

Abstract
Triple negative breast cancer is an aggressive, heterogeneous disease with high recurrence and metastasis rates even with modern chemotherapy regimens and thus is in need of new therapeutics. Here, three novel synthetic analogues of chalcones, plant-based molecules that have demonstrated potency against a wide variety of cancers, were investigated as potential therapeutics for triple negative breast cancer. These compounds exhibit IC50 values of ~ 5 μM in triple negative breast cancer cell lines and are more potent against triple negative breast cancer cell lines than against non-tumor breast cell lines according to viability experiments. Tandem mass tag-based quantitative proteomics followed by gene set enrichment analysis and validation experiments using flow cytometry, apoptosis, and western blot assays revealed three different anti-cancer mechanisms for these compounds. First, the chalcone analogues induce the unfolded protein response followed by apoptosis. Second, increases in the abundances of MHC-I pathway proteins occurs, which would likely result in immune stimulation in an organism. And third, treatment with the chalcone analogues causes disruption of the cell cycle by interfering with microtubule structure and by inducing G1 phase arrest. These data demonstrate the potential of these novel chalcone derivatives as treatments for triple negative breast cancer, though further work evaluating their efficacy in vivo is needed.
Keywords: Chalcone, triple negative breast cancer, mechanism of action, unfolded protein response, tandem mass tag (TMT)
Graphical Abstract
Introduction
Triple negative breast cancer (TNBC) accounts for 15-20% of all breast cancer diagnoses, and it is the most aggressive phenotype of this disease, with high metastasis and recurrence rates and low five-year survival rates (1-3). TNBC is distinguished by its lack of estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 (HER2) expression, meaning these targets for treatment are not available. Although there are some common mutations (e.g., in BRCA1/2 (4, 5) and TP53 (5)) and overexpressed proteins (e.g., programmed death-ligand one (PD-L1) (6) and epidermal growth factor receptor (EGFR) (5)), TNBC is highly heterogeneous, making treatment challenging. Though new inroads are being made in TNBC treatment with immunotherapies (7), currently the main treatment for TNBC patients is chemotherapy with small-molecule drugs in addition to radiotherapy and surgery. Thus, it is valuable to develop novel chemotherapeutics that can broadly target TNBCs to render this highly deadly disease subtype curable.
Plant-based natural products have been the basis for many successful chemotherapy drugs from plants as notorious as the wild mandrake to those as delightful as the periwinkle (8). Chalcones – trans-1,3-diphenyl-2-propene-1-ones or open chain flavonoids – are found in a wide variety of plants and, depending on their structures, can have many different advantageous effects in the body, including anti-inflammatory, anti-oxidant, anti-viral, and anti-bacterial in addition to anti-cancer effects (for a review, see (9)). Natural chalcones and synthetic derivatives exert their anti-cancer properties through a diversity of molecular mechanisms, such as anti-angiogenesis, anti-metastasis, pro-apoptosis, cell cycle inhibition, and anti-inflammation (for reviews, see (10, 11)). The effect of chalcones on TNBC has not been well-explored, but some chalcones derived from plants have been shown to be effective against TNBC in vitro (12-15). For example, in TNBC cell lines, cardamonin, which is found in cardamom and several other plants (16), induces cell cycle arrest in the G2/M phase and reverses the epithelial-to-mesenchymal transition (EMT) by downregulation of the Wnt/β-catenin signaling pathway (14). And xanthohumol from hops has been shown to inhibit cell invasion by downregulating the expression of matrix metallopeptidase 9 (MMP-9) and to induce apoptosis by decreasing expression of apoptosis regulator (Bcl-2) in the TNBC cell line MDA-MB-231 (13). Furthermore, chalcones are easy to synthesize and modify, and are typically well tolerated in the body (17), making them attractive molecules for drug development. Earlier, we reported novel chalcone derivatives that showed potential anti-cancer activity in prostate cancer cell lines by inhibiting expression of prostate specific antigen (PSA) and nuclear translocation of androgen receptor (18).
Here, we report proteomic studies to identify the mechanism of action of three novel chalcone analogues in TNBC cell lines. With quantitative proteomic and gene set enrichment analyses followed by pathway validation, we show that TNBC treatment with these three molecules results in induction of the unfolded protein response and cell cycle arrest in G1 phase, resulting in apoptosis. Additionally, treatment with these three drug candidates results in an increase in the abundances of proteins involved in the major histocompatibility complex class I (MHC-I) pathway, which would likely lead to increased efficacy of these molecules in the body via immune stimulation against TNBC cells. Thus, these chalcone derivatives represent promising new therapeutics for TNBC that deserve further exploration.
Experimental Section
Synthesis of chalcone derivatives.
Chalcone derivatives (labeled CH-1 to CH-3, Figure 1) were synthesized and characterized as described previously (18). Chalcone derivative stock solutions were made by dissolving each derivative in dimethyl sulfoxide (DMSO) to 20 mM concentration, and they were stored at −20 °C.
Figure 1.

General workflow for studying the effects of novel chalcone derivatives CH-1, CH-2, and CH-3 on TNBC cell lines and non-tumor breast cell lines. Potency assays (viability and clonogenic assays) were performed for all cell lines with each derivative. Each cell line was then treated with DMSO (control) or 5 μM derivative for 12 hours before harvesting and labeling with TMT labels. Each TMT six-plex experiment contained one cell line treated with each derivative and DMSO, and contained two biological replicates of the DMSO-treated and CH-2-treated cells. The samples were analyzed by LC/MS-MS and processed using MaxQuant, Perseus, and GSEA in order to determine the main proteins, pathways, and processes affected by these derivatives. Validation in the form of cell cycle, apoptosis, and Western blot assays were then performed.
Cell culture.
The optimal growth conditions recommended by ATCC were used for each cell line. TNBC cell lines MDA-MB-231 and MDA-MB-468 were grown in L15 media supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (ATCC, Manassas, VA, for all) and were grown in 100% air at 37 °C. Non-tumor fibrocystic breast epithelial cell lines MCF-12A and MCF-10A were grown in MEBM media (Lonza, Walkersville, MD) and were grown in 95% air, 5% carbon dioxide at 37 °C. These different growth conditions for the various cell types may result in differences in the effects of the chalcone derivatives on the cells compared to if the cells were all grown under the same conditions. However, using optimal growth conditions for the different cell types helps ensure that the effects of treatment that are observed are due primarily to perturbations to the cells by chalcone treatment rather than an artifact of suboptimal growth conditions.
Viability assays.
A CellTiter-Blue viability assay (Promega, Madison, WI) was performed according to the manufacturer’s protocol to determine the potency of the three chalcone analogues with each cell line. Briefly, 20,000 cells in 100 μL media per well were plated on 96-well, tissue culture-treated, flat-bottom plates (Corning, Corning, NY) and incubated overnight. Media was removed, and 100 μL fresh media per well with no chalcone additive or each chalcone derivative at concentrations ranging from 0.005 to 50 μM was added. Cells were incubated with the chalcone derivatives for 48 hours. 20 μL CellTiter-Blue Reagent was added to each well, and the plates were incubated in the dark for two hours before viability was measured by fluorescence (560/590 nm excitation/emission) using an Infinite M1000 plate reader (Tecan, Morrisville, NC).
Colony formation assay.
Cells were seeded in 12-well plates (Corning, Corning, NY) at a density of 500 cells per well and incubated at 37 °C. After 24 hours, the media was replaced with fresh media containing DMSO (vehicle control) or a chalcone derivative at 1, 5, and 10 μM concentration, and the cells were cultured for 6 days in the same media. The colonies were fixed with ice-cold methanol:glacial acetic acid (3:1) for 10 minutes and stained with 1% crystal violet. The number of colonies was calculated after normalization to the plating efficiency for each sample. Each experiment was conducted in triplicate.
Sample preparation for quantitative proteomics.
