

Abstract
The oxidative photocatalytic method for phenol-phenol homo-coupling and cross-coupling is described and isolated yields of 16–97% are obtained. Measured oxidation potentials and computed nucleophilicity parameters support a mechanism of nucleophilic attack of one partner onto the oxidized neutral radical form of the other partner. Understanding of this model permitted development of cross-coupling reactions between nucleophilic phenols/arenes and easily oxidized phenols with high selectivity and efficiency. A highlight of this method is that one equivalent of each coupling partner is utilized. Building on these findings, a non-enzymatic, catalytic method for coupling tyrosine was also developed.
Keywords: Oxidative coupling, phenol coupling, photocatalysis, tyrosine dimerization, cross-coupling
Graphical Abstract
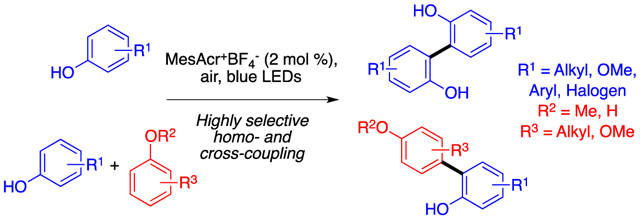
INTRODUCTION
The biphenol scaffold is a prevalent substructure in biologically active natural compounds (Chart 1).1–7 Since the discovery that phenol oxidation is the key step in the synthesis of several of these natural products, chemists have explored analogous means to effect such transformations.
Chart 1.

Coupled Phenol Natural Products
While many stoichiometric phenol homo- and cross-couplings have been reported,8–11 Uang and coworkers disclosed in 1999 one of the first catalytic reports of non-naphthol phenol homo-coupling using a vanadium catalyst under an oxygen atmosphere.12 In 2011, the Waldvogel group revealed the homo-coupling of select phenols by utilizing a Boron Doped Diamond (BDD) anode in yields up to 83%.13,14 In 2014, we reported the use of a metal salen/an catalyst library to achieve highly selective homo- and cross-couplings of several alkyl and alkoxy substituted phenols,15 while Lumb and coworkers revealed a copper catalyzed aerobic oxidation of phenols.16 Finally, more-recent reports of cross-coupling from Pappo and Kozlowski reveal selective, high-yielding phenol cross-couplings using iron porphyrin and chromium salen catalysts, respectively.17,18
Importantly, these catalytic oxidative phenolic coupling reactions proceed via distinct mechanisms that dictate the reactivity and selectivity. For example, the copper catalyzed oxidative homo-coupling of phenols reported by Lumb and coworkers is proposed to proceed via combination of two neutral phenoxy radicals.16 Kozlowski and coworkers proposed radical anion coupling via a high spin state in the mechanism for phenol cross-coupling a chromium catalyst.19 Pappo and coworkers also propose a radical-radical mechanism in cross-couplings with an iron porphyrin catalyst,17 but propose a distinct chelated radical-anion mechanism for phenolic cross-coupling with catalytic Fe(III) and (t-BuO)2.20 In the aforementioned cases, metal phenolates are often invoked as key intermediates that undergo inner sphere oxidation at a metal center. Under electrochemical conditions, Waldvogel and colleagues propose generation of a free phenoxyl radical by outer sphere electron transfer, followed by trapping with an unoxidized phenol or arene.13,14 Even though there are occasional similar outcomes, the mechanisms for each of those homo- and cross-coupling transformations is fundamentally different.21
While these advances in the field of oxidative coupling have facilitated the synthesis of a diverse array of ligands and natural products, they still require the use of high loadings of transition metal catalysts, diamond electrodes, or stoichiometric oxidants that are not atom economical.5,14,20,22,23 In addition, certain coupling patterns are difficult to access, and couplings of mono-substituted phenols are especially challenging.
In particular, efficacious non-enzymatic catalytic methods for the direct oxidative ortho-ortho coupling of L-tyrosine have remained an elusive goal for chemists (Scheme 1). Many attempts have been made to effect such a coupling, as it would engender a biomimetic approach to the dityrosine motif found in several natural products, including mycocyclosin3 and RP-66453.4 However, most efforts have culminated in Suzuki type cross-couplings to forge the biaryl bond, as is the case in all efforts to generate mycocyclosin (Scheme 1a),3,24 a natural product that is also a key intermediate to the herqulines.25–27 Notably, Baran deployed an intramolecular oxidative coupling of tyrosine and para-hydroxyphenylglycine by employing a stoichiometric CuIII-TMEDA complex (Scheme 1b).28 In spite of these efforts, reports of intermolecular tyrosine coupling are limited. In 2001, Rieker and coworkers disclosed that superstoichiometric [bis(trifluoroacetoxy)iodo]benzene (PIFA) couples tyrosine in less than 40% isolated yield (Scheme 1c).29 Additionally, they reported a 90% yield using horseradish peroxidase enzyme and H2O2, for which material throughput was an issue.
Scheme 1.

Previous Approaches to Inter- and Intramolecular Tyrosine Coupling
More recently, in 2020, the Pappo group leveraged the presence of a tert-butyl activating group on tyrosine, which had been reported by us and others as a removable group that activates phenols toward oxidative coupling and blocks additional reactive sites,30–33 to effect the synthesis of several arylomycin analogues via an intermolecular iron catalyzed tyrosine coupling (Scheme 1d).34 Herein, we disclose a photocatalytic process to accomplish such couplings without the aid of a directing group (Scheme 1e).35
Our initial approach involved screening our library of oxidizing transition metal catalysts in the coupling of tyrosine and related phenols but revealed no reactivity and recovery of starting material. Reasoning that these catalysts did not have sufficiently high oxidation potentials, we turned to photoredox catalysts, which have a wide range of reported oxidation potentials. By pairing a photoredox catalyst with a stoichiometric oxidant to generate controlled amounts of the oxidized intermediate, the coupling of phenols should be feasible. While net-oxidative photoredox processes are less common overall,36 we were encouraged by reports of the photo-crosslinking of tyrosineresidues in various polymers.37 Although there was no direct evidence of tyrosine-tyrosine couplings, it appeared that tyrosine was within the oxidation range of known photocatalysts.37,38
Notably, in 2017, König39 and Xia40 disclosed the photocatalytic cross-coupling of phenols with arenes and amines, respectively. In the former case, control of C-C vs C-O coupling was a challenge. In the latter case, a very electron-rich phenol was oxidized by persulfate and the amine was oxidized by the photocatalyst. While there are limited examples of photocatalytic naphthol-phenol cross-couplings,41 to the best of our knowledge, there are no reports describing photocatalytic phenol-phenol cross- or homo- couplings. Challenges associated with such processes include identifying an appropriate oxidant that does not act on phenols directly, preventing decomposition of the products, controlling cross-selectivity by selective oxidation, and controlling regioselectivity.
Herein, we disclose the transition-metal free photocatalytic oxidative homo- and cross-coupling of phenols. Use of high throughput experimentation (HTE) allowed for the rapid identification of optimal photocatalyst, oxidant, and solvent combination for the transformation. Hallmarks of the reaction include good scope for homo- and cross-coupling, use of a mild, readily available oxidant, a 1:1 ratio of coupling partners in cross-coupling, and excellent control of regioselectivity in cross-coupling based on nucleophilicity and oxidation potential parameters of phenol partners. Mechanism experiments support a mechanism wherein a neutral phenol radical is attacked by a nucleophilic phenol or arene, followed by subsequent oxidation by photocatalyst or H2O2 to furnish the biphenol product. Given the importance of the dityrosine motif, an additional HTE screen was undertaken to develop photocatalytic conditions for the coupling of tyrosine.
RESULTS AND DISCUSSION
Reaction Optimization.
