

ABSTRACT
Glioblastoma (GBM) is the most aggressive and lethal neoplasia of the central nervous system in adults. Based on the molecular signature genes, GBM has been classified in proneural, neural, mesenchymal and classical subtypes. The Metallophosphoesterase-domain-containing protein 2 (MPPED2) gene encodes a metallophosphodiesterase protein highly conserved throughout the evolution. MPPED2 downregulation, likely due to its promoter hypermethylation, has been found in several malignant neoplasias and correlated with a poor prognosis. In this study, we aimed to investigate the expression and the functional role of MPPED2 in GBM. TCGA and Gravendeel databases were employed to explore the MPPED2 expression levels in this type of tumor. We have found that MPPED2 expression is downregulated in GBM patients, showing a positive correlation with survival. Moreover, TCGA and Gravendeel data also revealed that MPPED2 expression negatively correlates with the most aggressive mesenchymal subtype. Additionally, the restoration of MPPED2 expression in U251 and GLI36 GBM cell lines decreases cell growth, migration and enhanced the sensitivity to the temozolomide, inducing apoptotic cell death, of GBM cells. These findings suggest that the restoration of MPPED2 function can be taken into consideration for an innovative GBM therapy.
KEYWORDS: MPPED2, tumor suppressor, glioblastoma, temozolomide
Introduction
Gliomas are the most predominant aggressive and lethal primary brain tumors in adults with constant unfavorable prognosis [1]. These tumors are divided into low-grade glioma (LGG), including astrocytoma and oligodendroglioma, and high-grade glioma (HGG), represented by glioblastoma (GBM), the most common brain neoplasia, accounting for 60–70% of all gliomas [2,3]. According to the World Health Organization (WHO), gliomas are further categorized into four grades: grade I, II and III gliomas are classified as LGG and characterized by a slow growth rate and better survival than HGG, which comprises grade IV GBM [4], representing the most malignant and lethal neoplasia of the central nervous system in adults with a median patient survival rate lower than two years [5–7]. Recently, through extensive analysis of the gene expression profile available in The Cancer Genome Atlas (TCGA) database, GBM has been further classified into four molecular subtypes, including Proneural (PN), Neural (N), Classical (CL) and Mesenchymal (MES) subtypes. This classification is useful for diagnosis, prognosis and development of specific clinical strategies [8]. Among these molecular subtypes, the MES subtype is the most aggressive, associated with negative response to therapy and characterized by the loss of the epithelial markers and, conversely, by high expression of mesenchymal hallmarks such as CHI3L1/YKL40, Vimentin (VIM) and Fibronectin (FN1) [9]. Additionally, MES subtype is characterized by the deregulation of NF-ĸB pathway and mutations or loss of neurofibromin 1 (NF1) gene that result in the activation of the downstream PI3K/AKT pathway [8,10,11]. Currently, the standard-of-care (SOC) in patients with GBM consists of its surgical resection followed by radiotherapy and treatment with the chemotherapeutic alkylating agent temozolomide (TMZ) [12,13]. TMZ induces cytotoxicity by transporting methyl group to guanine preferentially in O6-methylguanine (O6MeG) DNA position and represents the elective drug for the treatment of GBM [14,15]. Unfortunately, after some months, the patients develop resistance to TMZ treatment, thus evidencing an urgent need to search for new therapeutic approaches or efficient drug combinations to treat GBM [16–18]. Several studies report that upon the treatment, PN GBM cells acquire MES features through proneural-mesenchymal transition (PMT) (marked by aggressiveness and treatment-resistance) as a mechanism through which GBM acquired drug resistance [19]. Therefore, there is a need to unveil the molecular mechanisms underlying the development of GBM that could allow new therapeutic approach for GBM and MES phenotype, in particular.
Then, we focused on the role of the MPPED2 gene in the development of human GBM. Indeed, MPPED2 codes for a member of metallophosphodiesterase protein, and its expression has been found downregulated in several human cancer types, comprising neuroblastoma [20], oral squamous [21], cervical [22], thyroid [23] and breast cancer [24], underlining the loss of MPPED2 expression as a general event in carcinogenesis. Moreover, MPPED2 gene deletion is associated with WAGR syndrome, a rare genetic disorder that consists of Wilms tumor, aniridia, genitourinary anomalies and mental retardation [25–27].
Hypermethylation of MPPED2 promoter likely accounts for MPPED2 gene downregulation as already reported in breast [24] and colon cancer [28]. Noteworthy, a tumor suppressor role for MPPED2 in cancer progression has been validated by functional experiments demonstrating its role in cancer cell proliferation and migration [20,23,24].
In this study, we have analyzed MPPED2 expression through data available at the TCGA and Gravendeel datasets, evaluating its expression levels in different brain tumors and several GBM subtypes. This analysis revealed that MPPED2 expression is reduced in glioma, particularly in the most aggressive MES subtype, where its expression negatively correlates with MES signature genes. We also report that the restoration of MPPED2 inhibits GBM cell proliferation and migration and increases the cytotoxic effect of TMZ by enhancing apoptosis, thereby validating the contribution of MPPED2 downregulation to GBM development.
Materials and methods
Cell culture and transfections
Human GBM cell lines U87, U251, A172, GLI36, LN18 and LN226 were cultured in DMEM (Sigma-Aldrich, Saint Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS) (Euroclone, Milan, Italy), 1% L-glutamine, 1% penicillin/streptomycin (Sigma-Aldrich, Saint Louis, MO, USA). Cell lines were kept at 37°C under 5% CO2 atmosphere. Lipofectamine 2000 (Thermofisher, Waltham, MA, USA) reagent was used to transfect the cells according to the manufacturer’s instructions. The stable-expression cell lines, U251 and GLI36 were selected in a medium containing 25 µg/ml and 50 µg/ml of hygromycin (Sigma-Aldrich, Saint Louis, MO, USA), respectively.
Plasmids
The expression vector encoding human gene MPPED2 was obtained by cloning cDNA sequence in the vector pcDNA3.1/Hygro(+) (Thermofisher, Waltham, MA, USA) in the N-terminal region, using the restriction sites of the enzymes HindIII and NotI. The plasmid was checked by sequencing (Eurofins Genomics, Vimodrone, Italy) and MPPED2 expression was validated by qRT-PCR and Western blot analysis.
Bioinformatic analysis
The expression data for LGG and GBM samples used in this study were obtained from TCGA and Gravandeel datasets by using web-based software GlioVis (http://gliovis.bioinfo.cnio.es/) [29,30]. The whole TCGA cohort used for MPPED2 includes a total of 365 LGG samples, 141 GBM and 4 normal brain samples. Patients were classified based on the GBM subtype (CL, n = 59; PN, n = 46; MES, n = 51). Correlation analysis between MPPED2 expression and the subtype molecular signature genes was performed. The methylation data for LGG and GBM samples were obtained from TCGA database by using web-based software Wanderer [31]. For the methylation analysis, n = 2 normal brain tissues, n = 511 LGG and n = 129 GBM were examined. The Gravendeel cohort used for MPPED2 comprised a total of 80 LGG, 161 GBM and 6 normal brain surrounding tissues were considered for this study. Patients were classified on the basis of the GBM subtype (CL, n = 52; PN, n = 53; MES, n = 54).
