

ABSTRACT
Pancreatic cancer tends to be highly resistant to current therapy and remains one of the great challenges in biomedicine with very low 5-year survival rates. Here, we report that zalcitabine, an antiviral drug for human immunodeficiency virus infection, can suppress the growth of primary and immortalized human pancreatic cancer cells through the induction of ferroptosis, an iron-dependent form of regulated cell death. Mechanically, this effect relies on zalcitabine-induced mitochondrial DNA stress, which activates the STING1/TMEM173-mediated DNA sensing pathway, leading to macroautophagy/autophagy-dependent ferroptotic cell death via lipid peroxidation, but not a type I interferon response. Consequently, the genetic and pharmacological inactivation of the autophagy-dependent ferroptosis pathway diminishes the anticancer effects of zalcitabine in cell culture and animal models. Together, these findings not only provide a new approach for pancreatic cancer therapy but also increase our understanding of the interplay between autophagy and DNA damage response in shaping cell death.
Abbreviations: ALOX: arachidonate lipoxygenase; ARNTL/BMAL1: aryl hydrocarbon receptor nuclear translocator-like; ATM: ATM serine/threonine kinase; ATG: autophagy-related; cGAMP: cyclic GMP-AMP; CGAS: cyclic GMP-AMP synthase; ER: endoplasmic reticulum; FANCD2: FA complementation group D2; GPX4: glutathione peroxidase 4; IFNA1/IFNα: interferon alpha 1; IFNB1/IFNβ: interferon beta 1; MAP1LC3B/LC3: microtubule-associated protein 1 light chain 3 beta; MDA: malondialdehyde; mtDNA: mitochondrial DNA; NCOA4: nuclear receptor coactivator 4; PDAC: pancreatic ductal adenocarcinoma; POLG: DNA polymerase gamma, catalytic subunit; qRT-PCR: quantitative polymerase chain reaction; RCD: regulated cell death; ROS: reactive oxygen species; SLC7A11: solute carrier family 7 member 11; STING1/TMEM173: stimulator of interferon response cGAMP interactor 1; TFAM: transcription factor A, mitochondrial
KEYWORDS: Antiviral drug, autophagy, CGAS, DNA damage, ferroptosis, mitochondria, POLG, TFAM, STING1, tumor therapy
Introduction
Macroautophagy (hereafter referred to as autophagy) is a highly conserved intracellular degradation system that is activated in response to physiological or pathological stressors, such as starvation, hypoxia, oxidative stimulation, infection, and oncogene activation [1,2]. Autophagy-related (ATG) genes and proteins play an essential role in the regulation of the process and function of autophagy in human health and disease [3]. As a homeostatic control mechanism, an increased level of autophagy can protect healthy cells from environmental stress to promote survival [4]. However, the excessive or impaired autophagic activity can trigger cell injury and death [5,6]. The Nomenclature Committee on Cell Death has recently defined autophagy-dependent cell death as “a form of regulated cell death (RCD) that mechanistically depends on the autophagic machinery (or components thereof),” [7] which suggests that ATG genes are responsible for this process.
Autophagy-dependent cell death is highly contextual and exhibits different relationships with various forms of RCD [8]. Among them, ferroptosis, an iron-dependent RCD discovered in 2012 [9], was recently recognized as a type of autophagy-dependent cell death in multiple cells, including various cancer cells [10]. Iron is not only a chemical element for the modulation of enzyme activity, but can also oxidize a wide range of substrates, causing biological damage [11]. Iron-mediated oxidative stress is a core mechanism of ferroptosis [12,13]. Although increased autophagy promotes iron accumulation and subsequent lipid peroxidation via the selective degradation of anti-ferroptosis regulators (e.g. NCOA4 [nuclear receptor coactivator 4], GPX4 [glutathione peroxidase 4], and ARNTL/BMAL1 [aryl hydrocarbon receptor nuclear translocator like]) [14–20], the underlying molecular mechanism still remains largely unknown.
Mitochondria are the powerhouse of the cell that play a fundamental role in oxidative stress and cell death. While ferroptosis is associated with increased mitochondrial fragmentation and permeabilization [9], the mitochondrial regulation of ferroptosis remains controversial. On the one hand, voltage-dependent anion channels (also known as mitochondrial porins) were identified as a direct target of erastin responsible for ferroptotic cancer cell death [21]. Mitochondrial metabolism also facilitates cysteine deprivation-induced ferroptosis [22]. On the other hand, the mitochondrial DNA (mtDNA)-depleted cancer cell line (termed ρ0 cells) generated from a long-term culture with ethidium bromide displays the same sensitivity to the ferroptosis inducer compared to its parental line [9]. In contrast, certain ρ0 cancer cells are more sensitive to hydrogen peroxide-induced cell death via the promotion of lipid peroxidation [23]. Thus, further understanding the relationship between mitochondria and ferroptosis will help to develop effective cancer treatments.
In this study, we provided a novel mode of mtDNA stress-mediated ferroptotic cancer cell death. We demonstrated that the antiviral drug zalcitabine causes mtDNA depletion and oxidative DNA damage, which trigger cell death in primary and immortalized human pancreatic cancer cells through the activation of autophagy-dependent ferroptosis. We further demonstrated that the STING1/TMEM173 (stimulator of interferon response cGAMP interactor 1)-dependent DNA sensing pathway is required for this process. Consequently, the genetic and pharmacological inactivation of the autophagy-dependent ferroptosis pathway limits the anticancer activity of zalcitabine in vitro and in vivo. Together, these findings provide a framework to understand how autophagy promotes ferroptosis.
Results
Zalcitabine induces ALOX5-dependent ferroptosis in pancreatic cancer cells
The nucleoside analog zalcitabine, also known as 2ʹ, 3ʹ-dideoxycytidine, is used for the treatment of human immunodeficiency virus (HIV)-infected patients with acquired immune deficiency syndrome (AIDS). In addition to antiviral effects, a few studies have shown that relative long-term treatment with zalcitabine (≥3 d) can induce cell death in leukemia, melanoma, or brain cancer cells [24–27]. To determine whether zalcitabine has similar effects on pancreatic ductal adenocarcinoma (PDAC), one of the most deadly of all types of human cancer, we first used The Cancer Genome Atlas database to analyze the expression of the mtDNA replication and repair regulator POLG (DNA polymerase gamma, catalytic subunit), the drug target of zalcitabine [28]. The mRNA expression of POLG was upregulated in the tumor group compared with the control group (Figure 1A). Consistently, we observed the upregulation of POLG protein expression in human PDAC cell lines (PANC1 and Capan2) as well as in primary PDAC cells (pHsPDAC) compared to normal pancreatic ductal cells (Figure 1B).
Figure 1.