For proteomics experiments, cells were grown to 80% confluence and then incubated with 5 μM of each chalcone derivative or with a DMSO vehicle control for 12 hours. This time was chosen to minimize protein signal from apoptosis. Each 10 cm plate of cells was processed separately. Cells were washed twice with phosphate-buffered saline (PBS), harvested by scraping, and pelleted by centrifugation at 300 x g or 100 x g for TNBC and non-tumor breast cell lines, respectively. Samples were processed and labeled with Tandem Mass Tag (TMT) (19) six-plex reagents (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s protocol. Cells were lysed by vortexing in lysis buffer (100 mM triethylammonium bicarbonate (TEAB, Thermo Fisher Scientific), 1% sodium dodecyl sulfate (SDS)). The lysate was centrifuged at 16,000 x g at 4 °C for ten minutes, and the supernatant retained. 50 μg of protein per sample as determined by a bicinchoninic acid assay (BCA, Pierce, Rockford, IL) was reserved, and samples were reduced with 10 mM TCEP (Sigma Aldrich) for one hour at 55 °C and alkylated with 18.75 mM iodoacetamide (Sigma Aldrich) for 30 minutes at room temperature in the dark. Proteins were precipitated with acetone overnight at −20 °C. Proteins were pelleted by centrifugation at 8,000 x g at 4 °C for ten minutes, redissolved in 50 mM TEAB, and digested with trypsin (Pierce, Rockford, IL) overnight at 37 °C. Tandem Mass Tag (TMT) six-plex reagents were brought to room temperature immediately before use and dissolved in 41 μL anhydrous acetonitrile. 20.5 μL of reagent was added to each sample, and the labeling reaction proceeded for one hour at room temperature before being quenched with 8 μL of 5% hydroxylamine (Thermo Fisher Scientific). For each six-plex experiment, samples were combined in equal protein amounts. Each six-plex experiment contained two biological replicates of a vehicle control-treated sample, a CH-1-treated sample, two biological replicates of a CH-2-treated sample, and a CH-3-treated sample, all for a single cell line. This resulted in four separate TMT experiments, one for each of the four cell lines.
LC-MS/MS analysis.
Samples were analyzed in triplicate by LC-MS/MS on a LTQ-Orbitrap Elite mass spectrometer (Thermo Fisher Scientific) equipped with a Dionex Ultimate 3000 LC (Thermo Fisher Scientific). Three microliters of sample were injected first onto a 5 mm C18 PepMap100 column (ID: 300 μm, particle size: 5 μm, pore size: 100 Å, Thermo Fisher Scientific) for desalting, and then onto a PicoFrit self-pack analytical column (OD: 360 μm, ID: 75 μm, Tip: 15 ± 1 μm, no coating, New Objective, Woburn, MA) packed with 25 cm of MagicC18 AQ (particle size: 5 μm, pore size: 100 Å, C18 resin, Michrom, Auburn, CA). Mobile phase A was 0.1% formic acid in water, and mobile phase B was 0.1 % formic acid in acetonitrile. Using a flow rate of 0.6 μL/min, peptides were separated over 100 minutes using a gradient of 2-35% B, followed by two minutes with a gradient of 35-85% B, and seven minutes at a constant 85% B. Full scans were acquired in the Orbitrap with a resolution of 30,000, scan range of 400 – 1800 m/z, AGC setting of 1e6, and a maximum inject time of 100 ms. Ions were selected for fragmentation using a top-eight, data-dependent method with a charge state requirement of 2+ or higher, a 4 mlz isolation window, and a dynamic exclusion window of 30 s. High energy collision-induced dissociation (HCD) was performed on these isolated precursors with a normalized collision energy of 35, 0.1 ms activation time, 100 ms maximum inject time, and an AGC setting of 5e4. Fragment ions were detected over a mass range of 110 – 2000 mlz in the Orbitrap with a resolution of 30,000.
Protein identification and quantitation.
Peptide identification and quantitation was performed using MaxQuant (20) version 1.6.0.1 and Perseus (21) version 1.6.0.7 (Cox Lab, Max Planck Institute). Triplicate runs were analyzed together as fractions in MaxQuant against the human Swiss-Prot database (08/03/2017, 42,210 entries). The reporter ion MS2 method for TMT six-plex samples was used with a reporter ion mass tolerance of 0.003 Da. Specific digestion was selected with trypsin/P as the enzyme and a maximum of two missed cleavages allowed. The precursor and fragment mass tolerance was 20 ppm, and the minimum peptide length was five amino acids. Allowed variable modifications were oxidation at methionine, acetylation at the protein N-terminus, and glutamine or glutamic acid conversion to pyroglutamic acid, with a maximum of five modifications allowed per peptide, and the only fixed modification was carbamidomethylation at cysteine residues. A 1% FDR for peptide and protein IDs was used from a target-decoy search using reverse peptide sequences. The razor protein ID was used in cases where multiple protein IDs could be made.
Results were filtered to remove contaminants (as identified by MaxQuant), reverse sequence IDs, and IDs for which there were fewer than two peptides detected. To obtain fold change information for identified proteins, corrected reporter ion intensities for a given protein for biological replicates were averaged, and the ratio of corrected reporter ion intensities was calculated. The base two logarithm of these fold changes was calculated, and median centering was performed in Perseus.
Statistical analysis.
KEGG pathway and Gene Ontology-Biological Processes analysis was performed using Gene Set Enrichment Analysis (GSEA) (22). Proteins were pre-ranked for GSEA according to the average of log2(fold change upon treatment) for all chalcone derivative-treated samples. In cases for which there was more than one fold change reported for a single protein, the value corresponding to the largest change was used. Five or more genes were allowed in each set, and the p-value calculated in GSEA for each pathway or biological process represents the statistical significance of enrichment compared to 1000 permutations of the proteomics data according to multiple hypothesis testing. A detailed description of the statistics performed in GSEA can be found in the appendix of the original publication on GSEA (22).
Cell cycle phase distribution analysis.
The effect of treatment with CH-1 on the distribution of MDA-MB-231 and MDA-MB-468 in each phase of the cell cycle was determined by flow cytometry using propidium iodide (PI). Briefly, 1 × 105 cells/well were seeded in a 6-well plate (Corning) and incubated at 37 °C. After 24 hours the media was replaced with fresh media containing CH-1 at 1, 5, and 10 μM concentration for specified time periods (12 and 24 hours). Floating and adherent cells were collected, washed with PBS twice, and fixed in 70% ethanol overnight at 4 °C. The fixed cells were incubated with 80 μg/mL RNase A and 50 μg/mL propidium iodide in saponin-EDTA solution for 30 min at 37 °C and analyzed using a Guava easyCyte Flow Cytometer (MilliporeSigma, Burlington, MA). The percentage of cells in different phases of the cell cycle was calculated for control (DMSO) and CH-1 treated samples after gating to remove debris and aggregates. The significance of changes observed between control and treated samples was calculated using Tukey's multiple comparisons test.
Annexin V apoptosis assay.
Cells were seeded and treated with CH-1 as done for the cell viability assay. At the end of treatment for 24 or 48 hours, both floating and attached cells were collected and processed for annexin V and PI staining using a FITC annexin V apoptosis detection kit (BD Biosciences Pharmingen, San Jose, CA) following the manufacturer's protocol. The flow analysis for annexin V and PI stained cells was carried out using a Guava easyCyte Flow Cytometer (MilliporeSigma, Burlington, MA). The significance of changes observed between control and treated samples was calculated using Tukey's multiple comparisons test.
Western blot analysis.
MDA-MB-231 and MDA-MB-468 cells were seeded in 10 cm culture dishes (Corning) and incubated at 37 °C until the cells reached 60% confluency. The cells were then treated with CH-1 at 1, 5, and 10 μM concentration in fresh media for 24 hours. Both floating and adherent cells were collected and lysed on ice using a solution of 50 mM Tris, 1% Triton X-100, 0.1% SDS, and 150 mM NaCl containing Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, Waltham, MA). The lysates were centrifuged at 19,000 x g for 30 min at 4 °C, and lysate proteins were resolved by 10 or 12.5% SDS polyacrylamide gel electrophoresis and transferred onto a nitrocellulose membrane (Bio-Rad, Hercules, CA). The membrane was blocked using tris-buffered saline containing 0.05% Tween-20, and 5% (weight/volume) non-fat dry milk for 1 hour and incubated with the desired primary antibody overnight at 4 °C, followed by treatment with an appropriate HRP-conjugated secondary antibody. The immunoreactive bands were visualized using an enhanced chemiluminescence method. The blots were stripped and re-probed with an anti-β-actin antibody to normalize for differences in protein loading. The following antibodies were used for these studies: Bcl-2 (1:1000 dilution, Cat. #2876S), Cyclin-D1 (1:1000 dilution, Cat. #2978T), CDK4 (1:2000 dilution, Cat. #12790T), and Drp-1 (1:500 dilution, Cat. #8570S) purchased from Cell Signaling Technology (Danvers, MA), β-actin (1:10000 dilution, Cat. #NB600-501) purchased from Novus Biologicals (Littleton, CO), and Hsp40 (1:500 dilution, Cat. #sc-398766) purchased from Santa Cruz Biotechnology, Inc (Dallas, TX). The immunoreactive bands were visualized using a Luminata Crescendo western HRP substrate (EMD Millipore, Burlington, MA) for enhanced chemiluminescence on an IVIS Lumina Imaging System (Perkin Elmer, Waltham, MA), and quantified using ImageJ (v1.8.0, NIH).
