

Abstract
Understanding the diversity of cell types in the brain has been an enduring challenge and requires detailed characterization of individual neurons in multiple dimensions. To systematically profile morpho-electric properties of mammalian neurons, we established a single cell characterization pipeline using standardized patch clamp recordings in brain slices and biocytin-based neuronal reconstructions. We built a publicly-accessible online database, the Allen Cell Types Database, to display these data sets. Intrinsic physiological properties were measured from 1,938 neurons from the adult laboratory mouse visual cortex, morphological properties were measured from 461 reconstructed neurons, and 452 neurons had both measurements available. Quantitative features were used to classify neurons into distinct types using unsupervised methods. We established a taxonomy of morphologically- and electrophysiologically-defined cell types for this region of cortex with 17 e-types, 38 m-types, and 46 me-types and found good correspondence with previously-defined transcriptomic cell types and subclasses using the same transgenic mouse lines.
INTRODUCTION
Neurons of the mammalian neocortex exhibit diverse physiological and morphological characteristics. Classifying these neurons into cell types, following Plato’s dictum to “carve nature at its joints,” provides a useful abstraction when investigating how these neurons interact in neocortical circuits1. The delineation of neocortical cell types has benefited from a wealth of studies detailing the molecular, morphological, and physiological properties of excitatory and inhibitory neurons, leading to the characterization of three major populations (i.e., Pvalb+, Sst+, and Htr3a+) of neocortical interneurons, each with distinct types, and excitatory neuron populations with features often linked to their laminar locations as well as the locations to which they project their axons2–4. Historically, though, direct comparisons of morpho-electric properties across smaller studies have been constrained by differences in experimental protocols, analysis methods, and access to specific transgenic lines, limiting the construction of a systematic classification scheme. Recent studies performed at larger scales5,6, including the Blue Brain Project’s thorough classification of morpho-electric diversity in juvenile rat somatosensory cortex, have overcome many of these issues, using large, coherent data sets to identify principles of local connectivity and computation within a cortical column.
So far, morphological and physiological descriptions of cell types (including in these large-scale studies) have used quantitative feature extraction but often ultimately relied on expert annotation and categorization7. Such qualitative descriptions are valuable and can be consistent with statistical analyses of features8, but they can be limited by the number of criteria that can be simultaneously used to differentiate types. In addition, features used to distinguish types can be specific to the population being studied and may not be broadly applicable (or distinguishing) across all cortical neurons. Specific cell types have been characterized quantitatively9–11, but these studies frequently have used additional labels (individual gene markers) to pre-define cell types. In addition, these approaches may successfully identify heterogeneity in features between specific populations under investigation, but these differences may not necessarily co-vary with other features or display discontinuities when examined in the context of a broader set of cortical cells—significant criteria for cell-type classification1.
Recent studies using transcriptomic characterization based on single-cell RNA-sequencing techniques performed unsupervised classification to generate a taxonomy of transcriptomic types in the mouse neocortex12–14. These approaches rely on data from thousands of neurons generated in a standardized manner. Here we took a similar approach to classifying morpho-electric properties of adult mouse visual cortical neurons using electrophysiological data collected from 1,938 cells and morphological reconstructions of 461 cells, with both data types collected from 452 neurons. We characterized mouse visual cortex neurons with a uniform experimental protocol and developed an unsupervised classification scheme of cell types based on electrophysiology and morphology. Every aspect of the pipeline, from slice preparation, recording, and stimulation, to staining, imaging, reconstruction, and mapping of cells to a reference atlas, employed highly standardized and quality-controlled methodology. A diverse set of transgenic mouse lines14–18 were used to ensure broad coverage of excitatory and inhibitory classes across all cortical layers, to enable selective targeting of rare cell populations, and to link our study with other experimental approaches, such as transcriptomic characterization19. Unsupervised clustering methods mapped neurons of the adult mouse visual cortex to 17 distinct electrophysiological or e-types and 38 morphological or m-types. Joint clustering based on combined electrophysiological and morphological data resulted in 46 me-types. The use of transgenic labels established preliminary correspondences with major transcriptomic subclasses and specific transcriptomic types. The experimental data and analysis tools are made available as a public resource as part of the Allen Cell Types Database and the Allen Software Development Kit.
RESULTS
Creating the in vitro single cell characterization platform
We set out to characterize the diversity of intrinsic electrophysiological and morphological properties of mouse visual cortical neurons by establishing a platform for generating a standardized data set (Fig. 1). We made whole-cell patch clamp recordings from neurons labeled by fluorescent proteins with an expression pattern determined by a Cre-based driver. We used a variety of driver lines to sample broadly across the cortical circuit as well as to target specific neuronal populations (Supplementary Figs. 1 and 2). The recording pipette contained biocytin to enable filling, staining, imaging, and three-dimensional reconstruction of neuronal morphologies. Each neuron was localized within the mouse reference atlas using serial block-face images generated during tissue slicing. Most cells (n=1,599) were from primary visual cortex (VISp); the remainder were in nearby higher visual areas. For morphological analysis, we reconstructed a subset of recorded neurons, selected based on fill quality and data coverage. All recorded cells underwent the same workflow (Fig. 1a), and only cells that met pre-defined quality control (QC) standards were included in the final data set, which consisted of 1,938 cells passing electrophysiology QC. Of these, 928 were spiny neurons, which we assume to be excitatory, and 1,010 were aspiny or sparsely spiny, which we assume to be inhibitory; this determination was made by examining the image of each cell (Methods). We reconstructed the morphologies (Methods) of 461 cells (254 spiny and 207 aspiny/sparsely spiny). All but 9 of those 461 cells underwent electrophysiological classification; the primary reason for not including these nine was failure to elicit an action potential with the 25 pA/s ramp stimulus. Electrophysiological, imaging and morphology reconstruction data (when available) are accessible as part of the Allen Cell Types Database (http://celltypes.brain-map.org/) (Fig. 1b), including an interactive website as well as the opportunity to download data files. As of this writing, 1,861 out of 1,938 cells had electrophysiological data on the website, and 431 out of 461 reconstructions were on the website. The remainder will be published in the next scheduled data release.
Figure 1: A pipeline to generate and analyze standardized morpho-electric data at scale.
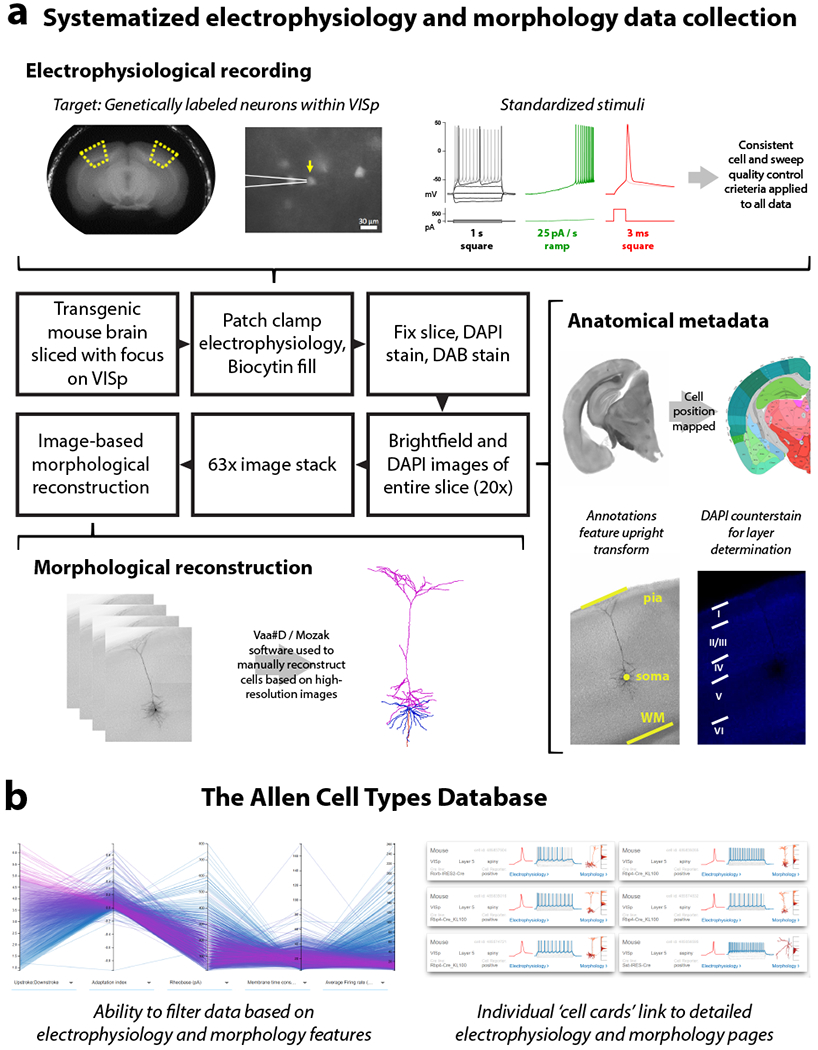
(a) An in vitro single cell characterization pipeline was established to generate standardized electrophysiological and morphological data from mouse cortical neurons. Mouse brains were imaged during vibratome sectioning to aid in cell localization to a common mouse reference atlas, Allen Mouse Common Coordinate Framework version 3 (CCF v3). Fluorescently labeled neurons from specific transgenic mouse lines were recorded by whole cell patch clamping to characterize each cell’s intrinsic electrical properties. Each cell was stimulated with a standard electrophysiological stimulation paradigm and underwent consistent cell and sweep quality control, allowing for routine feature extraction and alignment of data traces from diverse cell types. During the electrophysiology recording, cells were filled with biocytin, then tissue slices were fixed, stained and mounted, and imaged at 20× in a single plane. Cells were mapped to the reference atlas and layer determination was made using a DAPI counterstain. A subset of neurons were then selected to be imaged in a high-resolution 63× stack. High quality cells were then manually reconstructed based on the z-stack images by using the Vaa3D / Mozak software package. (b) Electrophysiology, imaging, and morphology data and metadata for each cell are made freely accessible through the Allen Cell Types Database. An interactive user interface allows users to filter thousands of cells by electrophysiology and morphology features, then each cell has a specific page with detailed electrophysiology and morphology data, when available.
Electrophysiology classification
We characterized the intrinsic electrophysiological properties with a standardized current-clamp protocol that contained a variety of stimuli, including square pulses, ramps, and noisy current injections (Fig. 1a, Supplementary Fig. 3). Our goal was to assess a diverse array of e-features while still enabling comparison across all cells.
We derived features from both membrane potential traces and specific characteristics of action potentials (APs). For example, waveforms of the first APs evoked from a 3 ms current step, a 1 s current step, and a ramp stimulus were collected from each cell and aligned on the time of their thresholds (Fig. 2a). Responses to a series of hyperpolarizing current steps were also extracted and aligned (Fig. 2b). For each AP evoked by depolarizing current steps, a set of features was calculated (Methods). Since the number of APs evoked varied over two orders of magnitude across stimulus amplitudes and cells, responses were compared by dividing the stimulus interval into 20 ms bins, calculating the average of the AP features within that bin, and interpolating values for bins without APs (Methods). An example of the threshold voltage feature is shown in Fig. 2c. The shape of the membrane potential trajectory during the interspike interval (ISI) also varied significantly across cells. To capture this, the trajectories were normalized by the duration of the interspike interval and calculated as a difference from the AP threshold voltage (Fig. 2d). Other features were collected in similar ways (Methods) forming 13 different subsets of data (Supplementary Table 1). Together, they represented multiple aspects of the electrophysiological responses that could be robustly extracted and compared across all the cells.
Figure 2: Classification of electrophysiological properties.