RNA extraction and qRT-PCR
Total RNA from GBM cell lines was extracted using Trizol reagent (Thermofisher, Waltham, MA, USA). 1 µg of total RNA from each sample was used to obtain single strand cDNA with the QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany). Quantitative Real-Time PCR (qRT-PCR) was performed with the CFX96 thermocycler (Bio-Rad, Hercules, CA, USA) in 96-well plates. For each of the PCR reaction, it was used 10 µl of 2X Sybr Green (Bio-Rad, Hercules, CA, USA), 200 nM of each primer, and 20 ng of the cDNA, previously generated. The oligonucleotides for qRT-PCR, comprising exon-exon junctions, were designed with Primer-BLAST software and purchased from Integrated DNA Technologies (San Diego, CA, USA).
MPPED2 Fw: 5ʹGCTTCAAAGAGTGGGCTGTG3, Rv: 5ʹGAGGGTTGGTCGGTTGAAAG
RP18S Fw: 5ʹTGCGAGTACTCAACACCAA, Rv: 5ʹTTGGTGAGGTCAATGTCTGC
FN1 Fw: 5ʹCTCTTCATGACGCTTGTGGA, Rv: 5ʹATGATGAGGTGCACGTGTGT
VIM Fw: 5ʹTGAGATTGCCACCTACAGGAA, Rv: 5ʹGAGGGAGTGAATCCAGATTAGTTT
CHI3L1/YKL40 Fw: 5ʹ AATTCGGCCTTCATTTCCTT, Rv: 5ʹ GATAGCCTCCAACACCCAGA
Relative gene expression was determined using comparative C(T) method, as described elsewhere [32]. RP18S was used as housekeeping gene.
Protein extraction and western blot
Cells were homogenized in RIPA lysis buffer (20 mM Tris-HCl pH 7.5, 5 mM EDTA, 150 mM NaCl, 1% Nonidet P40, and a mix of protease inhibitors). Proteins were then subjected to SDS/PAGE electrophoresis and transferred onto Immobilon-P Transfer membranes (Millipore, Billerica, MA, USA). Membranes were blocked with 5% nonfat milk proteins and probed with the indicated antibodies at the appropriate dilutions: MPPED2 (NBP1-80,499, Novus Biologicals, Centennial, CO, USA), PI3K (#4257S, Cell Signaling, Danvers, MA, USA), p-PI3K(#4228S, Cell Signaling), AKT (#92,725, Cell Signaling), p-AKT (#4051, Cell Signaling) and β-actin (A5441; Sigma-Aldrich, Saint Louis, MO, USA). Membranes were then incubated with horseradish peroxidase-conjugated secondary antibody for 60 min at room temperature and the signals were detected by western blot detection system (ECL) (Thermofisher, Waltham, MA, USA). Densitometric analyses of the Western blot bands were performed by using ImageJ 1.43 software (NIH, Bethesda, MD, USA) [33].
Colony formation assay
U251 and GLI36 cells at 80% confluency were transfected with pCDNA3.1-EV and pCDNA3.1-MPPED2 into 60 mm plate. 48 h later after transfection, 25 µg/mL and 50 µg/mL of hygromycin were added into the medium for U251 and GLI36, respectively. After 3 weeks of incubation with hygromycin selection, cells were fixed and stained with crystal violet.
CellTiter proliferation assay
Cell proliferation was estimated by seeding 5 × 103 U251 and GLI36 cells overexpressing MPPED2 and carrying the EV were plated in 96-well plates. Cell growth was assessed using CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) (Promega, Madison WI, USA), at 0, 24, 48, 72, and 96 h. Measures were performed at 490 nm using a microplate reader (LX800, BioTek Instruments, Inc., Winooski, VT, USA).
BrdU cell proliferation assay
Cell proliferation was assessed using the colorimetric BrdU Cell Proliferation ELISA Kit (Abcam, ab126556) according to manufacturer’s instruction. Briefly, 2 × 104 U251 and GLI36 cells were seeded in 96-well plate and were cultured overnight. Subsequently, 10 μM BrdU was added to each well, and samples were incubated for 12 h at 37°C. BrdU incorporation was determined by measuring the absorbance at 450 nm in a microplate reader (LX800, BioTek Instruments). All experiments were repeated at least three times and presented as mean ± SD.
Cell migration assay
Transwell migration assay was performed as previously described [34]. Briefly, 3 × 104 U251 and 5 × 104 GLI36 cells were seeded in the upper chamber in 0.2 ml of serum-free medium and 0.5 ml of complete medium was added in the lower chamber as chemoattractant. After 24 h of incubation, the migrated cells were fixed and stained with crystal violet solution (crystal violet 0.05%, methanol 20%). Then, crystal violet in the chamber was de-stained with PBS-0.1% SDS solution and was read at 590 nm in a microplate reader (LX800, Universal Microplate Reader, BioTek Instruments). Additionally, to normalize the number of the cells 3 × 104 U251 and 5 × 104 GLI36 cells were also seeded in a 96-well plate and, after 3 h, the absorbance at 490 nm was read using cell titer (Promega) in a microplate reader (LX800, BioTek Instruments). Results were obtained by normalizing the crystal violet values to cell titer ones.
5-Aza-2ʹ-deoxycytidine (5-Aza-dC) treatment
1 x 105 GBM cells were seeded into a 60 mm plate 24 h before treatment. Cells were treated with 5-Aza-2ʹ-deoxycytidine (A3656, Sigma-Aldrich) at a concentration of 2 µM in the growth medium. The growth medium and 5-Aza-dC treatment were refreshed every 24 h for a total of 96 h.
Cell viability assay
Drug-induced cytotoxicity was quantified by using CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) (Promega). U251 and GLI36 cells were seeded in 96-well plates at 5 × 103 cells/well, then exposed to serial dilutions of TMZ (25, 50 and 100 µM). After 48 h absorbance was measured at 490 nm in a microplate reader (LX800, BioTek Instruments).
Evaluation of caspase 3/7 activity
Caspases 3 and 7 activity was analyzed by using Caspase-Glo® 3/7 Assay (Promega, Madison WI, USA) according to the manufacturer’s instructions. Briefly, 3 × 103 U251 and GLI36 cells were seeded into white-walled 96-well plates in triplicate (Becton Dickinson, Milan, Italy), and after 24 h, U251 and GLI36 cells were treated with 100 μM and 50 μM of TMZ, respectively. Subsequently, 100 μl of Caspase-Glo® 3/7 reagent was dispensed to each well, and plates were incubated for 30 min at room temperature. Luminescence was estimated by employing a plate-reading luminometer (Bio-Tek Instruments) [35].
Statistical analysis
Two-sided unpaired Student’s t tests, Mann-Whitney and ANOVA tests were utilized to analyze data. p < 0.05 values were considered as statistically significant. The mean values ± SD were obtained from three independent experiments. GraphPad Prism Software 8.0 was used to obtain regression analyses and correlation coefficients.