Results and Discussion
All samples were processed according to the workflow in Fig. 1 in order to determine the effect of the chalcone derivatives on TNBC and non-tumor breast cell lines. All cell lines were treated with each of the three chalcone derivatives (CH-1, CH-2, and CH-3, which differ solely by substitution at a single site, Fig. 1) to evaluate the effect of the chalcones on cell viability and proliferation, as well as on changes in the cellular proteome in order to gain insight into the mechanism of action of these molecules. The effects on pathways or processes that were found in the proteomics analysis were then validated via Western blot and flow cytometry experiments. The cell lines chosen for these experiments were two TNBC cell lines, MDA-MB-231 and MDA-MB-468, and two non-tumor breast epithelial cell lines, MCF10A and MCF12A, which represent some of the most widely-studied TNBC and non-tumor breast cell lines. Both TNBC cell lines were derived from metastatic sites of adenocarcinomas and are basal-like, and both non-tumor cell lines were spontaneously immortalized breast cells derived from fibrocystic disease.
Effect on cell viability and colony formation.
The potency of CH-1, CH-2, and CH-3 was evaluated using the Cell Titer Blue (Promega) protocol for measuring cell viability under the culture conditions recommended by ATCC. The results of these assays are shown in Fig. 2a. The chalcone derivatives have average IC50 values on the order of 1-10 μM for the TNBC cell lines (see Table S-1 for all IC50 values). By contrast, for all three chalcone derivatives, there was less of an effect on the viability of non-tumor breast epithelial cell lines. MCF12A viability was unaffected with CH-2, and IC50 values with CH-1 and CH-3 were an order of magnitude higher than for the TNBC cell lines. IC50 values for MCF10A with CH-2 and CH-3 were, as for the TNBC cell lines, on the order of 1-10 μM, but with CH-2, the IC50 value was an order of magnitude higher. This data indicates the chalcone derivatives have a higher potency in TNBC cells and thus target them more effectively compared to non-tumor breast epithelial cells, an important quality in the development of chemotherapeutics in order to minimize the severity of treatment side effects at the therapeutic dose.
Figure 2.

(a) Percent cell viability, measured using a CellTiter-Blue assay, of two non-tumor breast (MCF12A and MCF10A) and two TNBC (MDA-MB-231 and MDA-MB-468) cell lines after 24 hours treatment with CH-1, CH-2, and CH-3 at concentrations between 50 and 0.005 μM. The percent cell viability is calculated with respect to DMSO vehicle control-treated samples, with 100% viability indicating the same number of viable cells as for the control sample and 0% viability indicating complete cell death. The dotted line at 5 μM analogue concentration indicates the analogue concentration chosen for proteomics experiments. At this concentration, the most significant differences between TNBC and non-tumor breast cell viability is observed. (b) Results of a clonogenic assay measuring the effect of all three chalcone analogues on colony formation by the non-tumor and TNBC cell lines. Cells were plated at 500 cells/ well in a 12-well plate and treated with DMSO vehicle control or 1, 5, or 10 μM of CH-1. Colonies were allowed to grow for six days and then were fixed and stained. The number of colonies containing >50 cells under each condition were counted, and the percent colony formation compared to the DMSO treated cells is plotted.
The anti-proliferative effect of the chalcone derivatives were also validated using a clonogenic assay at three different concentrations (1, 5, and 10 μM) (Fig. 2b). Results showed a decrease in colony formation for all cell lines with increasing derivative concentration. Consistent with the viability assay results, MCF12A displays the most resistance to chalcone treatment, for example, with a decrease of only 18 ± 2% in colony formation compared to control after treatment with 10 μM CH-1. The TNBC cell lines exhibited the greatest decrease in colony formation with chalcone treatment, with >90% inhibition after treatment with 10 μM chalcone as compared to the vehicle control treated sample.
Proteomic changes with candidate treatment.
TMT experiments were performed to quantify the relative abundances of proteins in cell lines treated with each small molecule candidate versus a vehicle control. From cell viability curves (Fig. 2a), 5 μM (dotted lines), was chosen as the optimal concentration for proteomic analysis. This concentration corresponds to the lowest amount at which a difference was observed in viability between non-tumor and cancer cells, facilitating the study of proteomic changes linked to the difference in drug potency between these two breast cell types. This concentration is also approximately the IC50 value of these three compounds in the TNBC cell lines (Fig. 2a). After stringent filtering (see Experimental Section), on average ~952 proteins were quantified in each TMT six-plex experiment. A summary of the top ten proteins for which an increase or decrease in abundance was observed under each treatment condition is given in Table S-2. The average coefficient of variation (CV%) for fold changes calculated from technical replicates, i.e., replicate LC-MS runs of each sample, was 11% (range 6-19%). The average CV% for fold changes calculated from biological replicates of the control and CH-2-treated cells was 26% (range 13-37%). These CV% values agree well with other CV% values reported in the literature for biological replicates of cell lines subjected to quantitative proteomic analysis using TMT labeling strategies (23, 24).
Gene ontology (biological processes) GSEA.
GSEA of the proteomics data using the gene ontology (GO) – biological processes database was performed to determine the enrichment of specific processes. GSEA was performed on the average log2(fold change) from the three chalcone analogues on each cell line. Sixteen processes, categorizable into ten sections due to the similarity in proteins between enriched processes, were found to be increased, while two processes were found to be decreased significantly (p-value < 0.005, Table 1). No processes were enriched in all four cell lines. Three processes, regulation of cellular response to heat, protein refolding, and de novo protein folding, increased in three out of four cell lines (MDA-MB-231, MCF10A, and MCF12A). The proteins for which there was an increase in abundance in these processes were mostly heat shock proteins (HSPs), chaperones which help fold, refold, or prevent the aggregation of proteins, especially during periods of cellular stress (25), or co-chaperones of HSPs. This data indicates that three of the four cell lines initiate a protective response after experiencing stress upon treatment with the chalcone analogues.
Table 1.
Most significant (average p < 0.005) processes found to be enriched after 12 hours treatment with CH-1, CH-2, and CH-3 according to GSEA using the gene ontology (GO) – biological processes database. Proteins shown were enriched in each pathway. A total list of proteins from each pathway that were quantified can be found in Table S-1.
| GO Biological Process | Average p-value |
Proteins |
|---|---|---|
| Increase in 3/4 cell lines | ||
| Regulation of cellular response to heat | 1.3E-03 | BAG2, BAG3, CRYAB, DNAJB1, FKBP4, HSP90AA1, HSP90AB1, HSPA1B, HSPA8, HSPH1, NUP205, NUP93, PTGES3, TPR, YWHAE |
| Protein refolding, De novo protein folding | 2.7E-03, 4.3E-03 | B2M, CCT2, DNAJB1, ERO1L, FKBP1A, HSP90AA1, HSPA1B, HSPA6, HSPA8, HSPD1, HSPE1, HSPH1, PTGES3, UGGT1 |
| Increase in cancer cell lines | ||
| Positive regulation of apoptotic signaling pathway, Regulation of apoptotic signaling pathway | 5.0E-04, 1.0E-03 | ATPIF1, BCAP31, BCLAF1, BID, CAV1, CD44, CTNNA1, CTTN, DYNLL1, FIS1, GCLM, GNB2L1, GSN, GSTP1, HMGB2, HMOX1, HNRNPK, HSPA1B, HSPH1, LGALS3, LMNA, MIF, NONO, P4HB, PARK7, PDCD5, PEA15, PPP1CA, PPP2R1A, PTPN1, RPS3, S100A8, SFN, SLC25A5, SLC9A3R1, SOD1, SOD2, TPD52L1, VDAC2, YWHAB, YWHAE, YWHAG, YWHAH, YWHAQ, YWHAZ |