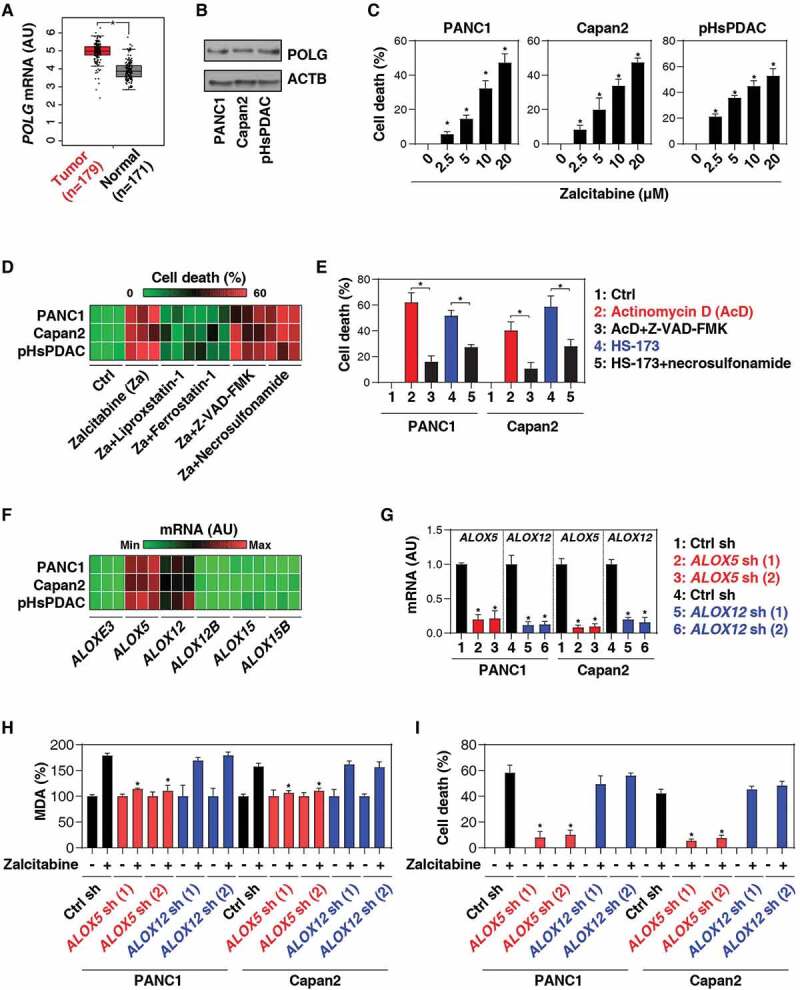
Zalcitabine induces ALOX5-dependent ferroptosis in pancreatic cancer cells. (A) Upregulation of POLG gene expression within the tumor from PDAC patients compared to normal controls (*P < 0.05). (B) Western blot analysis of POLG protein expression in the indicated human PDAC or normal ductal cells. (C) The indicated human PDAC cells were treated with zalcitabine (2.5–20 µM) for 72 h, and the level of cell death was assayed (n = 3, *P < 0.05 versus the control group). (D) Heat map of levels of cell death in the indicated human PDAC cells following treatment with zalcitabine (20 µM) in the absence or presence of Z-VAD-FMK (20 µM), necrosulfonamide (1 µM), ferrostatin-1 (1 µM), or liproxstatin-1 (1 µM) for 72 h (n = 3). (E) Analysis of cell death in PANC1 cells following treatment with actinomycin D (1 µM) or HS-173 (1 µM) in the absence or presence of Z-VAD-FMK (20 µM) or necrosulfonamide (1 µM) for 72 h (n = 3, *P < 0.05). (F) Heat map of levels of gene expression in the indicated PDAC cells (n = 3). (G) Levels of gene expression in the indicated gene knockdown PDAC cells (n = 3, *P < 0.05 versus control shRNA group). (H, I) Analysis of levels of MDA (H) and cell death (I) in the indicated PDAC cells following treatment with zalcitabine (20 µM) for 72 h (n = 3, *P < 0.05 versus control shRNA group)
To investigate whether POLG is a potential therapeutic target in human PDAC, we treated PANC1, Capan2, and pHsPDAC cells with zalcitabine for 72 h. Zalcitabine dose-dependently induced cell death (Figure 1C). Notably, the cytotoxicity of zalcitabine was mainly reduced by using the inhibitors of ferroptosis (liproxstatin-1 and ferrostatin-1), but not apoptosis (Z-VAD-FMK) and necroptosis (necrosulfonamide) (Figure 1D). As expected, Z-VAD-FMK and necrosulfonamide efficiently blocked cell death in PANC1 and Capan2 cells in response to their corresponding positive stimuli, actinomycin D [29] or HS-173 [30], respectively (Figure 1E).
We next determined whether the genetic inhibition of ferroptosis can block zalcitabine-induced cell death. The mammalian ALOX (arachidonate lipoxygenase) family, consisting of six members (ALOXE3, ALOX5, ALOX12, ALOX12B, ALOX15, and ALOX15B) with different cell and tissue expression patterns, is responsible for ferroptosis by lipid peroxidation [12,13]. A quantitative polymerase chain reaction (qRT-PCR) analysis demonstrated that the primary and immortalized PDAC cells have a higher mRNA expression of ALOX5 or ALOX12 compared to other lipoxygenase family members (Figure 1F). Functional assay further confirmed that the knockdown of ALOX5, but not ALOX12, by two different shRNAs (Figure 1G) blocked zalcitabine-induced malondialdehyde (MDA, a lipid peroxidation marker) production (Figure 1H) and cell death (Figure 1I) in PANC1 and Capan2 cells. These findings suggest that zalcitabine triggers ALOX5-dependent ferroptotic cancer cell death by lipid peroxidation.
Zalcitabine-induced TFAM degradation promotes ferroptosis
In addition to POLG, the replication of mtDNA also requires TFAM (transcription factor A, mitochondrial), a member of the high-mobility group protein family [31]. The interaction and feedback between POLG and TFAM decide mtDNA homeostasis in various types of cells [32]. We further explored the impact of inhibiting POLG by zalcitabine on the expression of TFAM. In fact, western blot analysis found that zalcitabine reduced TFAM protein expression in PANC1, Capan2, and pHsPDAC cells, and these processes were reversed by treatment with bortezomib (a proteasome inhibitor) or TMP (2,3,5,6-tetramethylpyrazine, a direct inhibitor of TFAM protein degradation and one of the major components of the Chinese medicine Ligusticum striatum/chuanxiong [33]) (Figure 2A). Bortezomib is also an inhibitor of mitochondrial protease LONP1/Lon (lon peptidase 1, mitochondrial), a major driver of TFAM protein degradation [34]. Moreover, TMP binds to TFAM and blocks its degradation by LONP1 [33]. To determine whether bortezomib-mediated proteasome inhibition is essential for blocking zalcitabine-induced TFAM protein degradation, we treated PANC1 cells with an additional proteasome inhibitor epoxymicin, which does not inhibit LONP1 activity [35]. Unlike bortezomib, epoxymicin failed to inhibit zalcitabine-induced TFAM protein degradation in PANC1 cells (Figure 2B). Moreover, knockdown of LONP1 by shRNA also reduced zalcitabine-induced TFAM protein degradation in PANC1 cells (Figure 2C). These findings are consistent with previous studies suggesting that the LONP1-dependent pathway is the principal mechanism for TFAM degradation during mtDNA stress [34].
Figure 2.
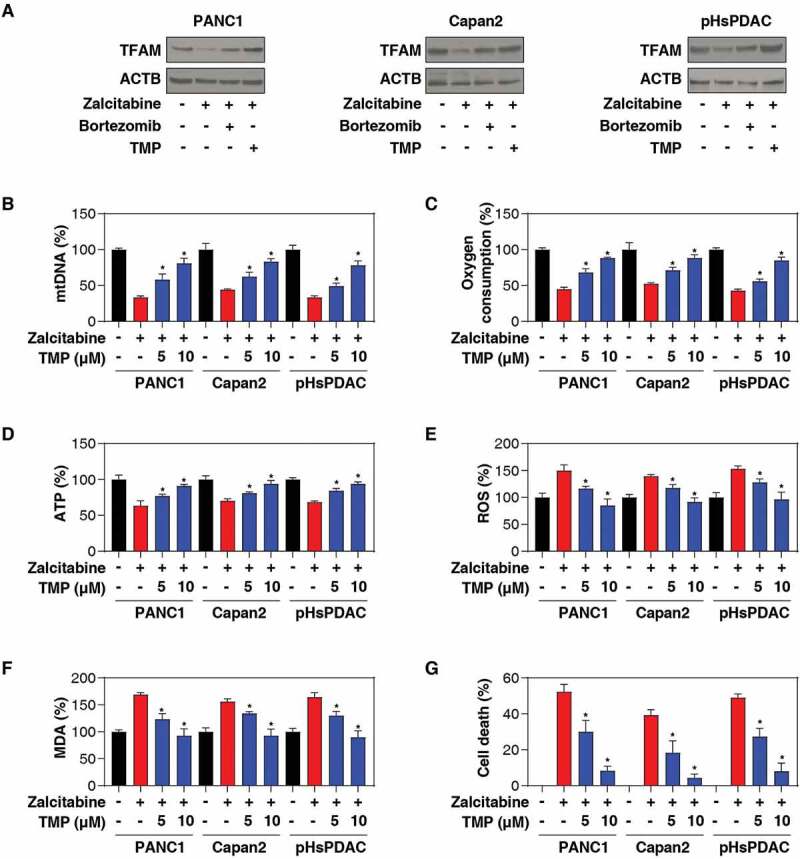
Zalcitabine-induced TFAM degradation promotes ferroptosis. (A-C) Western blot analysis of TFAM expression in the indicated human PDAC cells following treatment with zalcitabine (20 µM) in the absence or presence of bortezomib (500 nM) or TMP (10 µM) or epoxymicin (500 nM) for 72 h. (D-I) The indicated human PDAC cells were treated with zalcitabine (20 µM) in the absence or presence of TMP (5 or 10 µM) for 72 h. The relative levels of mtDNA copy number (D), oxygen consumption (E), ATP production (F), total ROS (G), MDA production (H), and cell death (I) were assayed using commercially available assay kits (n = 3, *P < 0.05 versus zalcitabine-alone group)
A deficiency in the POLG or TFAM gene can decrease the mtDNA copy number, leading to respiratory chain disorder, mitochondrial dysfunction, and oxidative stress [31,32,36,37]. We next sought to explore the relationship between mtDNA stress and ferroptosis. As expected, treatment with zalcitabine resulted in mitochondrial dysfunction, including a reduced mtDNA copy number (Figure 2D), decreased oxygen consumption (Figure 2E), limited ATP production (Figure 2F), and increased total ROS (reactive oxygen species) generation (Figure 2G), in PANC1, Capan2, and pHsPDAC cells. In contrast, TMP protected against zalcitabine-induced mitochondrial dysfunction in these primary and immortalized PDAC cells (Figure 2D-G). TMP also inhibited zalcitabine-induced MDA production (Figure 2H), and cell death (Figure 2I). These findings indicate that preventing TFAM degradation can limit zalcitabine-induced ferroptosis by restoring mitochondrial dysfunction.