Results
Downregulation of MPPED2 expression in GBM correlates with reduced overall survival of GBM patients
The MPPED2 expression was evaluated in LGG, comprising oligodendroglioma and astrocytoma tumors, GBM and non-tumoral brain tissues examining data available at the TCGA dataset. MPPED2 levels were reduced in brain tumors, particularly in GBM samples, compared with the non-tumoral brain tissues (Figure 1(a)). In accordance with these results, the lowest expression of MPPED2 was found in grade IV tumors (Figure 1(b)), indicating that MPPED2 downregulation could be associated with a poor prognosis. These findings were confirmed by examining the data from the Gravendeel database. As shown in Figure 1(c), the MPPED2 expression was significantly lower in GBM tissues with respect to LGG tumors (p < 0.0001) and in the grade IV with respect to the less aggressive grade II and III tumors (p < 0.0001) (Figure 1(d)), further confirming that the loss of MPPED2 correlates with an unfavorable prognosis. Then, we evaluated the methylation status of MPPED2 promoter in LGG and GBM tissues by using the TCGA dataset. Interestingly, a strong methylation level in the MPPED2 regulatory region in LGG and GBM was found when compared with non-tumoral brain samples. Specifically, the methylation levels were significantly higher in GBM samples when compared with LGG ones (p < 0.0001) (Figure 1(e)) and a significant inverse correlation was found between MPPED2 expression and its methylation in GBM samples (p < 0.0028, r = −0.4495) (Figure 1(f)). These results indicate that hypermethylation of MPPED2 promoter likely accounts for the loss of MPPED2 expression also in brain tumor progression, as previously reported in breast and colon cancer [24,28].
Figure 1.
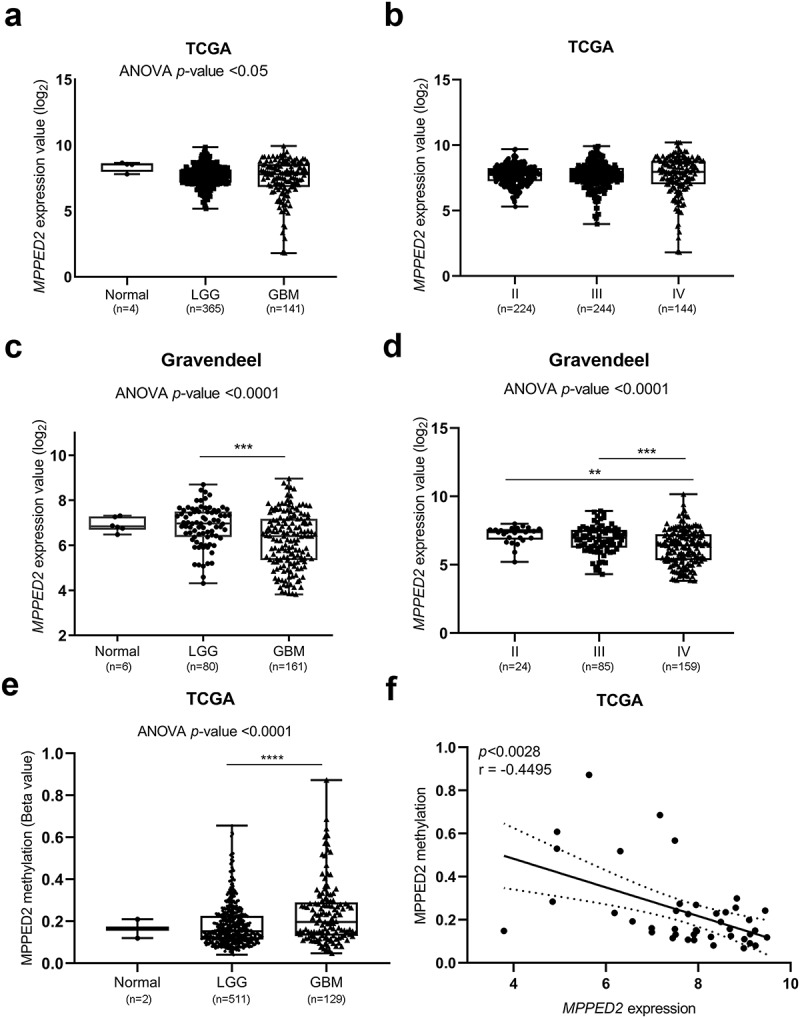
Expression and methylation analysis of MPPED2 in glioma patients. (a) MPPED2 expression profile analyzed in normal brain tissue (n= 4), LGG (n = 365) and GBM (n = 141) samples by using data from the RNA-seq of the TCGA database. One-way analysis of variance (ANOVA) test: *p < 0.05. (b) MPPED2 expression data from the RNA-seq of the TCGA database in the grade II (n = 224), III (n = 244) and IV (n = 144) of glioma patients. ANOVA test: not significant. (c) MPPED2 expression profile analyzed in normal brain tissue (n = 6), LGG (n = 80) and GBM (n = 161) samples by using data from the Gravendeel database. ANOVA test: ***p < 0.001, GBM vs LGG. (d) MPPED2 expression data from Gravendeel database in the grade II (n = 24), III (n = 85) and IV (n = 159) of glioma patients. ANOVA test: **p < 0.01, grade II vs grade IV, ***p < 0.001, grade III vs grade IV. (e) MPPED2 methylation levels were evaluated in a dataset available at the TCGA. ANOVA test: ****p < 0.0001, LGG vs GBM. (f) Correlation scatter plot (Spearman’s Rank) analysis between MPPED2 methylation values and expression levels in the TCGA GBM cohort (r = −0.4495; p < 0.0028)
Subsequently, we analyzed the survival data from the TCGA database and their association with MPPED2 expression in LGG and GBM samples by performing Kaplan-Meier analysis. Interestingly, patients with low MPPED2 expression exhibited a significantly reduced overall survival in glioma samples (p < 0.01) (Figure 2(a)). Similar findings were obtained for GBM patients, showing that higher MPPED2 expression was associated with a more favorable patient outcome with respect to that of patients showing lower MPPED2 expression (p < 0.01) (Figure 2(b)). Further, the correlation between MPPED2 expression and the response to chemo- and radiotherapy was also evaluated in accordance with the TCGA dataset. As shown in Figure 2(c), the GBM patients with lower MPPED2 expression receiving radiotherapy had a lower survival time than patients with high MPPED2 expression. Similarly, for the GBM patients treated with chemotherapy. As far as the GBM patients treated with chemotherapy are concerned, those showing high MPPED2 expression levels had a better prognosis than the low MPPED2 expression patients (Figure 2(d)).
Figure 2.