| Mitochondrial membrane organization, Positive regulation of mitochondrial outer membrane permeabilization involved in apoptotic signaling pathway, Regulation of mitochondrial outer membrane permeabilization involved in apoptotic signaling pathway, Regulation of protein insertion into mitochondrial membrane involved in apoptotic signaling pathway | 2.0E-03, 3.0E-03, 4.0E-03, 5.0E-03 | ATPIF1, BID, CHCHD3, DYNLL1, HSP90AA1, HSPA4, IMMT, MTCH2, SFN, SLC25A5, YWHAB, YWHAE, YWHAG, YWHAH, YWHAQ, YWHAZ |
| Increase in non-tumor cell lines | ||
| Positive regulation of protein metabolic process | 1.5E-03 | ACSL1, ANP32B, ANXA2, ARL6IP5, ASPH, BCAP31, BUB3, CDC42, CNPY2, CSNK2A1, CTBP1, CTNNB1, CYCS, DDX39B, DNAJA3, EIF5A, ERP29, GNAI2, GNL3, HMGB1, HMOX1, HNRNPD, HRAS, HSP90AB1, HSPA5, HSPB1, HSPD1, HSPE1, HSPH1, IQGAP1, ITGA5, ITGB1, KHDRBS1, MSN, MUC1, NDUFA13, NENF, NPTN, PHB2, PML, PRMT1, PSAP, PSMA1, PSMA4, PSMA5, PSMA6, PSMA7, PSMB2, PSMB7, PSMC1, PSMC3, PSMC4, PSMC5, PSMC6, PSMD11, PSMD12, PSMD13, PSMD14, PSMD2, PSMD3, PSME3, PTPN1, RAB1A, RAB1B, RAB7A, RAP2B, RHOA, RPS27A, RPS9, SEC22B, SKP1, SNW1, SOD1, SQSTM1, SUMO2, SYNCRIP, TXN, UBQLN1, VCP |
| Regulation of response to stress | 2.0E-03 | ACIN1, ACTL6A, ANXA1, ANXA2, ANXA5, APEX1, ARL6IP5, B2M, BAG3, BCAP31, CAPN1, CD59, CDC42, CRYAB, CTSB, DDX39B, DNAJA1, DNAJA3, DNAJB1, DNAJC3, EIF2S1, ERP29, FKBP4, GAPDH, GSTP1, HIST1H3A, HLA-A, HLA-B, HMGA1, HMGB1, HRAS, HSP90AA1, HSP90AB1, HSP90B1, HSPA1B, HSPA5, HSPA8, HSPB1, HSPD1, HSPH1, IFIT1, LAMP1, NT5E, NUP205, NUP93, OPA1, PAFAH1B2, PCBP2, PCNA, PHB2, PML, PRDX1, PRMT1, PSAP, PSMA1, PSMA4, PSMA5, PSMA6, PSMA7, PSMB2, PSMC1, PSMC3, PSMC4, PSMC5, PSMC6, PSMD11, PSMD13, PSMD14, PSMD2, PSMD3, PSME3, PTGES3, PTPN1, RAB1B, RAB7A, RPS27A, RUVBL1, SERPINB2, SKP1, SNRPB, SOD1, SOD2, SQSTM1, TPR, TSPO, TXN, VDAC1, YWHAE, ZC3HAV1 |
| Response to temperature stimulus, Response to heat | 3.0E-03, 4.0E-03 | DNAJA1, DNAJA3, EIF2S1, HMOX1, HSP90AA1, HSPA1B, HSPA6, HSPD1, MYOF, SOD1, TPR |
| Protein folding | 4.5E-03 | B2M, BAG2, BAG3, CALR, CANX, CCT2, CCT4, CCT5, CCT6A, CCT7, CRYAB, CSNK2A1, DNAJA1, DNAJA3, DNAJB1, ERP29, FKBP4, FKBP5, GNAI2, GNB2, GRPEL1, HSP90AA1, HSP90AB1, HSP90AB2P, HSP90B1, HSPA1B, HSPA5, HSPA6, HSPA8, HSPA9, HSPD1, HSPE1, HSPH1, LMAN1, P4HB, PDIA6, PPIA, PPIB, PPIF, PTGES3, TCP1, TXN, TXNDC5, UGGT1 |
| Decrease in cancer cell lines | ||
| Organic acid metabolic process | <1.0E-03 | AARS, ABCD3, ACADVL, ACAT1, ACLY, ACO1, ACO2, ACOT1, ACOT7, ACOX1, ACSL3, ACSL4, ADSS, AHCY, AIMP1, AIMP2, AKR1A1, AKR1C3, ALDH18A1, ALDH1A3, ALDH7A1, ALDH9A1, ALDOA, ALDOC, ASNS, ASS1, ATIC, ATPIF1, BCAT2, BPNT1, BSG, CAD, CARS, CBR1, CKMT1A, CNDP2, CRABP2, CRTAP, CS, CYB5R3, DARS, DDAH2, DLAT, DLD, DLST, DNMT1, ECI1, ECI2, EEF1E1, EIF4A3, ENO1, EPRS, ETFA, ETFB, FARSA, FASN, FH, GAPDH, GARS, GART, GCSH, GFPT1, GLO1, GOT1, GOT2, GPI, GPT2, GPX1, GRHPR, GSS, GSTO1, HADH, HADHA, HADHB, HARS, HIBADH, HK1, HSD17B10, HSD17B4, IARS, IARS2, IDH1, IDH2, KARS, KYNU, LARS, LDHB, LRRC47, LTA4H, LYPLA2, MAPK14, MARS, MDH1, MDH2, MTHFD1, NARS, NDUFAB1, OAT, OGDH, P4HA1, P4HB, PADI2, PARK7, PC, PCYOX1, PDHA1, PDHB, PFKL, PFKP, PGAM1, PGD, PGK1, PGM1, PHGDH, PNP, POR, PPA1, PPA2, PRPS1, PSAT1, PSPH, PTGES3, QARS, RARS, RBP1, RNPEP, SARS, SCD, SCP2, SDHA, SHMT2, SMS, SUCLG2, TARS, TPI1, UGDH, UGP2, VARS, WARS, YARS |
| Decrease in non-tumor cell lines | ||
| Establishment of protein localization to endoplasmic reticulum | <1.0E-03 | BCAP31, FAU, RAB10, RPL10, RPL10A, RPL11, RPL12, RPL13, RPL13A, RPL14, RPL15, RPL17, RPL18, RPL18A, RPL19, RPL21, RPL23A, RPL24, RPL26, RPL27, RPL27A, RPL28, RPL29, RPL3, RPL30, RPL31, RPL32, RPL34, RPL35A, RPL36, RPL36A, RPL37A, RPL38, RPL4, RPL5, RPL6, RPL7, RPL7A, RPL8, RPL9, RPLP0, RPLP2, RPS10, RPS11, RPS12, RPS13, RPS14, RPS15A, RPS16, RPS17, RPS18, RPS19, RPS2, RPS20, RPS21, RPS23, RPS24, RPS25, RPS3, RPS3A, RPS4X, RPS6, RPS7, RPS8, RPS9, RPSA, SEC61A1, SRP68, SRP9 |
In only TNBC-derived cell lines, proteins associated with apoptotic signaling increased, as evidenced by enrichment of several apoptosis regulatory and mitochondrial membrane permeability processes. Proteins that decreased in only the TNBC cell lines are related to metabolism, such as glycolytic enzymes (e.g., ALDOA, GAPDH, PFKL, PGM1) and amino acid metabolism enzymes (e.g., LARS, MARS, NARS, YARS). Together, this data indicates apoptosis mediated by the mitochondria. Increases in mitochondrial membrane permeability results in the secretion of apoptogenic factors into the cytosol, precipitating apoptosis (26). Additionally, during times of cellular stress, metabolism, regulated by the mitochondria, slows, which can result in cell cycle arrest and apoptosis (27). The detection of these apoptosis markers correlate with the higher potency of the chalcone analogues against the TNBC cell lines.
In only non-tumor cell lines, increases in stress response proteins, HSPs, and HSP co-chaperones were observed, consistent with the processes enriched for three of the four cell lines (see above). This data is consistent with that in Table S-2, which shows that greater than one-third of the top ten proteins that increased for the non-tumor cell lines were related to stress and heat responses. Decreases in proteins associated with protein localization to the endoplasmic reticulum were also observed for only the non-tumor cell lines, evidenced by a decrease in mainly ribosomal proteins, suggesting a decrease in protein synthesis likely in response to stress caused treatment with the chalcone analogues.
Overall, the GO – biological processes GSEA data suggest that the non-tumor cell lines are more resistant to chalcone treatment (as seen in the viability and clonogenic assays) because of the initiation of a stress response, whereas the TNBC cell lines are more susceptible to chalcone treatment and are already in the process of apoptosis after 12 hours treatment. GSEA was performed on the log2(fold change) data for each chalcone derivative separately, and the results were the same as for the GSEA using the average log2(fold change) for all analogues with each cell line. Thus, all derivatives seemed to act on the cells with the same pathways and proteins affected, indicating that the small structural differences in these derivatives likely have little effect on the mechanism of action of this group of chalcones.
KEGG pathway GSEA.
GSEA of the average log2(fold change) for all chalcone derivatives referencing the KEGG pathway database was performed to determine the enrichment of specific pathways. Two significantly (p-value < 0.1) enriched pathways due to increases and one due to decreases in protein abundances are shown in Table 2. No pathways were enriched in all four cell lines or in only the non-tumor cell lines. GSEA showed enrichment of the antigen processing and presentation pathway in MDA-MB-231, MDA-MB-468, and MCF10A due to the increases in the abundances of heat shock proteins (HSPs), the HLA-A, -B, and -C, members of the major histocompatibility complex (MHC), and several of binders and interaction partners of the MHC in the MHC-I pathway, in particular. The MHC-I pathway processes endogenous antigens produced by the proteasome and presents them to CD8+ T cells, thus playing a vital role in immune recognition of malignant cells (28, 29). A significant enrichment in this pathway was observed for all cell lines except MCF12A, which was also the cell line that showed the weakest response to chalcone analogue treatment (Fig. 2a). GSEA also showed enrichment of the cell cycle pathway in all cell lines except MCF10A. Increases in several cell cycle regulatory proteins were observed, mainly S-phase (e.g., MCM3-7, PCNA, and SMC1A) and G2/M –phase initiators (e.g., CDK1, SFN, and YWHA-B,E,G,H,Q,Z). Increases in these proteins compared to the untreated controls may suggest that the treated cells undergo cell cycle arrest.