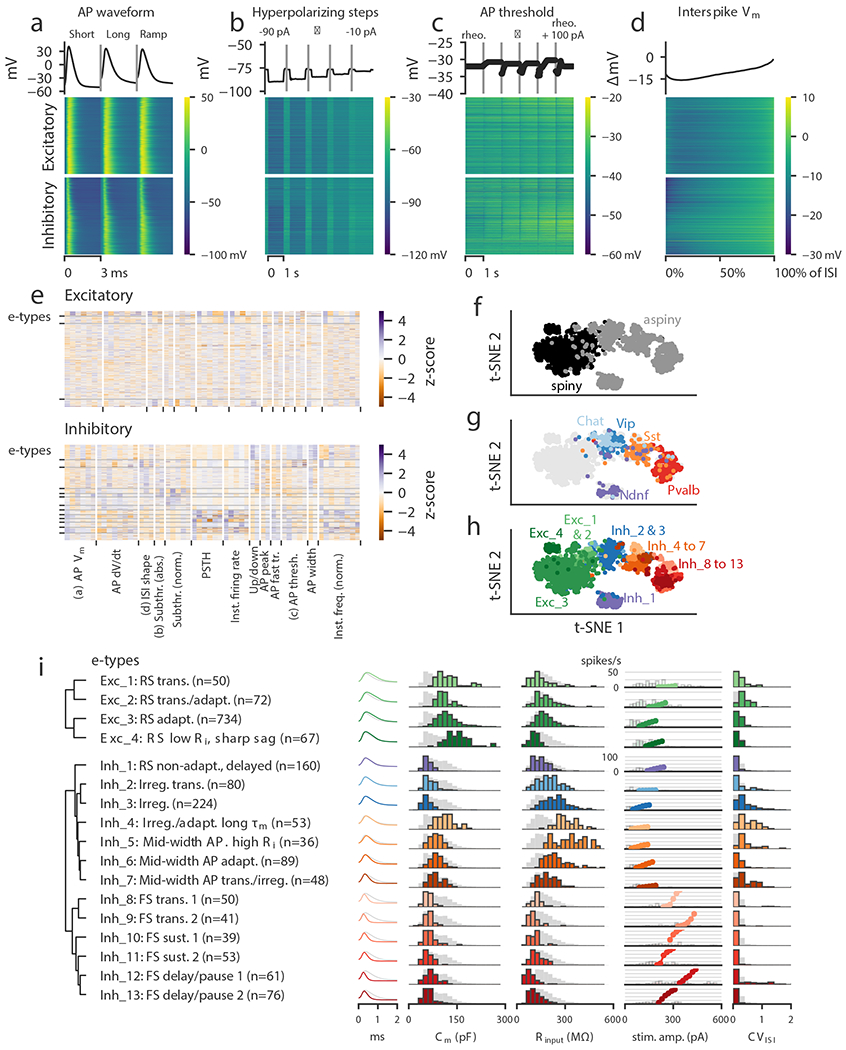
(a) Action potential waveforms of n=1,938 cells evoked by a short (3 ms) current pulse, a long square (one second) current step, and a slow current ramp (25 pA/s). Example trace (top) and heat map of all responses (bottom). The cells in the heatmap are split into excitatory (spiny) cells above and inhibitory (aspiny/sparsely spiny) cells below (as determined from the images of each cell), and ordered within each of those groups by their average upstroke/downstroke ratio during long square current steps. The order of cells is the same in the heat maps of (a)–(d). In (a)–(c), vertical lines shown within examples separate data collected from different sweeps. (b) Membrane potential responses to hyperpolarizing current steps. (c) Action potential threshold voltages of spikes evoked by a series of depolarizing current steps. (d) Interspike interval membrane potential trajectories. For a given sweep, each interspike interval duration was normalized, resampled to have a consistent number of points, aligned on the action potential threshold (set to 0 mV), and averaged together. (e) Sparse principal component values collected from each data type, indicated by labels at the bottom. Each component’s values were transformed into a z-score. Rows are sorted into clusters indicated by left tick marks. (f) t-SNE plot using the components shown in (e) with aspiny/sparsely spiny (collectively referred to as “aspiny”) and spiny neurons identified (n=1,938 cells). The same t-SNE plot is shown in (f-h). (g) t-SNE plot with selected inhibitory-dominant transgenic lines identified. Only aspiny neurons from those lines are shown. (h) t-SNE plot with electrophysiology types (e-types) identified. (i) E-types and specific features. Dendrogram on left was created by hierarchical clustering based on distances between each cluster’s centroid. For AP shape, e-type averages are shown as colors and the grand average across all cells is shown in gray. For histograms, e-type values are shown in colors and full population is shown in gray. All histograms are scaled to their highest value. The f-I curves were aligned on the rheobase value and averaged. The average curve is plotted starting at the median rheobase. Distribution of rheobase values for cells in the clusters are shown as histograms behind the average curve.
These 13 data sets provided the foundation for an unsupervised classification of e-cell types. Given the extensiveness and complexity of the data, we first reduced its dimensionality, then applied an unsupervised clustering algorithm to identify different e-types. We applied a sparse principal components analysis20,21 (sPCA) procedure to each collected subset of the data, which typically identified 1-8 components that exceeded an adjusted explained variance threshold per subset (Supplementary Fig. 4, Methods). These components were collected (Fig. 2e; 54 components for excitatory neurons, 56 for inhibitory neurons), and a Gaussian mixture model (GMM) was fit to the data to divide it into clusters21, followed by a merging step using an entropy criterion22. The stability of clusters was assessed by subsampling analysis23, and unstable clusters were merged with others (Supplementary Fig. 5, Methods). This procedure yielded 17 total clusters, or e-types: four e-types for the 928 excitatory neurons and thirteen for the 1,010 inhibitory neurons. We clustered these two classes separately; clustering them together resulted in a similar, though not identical, number of clusters (14 total), but it also produced one cluster that contained both excitatory and inhibitory neurons (Supplementary Fig. 6).
We visualized the entire data set by projecting sPCA features (re-calculated using the combined data from both excitatory and inhibitory cells, yielding 54 components) onto two dimensions with the t-distributed stochastic neighbor embedding (t-SNE) method24. This procedure performs a nonlinear embedding into two dimensions that attempts to preserve the local similarity structure of high-dimensional data. The projection separated the spiny excitatory neurons from the aspiny/sparsely spiny inhibitory neurons reasonably well (Fig. 2f). Furthermore, populations of cells defined by certain transgenic drivers were also relatively coherent in this projection (Fig. 2g, Supplementary Fig. 7). For example, Pvalb-labeled cells were adjacent to Sst cells, and Vip cells were found on the other side of Sst cells. These markers label largely mutually-exclusive subclasses of interneurons2,12, and we likewise saw relatively little overlap in the t-SNE projection. On the other hand, Chat cells, a subset of Vip cells12,25,26, were located within the broader region populated by the Vip cells near its border with excitatory cells. This cohesiveness suggests that there is similarity in e-features within genetically-defined populations of cortical cells, as well as distinctiveness between non-overlapping populations. The 17 e-types identified by the GMM in higher-dimensional sPCA spaces were largely coherent in the t-SNE projection as well (Fig. 2h).
We next examined basic electrophysiological characteristics of the 17 identified e-types, such as the AP shape, the estimated membrane capacitance and input resistance, the average f-I curve, and the coefficient of variation of the ISI durations (CVISI, Fig. 2i). We also examined several other firing pattern characteristics used to classify neurons in previous studies6,7, including the delay to the first spike, bursting (here referring to short periods of high-frequency firing amid lower-frequency firing), pausing (long interspike intervals amid higher-frequency firing), and spike frequency adaptation (Supplementary Figs. 8–11, see Methods for specific definitions). Note that these specific measures were not directly used in the classification; however, they were used in giving descriptive names to the e-types as shown in Fig. 2i. We also assessed the transgenic-line composition (Supplementary Fig. 12) and laminar distribution (Supplementary Fig. 13) of each e-type.
The excitatory e-types were relatively similar to each other, with wide APs and similar distributions of membrane capacitance and input resistance (Fig. 2i). Across cells, firing rates at 100 pA above rheobase rarely exceeded 30 spikes/s. Inhibitory e-types were more diverse than their excitatory counterparts. As expected, membrane capacitance values were nearly all lower than those of the excitatory e-types (though Inh_4 was a notable exception) since interneurons typically have smaller cell bodies, simpler dendritic arbors, and lack spines (Fig. 2i). Specific e-types differed from each other across a variety of characteristics, including firing patterns (e.g., adapting, irregular, transient), AP shape, subthreshold properties (membrane time constant, sag), and f-I curve location and shape.
We investigated which features were most informative about e-type by training a random-forest classifier on the data set (Supplementary Fig. 14), which found that components derived from AP width and instantaneous firing rate had higher relative feature importances. Feature importance was defined as the average decrease in the Gini index when a decision tree made a split using the given feature27,28 (the Gini index reflects the “purity” of the composition of final leaves in the tree and is small when each leaf only contains samples from a single type). Interestingly, the t-SNE projection was fairly robust to feature removal (Supplementary Fig. 14), suggesting that at least coarse e-type identity could be inferred from multiple electrophysiological features.
While the different e-types had distinguishing characteristics, it is important to note that each one was not entirely homogeneous. Properties may vary considerably within a type as long as gradual variation is present; the existence of these intermediates means that there is no identifiable place to subdivide the cluster. The t-SNE projection, by emphasizing local similarity, can illustrate these patterns within a cluster: for example, sets of cells labeled by a given transgenic lines in some cases appear near each other but separated from cells labeled by other lines, even within the same e-type (Supplementary Fig. 7).
Our results here were largely in agreement with cell-type classifications based on slice electrophysiology presented in previous studies in the same area and related areas and species6,8,29; however, we noted some particular differences. For example, we characterized bursts and pauses with a graded measure indicating the occurrence of short sets of ISIs with substantially higher- or lower-frequency firing among more regular firing. We used this measure for consistency across the wide range of firing patterns in our data set, but this differed from characterizations used in other studies (such as first-to-last ISI ratio, or expert annotations of bursts and pauses). While, for example, Markram et al.6 presented types explicitly categorized by bursting or delayed firing patterns, here certain e-types had higher proportions of neurons that exhibited these patterns (e.g., Exc_4 for bursting, Inh_1 for delayed firing) but were not strictly demarcated by these patterns. Additional observed differences in e-types could be due to differences in model systems (i.e., P14 rat somatosensory cortex in that study versus P56 mouse visual cortex here) and experimental conditions.
Morphology classification
To create a comparable standardized, objective m-type classification system, we generated morphological reconstructions for a subset of aspiny and spiny cells described above. Reconstructions were based on a digitized, brightfield image stack of single, biocytin-filled neurons using semi-automated and manual tools in Vaa3D software (Fig. 1a). We chose neurons for reconstruction to provide representation across broad classes, qualitatively assessed morphological types, layers, and transgenic lines. The apical dendrite is most often used to distinguish among spiny, excitatory neurons (e.g., star pyramids and thick tufted neurons)30,31 and so we reconstructed the apical and basal dendrites and only a small portion of the initial axon of spiny neurons. In contrast, the axon of aspiny neurons has been widely recognized as the most defining part of inhibitory cortical interneurons (e.g., layer 1-projecting Martinotti neurons and more layer restricted non-Martinotti neurons)2,7,15 and so we reconstructed the axon and dendrites of aspiny neurons to the maximal extent possible (within the confines of the slice).23,24
Drawing from the literature and existing analysis tool kits (e.g., L-measure25 and the Blue Brain Project5), a feature set was created that allowed extraction of multiple, well-known shape features from either dendritic or axonal branches in our reconstructions (e.g., branch number, total branch length) and new features, such as the intersection between different branch types (e.g., between apical and basal dendrites) were also introduced. Critically, all reconstructions were aligned to the pia, which allowed for additional features, such as the laminar distribution of axon (axon histogram) to be included in our clustering analysis. Features predominantly determined by differences in the z-dimension were not included in our morphological classification due to z-compression in the acute slice after processing32,33 (Methods). Due to the fundamentally different nature of the branch structures captured in the spiny and aspiny neuron reconstructions, unsupervised clustering analysis was run on spiny and aspiny neurons separately. Hierarchical clustering was performed on 254 spiny neurons using 39 features of the apical and basal dendrites (Supplementary Fig. 15, Supplementary Table 2), and 207 aspiny neurons using 45 features of the dendrites and axons (Supplementary Fig. 16, Supplementary Table 2). The number of clusters for each population was determined by maximizing between-cluster variation and minimizing within-cluster variation, then performing a co-clustering step11 to identify robust clusters (Fig. 3a, 4a, Supplementary Fig. 17). Next, neurons in small (n≤3) clusters were reassigned to the most highly correlated neighboring cluster, and clusters with no significantly different features between them were merged. For aspiny and spiny datasets, clusters showed good predictability by cross-validation with two supervised classifiers (support vector machine and random forest, see Methods).