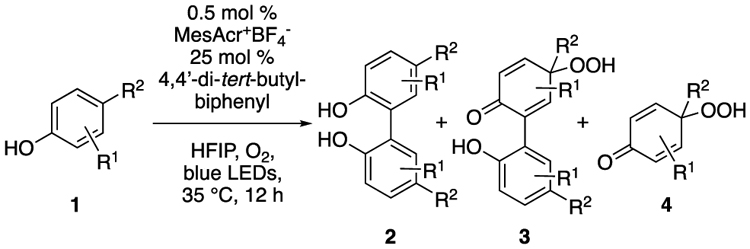
Our initial strategy to explore this nascent field focused on the use of Ru(bpy)3Cl2 and a persulfate oxidant.37 Initial efforts in coupling N-AcTyrOMe were unsuccessful, motivating us to screen a series of photocatalysts and oxidants with simpler phenols at microscale against an internal standard (full results found in Supporting Information). The screen revealed four potent photocatalysts, MesAcr+BF4−, 3,6-di-tert-butyl-MesAcr+BF4−, [Ir{dF(CF3)2ppy}2(dtbbpy)]PF6 and [Ru(bpm)3](PF6)2], for the homo-coupling transformation. Upon validation at larger scale, MesAcr+BF4− was identified as the most effective catalyst due to its ability to convert several phenols to their dimers in higher conversions (Table 1). For certain phenol substrates, use of MesAcr+BF4− in the presence of O2 led to the formation of oxidized byproducts, such as the para-peroxyquinols of the dimeric product (3) or the phenol monomer (4) (Table 2), the latter of which was confirmed with a crystal structure (see Supporting Information). Other oxidants (DDQ, persulfates, peroxides, CBrCl3) were also screened (not shown), but were less effective.
Table 1.
Benchtop Photocatalyst Screen
 | ||
|---|---|---|
| entry | photocatalyst | NMR yield |
| 1 | MesAcr+BF4− | 55% |
| 2 | 3,6-di-tert-butyl-MesAcr+BF4− | 50% |
| 3 | Ru(bpm)3[PF6]2 | 41% |
| 4 | [Ir{dF(CF3)2ppy}2(dtbbpy)]PF6 | 33% |
Table 2.
Phenolic Coupling Byproducts
 | ||||
|---|---|---|---|---|
| entry | substrate | 2 | 3 | 4 |
| 1 | 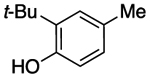 |
70% | 5% | 10% |
| 2 |  |
65% | 7% | 5% |
| 3 |  |
- | - | 33% |
A solvent screen revealed that halogenated solvents gave good conversion (ClCH2CH2Cl, CH2Cl2) of phenol monomer, with 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) providing the highest yields. The increased yield of the reaction using HFIP as a solvent was not unexpected, as recent literature reports on oxidative phenol coupling17,20,34,42 disclose the use of this solvent due to its ability to stabilize radical/charged intermediates.43 Even so, the conditions shown in Table 1 also generated undesired side products 3 and 4. Reasoning that lower oxygen concentrations would prevent direct oxygenation to yield para-peroxyquinol products, reactions under an atmosphere of air were attempted. This change indeed curtailed the formation of these side products considerably, as 2a was observed in 80% yield (vs 70% under O2; Table 2, entry 1).
When attempting to attain isolated yields of 2b on larger scale without the biphenyl internal standard included in the initial screen, a notable decrease in yield was observed (Table 3, entry 2 vs 1). Thus, 4,4’-di-tert-butylbiphenyl (BP) was included in all subsequent reactions. Notably, reactions conducted without the photocatalyst but with the internal standard resulted in significant amounts of product in the case of 1b (entry 3), suggesting that the internal standard can itself act as a photocatalyst. There is precedent that even simple arenes, such as dicyanoanthracene and benzophenone, can serve as photocatalysts.44 Controls without light (entry 4) and without MesAcr+BF4− or 4,4’-di-tert-butylbiphenyl (entry 5) confirmed that both light and a photocatalyst are needed to effect the coupling reaction.
Table 3.
Evidence of Biphenyl as a Photocatalyst
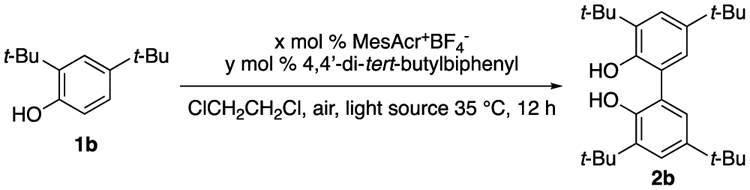 | ||||
|---|---|---|---|---|
| entry | x | y | light source | result |
| 1 | 2 | 25 | blue LEDs | 45% |
| 2 | 2 | 0 | blue LEDs | 25% |
| 3 | 0 | 25 | blue LEDs | 30% |
| 4 | 0 | 25 | none | 0% |
| 5 | 0 | 0 | blue LEDs | 0% |
Homo-Coupling Scope.
Under the optimized conditions, substrates with 2,4-, 2,4,5-, or 3,4,5-substitution patterns exhibited good reactivity, affording dimers in isolated yields up to 76% (Figure 1). There was limited tolerance for bromo-substitution, as 2d was achieved in only 16% isolated yield. The conditions were effective with aryl-substituted phenols (2j-2q). Remarkably, electron poor CF3-aryl-substituted phenols (2l, 2o) provided higher conversion and yield than electron rich MeO-aryl-substituted phenols (2j, 2m), an unexpected outcome as electron poor phenols are typically less easily oxidized (vide infra). Additionally, the ortho-tert-butyl substituted tyrosine substrate (2r) afforded the highest isolated yield, indicating the synthetic utility of this method as found independently by Pappo and coworkers.34 In certain cases, 1,2-dichloroethane was added as a cosolvent with HFIP to ensure complete solubility of reaction components over the time course of the reaction. Unfortunately, methoxy-substituted phenols, which are more easily oxidized, and substrates lacking a 4-substituent proved untenable for dimerization, as no significant yields (<5%) were achieved.
Figure 1.
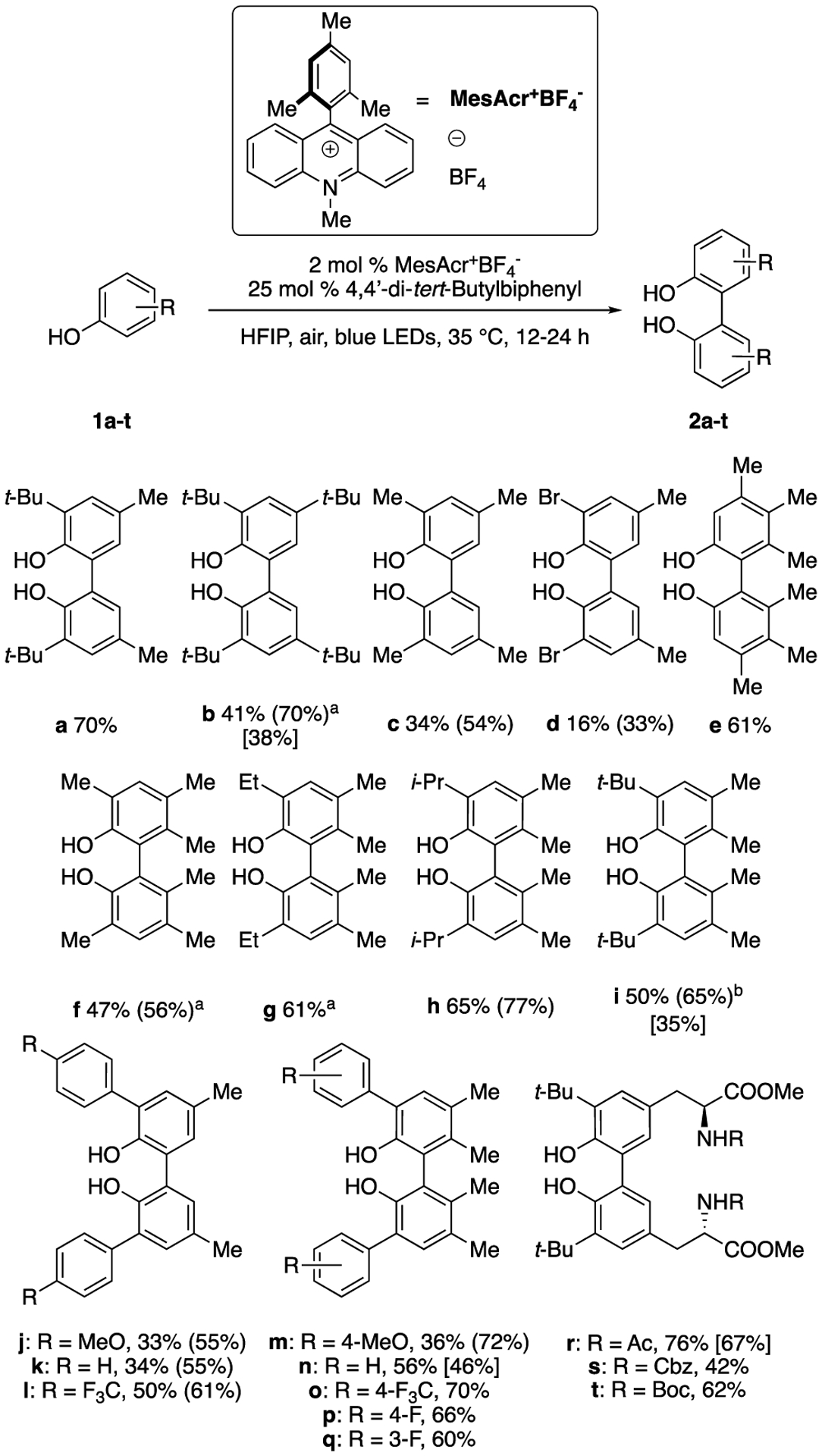
Scope of photocatalytic homo-coupling of phenols. Reported yields are of isolated material; yields in parentheses are based on recovery of starting material. Values in brackets are yields from reactions run without biphenyl. aSolvent = ClCH2CH2Cl; bSolvent = ClCH2CH2Cl: HFIP (1:2).
