto S, Mio C, Cifu A, Verardo R, Pipan C, Schneider C, et al. A streamlined approach to rapidly detect SARS-CoV-2 infection, avoiding RNA extraction. medRxiv [Preprint] 11 April 2020:1-10. [DOI: 10.1101/2020.04.06.20054114] [DOI] [PMC free article] [PubMed] [Google Scholar]
McCormick‐Baw 2020 {published data only}
- McCormick-Baw C, Morgan K, Gaffney D, Cazares Y, Jaworski K, Byrd A, et al. Saliva as an alternate specimen source for detection of SARS-CoV-2 in symptomatic patients using Cepheid Xpert Xpress SARS-CoV-2. Journal of Clinical Microbiology 2020;58(8):e01109-20. [DOI: 10.1128/JCM.01109-20] [DOI] [PMC free article] [PubMed] [Google Scholar]
McDonald 2020 {published data only}
- McDonald S, Courtney DM, Clark AE, Muthukumar A, Lee F, Balani J, et al. Diagnostic performance of a rapid point-of-care test for SARS-CoV-2 in an urban emergency department setting. Academic Emergency Medicine 2020;27(8):764-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
McRae 2020 {published data only}
- McRae MP, Simmons GW, Christodoulides NJ, Lu Z, Kang SK, Fenyo D, et al. Clinical decision support tool and rapid point-of-care platform for determining disease severity in patients with COVID-19. medRxiv [Preprint] 22 April 2020. [DOI: 10.1101/2020.04.16.20068411] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mei 2020 {published data only}
- Mei X, Lee HC, Diao K, Huang M, Lin B, Liu C, et al. Artificial intelligence-enabled rapid diagnosis of COVID-19 patients. medRxiv [Preprint] 7 May 2020. [DOI: 10.1101/2020.04.12.20062661] [DOI] [PMC free article] [PubMed] [Google Scholar]
Meyerson 2020 {published data only}
- Meyerson NR, Yang Q, Clark SK, Paige CL, Fattor WT, Gilchrist AR, et al. A community-deployable SARS-CoV-2 screening test using raw saliva with 45 minutes sample-to-results turnaround. medRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
Michel 2020 {published data only}
- Michel D, Danzer KM, Gross R, Conzelmann C, Muller JA, Freischmidt A, et al. Rapid, convenient and efficient kit-independent detection of SARS-CoV-2 RNA. Journal of Virological Methods 2020;286:113965. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mlcochova 2020 {published data only}
- Mlcochova P, Collier D, Ritchie A, Assennato SM, Hosmillo M, Goel N, et al. Combined point-of-care nucleic acid and antibody testing for SARS-CoV-2 following emergence of D614G spike variant. Cell Reports. Medicine 2020;1(6):100099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mohon 2020 {published data only}
- Mohon AN, Oberding L, Hundt J, Van Marle G, Pabbaraju K, Berenger BM, et al. Optimization and clinical validation of dual-target RT-LAMP for SARS-CoV-2. Journal of Virological Methods 2020;286:113972. [DOI] [PMC free article] [PubMed] [Google Scholar]
Moses 2020 {published data only}
- Moses SE, Warren C, Robinson P, Curtis J, Asquith S, Holme J, et al. Endpoint PCR Detection of Sars-CoV-2 RNA. medRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
Mostafa 2020 {published data only}
- Mostafa HH, Hardick J, Morehead E, Miller JA, Gaydos CA, Manabe YC. Comparison of the analytical sensitivity of seven commonly used commercial SARS-CoV-2 automated molecular assays. Journal of Clinical Virology 2020;130:104578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Muraoka 2020 {published data only}
- Muraoka M, Tanoi Y, Tada T, Mizukoshi M, Kawaguchi O. Quickly and simply detection for coronavirus including SARS-CoV-2 on the mobile real-time PCR device and without RNA Extraction. medRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
Nachtigall 2020 {published data only}
- Nachtigall FM, Pereira A, Trofymchuk OS, Santos LS. Detection of SARS-CoV-2 in nasal swabs using MALDI-MS. Nature Biotechnology 2020;38(10):1168-73. [DOI] [PubMed] [Google Scholar]
Newman 2020 {published data only}
- Newman CM, Dudley DM, Wiseman RW, McLaughlin MT, Karl JA, Stauss MR, et al. Initial evaluation of a mobile SARS-CoV-2 RT-LAMP testing strategy. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Noerz 2020 {published data only}
