

ABSTRACT
Possible phenotypic impairments associated with maternal stress during gestation in beef cattle may be explained by epigenetic effects. This study examined the impact of prenatal transportation stress on DNA methylation of lymphocytes of Brahman cows over the first 5 years of life. Methylation analysis through reduced representation bisulphite sequencing was conducted on DNA from lymphocytes from 28 paired samples from 6 prenatally stressed (PNS) and 8 control (Control) females obtained initially when they were 28 days of age and 5 years of age. There were 14,386 CpG (C = cytosine; p = phosphate; G = guanine) sites differentially methylated (P < 0.01) in 5-yr-old Control cows compared to their lymphocyte DNA at 28 days of age, this number was slightly decreased in 5-yr-old PNS with 13,378 CpG sites. Only 2,749 age-related differentially methylated CpG sites were seen within PNS females. There were 2,637 CpG sites differentially methylated (P < 0.01) in PNS cows relative to Controls at 5 years of age. There were differentially methylated genes in 5-yr-old cows that contributed similarly to altered gene pathways in both treatment groups. Canonical pathways altered in PNS compared to Control cows at 5 years of age were mostly related to development and growth, nervous system development and function, and immune response. Prenatal stress appeared to alter the epigenome in Brahman cows compared to Control at 5 years of age, which implies a persistent intervention in DNA methylation in lymphocytes, and may confer long-lasting effects on gene expression, and consequently relevant phenotypic changes.
KEYWORDS: Ageing, Brahman, cattle, DNA methylation, epigenetics, prenatal stress
Introduction
Epigenetic mechanisms contribute to regulation of gene expression that mediates all mammalian development [1], and enable intrinsic and environmental signals to alter the genome [2]. As environmentally sensitive components, epigenetic modifications have emerged
as a possible key mechanism by which prenatal stress affects foetal (and subsequent) development. The prenatal period through weaning is a time of maximal epigenetic plasticity in mammals [3]. In utero, with placental maintenance of the foetal environment, the foetus is vulnerable to a wide variety of external environmental exposures. Stress-triggering stimuli appear to initiate maternal hypothalamus–pituitary–adrenal (HPA) axis activity, resulting in increased glucocorticoid secretion that affects the foetus through the placenta [4]. During foetal development, exposure to elevated maternal cortisol induces dysregulation of the HPA axis in offspring that can have long-lasting behavioural and neurobiological effects [5]. However, the postnatal effects of adrenocorticotropic hormone injection to increase cortisol during gestation were not the same as observed with transportation stress on cattle [6], which demonstrates that elevated cortisol alone is not sufficient to induce the postnatal effects.
DNA methylation is a well-studied and documented epigenetic alteration. It is affected by the addition of a methyl group to cytosines, typically at cytosine-guanine dinucleotides in mammals; this modification modulates gene expression at the transcription level [2]. Evidence from animal and human studies has shown the regulation of gene expression through DNA methylation or other epigenetic mechanisms as a key factor regulating the link between prenatal stress and unfavourable outcomes. Tobi et al. [7] reported a correlation between altered methylation at CpG dinucleotides (C = cytosine; p = phosphate bond; G = guanine) in the DNA of whole blood cells and early gestation stress in the study conducted with adult individuals prenatally exposed to famine during the Dutch Hunger Winter of 1944 and 1945. The effects of prenatal transportation stress [8,9] on temperament have been shown in calves. Prenatal stress altered lymphocyte DNA methylation profiles early in life (28 days of age) in males (10; half-siblings to the females in the present study) and females (11; the same females in the present study). Littlejohn et al. [10] uncovered stress-response genes that were differentially methylated in prenatally stressed bull calves. Considering the possible consequences on animal welfare, health, and reproduction, prenatal stress in beef cattle may be a source of economic and welfare problems. The objective of this study was to examine the effects of prenatal transportation stress on lymphocyte DNA methylation changes of Brahman heifer calves during the first 5 years of life by investigating DNA methylation patterns of both PNS and Control groups at two postnatal time points (day 28 and year 5). We hypothesized that DNA methylation patterns would change from 28 days of age to 5 years of age when cows are mature. Furthermore, the normal pattern of methylation changes would be disturbed for some genes and pathways in animals exposed to transportation as a prenatal stressor.
Materials and methods
Animal procedures and sample acquisition
All procedures were in compliance with the Guide for the Care and Use of Agricultural Animals in Research and Teaching (FASS, 2010) and approved by the Texas A&M AgriLife Research Animal Use and Care Committee. Blood samples were obtained from 5-yr-old Brahman cows (females) whose dams either a) were exposed to transportation during gestation as prenatal stress treatment group or b) served as the non-transported control group. Details of the initial exposure were previously described by Littlejohn et al [12]. Briefly, pregnant Brahman cows (n = 48; these cows were artificially-inseminated and confirmed as conceived in the same week) were transported in trailers for 2-hour periods at 60 ± 5, 80 ± 5, 100 ± 5, 120 ± 5, and 140 ± 5 days of gestation as per our original prenatal transportation stress model [9]. Non-transported pregnant cows (n = 48) were designated as a Control group. The earliest time point to initiate transportation stress (day 60; the later portion of first trimester of gestation) was selected to be after placentation is completed in the cow in order to minimize the risk of inducing early embryonic or foetal loss [13]. The latest time point to apply transportation stress (day 140; the middle trimester of gestation) was selected to be before stress-induced elevation in secretion of foetal adrenal cortisol might induce premature parturition in the cow [14,15]. Transported and control cows were maintained together in the same pasture and nutritional conditions. Transported cows gave birth to 21 female and 20 male calves (prenatally stressed group; PNS), while cows that were maintained as controls gave birth to 18 female and 26 male calves (Control). Blood samples were obtained from all calves at 28 days of age and leukocytes stored at – 80°C. For the study reported herein, 28 matching blood samples were analysed from the same 6 PNS and 8 Control females when they attained 28 days and 5 years of age. The animals were selected at random when they averaged 5 years of age.
Sample analysis
Processing of blood samples and DNA extraction
White blood cells were isolated from 10-mL blood samples upon centrifugation. They were cleaned through repeated washes with blood cell lysis buffer to lyse red blood cells followed by centrifugations, while leaving leukocytes intact. Extraction of DNA was performed using a phenol-chloroform extraction protocol, and extracted DNA was suspended in 150-to-200 μL Tris-EDTA buffer and stored at – 80°C. Samples were transported on dry ice to Zymo Research Corp. (Irvine, CA) for reduced representation DNA methylation analysis, as previously described [12].
Library construction and alignment of DNA reads
Profiling of DNA methylation was conducted with the reduced representation bisulphite sequencing (RRBS) platform (Methyl-MiniSeq, Zymo Research Crop, Irvine, CA). For library preparation, 300 ng genomic DNA from 28 paired samples from the 14 subjects collected at 28 days and 5 years of age was digested with TaqαI and MspI restriction enzymes followed by adaptor ligation. Bisulphite conversion of processed DNA fragments was accomplished in conformity with the EZ DNA methylation-lightning kit by Zymo Research. After polymerase chain reaction (PCR) amplification, bisulphite-treated DNA fragments were sequenced using an Illumina HiSeq platform. Sequenced DNA reads were aligned to the UMD 3.1 Bos taurus genome assembly [16] with Bismark software [17] by Zymo Research to extract methylation information of cytosine nucleotides.
Preprocessing of data
The minimum read coverage was set to 5x for each CpG site to minimize the data loss; the recommended coverage to obtain satisfactory sensitivity is between 5x and 15x for ≤ 3 biological replicates for genome-wide DNA methylation analysis [18], and CpG sites with high coverage (> 500x) were removed to eliminate potential PCR bias. The median coverage normalization was applied to read coverages between samples to prevent introduction of bias. Methylation ratios were determined by dividing the number of methylated reads mapped to a CpG site by the total number of reads mapped to the same CpG site.
Differential DNA methylation analyses and annotation
Differentially methylated CpG sites were identified by analysis with the R/Bioconductor package methylKit version 1.8.1 [19]. Key comparisons included:
1. Age: day 28 and year 5 methylation ratios within PNS and Control groups.
2. Treatment: year 5 methylation ratios of PNS and Control groups.
Overdispersion corrections were performed by applying a scaling parameter to variance estimated by the model to account for within-condition variability and to avoid high false-positive rates [20]. Probability (P) values of age comparisons were corrected for multiple testing using a sliding linear model [21] but they were not in treatment comparisons because of the exploratory nature of this effort and the few detections. Differentially methylated CpG (dmCpG) sites were declared using two criteria: P < 0.01 from Fisher’s exact test and the absolute value of difference in methylation between treatment and control ratios > 0.10. More than 10% increase and decrease in methylation were considered hypermethylated and hypomethylated, respectively. Differentially methylated sites were annotated (located on the reference genome) using the R/Bioconductor package annotatr version 1.8.0 [22] to the reference genome UMD 3.1 Bos taurus assembly [16]. Promoter regions were defined as located within 1,000 bp upstream and 400 bp downstream of the transcription start sites of genes. Age methylation differences were calculated by subtracting the methylation ratio at 28 days of age from that at 5 years of age within the two treatment groups. Treatment methylation differences were estimated for samples obtained at 5 years of age by subtracting the methylation ratio in Control cows from that of PNS cows.
Pathway analysis
Differentially methylated genes were defined as genes that had at least one CpG site within their promoter region; differentially methylated genes within the age comparisons and the treatment comparison were subjected to Ingenuity Pathway Analysis (IPA; Redwood City, CA) to determine altered canonical pathways and biological functions that were potentially induced by methylation differences. Probability values for canonical pathways of enriched genes were calculated with right-tailed Fisher’s exact tests for each canonical pathway in which differentially methylated genes were enriched.
Results and discussion
Mapping summary
At 28 days of age the read pairs averaged 39,106,892 and 37,353,167 for samples from the Control and PNS heifers, respectively. The Control and PNS samples had 39,067,133 and 49,781,832 read pairs average at 5 years of age, respectively, in which variations are attributed to procedures associated with sample processing. Sodium bisulphite treatment successfully resulted in a 99% conversion rate for each sample in each treatment and age group. All CpG sites were mapped against the reference genome from 29 to 31% efficiency in samples from both treatments at both ages. Each unique CpG site averaged from 7x to 9x coverage. The mapping efficiency varies depending on sequence quality and read number, as well as the quality of the reference genome annotation [23].
