

ABSTRACT
Human parainfluenza virus type III (HPIV3) is a common respiratory pathogen that afflicts children and can be fatal in vulnerable populations, including the immunocompromised. There are currently no effective vaccines or therapeutics available, resulting in tens of thousands of hospitalizations per year. In an effort to discover a protective antibody against HPIV3, we screened the B cell repertoires from peripheral blood, tonsils, and spleen from healthy children and adults. These analyses yielded five monoclonal antibodies that potently neutralized HPIV3 in vitro. These HPIV3-neutralizing antibodies targeted two non-overlapping epitopes of the HPIV3 F protein, with most targeting the apex. Prophylactic administration of one of these antibodies, PI3-E12, resulted in potent protection against HPIV3 infection in cotton rats. Additionally, PI3-E12 could also be used therapeutically to suppress HPIV3 in immunocompromised animals. These results demonstrate the potential clinical utility of PI3-E12 for the prevention or treatment of HPIV3 in both immunocompetent and immunocompromised individuals.
KEYWORDS: Parainfluenza, respiratory viruses, fusion protein, neutralizing antibodies, immunosuppression
Introduction
Human parainfluenza virus type III (HPIV3) is a common cause of respiratory illness in infants and children. Over 11,000 hospitalizations per year in the US occur for fever or acute respiratory illness due to HPIV3.1 HPIV3, like respiratory syncytial virus (RSV), infects early in life and frequently causes severe bronchiolitis and pneumonia in infants under 6 months of age who are unable to mount a robust antibody response.2,3 HPIV3 is also an important cause of mortality, morbidity, and health-care costs in other vulnerable populations, such as immunocompromised hematopoietic stem cell transplant (HCT) recipients.4 Up to a third of HCT recipients acquire a respiratory viral infection within 6 months of transplant.5–11 In up to a third of those patients, the virus progresses from the upper to the lower respiratory tract.6,9 Once the virus gains a foothold in the lower tract, little can be done for most patients beyond supportive care, and up to 40% of patients with lower tract disease die within 3 months. HPIV3 is an important cause of serious respiratory viral infections after HCT, with a cumulative incidence of 18% post–transplant at the Fred Hutchinson Cancer Research Center.5,12–14 In the absence of any vaccine or therapy, there is a substantial need for preventive and therapeutic interventions against HPIV3.
Neutralizing monoclonal antibodies (mAbs) have been correlated with protection against several respiratory viruses, including RSV and influenza.15–19 Palivizumab, a humanized mAb targeting the Fusion (F) protein of RSV, is approved for use as immunoprophylaxis to prevent severe disease in high-risk infants.20 The F protein of RSV is an essential surface glycoprotein, and therefore a major neutralizing antibody target. As a class I fusion protein, F mediates viral entry by transitioning between a metastable prefusion (preF) conformation and a stable postfusion (postF) conformation. Since preF is the major conformation on infectious virus, antibodies to preF are the most potent at neutralizing virus, whereas antibodies targeting postF generally are not.21,22 Similar to RSV, the F protein of HPIV3 also adopts preF and postF conformations.23,24 HPIV3 F was recently stabilized in the preF conformation and induced higher serum neutralizing titers than the HPIV3 postF conformation.25 In an effort to isolate mAb candidates for prevention and therapy, we developed a high-throughput screening strategy that enabled the rapid selection and testing of human HPIV3 preF-specific B cells for the ability to neutralize HPIV3. We used this method to isolate several potent neutralizing mAbs, characterized their binding, and tested one of these antibodies in an in vivo challenge model.
Results
Identification and isotype of HPIV3-specific B cells within the human B cell repertoire
We biotinylated the HPIV3 F protein in either the preF or postF conformation and mixed each with fluorochrome-labeled streptavidin. We then enriched for HPIV3 preF- and postF-binding B cells using magnetic microbeads conjugated to antibodies targeting the fluorochrome. Using this approach, we identified B cells that specifically bound the preF conformation and not the postF conformation or fluorochrome-labeled streptavidin using flow cytometry (Figure 1). Since the seroprevalence to HPIV3 in humans is almost complete,26 we did not need to pre-screen donors for sero–positivity against HPIV3. Using this approach, we found on average 0.13% (95% confidence interval 0.10–0.15, N = 6 donors) of B cells in peripheral blood bound HPIV3 preF.
Figure 1.

Screening human PBMCs for HPIV3-specific B cells. HPIV3-specific B cells were labeled with APC-conjugated streptavidin tetramers of biotinylated HPIV3 prefusion (preF) protein followed by magnetic enrichment using microbeads against APC. Representative flow cytometry plot of enriched HPIV3-specific B cells after gating on live (fixable viability dye negative), CD3/CD14/CD16−, and CD19+ B cells. Cells in the red box of the enriched fraction are B cells that bind the preF but not postfusion (postF) conformation of the HPIV3 F protein. The percentage is of total cells shown in the flow plot. N = 6 donors
Binding and neutralization of HPIV3 within the human B cell repertoire
We next sought to determine how many of the HPIV3 preF-binding B cells identified by flow cytometry produced antibodies that could bind and neutralize HPIV3. For this, we sorted 92 HPIV3 preF-binding B cells from two donor peripheral blood mononuclear cell (PBMC) samples into individual wells of a 96-well plate and obtained 25 paired heavy and light chain sequences (Supplemental Table 1). We expressed all 25 as mAbs and confirmed binding to purified HPIV3 preF for 100% (25/25) of antibodies using bio-layer interferometry (BLI) (Figure 2a), highlighting the specificity of our approach. Among this group, two mAbs, PI3-E12 and PI3-C9, demonstrated the highest signal during the association phase, suggesting a possible difference in binding stoichiometry compared to the other mAbs. However, PI3-E12, but not PI3-C9, was capable of neutralizing live virus in vitro (Figure 2b). Together, these data indicated that a low frequency of HPIV3 preF tetramer-binding B cells express high-affinity antibodies capable of neutralizing HPIV3.
Figure 2.
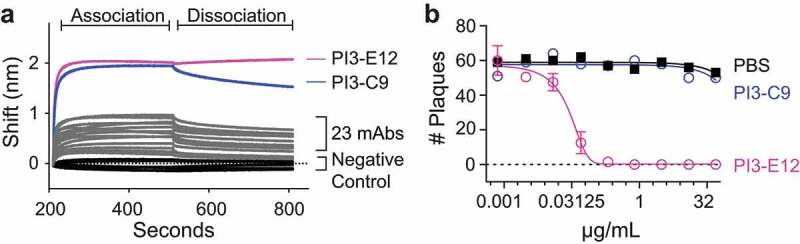
Identification of a potent HPIV3-neutralizing antibody from HPIV3 preF-binding B cells. (a) Bio-Layer Interferometry (BLI) measurements of association and dissociation between HPIV3 preF and 25 monoclonal antibodies (mAbs) cloned directly from individually sorted HPIV3 preF-binding B cells. (b) Plaque reduction neutralization test of the PI3-E12 mAb (N = 2 per group). Error bars in b represent standard deviation
To focus upon B cells producing neutralizing antibodies, we modified our assay to sort individual B cells onto irradiated 3T3 feeder cells expressing CD40L, IL-2, and IL-21 to allow for higher throughput screening of culture supernatants for neutralization prior to antibody cloning, as described.27 In general, over half of sorted B cells and 87% of sorted IgD− B cells produced antibody levels detectable by enzyme-linked immunosorbent assay (ELISA) (Figure 3a). Aiming to improve our yield for isolating B cells capable of producing neutralizing antibodies, we excluded IgD-expressing cells since these cells would be the least likely to have undergone the somatic hypermutation and affinity maturation necessary for potent neutralization. We also included tonsils and spleens from unmatched donors, since these secondary lymphoid organs might be enriched for B cells that had undergone affinity maturation for binding HPIV3. Using this approach, we found that 14% of IgD− HPIV3 preF-binding B cells sorted from tonsils produced HPIV3-neutralizing antibodies, as compared to 5% from the spleen and 2% from peripheral blood (Figure 3b).
Figure 3.