Mechanism.
Previous reports in the area of oxidative phenol photocatalysis propose two distinct types of intermediates (see Supporting Information for alternative mechanisms). In many electrochemical and photocatalytic reports,39,41,42,45,46 single electron oxidation of the phenol partner generates a radical cation, which then reacts to form a C-C bond either as the phenol radical cation or the neutral phenoxyl radical. Alternatively, it has been proposed that the phenol may undergo two serial single electron oxidations under photocatalytic or electrochemical conditions to generate a phenoxonium, which then reacts to form C-C bonds with the appropriate partner.47,48 Additionally, the identification of para-peroxyquinol products (4) in the reaction (Table 2) raised the possibility that these compounds may serve as intermediates in the overall transformation.49,50 As such, all three mechanistic possibilities were considered.
Reactions conducted in the absence of O2 resulted in monomer conversion consistent with photocatalyst loading (see Supporting Information). No para-peroxyquinol or para-quinol ether products were observed in absence of O2. To further probe whether an additional pathway under air involving para-peroxyquinols can occur, 2-tert-amyl-4-methyl phenol and para-peroxyquinol 4a were subjected to reaction conditions in a 1:1 ratio. This phenol substrate serves as a proxy for 1a, with similar electronic and steric features. Under the optimized conditions, the tert-amyl phenol partner completely decomposed, while the peroxyquinol 4a was largely unreacted. When light or photocatalyst were omitted, neither the phenol nor peroxyquinol reacted (see Supporting Information). Taken together, these experiments indicate that the para-peroxyquinol is not a putative intermediate in the overall homo- and cross-coupling transformation but is a consequence of an unproductive off-cycle reaction.
To further probe whether a phenoxonium was involved, two key experiments were undertaken. First, easily oxidized 2-tert-butyl-4-methoxyphenol (1u) was subjected to the reaction conditions in the presence of stoichiometric methanol as a nucleophile. When treated with hypervalent iodine and methanol, this same phenol undergoes trapping of methanol at the para-position to give a dimethoxy quinone ketal product via a two-electron oxidation to the phenoxonium.51 Under the optimized conditions with strongly oxidizing MesAcr+BF4− photocatalyst, none of the quinone ketal product was observed, pointing away from the intermediacy of a phenoxonium cation. Furthermore, the less oxidizing photocatalyst Eosin Y (EOx = 0.83 V),52 which operates below the oxidation potential needed to generate the phenoxonium,53,54 is effective in the coupling reaction (see Supporting Information). This finding further supports the intermediacy of radical cations or phenoxyl radicals arising from one-electron oxidation, as opposed to phenoxonium species arising from two-electron oxidation.
Analysis of the aqueous extract from a completed reaction revealed the presence of significant quantities of hydrogen peroxide (see Supporting Information), supporting a mechanism that proceeds with the generation of superoxide anion and peroxyl radicals. The introduction of an exogenous base (organic or inorganic) to the system also decreased the rate of reaction, indicating that a radical-anion coupling was not likely. In light of these findings, the reaction pathway most likely commences with the oxidation of the phenol by MesAcr+BF4− (EOx = 2.06 V)55 in the excited state (IVa to V, Scheme 2). The reduced photocatalyst can then be re-oxidized by dioxygen (III to I) to afford a superoxide anion and ground state MesAcr+BF4− (I). Thereafter, the oxidized phenol radical cation V(pKa ~ −2.0)56 is deprotonated by superoxide anion (pKa of HO2• → O2–• = 4.9)57 (V to VI) and attacked by a neutral phenol (VI to VII). A peroxyl radical or the excited state MesAcr+BF4− subsequently oxidizes intermediate VII to provide VIII. Tautomerization of VIII then affords the product IX. The superoxide/peroxyl radical pathway shown in Scheme 2 is used to illustrate a balanced chemical equation and to account for the formation of hydrogen peroxide in-situ.
Scheme 2.