Methylation at 28 days and 5 years of age in Control cows
A slight majority (59%) of dmCpG sites were hypomethylated in Control cows at 5 years of age compared to those females at 28 days of age (Table 1). There were more hypermethylated dmCpG within promoters and exons, while the opposite was the case for introns. Epigenetic changes are common with ageing [24]; age prediction models have been proposed based on changes in DNA methylation with high accuracy [25]. Overall DNA methylation declines gradually with increasing age [26]; however, age-associated hypermethylation or hypomethylation of DNA has been documented at specific loci [25]. Because of the large volume of results, the 30 sites with the smallest P-values, that are hypermethylated and hypomethylated within promoters and gene bodies in this study are included in Supplementary Tables 1a to 1d. Concise information from those is shown for comparison purposes in the appropriate tabular and text presentation of PNS results. Changes in DNA methylation induced by age in the Control group were detected across the entire bovine genome (Supplementary Figure 1).
Table 1.
Distribution of dmCpG sites (P < 0.01) across genomic regions
| |
|
|
Number of |
Hypomethylatedc |
Hypermethylatedd |
||
|---|---|---|---|---|---|---|---|
| Comparison/location | Numberb | Proportion | affiliated genes | Number | Proportion | Number | Proportion |
| Control year 5 – day 28 | |||||||
| Total | 14,386 | 1 | 2,164 | 8,454 | 1 | 5,932 | 1 |
| Introna | 3,002 | 0.21 | 1,045 | 1,840 | 0.22 | 1,162 | 0.20 |
| Exona | 1,296 | 0.09 | 639 | 256 | 0.03 | 1,040 | 0.17 |
| Promoter | 1,446 | 0.1 | 423 | 169 | 0.02 | 1,277 | 0.21 |
| Intergenic | 9,119 | 0.63 | 6,182 | 0.73 | 2,937 | 0.49 | |
| PNS year 5 – day 28 | |||||||
| Total | 13,378 | 1 | 2,823 | 6,447 | 1 | 6,931 | 1 |
| Introna | 2,976 | 0.22 | 1,545 | 1,596 | 0.25 | 1,380 | 0.20 |
| Exona | 1,617 | 0.12 | 924 | 298 | 0.05 | 1,319 | 0.19 |
| Promoter | 1,901 | 0.14 | 1,46 | 145 | 0.02 | 1,756 | 0.25 |
| Intergenic | 7,583 | 0.57 | 4,402 | 0.68 | 3,181 | 0.46 | |
| PNS year 5 – Control year 5 | |||||||
| Total | 2,637 | 1 | 182 | 930 | 1 | 1,707 | 1 |
| Introna | 86 | 0.03 | 77 | 34 | 0.04 | 52 | 0.03 |
| Exona | 87 | 0.03 | 73 | 16 | 0.02 | 71 | 0.04 |
| Promoter | 111 | 0.04 | 87 | 32 | 0.03 | 79 | 0.05 |
| Intergenic | 2,410 | 0.91 | 859 | 0.92 | 1,551 | 0.91 | |
aIntrons and exons together were considered to be the gene body region.
bDue to coannotation of sites, the sum of number of CpG sites differs from the totals slightly.
cDecrease in DNA methylation in females at 5 years of age relative to 28 days of age or of PNS relative to Control females.
dIncrease in DNA methylation in females at 5 years of age relative to 28 days of age or of PNS relative to Control females.
Methylation at 28 days and 5 years of age in PNS cows
There were 13,378 CpG sites identified as differentially methylated (P < 0.01) in DNA from PNS cows at 5 years of age compared to their DNA at 28 days of age with a slight tendency towards an increase in methylation (52% of the total dmCpG sites were hypermethylated). Most dmCpG sites were detected in intergenic regions, and the remaining dmCpG sites were distributed across introns, promoters, and exons with lower percentages in PNS. The distribution of dmCpG sites across the entire bovine genome (Figure 1) may suggest methylation events that are part of the dynamic regulation of CpG sites as a part of normal development.
Figure 1.
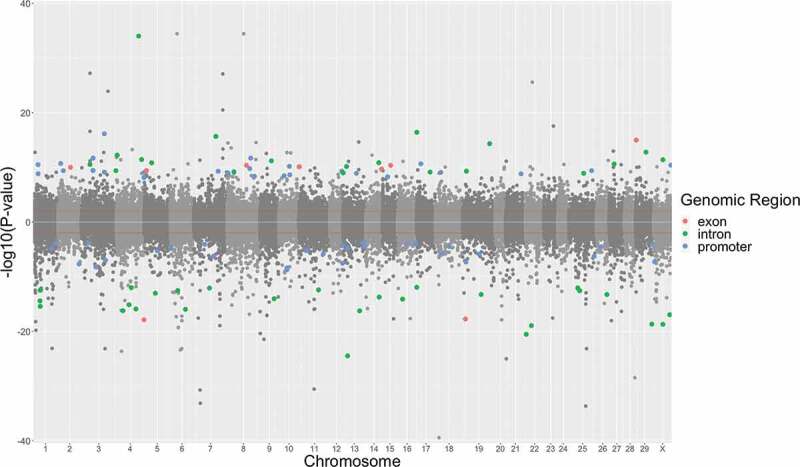
Differential methylation in promoter regions of PNS cows
There were 1,901 differentially methylated CpG sites (P < 0.01) located in promoter regions of DNA from cows at 5 years of age compared to their DNA at 28 days of age (Table 1). The 30 hypermethylated sites in promoter regions with the smallest P-values are listed in Table 2 along with the genes with which they are affiliated. Only two hypermethylated CpG sites were common in PNS and Control females at 5 years of age (Table 2). However, the majority of the genes (20 of 30) that had hypermethylated CpG sites within their promoters were the same in PNS and Control 5-yr-old cows; these may be downregulated (assuming that hypermethylation is suppressive [27]) as a normal response to ageing. Other (that is, not in the top 30 sites) hypermethylated CpG sites (P < 0.01) located in promoter regions of 5-yr-old PNS and Control cows are presented in Supplementary Tables 2a and 2b, respectively. Among those in Table 2, a single gene had some documented connection with stress in animals. The potassium voltage-gated channel subfamily C member 1 (KCNC1) gene had eight hypermethylated CpG sites (P < 0.01) in 5-yr-old PNS cows, but was not differentially methylated in 5-yr-old Control cows. The KCNC1 protein modulates potassium ion transport based on the voltage difference and is expressed in GABAergic interneurons [28]. In the offspring of pregnant mice exposed to valproic acid, which is a drug confirmed to induce seven-fold greater occurrence of autism spectrum disorders or related symptoms [29], brain expression levels of Kcnc1 were reduced by approximately 40% [30].
Table 2.
Top 30 hypermethylated CpG sites (P < 7.02E–09) located within promoter regions of genes in PNS cows at 5 years of age compared with 28 days of age
| |
|
|
|
Additional sitesd |
|
|---|---|---|---|---|---|
| BTAa | Mb | Differenceb | Genec | Control | PNS |
| 3 | 85.2 | 0.62 | Nuclear factor I A (NFIA) | 2 | 8 |
| 29 | 6.0 | 0.24 | Folate hydrolase 1B (FOLH1B) | 5 | 13 |
| 4 | 7.6 | 0.29 | Uridine phosphorylase 1 (UPP1) | 0 | 3 |
| 8 | 77.3 | 0.51 | Ciliary neurotrophic factor receptor (CNTFR) | 1 | 0 |
| 3 | 44.8 | 0.24 | Sorting nexin 7 (SNX7) | 3 | 2 |
| 2 | 20.9 | 0.27 | Homeobox D13 (HOXD13) | 16 | 2 |
| 17 | 45.2 | 0.38 | Zinc finger protein 891 (ZNF891) | 0 | 2 |
| 1 | 40.6 | 0.43 | EPH receptor A6 (EPHA6) | 19 | 20 |
| X | 145.4 | 0.25 | Anosmin 1 (ANOS1) | 0 | 3 |
| 10 | 57.7 | 0.21 | One cut homeobox 1 (ONECUT1) | 1 | 1 |
| 11 | 1.2 | 0.20 | BCL2 like 11 (BCL2L11) | 2 | 1 |
| 8 | 75.1 | 0.25 | PNMA family member 2 (PNMA2) | 0 | 1 |
| 14 | 79.0 | 0.28 | ATPase H+ transporting V0 subunit D2 (ATP6V0D2) | 0 | 1 |
| 3 | 44.8 | 0.20 | Sorting nexin 7 (SNX7) | 3 | 2 |
| 25 | 38.1 | 0.24 | Neuronal pentraxin 2 (NPTX2) | 5 | 0 |
| 2 | 41.5 | 0.33 | Potassium voltage-gated channel subfamily J member 3 (KCNJ3) | 2 | 3 |
| 12 | 90.0 | 0.26 | SRY-box 1 (SOX1) | 4 | 3 |
| 7 | 59.5 | 0.18 | POU class 4 homeobox 3 (POU4F3) | 2 | 0 |
| 3 | 85.2 | 0.48 | Nuclear factor I A (NFIA) | 2 | 8 |
| 5 | 1.9 | 0.30 | Thyrotropin releasing hormone degrading enzyme (TRHDE) | 2 | 8 |
| 18 | 13.2 | 0.21 | Junctophilin 3 (JPH3) | 0 | 1 |
| 8 | 5.8 | 0.39 | Heart and neural crest derivatives expressed 2 (HAND2) | 3 | 8 |
| 13 | 55.1 | 0.27 | Neurotensin receptor 1 (NTSR1) | 1 | 1 |
| 1 | 40.6 | 0.38 | EPH receptor A6 (EPHA6) | 19 | 20 |
| 21 | 66.3 | 0.13 | HHIP like 1 (HHIPL1) | 0 | 2 |
| 10 | 59.1 | 0.25 | Gliomedin (GLDN) | 0 | 1 |
| 10 | 21.4 | 0.37 | BCL2 like 2 (BCL2L2) | 4 | 0 |
| 8 | 101.8 | 0.19 | Sushi, von willebrand factor type A, EGF and pentraxin domain containing 1 (SVEP1) | 0 | 8 |
| 15 | 35.3 | 0.23 | Potassium voltage-gated channel subfamily C member 1 (KCNC1) | 0 | 7 |
| 4 | 120.5 | 0.24 | Vasoactive intestinal peptide receptor 2 (VIPR2) | 4 | 0 |
aBTA: Bos taurus autosome. X indicates Bos taurus X chromosome.
bMethylation difference was calculated by subtracting the mean methylation ratio in PNS at 28 days of age from the mean methylation ratio in PNS at 5 years of age.
cGenes in bold have the same CpG site differentially hypermethylated in Control (P < 0.01) as in PNS.
dTotal number of CpG sites differentially hypermethylated (P < 0.01; methylation ratio difference > 0.1) in the same gene’s promoter in Control cows, and additional sites in the promoter of the same gene in PNS, respectively.