- Noerz D, Fischer N, Schultze A, Kluge S, Mayer-Runge U, Aepfelbacher M, et al. Clinical evaluation of a SARS-CoV-2 RT-PCR assay on a fully automated system for rapid on-demand testing in the hospital setting. Journal of Clinical Virology 2020;128:104390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ogawa 2020 {published data only}
- Ogawa T, Fukumori T, Nishihara Y, Sekine T, Okuda N, Nishimura T, et al. Another false-positive problem for a SARS-CoV-2 antigen test in Japan. Journal of Clinical Virology 2020;131:104612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Osterdahl 2020 {published data only}
- Osterdahl MF, Lee KA, Ni LM, Wilson S, Douthwaite S, Horsfall R, et al. Detecting SARS-CoV-2 at point of care: preliminary data comparing Loop-mediated Isothermal Amplification (LAMP) to PCR. medRxiv [Preprint] 4 April 2020:1-9. [DOI: 10.1101/2020.04.01.20047357] [DOI] [PMC free article] [PubMed] [Google Scholar]
Paden 2020 {published data only}
- Paden CR, Tao Y, Queen K, Zhang J, Li Y, Uehara A, et al. Rapid, sensitive, full genome sequencing of severe acute respiratory syndrome virus coronavirus 2 (SARS-CoV-2). bioRxiv [Preprint] 24 April 2020:1-13. [DOI: 10.1101/2020.04.22.055897] [DOI] [PMC free article] [PubMed] [Google Scholar]
Patchsung 2020 {published data only}
- Patchsung M, Jantarug K, Pattama A, Aphicho K, Suraritdechachai S, Meesawat P, et al. Clinical validation of a Cas13-based assay for the detection of SARS-CoV-2 RNA. Nature Biomedical Engineering 2020;4(12):1140-9. [DOI] [PubMed] [Google Scholar]
Pellanda 2020 {published data only}
- Pellanda LC, Wendland EM, McBride AJ, Tovo-Rodrigues L, Ferreira MR, Dellagostin OA, et al. Sensitivity and specificity of a rapid test for assessment of exposure to SARS-CoV-2 in a community-based setting in Brazil. medRxiv [Preprint] 10 May 2020:1-10. [DOI: 10.1101/2020.05.06.20093476] [DOI] [Google Scholar]
Peto 2020 {published data only}
- Peto L, Rodger G, Carter DP, Osman KL, Yavuz M, Johnson K, et al. Diagnosis of SARS-CoV-2 infection with LamPORE, a high-throughput platform combining loop-mediated isothermal amplification and nanopore sequencing. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pfefferle 2020 {published data only}
- Pfefferle S, Reucher S, Norz D, Lutgehetmann M. Evaluation of a quantitative RT-PCR assay for the detection of the emerging coronavirus SARS-CoV-2 using a high throughput system. EuroSurveillance 2020;25(9):2000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pollock 2020a {published data only}
- Pollock NR, Savage TJ, Wardell H, Lee R, Mathew A, M Stengelin, et al. Correlation of SARS-CoV-2 nucleocapsid antigen and RNA concentrations in nasopharyngeal samples from children and adults using an ultrasensitive and quantitative antigen assay. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Qian 2020 {published data only}
- Qian J, Boswell SA, Chidley C, Lu ZX, Pettit ME, Gaudio BL, et al. An enhanced isothermal amplification assay for viral detection. bioRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rabe 2020 {published data only}
- Rabe BA, Cepko C. SARS-CoV-2 detection using isothermal amplification and a rapid, inexpensive protocol for sample inactivation and purification. Proceedings of the National Academy of Sciences of the United States of America 2020;117(39):24450-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rauch 2020 {published data only}
- Rauch JN, Valois E, Ponce-Rojas JC, Aralis Z, Lach RS, Zappa F, et al. CRISPR-based and RT-qPCR surveillance of SARS-CoV-2 in asymptomatic individuals uncovers a shift in viral prevalence among a university population. medRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
Rodel 2020 {published data only}
- Rodel J, Egerer R, Suleyman A, Sommer-Schmid B, Baier M, Henke A, et al. Use of the variplex SARS-CoV-2 RT-LAMP as a rapid molecular assay to complement RT-PCR for COVID-19 diagnosis. Journal of Clinical Virology 2020;132:104616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rodriguez‐Manzano 2020 {published data only}
- Rodriguez-Manzano J, Malpartida-Cardenas K, Moser N, Pennisi I, Cavuto M, Miglietta L, et al. A handheld point-of-care system for rapid detection of SARS-CoV-2 in under 20 minutes. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Seo 2020 {published data only}