Table 2.
Most significant (average p < 0.100) pathways found to be enriched after 12 hours treatment with CH-1, CH-2, and CH-3 according to GSEA using the KEGG pathway database. Proteins shown were enriched in each pathway. A total list of proteins from each pathway that were quantified can be found in Table S-2.
| KEGG Pathway | Average p-value |
Proteins |
|---|---|---|
| Increase in 3/4 cell lines | ||
| Antigen processing and presentation | 4.5E-02 | B2M, CALR, CANX, CTSB, HLA-A, HLA-B, HLA-C, HSP90AA1, HSP90AB1, HSPA1B, HSPA4, HSPA5, HSPA6, HSPA8, PDIA3, PSME1, PSME3, TAPBP |
| Cell cycle | 9.3E-02 | BUB3, CDK1, HDAC1, MCM3, MCM4, MCM5, MCM7, PCNA, SFN, SKP1, SMC1A, YWHAB, YWHAE, YWHAG, YWHAH, YWHAQ, YWHAZ |
| Decrease in both TNBC cell lines | ||
| Gap junction | 9.0E-02 | EGFR, GNAI1, GNAI2, MAP2K2, PRKACA, TUBA1B, TUBA1C, TUBA4A, TUBAL3, TUBB, TUBB3, TUBB4B, TUBB6, TUBB8 |
In only the two TNBC cell lines, enrichment of the gap junction pathway was observed due to decreases in the abundances of tubulins and other proteins associated with gap junction formation. Gap junctions are essential for intercellular communication (30), and a decrease in these structures can either indicate or induce the initiation of apoptosis. A decrease in tubulin abundances may also result in apoptosis due to cell cycle arrest because the cells are unable to form the microtubule spindle necessary for mitosis (31). This data complements the increase in cell cycle-associated proteins and provides further suggestion of cell cycle arrest resulting from treatment with the chalcone derivatives.
In total, the KEGG pathway GSEA data suggest that cell cycle arrest mediated by cell cycle checkpoint proteins and/or changes in microtubule structure or dynamics as well as signaling for immune stimulation occur in the TNBC cell lines with chalcone treatment, and to a more minor extent in the non-tumor cell lines. Similar to the GO-biological processes, GSEA was performed on the log2(fold change) data for each derivative separately, and the results were the same as for the average log2(fold change) GSEA, suggesting that the structural changes in these derivatives impart very little change in their activities.
Validation of key proteomic results.
Quantitative proteomic experiments suggest that the chalcone derivatives may have a significant effect on the cell cycle, seen in enrichment of the cell cycle KEGG pathway. Additionally, there is evidence that the chalcone derivatives cause apoptosis of TNBC cells, as observed by decreases in the abundances of tubulins and proteins essential to transcription and translation, as well as enrichment of apoptotic processes in the GSEA. The GSEA data also suggest that apoptosis in TNBC cells treated with the chalcone analogues may be regulated by changes in mitochondrial membrane permeability. To validate these observations and to understand the mechanism involved in the anti-proliferative activity of these chalcones, cell cycle analysis, an apoptosis assay, and western blot experiments were performed on TNBC cell lines treated with CH-1. CH-1 was chosen for validation experiments because in the clonogenic assay, the largest differences in proliferation were observed between the non-tumor and TNBC cell lines with this derivative. Thus, CH-1 was the compound with the highest cancer specificity when compared to non-tumor cells.
The cell cycle distribution of MDA-MB-231 and MDA-MB-468 after 24 hours treatment with CH-1 at 1-10 μM versus DMSO control is shown in Fig. 3. A dose-dependent and significant (p ≤ 0.0001 at 10 μM CH-1) increase in the percentage of cells in G0/G1 phase is observed with treatment of MDA-MB-231, whereas no significant change or a significant decrease (p ≤ 0.0001) is observed in the percentage of cells in S and G2/M phases. Similarly, for MDA-MB-468, a dose-dependent and significant (p ≤ 0.05 at 5 μM CH-1) increase in the percentage of cells in G0/G1 phase, and either no significant change or a significant decrease in the proportion of cells in S (p ≤ 0.0001) and G2/M (p ≤ 0.001) phases is observed with concentrations of CH-1 up to 5 μM. Dose-dependent and significant (p ≤ 0.0001 at 10 μM CH-1) increases in the fraction of cells in G0/G1 phase was also observed at 12 hours treatment (Fig. S1). This data indicates that CH-1 induces cell cycle arrest in G0/G1 phase. Treatment of MDA-MB-468 with 10 μM CH-1, however, results in a significant (p ≤ 0.0001) decrease in the fraction of cells in G0/G1 phase, likely due to cell death, as evidenced by a significant (p ≤ 0.0001) increase in the percentage of cells in sub-G0/G1 phase. This increase in the fraction of cells in sub-G0/G1 phase is also observed in MDA-MB-231 with 10 μM CH-1 treatment, indicating that cell death occurs in both TNBC cell lines at high CH-1 concentrations.
Figure 3.

(a) Representative histograms of the cell cycle distribution of MDA-MB-231 and MDA-MB-468 and (b) the average of triplicate sample measurements of the percentage of cells in the different phases after treatment with CH-1 or DMSO (control), as measured by flow cytometry. The cells were treated with DMSO (control) or 1, 5, or 10 μM CH-1 for 24 hours. RNA was digested with ribonuclease A, and DNA was stained with propidium iodide (PI) for analysis by flow cytometry. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, and **** p ≤ 0.0001 versus control.
Western blotting of key cell cycle and apoptosis regulation proteins and an annexin V assay were performed to confirm the cell cycle arrest and cell death findings from the cell cycle analysis. In cancer cells, G1 and S checkpoint regulators are often overexpressed and/or up-regulated, which is associated with excessive cell growth and division (32). Specifically, the cyclin-D1/CDK4 complex is associated with the progression of the cell cycle from G1 to S phase, and cyclin-D1 is overexpressed in 50% of all breast cancers (32); dynamin-1-like protein (Drp1) is a mediator of mitochondrial fission, leading to G1-to-S cell cycle progression and proliferation in cancer cells (33, 34). Treatment of TNBC cell lines with CH-1 (1-10 μM for 24 hours) causes a dose-dependent decrease in the expression of cyclin-D1, CDK4, and Drp1 (Fig. 5), further supporting the evidence for G0/G1 phase arrest with chalcone analogue treatment. This data also agrees with the proteomics finding that Drp1 (gene name DNM1L) exhibited the greatest decreases in abundance when MDA-MB-231 was treated with all three chalcone analogues (Table S-2), with an average decrease of ~1.4 fold.
Figure 5.

Western blot for cyclin-D1, CDK4, Drp1, Bel-2, and Hsp40 using lysates from MDA-MB-231 and MDA-MB-468 cells treated with CH-1 for 24 hours at concentrations between 1-10 μM. Membranes were stripped and re-probed with an anti-β-actin antibody to account for differences in protein loading. Fold changes calculated using ImageJ are given on top of each band.
Using an annexin V FITC apoptosis assay, dose-dependent and time-dependent enhancement of apoptosis in both TNBC cell lines was observed after 24 and 48 hours treatment with CH-1 (Fig. 4). Treatment with 10 μM CH-1 for 24 hours resulted in apoptosis in 25% and 36% of cells for MDA-MB-231 and MDA-MB-468, respectively, consistent with the presence of 28% and 40% of cells, respectively, in the sub-G0/G1 phase in the cell cycle analysis under the same conditions. Furthermore, a dose-dependent decrease in the abundance of Bcl-2, an anti-apoptotic protein overexpressed in cancer cells (35), is observed by western blot for both TNBC cell lines treated with CH-1 for 24 hours (Fig. 5). By contrast, when the apoptosis assay was performed on the non-tumor cell lines (Fig. S-2), even after 48 hours, only 11% of MCF12A and 42% of MCF10A cells were apoptotic, compared to 44% and 52% of MDA-MB-231 and MDA-MB-468 cells, respectively. This data is consistent with the viability and clonogenic assay results, which showed MCF12A to be the most resistant to chalcone derivative treatment. It is also consistent with the GSEA results, in which significant enrichment of apoptotic processes was found for only the TNBC cell lines, which had the greatest number of apoptotic cells according to the apoptosis assay. Together, these data indicate that CH-1 induces apoptosis-mediated cell death at higher concentrations and longer treatment times, and this effect is more pronounced in TNBC cells compared to the non-tumor cells.