There were 145 hypomethylated CpG sites detected in promoter regions of 5-yr-old PNS cows compared to their DNA at 28 days of age. The 30 hypomethylated CpG sites with the smallest P-values are shown in Table 3. There were 4 hypomethylated CpG sites common to 5-yr-old Control and PNS cows. Additional hypomethylated CpG sites (P < 0.01) within the promoter region of listed genes (Table 3) in Control cows as well as PNS cows show the differences in methylation patterns (Supplementary Tables 2c and 2d); while 23 of those listed genes had their promoter regions hypomethylated only in the 5-yr-old PNS group, the promoter regions of 6 genes were hypomethylated commonly. One hypomethylated CpG site was located within the promoter region of myelin-associated glycoprotein (MAG), which may have a role in mammalian response to stress. Schraut et al. [31] reported elevated expression of MAG in hippocampal DNA extracted from mice exposed to a restraint stress paradigm of prenatal stress. The MAG gene had 2 hypomethylated CpG sites in Control cows (one of those was common in Control and PNS cows); however, the common CpG site had a smaller P-value in PNS cows, which may indicate increased expression of this gene relative to Control cows.
Table 3.
Top 30 hypomethylated CpG sites (P < 1.76E–04) located within promoter regions of genes in PNS cows at 5 years of age compared with 28 days of age
| |
|
|
|
Additional sitesd |
|
|---|---|---|---|---|---|
| BTAa | Mb | Differenceb | Genec | Control | PNS |
| 10 | 41.9 | −0.24 | Melanoma-associated antigen 10-like (LOC618806) | 11 | 4 |
| 10 | 41.9 | −0.30 | Melanoma-associated antigen 10-like (LOC618806) | 11 | 4 |
| 3 | 68.1 | −0.46 | ST6 N-acetylgalactosaminide alpha-2,6-sialyltransferase 5 (ST6GALNAC5) | 1 | 0 |
| 2 | 133.4 | −0.34 | Phospholipase A2 group IIE (PLA2G2E) | 0 | 0 |
| 12 | 90.7 | −0.26 | Transcription factor Dp-1 (TFDP1) | 0 | 1 |
| 19 | 12.8 | −0.27 | Chromosome 19 C17ORF64 homolog (C19H17ORF64) | 0 | 0 |
| X | 14.1 | −0.26 | Zinc finger protein 280 C (ZNF280C) | 2 | 0 |
| 3 | 92.3 | −0.43 | acyl-CoA thioesterase 11 (ACOT11) | 0 | 0 |
| 25 | 41.4 | −0.18 | Kinesin-like protein (F1N4G9) | 0 | 0 |
| 7 | 45.5 | −0.18 | polo like kinase 5 (PLK5) | 0 | 0 |
| 19 | 39.2 | −0.40 | Proline rich 15 like (PRR15L) | 0 | 0 |
| 18 | 14.3 | −0.39 | acyl-CoA synthetase family member 3 (ACSF3) | 0 | 0 |
| 11 | 101.6 | −0.43 | Proline rich coiled-coil 2B (PRRC2B) | 0 | 0 |
| 5 | 91.8 | −0.32 | Phospholipase C zeta 1 (PLCZ1) | 0 | 0 |
| 5 | 91.8 | −0.32 | Capping actin protein of muscle Z-line subunit alpha 3 (CAPZA3) | 0 | 0 |
| 11 | 38.2 | −0.42 | Protein phosphatase 4 regulatory subunit 3B (PPP4R3B) | 1 | 0 |
| 1 | 143.6 | −0.30 | PR/SET domain 15 (PRDM15) | 0 | 1 |
| 13 | 30.2 | −0.29 | Non-coding RNA: RF00026 | 0 | 0 |
| 6 | 5.5 | −0.12 | Mitotic arrest deficient 2 like 1 (MAD2L1) | 1 | 0 |
| 26 | 22.9 | −0.11 | Nuclear factor kappa B subunit 2 (NFKB2) | 0 | 0 |
| 18 | 46.2 | −0.22 | Myelin-associated glycoprotein (MAG) | 2 | 0 |
| 13 | 79.4 | −0.29 | RIPOR family member 3 (RIPOR3) | 0 | 0 |
| 12 | 90.7 | −0.38 | Transcription factor Dp-1 (TFDP1) | 0 | 1 |
| 29 | 50.3 | −0.36 | Troponin I2, fast skeletal type (TNNI2) | 0 | 0 |
| 7 | 18.3 | −0.29 | Zinc finger protein 414 (ZNF414) | 0 | 0 |
| 16 | 49.7 | −0.25 | Leiomodin 1 (LMOD1) | 0 | 0 |
| 16 | 77.5 | −0.22 | CD46 molecule (CD46) | 0 | 0 |
| 1 | 147.4 | −0.35 | Collagen type VI alpha 1 chain (COL6A1) | 0 | 1 |
| 3 | 20.2 | −0.38 | Extracellular matrix protein 1 (ECM1) | 0 | 1 |
| 13 | 75.4 | −0.25 | Zinc finger SWIM-type containing 3 (ZSWIM3) | 0 | 0 |
aBTA: Bos taurus autosome. X indicates Bos taurus X chromosome.
bMethylation difference was calculated by subtracting the mean methylation ratio in PNS at 28 days of age from the mean methylation ratio in PNS at 5 years of age.
cGenes in bold have the same CpG site differentially hypomethylated in Control (P < 0.01) as in PNS.
dTotal number of CpG sites differentially hypomethylated (P < 0.01; methylation ratio difference > 0.1) in the same gene’s promoter in Control cows, and additional sites in the promoter of the same gene in PNS, respectively.
Differential methylation in gene body regions of PNS cows
As many as 4,593 CpG sites located within gene body regions were differentially methylated in 5-yr-old PNS cows, more frequently hypermethylated with 2,699 sites (Table 1). The 30 hypermethylated CpG sites with the smallest P-values are presented in Table 4. Two of those genes appear to have a stress-response role. Dipeptidyl peptidase like 6 (DPP6) encodes a subunit of voltage-gated potassium channel subfamily D member 2 that modulates neuronal excitability in the brain [32]. Associations have been shown between DPP6 gene variants and neurodevelopmental disorders, including autism spectrum disorder [33,34]. Three hypermethylated CpG sites within DPP6 were detected in both PNS and Control 5-yr-old cows. However, hypermethylation of these three sites was stronger in PNS as indicated by magnitudes of P-value and methylation differences (Supplementary Tables 3a and 3b). The number of hypermethylated sites in the homeobox protein lim-1 (LHX1) gene body in PNS (n = 7) is noteworthy compared to Control (n = 0). The LHX1 gene is integral for differentiation of GABAergic interneurons through modulating paired box gene 2 expression [35], and in the GABAergic inhibitory-neurotransmitter programming for appropriate development of interneurons [36].
Table 4.
Top 30 hypermethylated CpG sites (P < 1.19E–09) located within gene body regions in PNS cows at 5 years of age compared with 28 days of age
| BTAa | Mb | Differenceb | Genec |
Additional sitesd |
|
|---|---|---|---|---|---|
| Control | PNS | ||||
| 4 | 103.7 | 0.28 | ubinuclein 2 (UBN2) | 31 | 14 |
| 17 | 0.6 | 0.14 | Carboxypeptidase E (CPE) | 0 | 1 |
| 7 | 51.3 | 0.18 | GDNF family receptor alpha 3 (GFRA3) | 4 | 2 |
| 29 | 6.0 | 0.24 | Folate hydrolase 1B (FOLH1B) | 6 | 14 |
| 19 | 55.9 | 0.36 | Cytoglobin (CYGB) | 3 | 2 |
| 29 | 44.4 | 0.40 | Latent transforming growth factor beta binding protein 3 (LTBP3) | 7 | 11 |
| 4 | 7.6 | 0.29 | Uridine phosphorylase 1 (UPP1) | 1 | 3 |
| 3 | 44.8 | 0.24 | Sorting nexin 7 (SNX7) | 3 | 1 |
| 4 | 117.3 | 0.29 | Dipeptidyl peptidase like 6 (DPP6) | 3 | 2 |
| X | 83.2 | 0.15 | Phosphorylase kinase regulatory subunit alpha 1 (PHKA1) | 13 | 6 |
| 9 | 70.3 | 0.39 | A-kinase anchoring protein 7 (AKAP7) | 0 | 0 |
| 14 | 47.2 | 0.24 | MAL, T cell differentiation protein 2 (gene/pseudogene) (MAL2) | 2 | 0 |
| 5 | 54.1 | 0.19 | Solute carrier family 16 member 7 (SLC16A7) | 6 | 2 |
| 27 | 26.4 | 0.23 | WRN RecQ like helicase (WRN) | 8 | 9 |
| 3 | 24.2 | 0.27 | T-box 15 (TBX15) | 14 | 15 |
| X | 145.4 | 0.24 | Anosmin 1 (ANOS1) | 0 | 4 |
| 8 | 71.7 | 0.23 | NK2 homeobox 6 (NKX2-6) | 7 | 6 |
| 15 | 53.6 | 0.38 | Rho guanine nucleotide exchange factor 17 (ARHGEF17) | 0 | 0 |
| 13 | 24.5 | 0.21 | Pancreas associated transcription factor 1A (PTF1A) | 4 | 4 |
| 11 | 1.2 | 0.20 | BCL2 like 11 (BCL2L11) | 1 | 2 |
| 2 | 107.6 | 0.38 | WNT family member 10A (WNT10A) | 3 | 3 |
| 14 | 79.0 | 0.28 | ATPase H+ transporting V0 subunit D2 (ATP6V0D2) | 0 | 1 |
| 3 | 44.8 | 0.20 | Sorting nexin 7 (SNX7) | 3 | 1 |
| 5 | 26.2 | 0.40 | Homeobox C9 (HOXC9) | 0 | 1 |
| 3 | 121.1 | 0.34 | THAP domain containing 4 (THAP4) | 3 | 2 |
| 19 | 13.6 | 0.52 | LIM homeobox 1 (LHX1) | 0 | 6 |
| 8 | 30.0 | 0.53 | Nuclear factor I B (NFIB) | 0 | 1 |
| 17 | 71.7 | 0.60 | Pescadillo ribosomal biogenesis factor 1 (PES1) | 0 | 2 |
| 13 | 2.4 | 0.30 | Lysosomal associated membrane protein family member 5 (LAMP5) | 0 | 0 |
| 25 | 28.9 | 0.20 | Calneuron 1 (CALN1) | 1 | 3 |
aBTA: Bos taurus autosome. X indicates Bos taurus X chromosome. DNA methylation in bolded chromosomes was located within exons, otherwise within introns.
bMethylation difference was calculated by subtracting the mean methylation ratio in PNS at 28 days of age from the mean methylation ratio in PNS at 5 years of age.
cGenes in bold have the same CpG site differentially hypermethylated in Control (P < 0.01) as in PNS.
dTotal number of CpG sites differentially hypermethylated (P < 0.01; methylation ratio difference > 0.1) in the same gene’s gene body in Control cows, and additional sites in the gene body of the same gene in PNS, respectively.