- Seo G, Lee G, Kim MJ, Baek SH, Choi M, Ku KB, et al. Rapid detection of COVID-19 causative virus (SARS-CoV-2) in human nasopharyngeal swab specimens using field-effect transistor-based biosensor. ACS Nano 2020;14(4):5135-42. [DOI] [PubMed] [Google Scholar]
Shirato 2020 {published data only}
- Shirato K, Nao N, Matsuyama S, Takeda M, Kageyama T. An ultra-rapid real-time RT-PCR method using the PCR1100 to detect severe acute respiratory syndrome coronavirus-2. Japanese Journal of Infectious Diseases 2020 Jun 30 [Epub ahead of print]. [DOI: 10.7883/yoken.JJID.2020.324] [DOI] [PubMed] [Google Scholar]
Singh 2020a {published data only}
- Singh NK, Ray P, Carlin AF, Magallanes C, Morgan SC, Laurent LC, et al. Hitting the diagnostic sweet spot: point-of-care SARS-CoV-2 salivary antigen testing with an off-the-shelf glucometer. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Singh 2020b {published data only}
- Singh P, Chakraborty R, Marwal R, Radhakrishan VS, Bhaskar AK, Vashisht H, et al. A rapid and sensitive method to detect SARS-CoV-2 virus using targeted-mass spectrometry. medRxiv [Preprint] 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Smyrlaki 2020 {published data only}
- Smyrlaki I, Ekman M, Lentini A, Rufino de Sousa N, Papanicolaou N, Vondracek M, et al. Massive and rapid COVID-19 testing is feasible by extraction-free SARS-CoV-2 RT-PCR. Nature Communications 2020;11(1):4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyrlaki I, Ekman M, Lentini A, Vondracek M, Papanicoloau N, Aarum J, et al. Massive and rapid COVID-19 testing is feasible by extraction-free SARS-CoV-2 RT-qPCR. medRxiv [Preprint] 12 May 2020:1-18. [DOI: 10.1101/2020.04.17.20067348] [DOI] [PMC free article] [PubMed] [Google Scholar]
St Hilaire 2020 {published data only}
- St Hilaire BG, Durand NC, Mitra N, Pulido SG, Mahajan R Blackburn A, et al. A rapid, low cost, and highly sensitive SARS-CoV-2 diagnostic based on whole genome sequencing. bioRxiv [Preprint] 11 May 2020:1-29. [DOI: 10.1101/2020.04.25.061499] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tan 2020 {published data only}
- Tan X, Lin C, Zhang J, Khaing OM, Fan X. Rapid and quantitative detection of COVID-19 markers in micro-liter sized samples. bioRxiv [Preprint] 22 April 2020:1-17. [DOI: 10.1101/2020.04.20.052233] [DOI] [Google Scholar]
Tanida 2020 {published data only}
- Tanida K, Koste L, Koenig C, Wenzel W, Fritsch A, Frickmann H. Evaluation of the automated cartridge-based ARIES SARS-CoV-2 Assay (RUO) against automated Cepheid Xpert Xpress SARS-CoV-2 PCR as gold standard. European Journal of Microbiology and Immunology (Bp) 2020;10(3):156-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tibbetts 2020 {published data only}
- Tibbetts R, Callahan K, Rofoo K, Zarbo RJ, Samuel L. Comparison of the NeuMoDX, Diasorin Simplexa, Cepheid and Roche CDC SARS-CoV 2 EUA assays using nasopharyngeal/nasal swabs in universal transport media (UTM) and sputum and tracheal aspirates. bioRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
Tran 2020 {published data only}
- Tran DH, Cuong HQ, Tran HT, Le UP, Do HD, Kui LM, et al. A comparative study of isothermal nucleic acid amplification methods for SARS-CoV-2 detection at point of care. bioRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
Visseaux 2020 {published data only}
- Visseaux B, Le Hingrat Q, Collin G, Bouzid D, Lebourgeois S, Le Pluart D, et al. Evaluation of the QIAstat-Dx Respiratory SARS-CoV-2 Panel, the first rapid multiplex PCR commercial assay for SARS-CoV-2 detection. Journal of Clinical Microbiology 2020;58(8):e00630-20. [DOI: 10.1128/JCM.00630-20] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wang 2020a {published data only}
- Wang X, Zhong M, Liu Y, Ma P, Dang L, Meng Q, et al. Rapid and sensitive detection of COVID-19 using CRISPR/Cas12a-based detection with Naked Eye Readout, CRISPR/Cas12a-NER. Science Bulletin (Beijing) 5 May 2020 [Epub ahead of print]. [DOI: 10.1016/j.scib.2020.04.041] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wang 2020b {published data only}
- Wang X, Yao H, Xu X, Zhang P, Zhang M, Shao J, et al. Limits of detection of six approved RT-PCR kits for the novel SARS-coronavirus-2 (SARS-CoV-2). Clinical Chemistry 2020;66(7):977-9. [DOI: 10.1093/clinchem/hvaa099] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wang 2020c {published data only}
