

Abstract
PURPOSE
This trial assessed the utility of applying tumor DNA sequencing to treatment selection for patients with advanced, refractory cancer and somatic mutations in one of four signaling pathways by comparing the efficacy of four study regimens that were either matched to the patient's aberrant pathway (experimental arm) or not matched to that pathway (control arm).
MATERIALS AND METHODS
Adult patients with an actionable mutation of interest were randomly assigned 2:1 to receive either (1) a study regimen identified to target the aberrant pathway found in their tumor (veliparib with temozolomide or adavosertib with carboplatin [DNA repair pathway], everolimus [PI3K pathway], or trametinib [RAS/RAF/MEK pathway]), or (2) one of the same four regimens, but chosen from among those not targeting that pathway.
RESULTS
Among 49 patients treated in the experimental arm, the objective response rate was 2% (95% CI, 0% to 10.9%). One of 20 patients (5%) in the experimental trametinib cohort had a partial response. There were no responses in the other cohorts. Although patients and physicians were blinded to the sequencing and random assignment results, a higher pretreatment dropout rate was observed in the control arm (22%) compared with the experimental arm (6%; P = .038), suggesting that some patients may have had prior tumor mutation profiling performed that led to a lack of participation in the control arm.
CONCLUSION
Further investigation, better annotation of predictive biomarkers, and the development of more effective agents are necessary to inform treatment decisions in an era of precision cancer medicine. Increasing prevalence of tumor mutation profiling and preference for targeted therapy make it difficult to use a randomized phase II design to evaluate targeted therapy efficacy in an advanced disease setting.
INTRODUCTION
Targeting therapy to the molecular characteristics of an individual's tumor is a primary goal of precision cancer medicine.1-3 This approach hypothesizes that the presence of a mutation will render the tumor susceptible to an agent targeting that mutation. To explore this, we conducted a randomized, histology-agnostic clinical trial to examine whether patients with advanced, refractory cancer who had a tumor mutation in a gene in one of three signaling pathways (DNA repair, PI3K, or RAS/RAF/MEK) were more likely to derive clinical benefit if treated with regimens targeting that pathway (the experimental arm) than if they were treated with regimens that did not (the control arm). The treatment for each patient, whether experimental or control, was chosen from the same panel of four regimens: veliparib with temozolomide (TMZ), adavosertib with carboplatin, everolimus, or trametinib. For the experimental arm, the performance of each individual treatment cohort was to be compared with historical standards; the performance of the experimental arm, composed of all four targeted treatment cohorts, was to be compared with that of the control arm. Patients and treating physicians were blinded to the arm assignment and tumor sequencing data until the patient progressed.
CONTEXT
Key Objective
A molecular aberration in a patient's tumor is expected to render the tumor susceptible to a drug targeting that aberration, but randomized, controlled, blinded phase II trials confirming the efficacy of this precision medicine approach are both sparse and challenging to design.
Knowledge Generated
Efficacy in patients with study-defined actionable mutations in the DNA repair, RAS/RAF/MEK, or AKT/PI3K/MTOR pathways either did not achieve the target objective response rate (trametinib or adavosertib with carboplatin) or indicated futility despite accrual challenges (everolimus or veliparib with temozolomide). Patients randomly assigned to the nontargeted control arm had a higher pretreatment dropout rate than the experimental arm, suggesting a preference for targeted therapy based on prestudy genetic profiling.
Relevance
The prevalence of genetic data makes it challenging to randomly assign patients to a nontargeted control arm. Better gene- and variant-specific biomarkers that predict response to drugs are needed for patients with cancer.
MATERIALS AND METHODS
Participants
This study enrolled patients 18 years of age or older with histologically documented solid tumors whose disease had progressed following at least one line of standard therapy and/or for whom no standard treatment shown to improve survival was available. Patients were required to have measurable disease, be willing to undergo tumor biopsy to establish presence of a study-defined actionable mutation of interest (aMOI), and have tumor amenable to interventional radiology-guided percutaneous biopsy with a 16- to 18-gauge needle; excisional biopsy was allowed if indicated and evaluable. A Karnofsky performance status score ≥ 70% and adequate liver, kidney, and marrow function (as defined in the Data Supplement) were required. Previous anticancer therapy or surgery must have been completed at least 3 weeks prior to enrollment; patients with active brain metastases were ineligible. Patients who had prior treatment with any of the investigational agents were eligible to participate but were not assigned that same agent. Agent-specific eligibility criteria are included in the Data Supplement.
This trial was conducted under a National Cancer Institute (NCI)-sponsored Investigational New Drug Application with institutional review board approval. Protocol design and conduct followed all applicable regulations, guidances, and local policies (ClinicalTrials.gov identifier: NCT01827384). The investigators obtained informed consent from each participant.