There were 1,894 hypomethylated CpG sites detected within gene body regions. The most significant 30 hypomethylated CpG sites are listed in Table 5. Of those, 23 were hypomethylated in both 5-yr-old PNS and Control cows. The genes in Table 5 have many other hypomethylated CpG sites (P < 0.01 and methylation ratio difference of at least 0.1) that are listed in Supplementary Tables 3c and 3d. A hypomethylated CpG gene body site was located in the inner mitochondrial membrane peptidase subunit 2 (IMMP2L) gene on Bos taurus autosome (BTA) 4, which may have neurological roles related to stress. This gene has been reported to be associated with neurodevelopmental disorders including autism spectrum disorder [37] and schizophrenia [38]. The number of hypomethylated sites in the 5-yr-old PNS and Control cows (9 and 14, respectively) may suggest decreased IMMP2L gene expression associated with ageing in cattle.
Table 5.
Top 30 hypomethylated CpG sites (P < 1.17E–12) located within gene body regions in PNS cows at 5 years of age compared with 28 days of age
| BTAa | Mb | Differenceb | Genec |
Additional sitesd |
|
|---|---|---|---|---|---|
| Control | PNS | ||||
| 13 | 31.6 | −0.24 | Cubilin (CUBN) | 16 | 6 |
| 22 | 2.9 | −0.15 | Zinc finger CW-type and PWWP domain containing 2 (ZCWPW2) | 36 | 22 |
| 22 | 37.9 | −0.17 | Synaptoporin (SYNPR) | 27 | 7 |
| X | 83.2 | −0.21 | Phosphorylase kinase regulatory subunit alpha 1 (PHKA1) | 46 | 27 |
| 29 | 50.3 | −0.56 | Lymphocyte specific protein 1 (LSP1) | 0 | 1 |
| 4 | 120.6 | −0.17 | Ubiquitin carboxyl-terminal hydrolase 17-like protein 6 (LOC112446467) | 2 | 1 |
| 19 | 11.1 | −0.54 | Tubulin delta 1 (TUBD1) | 0 | 0 |
| X | 135.8 | −0.20 | Motile sperm domain containing 2 (MOSPD2) | 6 | 4 |
| 13 | 63.8 | −0.18 | Charged multivesicular body protein 4B (CHMP4B) | 5 | 1 |
| 4 | 30.6 | −0.16 | Dynein heavy chain 11, axonemal (LOC104968411) | 8 | 0 |
| 6 | 89.2 | −0.17 | ADAM metallopeptidase with thrombospondin type 1 motif 3 (ADAMTS3) | 48 | 23 |
| 4 | 95.1 | −0.13 | Coatomer protein complex subunit gamma 2 (COPG2) | 6 | 3 |
| 1 | 57.5 | −0.15 | Solute carrier family 9 member C1 (SLC9C1) | 66 | 36 |
| 4 | 57.4 | −0.17 | Inner mitochondrial membrane peptidase subunit 2 (IMMP2L) | 14 | 8 |
| 1 | 57.4 | −0.18 | Solute carrier family 9 member C1 (SLC9C1) | 66 | 36 |
| s | 36.6 | −0.18 | Regulator of G protein signalling 7 (RGS7) | 11 | 9 |
| 9 | 88.3 | −0.32 | Retinoic acid early transcript 1 L (RAET1G, ULBP21) | 4 | 0 |
| 14 | 61.1 | −0.55 | Zinc finger protein, FOG family member 2 (ZFPM2) | 0 | 2 |
| 26 | 47.1 | −0.17 | Dedicator of cytokinesis 1 (DOCK1) | 12 | 8 |
| 19 | 41.2 | −0.18 | Retinoic acid receptor alpha (RARA) | 7 | 2 |
| 5 | 75.9 | −0.52 | Potassium channel tetramerization domain containing 17 (KCTD17) | 0 | 1 |
| 25 | 19.0 | −0.24 | Dynein axonemal heavy chain 3 (DNAH3) | 113 | 67 |
| 6 | 27.0 | −0.15 | Methionyl aminopeptidase 1 (METAP1) | 5 | 1 |
| 1 | 57.5 | −0.15 | Solute carrier family 9 member C1 (SLC9C1) | 66 | 36 |
| 11 | 94.5 | −0.50 | Crumbs cell polarity complex component 2 (CRB2) | 1 | 0 |
| 1 | 57.5 | −0.14 | Solute carrier family 9 member C1 (SLC9C1) | 66 | 36 |
| 25 | 18.9 | −0.24 | Dynein axonemal heavy chain 3 (DNAH3) | 113 | 67 |
| 7 | 32.7 | −0.43 | Synuclein alpha interacting protein (SNCAIP) | 0 | 0 |
| 4 | 67.5 | −0.17 | Carboxypeptidase vitellogenic like (CPVL) | 3 | 2 |
| 17 | 0.6 | −0.19 | Carboxypeptidase E (CPE) | 6 | 2 |
aBTA: Bos taurus autosome. X indicates Bos taurus X chromosome. DNA methylation in bolded chromosomes was located within exons, otherwise within introns.
bMethylation difference was calculated by subtracting the mean methylation ratio in PNS at 28 days of age from the mean methylation ratio in PNS at 5 years of age.
cGenes in bold have the same CpG site differentially hypomethylated in Control (P < 0.01) as in PNS.
dTotal number of CpG sites differentially hypomethylated (P < 0.01; methylation ratio difference > 0.1) in the same gene’s gene body in Control cows, and additional sites in the gene body of the same gene in PNS, respectively.
Differential methylation and age: correspondence across treatments
Individual CpG sites that were differentially methylated in 5-yr-old Control and PNS cows were compared based on their chromosomal coordinates (Figure 2). There were over three times as many hypomethylated CpG sites that overlapped the two treatments than hypermethylated CpG sites. While the overlap of dmCpG sites can be attributed to the changes associated with ageing in both PNS and Control, dmCpG sites unique to PNS suggest that the ageing changes in dmCpG sites may be influenced by the stress imposed while these cows themselves were in utero. The majority of dmCpG sites were located in intergenic regions (Table 1); these may be benign or not influential on gene expression [39]. However, they may be indicative of genomic regions that are regulatory and(or) have an enhancement or inhibition effect on expression of nearby (or far away) genes [40,41].
Figure 2.
Differential methylation of prenatal stress vs. controls at 5 years of age
The distribution of CpG sites tested across the entire genome is shown in Figure 3, and significant CpG sites that were differentially methylated in PNS cows relative to Control cows at 5 years of age are depicted above the P-value threshold (unadjusted P < 0.01). Few dmCpG sites met significance criteria. Because of the limited number of differences, the results that follow are based upon unadjusted P-values. There were 2,637 dmCpG sites (Table 1) with a notable tendency towards an increase in methylation in PNS cows at 5 years of age (65% of CpG sites hypermethylated) compared to Control at 5 years of age.
Figure 3.
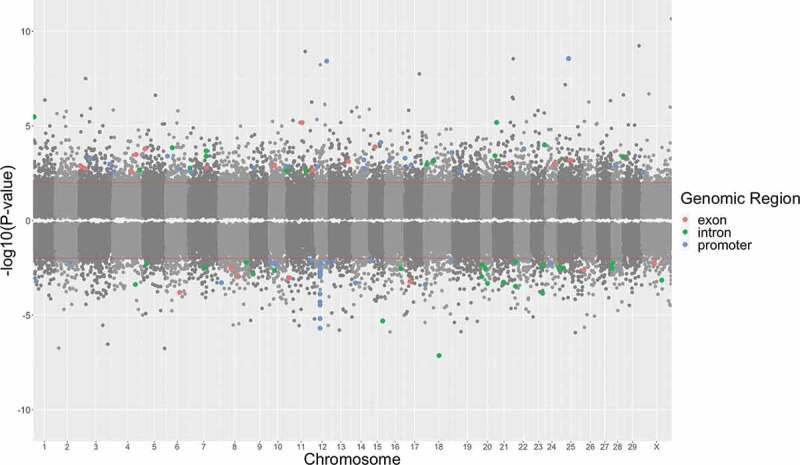
Promoter regions
A total of 111 dmCpG sites were located within promoter regions of genes. Hypermethylation of promoter regions results in heterochromatin formation that prevents transcription factors from binding promoter regions of genes and ultimately represses gene expression [42]. Genes with promoter regions that were hypermethylated in PNS compared to Control may be downregulated. There were 79 hypermethylated CpG sites within promoter regions. The 30 hypermethylated CpG sites with the lowest P-values within promoter regions are listed in Table 6. Several of those genes may have neural or stress-response involvement. One of those CpG sites was located within the promoter region of the Rho guanine nucleotide exchange factor 7 (ARHGEF7) gene on BTA 12. This gene produces a regulator of the number of γ-aminobutyric acid A [GABA(A)] receptors at the neuronal surface in rats. Knockdown of ARHGEF7 results in a diminished level of GABA(A) receptors [43]. The strength of inhibitory synaptic transmission depends on the surface levels of GABA(A) receptors at synaptic sites [44]. Interrupted inhibition resulting from a decreased number of GABA(A) receptors can disrupt the balance between excitatory and inhibitory neurotransmission (E/I balance) in the brain [43]. Changes in the E/I balance have been associated with neuropsychiatric diseases, including attention deficit hyperactivity disorder [45] and bipolar disorder [46]. The sorting nexin 7 (SNX7) gene harbours a dmCpG site within the promoter region. In humans, an allele related to reduced expression of SNX7 has been identified as a potential for increased risk for vulnerability for bipolar disorder and psychotic symptoms by means of elevated brain kynurenic acid generation [47]. The gene solute carrier family 30 member 10 (SLC30A10) is essential for maintaining Mn levels. Elevated levels of Mn induce neuropathologic defects [48]. Loss-of-function mutations in SLC30A10 were associated with neurodegenerative symptoms due to Mn accumulation in the brain and blood [49]. Aschner et al. [50] postulated that impaired norepinephrine (NE) neurotransmitter systems [51] altered GABA transporter and receptor expression and alleviated extracellular GABA levels [52] due to excessive Mn deposition in the brain. Adrenergic receptor alpha 2B (ADRA2B) had a hypermethylated CpG site within its promoter region; this gene encodes the α2B adrenergic receptor that modulates neurotransmitter release including NE from the noradrenergic neurons [53]. A deletion variant of ADRA2B was associated with elevated emotional memory [54]. Zoladz et al. [55] reported the effects of stress exposed just before learning on long-term memory in ADRA2B deletion carriers, suggesting increased risk for traumatic memory formation. Liberzon et al. [56] reported an association between a polymorphism in ADRA2B and increased vulnerability for development of post-traumatic stress disorder.