The CGAS-STING1 pathway promotes zalcitabine-induced ferroptosis
Since the discovery of TFAM, many studies have shown that TFAM deficiency is involved in the activation of the DNA damage and sensing pathway [31]. In particular, the loss of the TFAM gene increases mtDNA stress, injury, and cytosolic release, leading to an antiviral innate immune response via the activation of the CGAS (cyclic GMP-AMP synthase)-STING1 pathway during infection [38]. CGAS is a DNA sensor responsible for producing the second messenger cyclic GMP-AMP (cGAMP) to activate STING1 via the phosphorylation of STING1 at Ser366 [39,40]. However, it remains unknown whether similar signaling happens in zalcitabine-induced ferroptotic cancer cell death. To address this question, we assayed the levels of 8-hydroxydeoxyguanosine (8-OHdG; a marker of oxidative DNA damage) (Figure 3A), cytosolic mtDNA (Figure 3B), cGAMP (Figure 3C), and phosphorylated STING1 (Ser366) (Figure 3D). Indeed, the treatment of PDAC cells (PANC1, Capan2, and pHsPDAC) with zalcitabine resulted in increased levels of 8-OHdG (Figure 3A), cytosolic mtDNA (Figure 3B), cGAMP (Figure 3C), and phosphorylated STING1 (Ser366) (Figure 3D). This process was reversed by TMP, indicating that zalcitabine-induced TFAM degradation can trigger oxidative DNA damage, mtDNA release to the cytosol, and subsequent activation of the CGAS-STING1 pathway.
Figure 3.
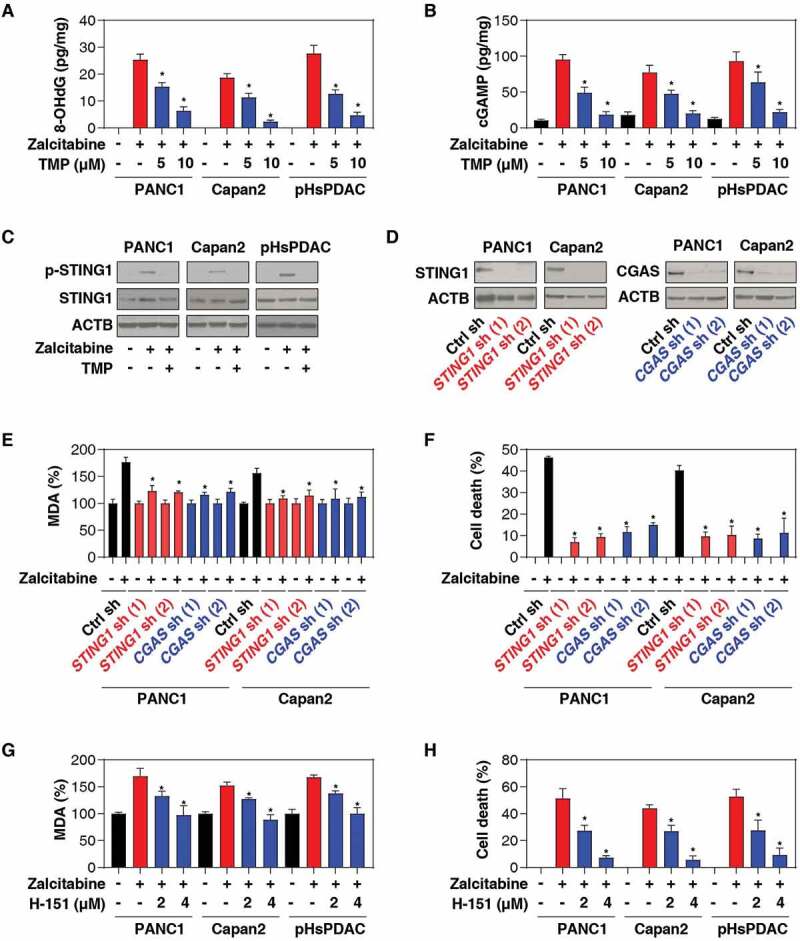
The CGAS-STING1 pathway promotes zalcitabine-induced ferroptosis. (A-C) The indicated human PDAC cells were treated with zalcitabine (20 µM) in the absence or presence of TMP (5 or 10 µM) for 72 h. The intracellular levels of 8-OHdG (A), cytosolic mtDNA (B), and cGAMP (C) were assayed with ELISA kits (n = 3, *P < 0.05 versus zalcitabine-alone group). (D) Western blot analysis of protein expression in human PDAC cells following treatment with zalcitabine (20 µM) in the absence or presence of TMP (10 µM) for 72 h. (E) Western blot analysis of protein expression in the indicated STING1- and CGAS-knockdown PDAC. (F, G) Analysis of levels of MDA (F) and cell death (G) in the indicated STING1- and CGAS-knockdown PDAC cells following treatment with zalcitabine (20 µM) for 72 h (n = 3, *P < 0.05 versus control shRNA group). (H-I) Analysis of levels of MDA (H) and cell death (I) in the indicated PDAC cells following treatment with zalcitabine (20 µM) in the absence or presence of the STING1 inhibitor H-151 (2 or 4 µM) for 72 h (n = 3, *P < 0.05 versus zalcitabine-alone group)
To determine the direct effects of the CGAS-STING1 pathway activation on ferroptosis, we suppressed CGAS or STING1 expression by shRNA in PANC1 and Capan2 cells (Figure 3E). Remarkably, the knockdown of CGAS or STING1 inhibited MDA production (Figure 3F) and the cytotoxicity (Figure 3G) of zalcitabine. Similarly, H-151, a direct inhibitor of STING1 [41], also blocked zalcitabine-induced MDA production (Figure 3H) and cell death (Figure 3I). These results suggest that the CGAS-STING1 DNA-sensing pathway promotes zalcitabine-induced lipid peroxidation and ferroptosis.
STING1-mediated autophagy promotes zalcitabine-induced ferroptosis
Considering that the main function of STING1 is to organize cell defenses against infection via the induction of type I interferon (IFNA1/IFNα [interferon alpha 1] and IFNB1/IFNβ [interferon beta 1]) production [40,42–44], we next determined whether a type I IFN response contributes to zalcitabine-induced ferroptosis. IFNA1 or IFNB1 did not appear to affect zalcitabine-induced cell death in PDAC cells (Figure 4A). Furthermore, the shRNA-mediated knockdown of IRF3 (interferon regulatory factor 3) (Figure 4B), a transcription factor responsible for STING1-induced type I IFN production, also failed to change zalcitabine-induced cell death in PANC1 or Capan2 cells (Figure 4C). These findings suggest that STING1-dependent type I IFN production is not required for zalcitabine-induced ferroptosis.
Figure 4.