Figure 3: Unsupervised classification of spiny neurons into morphological types.
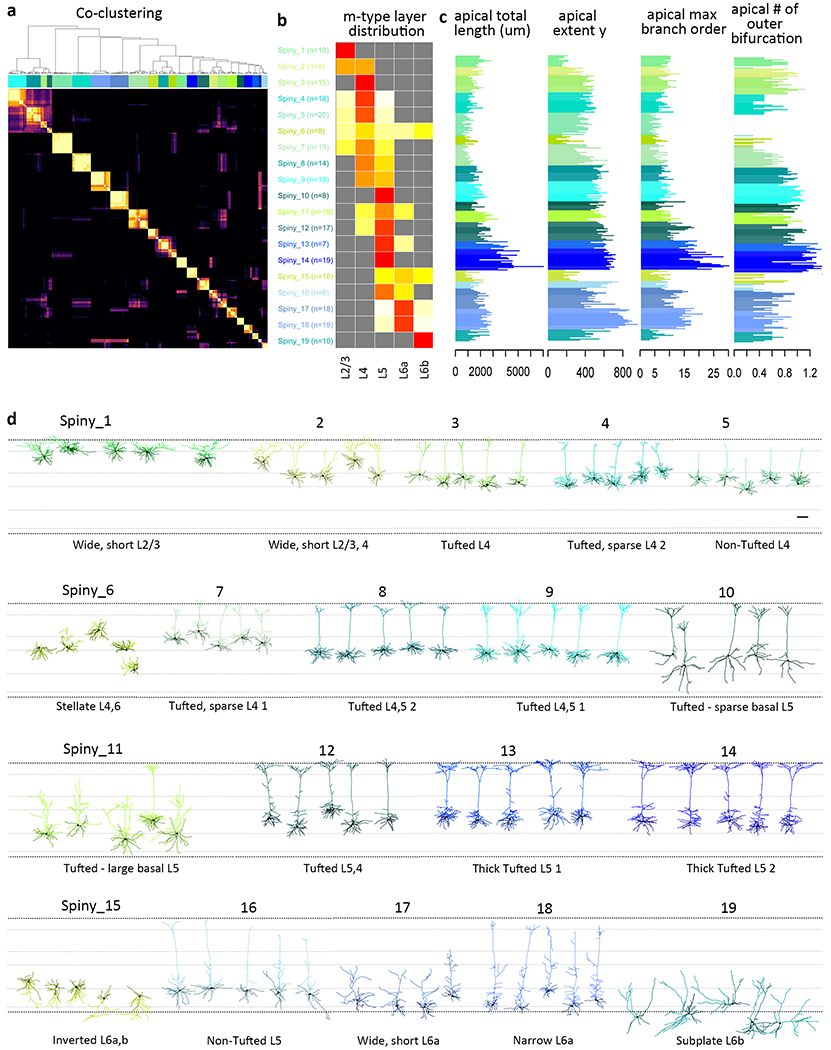
(a) Co-clustering diagram from 1000 runs of hierarchical clustering with 90% subsampled data. 19 morphological types (m-types) are identified by CutreeHybrid()*, which included a step to merge neurons in clusters with an n≤3 with their most highly correlated cluster and clusters with no significantly different features between them. Each m-type is assigned a color that is maintained throughout the figure (n=253 spiny neurons). (b) Laminar distribution of m-types across layers 2/3-6b. Relative proportion of each m-type per layer is shown. (c) Examples of apical dendrite features that vary systematically across m-types. See Supplementary Fig. 19 for all morphological features included in the analysis. (d) Representative examples of each m-type, roughly ordered by their location in layers 2/3-6b. Neurons in each m-type are shown with respect to averaged cortical layers (see methods for details). Each m-type has two names, a numbered name (e.g., Spiny_1) and a descriptive name (e.g., Non-Tufted L4). Apical dendrites are shown in the lighter color and basal dendrites in the darker color. Morphology scale bar: 100 μm. See Supplementary Fig. 18 for individual morphologies that went into this clustering analysis and Supplementary Fig. 22 for m-type representation across transgenic lines. All reconstructions and the corresponding images are available online (http://celltypes.brain-map.org/).
In total, 38 clusters or m-types were identified, 19 for spiny neurons (Fig. 3) and 19 for aspiny neurons (Fig. 4). The 19 spiny, excitatory m-types (Fig. 3, Supplementary Figs. 18-19, Supplementary Spreadsheet 1) were distributed across layers (L) 2/3-L6b (Fig. 3b). The majority of spiny m-types were distinguished based on their relative soma depth, the size and extent of their apical dendrite, particularly the apical tuft, and the relative distribution of the basal dendrites (Fig. 3c). Spiny m-types were predominantly found in a single layer, but were not exclusive to that layer, although see Spiny_1, 3, 10, 14, and 19 (Fig. 3b). This agrees with previous descriptions of excitatory m-type distribution in other brain regions8,21. The 19 m-types for inhibitory interneurons were distributed across all cortical layers (L1-L6b), with most m-types found in multiple layers with the exception of several L2/3-specific m-types (Fig. 4b). Relative soma depth, dendrite and axon size and distribution were all important for distinguishing among inhibitory m-types (Fig. 4, Supplementary Figs. 20-21, Supplementary Spreadsheet 1). The distribution of m-types varied across transgenic lines (Supplementary Fig. 22), and many, but not all, m-types were found in relatively consistent locations on the electrophysiology-based t-SNE projection (Supplementary Fig. 23).
Figure 4: Unsupervised classification of aspiny neurons into morphological types.
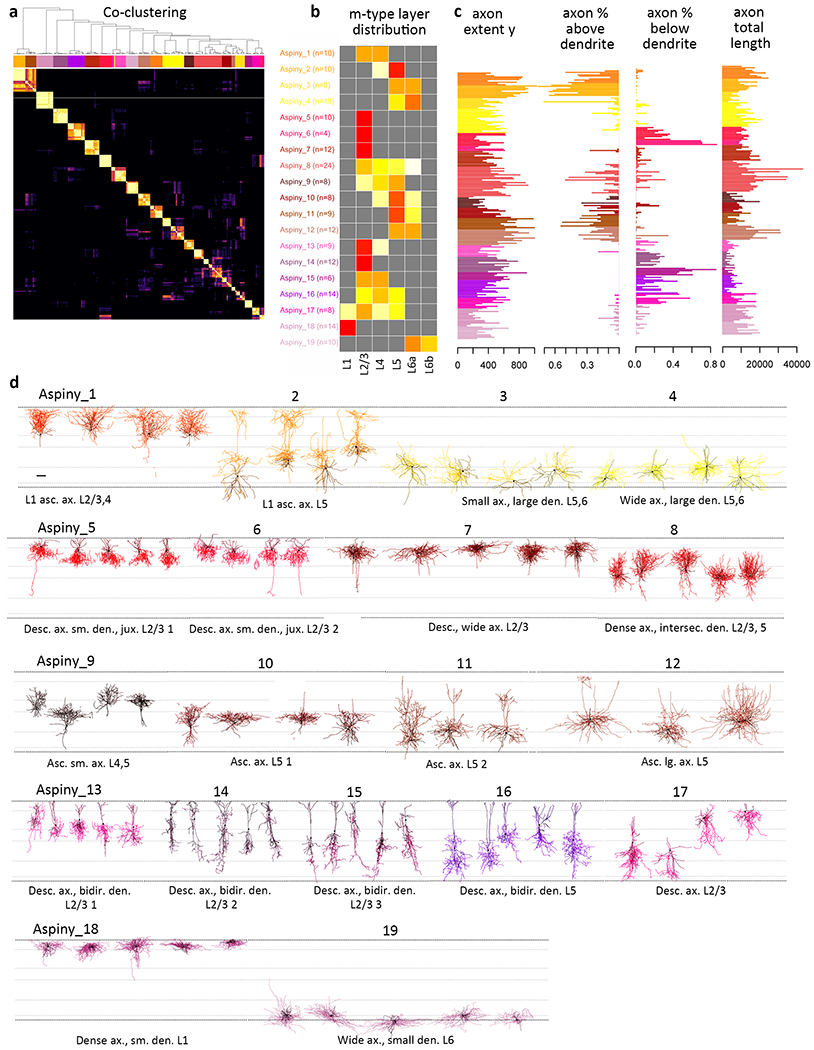
(a) Co-clustering diagram from 1000 runs of hierarchical clustering with 90% subsampled data. 19 clusters are identified by CutreeHybrid()*, which included a step to merge neurons in clusters with an n≤3 with their most highly correlated cluster and clusters with no significantly different features between them. Each m-type is assigned a color that is maintained throughout the figure (n=207 aspiny neurons). (b) Laminar distribution of m-types across layers 2/3-6b. Relative proportion of each m-type per layer is shown. (c) Examples of axonal features that vary systematically across m-types. See supplementary Fig. 19 for all morphological features included in the analysis. (c) Representative morphologies from each quantitatively defined aspiny type. Neurons are shown in their approximate laminar location with respect to averaged cortical layers. M-types are grouped by their most dominant molecular class (as defined by Cre line). Each m-type has two names, a numbered name (e.g., Aspiny_1) and a descriptive name (e.g., Dense ax., sm. den. L1). Axons are shown in the lighter color and dendrites in the darker color. Morphology scale bar: 100 μm. See Supplementary Fig. 20 for individual morphologies that went into the analysis and Supplementary Fig. 22 for m-type representation across transgenic lines. All reconstructions and the corresponding images are available online (http://celltypes.brain-map.org/).
To assess how transferable our morphological classification system is to other brain regions and/or species, we applied our feature extraction and morphological classification approach to the large dataset of juvenile rat somatosensory cortex morphologies available through the Neocortical Microcircuit Collaboration Portal of the Blue Brain Project (http://microcircuits.epfl.ch/#/main, Supplementary Figs. 24-26). Due to differences in the data collection scheme, after splitting the aspiny and spiny neurons (based on the absence or presence of an apical dendrite in the reconstruction), we further split the dataset into assigned layer groupings, then performed our clustering analysis. Results were evaluated with respect to their expert-defined m-types6. When we split cells into individual layer groupings, we identified spiny clusters that subdivided the BBP qualitative m-types (e.g., the single L2/3 PC m-type was split into 6 m-types), and those that merged m-types (e.g., L5 thick tufted 1 and 2, Supplementary Fig. 24), which agrees with recent analyses of these same data34. Aspiny m-types within a subclass (e.g., nest and small BCs) were frequently merged within a layer, but we also identified fairly distinct groupings of Martinotti cells, double-bouquet cells, and chandelier cells (Supplementary Fig. 25). Agreement between the two classifications may be further improved with the addition of information about cortical context (e.g., relative soma depth and aspiny axon layer distribution) that was used in this study but was unavailable for the rat dataset.
Joint classification with electrophysiology and morphology
We next investigated the extent to which electrophysiological and morphological characteristics co-varied by performing a joint clustering analysis using cells for which both sets of features were available (n=253 excitatory, 199 inhibitory). Clustering was again performed separately for excitatory and inhibitory cells using both the sPCA electrophysiology components and morphological feature sets described above. Since the relative importance of the different modalities was not known, we clustered the joint data using several different methods and relative weightings of the two data sets (Methods), then performed a cell-wise co-clustering analysis to identify and refine consensus clusters14. Cluster stability was also assessed through subsampling23. We found 20 excitatory clusters and 26 inhibitory clusters, or me-types (Fig. 5a-b) for which we also assessed the transgenic line composition (Supplementary Fig. 27) and laminar distributions (Supplementary Fig. 28). The number of me-types from this analysis (46 total) was substantially smaller than the number of e-type/m-type combinations observed by a simple intersection of both unimodal classifications (120 total, Supplementary Fig. 29).
Figure 5: Classification using paired electrophysiological and morphological data.