Trial Design
This was a multihistology, multicenter, randomized phase II study of four investigational drug regimens demonstrated to inhibit the DNA repair pathway, RAS/RAF/MEK pathway, or AKT/PI3K/MTOR pathway. Eligible patients with an aMOI detected were randomly assigned 2:1 to receive the recommended phase II dose of either (1) a predefined targeted regimen based on mutation status (experimental arm) or (2) a regimen, chosen from the four study regimens, that did not target their aMOIs (control arm). Treatment assignment was based on the presence of mutations in a panel of genes, each with a mutation frequency of > 5% in the Catalog of Somatic Mutations in Cancer database version 61,4 which were identified as direct or upstream targets for these drug combinations. The specific study aMOIs (Data Supplement) were selected on the basis of published functional evidence or implications for protein translation and pathway function; they were detected in patient samples via a Clinical Laboratory Improvement Amendments (CLIA) sequencing assay developed and validated by the Molecular Characterization Laboratory at the Frederick National Laboratory for Cancer Research, as previously described.5
Patient eligibility determination and randomized treatment assignment were performed with the GeneMed informatics system as described.6 If more than one aMOI was detected for a patient randomly assigned to the experimental arm, selection of the targeted treatment was based on the higher allele frequency; if allele frequencies were within 15%, the patient was assigned to the targeted treatment cohort with the fewest patients enrolled (with the option to receive the other treatment regimen upon disease progression). If a patient randomly assigned to the control arm had multiple nontargeted treatment options, their regimen was chosen based on the proportions of treatment assignments on the experimental arm, so as to balance the two arms with respect to proportions receiving each of the four regimens.
For the purpose of assessing the primary end point of this trial, only response to the first regimen was used. Adverse events (AEs) were graded according to NCI Common Toxicity Criteria version 4.0 until March 31, 2018, when version 5.0 was implemented; all AEs were mapped to version 5.0 before the final analysis. Doses of all drugs were reduced for grade ≥ 3 nonhematologic and grade 4 hematologic toxicities (except lymphopenia or leukopenia in the absence of neutropenia). Patients were allowed up to two dose reductions before being taken off treatment. Radiographic evaluation was performed at baseline and every two cycles to assess tumor response based on the RECIST version 1.1.7
Study Agents
Veliparib (ABT-888; NSC 737664), adavosertib (AZD1775; NSC 751084), trametinib (NSC 763093), and everolimus (NSC 733504) were supplied by the Division of Cancer Treatment and Diagnosis, NCI, under Collaborative Research and Development Agreements with AbbVie (North Chicago, IL), AstraZeneca (Cambridge, United Kingdom), GlaxoSmithKline (Brentford, United Kingdom), and Novartis (Basel, Switzerland), respectively. TMZ (NSC 362856) and carboplatin (NSC 241240) were obtained from commercial sources. Study drugs were administered at the recommended phase II doses and schedules (Data Supplement).
Statistical Study Design
The accrual ceiling for each regimen cohort of the experimental arm was set at 30 patients to discriminate between tumor response rates of 20% versus 5%. If at least four objective responses (at least 13%) were observed among the 30 patients, this regimen would have been considered promising for this mutation category. The design included an interim futility analysis; if no objective responses were observed among the initial 12 patients in each experimental cohort, the cohort was to be terminated early (with 54% likelihood under the null hypothesis), with 93% confidence that the response rate would be lower than the target 20% rate. This design yields at least 84% power to detect a true objective response rate of at least 20% and at least 0.94 probability of a negative result if the true objective response rate was no more than 5%. Four-month progression-free survival (PFS), defined as the time from random assignment to progression or death from any cause (whichever comes first), was evaluated as a secondary end point using Kaplan-Meier estimates and CIs calculated using Greenwood's formula. Twelve or more instances of 4-month PFS (at least 40%) among the 30 patients in an experimental cohort was to be considered promising; this would occur with 90% likelihood if the true 4-month PFS rate is 50% (median PFS of 4 months) and with 5% likelihood if the true 4-month PFS rate is 25% (median PFS of 2 months).
RESULTS
Enrollment and Treatment Assignment
One hundred ninety-eight patients who met study eligibility criteria were enrolled from January 2014 to April 2018 (Fig 1, Table 1, Data Supplement), 108 (55%) of whom underwent a tumor biopsy procedure and ultimately had a study-actionable mutation detected (Fig 2A). Ninety-six (89%) of the patients with an identified aMOI were randomly assigned to a treatment arm (Fig 1, Data Supplement); patients without aMOIs or with insufficient tumor or DNA were taken off study without treatment.
FIG 1.