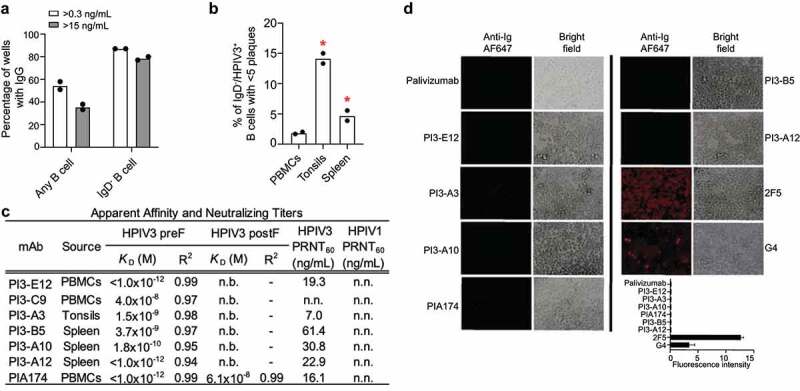
Higher throughput screening of human PBMCs, tonsils, and spleen for B cells capable of producing neutralizing antibodies against HPIV3. a) Detection of IgG by ELISA in supernatant from total B cells and IgD− B cells of unknown specificities (N = 2 donors) individually sorted and expanded on irradiated IL-21+/IL-2+/CD40L+ 3T3 feeder cells. (b) Plaque reduction neutralization screen of supernatant from HPIV3 preF-specific B cells individually sorted and expanded on feeder cells. N = 2 donors in each group with a total of 120 cells from PBMCs, 120 cells from tonsils, and 1,235 cells from spleen. (c) Analysis of apparent affinity (KD) and neutralization potency of monoclonal antibodies isolated from the neutralization screen. Binding to HPIV3 preF or postF at 0.5, 0.25, 0.125, and 0.0625 µM was measured by BLI. The dissociation step was extended to 600 s. Error bars represent standard deviation. R2 represents the coefficient of determination. N.b. indicates no binding detected. Neutralizing titers of HPIV3-specific monoclonal antibodies were determined by 60% plaque reduction neutralization tests on Vero cells using green fluorescent protein (GFP)-labeled HPIV3 or HPIV1. N.n. indicates non-neutralizing. (d) Auto-reactivity antibody assay in HEp-2 cells using mAbs targeting HPIV3 preF. Binding was detected using a secondary Alexa Fluor 647 (AF647)-conjugated goat anti-human antibody. The mAb palivizumab was used as a negative control and 2F5 and G4 as a positive control for autoreactivity. The average fluorescence intensity was calculated from two independent experiments and error bars represent standard deviation
From these cultures, we cloned four additional HPIV3-neutralizing mAbs named PI3-A3, PI3-B5, PI3-A10, and PI3-A12 (Figure 3c). None of these antibodies could neutralize the related virus HPIV1. Since we envisioned these antibodies potentially being administered clinically in the IgG1 format, similar to palivizumab, we measured apparent binding affinity, which incorporates avidity (Figure 3c, Supplemental Figure 1, and Supplemental Table 2). Under avid binding conditions, all antibodies bound with a high apparent binding affinity to HPIV3 preF (Supplemental Figure 1 and Supplemental Table 2). The neutralization potency of these antibodies ranged from 7.0 to 61.4 ng/mL. Each neutralizing mAb used different immunoglobulin heavy and light chain alleles except for PI3-A3 and PI3–B5, which both utilized the kappa allele 1–5*03 (Supplemental Table 3). None of the alleles matched those of the previously described HPIV3 antibody PIA174.25 The similarity to germline sequences of the variable genes from these neutralizing antibodies ranged from 90–97% (Supplemental Table 3). All of these newly described antibodies bound strongly to the preF conformation without any detectable binding to the postF conformation (Figure 3c), as expected given the exclusion of B cells binding postF during the sort. In contrast, the previously described antibody PIA174 bound weakly to the postF conformation. In anticipation of administering these antibodies in vivo, we confirmed that none bound to permeabilized HEp-2 cells (Figure 3d), a common assessment of autoreactivity.28,29
We next performed cross-competition binding experiments to gauge the antigenic sites on HPIV3 preF allowing for neutralization. Three of these five new neutralizing mAbs (PI3-E12, -A3, and -B5) fully competed with each other and the previously described antibody PIA174 (Figure 4a). PI3-A10 also competed with this group, but only partially with PI3-E12 (Figure 4a). Based on the known binding site of PIA174, this antigenic site is likely located at the apex of HPIV3 preF.25 We propose calling this antigenic site Ø on HPIV3 preF for consistency, since the apices of RSV and HMPV preF are also called antigenic site Ø.30,31 The fifth neutralizing monoclonal, PI3-A12, only weakly competed with PI3-A10 and not at all with the others, suggesting the presence of an antigenic site vulnerable to neutralization by antibodies outside of antigenic site Ø (Figure 4a).
Figure 4.

Structural analysis of monoclonal antibodies against HPIV3 preF. (a) Epitope binning of mAbs using the Octet system. Penta-HIS probes were coated with HIS-tagged HPIV3 preF. The mAb listed on the left–side of the chart was loaded first onto the coated probe followed by the mAb listed on the top of the chart. Values represent the level of competition between antibodies for the same binding site on HPIV3 preF. This is expressed as the percent drop in maximum signal of the top mAb in the presence of the left mAb compared to the maximum signal of the top mAb alone. Red boxes represent 80–100% competition for the same binding site, yellow boxes represent 40–80% competition, and white boxes represent 0–40% competition. (b, c) Negative stain electron microscopy (nsEM) 2D classifications of HPIV3 preF in complex with PI3-A12 Fab (b) and PI3-E12 Fab (c) with 3D reconstruction. Coordinates of HPIV3 preF trimer (blue, PDB ID 6MJZ), trimeric domain GCN4 (orange, PDB ID 4DME), and crystal structure of PI3-E12 Fab (green, this paper) were fitted in the 3D map. (d) Structure of PI3-E12 Fab with CDRs colored and labeled. Left, cartoon representation. Right, surface representation. (e) Structural alignment with PIA174 (shown in blue). (f) Sequence alignment of PI3-E12 and PIA174. Alignment with germline (gl) CDRL1 sequences are also shown with mutations from germline highlighted in red
We performed negative stain electron microscopy (nsEM) of PI3-E12 Fab, PI3-A12 Fab, and PI3-C9 Fab in complex with HPIV3 preF (Figure 4b, c) to confirm the binding location of these antibodies. Although we could form a complex by size exclusion chromatography (SEC) of HPIV3 preF with PI3-C9 Fab, nsEM did not show bound PI3-C9 Fab molecules. 2D classifications and 3D reconstruction indicated that PI3-A12 Fab was bound to the side of HPIV3 preF in a 3:1 ratio, confirming that its epitope does not overlap with that of previously described PIA174 antibody and defining a new site of neutralization on HPIV3 preF (Figure 4b). Of note, this site is reminiscent of site V on RSV F.22 As predicted earlier by our BLI and competition experiments, 2D classifications and 3D reconstruction showed that the PI3-E12 Fab bound at the apex of HPIV3 preF in a 1:1 ratio (Fab:trimer). We obtained a 2.1 Å structure of PI3-E12 Fab using X-ray crystallography (Figure 4d, Supplemental Table 4). We superimposed the structure of PI3-E12 and PIA174 Fabs (root mean standard deviation: 1.4 Å2) and the main differences in structural organization lay within the CDRL1, which is longer in PI3-E12 and CDRH3 (Figure 4e, f). Both CDRL1 sequences in PI3-E12 and PIA174 contained mutations from germline (Figure 4f).
Together, our results indicate that the HPIV3 preF apical antigenic site Ø is a common target of neutralizing antibodies that can be accessed by antibodies using different gene segments.
In vivo protection against HPIV3 infection
We next investigated the potential clinical utility of mAb administration in an animal challenge model of HPIV3 infection. We chose to focus our efforts on mAbs that bound site Ø, since a mAb against RSV targeting this homologous site, nirsevimab, is in late-stage clinical trials.32 Therefore, we selected PI3-E12 for in vivo testing because it was among the most potent neutralizing antibodies targeting site Ø and, unlike PIA174, did not bind to the post–fusion conformation of HPIV3 F. Although the human parainfluenza viruses do not replicate in mice, lower respiratory tract viral replication can be demonstrated in cotton rats infected intranasally with HPIV3.26,33 The cotton rat model was used in the past to predict not only the efficacy of antibody immunoprophylaxis, but also the exact dose of palivizumab, 15 mg/kg, that would be effective against RSV in human infants.20 Therefore, we adopted a similar experimental design and injected 0.625–5 mg/kg of PI3-E12 intramuscularly 1 day prior to intranasal infection of cotton rats with 105 plaque-forming units (pfu) of HPIV3 (Figure 5a).
Figure 5.