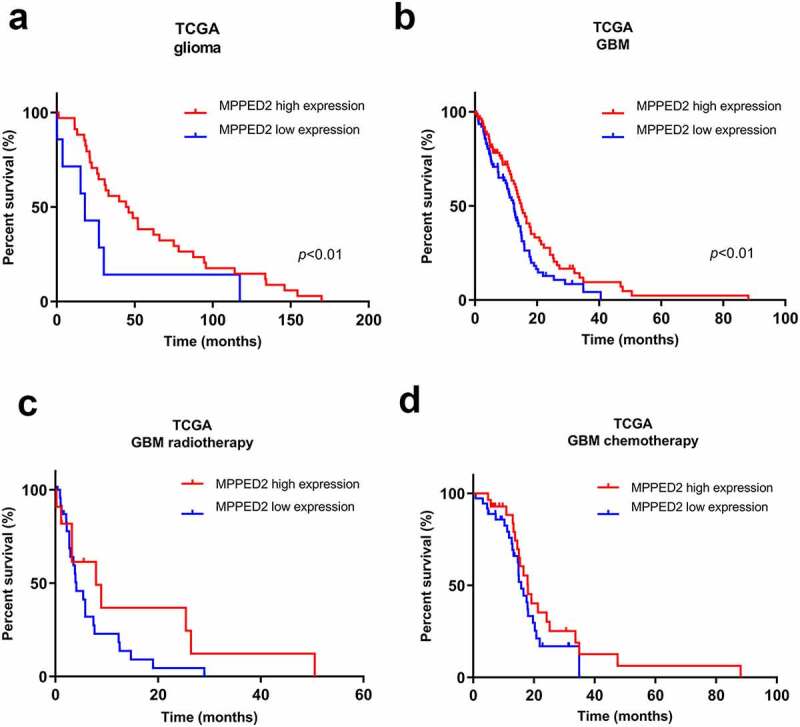
Correlation analysis of MPPED2 expression with glioma patient survival. Kaplan-Meier analysis showed the correlation between MPPED2 expression and the survival of (a) glioma and (b) GBM patients by using TCGA database. **p < 0.01. Kaplan-Meier analysis of survival of GBM patients treated with (c) radiotherapy and (d) chemotherapy from the TCGA according to MPPED2 expression
Overall, these findings indicate that MPPED2 downregulation could have a critical role in GBM progression.
MPPED2 expression negatively correlates with the MES subtype
Then, we further investigated MPPED2 expression in the different molecular GBM subtypes by analyzing the TCGA dataset. Interestingly, this analysis revealed that, among the GBM subtypes, MPPED2 expression was significantly lower in the most aggressive MES subtype with respect to the CL and PN ones (p < 0.0001) (Figure 3(a)). These findings were confirmed by examining the data from the Gravendeel database. Indeed, a drastic reduction of MPPED2 expression was found in the GBM of MES subtype (p < 0.0001) (Figure 3(b)), further confirming that the loss of MPPED2 correlates with a poor prognosis. Moreover, a significant negative correlation was observed between MPPED2 expression and that of the MES subtype-specific genes, such as FN1, VIM and CHI3L1/YKL40 as shown in Figure 3(c), and other MES signature genes reported in Supplementary Table 1. Conversely, a significant positive correlation was found with the genes specific of the PN subtype, such as Olig2, DLL3 and BCAN genes (Figure 3(d) and Supplementary Table 1), while no correlation was observed between MPPED2 and the CL signature gene expression (Supplementary Table 1).
Figure 3.
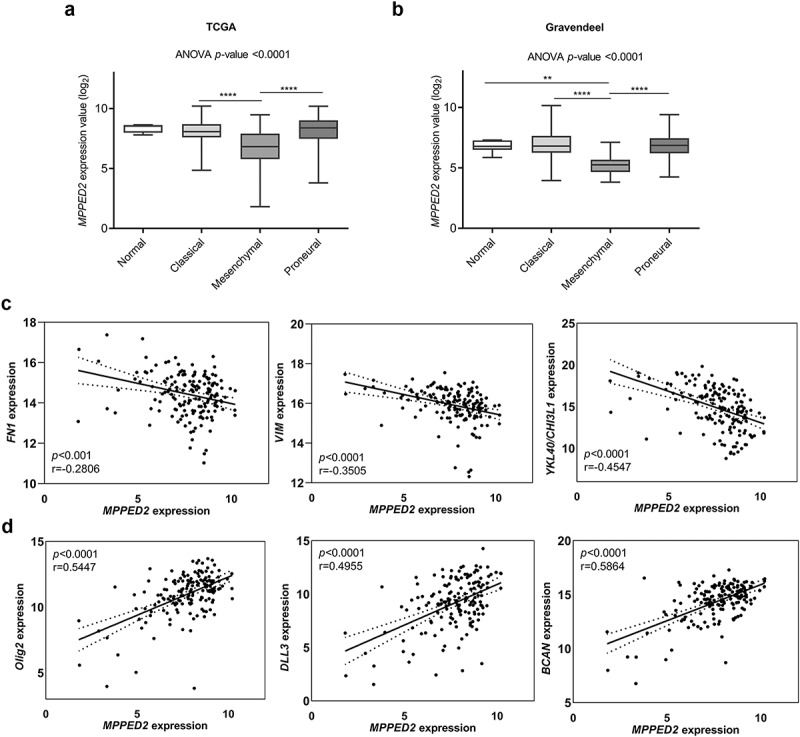
MPPED2 expression in GBM molecular subtypes. (a) Evaluation of MPPED2 expression values in the different GBM subtypes from RNA-seq of TCGA database. ****p < 0.0001, MES vs CL; ****p < 0.0001, MES vs PN. (b) Evaluation of MPPED2 expression values in the different GBM subtypes from the microarray data of the Gravendeel database. **p < 0.01, MES vs Normal; ****p < 0.0001, MES vs CL; ****p < 0.0001, MES vs PN. (c) Pearson correlation analysis was carried out to evaluate the link between MPPED2 expression and the MES specific genes FN1 (r = −0.2806, p < 0.001), VIM (r = −0.3505, p < 0.0001), CHI3L1/YKL40 (r = −0.4547, p < 0.0001). (d) Pearson correlation analysis was calculated to analyze the link between MPPED2 expression and the PN specific genes Olig2 (r = 0.5447, p < 0.0001), DLL3 (r = 0.4955, p < 0.0001), BCAN (r = 0.5864, p < 0.0001)
To further validate the contribution of MPPED2 downregulation to the development of GBM of the MES subtype, we evaluated the effects of MPPED2 expression on that of the target genes involved in the PI3K/AKT and NF-kB signaling pathways, known to be deregulated in MES subtype [8,10]. To achieve this aim, we first evaluated MPPED2 expression in U87, U251, A172, GLI36, LN18 and LN226 GBM cells by qRT-PCR analysis. As reported in Figure 4(a), the MPPED2 levels were lower in all GBM cells analyzed when compared with the mean of six non-tumoral brain tissues. We, then, transfected U251 and GLI36 cell lines that showed the lowest MPPED2 expression levels, with a pCDNA3.1-MPPED2 overexpressing vector to restore MPPED2 expression. Stable MPPED2 expressing cell clones were selected and the overexpression of MPPED2 in U251 and GLI36 cells was confirmed by qRT-PCR (Figure 4(b) and Figure S1) and Western blot analyses (Figure 4(c)). Interestingly, Western blot analysis revealed that MPPED2 overexpression reduces the phosphorylation levels of PI3K and AKT when compared with the corresponding empty vector (EV) (Figure 4(d)). We also observed a reduction of p65 expression in U251-MPPED2 and GLI36-MPPED2 overexpressing cells (Figure 4(d)).
Figure 4.