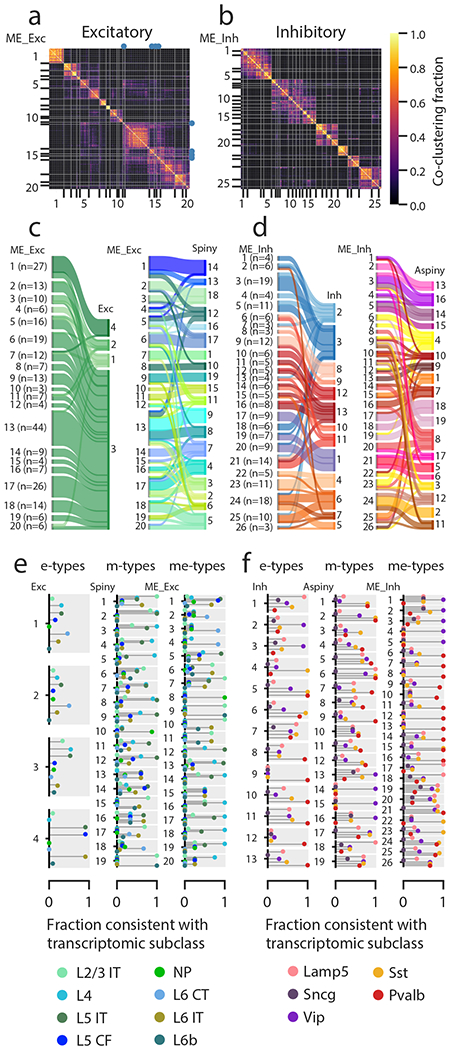
(a) Excitatory cellwise co-clustering matrix (across several clustering methods and weights) with consensus clusters (me-types) identified (n=253 excitatory cells with both electrophysiological data and morphological reconstructions). The me-types that were identified as unstable by subsampling analysis are identified with blue dots. (b) Same as (a) but for inhibitory cells (n=199). (c-d) Correspondences between me-types and either e-types or m-types for excitatory (c) and inhibitory (d) cells. Widths of bars are proportional to the numbers of cells in a type. (e) Consistency of excitatory e-types, m-types, and me-types with transcriptomic subclasses as inferred from transgenic line compositions. The plot for each type has separate bars for each transcriptomic subclass identified by different colors; each type has the same set of bars (see legend for labels). A consistency value close to 1 means that most of the cells with that type came from transgenic lines that were all consistent with labeling that subclass, while a value close to 0 means that cells came from lines that do not label that subclass (see Supplementary Fig. 1), based on FACS data from another study14. A single consistency value of 1 (e.g., ME_Exc_8) indicates that all the cells of that type are from the indicated transcriptomic subclass (by virtue of all cells being from one or more unambiguous transgenic lines). IT: intratelencephalic, CF: corticofugal, NP: near-projecting, CT: corticothalamic. (f) Same as (e) but for inhibitory cells.
When we examined the composition of these me-types in terms of unimodal e- or m-types, most me-types appeared relatively homogeneous. Unsurprisingly, the large excitatory e-type Exc_3 was divided across many excitatory me-types. Each of these me-types was usually consistent with an excitatory m-type (Fig. 5c), though in some cases multiple m-types were collapsed into fewer me-types. Inhibitory me-types were consistent with the different inhibitory e-types, although there was some mixing between, for example, the different fast-spiking e-types (Inh_8 through Inh_13, Fig. 5d). Certain inhibitory m-types (e.g. Aspiny_4, Aspiny_8) were distributed across multiple me-types due to differences in electrophysiological properties. Overall, we found that our joint clustering produced a set of me-types that reflected both experimental modalities measured without either dominating the classification.
Next, we investigated whether these me-types were consistent with other identifiers of cell type, using transgenic line labeling as a proxy of transcriptomic identity. While transgenic lines typically do not label individual transcriptomic types12,14, we evaluated each line for whether or not the labeling was consistent with each transcriptomic subclass (see Supplementary Fig. 1) based on FACS data from another study14. These include eight excitatory subclasses (L2/3 intratelencephalic (IT), L4, L5 IT, L5 corticofugal (CF), near-projecting (NP), L6 IT, L6 corticothalamic (CT), and L6b) and five inhibitory subclasses (Lamp5, Sncg, Vip, Sst and Pvalb).
We chose to make only binary classifications (i.e., a line was defined as consistent if at least 5% of cells in that line were found in a given transcriptomic subclass) due to sampling decisions and biases that prevented more graded analyses of composition. Then, we calculated consistency scores for each e-, m-, and me-type by counting each cell as consistent or not with each subclass based on the transgenic line that labeled it (Fig. 5e-f). For example, if all cells in an me-type came from Pvalb-IRES-Cre, which is only consistent with the Pvalb subclass, that me-type would have a score of 1 for the Pvalb subclass and 0 for all others. If, however, half came from Pvalb-IRES-Cre and the other half came from Nkx2-1-CreERT2, which is consistent with Pvalb, Lamp5, and Sst, the score would be 1 for Pvalb (since all the cells are from lines consistent with Pvalb), 0.5 for Lamp5 and Sst (since half the cells are from a line consistent with those subclasses), and 0 for the others (Vip and Sncg). Note that ambiguity in transgenic line labeling (i.e., lines labeling multiple subclasses) could lead to multiple subclasses having high scores for the same me-type; if all the cells had come from Nkx2-1-CreERT2, the scores would be 1 for Pvalb, Lamp5 and Sst and 0 for Vip and Sncg.
Among excitatory cells, we found that most excitatory e-types had low consistency scores (except Exc_4), while more m-types had higher scores, and even more me-types had high scores (Fig. 5e). On the inhibitory side, e-types showed relatively high consistency, m-types had lower scores, and most me-types were again highly consistent with transcriptomic subclasses—for many me-types, a single subclass had a high score while the rest were low (Fig. 5f). These results suggest that the joint morpho-electric clustering identifies a set of me-types that is better correlated with neuronal subclasses than each modality separately.
Comparison to transcriptomic classification via transgenic lines
Connecting transcriptional profiles with morphological and electrophysiological properties is a powerful way to understand the functional implications of diverse gene expression35. Recent studies have defined taxonomies of mouse neocortical cell types by single-cell RNA-seq transcriptomic profiling of isolated neurons and glia12–14. However, it remains an open question as to how well a cell’s specific transcriptomic identity corresponds to its electrophysiological and morphological phenotypes.
We used the transcriptomic subclass consistency results (Fig. 5e-f) to infer the most likely subclass associated with each me-type (Fig. 6a-b). In cases where an me-type had more than one high consistency score, we resolved the ambiguity using prior knowledge (e.g., linking ME_Exc_1, containing thick-tufted pyramidal cells, to the L5 CF subclass) and/or laminar location (e.g., ME_Exc_9, consistent with both L2/3 IT and L6b subclasses, contains only cells from L6b). Individual examples of morphologies and electrophysiological responses are shown for each me-type organized by putative transcriptomic subclasses (Fig. 6a-b). Detailed assessments of the me-types derived from our study and comparisons with prior cell type knowledge are provided in Supplemental Table 3 and the Supplemental Note.
Figure 6: Transcriptomic subclasses and me-types.
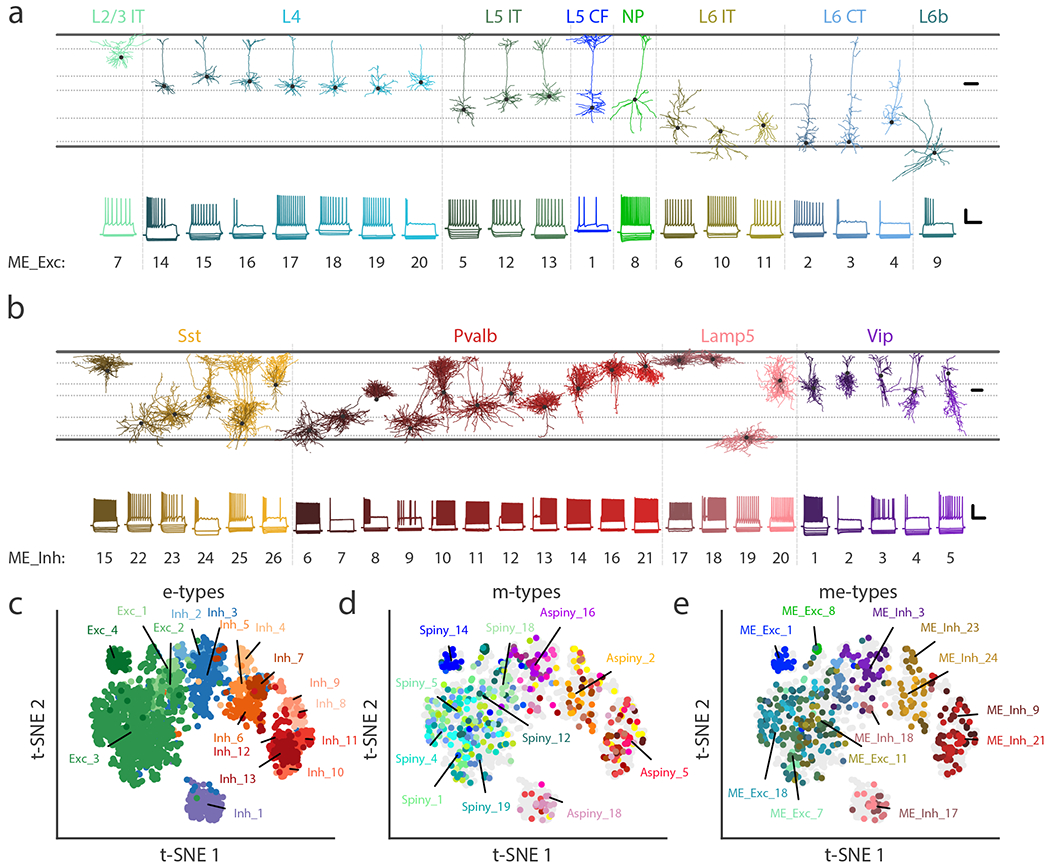
(a) Example cells from each excitatory me-type grouped by putative transcriptomic subclass. Top shows morphological reconstructions of dendrites and bottom shows electrophysiological responses from the same examplar cell. Some notable specific types include non-tufted (ME_Exc_18 & 20), thick-tufted (ME_Exc_1), and inverted (ME_Exc_10 & 11) pyramidal cells. See Supplementary Table 3 for detailed descriptions of each me-type. Morphology scale bar: 100 μm. Electrophysiology scale bar: vertical, 40 mV; horizontal, 500 ms. IT: intratelencephalic, CF: corticofugal, NP: near-projecting, CT: corticothalamic. (b) Same as (a) but for inhibitory me-types, except both axonal (lighter shades) and dendritic (darker shades) compartments are shown. Some notable specific types include deep non-Martinotti cells (ME_Inh_22 & 23), Martinotti cells (ME_Inh_15, 24, 25), chandelier cells (ME_Inh_21), and late-spiking L1 neurogliaform cells (ME_Inh_17). (c-e) t-SNE plots based on electrophysiological features (n=1,938 cells) showing e-types (c), m-types (d), and me-types (e). Colors correspond to Figs. 2-4 for e- and m-types and to (a-b) for me-types.
To determine which features were most informative for me-type classification, we trained a supervised classifier to predict me-types from the combined modalities, as well as from electrophysiological features alone and morphological features alone (Supplementary Fig. 30). This confirmed that morphological features were more distinguishing for excitatory me-type classification, while both electrophysiological and morphological features could accurately predict many inhibitory me-types. In addition, the classifiers generally made fewer errors predicting me-types associated with different transcriptomic subclasses, further supporting the proposed organization of me-types.
We also visualized these results together using the electrophysiology-based t-SNE projection using the larger set of 1,938 cells (Fig. 6c-e). The me-types and, to a lesser extent, m-types were found in consistent locations in the electrophysiological projection (Fig. 6d-e, Supplementary Figs. 23 and 31).
Correspondence with specific transcriptomic types
While transgenic lines are imperfect labels of transcriptomic cell types, we were nevertheless able to identify a subset of transgenic line and layer combinations that were each selective for a small number of transcriptomic types (t-types, Fig. 7). For example, the cells in L4 labeled by the Nr5a1-Cre line are predominantly of a single t-type (Fig 7a, 96% in one type), while cells in L5-L6 labeled by the Slc17a8-iCre line are mostly one of two related t-types (Fig 7b, 83% combined).
Figure 7: Correspondence of me-types with transcriptomic types.