Efficacy of prophylactic and therapeutic administration of the neutralizing HPIV3 mAb PI3-E12 in vivo. (a) Schematic of experiments in which cotton rats were injected intramuscularly with 0.625 – 5 mg/kg of PI3-E12 one day prior to intranasal challenge with 105 pfu HPIV3 (N = 4). (b) HPIV3 titers by plaque assay in nasal and lung homogenates at day four post-infection. Error bars represent standard deviation and asterisks indicate P < .05 by t-test compared to control mice injected with PBS
The amount of HPIV3 detected in the lung was reduced ~6-fold at the lowest tested dose of 0.625 mg/kg PI3-E12 and was below the limit of detection in 8/9 animals injected with 2.5 mg/kg or more (Figure 5b).
More modest reductions in HPIV3 replication in the nose were also detected (Figure 5b), which was expected given the relatively poor ability of IgG antibodies to enter this compartment.34,35 Together, the data indicate an EC50 of 0.35 mg/kg and an EC99 of 1.80 mg/kg for PI3-E12-mediated prevention of HPIV3 infection in the lungs.
Since patients receiving cytotoxic therapy for cancer or autoimmune diseases and other immunocompromised groups are at the highest risk for severe disease and mortality due to HPIV3 infection, we tested the efficacy of PI3–E12 as treatment in immunosuppressed animals. For this, we adopted a similar experimental design used to model RSV in immunocompromised cotton rats in which the drug Cytoxan (cyclophosphamide) is administered to deplete lymphocytes.36–38 Animals were treated with 5 mg/kg of Cytoxan injected every 3 days for 21 days prior to intranasal infection with 105 pfu of HPIV3 (Figure 6a). This regimen led to stable lymphopenia, with absolute lymphocyte counts decreasing from an average of 6,200 cells/µL to 2,000 cells/µL (Figure 6b). Five days after infection, ~104 pfu/g could be detected in the lungs and nose of control animals that did not receive PI3-E12 (Figure 6c). In contrast, viral titers were diminished 28-fold in the lungs and 2-fold in the nose when 5 mg/kg of PI3-E12 was injected 1 day after infection (Figure 6c).
Figure 6.

Efficacy of therapeutic administration of the neutralizing HPIV3 mAb PI3-E12 in vivo. (a) Schematic of experiments in which cotton rats were injected intramuscularly 5 mg/kg Cytoxan every three days for three weeks prior to infection with 105 PFU of HPIV3 with or without 5 mg/kg PI3-E12 one day later. (N = 5). (b) White blood cell counts (WBC) and absolute lymphocyte counts (ALC) prior to Cytoxan treatment and after 21 days of Cytoxan treatment. (c) HPIV3 titers by plaque assay in nasal and lung homogenates at day four post-infection. Error bars represent standard deviation and asterisks indicate P < .05 by t-test compared to pre-Cytoxan treatment in (b) or to control mice injected with PBS in (c)
Together, our data indicate that PI3-E12 can both prevent and treat HPIV3 infection.
Discussion
The ability to isolate neutralizing mAbs and to identify their antigenic-binding sites has revolutionized our ability to understand, prevent, and treat viral infections, some of which include HIV, Ebola, RSV, influenza, and the newly emerged SARS-CoV-2, responsible for the current COVID–19 pandemic.39–41 One of the goals of this study was to quantify the frequency of B cells capable of producing neutralizing antibodies against HPIV3. We designed our B cell probes and flow cytometry panel to allow the selection of B cells that bind specifically to the preF but not the postF conformation of HPIV3 F protein. Even with this selection strategy, we found that the majority of HPIV3 preF-specific B cells failed to neutralize virus. This is not surprising, since B cells undergo positive selection based on signals that stem from the affinity of binding between their immunoglobulin receptor and cognate antigen, regardless of neutralization.42,43 In the circulating peripheral blood of healthy individuals, the frequency of HPIV3 preF-binding B cells was only 0.13% of all B cells, and the frequency of HPIV3-neutralizing B cells was only 2% of IgD−, HPIV3 preF-binding B cells. As a result, our original strategy of sorting and directly cloning antibodies from individual HPIV3 preF-specific B cells from peripheral blood identified predominantly low-affinity naïve B cells, was laborious, expensive, and inefficient, and yielded only a single neutralizing mAb. Therefore, we switched to a higher-throughput neutralization screening strategy for HPIV3 based upon an assay developed for HIV.27,44 This allowed us to scan thousands of individual B cells from peripheral blood, tonsils, and spleens and select only those that produced neutralizing antibodies against HPIV3 for subsequent mAb cloning. Using this method, we confirmed the low frequency of HPIV3 preF-specific B cells able to produce neutralizing antibodies and isolated four additional potent HPIV3-neutralizing mAbs.
Many human-derived neutralizing mAbs are based on B cells found in peripheral blood.22,44,45 We decided to compare the frequency of B cells capable of producing neutralizing antibodies in readily accessible secondary lymphoid organs that might be enriched for B cells that had undergone affinity maturation. We therefore sampled human tonsils from children undergoing elective tonsillectomy and human spleens from previously healthy adult deceased organ donors. Since virtually all children by the age of three demonstrate serologic evidence of infection by parainfluenza virus, we did not need to screen donors for evidence of previous infection.26 We found that tonsils were significantly enriched with B cells capable of producing HPIV3-neutralizing mAbs. Although tonsillectomy is a common procedure and has long been thought to have negligible long-term health effects, more recent data suggest tonsillectomy may be associated with increased long-term risks for respiratory infections.46 Human spleens were also enriched for B cells capable of neutralizing HPIV3, although the magnitude of enrichment was much lower than in tonsils.
The majority of neutralizing antibodies against HPIV3 appeared to target the apex of the F protein in a similar fashion to neutralizing antibodies against RSV.30,47 The ability of PI3-E12 to bind the apex of HPIV3 preF in a ratio of 1 Fab: 1 trimer is also reminiscent of the binding mode of VRC26.25, the most potent HIV V1V2-recognizing antibody isolated to date.48 PI3-E12 showed high specificity for the pre-fusion conformation of HPIV3 F, unlike the previously isolated apical binding mAb PIA174, which also bound weakly to the post-fusion conformation. The ability of PIA174 to bind the post-fusion conformation was unexpected since the antigenic site at the apex is unique to the prefusion conformation. In the related fusion protein of RSV, the apical antigenic site consists of an unstructured region and an alpha-helix that are displaced by more than 5 Å in the post-fusion conformation.49 It is possible that PIA174 is able to bind weakly to small stretches of linear epitopes found in both the pre- and post-fusion conformations. Interestingly, the mAbs we isolated all used different immunoglobulin heavy and light chain alleles. A similar phenomenon was previously described in which human antibodies targeting the receptor binding site of hemagglutinin were found to arise from nearly unrestricted germ–line origins from multiple donors, and as a result viral resistance to one antibody did not confer resistance to all.50 This suggests a wide variety of evolved solutions to the problem of blocking viral attachment and binding are available in the general population, making this antigenic site an appealing vaccine target.51
Since HPIV types 1–4 are related, cross-neutralizing antibodies may exist in the B cell repertoire. Of the parainfluenza viruses, HPIV1 and HPIV3 are phylogenetically the most closely related, belonging to the same Respirovirus genus. However, immunity is type-specific and vaccination of non-human primates with one type does not induce cross-neutralization.25 The resolution of available structural maps has limited the analysis of potential epitope conservation between HPIV3 and HPIV1.25 None of the HPIV3-neutralizing antibodies described here neutralize HPIV1.
Given the in vitro potency of PI3-E12, we anticipated a low EC50. Similar to palivizumab for RSV, a dose of at least 2.5 mg/kg reduced HPIV3 levels in the lungs in virtually all animals. Additionally, 5 mg/kg of PI3-E12 significantly reduced HPIV3 replication in the nose, in contrast to palivizumab which failed to suppress RSV replication in the nose even at a dose of 8 mg/kg.52,53 Given the ability of PI3-E12 to suppress HPIV3 replication in the nose and lungs of immunocompromised animals when given after infection, PI3-E12 could play a role in both prophylaxis or therapy against HPIV3 infections in the HCT population. Testing the in vivo efficacy of potent HPIV3-neutralizing antibodies at additional, later time-points in cotton rats and non-human primates would provide further insights into the therapeutic window. We also identified PI3-A12, an mAb that binds a site of vulnerability to neutralization outside of site Ø on HPIV3 F. Antibodies binding this site could also hold potential clinical utility. We have recently described a method of engineering B cells using CRISPR/Cas9 to express palivizumab and conferred protection against RSV in naïve animals by adoptive transfer of these cells.54 In this revolutionary new age of cellular therapy and immunotherapy against cancer, it is conceivable that B cells could be engineered to produce a variety of protective antibodies against multiple pathogens and transferred along with the stem cell product during transplant as part of treatment for an underlying disease.