- Wang J, Cai K, He X, Shen X, Liu J, Xu J, et al. A multiple center clinical evaluation of an ultra-fast single-tube assay for SARS-CoV-2 RNA. Clinical Microbiology and Infection 2020;26(8):P1076-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wee 2020 {published data only}
- Wee SK, Sivalingam SP, Yap EP. Rapid direct nucleic acid amplification test without RNA extraction for SARS-CoV-2 using a portable PCR thermocycler. bioRxiv [Preprint] 20 April 2020:1-12. [DOI: 10.1101/2020.04.17.042366] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wu 2020 {published data only}
- Wu T, Ge Y, Zhao K, Zhu X, Chen Y, Wu B, et al. A reverse-transcription recombinase-aided amplification assay for the rapid detection of N gene of severe acute respiratory syndrome coronavirus 2(SARS-CoV-2). Virology 2020;549:1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Xue 2020 {published data only}
- Xue G, Li S, Zhang W, Du B, Cui J, Yan C, et al. Reverse-transcription recombinase-aided amplification assay for rapid detection of the 2019 novel coronavirus (SARS-CoV-2). Analytical Chemistry 2020;92(14):9699-705. [DOI: 10.1021/acs.analchem.0c01032] [DOI] [PubMed] [Google Scholar]
Yan 2020 {published data only}
- Yan C, Cui J, Huang L, Du B, Chen L, Xue G, et al. Rapid and visual detection of 2019 novel coronavirus (SARS-CoV-2) by a reverse transcription loop-mediated isothermal amplification assay. Clinical Microbiology and Infection 2020;26(6):773-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yang 2020b {published data only}
- Yang W, Dang X, Wang Q, Xu M, Zhao Q, Zhou Y, et al. Rapid detection of SARS-CoV-2 using reverse transcription RT-LAMP method. medRxiv [Preprint] 3 March 2020:1-25. [DOI: 10.1101/2020.03.02.20030130] [DOI] [Google Scholar]
Yu 2020a {published data only}
- Yu L, Wu S, Hao X, Dong X, Mao L, Pelechano V, et al. Rapid detection of COVID-19 coronavirus using a reverse transcriptional loop-mediated isothermal amplification (RT-LAMP) diagnostic platform. Clinical Chemistry 2020;66(7):975-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yu 2020b {published data only}
- Yu L, Wu S, Hao X, Li X, Liu X, Ye S, et al. Rapid colorimetric detection of COVID-19 coronavirus using a reverse transcriptional loop-mediated isothermal amplification (RT-LAMP) diagnostic platform: iLACO. medRxiv [Preprint] 24 February 2020:1-19. [DOI: 10.1101/2020.02.20.20025874] [DOI] [PMC free article] [PubMed] [Google Scholar]
Yu 2020c {published data only}
- Yu S, Nimse SB, Kim J, Song KS, Kim T. Development of a lateral flow strip membrane assay for rapid and sensitive detection of the SARS-CoV-2. Analytical Chemistry 2020;92(20):14139-44. [DOI] [PubMed] [Google Scholar]
Zamecnik 2020 {published data only}
- Zamecnik CR, Rajan JV, Yamauchi KA, Mann SA, Sowa GM, Zorn KC, et al. ReScan, a multiplex diagnostic pipeline, pans human sera for SARS-CoV-2 antigens. medRxiv [Preprint] 13 May 2020:1-21. [DOI: 10.1101/2020.05.11.20092528] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zeng 2020 {published data only}
- Zeng W, Liu G, Ma H, Zhao D, Yang Y, Liu M, et al. Biochemical characterization of SARS-CoV-2 nucleocapsid protein. Biochemical and Biophysical Research Communications 2020;527(3):618-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2020 {published data only}
- Zhang Y, Odiwuor N, Xiong J, Sun L, Nyaruaba RO, Wei H, et al. Rapid molecular detection of SARS-CoV-2 (COVID-19) virus RNA using colorimetric LAMP. medRxiv [Preprint] 29 February 2020:1-14. [DOI: 10.1101/2020.02.26.20028373] [DOI] [Google Scholar]
Zhao 2020 {published data only}
- Zhao Z, Cui H, Song W, Ru X, Zhou W, Yu X. A simple magnetic nanoparticles-based viral RNA extraction method for efficient detection of SARS-CoV-2. bioRxiv [Preprint] 27 February 2020:1-18. [DOI: 10.1101/2020.02.22.961268] [DOI] [Google Scholar]
Zhu 2020 {published data only}
- Zhu X, Wang X, Han L, Chen T, Wang L, Li H, et al. Multiplex reverse transcription loop-mediated isothermal amplification combined with nanoparticle-based lateral flow biosensor for the diagnosis of COVID-19. Biosensors and Bioelectronics 2020;166:112437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Additional references