Table 6.
Top 30 hypermethylated CpG sites (P < 2.82E–03) located within promoter regions of genes in PNS cows compared with Control cows at 5 years of age
| BTAa | Mb | Differenceb | Gene |
|---|---|---|---|
| 25 | 20.2 | 11.58 | Cerebellar degeneration related protein 2 (CDR2) |
| 12 | 89.5 | 14.61 | Rho guanine nucleotide exchange factor 7 (ARHGEF7) |
| 21 | 10.8 | 15.93 | Nuclear receptor subfamily 2 group F member 2 (NR2F2) |
| 15 | 66.9 | 17.42 | Four-jointed box kinase 1 (FJX1) |
| 15 | 34.3 | 15.72 | CXADR like membrane protein (CLMP) |
| 22 | 51.5 | 30.43 | Laminin subunit beta 2 (LAMB2) |
| 5 | 26.2 | 21.41 | Homeobox C12 (HOXC12) |
| 18 | 64.9 | 12.84 | Zinc finger protein 772 (ZNF772) |
| 4 | 101.8 | 15.80 | Pleiotrophin (PTN) |
| 6 | 8.9 | 14.73 | RNA gene (RF00001) |
| 17 | 1.5 | 18.60 | Tolloid like 1 (TLL1) |
| 3 | 44.8 | 10.73 | Sorting nexin 7 (SNX7) |
| 14 | 34.4 | 11.97 | Chromosome 8 open reading frame 34 (C14H8ORF34) |
| 16 | 24.1 | 11.24 | Solute carrier family 30 member 10 (SLC30A10) |
| 3 | 118.6 | 14.68 | Twist family BHLH transcription factor 2 (TWIST2) |
| 18 | 8.8 | 11.69 | M-phase phosphoprotein 6 (MPHOSPH6) |
| 17 | 54.4 | 14.85 | Rab interacting lysosomal protein like 1 (RILPL1) |
| 28 | 26.1 | 16.41 | Neurogenin 3 (NEUROG3) |
| 3 | 3.6 | 15.36 | Retinoid X receptor gamma (RXRG) |
| 11 | 2.2 | 14.29 | Adrenoceptor alpha 2B (ADRA2B) |
| 12 | 25.4 | 13.97 | Doublecortin like kinase 1 (DCLK1) |
| 7 | 48.7 | 11.05 | C-X-C motif chemokine ligand 14 (CXCL14) |
| 9 | 40.3 | 15.10 | Methyltransferase like 24 (METTL24) |
| 14 | 37.6 | 13.63 | Musculin (MSC) |
| 6 | 97.6 | 11.88 | Bone morphogenetic protein 3 (BMP3) |
| 4 | 76.7 | 14.89 | Insulin like growth factor binding protein 1 (IGFBP1) |
| 19 | 27.2 | 17.48 | Zinc finger MYND-type containing 15 (ZMYND15) |
| 4 | 6.0 | 13.43 | Von willebrand factor C domain containing 2 (VWC2) |
| 7 | 5.1 | 13.27 | Arrestin domain containing 2 (ARRDC2) |
| X | 7.2 | 22.37 | Glutamate ionotropic receptor AMPA type subunit (GRIA3) |
aBTA: Bos taurus autosome. X indicates Bos taurus X chromosome.
bMethylation difference was calculated by subtracting the mean methylation ratio in Control at 5 years of age from the mean methylation ratio in PNS at 5 years of age.
Hypomethylation of CpG sites located within promoter regions is often associated with activation of gene expression. Overall, 32 CpG sites (P < 0.01) located within promoter regions were hypomethylated in PNS compared to Control cows at 5 years of age. The 30 hypomethylated CpG sites with the lowest P-values within promoter regions along with the corresponding genes are listed in Table 7. A number of hypomethylated CpG sites (n = 13) were located within the promoter region of ubiquitin-specific peptidase 12 (USP12). There were 4 CpG sites in the promoter region (near BTA 12, 32.9 Mb) for this gene that were hypomethylated in lymphocytes of these PNS females at 28 days of age [11]. The differentially methylated CpG site in the promoter region of transmembrane protein 215 (TMEM15) was also hypomethylated in lymphocyte DNA of the PNS females at 28 days of age [11]. Aron et al. [57] showed that USP12 deubiquitinase suppresses neurodegeneration caused by the mutant huntingtin gene by inducing neuronal autophagy in Huntington’s disease. Ephrin receptor A4 (Epha4) regulates neurogenesis by modulating proliferation of hippocampal neurogenic precursors in the adult mouse brain [58].
Table 7.
Top 30 hypomethylated CpG sites (P < 9.03E–03) located within promoter regions of genes in PNS cows compared with Control cows at 5 years of age
| BTAa | Mb | Differenceb | Gene |
|---|---|---|---|
| 12 | 32.9 | −27.81 | Ubiquitin specific peptidase 12 (USP12) |
| 12 | 32.9 | −26.70 | Ubiquitin specific peptidase 12 (USP12) |
| 12 | 32.9 | −23.52 | Ubiquitin specific peptidase 12 (USP12) |
| 12 | 32.9 | −24.07 | Ubiquitin specific peptidase 12 (USP12) |
| 12 | 32.9 | −23.36 | Ubiquitin specific peptidase 12 (USP12) |
| 18 | 2.6 | −10.89 | Chymotrypsinogen B2 (CTRB2) |
| 14 | 2.3 | −21.66 | Zinc finger protein 485 (ZNF485) |
| 8 | 11.2 | −13.18 | Transmembrane protein 215 (TMEM215) |
| 1 | 8.8 | −16.85 | ADAM metallopeptidase with thrombospondin type 1 motif 5 (ADAMTS5) |
| 12 | 32.9 | −27.42 | Ubiquitin specific peptidase 12 (USP12) |
| 12 | 32.9 | −28.53 | Ubiquitin specific peptidase 12 (USP12) |
| 12 | 32.9 | −26.50 | Ubiquitin specific peptidase 12 (USP12) |
| 12 | 32.9 | −26.50 | Ubiquitin specific peptidase 12 (USP12) |
| 12 | 32.9 | −25.39 | Ubiquitin specific peptidase 12 (USP12) |
| 12 | 32.9 | −26.50 | Ubiquitin specific peptidase 12 (USP12) |
| 26 | 11.0 | −12.85 | Cholesterol 25-hydroxylase (CH25H) |
| 12 | 32.9 | −24.97 | Ubiquitin specific peptidase 12 (USP12) |
| 10 | 38.5 | −10.36 | Transmembrane protein 62; (TMEM62) |
| 24 | 32.6 | −24.09 | Impact RWD domain protein (IMPACT) |
| 12 | 32.9 | −25.39 | Ubiquitin specific peptidase 12 (USP12) |
| X | 56.9 | −22.06 | Ring finger protein 128 (RNF128) |
| 8 | 107.6 | −13.59 | Tripartite motif containing 32 (TRIM32) |
| 2 | 110.4 | −12.47 | EPH receptor A4 (EPHA4) |
| 12 | 90.0 | −18.99 | SRY-box 1 (SOX1) |
| 10 | 4.8 | −12.17 | Laeverin (LVRN) |
| 13 | 38.2 | −14.76 | Solute carrier family 6 member 9-like (LOC515755) |
| 15 | 17.4 | −12.90 | ELMO domain containing 1 (ELMOD1) |
| 11 | 104.9 | −14.75 | U6atac minor spliceosomal RNA (LOC112448934) |
| 18 | 64.9 | −12.45 | Zinc finger protein 772 (ZNF772) |
| 18 | 64.7 | −17.03 | Zinc finger protein 304 (ZNF304) |
aBTA: Bos taurus autosome. X indicates Bos taurus X chromosome.
bMethylation difference was calculated by subtracting the mean methylation ratio in Control at 5 years of age from the mean methylation ratio in PNS at 5 years of age.
Gene body regions
There were 173 differentially methylated CpG sites within gene body regions in PNS relative to Control cows. Those were almost equally divided between exons and introns. DNA methylation levels in transcribed gene regions have been positively correlated with gene expression levels [59]. Methylation within gene body regions has been positively correlated with transcription levels, possibly due to enhanced efficiency of transcription elongation by blocking alternate promoters and regulatory regions [60].
The 30 hypermethylated CpG sites with the lowest P-values within gene body regions in PNS cows are listed in Table 8 along with the corresponding genes. Genes having those CpG sites could potentially be upregulated [59]. At least two of those genes may have a role in neural activity or response to stress. A dmCpG site was detected in the gene body of solute carrier family 39 member 8 (SLC39A8), which encodes a Mn and Zn influx transporter. A site in this gene was differentially methylated in lymphocyte DNA of the PNS females at 28 days of age [11]. Mutations in SLC39A8 resulted in decreased Mn uptake, and ultimately caused severe Mn deficiency, resulting in neurological disorders [61]. Allelic variations in SLC39A8 caused inefficient Mn regulation and constituted neurodevelopmental consequences and increased risk for behavioural problems from attention deficit hyperactivity disorder in children [62]. Another significantly hypermethylated CpG site was detected in the gene body of DPP6, which had hypermethylated CpG sites detected in PNS and Control cows at 5 years of age compared with 28 days of age [11]. The DPP6 gene, encoding an important component of A-type potassium (K) channels in the brain [63], has been implicated in neurodevelopmental disorders. Lin et al. [34] demonstrated that Dpp6 deficiency resulted in impaired behaviours regarding memory, learning, and recognition in mice.
Table 8.