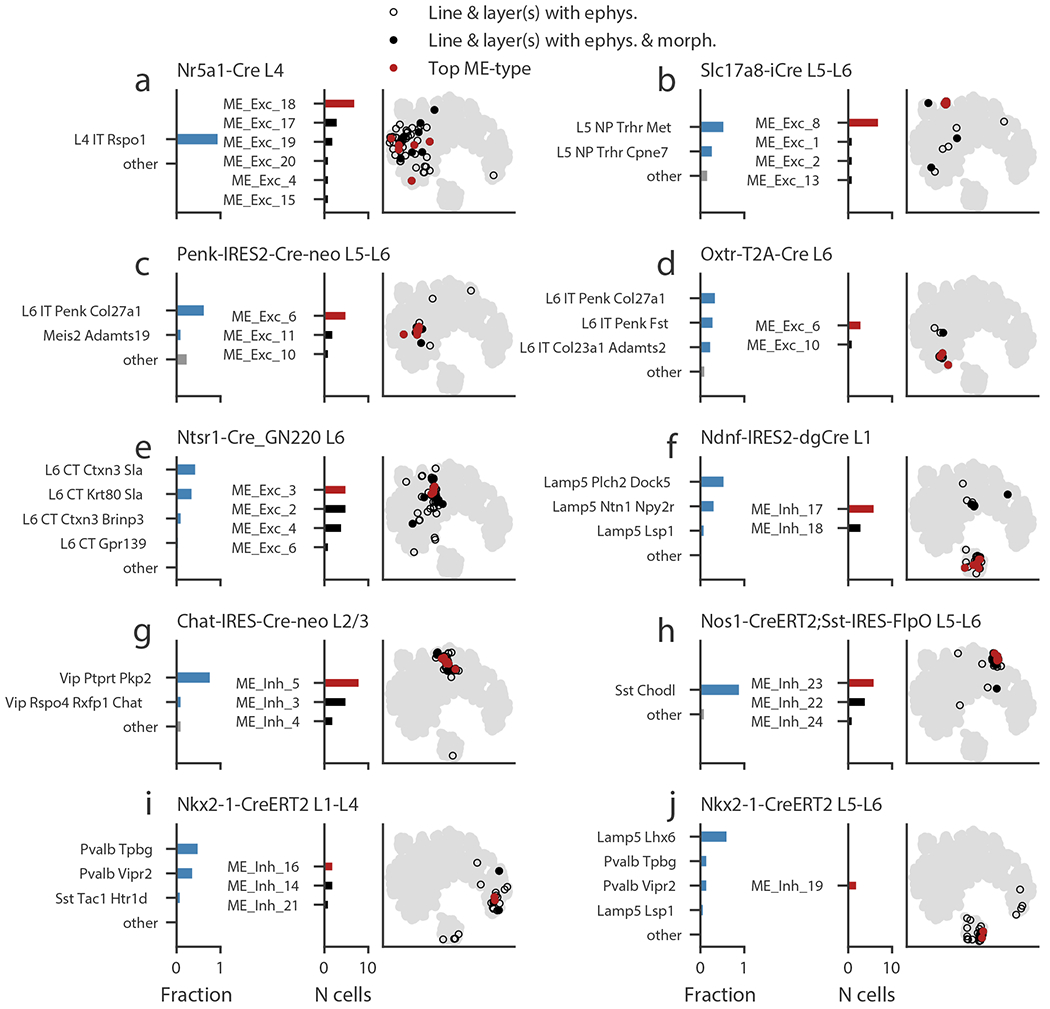
(a-j) Transgenic line/layer combinations that label transcriptomic types (t-types) and the corresponding me-types. Left bar chart shows the fraction of cells in a t-type identified by another study from FACS data collected with similar transgenic line and layer sampling. The t-types with fractions of 0.05 or more are shown individually; the remaining are grouped as “other.” Right bar chart shows the me-type distribution of cells in this study collected from the specified transgenic line/layer combination. The most common me-type is shown in red. Note that only cells with both electrophysiological data and a morphological reconstruction appear in this plot. The t-SNE plot based on electrophysiological features shows all cells with electrophysiology (gray), cells from the specified line/layer combination (hollow black circles), cells from the line/layer combination that also have morphological reconstructions (filled black circles), and those cells with the most-common me-type (red circles).
Using these selective transgenic line/layer combinations, we examined the me-type diversity associated with a small set of t-types. In some cases, there appeared to be a one-to-several correspondence between t-types and me-types (and vice versa). For example, several me-types were observed among L4 Nr5a1-labeled neurons, suggesting some heterogeneity within the single t-type L4-IT-Rspo1 (Fig. 7a). However, other combinations labeling between one and three t-types predominantly had a single me-type (Fig. 7b-d), which were also tightly clustered in the electrophysiological t-SNE projection. Interestingly, Penk-IRES2-Cre-neo in L5-L6 and Oxtr-T2A-Cre in L6, which labeled overlapping sets of t-types, both identified the same me-type (ME_Exc_6); the cells labeled were in neighboring yet offset locations in the t-SNE projection (see also Supplementary Fig. 31), suggesting some graded electrophysiological differences between those cells. L6 Ntsr1-labeled neurons (linked to four related L6 CT t-types) labeled a set of me-types with similar electrophysiological features (Fig. 7e).
Among inhibitory neurons, neurons labeled by Ndnf in L1, associated with a handful of Lamp5 t-types, had one of two me-types (ME_Inh_17 and 18, Fig. 7f). The L2/3 Chat-labeled neurons, mostly associated with the Vip-Ptprt-Pkp2 t-type, were in a few similar me-types (Fig. 7g). The L5-L6 neurons labeled by a Nos1/Sst intersectional strategy were quite coherent in the electrophysiological t-SNE projection and had mostly one of two similar me-types; these cells are expected to have the Sst-Chodl t-type, linked to deep long-range projecting interneurons12,14,36,37 (Fig. 7h). The Nkx2-1 line labeled several relatively specific populations. In L1-L4, the line is associated mostly with the Pvalb-Vipr2 type (expected to be chandelier cells14) and the Pvalb-Tpbg type (Fig. 7i). Accordingly, we found that these neurons had me-types that contained fast-spiking cells and had basket-cell and chandelier-cell morphologies (see Figs. 5d and 6b). In L5-L6, the Nkx2-1-labeled cells are mostly associated with the Lamp5-Lhx6 t-type, and while we had a small number of such cells with both morphology and electrophysiological data, the location in the t-SNE projection was tight and consistent with the me-type observed (Fig. 7j). While we were only able to perform this analysis for a limited set of t-types, we found that we could make relatively specific correspondences with me-types in most of those cases.
DISCUSSION
Obtaining the cell type composition, the “parts list,” of neural circuits is foundational to understanding circuit function. To do so it is essential to take a systematic, unbiased, and quantitative approach towards cell type classification using multi-dimensional criteria, in order to resolve debates and enable the field to adopt a common set of standards. Here we describe such an effort in the morpho-electric domain in the adult mouse visual cortex. We acquired data using a standardized pipeline and uniform quality control checks such that each cell was subject to an identical process; this data production enabled the combined analysis of over 1,900 patch-clamp recordings. We developed unsupervised classification methods that were consistently applied across all recorded cells, and we used both electrophysiological and morphological features measured from the same cell together when possible. This led to the identification of 17 e-, 38 m-, and 46 me-types that showed strong correlation with transcriptomically defined neuronal subclasses and types.
Conventional electrophysiological and morphological classification often relies on predetermined, cell-specific feature selection and/or qualitative assessments of firing pattern or cell shape5,7. Our approach, while certainly not free of biases (e.g., choice of stimuli delivered to the cells, morphological features quantified), has the advantage of being quantitative, reproducible, transferable to new data (e.g., Supplementary Fig. 24-26), and robust across the varied characteristics of the entire population of neurons studied. This mouse morpho-electric visual cortical dataset is large and diverse, and our use of standardized protocols and equipment throughout yields a classification that does not need to be adjusted for interlaboratory variability38. At the same time, we note that our electrophysiological results reflect the specific experimental protocols used here; different recording conditions and methods affect electrophysiological responses39–41 and could amplify or diminish differences in particular features across neurons. In addition, we anticipate that other protocols or measures outside the scope of this data set could reveal new features that distinguish particular cell types; these findings would test and potentially revise the classifications presented here. It should also be noted that our use of a variety of transgenic lines to capture as many neuronal types as we could does not allow us to assign relative proportions of the identified cell types in the visual cortical circuit – an issue to be addressed in future studies likely through a different approach.
Our methods and data are publicly accessible as part of the Allen Cell Types Database at celltypes.brain-map.org, allowing other investigators to build upon or independently evaluate this classification scheme. These data can also be used for investigations beyond what we describe here; for example, models of varying levels of complexity have been built using the electrophysiological and morphological data42–44, tools have been created to integrate the data into an automated analysis workflow45, and genetic/electrophysiological correlations have been inferred using these data in combination with single-cell transcriptomic data46, which were also publicly released via the Allen Cell Types Database.
A novel aspect of our approach is the dual-modality, joint clustering analysis combining electrophysiological and morphological features to define me-types across a diverse array of cortical neurons. A substantial improvement over previous attempts in deriving me-types from the simple intersectional overlay of m- and e-types, the joint clustering analysis is a more principled approach to extract co-varying features from each modality out of their respective diverse ranges of heterogeneity, resulting in more coherent and meaningful grouping of cells. For example, we identify 46 me-types by joint clustering, in contrast to the 120 observed combinations by just intersecting e- and m-types (Supplementary Fig. 29). The me-types we identified are better correlated with excitatory and inhibitory subclasses and types defined conventionally or by molecular identities than each modality separately (Figs. 5-7), and thus provide a more robust description of cortical cell types than single-modality data alone. We find that the ability of each modality to predict me-types varies across subclasses (Supplementary Fig. 31); morphological features capture the diversity of most excitatory cells more clearly than electrophysiological ones, while electrophysiological features clearly identify different L6 interneurons from Lamp5, Pvalb, and Sst subclasses that have similar dendritic and axonal arbors. Therefore, clustering with both modalities allows the most distinct features to be used across different types. It will be important to continue to apply this kind of multi-modal co-clustering approach to uncover meaningful patterns of correspondence across molecular, physiological, morphological, connectional and/or functional characteristics of cells, ultimately connecting cell type classification with the functional properties of the circuit.
Large-scale transcriptomic studies have provided informative taxonomies of cortical cell types12–14. Relating morphological and physiological properties to those results will refine our understanding of cell type hierarchies and the potential functions of those cell types in cortical circuits. The preliminary correspondences we find here (Fig. 6) support nearly all of the major transcriptomic subclasses identified in the mouse visual cortex. We note that we are not yet able to establish clear links to the recently described Sncg inhibitory subclass14; we presume that these cells are underrepresented in our data set and that adjustments to the sampling strategy could increase the rate of collection for additional study.
Interestingly, we also observe some degree of heterogeneity in me-types even among cells putatively from a single t-type (see, for example, Fig. 7a). It is known that individual t-types can exhibit substantial continuous variation within a type14; it remains to be seen if these variations correlate with morphological and physiological differences. The recent finding of shared t-types among inhibitory cells but different t-types among excitatory cells from separate cortical areas14 suggests that applying the methods of this study to a different cortical area may identify the same inhibitory me-types. How excitatory me-types vary across areas is more of an open question, since the divergent excitatory t-types could correspond to differences in long-range projection targets or other characteristics besides electrophysiological and morphological properties. It will be of considerable significance for future studies to investigate how molecular, physiological, and morphological features co-vary on a cell-by-cell basis in order to understand the relationships among these three modalities as they pertain to cell type definition or cell state-dependent variations. Molecular information can then be used to develop cell type-targeting tools to enable the study of the roles of these cell types in circuit function.
METHODS
Detailed descriptions of all experimental data collection methods in the form of technical white papers can also be found under ‘Documentation’ at http://celltypes.brain-map.org.