Materials and methods
Study design
The size of experimental groups is specified in figure legends. Peripheral blood was obtained by venipuncture from healthy, HIV-seronegative adult volunteers enrolled in the Seattle Area Control study, which was approved by the Fred Hutchinson Cancer Research Center institutional review board (protocol 5567). PBMCs were isolated from whole blood using Accuspin System Histopaque-1077 (Sigma-Aldrich, cat#10771). Institutional review board approval for studies involving human tonsils was obtained from Seattle Children’s Hospital. Studies involving human spleens were deemed non-human subjects research since tissue was de-identified, otherwise discarded, and originated from deceased individuals. Tissue fragments were passed through a basket screen, centrifuged at 300 × g for 7 minutes, incubated with ACK lysis buffer (Thermo Fisher, cat#A1049201) for 3.5 minutes, resuspended in RPMI (Gibco, cat#11875093), and passed through a stacked 500 µm and 70 µm cell strainer. Cells were resuspended in 10% dimethylsulfoxide in heat-inactivated fetal calf serum (Gibco, cat#16000044) and cryopreserved in liquid nitrogen before use.
Cell lines
293 F cells (Thermo Fisher, cat#R79007) were cultured in Freestyle 293 media (Thermo Fisher, cat#12338026). Vero cells (ATCC CCL-81), LLC-MK2 cells (ATCC CCL-7.1), and HEp-2 (ATCC CCL-23) were cultured in DMEM (Gibco, cat#12430054) supplemented with 10% fetal calf serum and 100 U/ml penicillin plus 100 μg/mL streptomycin (Gibco, cat#15140122). 3T3 CD40L/IL-2/IL-21 feeder cells were cultured in DMEM supplemented with 10% fetal calf serum, penicillin and streptomycin, plus 0.4 mg/mL geneticin as described.27 Irradiation was performed with 5,000 rads.
Viruses
Wild-type rHPIV3 was a recombinant version of strain JS (GenBank accession number Z11575) and modified as previously described to express enhanced GFP.55 Virus was cultured on LLC-MK2 cells and subsequently purified by centrifugation in a discontinuous 30%/60% sucrose gradient with 0.05 M HEPES and 0.1 M MgSO4 (Sigma-Aldrich, cat#H4034 and 230391, respectively) at 120,000 × g for 90 min at 4°C. Virus titers were determined by infecting Vero cell monolayers in 24-well plates with serial 10-fold dilutions of virus, overlaying with DMEM containing 4% methylcellulose (Sigma-Aldrich, cat#M0387), and counting fluorescent plaques using a Typhoon scanner at 5 days post-infection (GE Life Sciences).
Expression and purification of antigens
Expression plasmids for His-tagged HPIV3 preF and postF antigens are previously described.25 HPIV3 preF contained the following mutations including two disulfide linkages, Q162C-L168C, I213C-G230C, A463V, and I474V.25 293 F cells were transfected at a density of 106 cells/mL in Freestyle 293 media using 1 mg/mL PEI Max (Polysciences, cat#24765). Transfected cells were cultured for 7 days with gentle shaking at 37°C. Supernatant was collected by centrifuging cultures at 2,500 × g for 30 minutes followed by filtration through a 0.2 µM filter. The clarified supernatant was incubated with Ni Sepharose beads overnight at 4°C, followed by washing with wash buffer containing 50 mM Tris, 300 mM NaCl, and 8 mM imidazole. His-tagged protein was eluted with an elution buffer containing 25 mM Tris, 150 mM NaCl, and 500 mM imidazole. The purified protein was run over a 10/300 Superose 6 size-exclusion column (GE Life Sciences, cat#17–5172–01). Fractions containing the trimeric HPIV3 F proteins were pooled and concentrated by centrifugation in an Amicon ultrafiltration unit (Millipore, cat#UFC805024) with a 50 kDa molecular weight cutoff. Two units of biotinylated thrombin (Millipore, cat#69022) were mixed with each 1 mg of protein overnight to cleave off tags, streptavidin agarose (Millipore, cat#69022) was added for another hour to remove thrombin and the cleaved tags, and the mixture was centrifuged through a PVDF filter (Millipore, cat#UFC40GV25) to remove the streptavidin agarose. The concentrated sample was stored in 50% glycerol at −20°C.
Tetramerization of antigens
Purified HPIV3 F was biotinylated using an EZ-link Sulfo-NHS-LC-Biotinylation kit (Thermo Fisher, cat#A39257) using a 1:1.3 molar ratio of biotin to F. Unconjugated biotin was removed by centrifugation using a 50 kDa Amicon Ultra size exclusion column (Millipore). To determine the average number of biotin molecules bound to each molecule of F, streptavidin-PE (ProZyme, cat#PJRS25) was titrated into a fixed amount of biotinylated F at increasing concentrations and incubated at room temperature for 30 minutes. Samples were run on an SDS-PAGE gel (Invitrogen, cat#NW04127BOX), transferred to nitrocellulose, and incubated with streptavidin–Alexa Fluor 680 (Thermo Fisher, cat#S32358) at a dilution of 1:10,000 to determine the point at which there was excess biotin available for the streptavidin–Alexa Fluor 680 reagent to bind. Biotinylated F was mixed with streptavidin-allophycocyanin (APC) at the ratio determined above to fully saturate streptavidin and incubated for 30 min at room temperature. Unconjugated F was removed by centrifugation using a 300 K Nanosep centrifugal device (Pall Corporation, cat#OD300C33). APC/DyLight755 tetramers were created by mixing F with streptavidin-APC pre–conjugated with DyLight755 (Thermo Fisher, cat#62279) following the manufacturer’s instructions. On average, APC/DyLight755 contained 4–8 DyLight molecules per APC. The concentration of each F tetramer was calculated by measuring the absorbance of APC (650 nm, extinction coefficient = 0.7 µM−1 cm−1).
Tetramer enrichment
100–200 × 106 frozen PBMCs, 20–50 × 106 frozen tonsil cells, or 40–80 × 106 frozen spleen cells were thawed into DMEM with 10% fetal calf serum and 100 U/ml penicillin plus 100 µg/ml streptomycin. Cells were centrifuged and resuspended 50 µL of ice-cold fluorescence-activated cell sorting (FACS) buffer composed of phosphate-buffered saline (PBS) and 1% newborn calf serum (Thermo Fisher, cat#26010074). PostF APC/DyLight755 conjugated tetramers were added at a final concentration of 25 nM in the presence of 2% rat and mouse serum (Thermo Fisher) and incubated at room temperature for 10 min. PreF APC tetramers were then added at a final concentration of 5 nM and incubated on ice for 25 min, followed by a 10 mL wash with ice-cold FACS buffer. Next, 50 μL of anti-APC-conjugated microbeads (Miltenyi Biotec, cat#130–090–855) were added and incubated on ice for 30 min, after which 3 mL of FACS buffer was added and the mixture was passed over a magnetized LS column (Miltenyi Biotec, cat#130–042–401). The column was washed once with 5 mL ice-cold FACS buffer and then removed from the magnetic field and 5 mL ice-cold FACS buffer was pushed through the unmagnetized column twice using a plunger to elute the bound cell fraction.
Flow cytometry
Cells were incubated in 50 μL of FACS buffer containing a cocktail of antibodies for 30 minutes on ice prior to washing and analysis on a FACS Aria (BD). Antibodies included anti-IgM FITC (G20–127, BD), anti-CD19 BUV395 (SJ25C1, BD), anti-CD3 BV711 (UCHT1, BD), anti-CD14 BV711 (M0P-9, BD), anti-CD16 BV711 (3G8, BD), anti-CD20 BUV737 (2H7, BD), anti-IgD BV605 (IA6–2, BD), and a fixable viability dye (Tonbo Biosciences, cat#13–0870–T500). Absolute counts within each specimen were calculated by adding a known amount of AccuCheck Counting Beads (Thermo Fisher, cat#PCB100). B cells were individually sorted into either 1) empty 96-well PCR plates and immediately frozen, or 2) flat-bottom 96-well plates containing feeder cells that had been seeded at a density of 28,600 cells/well 1 day prior in 100 µL of IMDM media (Gibco, cat#31980030) containing 10% fetal calf serum, 100 U/ml penicillin plus 100 µg/ml streptomycin, and 2.5 µg/mL amphotericin. B cells sorted onto feeder cells were cultured at 37°C for 13 days.