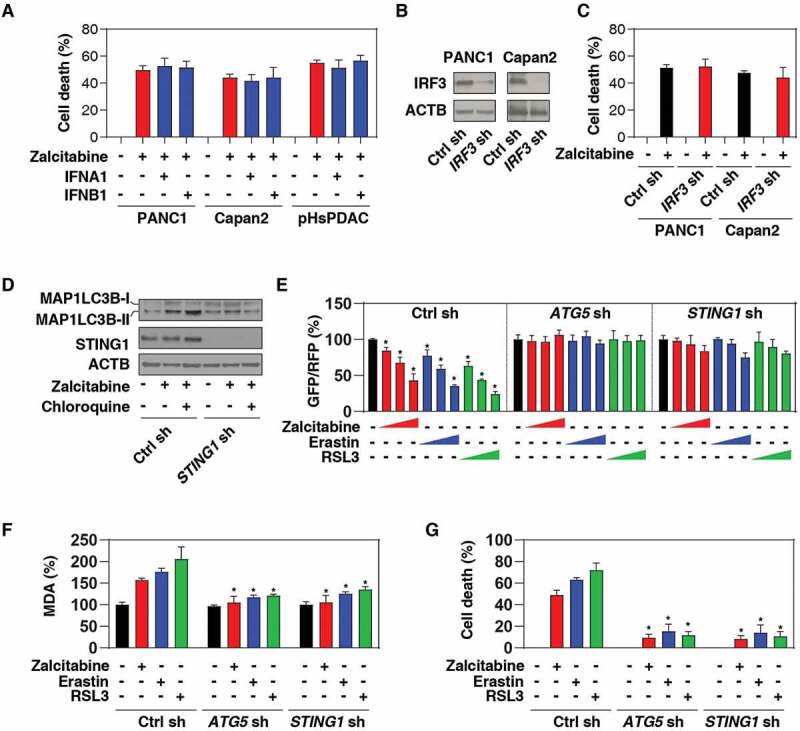
STING1-mediated autophagy promotes zalcitabine-induced ferroptosis. (A, B) Analysis of cell death in the indicated human PDAC cells following treatment with zalcitabine (20 µM) in the absence or presence of IFNA1 (5 ng/ml) or IFNB1 (5 ng/ml) for 72 h (n = 3). (B) Western blot analysis of IRF3 expression in the indicated IRF3-knockdown PDAC cells. (C) Analysis of cell death in the indicated human PDAC cells following treatment with zalcitabine (20 µM) for 72 h (n = 3). (D) Western blot analysis of MAP1LC3B in the indicated PANC1 cells following treatment with zalcitabine (20 µM) in the absence or presence of chloroquine (20 µM) for 72 h. (E) Analysis of the time course of relative GFP/RFP ratio in the indicated PANC1 cells expressing GFP-RFP-LC3ΔG in response to zalcitabine (20 µM; 24, 48, and 72 h), erastin (10 µM; 6, 12, and 24 h), or RSL3 (0.5 µM; 6, 12, and 24 h) (n = 3, *P < 0.05 versus untreated group). (F, G) Analysis of levels of MDA (F) and cell death (G) in the indicated PANC1 cells in response to zalcitabine (20 µM, 72 h), erastin (10 µM, 24 h), or RSL3 (0.5 µM, 24 h) (n = 3, *P < 0.05 versus control shRNA group)
Recent studies demonstrate that STING1 can activate autophagy through a mechanism that is independent of IFN induction [45,46]. Indeed, excessive autophagy mediates ferroptotic death in certain types of cancer cells, including human PDAC [15]. We, therefore, raise the hypothesis that STING1-mediated autophagy may promote zalcitabine-induced ferroptosis. We confirmed that the production of lipidated MAP1LC3B/LC3 (microtubule associated protein 1 light chain 3 beta)-II, a marker for autophagosomes, was significantly reduced in STING1-knockdown PANC1 cells following zalcitabine treatment (Figure 4D). Moreover, the lysosome inhibitor chloroquine further increased zalcitabine-induced expression of MAP1LC3B-II in control cells, but not in STING1-knockdown PANC1 cells (Figure 4D), indicating a potential role of STING1 in the regulation of autophagic flux.
To further evaluate the effects of STING1 on autophagic flux, we expressed GFP-LC3-RFP-LC3ΔG, a novel autophagic flux probe [47], in PANC1 cells. Upon autophagy induction, the GFP-LC3 signal is quenched within the acidic lysosome following autophagosome-dependent delivery, whereas the RFP-LC3ΔG signal remains unquenched (LC3∆G cannot be conjugated to phosphatidylethanolamine and hence does not associate with the autophagosome and remains in the cytosol). Thus, the GFP:RFP signal ratio inversely correlates with autophagic activity [47]. Using a microplate reader, we observed that the GFP:RFP ratio was time-dependent and decreased in response to zalcitabine, as well as the classical ferroptosis inducers (e.g. erastin and RSL3) (Figure 4E). Like the knockdown of ATG5 (a core regulator of autophagosome formation), the knockdown of STING1 also prevented zalcitabine-, erastin-, and RSL3-induced downregulation of the ratio between GFP and RFP in PANC1 cells (Figure 4E), supporting the idea that STING1 is a positive regulator of autophagic flux during ferroptosis. Consequently, the suppression of ATG5 or STING1 expression by shRNA inhibited zalcitabine-, erastin-, and RSL3-induced MDA production (Figure 4F) and cell death (Figure 4G). Thus, STING1-mediated autophagy contributes to zalcitabine-induced ferroptosis via lipid peroxidation.
Zalcitabine suppresses pancreatic tumor growth via autophagy-dependent ferroptosis in vivo
To determine whether zalcitabine suppresses pancreatic tumor growth via autophagy-dependent ferroptosis in vivo, wild-type, stable ALOX5-knockdown, ATG5-knockdown, or STING1-knockdown PANC1 cell lines were implanted into the subcutaneous space of the right flank of nude mice. Beginning on day 7, these mice were treated with zalcitabine. Compared with the control shRNA group, zalcitabine treatment failed to reduce the size of tumors formed (Figure 5A) with increased MDA levels (Figure 5B) and upregulated ACSL4 (acyl-CoA synthetase long chain family member 4) expression (Figure 5C), a biomarker and contributor to ferroptosis [48,49], by ALOX5- or ATG5- or STING1-knockdown cells observed within the tumor.
Figure 5.
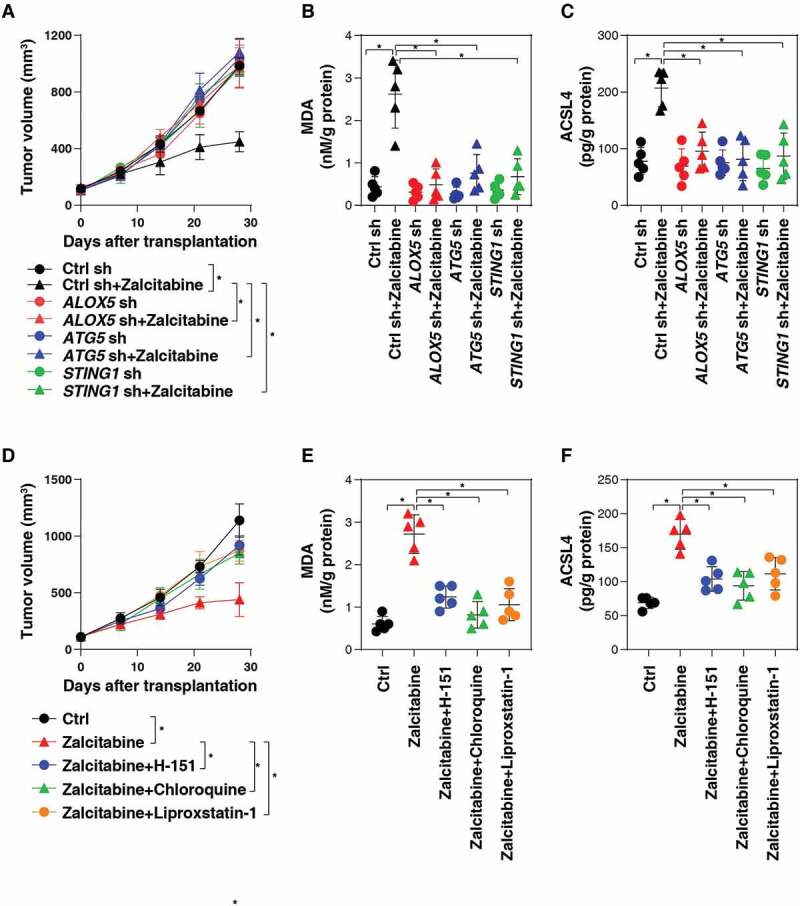
Zalcitabine suppresses pancreatic tumor growth via autophagy-dependent ferroptosis in vivo. (A) NOD SCID mice were injected subcutaneously with the indicated PANC1 cells (5 × 106/mouse) and treated with zalcitabine (50 mg/kg per day by i.p.) at day 7 for 2 weeks. Tumor volume was calculated weekly (n = 5 mice/group, data expressed as means ± SD, *P < 0.05, ANOVA LSD test). (B, C) In parallel, the levels of MDA (B) and ACSL4 (C) in the isolated tumors at day 28 were assayed by ELISA (n = 5 mice/group, *P < 0.05, ANOVA LSD test). (D) NOD SCID mice were injected subcutaneously with PANC1 cells (5 × 106/mouse) and treated with zalcitabine (50 mg/kg per day by i.p.) in the absence or presence of H-151 (750 nM per mouse, once every other day by i.p.), chloroquine (50 mg/kg once every other day by i.p.), or liproxstatin-1 (10 mg/kg once every other day by i.p.) at day 7 for 2 weeks. Tumor volume was calculated weekly (n = 5 mice/group, data expressed as means ± SD, *P < 0.05, ANOVA LSD test). (E, F) In parallel, the levels of MDA (E) and ACSL4 (F) in the isolated tumors at day 28 were assayed by ELISA (n = 5 mice/group, *P < 0.05, ANOVA LSD test)
Similarly, the STING1 inhibitor H-151, the autophagic flux inhibitor chloroquine, or the ferroptosis inhibitor liproxstatin-1 also reduced the anticancer activity of zalcitabine in subcutaneous tumor models (Figure 5D), which was associated with decreased MDA levels (Figure 5E) and ACSL4 expression (Figure 5F) within tumors. Collectively, these animal studies demonstrate that STING1-mediated autophagy promotes zalcitabine-induced ferroptosis in vivo.