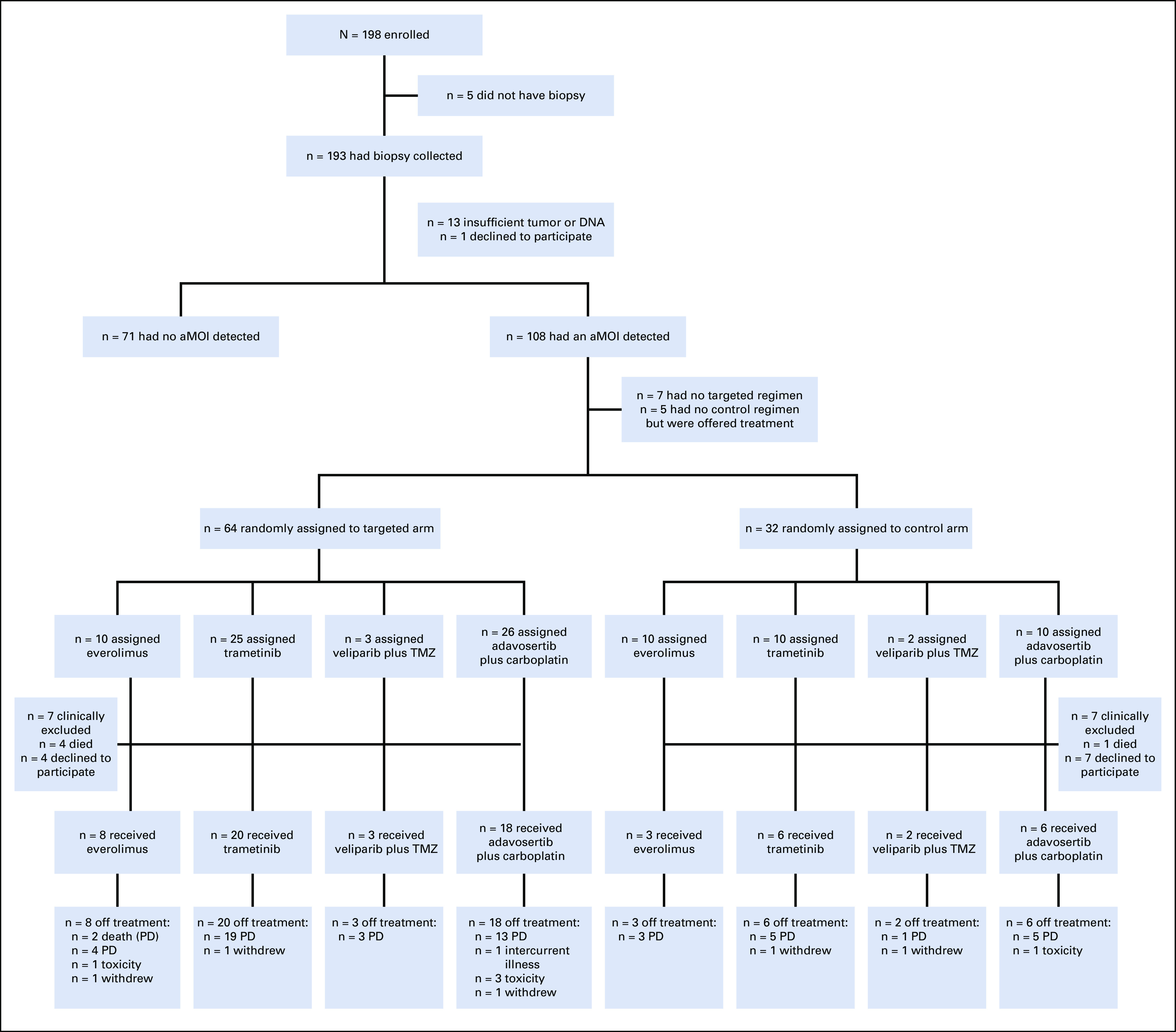
CONSORT diagram of the randomized portion of the NCI-MPACT study. Fifty percent of biopsied patients had an aMOI and were randomly assigned 2:1 to the experimental or control treatment arms, as outlined. Seven patients had an aMOI but could not be randomly assigned because they were ineligible for the targeted treatment (six patients: pancreatic cancer with an RAS mutation; one patient: unknown reason). Five patients had aMOIs in all three pathways and therefore had no control treatment available. These patients were not randomly assigned or considered evaluable for the study's primary end point, but were offered targeted treatment (Data Supplement). All 70 patients who initiated treatment have come off study. aMOI, actionable mutation of interest; PD, progressive disease; TMZ, temozolomide.
TABLE 1.