Arevalo‐Rodriguez 2020
- Arevalo-Rodriguez I, Buitrago-Garcia D, Simancas-Racines D, Zambrano-Achig P, Campo R, Ciapponi A, et al. False-negative results of initial RT-PCR assays for COVID-19: a systematic review. medRxiv [Preprint] 2020:1-26. [DOI: 10.1101/2020.04.16.20066787] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bossuyt 2015
- Bossuyt PM, Reitsma JB, Bruns DE, Gatsonis CA, Glasziou PP, Irwig L, et al. STARD 2015: an updated list of essential items for reporting diagnostic accuracy studies. BMJ 2015;351:h5527. [DOI: 10.1136/bmj.h5527] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Boyce 2018
- Boyce MR, Menya D, Turner EL, Laktabai J, Prudhomme-O'Meara W. Evaluation of malaria rapid diagnostic test (RDT) use by community health workers: a longitudinal study in western Kenya. Malaria Journal 2018;17(1):206. [DOI: 10.1186/s12936-018-2358-6] [DOI] [PMC free article] [PubMed] [Google Scholar]
Carter 2020
- Carter LJ, Garner LV, Smoot JW, Li Y, Zhou Q, Saveson CJ, et al. Assay techniques and test development for COVID-19 diagnosis. ACS Central Science 2020;6(5):591-605. [DOI: 10.1021/acscentsci.0c00501] [DOI] [PMC free article] [PubMed] [Google Scholar]
CDC 2020
- Centers for Disease Control and Prevention (CDC). Interim Guidance for Antigen Testing for SARS-CoV-2. Available from: www.cdc.gov/coronavirus/2019-ncov/lab/resources/antigen-tests-guidelines.html 2020.
Cevik 2021
- Cevik M, Tate M, Lloyd O, Maraolo AE, Schafers J, Ho A. SARS-CoV-2, SARS-CoV, and MERS-CoV viral load dynamics, duration of viral shedding, and infectiousness: a systematic review and meta-analysis. Lancet Microbe 2021;2(1):e13-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chen 2020
- Chen Q, He Z, Mao F, Pei H, Cao H, Liu X. Diagnostic technologies for COVID-19: a review. RSC Advances 2020;10(58):35257-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cheng 2020
- Cheng MP, Yansouni CP, Basta NE, Desjardins M, Kanjilal S, Paquette K, et al. Serodiagnostics for severe acute respiratory syndrome-related coronavirus-2: a narrative review. Annals of Internal Medicine 2020;15(173):450-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Corman 2020
- Corman V, Bleicker T, Brünink S, Drosten C, Landt O, Koopmans M, et al. Diagnostic detection of Wuhan coronavirus 2019 by real-time RT-PCR - protocol and preliminary evaluation as of Jan 13, 2020. Available from www.who.int/docs/default-source/coronaviruse/wuhan-virus-assay-v1991527e5122341d99287a1b17c111902.pdf?sfvrsn=d381fc88_2 2020.
Covidence [Computer program]
- Veritas Health Innovation Covidence. Version accessed 27 April 2020. Melbourne, Australia: Veritas Health Innovation. Available at covidence.org.
Crozier 2021
- Crozier A, Rajan S, Buchan I, McKee M. Put to the test: use of rapid testing technologies for COVID-19. BMJ 2021;372:n208. [DOI] [PubMed] [Google Scholar]
Deeks 2020a
- Deeks JJ, Raffle AE. Lateral flow tests cannot rule out SARS-CoV-2 infection. BMJ 2020;371:m4787. [DOI] [PubMed] [Google Scholar]
Deeks 2020b
- Deeks JJ, Dinnes J, Takwoingi Y, Davenport C, Leeflang MM, Spijker R, et al. Diagnosis of SARS-CoV-2 infection and COVID-19: accuracy of signs and symptoms; molecular, antigen, and antibody tests; and routine laboratory markers. Cochrane Database of Systematic Reviews 2020, Issue 4. Art. No: CD013596. [DOI: 10.1002/14651858.CD013596] [DOI] [Google Scholar]
Deeks 2020c
- Deeks JJ, Dinnes J, Takwoingi Y, Davenport C, Spijker R, Taylor-Phillips S, et al. Antibody tests for identification of current and past infection with SARS-CoV-2. Cochrane Database of Systematic Reviews 2020, Issue 6. Art. No: CD013652. [DOI: 10.1002/14651858.CD013652] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ferguson 2020
- Ferguson J, Dunn S, Best A, Mirza J, Percival B, Mayhew M, et al. Validation testing to determine the effectiveness of lateral flow testing for asymptomatic SARS-CoV-2 detection in low prevalence settings. medRxiv [Preprint] 2020:2020.12.01.20237784. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
FIND 2020
- FIND. SARS-COV-2 Diagnostic pipeline. www.finddx.org/covid-19/pipeline/ (accessed 5 January 2021).