Mouse breeding and husbandry
All procedures were carried out in accordance with Institutional Animal Care and Use Committee at the Allen Institute for Brain Science. Animals (< 5 mice per cage) were provided food and water ad libitum and were maintained on a regular 12-h light/dark cycle. Animals were maintained on the C57BL/6J background, and newly received or generated transgenic lines were backcrossed to C57BL/6J. Experimental animals were heterozygous for the recombinase transgenes and the reporter transgenes. Transgenic lines used in this study are summarized in Supplementary Spreadsheet 2. The numbers of experimental animals used across transgenic lines are reported in Supplementary Fig. 2. Standard tamoxifen treatment for CreER lines included a single dose of tamoxifen (40 μl of 50 mg ml−1) dissolved in corn oil and administered via oral gavage at postnatal day (P)10–14. Tamoxifen treatment for Nkx2.1-CreERT2;Ai14 was performed at embryonic day (E)17 (oral gavage of the dam at 1 mg per 10 g of body weight), pups were delivered by cesarean section at E19 and then fostered. Cux2-CreERT2;Ai14 mice received tamoxifen treatment at P35 ± 5 for five consecutive days. Trimethoprim was administered to animals containing Ctgf-2A-dgCre by oral gavage at P40 ± 5 for three consecutive days (0.015 ml per g of body weight using 20 mg ml−1 trimethoprim solution). Ndnf-IRES2-dgCre animals did not receive trimethoprim induction, since the baseline dgCre activity (without trimethoprim) was sufficient to label the cells with the Ai14 reporter12.
Tissue processing.
Mice (male and female) between the ages of P45-P70 were anesthetized with 5% isoflurane and intracardially perfused with 25 or 50 ml of ice cold slicing artificial cerebral spinal fluid (0.5mM calcium chloride (dehydrate), 25 mM D-glucose, 20 mM HEPES, 10 mM magnesium sulfate, 1.25 mM sodium phosphate monobasic monohydrate, 3mM myoinositol, 12 mM N-acetyl-L-cysteine, 96 mM N-methyl-d-glucamine chloride (NMDG-Cl), 2.5 mM potassium chloride, 25 mM sodium bicarbonate, 5 mM sodium L-ascorbate, 3 mM sodium pyruvate, 0.01 mM taurine, and 2 mM thiourea, pH 7.3, continuously bubbled with 95% O2 / 5% CO2). Slices (350μm) were generated (Compresstome VF-300 vibrating microtome, Precisionary Instruments), with a block-face image acquired (Mako G125B PoE camera with custom integrated software) before each section to aid in registration to the common mouse reference atlas.
Brains were mounted for slicing at an angle appropriate for the specific part of VISp being targeted. 90 (coronal), 107, and 124 degree angles were used for the rostral, mid, and caudal portions of VISp. Although only spiny neurons with an intact apical dendrite were included in morphological analysis, all neurons were included in electrophysiology analysis, though the truncation of the apical dendrite was annotated. Electrophysiologically, truncated neurons were distributed in each type and found throughout the t-SNE representation of electrophysiology space. However, Exc_4 has the lowest fraction of neurons with a truncated apical dendrite, and the region of the t-SNE projection where Exc_4 is primarily represented appears to have fewer truncated neurons (Supplementary Fig. 32).
Slices were transferred to an oxygenated and warmed (34ºC) slicing (group A) or incubation solution (group B, 2 mM calcium chloride (dehydrate), 25 mM D-glucose, 20 mM HEPES, 2 mM magnesium sulfate, 1.25 mM sodium phosphate monobasic monohydrate, 3 mM myo inositol, 12.3 mM N-acetyl-L-cysteine, 2.5 mM potassium chloride, 25 mM sodium bicarbonate, 94 mM sodium chloride, 5 mM sodium L-ascorbate, 3 mM sodium pyruvate, 0.01 mM taurine, and 2 mM thiourea, pH 7.3, continuously bubbled with 95% O2 / 5% CO2) for 10 minutes then transferred to room temperature incubation solution (group A), or allowed to cool gradually to room temperature (group B).
Patch clamp recording.
Slices were bathed in warm (34ºC) recording ACSF (2 mM calcium chloride (dehydrate), 12.5 mM D-glucose, 1 mM magnesium sulfate, 1.25 mM sodium phosphate monobasic monohydrate, 2.5 mM potassium chloride, 26 mM sodium bicarbonate, and 126 mM sodium chloride, pH 7.3, continuously bubbled with 95% O2 / 5% CO2). The bath solution contained blockers of fast glutamatergic and GABAergic synaptic transmission, 1 mM kynurenic acid and 0.1 mM picrotoxin, respectively. Thick walled borosilicate glass (Sutter BF150-86-10) electrodes were manufactured (Sutter P1000 electrode puller) with a resistance of 3 to 7 MΩ (most 3 – 5 MΩ). Prior to recording, the electrodes were filled with 20 μl of Internal Solution with Biocytin (126 mM potassium gluconate, 10.0 mM HEPES, 0.3 mM ethylene glycol-bis (2-aminoethylether)-N,N,N’,N’-tetraacetic acid, 4 mM potassium chloride, 0.3 mM guanosine 5’-triphosphate sodium salt hydrate, 10 mM phosphocreatine disodium salt hydrate, 4 mM adenosine 5′-triphosphate magnesium salt, and 0.5% biocytin (Sigma B4261), pH 7.3). The pipette was mounted on a Multiclamp 700B amplifier headstage (Molecular Devices) fixed to a micromanipulator (PatchStar, Scientifica).
The composition of bath and internal solution as well as preparation methods were made to a) maximize the tissue quality of slices from adult mice, and b) align with solution compositions typically used in the field (to maximize the chance of comparison to previous studies). Despite these efforts, direct comparisons with previous studies should take into account the fact that specific protocols and solution composition vary within the literature. An advantage of the present study is that the same protocols / conditions were used for each cell type targeted, making it an ideal dataset to bridge data collected in different laboratories, targeting different neurons38.
Electrophysiology signals were recorded using an ITC-18 Data Acquisition Interface (HEKA). Commands were generated, signals processed, and amplifier metadata was acquired using MIES (https://github.com/AllenInstitute/MIES/), written in Igor Pro (Wavemetrics). Data were filtered (Bessel) at 10 kHz and digitized at 50 or 200 KHz. Data were reported uncorrected for the measured47 –14 mV liquid junction potential between the electrode and bath solutions.
After formation of a stable seal and break-in, the resting membrane potential of the neuron was recorded (typically within the first minute and not more than 3 minutes after break-in). A bias current was injected, either manually or automatically using algorithms within the MIES data acquisition package, for the remainder of the experiment to maintain that initial resting membrane potential. Bias currents remained stable for a minimum of 1 second prior to each stimulus current injection.
To be included in analysis, a > 1 GΩ seal was recorded prior to break-in and the initial access resistance < 20 MΩ and < 15% of the Rinput. To stay below this access resistance cut-off, cells with a low input resistance were successfully targeted with larger electrodes. For an individual sweep to be included: (1) the bridge balance was < 20 MΩ and < 15% of the Rinput, (2) bias (leak) current 0 +/- 100 pA, (3) root mean square (RMS) noise measurements in a short window (1.5 ms, to gauge high frequency noise) and longer window (500 ms, to measure patch instability) < 0.07 mV and 0.5 mV, respectively and (4) the difference in the voltage at the end of the data sweep (measured over 500 ms of rest) and the voltage measured immediately prior to the stimulus onset < 1 mV.
Biocytin histology.
A horseradish peroxidase (HRP) enzyme reaction using diaminobenzidine (DAB) as chromogen was used to visualize the filled cells after electrophysiological recording. Following electrophysiology recording, slices were fixed in 4% PFA +/- 2.5% Glutaraldehyde, then kept in PBS (4ºC) until staining. Slices were stained with DAPI, then incubated in 1% hydrogen peroxide (H2O2) for 30 min to block endogenous peroxidases. Following permeabilization (2% or 5% Triton-X 100 detergent in PBS, 60 min, RT) slices were incubated in ABC (Vectastain, Vector Laboratories) with 0.1% Triton at 4ºC overnight to 2 days.
After a final series of three washes in 1X PBS, tissue slices were mounted on gelatin coated slides and coverslipped with glycerol-based Mowiol mounting media or Aqua- Poly/Mount Coverslipping Medium. Slides were dried for approximately 2 days prior to imaging. Mowiol mounting media was made in batches of 100ml and contained: 24g glycerol, 9.6g Mowiol 4-88 (Calbiochem 475904), 24ml MilliQ water, and 48ml 0.2M Tris base (pH 8.5). Slides were dried prior to imaging.
Imaging.
Mounted sections were imaged on an upright bright-field AxioImager Z2 microscope (Zeiss, Germany) equipped with an Axiocam 506 monochrome camera (6 megapixels with a 4.54 μm per pixel size). Two-dimensional (2D) images were captured with a 20X objective lens (Zeiss Plan-NEOFLUAR 20X/0.5) using the Tile & Position Zeiss Efficient navigation (ZEN) 2012 SP2 software module (Zeiss). Image quality evaluation included a qualitative evaluation of signal to noise for the imaged object (with high signal apparent in the cell body and dendrites, as opposed to background stain in the surrounding tissue, which can occur when cell filling leaks), in-focus cell body, and absent or negligible tessellation (tiling and stitching edge artifact). Overall evenness of section illumination and bounding box region for target tissue inclusion was evaluated.
Individual cells were imaged at higher resolution for the purpose of automated and manual reconstruction, quantitation and display. Light was transmitted using an oil-immersion condenser (1.4 NA). Series of 2D images of single neurons were captured with a 63X objective lens (Zeiss Plan APOCHROMAT 63X/1.4 oil), using the Tile & Position and Z-stack ZEN 2012 SP2 software modules (Zeiss). The composite 2D tiled images (X-Y resolution was set to 0.114 x 0.114 micron) were acquired at an interval of 0.28 μm along the Z-axis. Images were exported as 8-bit TIFF. Image series from individual slices or cells were processed and managed via a custom Laboratory Information Management System (LIMS).
Full dynamic range was achieved with a 20 ms exposure time and an optimal Tl VIS-LED lamp voltage control adjustment. Tiles were stitched with a minimum of 5% overlap and a 10% maximum shift. Image quality control included a z-stack plane count, a visual check for proper stitching alignment and even illumination throughout the images. 63X Z-stacks were evaluated based on quality metrics that would impact cell reconstruction, as opposed to aesthetic quality.
Electrophysiological feature analysis.
Electrophysiological features were measured from responses elicited by short (3 ms) current pulses, long (1 s) current steps, and slow (25 pA / s) current ramps. The code for feature analysis is publicly available as part of the Allen SDK. APs were detected by first identifying locations where the smoothed derivative of the membrane potential (dV/dt) exceeded 20 mV/ms. Putative AP peaks were identified as the maximum potential between detected events, and the putative AP threshold was identified by the point before the peak where the dV/dt was 5% of the maximum dV/dt. Putative APs were refined by several criteria: threshold-to-peak voltage difference must exceed 2 mV, threshold-to-peak time difference must be below 2 ms, and putative peak must be above -30 mV. The threshold was then re-calculated by finding the point for each AP where the dV/dt was 5% of the average maximal dV/dt across all APs. For each AP, several features were calculated: threshold, peak, fast trough (defined as where the dV/dt was 1% of the peak downstroke), and the width (defined as the width at half-height, where height was the difference between peak and fast trough)39. The ratio of the peak upstroke dV/dt to the peak downstroke dV/dt was also calculated (“upstroke/downstroke ratio”). In addition, the waveforms of the first APs elicited by the lowest-amplitude current pulses, steps, and ramps were analyzed by concatenating the 3 ms-long intervals following the AP threshold for the three conditions. The derivatives of these waveforms were also analyzed in this way.
The voltage trajectory of the ISI was also characterized to allow comparison across cells. For each cell, the sweep with the lowest stimulus amplitude that had at least five APs was identified (if a cell never fired at least five APs, the highest amplitude step was chosen instead). For each ISI, the voltage trajectory between the fast trough of the initial AP and the threshold of the following AP was extracted, and the threshold level of the initial AP was subtracted from it. The durations were normalized, then the traces were subsampled to 100 data points and averaged together. If the highest-amplitude step only elicited a single AP, a 100 ms interval following the fast trough was used in place of an ISI.