Patient Characteristics
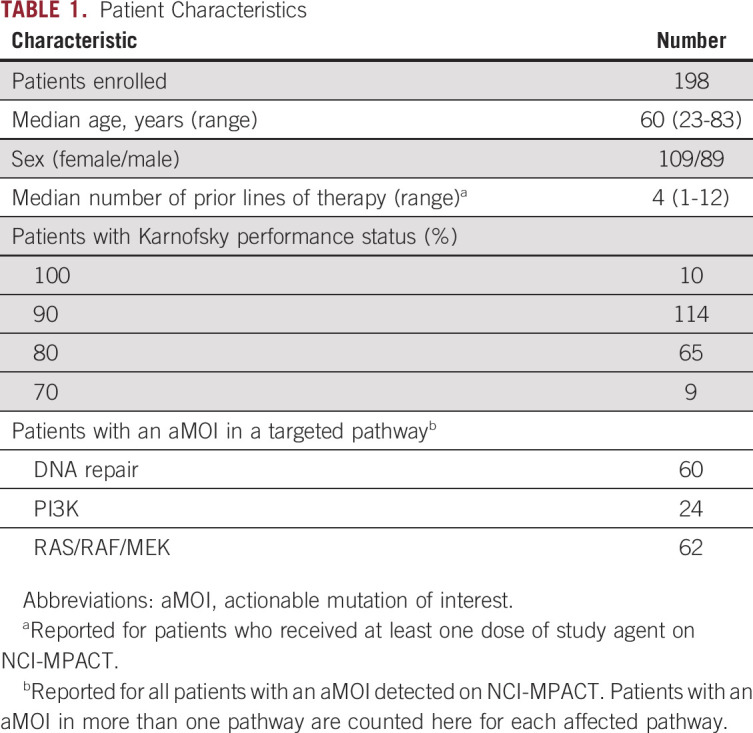
FIG 2.
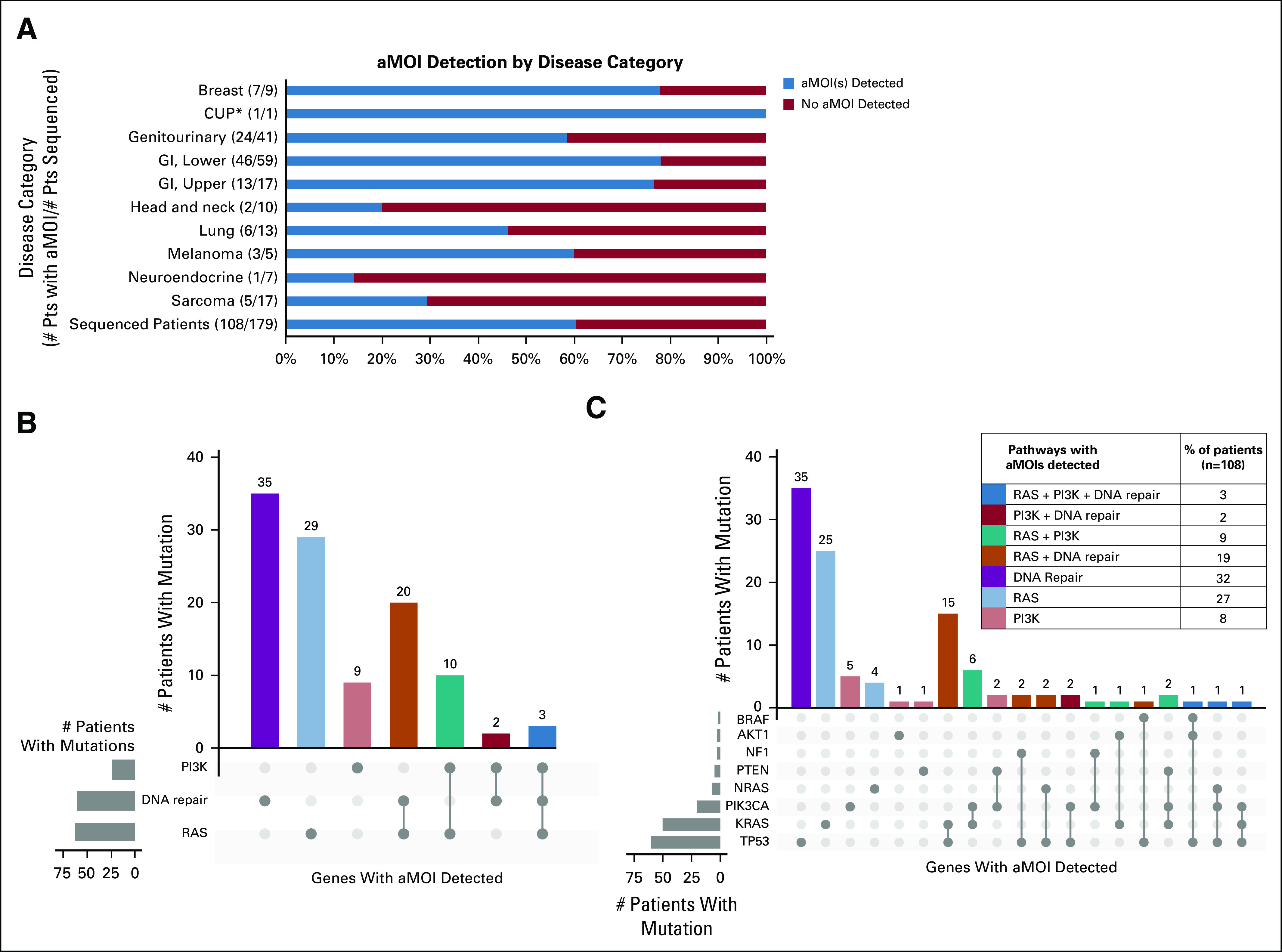
Prevalence of NCI-MPACT actionable mutations of interest. (A) Number and percentage of patients with NCI-MPACT sequencing results available for whom an aMOI was detected, presented by disease category. (B) Number of patients with an aMOI in the PI3K, DNA repair, and/or RAS pathway(s). aMOIs in the DNA-repair pathway were most common. Thirty-five patients had an aMOI in more than one pathway of interest. (C) Number and percentage of patients with an aMOI in the indicated gene(s). #, number; aMOI, actionable mutation of interest; CUP, cancer of unknown primary (*unknown primary adenocarcinoma); n, number of patients with ≥ 1 aMOI detected; Pts, patients.