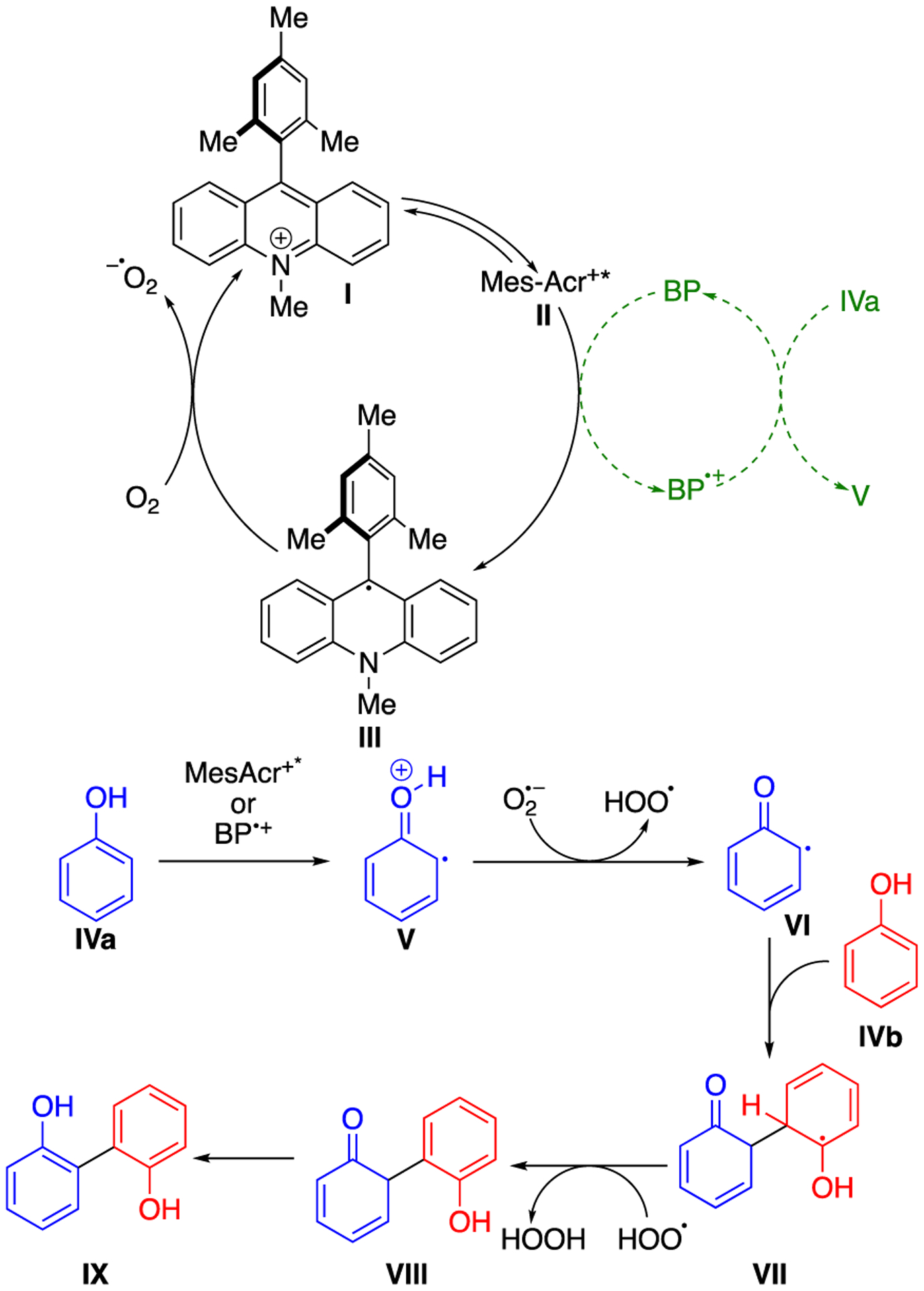
Proposed Mechanism for the Photocatalytic Coupling of Phenols
The proposed mechanism for C-C bond formation can be classified as a radical-neutral coupling between a neutral phenoxyl radical (VI) and a neutral nucleophilic phenol partner (IVb). This mechanism is akin to phenol cross-coupling under electrochemical conditions, but is unique in that the photocatalyst serves two roles: 1) from the excited state, it is a single electron oxidant that acts on the phenol and 2) it acts as the reductant for the in-situ production of a second stoichiometric oxidant (hydrogen peroxyl radical). One critical implication of the mechanism in Scheme 2 is that the coupling will proceed best when a monomer can be readily oxidized (IVa to V) and can act as a nucleophile (IVb to VII).
Experiments conducted in the absence of the biphenyl internal standard revealed its influence on the conversion of phenol monomer to dimer (Figure 1, yields in brackets). While the biphenyl can act as a photocatalyst to some extent and effect the coupling transformation in the absence of MesAcr+BF4− (Table 3, entry 3), a more compelling argument suggests that the biphenyl serves as a radical mediator or co-sensitizer in the presence of MesAcr+BF4−.36,44 Previous reports propose a mechanism wherein a biphenyl compound rapidly quenches the photocatalyst, inducing a biphenyl radical cation that can then serve as an oxidant.58,59 Interestingly, these reports indicate that the biphenyl may have a longer lifetime in its oxidized state than a photocatalyst in its excited state.44
Stern Volmer fluorescence quenching experiments revealed that the biphenyl does in-fact participate in photocatalyst quenching (Figure 2).60 Therefore, two separate pathways are likely: 1) direct oxidation of the phenol occurs by excited state MesAcr+BF4− (Scheme 2) or 2) the biphenyl quenches the photocatalyst to yield a biphenyl radical cation, which then oxidizes the phenol monomer. The advantages afforded by the latter pathway likely arise from the longer lifetime of the biphenyl radical cation. This proposed case of redox mediation is reminiscent of other oxidative processes,61 both biological and synthetic, and could potentially be leveraged in the development of more effective redox mediation pairs by altering biphenyl steric and electronic parameters.
Figure 2.

Steady-state Stern-Volmer plot for the emission quenching of excited state Mes-Acr+BF4− (λex = 450 nm, λmax = 534 nm) by two phenol monomers and 4,4’-di-tert-butylbiphenyl. I0 and I are the luminescence intensities in the absence and presence of the specific quencher at variable concentrations, respectively.
An additional implication of the mechanism proposed in Scheme 2 is that a cross-coupling should be feasible provided that one phenol is more readily oxidized(intermediate IVa, outlined in blue), while the other possesses a reactive site that is more nucleophilic (intermediate IVb, outlined in red). Stern Volmer fluorescence quenching experiments (Figure 2) with two phenol monomers, 2-tert-butyl-4-methoxyphenol (more oxidizable, less nucleophilic) and 2-tert-butyl-5-methylphenol (less oxidizable, more nucleophilic) revealed that both are capable of quenching the excited state photocatalyst, which is expected, as both substrates have oxidation potentials lying within the oxidizing range of the excited state of MesAcr+BF4− (Figure 2). Furthermore, the steeper slope for the more oxidizable 2-tert-butyl-4-methoxyphenol is consistent with more effective quenching. When both substrates are combined with the biphenyl additive and MesAcr+BF4−, differential quenching is likely. Due to the relative abundance of phenols vs biphenyl additive, the differences in oxidation potentials, and the differences in lifetimes, it is most probable that the blue phenol is oxidized by either the excited photocatalyst or the biphenyl radical cation before the red phenol.
Cross-Coupling Scope.
A number of recent reports have centered on phenolic cross-couplings;17,18,20,42,43 limitations include a reliance on an excess of one partner17,39,42,62,63 and/or poor selectivity due to competitive homo-coupling, as well as mixtures of ortho-vs para-coupled products.17,39 The homo-coupling reaction (Figure 1) requires specific substitution patterns, which include an alkyl group at the para-position. In our recent report on chromium catalyzed phenolic couplings, we leveraged site nucleophilicity to predict the regioselectivity of coupling.18 The calculated values revealed that open para-positions are significantly more nucleophilic than open ortho-positions. Consequently, a high yielding cross-coupling can be induced by selecting one substrate without a para-substituent (red phenol). The poor homo-coupling of such substrates (vide supra) and the high nucleophilicity of the para-position make these substrates excellent candidates for cross-coupling
Based on the cross-coupling model discussed above, it was anticipated that different di- and tri-substitution patterns on both the nucleophilic (red phenol) and more readily oxidized (blue phenol) partners would be effective (Figure 3) in cross-coupling. Remarkably, mono-substituted phenols, which are difficult to couple due to multiple reactive sites and high oxidation potentials,34 were compatible (6a-u through 6d-u).15,17,18 In contrast to the homo-coupling, a high level of success was also achieved with a halogenated substrate, as 6h-y was isolated in 73% yield. Furthermore, a more-easily oxidized (blue phenol) 3,4-di-substituted phenol also provided both good reactivity and regioselectivity, perhaps due to steric effects, with 6h-z being isolated in 71% yield. As in the case of homo-coupling, cross-coupling reactions conducted in the absence of the biphenyl cosensitizer revealed that the additive either had little effect (6e-u) or facilitated conversion (6h-u, 6h-aa, 6i-u) in the cross-coupling reaction (Figure 3, result in brackets).
Figure 3.

Scope of the photocatalytic phenol cross-coupling. Conditions: 2.0 mol % MesAcr+BF4–, 25 mol % 4,4’-di-tert-butylbiphenyl, HFIP, air, blue LEDs, 35 °C, 48 h. Reported yields are of isolated material; yields in parentheses are based on recovered starting material. Values in brackets are isolated yields in the absence of the biphenyl. aReaction stopped at 24 h; bSolvent = ClCH2CH2Cl: HFIP (1:1); cortho-ortho product recovered in 9% yield.