To enable comparison of AP features across the responses to long current steps given different numbers of APs across stimulus amplitudes and cells, the 1 s-long response was divided into 20 ms bins, and feature values of all APs falling within a bin were averaged. If no APs fell within a bin, the value was interpolated from neighboring bins that had APs. This was done for stimulus amplitudes starting at a given cell’s rheobase up to values +100 pA above rheobase, with a difference between amplitudes of +20 pA. If a sweep of an expected amplitude was unavailable (for example, if it failed one of the QC criteria), the missing values were interpolated from neighboring QC-passing sweeps. The instantaneous firing frequency (defined as the inverse of the ISI) was also binned and interpolated with 20 ms bins. In addition, a “PSTH” was estimated by counting APs in 50 ms bins, then converting to a firing rate by dividing by the bin duration. These two measures yield similar, but not identical, profiles of the firing pattern during a long current step response. The instantaneous firing frequency was also analyzed by normalizing to the maximum rate observed during the step to emphasize features like the adaptation of the firing frequency during the response. Though not used in the clustering analysis, the adaptation index was measured for each long step response by averaging the differences between consecutive ISIs normalized by their sums. The latency between the start of the current step and the first AP elicited was also measured.
To identify brief periods of high-frequency firing (“bursts”) amid longer intervals of lower-frequency firing as well as periods where firing temporarily but substantially slowed (“pauses”), the following procedure was used. The coefficient of variation of the instantaneous frequency was calculated for all sets of five consecutive ISIs observed during long current steps across all cells in the data set. The distribution of these CVs was bimodal, with a large, narrow peak at low CV values (considered to represent firing at a relatively constant rate) and a wide peak at higher CV values. The minimum value of between these peaks was found at CV = 0.18. This value was used as a threshold to define segments during a response where the firing rate was relatively stable. For each sweep, the instantaneous firing rate was analyzed using a change-point detection algorithm48 to identify locations where the mean firing rate changed. The CV of the instantaneous firing rate for each segment was compared to the threshold, and if all were below the threshold, the segmentation was accepted. If not, the change-point detection penalty was lowered and the analysis was repeated until all segments passed. Once this was completed, the segment with the most APs was identified, and the firing rate ratios between all segments and that largest segment were calculated. If more than one segment was tied for the most APs, the ratios were calculated using the median of the tied segments. Segments with high ratios were considered putative bursts, and segments with low ratios were putative pauses. The measures were constructed in this way to both reflect the gradual variations observed in firing patterns with increasing stimulus amplitudes in the same cell, as well as to be robust to the substantial differences in ISI shapes and firing rates across all cells analyzed.
Subthreshold responses to hyperpolarizing current steps were analyzed using downsampled (to averages in 10 ms bins) membrane potential traces that was concatenated together. Responses from -10 pA to -90 pA steps (at a -20 pA interval) were used, and 200 ms of the time before and after the step were included as well. In addition, the largest amplitude hyperpolarizing step response was analyzed by normalizing to the minimum membrane potential reached and the baseline membrane potential. This emphasized the “sag” in the membrane potential due to the activation of Ih observed in some cells. Though not used for clustering analysis, the input resistance was calculated by the slope of a linear fit to the minimum membrane potentials during these hyperpolarizing step responses, and the membrane time constant was estimated by exponential fits between 10% of the maximum voltage deflection and that maximum deflection. The membrane capacitance was estimated by dividing the membrane time constant by the input resistance.
Electrophysiological classification.
Data sets were built by accumulating the feature vectors in each category (e.g. AP waveform, each AP feature across long steps, subthreshold response waveforms, etc.; see Supplementary Table 1). Data from putatively excitatory cells and inhibitory cells (determined by the presence and type of dendritic spines) were analyzed separately, though similar results were observed when all cells were analyzed together (see Supplementary Fig. 6). Sparse principal component analysis20 was performed separately on each data set. Principal components with an adjusted explained variance exceeding 1% were kept (typically 1 to 8 components from a given data set). Analysis of inhibitory neurons yielded 56 total components, excitatory neurons yielded 54 components, and all neurons combined yielded 54 components. The components were then z-scored to standardize the scale and combined to form a reduced dimension feature matrix. The matrix was then fit with a series of Gaussian mixture models (GMMs) with a diagonal covariance matrix using different numbers of components; the GMM that minimized the Bayes information criterion was chosen as the best representation for the data21. Next, components of the selected GMM were iteratively merged22 to identify clusters that may have had non-Gaussian structure (and therefore would have been fit by the GMM with multiple components). At each step, the merge that maximized the change in entropy was identified, and the number of cells affected by the merge was recorded. The point where the rate of entropy decrease versus number of cells merged slowed was identified by a two-part linear fit, and the merges up to that point were used to define the final clusters. Cluster stability was then evaluated by subsampling analysis23. Random subsamples containing 90% of the data set were generated 100 times, and the subsamples were fit with a GMM using the number of components of the best GMM fit to the full data set, then merged as described above. The Jaccard similarity was calculated between each original cluster and each cluster from the subsample, and the maximum was found for each original cluster. These Jaccard similarities were averaged across all subsample runs, and clusters that had average Jaccard similarities below 0.5 were dissolved by merging into other clusters (using the entropy criterion above), resulting in a final set of clusters, or e-types. The entire procedure (including the robustness evaluation) was repeated on 90% subsets of the dataset to generate a cellwise co-clustering matrix, which supported the assessment of cluster stability. In addition, the procedure was used on 20%, 40%, 60%, and 80% random subsamples of the data (each drawn 10 times) to evaluate how the number of clusters identified varied with the number of samples used in the analysis.
The electrophysiological feature matrix used in the clustering analysis was also visualized with a two-dimensional projection using the t-distributed stochastic neighbor embedding (t-SNE) technique24. Cluster identities and other features of the cells were visualized using this projection throughout this study. Supervised classification by random forest (500 trees, balanced class weights) was used to assess relative feature importance by average decrease in the Gini index for splits on a given feature27,28.
Anatomical location.
To characterize the position of biocytin-labled cells in the mouse brain, a 20x brightfield and/or fluorescent image of DAPI (4’,6-diamidino-2-phenylindole) stained tissue was captured and analyzed to determine layer position and region. Soma position was annotated and used to calculate soma depth relative to drawings of the pia and white matter. Individual cells were then manually placed in the appropriate cortical region and layer within the Allen Mouse Common Coordinate Framework (CCF) by matching the 20x image of the slice with a “virtual” slice at an appropriate location and orientation within the CCF. Using the DAPI image, laminar borders were also drawn for all reconstructed inhibitory neurons.
Dendrite type assignment.
The dendritic morphology of each neuron (N=1938) was identified as either aspiny, sparsely spiny or spiny49 based on a qualitative assessment of the neuron’s dendrites by viewing the slides under the microscope and/or in the 63X image. These different dendritic types roughly equate to interneurons (aspiny and sparsely spiny) and pyramidal or spiny stellate neurons (spiny). Aspiny dendrites were defined by the absence of spiny protrusions and checked against a lack of a pronounced apical dendrite and/or the presence of an axon emerging from the soma or dendrite at odd angles that branches extensively. Sparsely spiny dendrites were defined by these same features, except that spines appeared with infrequent to moderately frequent expression (~ 1 spine/10 microns). Spiny dendrites were defined by the presence of frequent spiny protrusions (approximately one spine per 1-2 microns), and spiny cells were validated by an axon that descended directly toward the white matter with sparse, proximal branching at right angles to the primary axonal branch and/or a pronounced primary apical dendrite.
Morphological reconstruction.
Reconstructions of the dendrites and the initial part of the axon (spiny neurons) and/or the full axon (aspiny/sparsely spiny neurons) were generated for a subset of neurons with good quality electrophysiology and biocytin fill. The neurons in our electrophysiology dataset that were not reconstructed were excluded from the analysis due to 1) lack of healthy and/or intact axon (aspiny neurons only) or 2) already having a large number of neurons (n>30) with a similar morphology (spiny neurons only). Reconstructions were generated based on a 2D image stack that was run through a Vaa3D-based image processing and reconstruction pipeline15. The process included a variable enhancement of the signal to noise ratio in the image50. The enhanced image was then used to generate an automated reconstruction of the neuron using Neuron Crawler51 or TReMAP52. Automated reconstructions were then extensively manually corrected and curated using a range of tools (e.g., virtual finger, polyline) in the Mozak extension (Zoran Popovic, Center for Game Science, University of Washington) of Terafly tools50,53 in Vaa3D. Every attempt was made to generate a completely connected neuronal structure while remaining faithful to image data. If axonal processes could not be traced back to the main structure of the neuron, they were left unconnected. Using the most updated version of the Mozak-Terafly-Vaa3d tools, on average, dendrite only reconstructions of spiny neurons took 4.5 hours and full reconstructions of the axon and dendrites of aspiny neurons took 16 hours. Connected and disconnected axon components (axon cloud) were used in the quantitative analysis. As a final step in the manual correction and curation process, an alternate analyst checked for missed branches or inappropriate connections. Once the reconstruction was deemed complete, multiple plugins were used to prepare neurons (saved as SWC file) for qualitative and quantitative morphological analyses.
Morphology feature design and clustering.
Drawing from the literature and existing analysis tool kits (e.g., L-measure25 and the Blue Brain Project5), a feature set was created, which included well-known shape features from either dendritic or axonal branches in our reconstructions (e.g., branch number, total branch length) and new features such as the intersection between different branch types (e.g., between apical and basal dendrites). Since all reconstructions are aligned in the XY dimension to the pia, we introduced additional features, such as the laminar distribution of axon (e.g., axon histogram PC 1).Features predominantly determined by differences in the z-dimension were not included in our morphological classification due to z-compression in the acute slice after processing32,33, and the variable tilt angles of sampled visual cortical neurons. However, to test the effects of z-shrinkage and tilt angle on the classification results, we restored reconstructions to their original volumes and aligned them to the perpendicular cortical axis (based on registration to the common mouse reference atlas), and re-ran our clustering analysis (Supplementary Fig. 33). We found good agreement between the clustering results before and after adjustment. The full list of features we calculated for spiny and aspiny neurons and their descriptions are in Supplementary Table 2, and the morphological adjustments are provided in Supplementary Spreadsheet 3.
From this feature set, features with low variance (coefficient of variance < 0.25) were removed and a representative feature was chosen among highly correlated features (correlation > 0.95). These features were scaled by z-transform to form a feature set on which a standard hierarchical clustering with Ward’s agglomeration method using Euclidean distance was applied. The initial cluster assignment was made by cutting this hierarchical tree using the cutreehybrid() function in R package WGCNA54, which determined the number of clusters by maximizing between-cluster variation and minimizing within-cluster variation while considering branching structures of the dendrogram. In order to identify robust clusters, a co-clustering analysis was done55. This clustering step was run 1000 times with a randomly subsampled 90% of the data in a 10 fold manner and the proportion of all runs that every pair were grouped in the same cluster was recorded in the co-clustering diagram (Fig. 3a and 4a). Consensus clusters were found by building a Ward linkage hierarchical tree and cutting the tree by the same cutreehybrid() function. If the size of cluster is n≤3, individual samples were assigned to the highest correlated cluster. If any pair of clusters did not have features significantly different (adjusted t-test p-value <0.01 & fold change > 1.25) between the two, these two clusters were merged.
With identified clusters (m-types), we checked their predictability by designing two types of classifiers. Random forest classifier gave 79/70% out-of-bag prediction accuracy and support vector machine showed 89/79% prediction accuracy in leave-one-out cross validation for spiny/aspiny neurons. Each individual cluster’s out-of-bag prediction accuracy is listed in Supplementary Fig. 34. The importance of each feature predicting these clusters are listed in terms of Gini index28 in Supplementary Table 4.
Joint electrophysiological and morphological clustering.
To identify types that had both consistent electrophysiological and morphological characteristics, the electrophysiological feature matrix was combined with the morphological feature matrix for cells that had both types of data available (n=253 excitatory, 199 inhibitory). Several combined matrices with various relative weights of the electrophysiological features (0.5, 0.75, 1, 1.5) were generated. These matrices were used for unsupervised classification with several methods (GMM, Ward hierarchical clustering, and spectral clustering) and numbers of components (10, 15, 20, 25). In addition, a variant of Ward hierarchical clustering was performed on the morphological features with a connectivity constraint determined by nearest-neighbors in electrophysiological feature space (using neighborhoods of 4, 7, and 10 cells). In all, 60 clustering variants were used.
A cell-wise co-clustering matrix was calculated from these variants, and consensus clusters were determined14. The co-clustering matrix was iteratively divided by Ward hierarchical clustering using co-clustering fractions as the distance measure, stopping when a cluster would be below a minimum size set to n=3. Then, co-clustering rates between clusters were evaluated, and pairs were merged if the higher within-cluster rate did not exceed the between-cluster rate by 25%. Cells were reassigned to the best-matched cluster, and the process was repeated until convergence.