ELISA
Nunc maxsorp 96-well plates (Thermo Fisher, cat#442404) were coated with 100 ng of goat anti-human Fab (Jackson ImmunoResearch, cat#109–005–097) for 90 minutes at 4°C. Wells were washed three times with 1xDulbecco’s phosphate-buffered saline (DPBS) and then blocked with1xDPBS containing 1% bovine serum albumin (Sigma-Aldrich, cat#A2153) for 1 hour at room temperature. Antigen coated plates were incubated with culture supernatants for 90 minutes at 4°C. A standard curve was generated with serial two-fold dilutions of palivizumab. Wells were washed three times with 1xDPBS followed by a one-hour incubation with horseradish peroxidase-conjugated goat anti–human total Ig at a dilution of 1:6000 (Invitrogen, cat#31412). Wells were then washed four times with 1xDPBS followed by a 5–15 minute incubation with TMB substrate (SeraCare, cat#5120–0053). Absorbance was measured at 405 nm using a Softmax Pro plate reader (Molecular Devices). The concentration of antibody in each sample was determined by reference to the standard curve and dilution factor.
Neutralization assays
For neutralization screening of culture supernatants, Vero cells were seeded in 96-well flat bottom plates and cultured for 48 hours. After 13 days of culture, 40 µL of B cell culture supernatant was mixed with 25 µL of sucrose-purified GFP-HPIV3 diluted to 2,000 plaque forming units (pfu)/mL for 1 hour at 37°C. Vero cells were then incubated with 50 µL of the supernatant/virus mixture for 1 hour at 37°C to allow viral adsorption. Next, each well was overlaid with 100 µL DMEM containing 4% methylcellulose. Fluorescent plaques were counted at 5 days post-infection using a Typhoon imager. Titers of HPIV3-specific mAbs were determined by a 60% plaque reduction neutralization test (PRNT60). Vero cells were seeded in 24-well plates and cultured for 48 hours. MAbs were serially diluted 1:4 in 120 µL DMEM and mixed with 120 µL of sucrose-purified HPIV3 diluted to 2,000 pfu/mL for 1 hour at 37°C. Vero cells were incubated with 100 µL of the antibody/virus mixture for 1 hour at 37°C to allow viral adsorption. Each well was then overlaid with 500 µL DMEM containing 4% methylcellulose. Fluorescent plaques were counted at 5 days post-infection using a Typhoon imager. PRNT60 titers were calculated by linear regression analysis.
B cell receptor sequencing and cloning
For individual B cells sorted and frozen into empty 96-well PCR plates, reverse transcription (RT) was directly performed after thawing plates using SuperScript IV (Thermo Fisher, cat#18090200) as previously described.45,56 Briefly, 3 µL RT reaction mix consisting of 3 µL of 50 µM random hexamers (Thermo Fisher, cat#48190011), 0.8 µL of 25 mM deoxyribonucleotide triphosphates (dNTPs; Thermo Fisher, cat#N8080261), 1 µL (20 U) SuperScript IV RT, 0.5 µL (20 U) RNaseOUT (Thermo Fisher, cat#10777019), 0.6 µL of 10% Igepal (Sigma-Aldrich, cat#I8896), and 15 µL RNase-free water was added to each well containing a single sorted B cell and incubated at 50°C for 1 hour. For individual B cells sorted onto feeder cells, supernatant was removed after 13 days of culture, plates were immediately frozen on dry ice, stored at −80°C, thawed, and RNA was extracted using the RNeasy Micro Kit (Qiagen, cat#74034). The entire eluate from the RNA extraction was used instead of water in the RT reaction. Following RT, 2 µL of cDNA was added to 19 µl PCR reaction mix so that the final reaction contained 0.2 µL (0.5 U) HotStarTaq Polymerase (Qiagen, cat#203607), 0.075 µL of 50 µM 3′ reverse primers, 0.115 µL of 50 µM 5′ forward primers, 0.24 µL of 25 mM dNTPs, 1.9 µL of 10X buffer (Qiagen), and 16.5 µL of water. The PCR program was 50 cycles of 94°C for 30 s, 57°C for 30 s, and 72°C for 55 s, followed by 72°C for 10 min for heavy and kappa light chains. The PCR program was 50 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 55 s, followed by 72°C for 10 min for lambda light chains. After the first round of PCR, 2 µL of the PCR product was added to 19 µL of the second-round PCR reaction so that the final reaction contained 0.2 µL (0.5 U) HotStarTaq Polymerase, 0.075 µL of 50 µM 3′ reverse primers, 0.075 µL of 50 µM 5′ forward primers, 0.24 µL of 25 mM dNTPs, 1.9 µL 10X buffer, and 16.5 µL of water. PCR programs were the same as the first round of PCR. 4 μL of the PCR product was run on an agarose gel to confirm the presence of a ~500-bp heavy chain band or 450-bp light chain band. 5 μL from the PCR reactions showing the presence of heavy or light chain amplicons was mixed with 2 µL of ExoSAP-IT (Thermo Fisher, cat#78201) and incubated at 37°C for 15 min followed by 80°C for 15 min to hydrolyze excess primers and nucleotides. Hydrolyzed second-round PCR products were sequenced by Genewiz with the respective reverse primer used in the second-round PCR, and sequences were analyzed using IMGT/V-Quest to identify V, D, and J gene segments. Paired heavy chain VDJ and light chain VJ sequences were cloned into pTT3-derived expression vectors containing the human IgG1, IgK, or IgL constant regions using In-Fusion cloning (Clontech, cat#638911) as previously described.57
Monoclonal antibody production
Secretory IgG was produced by co-transfecting 293 F cells at a density of 106 cells/mL with the paired heavy and light chain expression plasmids at a ratio of 1:1 in Freestyle 293 media using 1 mg/mL PEI Max. Transfected cells were cultured for 7 days with gentle shaking at 37°C. Supernatant was collected by centrifuging cultures at 2,500 × g for 15 minutes followed by filtration through a 0.2 µM filter. Clarified supernatants were then incubated with Protein A agarose (Thermo Scientific, cat#22812) followed by washing with IgG binding buffer (Thermo Scientific, cat#21007). Antibodies were eluted with IgG Elution Buffer (Thermo Scientific, cat#21004) into a neutralization buffer containing 1 M Tris-base pH 9.0. Purified antibody was concentrated and buffer exchanged into 1xDPBS using an Amicon ultrafiltration unit with a 50 kDa molecular weight cutoff.
Bio-layer interferometry
BLI assays were performed on the Octet.Red instrument (ForteBio) at room temperature with shaking at 500 rpm. Anti-human IgG capture sensors (ForteBio, cat#18–5060) were loaded in kinetics buffer (PBS with 0.01% bovine serum albumin, 0.02% Tween 20, and 0.005% NaN3, pH 7.4) containing 40 µg/mL purified mAb for 150 s. After loading, the baseline signal was recorded for 60 s in kinetics buffer. The sensors were then immersed in kinetics buffer containing 1 µM purified HPIV3 F for a 300 s association step followed by immersion in kinetics buffer for an additional 300 s dissociation phase. For apparent affinity (KD) analyses, the dissociation step was extended to 600 s. The maximum response was determined by averaging the nanometer shift over the last 5 s of the association step after subtracting the background signal from each analyte-containing well using a negative control mAb at each time point. Curve fitting was performed using a 1:1 binding model and ForteBio data analysis software. For competitive binding assays, penta-His capture sensors (ForteBio, cat#18–5120) were loaded in kinetics buffer containing 1 µM His-tagged HPIV3 F for 300 s. After loading, the baseline signal was recorded for 30 s in kinetics buffer. The sensors were then immersed for 300 s in kinetics buffer containing 40 µg/mL of the first antibody followed by immersion for another 300 s in kinetics buffer containing 40 µg/mL of the second antibody. Percent competition was determined by dividing the maximum increase in signal of the second antibody in the presence of the first antibody by the maximum signal of the second antibody alone.
Autoreactivity assay
HEp-2 cells were seeded into 96-well plates at a density of 50,000 cells/well 1 day prior to fixation with 50% acetone and 50% methanol for 10 minutes at −20°C. Cells were then permeabilized and blocked with 1xDPBS containing 1% Triton X-100 (Sigma-Aldrich, cat#T8787) and 1% bovine serum albumin for 30 minutes at room temperature. 100 µL of each mAb at 0.1 mg/mL was added for 30 minutes at room temperature. The 2F5 positive control was obtained from the National Institutes of Health AIDS Reagent Program. The G4 positive control was isolated from an auto-reactive B cell obtained from the spleen of a human donor. Wells were then washed four times in 1xDPBS followed by incubation with goat anti-human IgG Alexa Fluor 594 (Thermo Fisher, cat#A11014) at a dilution of 1:200 in 1xDPBS for 30 minutes at room temperature in the dark. After washing four times with 1× DPBS, images were acquired using the EVOS Cell Imaging System (Thermo Fisher).