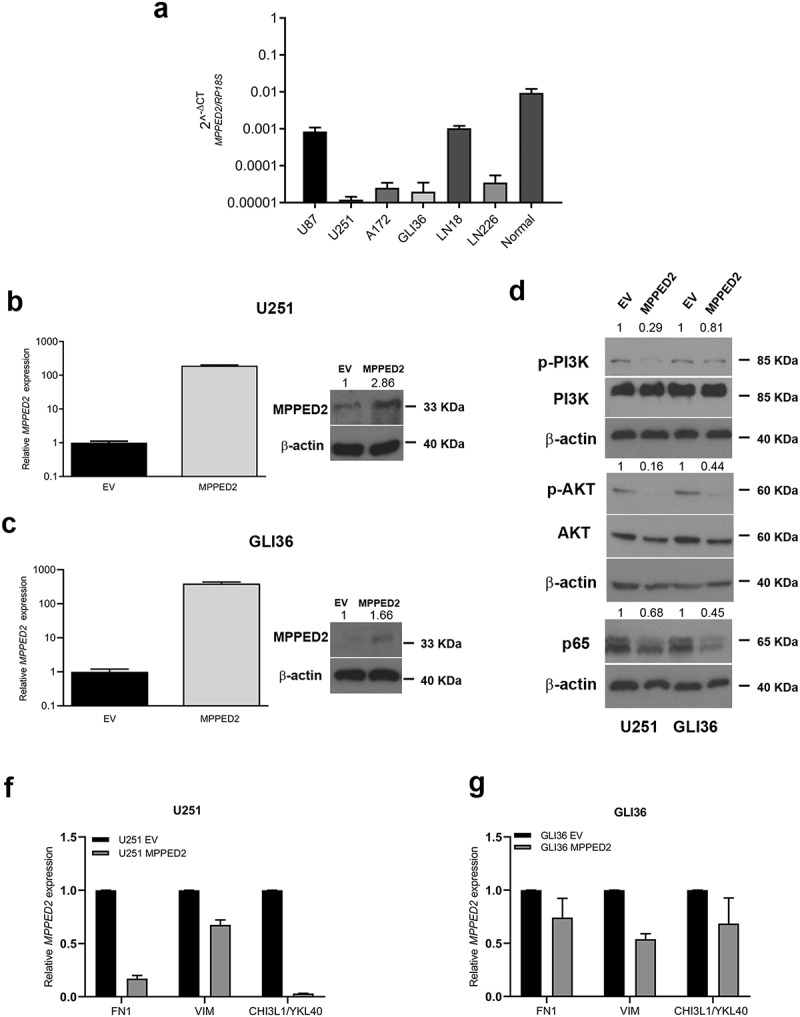
MPPED2 negatively correlates with MES subtype. (a) qRT-PCR analysis performed in U87, U251, A172, GLI36, LN18, LN226 GBM cell lines and normal brain tissue samples to evaluate MPPED2 expression. Data are reported as 2−ΔΔCt values ± SD for three independent experiment performed in triplicate. (b, c) qRT-PCR analysis (left panel) performed in U251 and GLI36 cell lines stably expressing MPPED2 or carrying the corresponding empty vector (EV). Data are reported as 2−ΔΔCt values ± SD and were compared to EV, set equal to 1. Western blot analysis (right panel) confirming the expression of MPPED2. β-actin was used to normalize the amount of loaded protein. Densitometric analysis was performed by using ImageJ software and normalizing to β-actin. (d) Western blot analysis of p-PI3K, PI3K, p-AKT, AKT, p65 in U251 and GLI36 cells stably expressing MPPED2 or carrying the corresponding EV. β-actin was used to normalize the amount of loaded protein. Densitometric analysis was performed by using ImageJ software and normalizing to β-actin. (f, g) The expression levels of the mesenchymal marker genes were analyzed by qRT-PCR in U251 and GLI36 cell lines stably expressing MPPED2 or carrying the EV. Data are reported as 2−ΔΔCt values ± SD for three independent experiment performed in triplicate and were compared to EV, set equal to 1
Additionally, to strengthen the negative correlation observed between MPPED2 and MES subtype, we evaluated the levels of the MES marker genes in U251- and GLI36-MPPED2 overexpressing cells by qRT-PCR. Interestingly, we found that FN1, VIM and CHI3L1/YKL40 genes were decreased in both cell lines stably expressing MPPED2 (Figure 4f and g), indicating once more the negative association of MPPED2 with the most aggressive MES subtype.
Restoration of MPPED2 attenuates proliferation and migration of GBM cells
Next, the functional role of MPPED2 in GBM was investigated in U251- and GLI36-MPPED2 overexpressing cells. First, the MPPED2 ability to regulate cell proliferation was examined by performing the MTS assay. The obtained results demonstrated that MPPED2-expressing GBM cells grew at a significantly slower rate with respect to the corresponding EV-transfected cells (Figure 5a and b). To strengthen these results, we performed BrdU incorporation assay. We observed that both U251- and GLI36-MPPED2 showed a significant reduction in cell proliferation compared with the respective EV control (Figure 5c and d). This result was, then, confirmed by colony-formation assay performed by transiently transfecting GBM cells with MPPED2-expressing vector or the relative EV control. Indeed, as shown in Figure 5(e), both U251- and GLI36-MPPED2 overexpressing cells gave rise to a significantly lower colony number in comparison with the controls. Thus, these findings demonstrate the involvement of MPPED2 in GBM cell growth.
Figure 5.