Notably, no homo-coupling products were observed and only trace amounts (0–5%) of the ortho-ortho coupling were found in all but one case (6l-u; 9% ortho-ortho vs 52% ortho-para). The greater site nucleophilicity of the para-position accounts for these observed differences. These results also accentuate the difference between the mechanisms proposed in this and recent reports.17,18 For instance, the iron porphyrin coupling reported by Pappo in 2017 proceeds via a radical-radical pathway, which does not rely on site nucleophilicity and exhibits lower regioselectivity.17 Specifically, that report revealed ~1:1 ratio of the ortho-ortho and ortho-para adducts for the substrates corresponding to 6j-u and 6k-u. Here, 6j-u and 6k-u form with high ortho-para selectivity (Figure 3). Furthermore, reactions of other known compounds proceed in similar or higher yields than other reports and use a 1:1 molar ratio of phenols, rather than an excess of one phenol. For example, the formation of 6h-u with our prior reported Cr catalyst18 gave 50% yield, whereas the protocol herein provided 97% of the same product.
Additionally, selective modifications can be made to the biphenol products obtained from the photocatalytic coupling reaction (Figure 4). For example, the methoxy group of 6a-u can be selectively deprotected to generate para-dihydroxy compound 7a-u using BBr3. Alternatively, the tert-butyl groups of the same compound can be selectively removed in the presence of the methoxy group using AlCl3 to generate monosubstituted biphenol compound 8a-u, a motif which would be difficult to access via conventional oxidative coupling methods.33 Furthermore, the tert-butyl and methyl ether groups can be simultaneously cleaved with AlCl3 at longer reaction times (7h-v). Therefore, the abundance of tert-butyl and methoxy-substituted substrates could be leveraged to access a more diverse array of biphenyl compounds.
Figure 4.
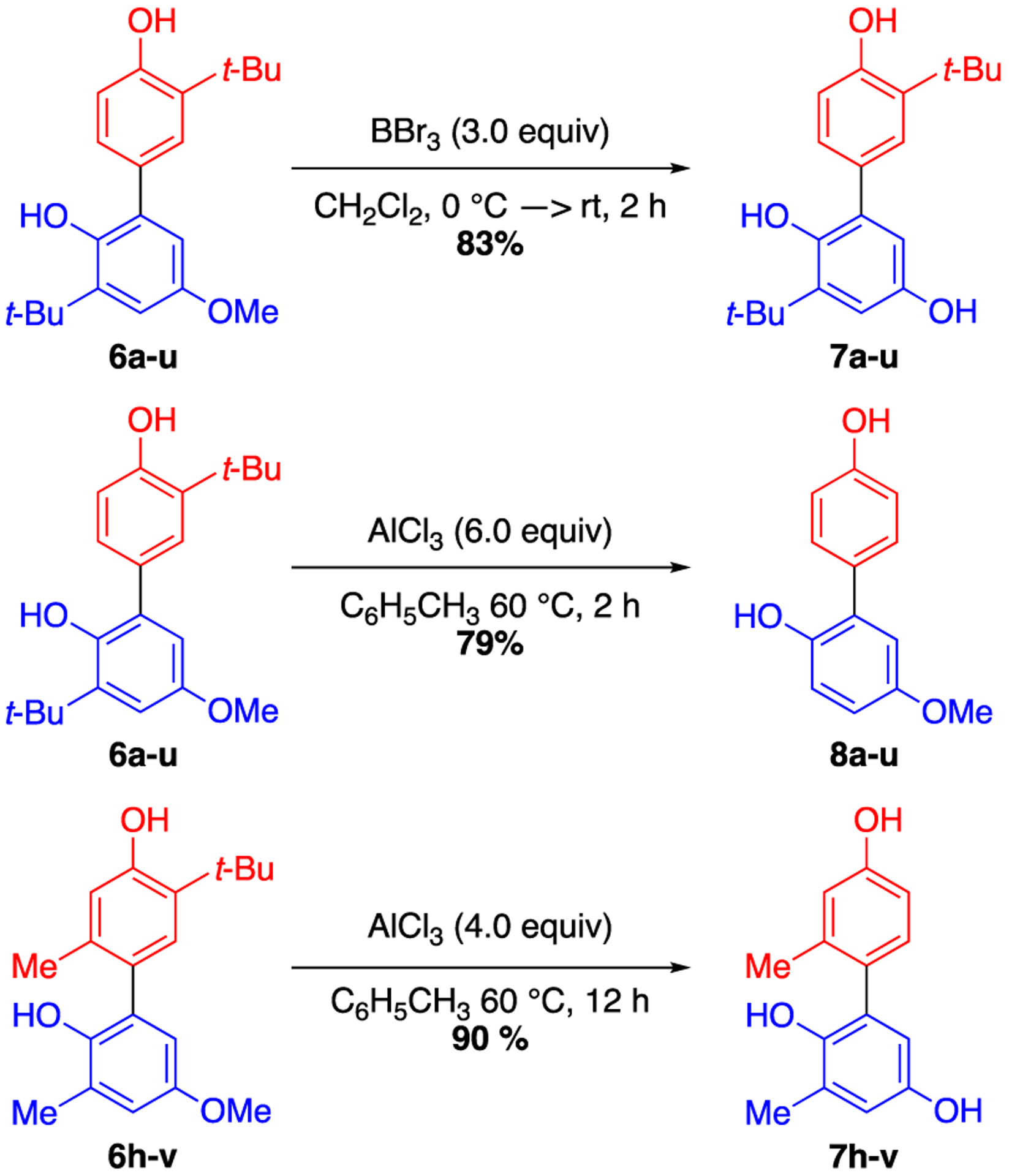
Selective Transformations of Dimeric Products.
Tyrosine Coupling.
Our final endeavor aimed to achieve the direct catalytic coupling of tyrosine. Although a highly efficient oxidative and catalytic method for coupling tyrosine derivatives was recently reported, a tert-butyl group positioned ortho- to the phenol was required to obtain a high conversion,34 which we had also observed independently (Figure 1, 2r, 2s, 2t). While this group can be readily removed in excellent yield, its use is not ideal due to the decrease in step-economy. Tyrosine derivatives lacking the activating tert-butyl substituent achieved isolated yields of only 10% under the optimized photocatalytic conditions described above, with significant amounts of byproducts observed. Specifically, when 1ee was subjected to the photocatalytic conditions reported in Figure 1, only the para-peroxyquinol 4ee was observed. A further HTE screen of photocatalysts was thus undertaken to determine if different photocatalysts could be more effective in producing the important dityrosine derivative 2ee. This screen revealed that Ru(bpz)3[PF6]2 paired with O2 converts 1ee to 2ee while attenuating the formation of byproducts. A larger scale reaction (Figure 5) confirmed this result, affording a 40% isolated yield of 2ee with no detected by-products (73% based on recovered starting material).
Figure 5.