Green 2020
- Green K, Graziadio S, Turner P, Fanshawe T, Allen J, on behalf of the Oxford COVID-19 Evidence Service Team Centre. Molecular and antibody point-of-care tests to support the screening, diagnosis and monitoring of COVID-19. Available at www.cebm.net/oxford-covid-19/ 7 April 2020.
Hadgu 1999
- Hadgu A. Discrepant analysis: a biased and an unscientific method for estimating test sensitivity and specificity. Journal of Clinical Epidemiology 1999;52(12):1231-7. [DOI: 10.1016/s0895-4356(99)00101-8] [DOI] [PubMed] [Google Scholar]
Healy 2020
- Healy B, Khan A, Metezai H, Blyth I, Asad H. The impact of false positive COVID-19 results in an area of low prevalence. Clinical Medicine Journal 2020. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jaafar 2020
- Jaafar R, Aherfi S, Wurtz N, Grimaldier C, Van Hoang T, Colson P, et al. Correlation between 3790 quantitative polymerase chain reaction–positives samples and positive cell cultures, including 1941 severe acute respiratory syndrome coronavirus 2 isolates. Clinical Infectious Diseases 2020:ciaa1491. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kozel 2017
- Kozel TR, Burnham-Marusich AR. Point-of-care testing for infectious diseases: past, present, and future. Journal of Clinical Microbiology 2017;55(8):2313-20. [DOI: 10.1128/JCM.00476-17] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kretzschmar 2020
- Kretzschmar ME, Rozhnova G, Bootsma MC, Van Boven M, Van de Wijgert JH, Bonten MJ. Impact of delays on effectiveness of contact tracing strategies for COVID-19: a modelling study. Lancet Public Health 2020;5(8):e452-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kucirka 2020
- Kucirka LM, Lauer SA, Laeyendecker O, Boon D, Lessler J. Variation in false-negative rate of reverse transcriptase polymerase chain reaction–based SARS-CoV-2 Tests by time since exposure. Annals of Internal Medicine 2020;173(4):262-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Landier 2018
- Landier J, Haohankhunnatham W, Das S, Konghahong K, Christensen P, Raksuansak J, et al. Operational performance of a Plasmodium falciparum ultrasensitive rapid diagnostic test for detection of asymptomatic infections in Eastern Myanmar. Journal of Clinical Microbiology 2018;56(8):e00565-18. [DOI: 10.1128/JCM.00565-18] [DOI] [PMC free article] [PubMed] [Google Scholar]
Larremore 2020
- Larremore DB, Wilder B, Lester E, Shehata S, Burke JM, Hay JA, et al. Test sensitivity is secondary to frequency and turnaround time for COVID-19 surveillance. medRxiv [Preprint] 2020:2020.06.22.20136309. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lee 2021
- Lee LY, Rozmanowski S, Pang M, Charlett A, Anderson C, Hughes GJ, et al. An observational study of SARS-CoV-2 infectivity by viral load and demographic factors and the utility lateral flow devices to prevent transmission. modmedmicro.nsms.ox.ac.uk/wp-content/uploads/2021/01/infectivity_manuscript_20210119_merged.pdf (accessed before March 2021).
Marks 2021
- Marks M, Millat-Martinez P, Ouchi D, Roberts CH, Alemany A, Corbacho-Monné M, et al. Transmission of COVID-19 in 282 clusters in Catalonia, Spain: a cohort study. Lancet Infectious Diseases 2021. [DOI: 10.1016/S1473-3099(20)30985-3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mayers 2020
- Mayers C, Baker K, Government Office for Science. Impact of false-positives and false-negatives in the UK’s COVID-19 RT-PCR testing programme. Available from assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/895843/S0519_Impact_of_false_positives_and_negatives.pdf 2020.