Discussion
There is a vital need to develop effective therapies for PDAC, the seventh-leading cause of cancer death in the world, with a very low 5-year survival rate [50]. In this study, we provided the first evidence that the antiviral drug zalcitabine can suppress PDAC cell growth through the induction of autophagy-dependent ferroptotic death in vitro and in vivo. Mechanistically, these effects are dependent on mtDNA stress-induced activation of the CGAS-STING1 pathway (Figure 6). These findings not only reveal a potential antitumor activity of zalcitabine in PDAC but also shine a light on the mechanism of autophagy-dependent cell death that links to ferroptosis and the cytosolic DNA-sensing pathway.
Figure 6.
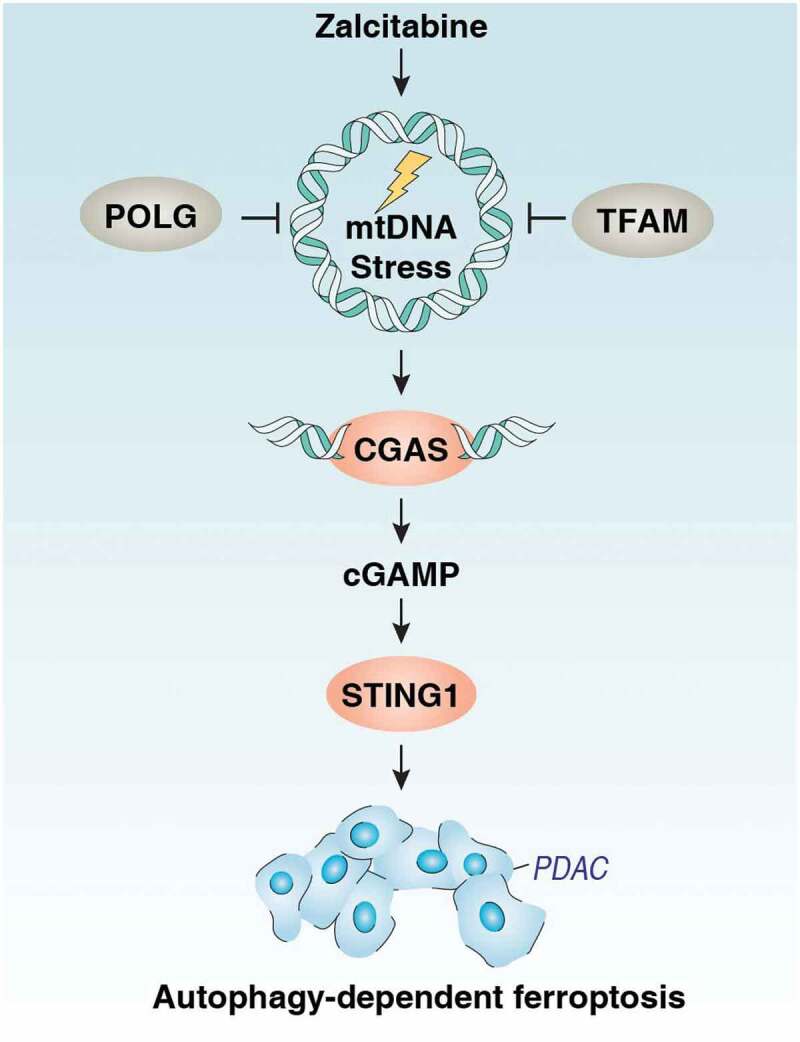
Schematic depicting the CGAS-STING1 pathway that is implicated in mtDNA stress-induced ferroptotic cancer cell death via the activation of autophagy. Zalcitabine induces oxidative mtDNA damage and decreased mitochondrial function as well as degradation of the mitochondrial transcription factor TFAM. These effects result in the activation of the DNA damage sensing CGAS-STING1 pathway, inducing autophagy and subsequently causing autophagy-dependent ferroptosis. See the text for details.`
Dysfunctional mitochondria are implicated in tumor development, progression, and resistance to therapy [51]. Mitochondrial alterations, such as mtDNA damage and mutation, metabolic reprogramming, fission and fusion dynamics, and cell death susceptibility, could provide unique therapeutic targets in cancer [51]. Zalcitabine, a nucleoside reverse transcriptase inhibitor for treating human immunodeficiency virus-infected patients displaying acquired immune deficiency syndrome, can cross-react with intracellular POLG, resulting in increased mtDNA depletion and oxidative DNA damage [52–55]. As a nuclear-encoded protein, POLG encodes the DNA polymerase responsible for mtDNA replication and repair, which is essential for mammalian embryogenesis, as demonstrated by an early developmental arrest of polg−/- mice [37,56]. In addition, POLG mutation drives growth, proliferation, and invasion of human breast cancer cells [57], and our current study indicates that POLG expression is upregulated in PDAC patients and serves as a cell death target to induce ferroptosis by zalcitabine. Thus, functional mitochondria exert multiple roles in the context of tumor biology.
There is an increasing context-dependent relationship between mtDNA depletion and cell death using mtDNA‑depleted ρ0 cell lines, which were originally generated by culture via ethidium bromide. The ρ0 human cancer cells have decreased proliferation rates and markedly reduced tumor formation in nude mice [58,59]. The ρ0 cell lines driven from SK-Hep1 (a human hepatocellular carcinoma cell line) or 143B (a human osteosarcoma cell line) are resistant to apoptosis due to increased SOD2/MnSOD (superoxide dismutase 2) expression, decreased ROS production, decreased mitochondrial respiration, or increased ABCB/P-glycoprotein expression [60–62]. In contrast, ρ0 cell lines driven from 143B are sensitive to death receptor signaling (e.g. TNF [tumor necrosis factor] and anti-FAS antibody)-mediated apoptosis [63]. Upon ferroptosis, ρ0 cancer cell lines have similar sensitivity to erastin- and RSL3-induced growth inhibition [9], although they may express higher levels of lipoxygenase for lipid peroxidation during oxidative injury [23]. It is worth noting that such long treatments with ethidium bromide (≥ 4 weeks) to generate ρ0 cell lines not only deplete mtDNA but also cause mutations in nuclear DNA and specific gene expression profiles, which may affect the phenotype occurrence [64–66]. Unlike the phenotype observed using ρ0 cell lines, we saw that zalcitabine-mediated mtDNA depletion led to ferroptosis, which can be rescued by TFAM, a core mtDNA packaging protein and damage-associated molecular pattern in cell death [67,68]. LONP1-mediated TFAM degradation is required for fine-tuning of mitochondrial quality control, which has been implicated in tumorigenesis and tumor therapy [69–71]. Our observations that the dysfunction of the LONP1-TFAM pathway promotes ferroptotic cancer cell death will be crucial to unravel the complexity of the mitochondrial network, resulting in metabolic stress and cell death. In addition to binding to TFAM, it remains unclear whether the TMP causes off-target effects on mitochondrial function per se during ferroptosis.
Additionally, the mitochondrial tricarboxylic acid cycle is required for ferroptosis during cysteine deprivation [22], whereas mitochondrial iron overload-induced oxidative stress accelerates ferroptotic cell death [72,73]. These findings also suggest an important role of mitochondria in ferroptosis involved in mtDNA biogenesis, energy metabolism, and mitochondrial iron transport and storage.
Our findings highlight the importance of STING1, an endoplasmic reticulum (ER)-associated protein, connecting DNA damage to ferroptosis and autophagy. The detection of foreign or self-DNA is an evolutionarily conserved mechanism in response to infection or tissue injury. Although many pathways have been described during the last 25 years, the CGAS-STING1 pathway could play a central role in sensing host DNA damage from mitochondria and the nucleus [43,44]. In addition, DNA damage response regulators, such as FANCD2 (FA complementation group D2) and ATM (ATM serine/threonine kinase), can regulate ferroptosis sensitivity in both DNA damage-dependent and -independent manners [74,75]. We demonstrated that zalcitabine-induced mtDNA depletion and oxidative DNA damage could activate the CGAS-STING1 pathway, resulting in ferroptosis. Interestingly, a STING1-mediated type I interferon response is not essential for zalcitabine-induced ferroptosis. Alternatively, STING1-mediated autophagy is responsible for zalcitabine-induced ferroptosis. The excessive activation of STING1 is implicated in various forms of RCD and stress responses, such as apoptosis, pyroptosis, necroptosis, senescence, ER stress, and autophagy [76,77]. The STING1-associated ER-Golgi intermediate compartment can serve as a membrane source for autophagosome formation in response to DNA damage [46]. In addition to the removal of pathogens by autophagy, impaired STING1-mediated autophagy flux promotes cell death in breast cancer cells [78]. Thus, the STING1 signaling pathway is responsible for not only generating a robust innate immune response but also triggering a cell death response upon DNA damage.