Structural analysis
Fab preparation
PI3-E12, PI3-C9, and PI3-A12 Fab were produced by incubating each 10 mg of IgG with 10 µg of LysC (New England Biolabs, cat#P8109S) overnight at 37°C followed by incubating with protein A for 1 hour at room temperature. The mixture was then centrifuged through a PVDF filter, concentrated in PBS with a 30 kDa Amicon Ultra size exclusion column, and purified further by SEC using Superdex 200 (GE Healthcare Life Sciences, cat#17–5175–01) in 5 mM Hepes and 150 mM NaCl.
Crystallization, data collection, and refinement
Crystals of PI3-E12 Fab were obtained using a NT8 dispensing robot (Formulatrix), and screening was done using commercially available screens (Rigaku Wizard Precipitant Synergy block #2, Molecular Dimensions Proplex screen HT-96, Hampton Research Crystal Screen HT) by mixing 0.1 µL/0.1 µL (protein/reservoir) by the vapor diffusion method. Crystals used for diffraction data were grown in the following conditions in solution containing 0.2 M ammonium phosphate monobasic, 0.1 M Tris, pH 8.5, and 50% (+/−) 2-methyl-2,4-pentanediol. Crystals were cryoprotected in Parabar Oil (Hampton, cat#HR2–643). Crystals diffracted to 2.1 Å (Supplemental Table 4). Data was collected on the Fred Hutchinson Cancer Research Center X-ray home source and processed using HKL2000.58 The structure was solved by molecular replacement using Phaser in CCP4 (Collaborative Computational Project, Number 4), and the Fab portion of PDB accession number 6MFT was used as a search model.25,59 Iterating rounds of structure building and refinement were performed in COOT60 and Phenix.61 Structural figures were made with Pymol62 and Chimera.63
Negative stain electron microscopy
The complex of PI3-E12 Fab + HPIV3 preF was formed by mixing both components at a 1:1 molar ratio and incubating overnight at 4°C. Complexes were purified by SEC using Superdex 200 in 5 mM Hepes and 150 mM NaCl at pH 7.4. Negative staining was performed using Formvar/carbon grids (Electron Microscopy Sciences) of 300 mesh size. 3 µL of HPIV3-PI3-E12 Fab complex protein was negatively stained at a concentration of 50 ug/mL on the grids using 1% uranyl formate staining solution.
Data were collected using a FEI Tecnai T12 electron microscope operating at 120 keV equipped with a Gatan Ultrascan 4000 CCD camera. The images were collected using an electron dose of 45.05 e− Å−2 and a magnification of 67,000× that resulted in a pixel size of 1.6 Å. The defocus range used was −1.00 µm to −2.00 µm. The data were collected using Leginon interface.64 Image processing was carried out using cisTEM.65 The final reconstruction was performed using ~12,000 unbinned particles, refining for 20 iterations with C1 symmetry applied. PI3–E12 Fab (as described here; PDB ID 6WRP), HPIV3 (PDB ID 6MJZ)25 and GCN4 (PDB ID 4DME)66 fitting was carried out using the Fit function in Chimera.63
Animals and HPIV3 challenge
Cotton rat challenge experiments were performed by Sigmovir. Animals in groups of N = 4–5 were infected intranasally with 100 µL of 105 pfu HPIV3. This sample size is consistent with previously published experiments testing the efficacy of RSV mAbs in the cotton rat model.36,37,52,67 MAb was either administered intramuscularly 1 day prior to infection or 1 day after infection. Cyclophosphamide (50 mg/kg) was administered intramuscularly at 21 days prior to infection and re-administered every 3 days for the duration of experiments involving immunosuppression. Nasal turbinates were removed for viral titration by plaque assay at day four post-infection. Lungs were removed for viral titration by plaque assay at day four post-infection. Lung and nose homogenates were clarified by centrifugation in EMEM (Gibco, cat#670086). Confluent HEp-2 monolayers were inoculated in duplicate with diluted homogenates in 24-well plates. After incubating for 2 hours at 37°C, wells were overlaid with 0.75% methylcellulose. After 4 days, the cells were fixed and stained with 0.1% crystal violet for 1 hour, and plaques were counted to determine titers as pfu per gram of tissue.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7. Pairwise statistical comparisons were performed using unpaired two-tailed t-test. P < .05 was considered statistically significant. Data points from individual samples are displayed.
Supplementary Material
Acknowledgments
We thank Julie McElrath for PBMCs from the Seattle Area Control cohort; LifeCenter Northwest for providing de-identified spleen remnants; Ursula Buchholz, Shirin Munir, and Peter Collins for providing the GFP-expressing HPIV3 for neutralization assays; Aliaksandr Druz for expression of preF HPIV3 F; Marina Boukhalova, Kevin Yim, and Jorge Blanco at Sigmovir for their expertise with cotton rat experiments; Steve Voght and Jessica Schembri for proof-reading the manuscript; Rebecca Putnam, Paula Culver, Russell Eberts and Laura Yates for administrative support; the Taylor lab for helpful discussions.
Funding Statement
This study was supported by the Vaccine and Infectious Disease Division Faculty Initiative (J.B. and J.T.) and the Joel Meyers Endowment Scholarship (J.B.) from the Fred Hutchinson Cancer Research Center, a New Investigator Award from the American Society for Transplantation and Cellular Therapy (J.B.), and by the National Institutes of Health under award number T32AI118690 (J.B.). Funding was also provided by the Intramural Research Program of the Vaccine Research Center, National Institute of Allergy and Infectious Diseases, National Institute of Health (G.B.E.S.-J and P.D.K.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health;Fred Hutchinson Cancer Research Center [Joel Meyers Endowment Scholarship];Fred Hutchinson Cancer Research Center [VIDD Faculty Initiative]; ;National Institutes of Health [T32AI118690]; American Society for Transplantation and Cellular Therapy [New Investigator Award];
Author contributions
J.B. conceived the study, designed and conducted the experiments, analyzed the data, and wrote the manuscript. S.S., C.W., J.R. and M.P. coordinated and performed the structural analysis. A.M. provided 3T3 CD40L/IL-2/IL-21 cells. R.B. provided spleens. J.P. provided tonsils. G.B.E.S.-J. and P.D.K. provided preF-stabilized HPIV3 F. J.T. conceived the study, designed experiments, analyzed the data, and edited the manuscript.
Competing interests
Work described in this manuscript has been included in a provisional patent application. The authors have no other competing financial interests in relation to the work described.