Figure 4.

Effect of CH-1 on induction of apoptotic death for TNBC cells (a) MDA-MB-231 and (b) MDA-MB-468. Cells were treated with either DMSO (control) or 1, 5, or 10 μM CH-1 for 24 and 48 hours. Cells were then harvested and stained with annexin V and PI, and the percentage of apoptotic cells (annexin V-positive) were measured by flow cytometry, with triplicate samples for each condition. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, and **** p ≤ 0.0001 versus control.
The proteomics experiments also suggest initiation of a stress response with chalcone analogue treatment in both non-tumor breast and TNBC cell lines, seen in increases in the abundances of HSPs (Table S-2) and enrichment of protein folding and stress response processes in GSEA (Table 1). One class of HSP, the Hsp40 family, plays a role in the unfolded protein response (UPR), regulates protein translation when in complex with Hsp70, and can play a role in cancer cell apoptosis (36). Western blots of both TNBC cells treated with CH-1 showed enhanced expression of Hsp40 only with treatment with 5 μM CH-1 (Fig. 5). However, no significant expression of Hsp40 was observed at 1 and 10 μM CH-1 concentration. This may be attributed to the fact that treatment of TNBC cells with CH-1 at 10 μM concentration for 24 hours leads to apoptosis of 25% and 36% of MDA-MB-231 and MDA-MB-468 cells, respectively (Fig. 4). This data provides further evidence that a stress response results from treatment with CH-1, at least at 5 μM concentration, the IC50 value for the TNBC cell lines, and the concentration of chalcone analogue used in the proteomics experiments.
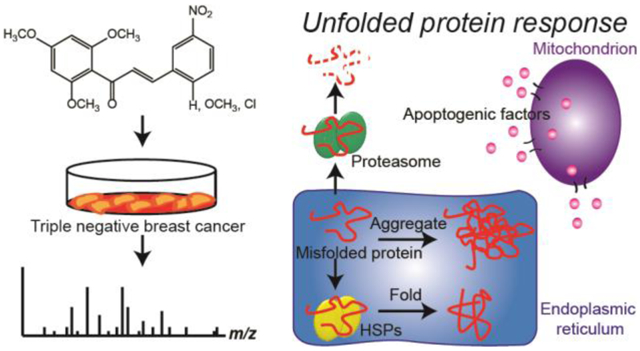
Overall, the proteomic and pathway validation data presented here convey three main mechanisms of anti-cancer action for the three chalcone derivatives used in these experiments (Fig. 6). The first mechanism is induction of the unfolded protein response (UPR) (37, 38), a response to stress placed on the endoplasmic reticulum (ER) in the form of an abundance of unfolded or misfolded proteins, resulting in apoptosis. Targeting the UPR is of increasing interest in cancer therapy, both for the purposes of causing apoptosis in cancer cells and for making resistant cells more susceptible to chemotherapies (39, 40). There are three main stages of the UPR. The first stage is reduction in the amount of new unfolded or misfolded proteins in the ER by decreasing protein synthesis and protein localization to the ER. Signatures of this stage were observed in the proteomics data as decreases in the abundances of transcription- and translation-related proteins (Table S-2) in all four cell lines, as well as decreases in the abundances of several enzymes related to sugar and amino acid metabolism (Tables S-2). Furthermore, in GSEA of the proteomics data, decreases in the abundances of specific proteins resulted in enrichment of the "organic acid metabolic process" in the GO – biological processes analysis for the TNBC cell lines and enrichment of the "establishment of protein localization to the endoplasmic reticulum" in the GO – biological processes analysis for non-tumor cell lines (Table 1). These decreases in markers of metabolism, protein synthesis, and protein localization to the ER resulted in cell cycle arrest in the G1 phase, as measured by western blotting of key G1/S checkpoint regulators (Fig. 5) and as confirmed by flow cytometry (Fig. 3), which is a typical consequence of UPR induction (38).
Figure 6.

Summary of the effects of chalcone derivatives on TNBC cells. Chalcone derivatives likely induce the unfolded protein response, in which stress placed on the endoplasmic reticulum by high amounts of unfolded or misfolded proteins causes an increase in heat shock proteins (HSPs) and proteasome proteins followed eventually by apoptosis. Chalcone derivatives may also induce immune activation by increasing the abundance of MHC-I associated proteins, and may be involved in cell cycle disruption via decreases in the abundances of tubulins, which play important roles in several different stages of the cell cycle, including the formation of the mitotic spindle.
The second stage of the UPR is reduction in the amount of unfolded or misfolded proteins that are already in the ER by folding, refolding, or degrading them. Degradation of unfolded or misfolded proteins is accomplished, as seen in the proteomics data for the non-tumor cell lines, by increases in proteins that form the proteasome, the cell's protein degradation machinery. Increased expression of proteins that fold unfolded proteins or refold misfolded proteins, i.e. HSPs and their co-chaperones, was also observed in the proteomics data for all four cell lines (Table S-2), as well as in enrichment of several heat and stress response and protein folding pathways in the GSEA GO – biological processes analysis (Table 1). In fact, several HSPs were among the top ten proteins with the greatest fold changes for each cell line (Table S-2, red). Western blot analysis of TNBC cells treated with chalcone CH-1 also showed significant enhancement in the expression of Hsp40 (Fig. 5).
The UPR confers a protective effect in times of stress, but if it is prolonged, it eventually leads its third stage, apoptosis. Signatures of apoptosis mediated by mitochondrial changes were observed in the TNBC cell lines in the GO – biological processes data, in which enrichment of processes related to mitochondrial membrane permeability regulation of apoptosis was observed (Table 1). Also consistent with mitochondria-mediated apoptosis is the observed reduction in metabolic enzymes in the TNBC cell lines in the proteomics data (Table S-2). Western blot experiments showed a dose-dependent decrease in the expression of Drp1, a protein that mediates mitochondrial division and is involved in regulating apoptosis, in both TNBC cell lines treated with chalcone CH-1 (Fig. 5). Apoptosis was confirmed in an annexin V apoptosis assay, in which the treatment of TNBC cell lines with CH-1 resulted in a dose-dependent enhancement in the percentage of apoptotic cells (Fig. 4). A dose-dependent decrease in the expression of the anti-apoptotic protein Bcl-2 was also observed in the western blot analysis of TNBC cells treated with CH-1 (Fig. 5).
Overall, this data suggests that the non-tumor cell lines after 12 hours treatment with the chalcone derivatives in this work are in the early stages of the UPR, initiating processes that result in the decrease of unfolded or misfolded proteins in the ER, but the TNBC cell lines have already passed through to the final stage of UPR, apoptosis. In recent work, two different trimethoxychalcone derivatives (41, 42) were also found to induce UPR-mediated apoptosis on TNBC cell lines but not on non-tumor breast epithelial cell lines, and xanthohumol, a natural chalcone found in hops that helps give hops its bitter taste, also causes UPR-mediated apoptosis (43).
The second theme displayed in the proteomics data is immune activation. Enrichment in the "antigen processing and presentation" pathway was observed in both TNBC cell lines and MCF10A, as evidenced by increases in the abundances of HLA-A, -B, and -C, members of the major histocompatibility complex (MHC), and several of its binders and interaction partners in the MHC-I pathway (Table 2), which processes cytosolic/endogenous antigens produced by the proteasome and present them to CD8+ T cells (44). Loss of antigen expression on cells is present in multiple cancer types, a mechanism by which tumor cells can evade immune recognition (45). One mechanism of the loss of antigen expression is decreased MHC-I protein expression, and lack of expression of MHC-I proteins has been shown to correlate with more aggressive disease (29). Several different classes of chemotherapeutics and radiotherapy have been shown to increase expression of or upregulate MHC-I proteins (summary (45)). Although increased expression of MHC-I proteins likely does not contribute to the toxicity of these chalcone analogues observed on cells in culture in our work, it would likely increase the efficacy of these drugs in the body, where the immune system would be enabled to recognize and kill tumor cells.