The genes with highest frequency of study aMOIs were TP53 and KRAS (56% and 46% of patients with an aMOI, respectively) (Fig 2C, Data Supplement). All patients assigned to the two DNA repair pathway cohorts of the experimental arm had a TP53 mutation and 75% of patients assigned to the trametinib experimental cohort had a KRAS mutation. The majority of aMOIs were nonsynonymous single-nucleotide variants (Data Supplement).
Toxicity
Thirty (45%) of the 66 patients who received at least one dose of study agent(s) experienced an AE of grade 3 or greater that was considered at least possibly related to study treatment (Data Supplement). The toxicities reported were consistent with those previously observed for the study drugs. Five patients came off treatment because of toxicity. Seven patients died within 30 days of their last dose of study drugs but none of the on-study deaths were considered related or likely related to the study treatments.
Treatment Compliance
Interim analysis revealed unanticipated differences in compliance for patients who were assigned to targeted versus nontargeted therapy (Fig 3A). The overall percentage of patients assigned to the control arm who never initiated treatment was 47% (15/32), significantly higher than the rate for patients assigned to the experimental arm (23%; 15/64; P = .034, two-sided, by Fisher's exact test). Evaluation of the reasons why patients came off study, as documented in the clinical database, reveals that a significantly higher percentage of the patients randomly assigned to the control arm chose not to start treatment (22% [7/32]) compared with the percentage of patients in the experimental arm who chose not to start treatment (6% [4/64]; P = .038, two-sided, by Fisher's exact test).
FIG 3.
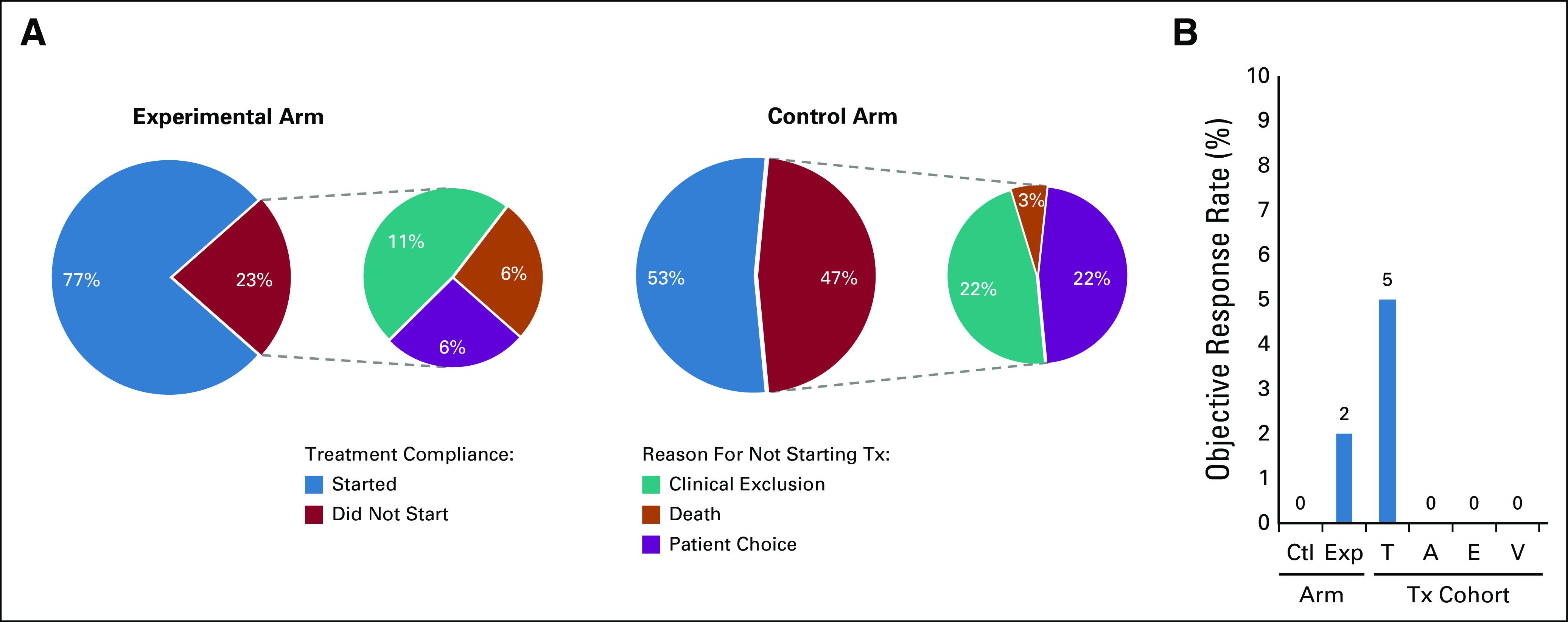
Interim futility analysis results. (A) NCI-MPACT treatment compliance rates: the percent of randomly assigned patients who initiated their assigned study treatment or did not start treatment because of death, clinical exclusion that developed after enrollment, or patient choice. (B) The proportions of randomly assigned and treated patients who experienced objective response (complete response or confirmed partial response) are presented by arm or targeted treatment cohort. A, adavosertib plus carboplatin; Ctl, control arm; E, everolimus; Exp, experimental targeted treatment arm; T, trametinib; Tx, treatment; V, veliparib.