Although the original study on ferroptosis indicates that it is a new type of regulated cell death distinct from apoptosis, necrosis, and autophagy [9], studies from autophagy-deficient cells suggest that ferroptosis is a type of autophagy-dependent cell death in some conditions [10]. Autophagy, especially certain types of selective autophagy, such as ferritinophagy [14,15,79,80], lipophagy [81], clockophagy [17,19], and chaperone-mediated autophagy [16], promotes ferroptosis through lipid peroxidation via multiple mechanisms, including increasing toxic iron accumulation and the degradation of antioxidant proteins. Moreover, BECN1, a mediator of autophagy, can promote ferroptosis through binding to SLC7A11 (solute carrier family 7 member 11) to limit cystine/glutamate transporter-mediated glutathione production [18,20]. Our current findings indicate that zalcitabine-induced mtDNA depletion triggers STING1-dependent autophagy and subsequent lipid peroxidation-mediated ferroptosis. This observation may explain the frequent activation of the DNA damage response and autophagy during oxidative injury [82].
In summary, we report the STING1 regulated autophagy-dependent ferroptosis pathway as a potential target for therapeutic intervention for PDAC. Given that autophagy plays a different role in the early versus later stage of tumor formation, further studies are needed to explore the impact of autophagy-dependent ferroptosis in tumorigenesis using autophagy- or ferroptosis-deficient mice [83].
Materials and methods
Reagents
Chloroquine (C6628), Z-VAD-FMK (V116), actinomycin D (A1410), HS-173 (5323840001), puromycin (P9620), and 2,3,5,6-tetramethylpyrazine (W323713) were obtained from Sigma-Aldrich. Ferrostatin-1 (S7243), liproxstatin-1 (S7699), zalcitabine (S1719), erastin (S7242), RSL3 (S8155), epoxymicin (S7038), and bortezomib (S1013) were obtained from Selleck Chemicals. H-151 (inh-h151) was obtained from InvivoGen. The antibodies to STING1 (13647), p-STING1 (Ser366; 50907), CGAS (15102), IRF3 (4302), ACTB (3700), POLG (13609), TFAM (8076), and MAP1LC3B (3868) were obtained from Cell Signaling Technology.
Cell culture
The PANC1 (CRL-1469) and Capan2 (HTB-80) cell lines were obtained from the American Type Culture Collection. These cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Thermo Fisher Scientific, 11995073) or McCoy’s 5a Medium Modified (Thermo Fisher Scientific, 16600082) supplemented with 10% heat-inactivated fetal bovine serum (Thermo Fisher Scientific, A3840001) and 1% penicillin and streptomycin (Thermo Fisher Scientific, 15070–063) at 37°C, 95% humidity, and 5% CO2. Cell line identity was validated by Short Tandem Repeat profiling, and routine mycoplasma testing was negative for contamination.
Human clinical samples from patients with pancreatic cancer who underwent surgery were collected from the China-Japan Union Hospital of Jilin University. The collection of samples was approved by its institutional review board. Primary PDAC cells were isolated by enzymatic digestion, as described previously [84].
Animal study
NOD SCID mice (394) were purchased from Charles River Laboratories. The mice were housed with their littermates in groups of 4–5 animals per cage and kept on a regular 12-h light and dark cycle (7:00–19:00 light period). Food and water were available ad libitum. Experiments were carried out under pathogen-free conditions and the health status of mouse lines was routinely checked by veterinary staff. Experiments were carried out with randomly chosen littermates of the same sex and matched by age and body weight. Indicated wild-type or gene knockdown PANC1 cells (5 × 106 cells) were subcutaneously injected into the dorsal side of NOD SCID mice. At day 7, these mice were administrated with the indicated drug (zalcitabine [50 mg/kg, per day by i.p.], H-151 [750 nM per mouse, once every other day by i.p.], chloroquine [50 mg/kg, once every other day by i.p.], or liproxstatin-1 [10 mg/kg, once every other day by i.p.]) for 2 weeks. We conducted all animal care and experimentation in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care guidelines (http://www.aaalac.org) and with approval from institutional animal care and use committees.
Cell death assay
Trypan blue staining was used to determine the number of viable cells present in a cell suspension. Only cells with intact membranes could effectively exclude the dye, so dead cells with compromised membranes became stained. A Countess II FL Automated Cell Counter (Thermo Fisher Scientific, AMQAF1000) was used to assay the percentages of viable and dead cells after cell staining with 0.4% trypan blue solution (Thermo Fisher Scientific, T10282).
RNAi
The following human shRNAs were obtained from Sigma-Aldrich in a lentiviral format: ALOX5-shRNA-1 (sequence: CCGGCCCGTGATATCCAGTTTGATACTCGAGTATCAAACTGGATATCACGGGTTTTTG), ALOX5-shRNA-2 (sequence: CCGCTCAAGATCAGCAACACTATTTCTCGAGAAATAGTGTTGCTGATCTTGATTTTTTG), ALOX12-shRNA-1 (sequ--ence: CCGGGCTGACTTCATCCTTCTGGATCTCGAGATCCAGAAGGATGAAGTCAGCTTTTTG), ALOX12-shRNA-2 (sequence: CCGGCCCAAAGCTGTGCTAAACCAACTCGAGTTGGTTTAGCACAGCTTTGGGTTTTTG), STING1-shRNA-1 (sequence: CCGGGCTGGCATGGTCATATTACATCTCGAGATGTAATATGACCATGCCAGCTTTTTTG), STING1-shRNA-2 (sequence: CCGGGCCCGGATTCGAACTTACAATCTCGAGATTGTAAGTTCGAATCCGGGCTTTTTTG), CGAS-shRNA-1 (sequence: CCGGCAACTACGACTAAAGCCATTTCTCGAGAAATGGCTTTAGTCGTAGTTGTTTTTTG), CGAS-shRNA-2 (sequence: CCGGCTTTGATAACTGCGTGACATACTCGAGTATGTCACGCAGTTATCAAAGTTTTTTG), IRF3-shRNA (sequence: CCGGGATCTGATTACCTTCACGGAACTCGAGTTCCGTGAAGGTAATCAGATCTTTTT), ATG5-shRNA (seq-uence: CCGGCCTGAACAGAATCATCCTTAACTCGAGTTAAGGATGATTCTGTTCAGGTTTTTTG), and LONP1-shRNA (sequence: CCGGCCAGCCTTATGTCGGCGTCTTCTCGAGAAGACGCCGACATAAGGCTGGTTTTTG). We seeded 5 × 105 cells in each well of a 12-well plate in 500 μl of complete medium and transduced by lentiviral vectors at a multiplicity of infection of 10:1. Transduction was carried out in the presence of polybrene (8 μg/ml; Sigma-Aldrich, TR-1003-G). After re-covering with complete culture medium, puromycin was used for the selection of transduced cells.
qRT-PCR assay
Total RNA was extracted using a QIAGEN RNeasy Plus Micro Kit (74034) according to the manufacturer’s instructions. Briefly, cell lysates were spun through gDNA Eliminator spin columns to remove genomic DNA. Total RNA was purified using RNeasy MinElute spin columns. First-strand cDNA was then synthesized from 1 µg of RNA using the iScript cDNA Synthesis kit (Bio-Rad, 1708890). Briefly, 20-μl reactions were prepared by combining 4 μl of iScript Select reaction mix, 2 μl of gene-specific enhancer solution, 1 μl of reverse transcriptase, 1 μl of gene-specific assay pool (20×, 2 μM), and 12 μl of RNA diluted in RNase-, DNase-, and genomic DNA-free water (Thermo Fisher Scientific, AM9935). We carried out a qRT-PCR using synthesized cDNA, primers (ALOXE3: 5ʹ- GCTGCTCTTCAATGCCATCCCT-3ʹ and 5ʹ- TGTCGTGAAGGTCTTATGGCACC-3ʹ; ALOX5: 5ʹ- GGAGAACCTGTTCATCAACCGC-3ʹ and 5ʹ-CAGGTCTTCCTGCCAGTGATTC-3ʹ; ALOX12: 5ʹ-CCTCGTTATGCTGAAGATGGAGC-3ʹ and 5ʹ- ATTTCGGACCCAGGACTTTGCC-3ʹ; ALOX12B: 5ʹ- TGCTGGAGACACACCTCATTGC-3ʹ and 5ʹ-GCCAATGCTGTTGATCTGGACG-3ʹ; ALOX15: 5ʹ-ACCTTCCTGCTCGCCTAGTGTT-3ʹ and 5ʹ- GGCTACAGAGAATGACGTTGGC-3ʹ; and ALOX15B: 5ʹ- CAATGCCGAGTTCTCCTTCCATG-3ʹ and 5ʹ-TGATGTGCAGGGTGTATCGGGT-3ʹ). The data were normalized to RNA18 S (5ʹ-CTACCACATCCAAGGAAGCA-3ʹ and 5ʹ-TTTTTCGTCACTACCTCCCCG-3ʹ) and the fold change was calculated via the 2−ΔΔCt method. The relative concentrations of mRNA were expressed in arbitrary units based on the untreated group, which was assigned a value of 1.