Clusters were evaluated for stability by repeating the entire procedure on 90% subsamples and calculated the average Jaccard similarity, as described above. Clusters with average Jaccard similarities of below 0.5 were considered unstable and were merged with another cluster with the highest between-cluster rate if it exceeded 35% (chosen because it was similar to the 5th percentile of the within-cluster rates of all cells in stable clusters). Unstable clusters that could not be merged were marked as such in Fig. 5.
Supervised classification by random forest (500 trees, balanced class weights) was performed to predict me-types by all features, only electrophysiological features, and only morphological features27,28. Confusion matrices were calculated by comparing actual me-types to the out-of-bag predictions of the classifier. The e-types, and m-types, and me-types for each cell are provided in Supplementary Spreadsheet 3.
Transcriptomic correspondences.
The associations between transgenic lines and transcriptomic types (t-types) were investigated using data from Tasic et al.14 to establish preliminary correspondences with the results here. Specific transgenic line/layer combinations that labeled a small number of t-types, defined as having five or fewer t-types containing at least 5% of the Tasic et al. cells from that line and layer set, were identified.
Correspondences between transgenic lines and transcriptomic subclasses defined by that study were also analyzed. A transgenic line was considered to be consistent with a given subclass if at least 5% of the Tasic et al. cells labeled by that line were found in that subclass. Excitatory and inhibitory cells were evaluated separately.
Statistics and research design.
Morphological features were compared between m-types and groups of m-types using the two-tailed Student’s t-test. Morphological feature distributions were assumed to be normal for this method, but this was not formally tested (see Supplementary Figs. 19 and 21 for plots of distributions). The p-values were adjusted for multiple testing using the two-stage Benjamini and Hochberge step-up procedure to control the false discovery rate. No statistical methods were used to pre-determine sample sizes, but the sample sizes here are similar to those reported in previous publications5,6,12. No randomization was used during data collection as there was a single experimental condition for all acquired data. The different stimulus protocols were not presented in a randomized order. Data collection and analysis were not performed blind to the conditions of the experiments as there was a single experimental condition for all acquired data. Addition research design information can be found in the Life Sciences Reporting Summary accompanying this study.
Data availability.
Electrophysiological and morphological data supporting the findings of this study are available in the Allen Cell Types Database, celltypes.brain-map.org. Morphological data are also available through the NeuroMorpho.org repository56, neuromorpho.org.
Code availability.
The custom electrophysiology data acquisition software (MIES) is available at github.com/alleninstitute/mies. The Vaa3D morphological reconstruction software, including the Mozak extension, is freely available at www.vaa3d.org and its code is available at github.com/Vaa3D. The code for electrophysiological and morphological feature analysis is available as part of the open-source Allen SDK repository (github.com/alleninstitute/allensdk) and IPFX repository (github.com/alleninstitute/ipfx). Clustering analysis code will be made available at github.com/alleninstitute/drcme.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Zoran Popovich for creating the Mozak custom user interface for the 3D reconstruction software, Terafly-Vaa3D. We thank Barb Berg, Sil Coulter, Chinh Dang, and Allan Jones for leadership and guidance. This work was funded by the Allen Institute for Brain Science, and by National Institutes of Health grant U01MH105982 to H.Z.
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Zeng H & Sanes JR Neuronal cell-type classification: challenges, opportunities and the path forward. Nat. Rev. Neurosci. 18, 530–546 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Tremblay R, Lee S & Rudy B GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron 91, 260–292 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harris KD & Shepherd GMG The neocortical circuit: themes and variations. Nat. Neurosci. 18, 170–181 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lodato S & Arlotta P Generating Neuronal Diversity in the Mammalian Cerebral Cortex. Annu. Rev. Cell Dev. Biol. 31, 699–720 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang X et al. Principles of connectivity among morphologically defined cell types in adult neocortex. Science 350, aac9462 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Markram H et al. Reconstruction and Simulation of Neocortical Microcircuitry. Cell 163, 456–492 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Ascoli GA et al. Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat. Rev. Neurosci. 9, 557–568 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Druckmann S, Hill S, Schürmann F, Markram H & Segev I A Hierarchical Structure of Cortical Interneuron Electrical Diversity Revealed by Automated Statistical Analysis. Cereb. Cortex 23, 2994–3006 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Bortone DS, Olsen SR & Scanziani M Translaminar Inhibitory Cells Recruited by Layer 6 Corticothalamic Neurons Suppress Visual Cortex. Neuron 82, 474–485 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim EJ, Juavinett AL, Kyubwa EM, Jacobs MW & Callaway EM Three Types of Cortical Layer 5 Neurons That Differ in Brain-wide Connectivity and Function. Neuron 88, 1253–1267 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dehorter N et al. Tuning of fast-spiking interneuron properties by an activity-dependent transcriptional switch. Science 349, 1216–1220 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tasic B et al. Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci. 19, 335–346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeisel A et al. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Tasic B et al. Shared and distinct transcriptomic cell types across neocortical areas. Nature 563, 72–78 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peng H, Ruan Z, Long F, Simpson JH & Myers EW V3D enables real-time 3D visualization and quantitative analysis of large-scale biological image data sets. Nat. Biotechnol. 28, 348–353 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harris JA et al. Anatomical characterization of Cre driver mice for neural circuit mapping and manipulation. Front. Neural Circuits 8, 76 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Madisen L et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daigle TL et al. A Suite of Transgenic Driver and Reporter Mouse Lines with Enhanced Brain-Cell-Type Targeting and Functionality. Cell 174, 465–480.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bernard A, Sorensen SA & Lein ES Shifting the paradigm: new approaches for characterizing and classifying neurons. Curr. Opin. Neurobiol. 19, 530–536 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Zou H, Hastie T & Tibshirani R Sparse Principal Component Analysis. J. Comput. Graph. Stat. 15, 265–286 (2006). [Google Scholar]
- 21.Baden T et al. The functional diversity of retinal ganglion cells in the mouse. Nature 529, 345–350 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baudry J-P, Raftery AE, Celeux G, Lo K & Gottardo R Combining Mixture Components for Clustering. J. Comput. Graph. Stat. 19, 332–353 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hennig C Cluster-wise assessment of cluster stability. Comput. Stat. Data Anal. 52, 258–271 (2007). [Google Scholar]
- 24.van der Maaten L & Hinton G Visualizing Data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008). [Google Scholar]
- 25.Gonchar Y, Wang Q & Burkhalter A Multiple distinct subtypes of GABAergic neurons in mouse visual cortex identified by triple immunostaining. Front. Neuroanat. 1, 3 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.von Engelhardt J, Eliava M, Meyer AH, Rozov A & Monyer H Functional Characterization of Intrinsic Cholinergic Interneurons in the Cortex. J. Neurosci. 27, 5633–5642 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.James G, Witten D, Hastie T & Tibshirani R An Introduction to Statistical Learning. (Springer; New York, 2013). [Google Scholar]
- 28.Breiman L, Friedman J, Stone CJ & Olshen RA Classification and Regression Trees. (Chapman & Hall/CRC, 1984). [Google Scholar]
- 29.Markram H et al. Interneurons of the neocortical inhibitory system. Nat. Rev. Neurosci. 5, 793–807 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Oberlaender M et al. Cell Type–Specific Three-Dimensional Structure of Thalamocortical Circuits in a Column of Rat Vibrissal Cortex. Cereb. Cortex 22, 2375–2391 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hattox AM & Nelson SB Layer V Neurons in Mouse Cortex Projecting to Different Targets Have Distinct Physiological Properties. J. Neurophysiol. 98, 3330–3340 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Deitcher Y et al. Comprehensive Morpho-Electrotonic Analysis Shows 2 Distinct Classes of L2 and L3 Pyramidal Neurons in Human Temporal Cortex. Cereb. Cortex 27, 5398–5414 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Egger V, Nevian T & Bruno RM Subcolumnar Dendritic and Axonal Organization of Spiny Stellate and Star Pyramid Neurons within a Barrel in Rat Somatosensory Cortex. Cereb. Cortex 18, 876–889 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Kanari L et al. Objective Classification of Neocortical Pyramidal Cells. bioRxiv 349977 (2018). doi: 10.1101/349977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toledo-Rodriguez M Correlation Maps Allow Neuronal Electrical Properties to be Predicted from Single-cell Gene Expression Profiles in Rat Neocortex. Cereb. Cortex 14, 1310–1327 (2004). [DOI] [PubMed] [Google Scholar]
- 36.He M et al. Strategies and Tools for Combinatorial Targeting of GABAergic Neurons in Mouse Cerebral Cortex. Neuron 92, 555 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Paul A et al. Transcriptional Architecture of Synaptic Communication Delineates GABAergic Neuron Identity. Cell 171, 522–539.e20 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tebaykin D et al. Modeling sources of interlaboratory variability in electrophysiological properties of mammalian neurons. J. Neurophysiol. 119, 1329–1339 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bean BP The action potential in mammalian central neurons. Nat. Rev. Neurosci. 8, 451–465 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Zhang L et al. Whole-cell recording of the Ca(2+)-dependent slow afterhyperpolarization in hippocampal neurones: effects of internally applied anions. Pflugers Arch. 426, 247–53 (1994). [DOI] [PubMed] [Google Scholar]
- 41.Kaczorowski CC, Disterhoft J & Spruston N Stability and plasticity of intrinsic membrane properties in hippocampal CA1 pyramidal neurons: effects of internal anions. J. Physiol. 578, 799–818 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Teeter C et al. Generalized leaky integrate-and-fire models classify multiple neuron types. Nat. Commun. 9, 709 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gouwens NW et al. Systematic generation of biophysically detailed models for diverse cortical neuron types. Nat. Commun. 9, 710 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arkhipov A et al. Visual physiology of the layer 4 cortical circuit in silico. PLOS Comput. Biol. 14, e1006535 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stockton DB & Santamaria F Integrating the Allen Brain Institute Cell Types Database into Automated Neuroscience Workflow. Neuroinformatics 15, 333–342 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tripathy SJ et al. Transcriptomic correlates of neuron electrophysiological diversity. PLOS Comput. Biol. 13, e1005814 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neher E Correction for liquid junction potentials in patch clamp experiments. in Methods in Enzymology 207, 123–131 (1992). [DOI] [PubMed] [Google Scholar]
- 48.Killick R, Fearnhead P & Eckley IA Optimal Detection of Changepoints With a Linear Computational Cost. J. Am. Stat. Assoc. 107, 1590–1598 (2012). [Google Scholar]
- 49.Braitenberg V & Schüz A Cortex: Statistics and Geometry of Neuronal Connectivity. (Springer Berlin Heidelberg, 1998). doi: 10.1007/978-3-662-03733-1 [DOI] [Google Scholar]
- 50.Peng H, Bria A, Zhou Z, Iannello G & Long F Extensible visualization and analysis for multidimensional images using Vaa3D. Nat. Protoc. 9, 193–208 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Zhou Z, Sorensen S, Zeng H, Hawrylycz M & Peng H Adaptive Image Enhancement for Tracing 3D Morphologies of Neurons and Brain Vasculatures. Neuroinformatics 13, 153–166 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Zhou Z, Liu X, Long B & Peng H TReMAP: Automatic 3D Neuron Reconstruction Based on Tracing, Reverse Mapping and Assembling of 2D Projections. Neuroinformatics 14, 41–50 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Bria A, Iannello G, Onofri L & Peng H TeraFly: real-time three-dimensional visualization and annotation of terabytes of multidimensional volumetric images. Nat. Methods 13, 192–194 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Langfelder P, Zhang B & Horvath S Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics 24, 719–720 (2008). [DOI] [PubMed] [Google Scholar]
- 55.Boldog E et al. Transcriptomic and morphophysiological evidence for a specialized human cortical GABAergic cell type. Nat. Neurosci. 21, 1185–1195 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ascoli GA, Donohue DE & Halavi M NeuroMorpho.Org: A Central Resource for Neuronal Morphologies. J. Neurosci. 27, 9247–9251 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Electrophysiological and morphological data supporting the findings of this study are available in the Allen Cell Types Database, celltypes.brain-map.org. Morphological data are also available through the NeuroMorpho.org repository56, neuromorpho.org.