McInnes 2018
- McInnes MD, Moher D, Thombs BD, McGrath TA, Bossuyt PM, PRISMA-DTA Group. Preferred reporting items for a systematic review and meta-analysis of diagnostic test accuracy studies: the PRISMA-DTA Statement. JAMA 2018;319(4):388-96. [DOI: 10.1001/jama.2017.19163] [PMID: ] [DOI] [PubMed] [Google Scholar]
McInnes 2020
- McInnes M, Leeflang MM, Salameh J-P, McGrath T, Van der Pol CB, Frank RA, et al. Imaging tests for the diagnosis of COVID-19. Cochrane Database of Systematic Reviews 2020, Issue 6. Art. No: CD013639. [DOI: 10.1002/14651858.CD013639] [DOI] [PubMed] [Google Scholar]
Meyerowitz 2020
- Meyerowitz EA, Richterman A, Bogoch II, Low N, Cevik M. Towards an accurate and systematic characterisation of persistently asymptomatic infection with SARS-CoV-2. Lancet Infectious Diseases 2020. [DOI: 10.1016/S1473-3099(20)30837-9] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA Statement. PLoS Medicine 2009;6(7):1000097. [DOI: 10.1371/journal.pmed1000097] [DOI] [PMC free article] [PubMed] [Google Scholar]
Nabavi 2021
- Nabavi N, Dobson J. Testing asymptomatic individuals for SARS-CoV-2—known unknowns. blogs.bmj.com/bmj/2021/02/19/testing-asymptomatic-individuals-for-sars-cov-2-known-unknowns/ 2021. [Google Scholar]
ONS 2020
- Office for National Statistics. Coronavirus (COVID-19) Infection Survey, UK: 21 August 2020. Available from: ons.gov.uk/peoplepopulationandcommunity/healthandsocialcare/conditionsanddiseases/bulletins/coronaviruscovid19infectionsurveypilot/englandandwales21august2020 2020.
Pai 2012
- Pai NP, Vadnais C, Denkinger C, Engel N, Pai M. Point-of-care testing for infectious diseases: diversity, complexity, and barriers in low- and middle-income countries. PLoS Medicine 2012;9(9):e1001306. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pilarowski 2021
- Pilarowski G, Lebel P, Sunshine S, Liu J, Crawford E, Marquez C, et al. Performance characteristics of a rapid severe acute respiratory syndrome coronavirus 2 antigen detection assay at a public plaza testing site in San Francisco. Journal of Infectious Diseases 2021:jiaa802. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pollock 2020
- Pollock NR, Jacobs JR, Tran K, Cranston A, Smith S, O’Kane C, et al. Performance and implementation evaluation of the Abbott BinaxNOW Rapid Antigen Test in a high-throughput drive-through community testing site in Massachusetts. medRxiv [Preprint] 2021. [DOI: 10.1101/2021.01.09.21249499] [DOI] [PMC free article] [PubMed] [Google Scholar]
Reitsma 2005
- Reitsma JB, Glas AS, Rutjes AW, Scholten RJ, Bossuyt PM, Zwinderman AH. Bivariate analysis of sensitivity and specificity produces informative summary measures in diagnostic reviews. Journal of Clinical Epidemiology 2005;58(10):982-90. [DOI: 10.1016/j.jclinepi.2005.02.022] [DOI] [PubMed] [Google Scholar]
Review Manager 2020 [Computer program]
- The Cochrane Collaboration Review Manager 5 (RevMan 5). Version 5.4. Copenhagen: The Cochrane Collaboration, 2020.
Riley 2020
- Riley S, Ainslie KE, Eales O, Walters CE, Wang H, Atchison C, et al. Transient dynamics of SARS-CoV-2 as England exited national lockdown. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.08.05.20169078] [DOI] [Google Scholar]
Salameh 2020
- Salameh J-P, Leeflang MM, Hooft L, Islam N, McGrath TA, Pol CB, et al. Thoracic imaging tests for the diagnosis of COVID-19. Cochrane Database of Systematic Reviews 2020, Issue 9. Art. No: CD013639. [DOI: 10.1002/14651858.CD013639.pub2] [DOI] [PubMed] [Google Scholar]
Singanayagam 2020
- Singanayagam A, Patel M, Charlett A, Lopez BJ, Saliba V, Ellis J, et al. Duration of infectiousness and correlation with RT-PCR cycle threshold values in cases of COVID-19, England, January to May 2020. Eurosurveillance 2020;25(32):2001483. [DOI] [PMC free article] [PubMed] [Google Scholar]
Stata [Computer program]
- Stata. Version 15. College Station, TX, USA: StataCorp, 2017. Available at www.stata.com.