Interim Futility Analysis
Accrual to the veliparib plus TMZ and everolimus cohorts was slow such that accrual did not reach the 12-patient threshold for interim analysis of the primary end point, objective response rate; no responses were measured on either regimen (Fig 3B). Slow accrual to the veliparib plus TMZ arm was because of the fact that every DNA repair pathway aMOI detected on study was within TP53; on the basis of preclinical evidence that loss-of-function mutations in TP53 affect regulation of cell cycle progression, patients who were randomly assigned to the experimental arm with these mutations were assigned adavosertib plus carboplatin as their first-line study treatment if eligible, not veliparib plus TMZ. Accrual to the adavosertib plus carboplatin and trametinib experimental cohorts exceeded the 12-patient analysis threshold because of inadvertent delays in reporting. There were no confirmed responses in the 18 treated patients in the adavosertib plus carboplatin experimental cohort, indicating futility. One (5%) of 20 patients in the experimental trametinib cohort had a confirmed partial response (PR), an outcome that did not support further accrual to that cohort. The other cohorts were also closed because of the lack of activity. All patients are now off study.
Clinical Outcomes
Seventeen randomly assigned patients were treated in the control arm, none of whom experienced an objective response (0%; 95% exact binomial CI, 0% to 19.5%). There was one objective response among the 49 randomly assigned patients treated in the experimental arm (2%; 95% exact binomial CI, 0% to 10.9%). The planned comparison of objective response rate and PFS between the control arm and the experimental arm is precluded by the premature arm closures and the high dropout rate of the control arm. The Kaplan-Meier estimate of 4-month PFS among the randomly assigned and treated patients was 38.1% (95% CI, 26.0% to 56.0%) in the experimental arm and 22.7% (95% CI, 7.7% to 67.1%) in the control arm (Figs 4A and 4B, respectively). PFS events were experienced by 12/17 randomly assigned and treated patients in the control arm; among the patients randomly assigned to the experimental cohorts, PFS events were documented in 6 of 8 treated with everolimus, 20 of 20 treated with trametinib, 13 of 18 treated with adavosertib plus carboplatin, and 3 of 3 treated with veliparib plus TMZ. Caution must be used in comparing the individual experimental cohorts to the control arm as the restriction of an experimental cohort to a particular target may be prognostic of better (or worse) PFS. The estimate of 4-month PFS for the experimental everolimus cohort (50%) was significantly higher than the protocol-specified standard of 25% (P = .045; 95% CI, 22.5% to 100.0%) (Fig 4C), as was the 45% estimated 4-month PFS for the experimental trametinib cohort (P = .01; 95% CI, 27.7% to 73.1%) (Fig 4D). The estimate of 4-month PFS for the experimental adavosertib plus carboplatin cohort (30.8%) was not found to differ significantly from the 25% standard (P = .32; 95% CI, 13.2% to 71.9%) (Fig 4E). None of the three patients in the experimental veliparib plus TMZ cohort were progression-free at 4 months (CI not provided because of small sample size).
FIG 4.

Progression-free survival (PFS). Kaplan-Meier estimates of PFS for randomized NCI-MPACT patients who received at least one dose of study treatment on the (A) experimental arm (eight patients censored); (B) control arm, regardless of agent (five patients censored); (C) everolimus experimental cohort (two patients censored); (D) trametinib experimental cohort (zero patients censored); and (E) adavosertib plus carboplatin experimental cohort (five patients censored).