Top 30 hypermethylated CpG sites (lowest P < 2.42E–03) located within gene body regions in PNS cows compared with Control cows at 5 years of age
| BTAa | Mb | Differenceb | Gene |
|---|---|---|---|
| 25 | 20.2 | 11.58 | Transmembrane protein 180 (LOC101905757) |
| 1 | 2.9 | 11.32 | Hormonally up-regulated neu-associated kinase (HUNK) |
| 21 | 10.8 | 15.93 | Nuclear receptor subfamily 2 group F member 2 (NR2F2) |
| 11 | 83.7 | 14.94 | LRAT domain containing 1 (LRATD1) |
| 23 | 51.6 | 11.87 | Forkhead box F2 (FOXF2) |
| 15 | 34.3 | 15.72 | CXADR like membrane protein (CLMP) |
| 6 | 23.8 | 14.77 | Solute carrier family 39 member 8 (SLC39A8) |
| 5 | 26.2 | 21.41 | Homeobox C12 (HOXC12) |
| 7 | 48.1 | 21.11 | Paired like homeodomain 1 (PITX1) |
| 18 | 64.9 | 12.84 | Zinc finger protein 772 (ZNF772) |
| 4 | 101.8 | 15.80 | Pleiotrophin (PTN) |
| 21 | 3.0 | 23.12 | ATPase phospholipid transporting 10A (Putative) (ATP10A) |
| 7 | 48.1 | 16.37 | Paired like homeodomain 1 (PITX1) |
| 28 | 45.4 | 15.89 | C-X-C motif chemokine ligand 12 (CXCL12) |
| 25 | 27.8 | 16.24 | Solute carrier family 5 member 2 (SLC5A2) |
| 18 | 19.7 | 13.90 | Spalt like transcription factor 1 (SALL1) |
| 13 | 73.3 | 29.53 | Junctophilin 2 (JPH2) |
| 24 | 46.0 | 13.84 | Sialic acid binding Ig like lectin 15 (SIGLEC15) |
| 21 | 61.2 | 12.40 | Goosecoid homeobox (GSC) |
| 18 | 8.8 | 11.69 | M-phase phosphoprotein 6 (MPHOSPH6) |
| 10 | 32.9 | 13.70 | Meis homeobox 2 (MEIS2) |
| 3 | 3.6 | 15.36 | Retinoid X receptor gamma (RXRG) |
| 7 | 48.7 | 11.05 | C-X-C motif chemokine ligand 14 (CXCL14) |
| 7 | 4.2 | 14.99 | Ceramide synthase 1 (CERS1) |
| 3 | 16.8 | 18.36 | Natriuretic peptide receptor 1 (NPR1) |
| 4 | 117.3 | 22.39 | Dipeptidyl peptidase like 6 (DPP6) |
| 11 | 105.9 | 16.92 | Glutamate ionotropic receptor NMDA type subunit 1 (GRIN1) |
| 11 | 98.1 | 10.29 | GTPase activating Rap/RanGAP domain like 3 (GARNL3) |
| 10 | 103.3 | 21.83 | Tetratricopeptide repeat domain 7B (TTC7B) |
| 4 | 76.7 | 14.89 | Insulin-like growth factor binding protein 1 (IGFBP1) |
aBTA: Bos taurus autosome. DNA methylation in bolded chromosomes was located within exons, otherwise within introns.
bMethylation difference was calculated by subtracting the mean methylation ratio in Control at 5 years of age from the mean methylation ratio in PNS at 5 years of age.
There were 50 hypomethylated CpG sites detected within gene body regions. The 30 hypomethylated CpG sites with the lowest P-values that were located within gene body regions are listed in Table 9. A single dmCpG was detected in the transcriptional corepressor chromodomain Y like (CDYL); a gene body region CpG was hypomethylated in lymphocytes in females at 28 days of age [11]. Qi et al. [64] reported that Cdyl modulates branching of hippocampal neurons. Later, those same researchers showed that Cdyl controls neuronal migration by suppressing the expression of the Rho family of GTPases A in Cdyl knockout mice, and loss of Cdyl enhances excitability of cortical pyramidal neurons [65]. Expression of this gene product (CDYL) in the brain (prelimbic cortex) appears to be influential in depressive behaviours associated with stress in mice [66].
Table 9.
Top 30 hypomethylated CpG sites (P < 6.50E–03) located within gene body regions in PNS cows compared with Control cows at 5 years of age
| BTAa | Mb | Differenceb | Gene |
|---|---|---|---|
| 18 | 46.4 | −18.75 | ATPase H+/K+ transporting subunit alpha (ATP4A) |
| 15 | 78.4 | −10.13 | SPI-1 proto-oncogene (SPI1) |
| 23 | 49.5 | −15.36 | Chromodomain Y like (CDYL) |
| 6 | 91.1 | −13.77 | Amphiregulin (AREG) |
| 22 | 2.9 | −17.73 | Zinc finger CW-type and PWWP domain containing 2 (ZCWPW2) |
| 4 | 98.8 | −19.07 | Leucine rich repeats and guanylate kinase domain containing (LRGUK) |
| 20 | 41.0 | −17.88 | Natriuretic peptide receptor 3 (NPR3) |
| 21 | 34.8 | −11.25 | Coiled-coil domain containing 33 (CCDC33) |
| 17 | 47.1 | −45.21 | Adhesion G protein-coupled receptor D1 (ADGRD1) |
| X | 100.1 | −10.72 | Moesin (MSN) |
| 11 | 10.1 | −17.24 | Ladybird homeobox 2 (LBX2) |
| 20 | 2.1 | −11.14 | Lymphocyte cytosolic protein 2 (LCP2) |
| 8 | 71.7 | −11.97 | NK2 homeobox 6 (NKX2-6) |
| 9 | 26.4 | −17.24 | TPD52 like 1 (TPD52 L1) |
| 10 | 32.9 | −10.81 | Meis homeobox 2 (MEIS2) |
| 26 | 11.0 | −12.85 | Cholesterol 25-hydroxylase (CH25H) |
| 16 | 69.6 | −23.49 | Phospholipase A2 group IVA (PLA2G4A) |
| 20 | 23.5 | −11.91 | Solute carrier family 38 member 9 (SLC38A9) |
| 8 | 53.3 | −15.60 | Forkhead box B2 (FOXB2) |
| 25 | 4.0 | −17.88 | Periplakin (PPL) |
| 25 | 0.5 | −17.94 | RAB40C, member RAS oncogene family (RAB40C) |
| 28 | 1.3 | −38.23 | Polypeptide N-acetylgalactosaminyltransferase 2 (GALNT2) |
| 7 | 45.7 | −11.43 | One cut homeobox 3 (ONECUT3) |
| 23 | 49.2 | −41.33 | Phenylalanyl-TRNA synthetase 2, mitochondrial (FARS2) |
| 20 | 3.0 | −17.19 | RAN binding protein 17 (RANBP17) |
| 5 | 30.7 | −29.53 | Complement C1q like 4 (C1QL4) |
| X | 56.9 | −22.06 | Ring finger protein 128 (RNF128) |
| 28 | 7.0 | −11.04 | Solute carrier family 35 member F3 (SLC35F3) |
| 8 | 107.6 | −13.59 | Astrotactin 2 (ASTN2) |
| 22 | 2.9 | −14.56 | Zinc finger CW-type and PWWP domain containing 2 (ZCWPW2) |
aBTA: Bos taurus autosome. X indicates Bos taurus X chromosome. DNA methylation in bolded chromosomes was located within exons, otherwise within introns.
bMethylation difference was calculated by subtracting the mean methylation ratio in Control at 5 years of age from the mean methylation ratio in PNS at 5 years of age.
Pathway analysis
Age – control cows
There were 64 canonical pathways enriched in Control cows at 5 years of age relative to 28 days of age (Supplementary Table 4a); 44 of those were also identified in PNS cows. Genes with dmCpG in promoters were enriched in the pathways of two main categories: ‘Organismal/Cellular Growth, Proliferation and Development’ and ‘Neurotransmitters and Other Nervous System Signaling’. Of 19 enriched pathways related to ‘Organismal/Cellular Growth, Proliferation and Development’, 14 canonical pathways were enriched in PNS as well. This was exactly the case in terms of numbers (14 canonical pathways out of 19 enriched in PNS females) for ‘Neurotransmitters and Other Nervous System Signaling’.
Age – PNS cows
There were 77 canonical pathways enriched in PNS cows at 5 years of age in comparison to their own DNA at 28 days of age (Supplementary Table 4b). In PNS, the top ten altered canonical pathways were involved in ‘Organismal/Cellular Growth, Proliferation and Development’ and ‘Neurotransmitters and Other Nervous System Signaling’ which make up two main signalling pathway categories.
The majority of the canonical pathways (n = 20) were related to ‘Organismal/Cellular Growth, Proliferation and Development’. Since DNA methylation is a key method of regulation of gene expression throughout mammalian development and cell differentiation [67], detection of a number of pathways associated with this functional group is not surprising and suggests changes in developmental dynamics of DNA methylation. Four enriched pathways were common to PNS and Control cows and associated with stem cell differentiation and pluripotency, including the ‘Role of NANOG in Mammalian Embryonic Stem Cell Pluripotency’ and the ‘Human Embryonic Stem Cell Pluripotency’ pathways. Massart et al. [68] reported differential methylation and distinct genes and enriched pathways corresponding to age (birth and 2 years of age) in T lymphocytes of rhesus monkeys (that is, postnatal DNA continued to evolve as the monkeys progressed through weaning phase through adolescence). Littlejohn et al. [12] reported that these two pathways were enriched with genes differentially methylated in 28-d-old bull calves (several sets of half-siblings to the females evaluated in the present study) exposed to prenatal transportation stress as compared to control animals. Another enriched pathway was the ‘cAMP-Response Element Binding Protein (CREB)’ which is an intracellular protein, and plays a critical role in the regulation of expression of the dopaminergic neuron-related gene [69]. Gene expression analyses in humans suggested that the transcriptional potential of CREB might be regulated at the epigenetic level via methylation as well as phosphorylation [70,71]. Among the enriched pathways of the present study, the role of ‘Wnt/β-catenin Signaling’ in skeletal muscle fibrosis and neural development were reported in detail [72,73]. These signalling pathways might be related to a set of critical growth and developmental processes in offspring exposed to prenatal stress.