Data availability
Sequencing and structural data that support the findings of this study have been deposited in the Protein Data Bank (PDB) and are accessible through PDB accession number 6WRP.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Abbreviations:
| ALC | Absolute Lymphocyte Count |
| BLI | Biolayer Interferometry |
| F | Fusion |
| gl | germline |
| HCT | Hematopoietic Stem Cell Transplant |
| HPIV3 | Human Parainfluenza Virus Type III |
| mAb | Monoclonal Antibody |
| nsEM | Negative Stain Electron Microscopy |
| pfu | Plaque-forming Unit |
| postF | Postfusion |
| preF | Prefusion |
| PRNT | plaque reduction neutralization test |
| RSV | Respiratory Syncytial Virus |
| RT | Reverse Transcription |
| SEC | Size Exclusion Chromatography |
| WBC | White Blood Cell |
References
- 1.Weinberg GA, Hall CB, Iwane MK, Poehling KA, Edwards KM, Griffin MR, Staat MA, Curns AT, Erdman DD, Szilagyi PG, et al. Parainfluenza virus infection of young children: estimates of the population-based burden of hospitalization. J Pediatr. 2009;154(5):694–453. doi: 10.1016/j.jpeds.2008.11.034. [DOI] [PubMed] [Google Scholar]
- 2.Karron RA, Wright PF, Belshe RB, Thumar B, Casey R, Newman F, Polack FP, Randolph VB, Deatly A, Hackell J, et al. Identification of a recombinant live attenuated respiratory syncytial virus vaccine candidate that is highly attenuated in infants. J Infect Dis. 2005;191(7):1093–104. doi: 10.1086/427813. [DOI] [PubMed] [Google Scholar]
- 3.Wright PF, Karron RA, Belshe RB, Thompson J, Crowe JE Jr., Boyce TG, Halburnt LL, Reed GW, Whitehead SS, Anderson EL, et al. Evaluation of a live, cold-passaged, temperature-sensitive, respiratory syncytial virus vaccine candidate in infancy. J Infect Dis. 2000;182(5):1331–42. doi: 10.1086/315859. [DOI] [PubMed] [Google Scholar]
- 4.Ison MG, Hirsch HH.. Community-acquired respiratory viruses in transplant Patients: diversity, impact, unmet clinical needs. Clin Microbiol Rev. 2019;32(4):32. doi: 10.1128/CMR.00042-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boeckh M. The challenge of respiratory virus infections in hematopoietic cell transplant recipients. Br J Haematol. 2008;143(4):455–67. doi: 10.1111/j.1365-2141.2008.07295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Renaud C, Xie H, Seo S, Kuypers J, Cent A, Corey L, Leisenring W, Boeckh M, Englund JA. Mortality rates of human metapneumovirus and respiratory syncytial virus lower respiratory tract infections in hematopoietic cell transplantation recipients. Biol Blood Marrow Transplant. 2013;19(8):1220–26. doi: 10.1016/j.bbmt.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chemaly RF, Shah DP, Boeckh MJ. Management of respiratory viral infections in hematopoietic cell transplant recipients and patients with hematologic malignancies. Clin Infect Dis. 2014;59(Suppl 5):S344–51. doi: 10.1093/cid/ciu623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campbell AP, Guthrie KA, Englund JA, Farney RM, Minerich EL, Kuypers J, Corey L, Boeckh M. Clinical outcomes associated with respiratory virus detection before allogeneic hematopoietic stem cell transplant. Clin Infect Dis. 2015;61(2):192–202. doi: 10.1093/cid/civ272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hutspardol S, Essa M, Richardson S, Schechter T, Ali M, Krueger J, Fujii H, Egeler RM, Gassas A. Significant Transplantation-related mortality from respiratory virus infections within the first one hundred days in children after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2015;21(10):1802–07. doi: 10.1016/j.bbmt.2015.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chu HY, Chin J, Pollard J, Zerr DM, Englund JA. Clinical outcomes in outpatient respiratory syncytial virus infection in immunocompromised children. Influenza Other Respir Viruses. 2016;10(3):205–10. doi: 10.1111/irv.12375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boonyaratanakornkit J, Vivek M, Xie H, Pergam SA, Cheng GS, Mielcarek M, Hill JA, Jerome KR, Limaye AP, Leisenring W, et al. Predictive value of respiratory viral detection in the upper respiratory tract for infection of the lower respiratory tract with hematopoietic stem cell transplantation. J Infect Dis. 2019. doi: 10.1093/infdis/jiz470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erard V, Chien JW, Kim HW, Nichols WG, Flowers ME, Martin PJ, Corey L, Boeckh M. Airflow decline after myeloablative allogeneic hematopoietic cell transplantation: the role of community respiratory viruses. J Infect Dis. 2006;193(12):1619–25. doi: 10.1086/504268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peck AJ, Englund JA, Kuypers J, Guthrie KA, Corey L, Morrow R, Hackman RC, Cent A, Boeckh M. Respiratory virus infection among hematopoietic cell transplant recipients: evidence for asymptomatic parainfluenza virus infection. Blood. 2007;110(5):1681–88. doi: 10.1182/blood-2006-12-060343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shah DP, Shah PK, Azzi JM, Chemaly RF. Parainfluenza virus infections in hematopoietic cell transplant recipients and hematologic malignancy patients: a systematic review. Cancer Lett. 2016;370(2):358–64. doi: 10.1016/j.canlet.2015.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glezen WP, Taber LH, Frank AL, Kasel JA. Risk of primary infection and reinfection with respiratory syncytial virus. Am J Dis Child. 1986;140(6):543–46. doi: 10.1001/archpedi.1986.02140200053026. [DOI] [PubMed] [Google Scholar]
- 16.Habibi MS, Jozwik A, Makris S, Dunning J, Paras A, DeVincenzo JP, de Haan CA, Wrammert J, Openshaw PJ, Chiu C, et al. Impaired Antibody-mediated protection and defective IgA B-cell memory in experimental infection of adults with respiratory syncytial virus. Am J Respir Crit Care Med. 2015;191(9):1040–49. doi: 10.1164/rccm.201412-2256OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hall CB, Walsh EE, Long CE, Schnabel KC. Immunity to and frequency of reinfection with respiratory syncytial virus. J Infect Dis. 1991;163(4):693–98. doi: 10.1093/infdis/163.4.693. [DOI] [PubMed] [Google Scholar]
- 18.Hampson AW, Osterhaus AD, Pervikov Y, Kieny MP. Report of the second meeting on the development of influenza vaccines that induce broad-spectrum and long-lasting immune responses, World Health Organization, Geneva, Switzerland, 6–7 December 2005. Vaccine. 2006;24(23):4897–900. doi: 10.1016/j.vaccine.2006.03.035. [DOI] [PubMed] [Google Scholar]
- 19.Cox MM, Patriarca PA, Treanor J. FluBlok, a recombinant hemagglutinin influenza vaccine. Influenza Other Respir Viruses. 2008;2(6):211–19. doi: 10.1111/j.1750-2659.2008.00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson S, Oliver C, Prince GA, Hemming VG, Pfarr DS, Wang SC, et al . Development of a humanized monoclonal antibody (MEDI-493) with potent in vitro and in vivo activity against respiratory syncytial virus. J Infect Dis. 1997;176(5):1215–24. doi: 10.1086/514115. [DOI] [PubMed] [Google Scholar]
- 21.Ngwuta JO, Chen M, Modjarrad K, Joyce MG, Kanekiyo M, Kumar A, Yassine HM, Moin SM, Killikelly AM, Chuang GY, et al. Prefusion F-specific antibodies determine the magnitude of RSV neutralizing activity in human sera. Sci Transl Med. 2015;7(309):7:309ra162. doi: 10.1126/scitranslmed.aac4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilman MS, Castellanos CA, Chen M, Ngwuta JO, Goodwin E, Moin SM, Mas V, Melero JA, Wright PF, Graham BS, et al. Rapid profiling of RSV antibody repertoires from the memory B cells of naturally infected adult donors. Sci Immunol. 2016;1(6):1. doi: 10.1126/sciimmunol.aaj1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yin HS, Paterson RG, Wen X, Lamb RA, Jardetzky TS. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc Natl Acad Sci U S A. 2005;102(26):9288–93. doi: 10.1073/pnas.0503989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moscona A. Interaction of human parainfluenza virus type 3 with the host cell surface. Pediatr Infect Dis J. 1997;16(10):917–24. doi: 10.1097/00006454-199710000-00003. [DOI] [PubMed] [Google Scholar]
- 25.Gbe S-J, Chuang GY, Xu K, Zhou T, Acharya P, Tsybovsky Y, Ou L, Zhang B, Fernandez-Rodriguez B, Gilardi V, et al. Structure-based design of a quadrivalent fusion glycoprotein vaccine for human parainfluenza virus types 1–4. Proc Natl Acad Sci U S A. 2018;115(48):12265–70. doi: 10.1073/pnas.1811980115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henrickson KJ. Parainfluenza viruses. Clin Microbiol Rev. 2003;16(2):242–64. doi: 10.1128/CMR.16.2.242-264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whaley RE, Ameny S, Arkatkar T, Seese A, Wall A, Khan I, Carter JJ, Scherer EM, Rawlings DJ, Galloway DA, et al. Generation of a cost-effective cell line for support of high-throughput isolation of primary human B cells and monoclonal neutralizing antibodies. J Immunol Methods. 2020;488:112901. doi: 10.1016/j.jim.2020.112901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steach HR, DeBuysscher BL, Schwartz A, Boonyaratanakornkit J, Baker ML, Tooley MR, Pease NA, Taylor JJ. Cross-reactivity with Self-antigen tunes the functional potential of naive B cells specific for foreign antigens. J Immunol. 2020;204(3):498–509. doi: 10.4049/jimmunol.1900799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bancroft T, DeBuysscher BL, Weidle C, Schwartz A, Wall A, Gray MD, Feng J, Steach HR, Fitzpatrick KS, Gewe MM, et al. Detection and activation of HIV broadly neutralizing antibody precursor B cells using anti-idiotypes. J Exp Med. 2019;216(10):2331–47. doi: 10.1084/jem.20190164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McLellan JS, Chen M, Leung S, Graepel KW, Du X, Yang Y, Zhou T, Baxa U, Yasuda E, Beaumont T, et al. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science. 2013;340(6136):1113–17. doi: 10.1126/science.1234914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Battles MB, Mas V, Olmedillas E, Cano O, Vazquez M, Rodriguez L, Melero JA, McLellan JS. Structure and immunogenicity of pre-fusion-stabilized human metapneumovirus F glycoprotein. Nat Commun. 2017;8(1):1528. doi: 10.1038/s41467-017-01708-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Griffin MP, Yuan Y, Takas T, Domachowske JB, Madhi SA, Manzoni P, Simoes EAF, Esser MT, Khan AA, Dubovsky F, et al. Single-dose nirsevimab for prevention of RSV in preterm infants. N Engl J Med. 2020;383(5):415–25. doi: 10.1056/NEJMoa1913556. [DOI] [PubMed] [Google Scholar]