Stegeman 2020
- Stegeman I, Ochodo EA, Guleid F, Holtman G, Yang B, Cunningham J, et al. Routine laboratory testing to determine if a patient has COVID-19. Cochrane Database of Systematic Reviews 2020, Issue 11. Art. No: CD013787. [DOI: 10.1002/14651858.CD013787] [DOI] [PMC free article] [PubMed] [Google Scholar]
Struyf 2021
- Struyf T, Deeks JJ, Dinnes J, Takwoingi Y, Davenport C, Leeflang MM, et al. Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19. Cochrane Database of Systematic Reviews 2021, Issue 2. Art. No: CD013665. [DOI: 10.1002/14651858.CD013665.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Takwoingi 2017
- Takwoingi Y, Guo B, Riley RD, Deeks JJ. Performance of methods for meta-analysis of diagnostic test accuracy with few studies or sparse data. Statistical Methods in Medical Research 2017;26(4):1896-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
University of Liverpool 2020
- University of Liverpool. Liverpool COVID-19 community testing pilot. Interim evaluation report. Available from liverpool.ac.uk/media/livacuk/coronavirus/Liverpool,Community,Testing,Pilot,Interim,Evaluation.pdf 2020.
Vogels 2020
- Vogels CB, Brito AF, Wyllie AL, Fauver JR, Ott IM, Kalinich CC, et al. Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT–qPCR primer–probe sets. Nature Microbiology 2020;5(10):1299-305. [DOI: 10.1038/s41564-020-0761-6] [DOI] [PMC free article] [PubMed] [Google Scholar]
Whiting 2011
- Whiting PF, Rutjes AW, Westwood ME, Mallett S, Deeks JJ, Reitsma JB, et al. QUADAS‐2: a revised tool for the quality assessment of diagnostic accuracy studies. Annals of Internal Medicine 2011;155(8):529-36. [DOI] [PubMed] [Google Scholar]
WHO 2018
- World Health Organization (WHO). Diagnostic assessment: in vitro diagnostic medical devices (IVDs) used for the detection of high-risk human papillomavirus (HPV) genotypes in cervical cancer screening. Licence: CC BY-NC-SA 3.0 IGO. Available at apps.who.int/iris/handle/10665/272282 2018.
WHO 2020a
- World Health Organization. WHO information notice for IVD users: nucleic acid testing (NAT) technologies that use real-time polymerase chain reaction (RT-PCR) for detection of SARS-CoV-2. Available from who.int/news/item/14-12-2020-who-information-notice-for-ivd-users 2020.
WHO 2020b
- World Health Organization. Antigen-detection in the diagnosis of SARS-CoV-2 infection using rapid immunoassays. Interim guidance 11 September 2020. Available from: apps.who.int/iris/bitstream/handle/10665/334253/WHO-2019-nCoV-Antigen_Detection-2020.1-eng.pdf?sequence=1&isAllowed=y 2020.
WHO 2020c
- World Health Organization. COVID-19 Target product profiles for priority diagnostics to support response to the COVID-19 pandemic v.1.0. Available from who.int/publications/m/item/covid-19-target-product-profiles-for-priority-diagnostics-to-support-response-to-the-covid-19-pandemic-v.0.1 2020.
WHO 2020d
- World Health Organization. Laboratory testing of 2019 novel coronavirus (2019-nCoV) in suspected human cases: interim guidance. Available from www.who.int/publications/i/item/laboratory-testing-for-2019-novel-coronavirus-in-suspected-human-cases-20200117 2020.
WHO 2020e
- Global surveillance for COVID-19 caused by human infection with COVID-19 virus: interim guidance 20 March 2020. Available from apps.who.int/iris/bitstream/handle/10665/331506/WHO-2019-nCoV-SurveillanceGuidance-2020.6-eng.pdf 2020.
Yang 2020a
- Yang Y, Yang M, Yuan J, Wang F, Wang Z, Li J, et al. Laboratory diagnosis and monitoring the viral shedding of SARS-CoV-2 infection. Innovation 2020;1(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Dinnes 2020
- Dinnes J, Deeks JJ, Adriano A, Berhane S, Davenport C, Dittrich S, et al. Rapid, point‐of‐care antigen and molecular‐based tests for diagnosis of SARS‐CoV‐2 infection. Cochrane Database of Systematic Reviews 2020, Issue 8. Art. No: CD013705. [DOI: 10.1002/14651858.CD013705] [DOI] [PMC free article] [PubMed] [Google Scholar]