Detection of mtDNA
Total DNA was isolated from whole-cell extracts using the QIAquick Nucleotide Removal Kit (QIAGEN, 28304). A Human Mitochondrial DNA Copy Number Assay Kit (Detroit R&D, MCN1) was then used to measure mtDNA copy using qRT-PCR according to the manufacturer’s instructions. In brief, the following procedure was used for each 20 μL reaction: 1 μl forward primer, 1 μl reverse primer, 8 μl DNA samples between 0.5 to 3.75 ng/μl, and 10 μl rtPCR reaction mix. The validated primers to quantify mtDNA and rtPCR reaction mix were provided by this assay kit.
Western blot analysis
Cells were lysed in Cell Lysis Buffer (Cell Signaling Technology, 9803) with protease inhibitor cocktail (Promega, G6521), phosphatase inhibitor cocktail (Sigma-Aldrich, P0044), and 1 mM Na3VO4 (New England Biolabs, P0758 S). Cleared lysates were resolved by SDS-PAGE (Bio-Rad, 3450124) and then transferred onto PVDF membranes (Bio-Rad, 1704273) [85]. The membranes were blocked with Tris-buffered saline Tween 20 (TBST; Cell Signaling Technology, 9997) containing 5% skim milk (Cell Signaling Technology, 9999) for 1 h at room temperature and then incubated with the indicated primary antibodies (1:1000–1:2000) overnight at 4°C. After being washed with TBST, the membranes were incubated with an HRP-linked anti-mouse IgG secondary antibody (Cell Signaling Technology, 7076) or HRP-linked anti-rabbit IgG secondary antibody (Cell Signaling Technology, 7074) for 1 h at room temperature. The membranes were washed three times in TBST and then visualized and analyzed with a ChemiDoc Touch Imaging System (Bio-Rad, 1708370). The intensities of bands were analyzed with Image Lab software (Bio-Rad, 1709690).
Biochemical assay
Commercially available assay kits were used to measure the concentrations or activity of MDA (Abcam, ab118970), ATP (Abcam, ab83355), oxygen consumption (Abcam, ab197243), ROS (Abcam, ab113851), 8-OHdG (Abcam, ab201734), cGAMP (Cayman Chemical, 501700), and ACSL4 (MyBioSource, MBS9331516) in the indicated samples.
Autophagic flux analysis
The GFP-LC3-RFP-LC3ΔG probe was a gift from Dr. Noboru Mizushima (Addgene, 84572), which is a simple and quantitative method to evaluate autophagic flux in vitro or in vivo [47]. In brief, PANC1 cells (5000 cells/well) expressing GFP-RFP-LC3ΔG in a black 96-well plate with a clear bottom (Corning, 3904) were treated with zalcitabine (20 µM), erastin (10 µM), or RSL3 (0.5 µM) at the indicated times and the signals of GFP and RFP were analyzed using a microplate reader (Tecan).
Statistical analysis
Data are presented as mean ± SD except where otherwise indicated. Unpaired Student’s t-tests were used to compare the means of two groups. A one-way analysis of variance (ANOVA) was used for comparison among the different groups. When the ANOVA was significant, post hoc testing of differences between groups was performed using the least significant difference (LSD) test. The log-rank test was used to compare differences in survival rates between groups. A P value of <0.05 was considered statistically significant.
Acknowledgments
We thank Dave Primm (Department of Surgery, University of Texas Southwestern Medical Center) for his critical reading of the manuscript. D.T. is supported by a grant from the American Cancer Society (research scholar grant RSG-16-014-01-CDD). D.J.K. is supported by a grant from the U.S. National Institutes of Health (R35GM131919). C.L. is supported by a grant from the National Natural Science Foundation of China (81872323). J.L. is supported by grants from the National Natural Science Foundation of China (31671435, 81400132, and 81772508).
Funding Statement
D.T. is supported by a grant from the American Cancer Society (RSG-16-014-01-CDD). D.J.K. is supported by a grant from the U.S. National Institutes of Health (R35GM131919). C.L. is supported by a grant from the National Natural Science Foundation of China (81872323). J.L. is supported by grants from the National Natural Science Foundation of China (31671435, 81400132, and 81772508).
Disclosure statement
No potential conflicts of interest were disclosed.
References
- [1].Yang Z, Klionsky DJ.. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12(9):814–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Klionsky DJ. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290(5497):1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176(1–2):11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Denton D, Kumar S. Autophagy-dependent cell death. Cell Death Differ. 2019;26(4):605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kriel J, Loos B. The good, the bad and the autophagosome: exploring unanswered questions of autophagy-dependent cell death. Cell Death Differ. 2019;26(4):640–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25:486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tang D, Kang R, Berghe TV, et al. The molecular machinery of regulated cell death. Cell Res. 2019;29:347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhou B, Liu J, Kang R, et al. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. 2019. DOI: 10.1016/j.semcancer.2019.03.002 [DOI] [PubMed] [Google Scholar]
- [11].Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer. 2013;13(5):342–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gao M, Monian P, Pan Q, et al. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26(9):1021–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wu Z, Geng Y, Lu X, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci U S A. 2019;116(8):2996–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yang M, Chen P, Liu J, et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv. 2019;5(7):eaaw2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kang R, Zhu S, Zeh HJ, et al. BECN1 is a new driver of ferroptosis. Autophagy. 2018;14(12):2173–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Liu J, Yang M, Kang R, et al. Autophagic degradation of the circadian clock regulator promotes ferroptosis. Autophagy. 2019;15(11):2033–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Song X, Zhu S, Chen P, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc(-) activity. Curr Biol. 2018;28(15):2388–99 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yagoda N, von Rechenberg M, Zaganjor E, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447(7146):864–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gao M, Yi J, Zhu J, et al. Role of mitochondria in ferroptosis. Mol Cell. 2019;73(2):354–63 e3. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tomita K, Takashi Y, Ouchi Y, et al. Lipid peroxidation increases hydrogen peroxide permeability leading to cell death in cancer cell lines that lack mtDNA. Cancer Sci. 2019;110(9):2856–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Louat T, Servant L, Rols MP, et al. Antitumor activity of 2ʹ,3ʹ-dideoxycytidine nucleotide analog against tumors up-regulating DNA polymerase beta. Mol Pharmacol. 2001;60(3):553–558. [PubMed] [Google Scholar]
- [25].Liyanage SU, Hurren R, Voisin V, et al. Leveraging increased cytoplasmic nucleoside kinase activity to target mtDNA and oxidative phosphorylation in AML. Blood. 2017;129(19):2657–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chen CH, Cheng YC. Delayed cytotoxicity and selective loss of mitochondrial DNA in cells treated with the anti-human immunodeficiency virus compound 2ʹ,3ʹ-dideoxycytidine. J Biol Chem. 1989;264(20):11934–11937. [PubMed] [Google Scholar]
- [27].Ullyatt E, Liang BC. 2ʹ,3ʹ-dideoxycytidine is a potent inducer of apoptosis in glioblastoma cells. Anticancer Res. 1998;18(3A):1859–1863. [PubMed] [Google Scholar]
- [28].Szymanski MR, Kuznetsov VB, Shumate C, et al. Structural basis for processivity and antiviral drug toxicity in human mitochondrial DNA replicase. Embo J. 2015;34(14):1959–1970. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kleeff J, Kornmann M, Sawhney H, et al. Actinomycin D induces apoptosis and inhibits growth of pancreatic cancer cells. Int J Cancer. 2000;86(3):399–407. [DOI] [PubMed] [Google Scholar]
- [30].Park JH, Jung KH, Kim SJ, et al. HS-173 as a novel inducer of RIP3-dependent necroptosis in lung cancer. Cancer Lett. 2019;444:94–104. [DOI] [PubMed] [Google Scholar]
- [31].Kang D, Kim SH, Hamasaki N. Mitochondrial transcription factor A (TFAM): roles in maintenance of mtDNA and cellular functions. Mitochondrion. 2007;7(1–2):39–44. [DOI] [PubMed] [Google Scholar]
- [32].Amaral A, Ramalho-Santos J, St John JC. The expression of polymerase gamma and mitochondrial transcription factor A and the regulation of mitochondrial DNA content in mature human sperm. Hum Reprod. 2007;22(6):1585–1596. [DOI] [PubMed] [Google Scholar]
- [33].Lan L, Guo M, Ai Y, et al. Tetramethylpyrazine blocks TFAM degradation and up-regulates mitochondrial DNA copy number by interacting with TFAM. Biosci Rep. 2017;37(3). DOI: 10.1042/BSR20170319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lu B, Lee J, Nie X, et al. Phosphorylation of human TFAM in mitochondria impairs DNA binding and promotes degradation by the AAA+ lon protease. Mol Cell. 2013;49(1):121–132. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Granot Z, Kobiler O, Melamed-Book N, et al. Turnover of mitochondrial steroidogenic acute regulatory (StAR) protein by lon protease: the unexpected effect of proteasome inhibitors. Mol Endocrinol. 2007;21(9):2164–2177. . [DOI] [PubMed] [Google Scholar]