‘Neurotransmitters and Other Nervous System Signaling’ consisted of 17 canonical pathways including ‘Axonal Guidance Signaling’, ‘GABA Receptor Signaling’ and ‘Corticotropin Releasing Hormone Signaling’. Out of those, 14 canonical pathways were also altered in 5-yr-old Control females, but with distinct sets of methylated genes. The most significantly altered canonical pathway in PNS was ‘Axonal Guidance Signaling’ (P = 8.54E–14) with 55 differentially methylated genes, and in Control (P = 1.71E–6) with 32 differentially methylated genes. The different set of genes would imply changes in ‘Axonal Guidance Signaling’ induced by prenatal stress, which appears to be consistent with altered expression levels of genes associated with neuropathology and axonal guidance in the brain through epigenetic changes in rats exposed to stress in utero [74]. There were different ‘Neurotransmitters and Other Nervous System Signaling’ pathways enriched in PNS compared to Control cows, which suggest prenatal stress may induce impairments in nervous system development and function.
Treatment – PNS vs. control cows
Potential canonical pathways altered in PNS compared to Control cows at 5 years of age were analysed by using genes harbouring dmCpG sites within their promoter regions. There were 32 pathways enriched (P < 0.05); those pathways and genes affiliated with the dmCpG are presented in Supplementary Table 5. Altered canonical pathways were mainly clustered into 3 categories; ‘Neurotransmitters and Other Nervous System Signaling’, ‘Organismal/Cellular Growth and Development’, and ‘Immune Response’.
The majority of the altered pathways (n = 11) including ‘GABA Receptor Signaling’, ‘CREB Signaling in Neurons’, ‘Opioid Signaling Pathway’, ‘Endocannabinoid Neuronal Synapse Pathway’ and ‘Glutamate Receptor Signaling’, were involved in neurotransmitters and other nervous system signalling. Prenatal stress altered the methylation patterns of genes associated with ‘GABA Receptor Signaling’ in heifer calves (the same females in the present study) at 28 days of age [11]. In this signalling pathway, GABA is the main inhibitory neurotransmitter in the brain, and interruption of GABAergic inhibition can result in emotional imbalance, depression, and anxiety [75].
Pathway analysis indicated the importance of genes involved in ‘Organismal/Cellular Growth and Development’ as in ‘Netrin Signaling’, ‘BMP Signaling Pathway’, and ‘Corticotropin-Releasing Hormone (CRH) Signaling’. Corticotropin-releasing hormone modulates neuroendocrine system activity through adjusting the hypothalamic-pituitary-adrenal axis and plays an instrumental role in the adjustment of autonomic and behavioural adaptive response to stress [76]. Prenatal stress-induced differential methylation of the CRH signalling pathway in the hypothalamus of rats [77] and in lymphocytes of bull calves that were paternal half-siblings (not all by one sire) to the females in the present study [12]. The comparison of PNS and Control lymphocyte methylation of these females at 28 days of age [11] also highlighted differentially methylated genes associated with the CRH pathway. As such, potential changes in neurological function date back to early stages in life and may persist into adulthood.
Results suggest a possible effect of prenatal stress-induced methylation on the bovine immune system and thus animal health and welfare. Enriched immune response pathways included ‘FcγRIIB Signaling in B Lymphocytes’ that negatively regulates B cell activation, cellular proliferation and antibody secretion [78], and ‘PKCθ Signaling in T Lymphocytes’, which is an important mediator of mature T cell activation and IL-2 production [79]. Methylation status of DNA in T cells corresponded to the effect of prenatal stress in humans [80].
Conclusions
Genome-wide DNA methylation differences in lymphocytes of female Brahman cattle due to age and prenatal stress were substantial. Detected methylation differences highlighted biological pathways related to organismal and cellular development and growth and nervous system development and function. Diversity in the sets of genes altering the same pathways as well as different sets of pathways suggest some influence of stress in utero. Prenatally stressed cows developed unique methylation patterns compared to Control cows over the course of their initial 5 years of life, which suggests an interaction of ageing with prenatal stress. It was not surprising to observe similar changes in the enriched pathways of PNS and Control females that correspond with ageing. Prenatally stressed cows at 5 years of age compared to 28 days of age had differential DNA methylation of genes biologically relevant to neuropathology and behaviour and pathways related to development, nervous system development and function, and immune response. Results suggest that there are potential gene expression level changes induced by prenatal stress; these may in part account for phenotypic differences later in life. Methylation differences of lymphocyte DNA should be compared to those of DNA of different tissue lineages, especially those with confirmed physiological roles in response to stress.
Supplementary Material
Acknowledgments:
This work was supported by Texas A&M AgriLife Research, Western Regional project TEX03212, Hatch projects H-9022 and H-TEX09377 the TAMU One Health Initiative, and NIFA Award #2018-67015-28131.
Funding Statement
This work was supported by the USDA-NIFA [2018-67015-28131].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447(7143):425–432. PMID:17522676. [DOI] [PubMed] [Google Scholar]
- [2].Jaenisch R, Bird A.. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33:245–254. PMID:12610534. [DOI] [PubMed] [Google Scholar]
- [3].Reynolds RM, Labad J, Buss C, et al. Transmitting biological effects of stress in utero: implications for mother and offspring. Psychoneuroendocrinology. 2013;38:1843–1849. PMID:23810315. [DOI] [PubMed] [Google Scholar]
- [4].Glover V. Prenatal stress and its effects on the fetus and the child: possible underlying biological mechanisms. Adv Neurobiol. 2015;10:269–283. PMID:25287545. [DOI] [PubMed] [Google Scholar]
- [5].Weinstock M. The potential influence of maternal stress hormones on development and mental health of the offspring. Brain Behav Immun. 2005;19:296–308. PMID:15944068. [DOI] [PubMed] [Google Scholar]
- [6].Lay DC, Randel RD Jr, Friend TH, et al. Effects of prenatal stress on suckling calves. J Anim Sci. 1997;75:3143–3151. PMID:9419987. [DOI] [PubMed] [Google Scholar]
- [7].Tobi EW, Slieker RC, Stein AD, et al. Early gestation as the critical time-window for changes in the prenatal environment to affect the adult human blood methylome. Int J Epidemiol. 2015;44:1211–1223. PMID:25944819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Littlejohn BP, Price DM, Banta JP, et al. Prenatal transportation stress alters temperament and serum cortisol concentrations in suckling Brahman calves. J Anim Sci. 2016;94:602–609. PMID:27065130. [DOI] [PubMed] [Google Scholar]
- [9].Price DM, Lewis AW, Neuendorff DA, et al. Physiological and metabolic responses of gestating Brahman cows to repeated transportation. J Anim Sci. 2015;93:737–745. PMID:26020755. [DOI] [PubMed] [Google Scholar]
- [10].Littlejohn BP, Price DM, Neuendorff DA, et al. Influence of prenatal transportation stress-induced differential DNA methylation on the physiological control of behavior and stress response in suckling Brahman bull calves. J Anim Sci. 2020;98(1). PMID:31807776. DOI: 10.1093/jas/skz368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Baker EC, Cilkiz KZ, Riggs PK, et al. Effect of prenatal transportation stress on DNA methylation in Brahman heifers. Livest Sci. 2020;240:104116. DOI: 10.1016/j.livsci.2020.104116. [DOI] [Google Scholar]
- [12].Littlejohn BP, Price DM, Neuendorff DA, et al. Prenatal transportation stress alters genome-wide DNA methylation in suckling Brahman bull calves. J Anim Sci. 2018;96:5075–5099. PMID:30165450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Schlafer DH, Fisher PJ, Davies CJ. The bovine placenta before and after birth: placental development and function in health and disease. Anim Reprod Sci. 2000;60:145–160. PMID:10844191. [DOI] [PubMed] [Google Scholar]
- [14].Senger PL. Pathways to pregnancy and parturition. 2nd ed. Pullman; Washington: Current Conceptions; 2003. [Google Scholar]
- [15].Hopper RM, ed. “Inducing parturition or abortion in cattle” Chapter 44 in bovine reproduction. Ames: John Wiley & Sons; 2015. [Google Scholar]
- [16].Zimin AV, Delcher AL, Florea L, et al. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 2009;10(4):R42. PMID:19393038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2017;27:1571–1572. PMID: 21493656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ziller MJ, Hansen KD, Meer A, et al. Coverage recommendations for methylation analysis by whole genome bisulfite sequencing. Nat Methods. 2015;12:230–232. PMID:25362363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Akalin A, Kormaksson M, Li S, et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13:R87. PMID:23034086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].McCullagh P, Nelder JA. Generalized linear models. New York: Routledge; 1989. [Google Scholar]
- [21].Wang HQ, Tuominen LK, Tsai CJ. SLIM: a sliding linear model for estimating the proportion of true null hypotheses in datasets with dependence structures. Bioinformatics. 2011;27:225–231. PMID:21098430. [DOI] [PubMed] [Google Scholar]
- [22].Cavalcante RG, Sartor MA. annotatr: genomic regions in context. Bioinformatics. 2017;33:2381–2383. PMID:28369316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Doherty R, Couldrey C. Exploring genome wide bisulfite sequencing for DNA methylation analysis in livestock: a technical assessment. Front Genet. 2014;5:126. PMID:24860595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Oberdoerffer P, Sinclair DA. The role of nuclear architecture in genomic instability and ageing. Nat Rev Mol Cell Biol. 2007;8:692–702. PMID:17700626. [DOI] [PubMed] [Google Scholar]
- [25].Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. PMID: 24138928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bollati V, Schwartz J, Wright R, et al. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech Ageing Dev. 2009;130:234–239. PMID:19150625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. PMID:11782440. [DOI] [PubMed] [Google Scholar]
- [28].Chow A, Erisir A, Farb C, et al. K(+) channel expression distinguishes subpopulations of parvalbumin- and somatostatin-containing neocortical interneurons. J Neurosci. 1999;19:9332–9345. PMID:10531438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Vinten J, Bromley RL, Taylor J, et al. The behavioral consequences of exposure to antiepileptic drugs in utero. Epilepsy Behav. 2009;14:197–201. PMID:18992367. [DOI] [PubMed] [Google Scholar]