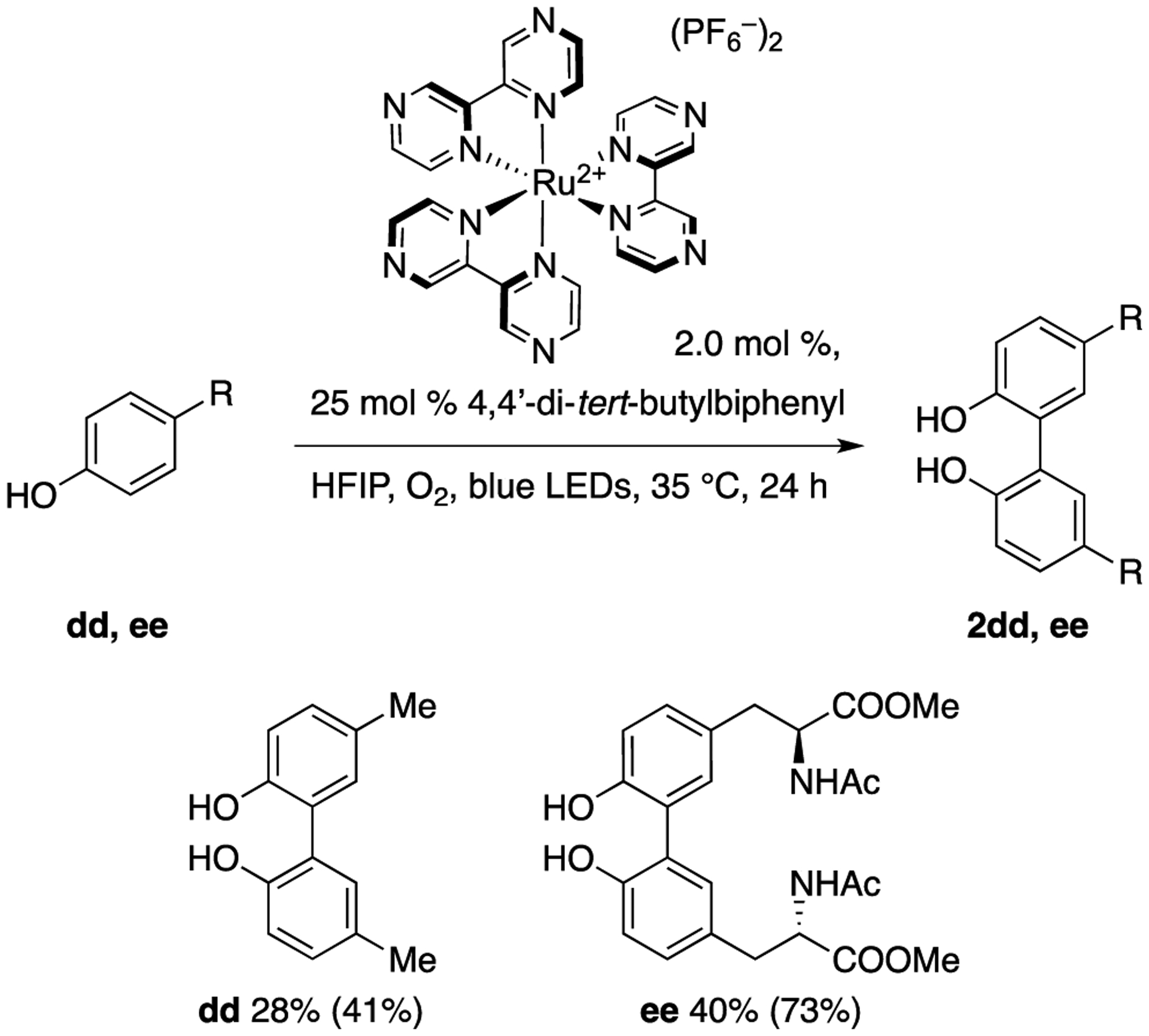
Homo-coupling of para-substituted phenols. Yields in parentheses are based on recovery of starting material. Conditions: 0.5 mol % Ru(bpz)3[PF6]2, 25 mol % 4,4’-di-tert-Butylbiphenyl, HFIP, O2, blue LEDs, 35 °C, 24 h.
The proposed radical-nucleophile coupling paradigm (Scheme 2), which implies reactivity relies solely on oxidation potentials and site nucleophilicity parameters, fails to explain the modest yields observed in the tyrosine homo-coupling. In the chromium and iron catalyzed couplings reported by Kozlowski and Pappo,17,18 covalently bound metal phenolates are proposed and the product, which is more sterically hindered, binds to the catalyst less readily. In contrast, photooxidative processes do not proceed via inner sphere oxidation, but through outer sphere electron transfer. Therefore, we postulate that differences in oxidation potentials between phenol monomers (1) and their corresponding dimers (2) may lead to reaction inhibition. Cyclic voltammetry (Table 4) reveals that the tyrosine dimer 2ee (1.07 V) is more readily oxidized than the tyrosine monomer 1ee (1.13 V), suggesting that the dimer can selectively quench the photocatalyst, accounting for lower conversion upon product formation. Further, in the oxidation return sweep, the dimer shows poor reversibility indicating that decomposition likely occurs via over-oxidation.
Table 4.
Selected Oxidation Potentials of Phenol Monomers and Dimers
| monomer | Eox (V)a | dimer | Eox (V)a | yield |
|---|---|---|---|---|
| 1ee | 1.13 | 2ee | 1.07 | 40% |
| 1m | 0.78 | 2m | 0.71 | 36% |
| 1n | 0.84 | 2n | 0.96 | 56% |
| 1o | 0.92 | 2o | 1.03 | 70% |
| 5h | 0.89 | 6h-u | 0.56 | 97% |
| 1u | 0.52 |
Oxidation potentials obtained in HFIP; defined by the potential at half peak height of local minimum for first oxidation wave.
The oxidation potentials of substrates from Figure 1 further suggest a correlation between the difference in oxidation potentials of monomer/dimer and conversion. For example, 1o, which is more readily oxidized than its dimeric product, affords 2o in 70% isolated yield. Conversely, 1m, which is less readily oxidized than its dimeric product, provides 2m in only 36% yield. This model likely also accounts for the observation that 1u, while more readily oxidized and nucleophilic at the ortho-position than 1a, does not form dimer under the coupling conditions described in Figure 1. However, 1u (0.52 V) is more readily oxidized than coupling partner 5h (0.89 V) and the corresponding cross-coupling dimer 6h-u (0.56 V), allowing for a high isolated yield of 6h-u (97%). Based on these findings, it is evident that phenol nucleophilicity and oxidation potential are key factors in determining overall reactivity and selectivity, but the oxidation potential of the biphenol products should also be considered.
CONCLUDING REMARKS
In summary, a photocatalytic method for coupling phenols was developed using MesAcr+BF4−. Mechanism studies suggest that an oxidation of the phenol followed by the nucleophilic addition of a neutral phenol is a plausible pathway. This mechanistic paradigm may be relevant to biosynthetic phenol coupling and is more plausible than two phenol radicals reacting in a termination event, as is often proposed.64,65 With the additional mechanistic understanding of how product oxidation potential factors into the outcome, the design of new catalytic systems can be undertaken.
The mechanistic model provided insight into the development of a cross coupling method with the same photocatalyst. The resultant process afforded a range of new compounds in high yields and selectivities, as well as known compounds with higher yields and selectivities. The method permitted more diverse substitution patterns on the nucleophilic coupling partner. Finally, the first non-enzymatic, catalytic coupling of tyrosine was achieved with a ruthenium photocatalyst.
Supplementary Material
ACKNOWLEDGMENT
We are grateful to the NSF (CHE1764298) and the NIH (R35 GM131902) for financial support of this research. Partial instrumentation support was provided by the NIH and NSF (1S10RR023444, 1S10RR022442, CHE-0840438, CHE-0848460, 1S10OD011980, CHE-1827457). P.H.G. thanks NSF for fellowship support (DGE-1845298). Dr. Charles W. Ross III (UPenn) is acknowledged for obtaining accurate mass data. We thank Dr. Patrick Carroll (UPenn) for X-ray analysis and Dr. Sergei Tcyrulnikov (UPenn) for calculations of site nucleophilicity values. We are grateful to William Neuhaus (UPenn) for helpful discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental protocols and spectroscopic data (pdf).