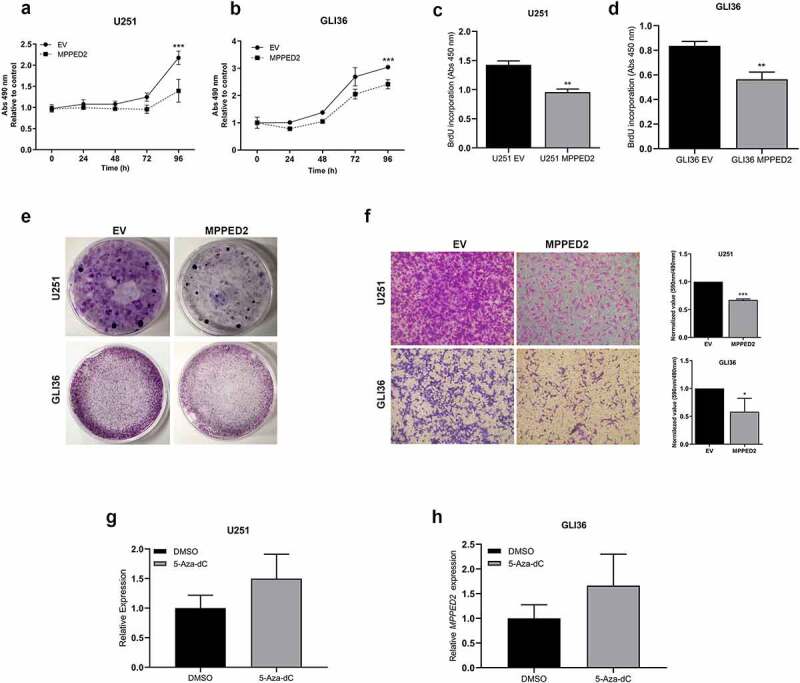
MPPED2 reduces cell proliferation and migration of GBM cells. Cell proliferation analysis of (a) U251 and (b) GLI36 stably expressing MPPED2 or carrying the EV. Cell numbers was evaluated by reading the absorbance at 490 nm at 24, 48, 72 and 96 h, after plating. The mean values ± SD deriving from three independent experiments performed in triplicate are reported in the graph. 2-way ANOVA test (Bonferroni post-test: ***p < 0.001, MPPED2 vs EV, 96 h, either U251 and GLI36). Cell proliferation was assessed using BrdU incorporation assay in (c) U251- and (d) GLI36-MPPED2 and those carrying EV. Data are reported as mean ± SD of three independent experiment performed in triplicate. ** p < 0.01, MPPED2 vs EV, either U251 and GLI36 (e) Representative images of colony assay carried out in U251 and GLI36 transiently transfected with MPPED2 or EV. After 3 weeks of selection with hygromycin the cells were stained with crystal violet solution. (f) In the left panel the representative images of migration assay carried out in U251 and GLI36 stably expressing MPPED2 or the corresponding EV was reported. Magnification 40 ×. Right panel: values from three independent experiments were obtained by reading the absorbance of migrating cells at 590 nm and divided by the corresponding cell titer values. The obtained values were normalized to EV, set equal to 1, and reported as ± SD. t-test: ***p < 0.001 (MPPED2 vs EV, U251 cells), *p < 0.05 (MPPED2 vs EV, GLI36 cells). (f, g) MPPED2 levels were evaluated by qRT-PCR in U251 and GLI36 cell lines after 2 µM 5-Aza-dC or DMSO (vehicle) treatment for 96 hours. Data were reported as 2−ΔΔCt ± SD for three independent experiments performed in triplicate and were compared to DMSO vehicle, set equal to 1
Then, to investigate whether MPPED2 restoration was able to inhibit migratory properties, the cell migration rate of MPPED2-overexpressing GBM cells was analyzed through transwell assay. As shown in Figure 5(f), U251- and GLI36-MPPED2 cell clones had a significantly reduced ability to migrate through the transwell chamber with respect to the controls in both GBM cell lines. Therefore, our findings support the tumor suppressor role of MPPED2 in the development of GBM.
Furthermore, to verify whether also in GBM the epigenetic mechanism accounts for MPPED2 reduction, we treated U251 and GLI36 cell lines with 2 µM of the demethylating agent 5-Aza-dC for 96 hours. After the treatment, the expression of MPPED2 was evaluated by qRT-PCR analysis. Consistently, increased MPPED2 expression was obtained in U251 and GLI36 cells treated with 5-Aza-dC when compared with those treated with the DMSO vehicle (Figure 5g and h), strongly suggesting that also in GBM the MPPED2 reduction could be due to its methylation status.
Restoration of MPPED2 enhances TMZ sensitivity of GBM cells
The next step has been to assess whether MPPED2 expression affects the sensitivity to TMZ of U251 and GLI36 GBM cells since TMZ is a chemotherapeutic drug widely used for the treatment of GBM patients. To this aim, we treated MPPED2-overexpressing GBM cell clones with increasing amounts of TMZ drug (25, 50 and 100 µM) and we evaluated cell viability after 48 h of treatment. As shown in Figure 6(a), U251 cells overexpressing MPPED2 displayed a significant increase in mortality after treatment with 100 µM of TMZ with respect to the control (p < 0.01). Similarly, GLI36-MPPED2 cells also showed a significant reduction in the viability of the cells treated with 50 and 100 µM of TMZ, if compared with the EV control cells (p < 0.05) (Figure 6(b)). Thereby, these findings indicate that high MPPED2 expression enhances the sensitivity to TMZ treatments.
Figure 6.
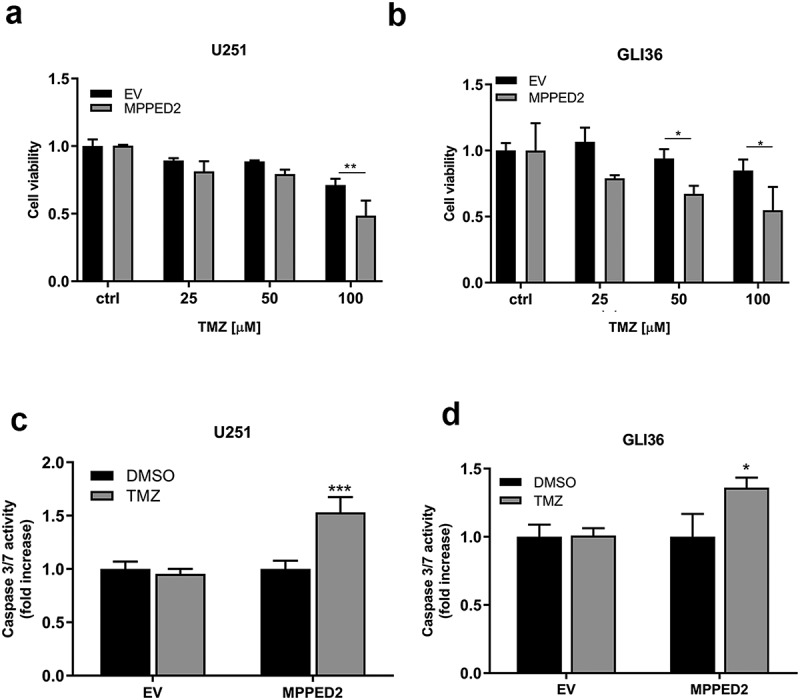
MPPED2 expression enhances the sensitivity of GBM cells to TMZ. (a,b) Cell viability assay was performed in U251 and GLI36 transiently transfected with MPPED2 or the corresponding EV and treated with increasing amount of TMZ for 48 h. Values are reported as mean ± SD of three independent experiments performed in triplicate. 2-way ANOVA test (Bonferroni post-test: **p < 0.01, U251; *p < 0.05, GLI36). (c, d) Evaluation of caspases 3 and 7 activity was performed in U251 and GLI36 cells transiently transfected with MPPED2 or with the corresponding EV and treated with 100 μM and 50 μM of TMZ for 24 h, respectively. Results were expressed as fold change relative to the cells treated with DMSO, used as control. Each column represents mean ± SD of three independent experiments performed in triplicate. t-test: ***p < 0.001, U251 cells; *p < 0.05, GLI36 cells
Subsequently, to investigate whether MPPED2 expression triggers apoptosis after TMZ treatment in GBM cells, the activation of caspase 3 and 7 was evaluated through an in vitro assay. Following treatment with 100 µM TMZ and DMSO, as vehicle in U251-MPPED2 and U251-EV cells, a significant increase of caspase 3 and 7 levels was found in cells overexpressing MPPED2 and treated with TMZ (Figure 6(c)). Similar results were achieved in GLI36-MPPED2 cells treated with 50 µM TMZ. In fact, as reported in Figure 6(d), high levels of caspase 3 and 7 were detected in GLI36-MPPED2 treated with TMZ at 24 h. Together, these results indicate that the restoration of MPPED2 expression plays a crucial role in response to the treatment of GBM by affecting the apoptotic response.