- [30].Lauber E, Filice F, Schwaller B. Prenatal valproate exposure differentially affects parvalbumin-expressing neurons and related circuits in the cortex and striatum of mice. Front Mol Neurosci. 2016;9:150. PMID:28066177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Schraut KG, Jakob SB, Weidner MT, et al. Prenatal stress-induced programming of genome-wide promoter DNA methylation in 5-HTT-deficient mice. Transl Psychiatry. 2014;4:e473. PMID:25335169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sun W, Maffie JK, Lin L, et al. DPP6 establishes the A-type K(+) current gradient critical for the regulation of dendritic excitability in CA1 hippocampal neurons. Neuron. 2011;71:1102–1115. PMID:21943606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Maussion G, Cruceanu C, Rosenfeld JA, et al. Implication of LRRC4C and DPP6 in neurodevelopmental disorders. Am J Med Genet A. 2017;173:395–406. PMID:27759917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lin L, Murphy JG, Karlsson RM, et al. DPP6 loss impacts hippocampal synaptic development and induces behavioral impairments in recognition, learning and memory. Front Cell Neurosci. 2018;12:84. DOI: 10.3389/fncel.2018.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Pillai A, Mansouri A, Behringer R, et al. Lhx1 and Lhx5 maintain the inhibitory-neurotransmitter status of interneurons in the dorsal spinal cord. Development. 2007;134:357–360. PMID:17166926. [DOI] [PubMed] [Google Scholar]
- [36].Symmank J, Zimmer-Bensch G. LHX1-a multifunctional regulator in preoptic area-derived interneuron development. Neural Regen Res. 2019;14:1213–1214. PMID:30804249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Leblond CS, Cliquet F, Carton C, et al. Both rare and common genetic variants contribute to autism in the Faroe Islands. NPJ Genom Med. 2019;4:1. PMID:30675382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Goes FS, McGrath J, Avramopoulos D, et al. Genome‐wide association study of schizophrenia in Ashkenazi Jews. Am J Med Genet B Neuropsychiatr Genet. 2015;168:649–659. PMID:26198764. [DOI] [PubMed] [Google Scholar]
- [39].Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008;9:465–476. PMID:18463664. [DOI] [PubMed] [Google Scholar]
- [40].Aran D, Sabato S, Hellman A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol. 2013;14:R21. PMID:23497655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Almamun M, Kholod O, Stuckel AJ, et al. Inferring a role for methylation of intergenic DNA in the regulation of genes aberrantly expressed in precursor B-cell acute lymphoblastic leukemia. Leuk Lymphoma. 2017;58:1–12. PMID: 28094574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lister R, Pelizzola M, Dowen RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Smith KR, Davenport EC, Wei J, et al. GIT1 and betaPIX are essential for GABA(A) receptor synaptic stability and inhibitory neurotransmission. Cell Rep. 2014;9:298–310. PMID:25284783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Smith KR, Muir J, Rao Y, et al. Stabilization of GABA(A) receptors at endocytic zones is mediated by an AP2 binding motif within the GABA(A) receptor β3 subunit. J Neurosci. 2012;32:2485–2498. PMID:22396422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Won H, Mah W, Kim E, et al. GIT1 is associated with ADHD in humans and ADHD-like behaviors in mice. Nat Med. 2011;17:566–572. PMID:21499268. [DOI] [PubMed] [Google Scholar]
- [46].Craddock N, Jones L, Jones IR, et al. Strong genetic evidence for a selective influence of GABAA receptors on a component of the bipolar disorder phenotype. Mol Psychiatry. 2010;15:146–153. PMID:19078961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sellgren CM, Kegel ME, Bergen SE, et al. A genome-wide association study of kynurenic acid in cerebrospinal fluid: implications for psychosis and cognitive impairment in bipolar disorder. Mol Psychiatry. 2016;21:1342–1350. PMID:26666201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Horning KJ, Caito SW, Tipps KG, et al. Manganese is essential for neuronal health. Annu Rev Nutr. 2015;35:71–108. PMID:25974698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Quadri M, Federico A, Zhao T, et al. Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am J Hum Genet. 2012;90:467–477. PMID:22341971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Aschner M, Erikson KM, Hernández EH, et al. Manganese and its role in Parkinson’s disease: from transport to neuropathology. Neuromol Med. 2009;11:252–266. PMID:19657747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Autissier N, Rochette L, Dumas P, et al. Dopamine and norepinephrine turnover in various regions of the rat brain after chronic manganese chloride administration. Toxicology. 1982;24:175–182. PMID:7135412. [DOI] [PubMed] [Google Scholar]
- [52].Anderson JG, Fordahl SC, Cooney PT, et al. Manganese exposure alters extracellular GABA, GABA receptor and transporter protein and mRNA levels in the developing rat brain. Neurotoxicology. 2008;29:1044–1053. PMID:18771689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Small DM, Zatorre RJ, Dagher A, et al. Changes in brain activity related to eating chocolate: from pleasure to aversion. Brain. 2001;124:1720–1733. PMID:11522575. [DOI] [PubMed] [Google Scholar]
- [54].de Quervain DJ, Kolassa IT, Ertl V, et al. A deletion variant of the α2b-adrenoceptor is related to emotional memory in Europeans and Africans. Nat Neurosci. 2007;10:1137–1139. PMID:17660814. [DOI] [PubMed] [Google Scholar]
- [55].Zoladz PR, Dailey AM, Nagle HE, et al. ADRA2B deletion variant influences time-dependent effects of pre-learning stress on long-term memory. Neurobiol Learn Mem. 2017;140:71–81. PMID:28254464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Liberzon I, King AP, Ressler KJ, et al. Interaction of the ADRB2 gene polymorphism with childhood trauma in predicting adult symptoms of posttraumatic stress disorder. JAMA Psychiatry. 2014;71:1174–1182. PMID:25162199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Aron R, Pellegrini P, Green EW, et al. Deubiquitinase Usp12 functions noncatalytically to induce autophagy and confer neuroprotection in models of Huntington’s disease. Nat Commun. 2018;9:3191. PMID:30266909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhao J, Taylor CJ, Newcombe EA, et al. EphA4 regulates hippocampal neural precursor proliferation in the adult mouse brain by d-serine modulation of n-methyl-d-aspartate receptor signaling. Cereb Cortex. 2018;29:4381–4397. PMID:30590507. [DOI] [PubMed] [Google Scholar]
- [59].Bender CM, Gonzalgo ML, Gonzales FA, et al. Roles of cell division and gene transcription in the methylation of CpG islands. Mol Cell Biol. 1999;19:6690–6698. PMC84656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Maunakea AK, Nagarajan RP, Bilenky M, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–257. PMID:20613842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Carrera N, Arrojo M, Sanjuán J, et al. Association study of nonsynonymous single nucleotide polymorphisms in schizophrenia. Biol Psychiatry. 2012;71:169–177. PMID:22078303. [DOI] [PubMed] [Google Scholar]
- [62].Wahlberg KE, Guazzetti S, Pineda D, et al. Polymorphisms in manganese transporters SLC30A10 and SLC39A8 are associated with children’s neurodevelopment by influencing manganese homeostasis. Front Genet. 2018;9:664. PMID:30619481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Nadal MS, Ozaita A, Amarillo Y, et al. The CD26-related dipeptidyl aminopeptidase-like protein DPPX is a critical component of neuronal A-type K+ channels. Neuron. 2003;37:449–461. PMID:12575952. [DOI] [PubMed] [Google Scholar]
- [64].Qi C, Liu S, Qin R, et al. Coordinated regulation of dendrite arborization by epigenetic factors CDYL and EZH2. J Neurosci. 2014;34:4494–4508. PMID:24671995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Qin R, Cao S, Lyu T, et al. CDYL deficiency disrupts neuronal migration and increases susceptibility to epilepsy. Cell Rep. 2017;18:380–390. PMID:28076783. [DOI] [PubMed] [Google Scholar]
- [66].Liu Y, Li M, Fan M, et al. Chromodomain y-like protein–mediated histone crotonylation regulates stress-induced depressive behaviors. Biol Psychiatry. 2019;85:635–649. PMID:30665597. [DOI] [PubMed] [Google Scholar]
- [67].Lei H, Oh SP, Okano M, et al. De-novo DNA cytosine methyltransferase activities in mouse embryonic stem cells. Development. 1996;122:3195–3205. PMID:8898232. [DOI] [PubMed] [Google Scholar]
- [68].Massart R, Nemoda Z, Suderman MJ, et al. Early life adversity alters normal sex-dependent developmental dynamics of DNA methylation. Dev Psychopathol. 2016;28:1259–1272. PMID:27687908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wang H, Xu J, Lazarovici P, et al. cAMP response element-binding protein (CREB): A possible signaling molecule link in the pathophysiology of schizophrenia. Front Mol Neurosci. 2018;11:255. PMID:30214393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Iguchi-Ariga SM, Schaffner W. CpG methylation of the cAMP-responsive enhancer/promoter sequence TGACGTCA abolishes specific factor binding as well as transcriptional activation. Genes Dev. 1989;3:612–619. PMID:2545524. [DOI] [PubMed] [Google Scholar]
- [71].Zhang X, Odom DT, Koo SH, et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci. 2005;102:4459–4464. PMID:15753290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Cisternas P, Henriquez JP, Brandan E, et al. Wnt signaling in skeletal muscle dynamics: myogenesis, neuromuscular synapse and fibrosis. Mol Neurobiol. 2014;49:574–589. PMID:24014138. [DOI] [PubMed] [Google Scholar]
- [73].Brafman D, Willert K. Wnt/β-catenin signaling during early vertebrate neural development. Dev Neurobiol. 2017;77:1239–1259. PMID:28799266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Zucchi FC, Yao Y, Ward ID, et al. Maternal stress induces epigenetic signatures of psychiatric and neurological diseases in the offspring. PloS One. 2013;8:e56967. PMID:23451123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Prager EM, Bergstrom HC, Wynn GH, et al. The basolateral amygdala γ-aminobutyric acidergic system in health and disease. J Neurosci Res. 2016;94:548–567. PMID: 26586374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].de Kloet ER, Joëls M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6:463–475. PMID:15891777. [DOI] [PubMed] [Google Scholar]
- [77].Xu L, Sun Y, Gao L, et al. Prenatal restraint stress is associated with demethylation of corticotrophin releasing hormone (CRH) promoter and enhances CRH transcriptional responses to stress in adolescent rats. Neurochem Res. 2014;39:1193–1198. PMID:24682755. [DOI] [PubMed] [Google Scholar]
- [78].Daëron M, Latour S, Malbec O, et al. The same tyrosine-based inhibition motif, in the intra-cytoplasmic domain of FcγRIIB, regulates negatively BCR-, TCR-, and FcR-dependent cell activation. Immunity. 1995;3:635–646. PMID:7584153. [DOI] [PubMed] [Google Scholar]
- [79].Isakov N, Altman A. Protein kinase Cθ in T cell activation. Annu Rev Immunol. 2002;20:761–794. PMID:11861617. [DOI] [PubMed] [Google Scholar]
- [80].Cao-Lei L, Massart R, Suderman MJ, et al. DNA methylation signatures triggered by prenatal maternal stress exposure to a natural disaster: project ice storm. PLoS One. 2014;9:e107653. PMID:25238154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.