REFERENCES
- (1).Fukuyama Y; Asakawa Y Novel Neurotrophic Isocuparane-Type Sesquiterpene Dimers, Mastigophorenes A, B, C and D, Isolated from the Liverwort Mastigophora Diclados. J. Chem. Soc. Perkin Trans. 1 1991, 11, 2737–2741. [Google Scholar]
- (2).Degnan AP; Meyers AI Total Syntheses of (−)-Herbertenediol, (−)-Mastigophorene A, and (−)- Mastigophorene B. Combined Utility of Chiral Bicyclic Lactams and Chiral Aryl Oxazolines. J. Am. Chem. Soc 1999, 121 (12), 2762–2769. [Google Scholar]
- (3).Cochrane JR; White JM; Wille U; Hutton CA Total Synthesis of Mycocyclosin. Org. Lett 2012, 14 (9), 2402–2405. [DOI] [PubMed] [Google Scholar]
- (4).Boisnard S; Carbonnelle AC; Zhu J Studies on the Total Synthesis of RP 66453: Synthesis of Fully Functionalized 15-Membered Biaryl-Containing Macrocycle. Org. Lett 2001, 3 (13), 2061–2064. [DOI] [PubMed] [Google Scholar]
- (5).Bringmann G; Gulder T; Gulder TAM; Breuning M Atroposelective Total Synthesis of Axially Chiral Biaryl Natural Products. Chem. Rev 2011, 111 (2), 563–639. [DOI] [PubMed] [Google Scholar]
- (6).Pal T; Anjali P Oxidative Phenol Coupling: A Key Step for the Biomimetic Synthesis of Many Important Natural Products. Curr. Sci 1996, 106–108. [Google Scholar]
- (7).Rauf A; Uddin G; Siddiqui BS; Molnár J; Csonka Á; Ahmad B; Szabó D; Farooq U; Khan A A Rare Class of New Dimeric Naphthoquinones from Diospyros Lotus Have Multidrug Reversal and Antiproliferative Effects. Front. Pharmacol 2015, 6, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Barton DHR; Cohen T In Festschrift Prof. Dr. Arthur Stoll Zum Siebzigsten Geburstag, 8; 1957. [Google Scholar]
- (9).Armstrong DR; Cameron C; Nonhebel DC; Perkins PG Oxidative Coupling of Phenols. Part 6. A Study of the Role of Spin Density Factors on the Product Composition in the Oxidations of 3,5-Dimethylphenol and Phenol. J. Chem. Soc. Perkin Trans. 2 1983, 5, 563–568. [Google Scholar]
- (10).Morimoto K; Sakamoto K; Ohnishi Y; Miyamoto T; Ito M; Dohi T; Kita Y Metal-Free Oxidative Para Cross-Coupling of Phenols. Chem. Eur. J 2013, 19 (27), 8726–8731. [DOI] [PubMed] [Google Scholar]
- (11).More NY; Jeganmohan M Solvent-Controlled Selective Synthesis of Biphenols and Quinones: Via Oxidative Coupling of Phenols. Chem. Commun 2017, 53 (69), 9616–9619. [DOI] [PubMed] [Google Scholar]
- (12).Hwang DR; Chen CP; Uang BJ Aerobic Catalytic Oxidative Coupling of 2-Naphthols and Phenols by VO(Acac)2. Chem. Commun 1999, 13, 1207–1208. [Google Scholar]
- (13).Kirste A; Schnakenburg G; Stecker F; Fischer A; Waldvogel SR Anodic Phenol-Arene Cross-Coupling Reaction on Boron-Doped Diamond Electrodes. Angew. Chemie - Int. Ed 2010, 49 (5), 971–975. [DOI] [PubMed] [Google Scholar]
- (14).Kirste A; Schnakenburg G; Waldvogel SR Anodic Coupling of Guaiacol Derivatives on Boron-Doped Diamond Electrodes. Org. Lett 2011, 13 (12), 3126–3129. [DOI] [PubMed] [Google Scholar]
- (15).Lee YE; Cao T; Torruellas C; Kozlowski MC Selective Oxidative Homo-and Cross-Coupling of Phenols with Aerobic Catalysts. J. Am. Chem. Soc 2014, 136 (19), 6782–6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Esguerra KVN; Fall Y; Petitjean L; Lumb JP Controlling the Catalytic Aerobic Oxidation of Phenols. J. Am. Chem. Soc 2014, 136 (21), 7662–7668. [DOI] [PubMed] [Google Scholar]
- (17).Shalit H; Libman A; Pappo D Meso-Tetraphenylporphyrin Iron Chloride Catalyzed Selective Oxidative Cross-Coupling of Phenols. J. Am. Chem. Soc 2017, 139 (38), 13404–13413. [DOI] [PubMed] [Google Scholar]
- (18).Nieves-Quinones Y; Paniak TJ; Lee YE; Kim SM; Tcyrulnikov S; Kozlowski MC Chromium-Salen Catalyzed Cross-Coupling of Phenols: Mechanism and Origin of the Selectivity. J. Am. Chem. Soc 2019, 141 (25), 10016–10032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Nieves-Quinones Y; J. Paniak T; Eun Lee Y; Min Kim S; Tcyrulnikov S; C. Kozlowski M; Paniak TJ; Lee YE; Kim SM; Tcyrulnikov S; Kozlowski MC Chromium-Salen Catalyzed Cross-Coupling of Phenols: Mechanism and Origin of the Selectivity. J. Am. Chem. Soc 2019, 141 (25), 10016–10032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Libman A; Shalit H; Vainer Y; Narute S; Kozuch S; Pappo D Synthetic and Predictive Approach to Unsymmetrical Biphenols by Iron-Catalyzed Chelated Radical-Anion Oxidative Coupling. J. Am. Chem. Soc 2015, 137 (35), 11453–11460. [DOI] [PubMed] [Google Scholar]
- (21).Huang Z; Lumb JP Phenol-Directed C-H Functionalization. ACS Catal. 2019, 9 (1), 521–555. [Google Scholar]
- (22).Kozlowski MC; Morgan BJ; Linton EC Total Synthesis of Chiral Biaryl Natural Products by Asymmetric Biaryl Coupling. Chem. Soc. Rev 2009, 38 (11), 3193–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kozlowski MC Oxidative Coupling in Complexity Building Transforms. Acc. Chem. Res 2017, 50 (3), 638–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhu X; McAtee CC; Schindler CS Scalable Synthesis of Mycocyclosin. Org. Lett 2018, 20 (10), 2862–2866. [DOI] [PubMed] [Google Scholar]
- (25).Cox JB; Kimishima A; Wood JL Total Synthesis of Herquline B and C. J. Am. Chem. Soc 2019, 141 (1), 25–28. [DOI] [PubMed] [Google Scholar]
- (26).He C; Stratton TP; Baran PS Concise Total Synthesis of Herqulines B and C. J. Am. Chem. Soc 2019, 141 (1), 29–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhu X; McAtee CC; Schindler CS Total Syntheses of Herqulines B and C. J. Am. Chem. Soc 2019, 141 (8), 3409–3413. [DOI] [PubMed] [Google Scholar]
- (28).Peters DS; Romesberg FE; Baran PS Scalable Access to Arylomycins via C-H Functionalization Logic. J. Am. Chem. Soc 2018, 140 (6), 2072–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Eickhoff H; Jung G; Rieker A Oxidative Phenol Coupling - Tyrosine Dimers and Libraries Containing Tyrosyl Peptide Dimers. Tetrahedron 2001, 57 (2), 353–364. [Google Scholar]
- (30).Hay ASP,P′-Biphenols. J. Org. Chem 1969, 34 (4), 1160–1161. [Google Scholar]
- (31).Matsuura BS; Keylor MH; Li B; Lin Y; Allison S; Pratt DA; Stephenson CRJ A Scalable Biomimetic Synthesis of Resveratrol Dimers and Systematic Evaluation of Their Antioxidant Activities. Angew. Chemie Int. Ed 2015, 54 (12), 3754–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Keylor MH; Matsuura BS; Griesser M; Chauvin JPR; Harding RA; Kirillova MS; Zhu X; Fischer OJ; Pratt DA; Stephenson CRJ Synthesis of Resveratrol Tetramers via a Stereoconvergent Radical Equilibrium. Science 2016, 354 (6317), 1260–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Kozlowski MC; Lee YE; Kim SM; Stanislav J Synthesis of Honokiol. US2017113989, WO2017070568A1, 2017. [Google Scholar]
- (34).Ben-Lulu M; Gaster E; Libman A; Pappo D Synthesis of Biaryl-Bridged Cyclic Peptides via Catalytic Oxidative Cross-Coupling Reactions. Angew. Chemie Int. Ed 2020, 59 (12), 4835–4839. [DOI] [PubMed] [Google Scholar]