Discussion
MPPED2 gene encodes a metallophosphodiesterase protein that belongs to III cyclic nucleotide phosphodiesterases (PDE) family, a group of enzymes able to breakdown the phosphodiester bond of the cyclic adenosine monophosphate (cAMP) and/or cyclic guanine monophosphate (cGMP) [36]. However, extensive biochemically and structurally studies reported that due to an aminoacidic substitution in the MPPED2 active site, its role may not be restricted only to hydrolyze phosphodiester substrates but MPPED2 may act also as a scaffold protein. Also, it has been recently demonstrated that the loss of MPPED2 expression is an event that occurs in several malignant neoplasias originating from different tissues. Moreover, its restoration in cancer cell lines induces apoptosis and negatively modulates cell proliferation [20–22], thus proposing MPPED2 as a novel potential candidate tumor suppressor gene.
More recently, our research group has demonstrated that the hypermethylation of MPPED2 promoter, found in the large majority of breast cancer samples, accounts for its downregulation since the treatment of breast cancer cells with 5-Aza-dC, a demethylating agent, restored its expression [24]. These findings are consistent with previous studies showing a strict correlation between the hypermethylation of MPPED2 promoter and colorectal neoplastic progression [28], then supporting the idea that this epigenetic alteration is one of the main regulatory mechanisms responsible for the loss of MPPED2 in cancer. Therefore, we investigated whether MPPED2 downregulation might be associated with other cancer types, and particularly, we evaluated the potential role of MPPED2 in GBM. This neoplasia represents the main aggressive and lethal brain tumor of grade IV with a worse prognosis compared with the LGG category of tumors. Extensive analysis of molecular signature genes of TCGA data has allowed the classification of GBM in MES, CL, PN, and NL subtypes. The MES phenotype showed more malignant features when compared with the other subtypes and the elevated expression of MES signature genes and the loss of PN specific-genes are strictly linked with a poor prognosis of glioma patients [19]. Consequently, we analyzed MPPED2 expression in LGG and GBM by using data at the TCGA and Gravendeel datasets. These results revealed that the lowest MPPED2 expression was observed in GBM samples, evidencing a correlation between the loss of MPPED2 and the progression of brain neoplasias. Intriguingly, through in silico analysis, we evaluated the methylation status of MPPED2 promoter, and we found that MPPED2 regulatory region was hypermethylated in both LGG and GBM compared with the non-tumoral brain. Its methylation status was more pronounced in GBM samples, revealing that even in brain tumors the loss of its expression is mainly regulated through this epigenetic regulation, as previously demonstrated in breast and colon cancer [24,28]. Then, Kaplan-Meier analysis carried out using TCGA data reported that patients with elevated MPPED2 expression showed a more favorable outcome compared to those with low expression in both LGG and GBM. Particularly, we observed that GBM patients with higher MPPED2 levels treated with radio- and chemotherapy, had a better prognosis than those with lower MPPED2 expression, evidencing once more the important role of MPPED2 downregulation in GBM tumorigenesis. Further, extensive analysis of MPPED2 levels in the TCGA and Gravendeel datasets demonstrated that MPPED2 expression levels are significantly reduced in the most aggressive MES subtype, and a significant negative correlation between MPPED2 and the MES signature genes was found. On the contrary, MPPED2 expression was positively associated with that of PR markers. Moreover, since several pathways, such as NF-kB and PI3K-AKT, are deregulated in MES subtype [8,10], we investigated the involvement of MPPED2 in the modulation of these targets. Interestingly, we observed that MPPED2 overexpression is able to reduce the phosphorylation levels of PI3K and AKT and affects the levels of p65 in U251 and GLI36 cells. Although further studies are needed, this molecular modulation may be triggered by MPPED2 upregulation (that regulates cAMP levels) due to its involvement in cAMP regulation. Indeed, several studies have reported that also a subtle regulation of cAMP levels is able to modulate the phosphorylation levels of both p65 and PI3K/AKT [37–39], and also other PDE superfamily members have been reported to act in this way [40,41]. Thus, MPPED2 may have a great research potential in order to clarify its molecular role as fine-tuner of cAMP or, as already proposed by Swarbrick et al. [42], for other PDE members, acting as scaffold protein. Thereafter, functional assays in U251 and GLI36 cells overexpressing MPPED2 showed that MPPED2 negatively affected cell proliferation and migration ability. These findings demonstrate the contribution of MPPED2 reduction to GBM progression. Next, we examined how MPPED2 expression could influence the sensitivity of GBM cells to TMZ, the drug commonly used for the therapy of GBM. We found that MPPED2 overexpression significantly increased the sensitivity to TMZ treatment in U251 and GLI36 cell systems. Additionally, we observed that MPPED2 expression increased cell death inducing the activation of caspase 3 and 7, indicating that the reduction of cell viability could be due to the increase of the apoptosis, which in turn could be likely due to the ability of MPPED2 to inhibit PI3K/AKT and NF-kB pathways,
Taken together, the findings reported here evidencing the negative association of MPPED2 with MES subtype clearly indicate an important role of MPPED2 in GBM progression, and suggest that restoration of MPPED2 expression and/or function could be explored as a possible therapeutic approach for this malignant neoplasia, since its expression impairs proliferation and migration and enhances the sensitivity of GBM cells to TMZ.
Conclusion
In conclusion, the results reported here clearly demonstrate the involvement of MPPED2 in GBM progression. Indeed, we observed that its expression is strongly reduced in the main aggressive MES subtype. Further, we report that the restoration of MPPED2 expression impairs cell growth and cell migration ability, and that its expression could be important for the regulation of sensitivity to TMZ.
Supplementary Material
Acknowledgments
We would like to thank Mrs. Mariarosaria Montagna (IEOS-CNR) for the technical support to research and Mrs. Giulia Speranza, Mrs. Giuseppina Ippolito and Mrs. Daniela Rastelli (IEOS-CNR) for the administrative support.
Funding Statement
This work was supported by grants from: CNR Flagship Projects (Epigenomics-EPIGEN).
Disclosure statement
The authors declare no conflict of interest.
Author contributions
Conception and design: SP, AF and PP. Formal analysis: MDM, FE, CQ. Investigation: SP. Manuscript writing: SP, AF and PP. Final approval of the manuscript: All authors.
Supplementary materials
Supplemental material for this article can be accessed here.