The final theme in the proteomics and pathway validation data is interruption of the cell cycle with chalcone treatment, and this occurs through multiple mechanisms. As previously mentioned in the discussion of the UPR, cell cycle analysis by flow cytometry revealed that at least one of the chalcone derivatives, CH-1, causes G1 arrest in both TNBC cell lines (Fig. 3), which was further validated by western blots showing decreased protein levels of G1-phase regulators, i.e. cyclin-D1, CDK4, and Drp1 (Fig. 5). But the GSEA using the KEGG pathway database also revealed increased levels of cell cycle proteins involved in the S- and G2/M-phase checkpoints for both TNBC cell lines and MCF12A when treated with chalcone analogues, and decreased levels of gap junction proteins, including tubulins, in TNBC cell lines (Table 2). These two pieces of data point to the disruption of mitosis initiation. Tubulins form microtubules, which provide cytoskeletal structural support to cells and are essential to the formation and function of the mitotic spindle during cell division. Thus, disruption of microtubule assembly and function typically results in cell cycle arrest in G2/M phase (31). Some of the most effective chemotherapeutics, such as Taxol and the Vinca alkaloids, target tubulins to inhibit cell proliferation by causing cell cycle arrest in G2/M phase and inducing apoptosis (46-53). Because cancer cells are more proliferative, passing through mitosis more frequently than non-malignant cells, tubulin-targeting drugs are more potent to these cells. However, some studies have shown that microtubule disruption (MTD) also leads to cell cycle arrest during interphase at a point in G1 phase known as the G1 phase-MTD arrest point, and this arrest is p21-dependent (54). Chalcones are known to inhibit cell proliferation by influencing tubulin polymerization (55) and by causing cell cycle arrest (56). Combrestatin-like chalcones, which are similar in structure to the chalcone derivatives in this work, have been shown to inhibit microtubule assembly (57), and another trimethoxychalcone derivative has been shown to induce cell cycle arrest and apoptosis by disrupting microtubule structure (58). These data show that the chalcone analogues impact different stages of the cell cycle, but the effect is most profound on G1 phase, as it is in this phase that cell cycle arrest is observed.
Conclusions
Three novel chalcone derivatives were demonstrated to have potency (IC50 ~ 5 μM) against TNBC cell lines and were more potent in TNBC than in non-tumor breast cell lines. Quantitative proteomics was used to effectively identify multiple processes and pathways influenced by treatment with the chalcone analogues. The proteomics data indicated induction of an unfolded protein response, an increase in proteins involved in antigen presentation, and cell cycle disruption. The analogues arrested TNBC cells in G1 phase and thereby caused apoptosis. Further work is needed to understand the effects of these potential novel TNBC therapeutics in vivo, including overall toxicity, and the role of immune activation in response to therapy, and further exploration into chalcone structure optimization to gain the maximum therapeutic effect with minimal toxicity to other organs is warranted.
The mass spectrometry proteomics data have been deposited to the PRIDE Archive (http://www.ebi.ac.uk/pride/archive/) via the PRIDE partner repository with the data set identifier PXD009553. Username: reviewer13356@ebi.ac.uk Password: iQ5S61qm
Supplementary Material
Acknowledgements
DT would like to thank the Indo-US Science and Technology Forum (IUSSTF) for a SERB Indo-US postdoctoral fellowship. TS is supported by NIH/NCI R00CA184397.
Footnotes
Supporting Information
Table S-1: Table of IC50 values calculated from the viability assay data. Table S-2: Table of top ten proteins that increased or decreased under all treatment conditions. Tables S-2 and S-3: Expanded versions of Tables 1 and 2, showing all proteins detected in an individual process or pathway. Figure S-1: Flow cytometry cell cycle analysis for TNBC cell lines with CH-1 after 12 hours treatment. Figure S-2: Apoptosis assay data for non-tumor cell lines MCF10A and MCF12A after 48 hours treatment with CH-1.
References
- 1.Dent R; Trudeau M; Pritchard KI ; Hanna WM; Kahn HK; Sawka CA; Lickley LA; Rawlinson E; Sun P; Narod SA Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence. Clin. Cancer Res 2007, 73, 4429–4434. [DOI] [PubMed] [Google Scholar]
- 2.Perou CM; Sørlie T; Eisen MB; van de Rijn M; Jeffrey SS; Rees CA; Pollack JR; Ross DT; Johnsen H; Akslen LA; Fluge Ø; Pergamenschikov A; Williams C; Zhu SX; Lønning PE; Børresen-Dale A-L; Brown PO; Botstein D Molecular portraits of human breast tumours. Nature 2000, 406, 747. [DOI] [PubMed] [Google Scholar]
- 3.Baneijee S; Reis-Filho JS; Ashley S; Steele D; Ashworth A; Lakhani SR; Smith IE Basal-like breast carcinomas: clinical outcome and response to chemotherapy. J. Clin. Pathol 2006, 59, 729–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gonzalez-Angulo AM; Timms KM; Liu S; Chen H; Litton JK; Potter J; Lanchbury JS; Stemke-Hale K; Hennessy BT; Arun BK; Hortobagyi GN; Do KA; Mills GB; Meric-Bernstam F Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin. Cancer Res 2011, 17, 1082–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.The Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gatalica Z; Snyder C; Maney T; Ghazalpour A; Holterman DA; Xiao N; Overberg P; Rose I; Basu GD; Vranic S; Lynch HT; Von Hoff DD; Hamid O Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 2965–2970. [DOI] [PubMed] [Google Scholar]
- 7.Cimino-Mathews A; Foote JB; Emens LA Immune targeting in breast cancer. Oncology (Williston Park, N.Y.) 2015, 29, 375–385. [PubMed] [Google Scholar]
- 8.Anticancer Agents From Natural Products, Cragg GMK, I. DG; Newman DJ, Eds.; Taylor & Francis Group: Boca Raton, FL, 2005. [Google Scholar]
- 9.Dimmock JR; Elias DW; Beazely MA; Kandepu NM Bioactivities of chalcones. Curr. Med. Chem 1999, 6, 1125–1149. [PubMed] [Google Scholar]
- 10.Orlikova B; Tasdemir D; Golais F; Dicato M; Diederich M Dietary chalcones with chemopreventive and chemotherapeutic potential. Genes Nutr. 2011, 6, 125–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jandial DD; Blair CA; Zhang S; Krill LS; Zhang Y-B; Zi X Molecular Targeted Approaches to Cancer Therapy and Prevention Using Chalcones. Curr. Cancer Drug Targets 2014, 14, 181–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang TH; Seo JH; Oh H; Yoon G; Chae JI; Shim JH Licochalcone A Suppresses Specificity Protein 1 as a Novel Target in Human Breast Cancer Cells. J. Cell. Biochem 2017, 118, 4652–4663. [DOI] [PubMed] [Google Scholar]
- 13.Kim SY; Lee IS; Moon A 2-Hydroxychalcone and xanthohumol inhibit invasion of triple negative breast cancer cells. Chem. Biol. Interact 2013, 203, 565–572. [DOI] [PubMed] [Google Scholar]
- 14.Shrivastava S; Jeengar MK; Thummuri D; Koval A; Katanaev VL; Marepally S; Naidu VGM Cardamonin, a chalcone, inhibits human triple negative breast cancer cell invasiveness by downregulation of Wnt/β-catenin signaling cascades and reversal of epithelial-mesenchymal transition. BioFactors 2017, 43, 152–169. [DOI] [PubMed] [Google Scholar]
- 15.Abu N; Akhtar MN; Yeap SK; Lim KL; Ho WY; Abdullah MP; Ho CL; Omar AR; Ismail J; Alitheen NB Flavokawain B induced cytotoxicity in two breast cancer cell lines, MCF-7 and MDA-MB231 and inhibited the metastatic potential of MDA-MB231 via the regulation of several tyrosine kinases In vitro. BMC Complement. Altern. Med 2016, 16, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goncalves LM; Valente IM; Rodrigues JA An overview on cardamonin. J. Med. Food 2014, 17, 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gomes MN; Muratov EN; Pereira M; Peixoto JC; Rosseto LP; Cravo PVL; Andrade CH; Neves BJ Chalcone Derivatives: Promising Starting Points for Drug Design. Molecules (Basel, Switzerland) 2017, 22, 1210–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim YS; Kumar V; Lee S; Iwai A; Neckers L; Malhotra SV; Trepel JB Methoxychalcone Inhibitors of Androgen Receptor Translocation and Function. Bioorg. Med. Chem. Lett 2012, 22, 2105–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thompson A; Schäfer J; Kuhn K; Kienle S; Schwarz J; Schmidt G; Neumann T; Hamon C Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Anal. Chem 2003, 75, 1895–1904. [DOI] [PubMed] [Google Scholar]