Two patients experienced sustained stable disease (SD) for a noteworthy ≥24 cycles of targeted treatment (Fig 5). One patient with endometrial cancer and PIK3CA H1047L and PTEN R130* aMOIs received everolimus and experienced SD for 24 cycles before progressing. The other patient, a 52-year-old woman with low-grade ovarian cancer with an NRAS Q61R aMOI, experienced SD for 27 cycles of trametinib treatment before her disease progressed. A 79-year-old male patient with melanoma and an NRAS Q61R aMOI received six cycles of targeted trametinib and experienced a PR before progressing. In the experimental adavosertib plus carboplatin cohort, one patient with endometrial carcinoma and TP53 R213* and PIK3CA C420R aMOIs had an unconfirmed PR before coming off treatment because of toxicity. None of the patients who crossed over at disease progression from the control arm to a targeted treatment or from one targeted treatment to another experienced clinical benefit on the crossover regimen (Data Supplement).
FIG 5.

Clinical outcomes by NCI-MPACT aMOIs. Cycles of treatment, best response, and limited demographic information for each randomly assigned and treated patient. Each patient's detected NCI-MPACT aMOIs are listed and color-coded to indicate the level of evidence that the mutation is susceptible to the assigned NCI-MPACT treatment (based on the information in the OncoKB and CIViC precision oncology knowledge bases at the time of writing). Where available, the results of whole exome sequencing are presented as the number of genetic alterations detected that are annotated in OncoKB as either oncogenic (# oncogenic mutations) or as oncogenic and actionable with available therapeutic agents (# OncoKB mutations). aMOI, actionable mutation of interest; CUP, cancer of unknown primary; dx, diagnosis; Illness, intercurrent illness required patient come off study; MPACT, molecular profiling-based assignment of cancer therapy; NR, no response; PD, progressive disease; PR, partial response; Prior Tx, number of lines of prior therapy; SD, stable disease; TMZ, temozolomide; Toxicity, study agent toxicity required patient come off study; Tx, treatment; uPR, unconfirmed partial response.
DISCUSSION
Highly effective, tailored therapy targeting specific genetic aberrations is a primary goal of precision medicine.1,2 Reports of exceptional responders, retrospective studies, and several nonrandomized trials indicate clinical benefit for genome-driven treatments, prompting optimism and discussion about the appropriate evidence framework for precision oncology.3,8-24 The results of our study, which was designed in 2012, highlight several important considerations for randomized precision oncology studies in the advanced disease setting that have evolved since then.25-28 With the greater prevalence of genetic tests and emphasis on precision medicine, it may be very challenging to randomly assign patients to a nontargeted control arm. Our data suggest that some patients and physicians may have had prior tumor mutation profile knowledge and, when randomly assigned to the control arm, appeared to show a bias favoring the presumed precision medicine approach, declining to participate in the study. This suggestion was anecdotally confirmed by conversations with some of the study investigators. The low dropout rate on the similarly designed SHIVA trial may be explained by having the patients randomly assigned to a physician's choice control arm; results from the SHIVA trial also did not demonstrate the superiority of molecularly targeted agents over the control treatment.25 The results of the WINTHER and CoPPO trials, two nonrandomized studies, describe clinical benefit for a small number of patients with personalized treatment but the PFS-based end point was not met in either study.29,30 Our results are similar in that the two statistically evaluable experimental cohorts indicate that neither trametinib nor adavosertib plus carboplatin was more effective than standard therapy at achieving objective response when assigned to target NCI-MPACT-defined aMOIs in RAS/RAF/MEK or DNA repair pathways (eg, KRAS gain of function or TP53 loss of function mutations, respectively). Although presumed to be a superior, precision medicine approach, the lack of significant response on the experimental arm argues for need of better therapies and further validation. Extensive genetic sequencing and clinical trials evaluating broader aspects of pathway alterations may reveal additional information about actionable sensitivity markers.
The targeted treatment assignments in NCI-MPACT were made based on molecular aberrations that were expected to confer therapeutic sensitivity at the level of a signaling pathway. Since the trial's initiation, the cancer research community has been engaged in an ongoing effort to identify gene- and variant-specific biomarkers that predict response to drugs.31-37 Retrospective review of two precision oncology knowledge bases, OncoKB and CIViC, suggests that there is little curated evidence to support many of the aMOI-drug associations targeted in the NCI-MPACT trial, which is consistent with the lack of clinical activity. For example, TP53 mutations, which were detected in every NCI-MPACT patient randomly assigned to a targeted DNA repair inhibitor cohort, are understood to be oncogenic but have remained largely undruggable.38 The experimental trametinib cohort is the exception in that for each RAS/MEK/ERK pathway aMOI that was detected, there is evidence curated in OncoKB or CIViC suggesting effective inhibition by trametinib. Notably, all three patients who were treated with trametinib to target an NRAS Q61R mutation experienced clinical benefit, in line with published clinical evidence of MEK inhibitor activity in patients carrying an NRAS Q61 mutation.39 Most of the patients treated in the experimental trametinib arm carried an aMOI in KRAS. Although there is preclinical support for treating KRAS aMOIs with an MEK inhibitor, the results of that approach were modest in this study, perhaps because of reported mechanisms of KRAS mutant resistance to ATP-noncompetitive MEK inhibitors.40-42
Furthermore, the presence of a therapy-targeted aMOI in a patient tumor does not indicate whether the aMOI is a driver or passenger mutation.43,44 In NCI-MPACT, the most frequently detected aMOIs—those in TP53 and KRAS—were detected in all four treatment cohorts and might have contributed to disease progression in cases where treatment was assigned to target an aMOI in a different pathway (eg, AKT/PI3K/MTOR pathway). This may be one reason for the limited efficacy of targeted treatment in the everolimus cohort, where many patients had aMOIs in more than one signaling pathway. Treatment assignment in such cases was made based on the aMOI with the highest allele frequency but there is precedent for subclonal molecular alterations driving disease progression.45-48 Two of the three patients who tolerated everolimus treatment and had aMOIs confined to only the PI3K pathway experienced prolonged SD (≥ 10 cycles).