References
- [1].Lapointe S, Perry A, Butowski NA.. Primary brain tumours in adults. Lancet. 2018;392(10145):432–446. [DOI] [PubMed] [Google Scholar]
- [2].Wang Y, Jiang T. Understanding high grade glioma: molecular mechanism, therapy and comprehensive management. Cancer Lett. 2013;331(2):139–146. [DOI] [PubMed] [Google Scholar]
- [3].Wang H, Lathia JD, Wu Q, et al. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem Cells. 2009;27(10):2393–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–820. [DOI] [PubMed] [Google Scholar]
- [5].Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359(5):492–507. [DOI] [PubMed] [Google Scholar]
- [6].Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lin CL, Lieu AS, Lee KS, et al. The conditional probabilities of survival in patients with anaplastic astrocytoma or glioblastoma multiforme. Surg Neurol. 2003;60(5):402–406. discussion 406 . [DOI] [PubMed] [Google Scholar]
- [8].Verhaak RG, Hoadley KA, Purdom E, et al., Cancer Genome Atlas Research N . Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Carro MS, Lim WK, Alvarez MJ, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463(7279):318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yamini B. NF-kappaB, mesenchymal differentiation and glioblastoma. Cells. 2018;7(9):125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zanotto-Filho A, Goncalves RM, Klafke K, et al. Inflammatory landscape of human brain tumors reveals an NFkappaB dependent cytokine pathway associated with mesenchymal glioblastoma. Cancer Lett. 2017;390:176–187. [DOI] [PubMed] [Google Scholar]
- [12].Stupp R, Hegi ME. Brain cancer in 2012: molecular characterization leads the way. Nat Rev Clin Oncol. 2013;10(2):69–70. [DOI] [PubMed] [Google Scholar]
- [13].Stupp R, Mason WP, Van Den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. [DOI] [PubMed] [Google Scholar]
- [14].Yan Y, Xu Z, Dai S, et al. Targeting autophagy to sensitive glioma to temozolomide treatment. J Exp Clin Cancer Res. 2016;35(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kanzawa T, Bedwell J, Kondo Y, et al. Inhibition of DNA repair for sensitizing resistant glioma cells to temozolomide. J Neurosurg. 2003;99(6):1047–1052. [DOI] [PubMed] [Google Scholar]
- [16].Lee SY. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016;3(3):198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Perazzoli G, Prados J, Ortiz R, et al. Temozolomide resistance in glioblastoma cell lines: implication of MGMT, MMR, P-glycoprotein and CD133 expression. PLoS One. 2015;10(10):e0140131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Garros-Regulez L, Aldaz P, Arrizabalaga O, et al. mTOR inhibition decreases SOX2-SOX9 mediated glioma stem cell activity and temozolomide resistance. Expert Opin Ther Targets. 2016;20(4):393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Phillips HS, Kharbanda S, Chen R, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9(3):157–173. [DOI] [PubMed] [Google Scholar]
- [20].Liguori L, Andolfo I, De Antonellis P, et al. The metallophosphodiesterase Mpped2 impairs tumorigenesis in neuroblastoma. Cell Cycle. 2012;11(3):569–581. [DOI] [PubMed] [Google Scholar]
- [21].Shen L, Liu L, Ge L, et al. miR-448 downregulates MPPED2 to promote cancer proliferation and inhibit apoptosis in oral squamous cell carcinoma. Exp Ther Med. 2016;12(4):2747–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang RY, Shen CL, Zhao LJ, et al. Dysregulation of host cellular genes targeted by human papillomavirus (HPV) integration contributes to HPV-related cervical carcinogenesis. Int J Cancer. 2016;138(5):1163–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sepe R, Pellecchia S, Serra P, et al. The long non-coding RNA RP5-1024C24.1 and its associated-gene MPPED2 are down-regulated in human thyroid neoplasias and act as tumour suppressors. Cancers (Basel). 2018;10(5):146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pellecchia S, Sepe R, Federico A, et al. The metallophosphoesterase-domain-containing protein 2 (MPPED2) gene acts as tumor suppressor in breast cancer. Cancers (Basel). 2019;11(6):797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tyagi R, Shenoy AR, Visweswariah SS. Characterization of an evolutionarily conserved metallophosphoesterase that is expressed in the fetal brain and associated with the WAGR syndrome. J Biol Chem. 2009;284(8):5217–5228. [DOI] [PubMed] [Google Scholar]
- [26].Schwartz F, Eisenman R, Knoll J, et al. cDNA sequence, genomic organization, and evolutionary conservation of a novel gene from the WAGR region. Genomics. 1995;29(2):526–532. [DOI] [PubMed] [Google Scholar]
- [27].Schwartz F, Neve R, Eisenman R, et al. A WAGR region gene between PAX-6 and FSHB expressed in fetal brain. Hum Genet. 1994;94(6):658–664. [DOI] [PubMed] [Google Scholar]
- [28].Gu S, Lin S, Ye D, et al. Genome-wide methylation profiling identified novel differentially hypermethylated biomarker MPPED2 in colorectal cancer. Clin Epigenetics. 2019;11(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gravendeel LA, Kouwenhoven MC, Gevaert O, et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009;69(23):9065–9072. [DOI] [PubMed] [Google Scholar]
- [30].Bowman RL, Wang Q, Carro A, et al. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol. 2017;19(1):139–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Diez-Villanueva A, Mallona I, Peinado MA. Wanderer, an interactive viewer to explore DNA methylation and gene expression data in human cancer. Epigenetics Chromatin. 2015;8(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Penha RCC, Sepe R, De Martino M, et al. Role of Dicer1 in thyroid cell proliferation and differentiation. Cell Cycle. 2017;16(23):2282–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Puca F, Tosti N, Federico A, et al. HMGA1 negatively regulates NUMB expression at transcriptional and post transcriptional level in glioblastoma stem cells. Cell Cycle. 2019;18(13):1446–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Pellecchia S, Sepe R, Decaussin-Petrucci M, et al. The long non-coding RNA prader willi/angelman region RNA5 (PAR5) is downregulated in anaplastic thyroid carcinomas where it acts as a tumor suppressor by reducing EZH2 activity. Cancers (Basel). 2020;12(1):235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cacciola NA, Sepe R, Forzati F, et al. Restoration of CBX7 expression increases the susceptibility of human lung carcinoma cells to irinotecan treatment. Naunyn Schmiedebergs Arch Pharmacol. 2015;388(11):1179–1186. [DOI] [PubMed] [Google Scholar]
- [36].Sutherland EW, Rall TW. Fractionation and characterization of a cyclic adenine ribonucleotide formed by tissue particles. J Biol Chem. 1958;232(2):1077–1091. [PubMed] [Google Scholar]
- [37].Gerlo S, Kooijman R, Beck IM, et al. Cyclic AMP: a selective modulator of NF-kappaB action. Cell Mol Life Sci. 2011;68(23):3823–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Cosentino C, Di Domenico M, Porcellini A, et al. p85 regulatory subunit of PI3K mediates cAMP-PKA and estrogens biological effects on growth and survival. Oncogene. 2007;26(14):2095–2103. [DOI] [PubMed] [Google Scholar]
- [39].Kim D, Kim S, Koh H, et al. Akt/PKB promotes cancer cell invasion via increased motility and metalloproteinase production. Faseb J. 2001;15(11):1953–1962. [DOI] [PubMed] [Google Scholar]
- [40].Smith PG, Wang F, Wilkinson KN, et al. The phosphodiesterase PDE4B limits cAMP-associated PI3K/AKT-dependent apoptosis in diffuse large B-cell lymphoma. Blood. 2005;105(1):308–316. [DOI] [PubMed] [Google Scholar]
- [41].Kim DU, Nam J, Cha MD, et al. Inhibition of phosphodiesterase 4D decreases the malignant properties of DLD-1 colorectal cancer cells by repressing the AKT/mTOR/Myc signaling pathway. Oncol Lett. 2019;17(3):3589–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Swarbrick JD, Shaw DJ, Chhabra S, et al. VPS29 is not an active metallo-phosphatase but is a rigid scaffold required for retromer interaction with accessory proteins. PLoS One. 2011;6(5):e20420. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.