- 20.Cox J; Mann M MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotech 2008, 26, 1367. [DOI] [PubMed] [Google Scholar]
- 21.Tyanova S; Temu T; Sinitcyn P; Carlson A; Hein MY; Geiger T; Mann M; Cox J The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731. [DOI] [PubMed] [Google Scholar]
- 22.Subramanian A; Tamayo P; Mootha VK; Mukherjee S; Ebert BL; Gillette MA; Paulovich A; Pomeroy SL; Golub TR; Lander ES; Mesirov JP Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci., U. S. A 2005, 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roumeliotis TI; Williams SP; Gonçalves E; Alsinet C; Del Castillo Velasco-Herrera M; Aben N; Ghavidel FZ; Michaut M; Schubert M; Price S; Wright JC; Yu L; Yang M; Dienstmann R; Guinney J; Beltrao P; Brazma A; Pardo M; Stegle O; Adams DJ; Wessels L; Saez-Rodriguez J; McDermott U; Choudhary JS Genomic Determinants of Protein Abundance Variation in Colorectal Cancer Cells. Cell Rep. 20, 2201–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Welle KA; Zhang T; Hryhorenko JR; Shen S; Qu J; Ghaemmaghami S Time-resolved Analysis of Proteome Dynamics by Tandem Mass Tags and Stable Isotope Labeling in Cell Culture (TMT-SILAC) Hyperplexing. Mol. Cell. Proteomics 2016, 15, 3551–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kregel KC Invited Review: Heat shock proteins: modifying factors in physiological stress responses and acquired thermotolerance. J. Appl. Physiol 2002, 92, 2177–2186. [DOI] [PubMed] [Google Scholar]
- 26.Kroemer G; Galluzzi L; Brenner C Mitochondrial Membrane Permeabilization in Cell Death. Physiol. Rev 2007, 87, 99–163. [DOI] [PubMed] [Google Scholar]
- 27.Mason EF; Rathmell JC Cell metabolism: an essential link between cell growth and apoptosis. Biochim. Biophys. Acta 2011, 1813, 645–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Festenstein H; Garrido F Tumour immunology: MHC antigens and malignancy. Nature 1986, 322, 502. [DOI] [PubMed] [Google Scholar]
- 29.Garrido F; Cabrera T; Concha A; Glew S; Ruiz-Cabello F; Stern PL Natural history of HLA expression during tumour development. Immunol. Today 1993, 14, 491–499. [DOI] [PubMed] [Google Scholar]
- 30.Goodenough DA; Paul DL Gap Junctions. Cold Spring Harb. Perspect. Biol 2009, 1, a002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rudner AD; Murray AW The spindle assembly checkpoint. Curr. Opin. Cell Biol 1996, 8, 773–780. [DOI] [PubMed] [Google Scholar]
- 32.Casimiro MC; Crosariol M; Loro E; Li Z; Pestell RG Cyclins and Cell Cycle Control in Cancer and Disease. Genes Cancer 2012, 3, 649–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhan L; Cao H; Wang G; Lyu Y; Sun X; An J; Wu Z; Huang Q; Liu B; Xing J Drp1-mediated mitochondrial fission promotes cell proliferation through crosstalk of p53 and NF-kappaB pathways in hepatocellular carcinoma. Oncotarget 2016, 7, 65001–65011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lima AR; Santos L; Correia M; Soares P; Sobrinho-Simões M; Melo M; Máximo V Dynamin-Related Protein 1 at the Crossroads of Cancer. Genes 2018, 9, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yip KW; Reed JC Bcl-2 family proteins and cancer. Oncogene 2008, 27, 6398. [DOI] [PubMed] [Google Scholar]
- 36.Wu J; Liu T; Rios Z; Mei Q; Lin X; Cao S Heat Shock Proteins and Cancer. Trends Pharmacol. Sci 38, 226–256. [DOI] [PubMed] [Google Scholar]
- 37.Ron D; Walter P Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol 2007, 8, 519. [DOI] [PubMed] [Google Scholar]
- 38.Diehl JA; Fuchs SY; Koumenis C The Cell Biology of the Unfolded Protein Response. Gastroenterology 2011, 141, 38–41.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Backer MV; Backer JM; Chinnaiyan P Chapter Three - Targeting the Unfolded Protein Response in Cancer Therapy. Methods Enzymol. 2011, 491, 37–56. [DOI] [PubMed] [Google Scholar]
- 40.Nagelkerke A; Bussink J; Sweep FCGJ; Span PN The unfolded protein response as a target for cancer therapy. Biochim. Biophys. Acta 2014, 1846, 277–284. [DOI] [PubMed] [Google Scholar]
- 41.Lee DH; Jung Jung Y; Koh D; Lim Y; Lee YH; Shin SY A synthetic chalcone, 2'-hydroxy-2,3,5'-trimethoxychalcone triggers unfolded protein response-mediated apoptosis in breast cancer cells. Cancer Lett. 2015, 372, 1–9. [DOI] [PubMed] [Google Scholar]
- 42.Shin SY; Lee JM; Lee MS; Koh D; Jung H; Lim Y; Lee YH Targeting cancer cells via the reactive oxygen species-mediated unfolded protein response with a novel synthetic polyphenol conjugate. Clin. Cancer Res 2014, 20, 4302–4313. [DOI] [PubMed] [Google Scholar]
- 43.Lust S; Vanhoecke B; M VANG; Boelens J; H VANM; Kaileh M; Vanden Berghe W; Haegeman G; Philippe J; Bracke M; Offner F Xanthohumol activates the proapoptotic arm of the unfolded protein response in chronic lymphocytic leukemia. Anticancer Res. 2009, 29, 3797–3805. [PMC free article] [PubMed] [Google Scholar]
- 44.Townsend A; Öhlén C; Bastin J; Ljunggren H-G; Foster L; Kärre K Association of class I major histocompatibility heavy and light chains induced by viral peptides. Nature 1989, 340, 443. [DOI] [PubMed] [Google Scholar]
- 45.de Charette M; Marabelle A; Houot R Turning tumour cells into antigen presenting cells: The next step to improve cancer immunotherapy? Eur. J. Cancer 2016, 68, 134–147. [DOI] [PubMed] [Google Scholar]
- 46.Mukhtar E; Adhami VM; Mukhtar H Targeting Microtubules by Natural Agents for Cancer Therapy. Mol. Cancer Ther 2014, 13, 275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jordan MA; Wilson L Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [DOI] [PubMed] [Google Scholar]
- 48.Schiff PB; Fant J; Horwitz SB Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665. [DOI] [PubMed] [Google Scholar]
- 49.Derry WB; Wilson L; Jordan MA Substoichiometric Binding of Taxol Suppresses Microtubule Dynamics. Biochemistry 1995, 34, 2203–2211. [DOI] [PubMed] [Google Scholar]
- 50.Kelling J; Sullivan K; Wilson L; Jordan MA Suppression of Centromere Dynamics by Taxol® in Living Osteosarcoma Cells. Cancer Res. 2003, 63, 2794–2801. [PubMed] [Google Scholar]
- 51.Jordan MA; Thrower D; Wilson L Mechanism of inhibition of cell proliferation by Vinca alkaloids. Cancer Res. 1991, 51, 2212–2222. [PubMed] [Google Scholar]
- 52.Jordan MA Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr. Med. Chem. Anticancer Agents 2002, 2, 1–17. [DOI] [PubMed] [Google Scholar]
- 53.Tulub AA; Stefanov VE Cisplatin stops tubulin assembly into microtubules. A new insight into the mechanism of antitumor activity of platinum complexes. Int. J. Biol. Macromol 2001, 28, 191–198. [DOI] [PubMed] [Google Scholar]
- 54.Mantel CR; Gelfano VM; Kim Y-J; McDaniel A; Lee Y; Boswell H; Broxmeyer HE P21waf-1-Chk1 Pathway Monitors G1 Phase Microtubule Integrity and Is Crucial for Restriction Transition. Cell Cycle 2002, 1, 325–325. [PubMed] [Google Scholar]
- 55.Lawrence NJ; McGown AT; Ducki S; Hadfield JA The interaction of chalcones with tubulin. Anticancer Drug Des. 2000, 15, 135–141. [PubMed] [Google Scholar]
- 56.Ducki S; Forrest R; Hadfield JA; Kendall A; Lawrence NJ; McGown AT; Rennison D Potent antimitotic and cell growth inhibitory properties of substituted chalcones. Bioorg. Med. Chem. Lett 1998, 8, 1051–1056. [DOI] [PubMed] [Google Scholar]
- 57.Ducki S; Rennison D; Woo M; Kendall A; Chabert JF; McGown AT; Lawrence NJ Combretastatin-like chalcones as inhibitors of microtubule polymerization. Part 1: synthesis and biological evaluation of antivascular activity. Bioorg. Med. Chem 2009, 17, 7698–7710. [DOI] [PubMed] [Google Scholar]
- 58.Lee JM; Lee MS; Koh D; Lee YH; Lim Y; Shin SY A new synthetic 2'-hydroxy-2,4,6-trimethoxy-5',6'-naphthochalcone induces G2/M cell cycle arrest and apoptosis by disrupting the microtubular network of human colon cancer cells. Cancer Lett. 2014, 354, 348–354. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.