- [36].Miranda S, Foncea R, Guerrero J, et al. Oxidative stress and upregulation of mitochondrial biogenesis genes in mitochondrial DNA-depleted HeLa cells. Biochem Biophys Res Commun. 1999;258(1):44–49. [DOI] [PubMed] [Google Scholar]
- [37].Stumpf JD, Saneto RP, Copeland WC. Clinical and molecular features of POLG-related mitochondrial disease. Cold Spring Harb Perspect Biol. 2013;5(4):a011395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].West AP, Khoury-Hanold W, Staron M, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520(7548):553–557. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sun L, Wu J, Du F, et al. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339(6121):786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ablasser A, Chen ZJ. cGAS in action: expanding roles in immunity and inflammation. Science. 2019;363(6431):eaat8657. [DOI] [PubMed] [Google Scholar]
- [41].Haag SM, Gulen MF, Reymond L, et al. Targeting STING with covalent small-molecule inhibitors. Nature. 2018;559(7713):269–273. [DOI] [PubMed] [Google Scholar]
- [42].Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455(7213):674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Barber GN. STING: infection, inflammation and cancer. Nat Rev Immunol. 2015;15(12):760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet. 2019;20(11):657–674. [DOI] [PubMed] [Google Scholar]
- [45].Liu D, Wu H, Wang C, et al. STING directly activates autophagy to tune the innate immune response. Cell Death Differ. 2019;26(9):1735–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gui X, Yang H, Li T, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature. 2019;567(7747):262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Kaizuka T, Morishita H, Hama Y, et al. An autophagic flux probe that releases an internal control. Mol Cell. 2016;64(4):835–849. . [DOI] [PubMed] [Google Scholar]
- [48].Yuan H, Li X, Zhang X, et al. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478(3):1338–1343. [DOI] [PubMed] [Google Scholar]
- [49].Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. [DOI] [PubMed] [Google Scholar]
- [51].Vyas S, Zaganjor E, Haigis MC. Mitochondria and cancer. Cell. 2016;166(3):555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lewis W, Day BJ, Kohler JJ, et al. Decreased mtDNA, oxidative stress, cardiomyopathy, and death from transgenic cardiac targeted human mutant polymerase gamma. Lab Invest. 2007;87(4):326–335. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Walker UA, Bauerle J, Laguno M, et al. Depletion of mitochondrial DNA in liver under antiretroviral therapy with didanosine, stavudine, or zalcitabine. Hepatology. 2004;39(2):311–317. [DOI] [PubMed] [Google Scholar]
- [54].Reiss P, Casula M, de Ronde A, et al. Greater and more rapid depletion of mitochondrial DNA in blood of patients treated with dual (zidovudine+didanosine or zidovudine+zalcitabine) vs. single (zidovudine) nucleoside reverse transcriptase inhibitors. HIV Med. 2004;5(1):11–14. [DOI] [PubMed] [Google Scholar]
- [55].Benbrik E, Chariot P, Bonavaud S, et al. Cellular and mitochondrial toxicity of zidovudine (AZT), didanosine (ddI) and zalcitabine (ddC) on cultured human muscle cells. J Neurol Sci. 1997;149(1):19–25. [DOI] [PubMed] [Google Scholar]
- [56].Hance N, Ekstrand MI, Trifunovic A. Mitochondrial DNA polymerase gamma is essential for mammalian embryogenesis. Hum Mol Genet. 2005;14(13):1775–1783. [DOI] [PubMed] [Google Scholar]
- [57].Singh KK, Ayyasamy V, Owens KM, et al. Mutations in mitochondrial DNA polymerase-gamma promote breast tumorigenesis. J Hum Genet. 2009;54(9):516–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246(4929):500–503. [DOI] [PubMed] [Google Scholar]
- [59].Morais R, Zinkewich-Peotti K, Parent M, et al. Tumor-forming ability in athymic nude mice of human cell lines devoid of mitochondrial DNA. Cancer Res. 1994;54(14):3889–3896. [PubMed] [Google Scholar]
- [60].Chen H, Wang J, Liu Z, et al. Mitochondrial DNA depletion causes decreased ROS production and resistance to apoptosis. Int J Mol Med. 2016;38(4):1039–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Liu CY, Lee CF, Hong CH, et al. Mitochondrial DNA mutation and depletion increase the susceptibility of human cells to apoptosis. Ann N Y Acad Sci. 2004;1011(1):133–145. [DOI] [PubMed] [Google Scholar]
- [62].Ferraresi R, Troiano L, Pinti M, et al. Resistance of mtDNA-depleted cells to apoptosis. Cytometry A. 2008;73(6):528–537. [DOI] [PubMed] [Google Scholar]
- [63].Wang J, Silva JP, Gustafsson CM, et al. Increased in vivo apoptosis in cells lacking mitochondrial DNA gene expression. Proc Natl Acad Sci U S A. 2001;98(7):4038–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Spadafora D, Kozhukhar N, Chouljenko VN, et al. Methods for efficient elimination of mitochondrial DNA from cultured cells. PLoS One. 2016;11(5):e0154684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Schubert S, Heller S, Loffler B, et al. Generation of rho zero cells: visualization and quantification of the mtDNA depletion process. Int J Mol Sci. 2015;16(12):9850–9865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Magda D, Lecane P, Prescott J, et al. mtDNA depletion confers specific gene expression profiles in human cells grown in culture and in xenograft. BMC Genomics. 2008;9(1):521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kang I, Chu CT, Kaufman BA. The mitochondrial transcription factor TFAM in neurodegeneration: emerging evidence and mechanisms. FEBS Lett. 2018;592(5):793–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Yang M, Li C, Zhu S, et al. TFAM is a novel mediator of immunogenic cancer cell death. Oncoimmunology. 2018;7(6):e1431086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Araujo LF, Siena ADD, Placa JR, et al. Mitochondrial transcription factor A (TFAM) shapes metabolic and invasion gene signatures in melanoma. Sci Rep. 2018;8(1):14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Lu B. Mitochondrial lon protease and cancer. Adv Exp Med Biol. 2017;1038:173–182. [DOI] [PubMed] [Google Scholar]
- [71].Quiros PM, Espanol Y, Acin-Perez R, et al. ATP-dependent lon protease controls tumor bioenergetics by reprogramming mitochondrial activity. Cell Rep. 2014;8(2):542–556. [DOI] [PubMed] [Google Scholar]
- [72].Yuan H, Li X, Zhang X, et al. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem Biophys Res Commun. 2016;478(2):838–844. [DOI] [PubMed] [Google Scholar]
- [73].Wang YQ, Chang SY, Wu Q, et al. The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front Aging Neurosci. 2016;8:308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Song X, Xie Y, Kang R, et al. FANCD2 protects against bone marrow injury from ferroptosis. Biochem Biophys Res Commun. 2016;480(3):443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Chen PH, Wu J, Ding CC, et al. Kinome screen of ferroptosis reveals a novel role of ATM in regulating iron metabolism. Cell Death Differ. 2020;27(3):1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sun F, Liu Z, Yang Z, et al. The emerging role of STING-dependent signaling on cell death. Immunol Res. 2019;67(2–3):290–296. [DOI] [PubMed] [Google Scholar]
- [77].Liu S, Guan W. STING signaling promotes apoptosis, necrosis, and cell death: an overview and update. Mediators Inflamm. 2018;2018:1202797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Bhatelia K, Singh K, Prajapati P, et al. MITA modulated autophagy flux promotes cell death in breast cancer cells. Cell Signal. 2017;35:73–83. [DOI] [PubMed] [Google Scholar]
- [79].Zhang Z, Yao Z, Wang L, et al. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy. 2018;14(12):2083–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Zhang Z, Guo M, Li Y, et al. RNA-binding protein ZFP36/TTP protects against ferroptosis by regulating autophagy signaling pathway in hepatic stellate cells. Autophagy. 2019;1–24. doi: 10.1080/15548627.2019.1687985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Bai Y, Meng L, Han L, et al. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun. 2019;508(4):997–1003. . [DOI] [PubMed] [Google Scholar]
- [82].Eliopoulos AG, Havaki S, Gorgoulis VG. DNA Damage response and autophagy: a meaningful partnership. Front Genet. 2016;7:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Dai E, Han L, Liu J, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. 2020;1–15. DOI: 10.1080/15548627.2020.1714209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Marchesi F, Monti P, Leone BE, et al. Increased survival, proliferation, and migration in metastatic human pancreatic tumor cells expressing functional CXCR4. Cancer Res. 2004;64(22):8420–8427. [DOI] [PubMed] [Google Scholar]
- [85].Tang D, Kang R, Livesey KM, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190(5):881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]