- (35).A preliminary version of this work was deposited at ChemRxiv: Niederer KA; Gilmartin PH; Kozlowski MC Oxidative Photocatalytic Homo- and Cross-Coupling of Phenols: Non-Enzymatic, Catalytic Method for Coupling Tyrosine. ChemRxiv 2020, Preprint. 10.26434/chemrxiv.11813403.v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Romero NA; Nicewicz DA Organic Photoredox Catalysis. Chem. Rev 2016, 116 (17), 10075–10166. [DOI] [PubMed] [Google Scholar]
- (37).Ding Y; Li Y; Qin M; Cao Y; Wang W Photo-Cross-Linking Approach to Engineering Small Tyrosine-Containing Peptide Hydrogels with Enhanced Mechanical Stability. Langmuir 2013, 29 (43), 13299–13306. [DOI] [PubMed] [Google Scholar]
- (38).Bjork JW; Johnson SL; Tranquillo RT Ruthenium-Catalyzed Photo Cross-Linking of Fibrin-Based Engineered Tissue. Biomaterials 2011, 32 (10), 2479–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Eisenhofer A; Hioe J; Gschwind RM; König B Photocatalytic Phenol-Arene C-C and C-O Cross-Dehydrogenative Coupling. Eur. J. Org. Chem 2017, 2017 (15), 2194–2204. [Google Scholar]
- (40).Zhao Y; Huang B; Yang C; Li B; Gou B; Xia W Photocatalytic Cross-Dehydrogenative Amination Reactions between Phenols and Diarylamines. ACS Catal. 2017, 7 (4), 2446–2451. [Google Scholar]
- (41).Wang J; Zhao Y; Gao H; Gao G-L; Yang C; Xia W Visible-Light-Mediated Dehydrogenative Cross-Coupling: Synthesis of Nonsymmetrical Atropisomeric Biaryls. Asian J. Org. Chem 2017, 6 (10), 1402–1407. [Google Scholar]
- (42).Kirste A; Elsler B; Schnakenburg G; Waldvogel SR Efficient Anodic and Direct Phenol-Arene C,C Cross-Coupling: The Benign Role of Water or Methanol. J. Am. Chem. Soc 2012, 134 (7), 3571–3576. [DOI] [PubMed] [Google Scholar]
- (43).Colomer I; Chamberlain AER; Haughey MB; Donohoe TJ Hexafluoroisopropanol as a Highly Versatile Solvent. Nat. Rev. Chem 2017, 1 (11), 1–12. [Google Scholar]
- (44).Skubi KL; Blum TR; Yoon TP Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev 2016, 116 (17), 10035–10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).McManus JB; Nicewicz DA Direct C-H Cyanation of Arenes via Organic Photoredox Catalysis. J. Am. Chem. Soc 2017, 139 (8), 2880–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Zhao Q; Jin J-K; Wang J; Zhang F-L; Wang Y-F Radical α-Addition Involved Electrooxidative [3 + 2] Annulation of Phenols and Electron-Deficient Alkenes. Chem. Sci 2020, 11, 3909–3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Blum TR; Zhu Y; Nordeen SA; Yoon TP Photocatalytic Synthesis of Dihydrobenzofurans by Oxidative [3+2] Cycloaddition of Phenols. Angew. Chemie Int. Ed 2014, 53, 11056–11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Wang Y; Tian B; Ding M; Shi Z Electrochemical Cross-Dehydrogenative Coupling between Phenols and β-Dicarbonyl Compounds: Facile Construction of Benzofurans. Chem. Eur. J 2020, 26, 4297–4304. [DOI] [PubMed] [Google Scholar]
- (49).Kotani H; Ohkubo K; Fukuzumi S Photocatalytic Oxygenation of Anthracenes and Olefins with Dioxygen via Selective Radical Coupling Using 9-Mesityl-10-Methylacridinium Ion as an Effective Electron-Transfer Photocatalyst. J. Am. Chem. Soc 2004, 126 (49), 15999–16006. [DOI] [PubMed] [Google Scholar]
- (50).Carreño MC; González-López M; Urbano A Oxidative De-Aromatization of Para-Alkyl Phenols into Para-Peroxyquinols and Para-Quinols Mediated by Oxone as a Source of Singlet Oxygen. Angew. Chemie Int. Ed 2006, 45 (17), 2737–2741. [DOI] [PubMed] [Google Scholar]
- (51).Chittimalla SK; Bandi C Unanticipated Participation of HCl in Nucleophilic Chlorination Reaction: Expedient Route to Meta Chlorophenols. Tetrahedron Lett. 2016, 57 (1), 15–19. [Google Scholar]
- (52).Hari DP; König B Synthetic Applications of Eosin Y in Photoredox Catalysis. Chem. Commun 2014, 50, 6688–6699. [DOI] [PubMed] [Google Scholar]
- (53).Waters W Comments on the Mechanism of One-Electron Oxidation of Phenols: A Fresh Interpretation of Oxidative Coupling Reactions of Plant Phenols. J. Chem. Soc. B 1971, 2026–2029. [Google Scholar]
- (54).Steuber FW; Dimroth K Polarographische Bestimmung Der Halbstufenpotentiale von Arylierten Phenolen Mit Der Rotierenden Graphitelektrode. Chem. Ber 1966, 99 (1), 258–263. [Google Scholar]
- (55).Joshi-Pangu A; Lévesque F; Roth HG; Oliver SF; Campeu L-C; Nicewicz D; DiRocco DA Acridinium-Based Photocatalysts: A Sustainable Option in Photoredox Catalysis. J. Org. Chem 2016, 81, 7244–7249. [DOI] [PubMed] [Google Scholar]
- (56).Nicholas AMP; Arnold DR Thermochemical Parameters for Organic Radicals and Radical Ions. Part 1. The Estimation of the PKa of Radical Cations Based on Thermochemical Calculations. Can. J. Chem 1982, 60 (17), 2165–2179. [Google Scholar]
- (57).Sawyer DT; Roberts JL; Calderwood TS; Sugimoto H; McDowell MS Reactivity and Activation of Dioxygen-Derived Species in Aprotic Media (a Model Matrix for Biomembranes). Philos. Trans. R. Soc. London 1985, 311, 483–503. [DOI] [PubMed] [Google Scholar]
- (58).Schaap AP; Lopez L; Anderson SD; Gagnon SD Cosensitization by 9,10-Dicyanoanthracene and Biphenyl of the Electron-Transfer Photooxygenation of 1,1,2,2-Tetraphenylcyclopropane. Tetrahedron Lett. 1982, 23 (52), 5493–5496. [Google Scholar]
- (59).Gutenberger G; Steckhan E; Blechert S α-Silyl Ethers as Hydroxymethyl Anion Equivalents in Photoinduced Radical Electron Transfer Additions. Angew. Chemie - Int. Ed 1998, 37 (5), 660–662. [DOI] [PubMed] [Google Scholar]
- (60).Arias-Rotondo DM; McCusker JK The Photophysics of Photoredox Catalysis: A Roadmap for Catalyst Design. Chem. Soc. Rev 2016, 45 (21), 5803–5820. [DOI] [PubMed] [Google Scholar]
- (61).Piera J; Bäckvall JE Catalytic Oxidation of Organic Substrates by Molecular Oxygen and Hydrogen Peroxide by Multistep Electron Transfer - A Biomimetic Approach. Angew. Chemie - Int. Ed 2008, 47 (19), 3506–3525. [DOI] [PubMed] [Google Scholar]
- (62).Gao PC; Chen H; Grigoryants V; Zhang Q Oxidative Phenol-Arene and Phenol-Phenol Cross-Coupling Using Periodic Acid. Tetrahedron 2019, 75 (13), 2004–2011. [Google Scholar]
- (63).Elsler B; Schollmeyer D; Dyballa KM; Franke R; Waldvogel SR Metal- and Reagent-Free Highly Selective Anodic Cross-Coupling Reaction of Phenols. Angew. Chemie - Int. Ed 2014, 53 (20), 5210–5213. [DOI] [PubMed] [Google Scholar]
- (64).Aldemir H; Richarz R; Gulder TAM The Biocatalytic Repertoire of Natural Biaryl Formation. Angew. Chemie - Int. Ed 2014, 53 (32), 8286–8293. [DOI] [PubMed] [Google Scholar]
- (65).Tang MC; Zou Y; Watanabe K; Walsh CT; Tang Y Oxidative Cyclization in Natural Product Biosynthesis. Chem. Rev 2017, 117 (8), 5226–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.