- 33.Ottolini MG, Porter DD, Hemming VG, Hensen SA, Sami IR, Prince GA . Semi-permissive replication and functional aspects of the immune response in a cotton rat model of human parainfluenza virus type 3 infection. J Gen Virol. 1996;77(Pt 8):1739–43. doi: 10.1099/0022-1317-77-8-1739. [DOI] [PubMed] [Google Scholar]
- 34.Kirkeby L, Rasmussen TT, Reinholdt J, Kilian M. Immunoglobulins in nasal secretions of healthy humans: structural integrity of secretory immunoglobulin A1 (IgA1) and occurrence of neutralizing antibodies to IgA1 proteases of nasal bacteria. Clin Diagn Lab Immunol. 2000;7(1):31–39. doi: 10.1128/CDLI.7.1.31-39.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fisher RG, Crowe JE Jr., Johnson TR, Tang YW, Graham BS. Passive IgA monoclonal antibody is no more effective than IgG at protecting mice from mucosal challenge with respiratory syncytial virus. J Infect Dis. 1999;180(4):1324–27. doi: 10.1086/315037. [DOI] [PubMed] [Google Scholar]
- 36.Boukhvalova M, Blanco JC, Falsey AR, Mond J. Treatment with novel RSV Ig RI-002 controls viral replication and reduces pulmonary damage in immunocompromised Sigmodon hispidus. Bone Marrow Transplant. 2016;51(1):119–26. doi: 10.1038/bmt.2015.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson RA, Prince GA, Suffin SC, Horswood RL, Chanock RM. Respiratory syncytial virus infection in cyclophosphamide-treated cotton rats. Infect Immun. 1982;37(1):369–73. doi: 10.1128/IAI.37.1.369-373.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ottolini MG, Curtis SR, Mathews A, Ottolini SR, Prince GA. Palivizumab is highly effective in suppressing respiratory syncytial virus in an immunosuppressed animal model. Bone Marrow Transplant. 2002;29(2):117–20. doi: 10.1038/sj.bmt.1703326. [DOI] [PubMed] [Google Scholar]
- 39.Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256(5517):495–97. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 40.Carter PJ, Lazar GA. Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat Rev Drug Discov. 2018;17:197–223. [DOI] [PubMed] [Google Scholar]
- 41.Walker LM, Burton DR. Passive immunotherapy of viral infections: ‘super-antibodies’ enter the fray. Nat Rev Immunol. 2018;18(7):297–308. doi: 10.1038/nri.2017.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Victora GD, Wilson PC. Germinal center selection and the antibody response to influenza. Cell. 2015;163(3):545–48. doi: 10.1016/j.cell.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. .Nair N, Feng N, Blum LK, Sanyal M, Ding S, Jiang B, Sen A, Morton JM, He XS, Robinson WH, et al . VP4- and VP7-specific antibodies mediate heterotypic immunity to rotavirus in humans. Sci Transl Med. 2017;9(395). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang J, Doria-Rose NA, Longo NS, Laub L, Lin CL, Turk E, Kang BH, Migueles SA, Bailer RT, Mascola JR, et al. Isolation of human monoclonal antibodies from peripheral blood B cells. Nat Protoc. 2013;8(10):1907–15. doi: 10.1038/nprot.2013.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS, Zhou T, Schmidt SD, Wu L, Xu L, et al. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science. 2010;329(5993):856–61. doi: 10.1126/science.1187659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Byars SG, Stearns SC, Boomsma JJ. Association of Long-term risk of respiratory, allergic, and infectious diseases with removal of adenoids and tonsils in childhood. JAMA Otolaryngol Head Neck Surg. 2018;144(7):594–603. doi: 10.1001/jamaoto.2018.0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Graham BS. Vaccine development for respiratory syncytial virus. Curr Opin Virol. 2017;23:107–12. doi: 10.1016/j.coviro.2017.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gorman J, Chuang GY, Lai YT, Shen CH, Boyington JC, Druz A, Geng H, Louder MK, McKee K, Rawi R, et al. Structure of Super-potent antibody CAP256-VRC26.25 in complex with HIV-1 envelope reveals a combined mode of trimer-apex recognition. Cell Rep. 2020;31(1):107488. doi: 10.1016/j.celrep.2020.03.052. [DOI] [PubMed] [Google Scholar]
- 49.McLellan JS. Neutralizing epitopes on the respiratory syncytial virus fusion glycoprotein. Curr Opin Virol. 2015;11:70–75. doi: 10.1016/j.coviro.2015.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmidt AG, Therkelsen MD, Stewart S, Kepler TB, Liao HX, Moody MA, Haynes BF, Harrison SC. Viral receptor-binding site antibodies with diverse germline origins. Cell. 2015;161(5):1026–34. doi: 10.1016/j.cell.2015.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Crowe JE Jr. Principles of broad and potent antiviral human antibodies: insights for vaccine design. Cell Host Microbe. 2017;22(2):193–206. doi: 10.1016/j.chom.2017.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu Q, McLellan JS, Kallewaard NL, Ulbrandt ND, Palaszynski S, Zhang J, Moldt B, Khan A, Svabek C, McAuliffe JM, et al. A highly potent extended half-life antibody as a potential RSV vaccine surrogate for all infants. Sci Transl Med. 2017;9(388). [DOI] [PubMed] [Google Scholar]
- 53.Boukhvalova MS, Yim KC, Blanco J. Cotton rat model for testing vaccines and antivirals against respiratory syncytial virus. Antivir Chem Chemother. 2018;26:2040206618770518. doi: 10.1177/2040206618770518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moffett HF, Harms CK, Fitzpatrick KS, Tooley MR, Boonyaratanakornkit J, Taylor JJ. B cells engineered to express pathogen-specific antibodies protect against infection.. Sci Immunol. 2019;4(35):4. doi: 10.1126/sciimmunol.aax0644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu X, Liang B, Liu X, Amaro-Carambot E, Surman S, Kwong PD, Graham BS, Collins PL, Munir S. Human parainfluenza virus type 3 expressing the respiratory syncytial virus pre-fusion F protein modified for virion packaging yields protective intranasal vaccine candidates. PLoS One. 2020;15(2):e0228572. doi: 10.1371/journal.pone.0228572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tiller T, Meffre E, Yurasov S, Tsuiji M, Nussenzweig MC, Wardemann H. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J Immunol Methods. 2008;329(1–2):112–24. doi: 10.1016/j.jim.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McGuire AT, Gray MD, Dosenovic P, Gitlin AD, Freund NT, Petersen J, Correnti C, Johnsen W, Kegel R, Stuart AB, et al. Specifically modified Env immunogens activate B-cell precursors of broadly neutralizing HIV-1 antibodies in transgenic mice. Nat Commun. 2016;7:10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–26. [DOI] [PubMed] [Google Scholar]
- 59.Collaborative Computational Project N . The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(5):760–63. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 60.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(12):2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 61.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.DeLano WL. Unraveling hot spots in binding interfaces: progress and challenges. Curr Opin Struct Biol. 2002;12(1):14–20. doi: 10.1016/S0959-440X(02)00283-X. [DOI] [PubMed] [Google Scholar]
- 63.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 64.Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B. Automated molecular microscopy: the new Leginon system. J Struct Biol. 2005;151(1):41–60. Cambridge, UK: eLife Sciences Publications Ltd. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 65.Grant T, Rohou A, Grigorieff N. cisTEM, user-friendly software for single-particle image processing. Elife. 2018. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oshaben KM, Salari R, McCaslin DR, Chong LT, Horne WS. The native GCN4 leucine-zipper domain does not uniquely specify a dimeric oligomerization state. Biochemistry. 2012;51(47):9581–91. doi: 10.1021/bi301132k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu H, Pfarr DS, Johnson S, Brewah YA, Woods RM, Patel NK, et al. Development of motavizumab, an ultra-potent antibody for the prevention of respiratory syncytial virus infection in the upper and lower respiratory tract. J Mol Biol. 2007;368(3):652–65. doi: 10.1016/j.jmb.2007.02.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing and structural data that support the findings of this study have been deposited in the Protein Data Bank (PDB) and are accessible through PDB accession number 6WRP.