It is important to note that we were able to achieve a successful biopsy collection rate of > 90%.49 Even with a turnaround time of ≤ 10 days for sequencing results,5 19 biopsied patients (10%) progressed to the point of ineligibility or death before treatment initiation, reflecting the advanced disease status of this patient population. Biopsy sampling could not have identified spatially or temporally isolated tumor subclones that may have been driving tumor growth or conferring resistance in these patients,43,44,50,51 challenges that can be explored further in preclinical studies as can the presence of putative driver mutations in other signaling pathways.
We are completing the analysis of a preclinical study performed in parallel with the NCI-MPACT trial using xenograft models derived from the tumors of cancer patients to overcome the hurdles inherent to clinical investigations of molecularly targeted treatment response (manuscript in preparation). Our preliminary data suggest that functional in vivo evidence of activity in molecularly defined models should be the basis on which to evaluate clinical benefit of targeted agents in specified patient subgroups. Given the paradigm shift toward precision oncology, it is imperative that additional robust, comparative biomarker-targeted studies are conducted to identify new therapeutic options, inform personalized treatment decisions, and move the field forward.
ACKNOWLEDGMENT
The authors thank Naoko Takebe, Elad Sharon, Howard Streicher, Sheila Prindiville, and Barbara Conley (National Cancer Institute) for their clinical contributions, and Robin Harrington, Courtney Bouk, and Kneshay N. Harper (Molecular Characterization Laboratory, Frederick National Laboratory for Cancer Research) for their excellent technical assistance.
SUPPORT
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
AUTHOR CONTRIBUTIONS
Conception and design: Alice P. Chen, Shivaani Kummar, Lawrence V. Rubinstein, P. Mickey Williams, David Sims, Mel Simpson, Chih-Jian Lih, Eric Polley, Richard Simon, James H. Doroshow
Financial support: James H. Doroshow
Administrative support: P. Mickey Williams, Richard Piekarz, James H. Doroshow
Provision of study materials or patients: Shivaani Kummar, Geraldine O'Sullivan Coyne, Funda Meric-Bernstam, Stephen Leong, Chris Karlovich
Collection and assembly of data: Alice P. Chen, Shivaani Kummar, Nancy Moore, Yingdong Zhao, P. Mickey Williams, Alida Palmisano, David Sims, Christina L. Rosenberger, Mel Simpson, Kanwal P. S. Raghav, Funda Meric-Bernstam, Stephen Leong, Biswajit Das, Eric Polley, Richard Simon, James H. Doroshow
Data analysis and interpretation: Alice P. Chen, Shivaani Kummar, Nancy Moore, Lawrence V. Rubinstein, Yingdong Zhao, P. Mickey Williams, Alida Palmisano, David Sims, Geraldine O'Sullivan Coyne, Christina L. Rosenberger, Mel Simpson, Kanwal P. S. Raghav, Stephen Leong, Saiama Waqar, Jared C. Foster, Mariam M. Konaté, Biswajit Das, Chris Karlovich, Chih-Jian Lih, Eric Polley, Richard Simon, Ming-Chung Li, Richard Piekarz, James H. Doroshow
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Shivaani Kummar
Stock and Other Ownership Interests: PathomIQ, Arxeon
Consulting or Advisory Role: Corvus Pharmaceuticals, MedTree, Nodus Therapeutics, Genentech, ShangPharma Innovation, Seattle Genetics, Bayer, Boehringer Ingelheim, Mundipharma EDO GMBH, Harbour BioMed, Cadila Pharmaceuticals
Research Funding: Bristol Myers Squibb, Dynavax Technologies, Pfizer, Loxo, Corvus Pharmaceuticals, Plexxikon, Jounce Therapeutics, ADC Therapeutics, Advenchen Laboratories, Incyte, Taiho Pharmaceutical, Bayer, Astex Pharmaceuticals, Seattle Genetics, Amgen, Genome & Company
Travel, Accommodations, Expenses: Bayer
Nancy Moore
Patents, Royalties, Other Intellectual Property: Nestle Nutrition
P. Mickey Williams
Research Funding: Illumina
Patents, Royalties, Other Intellectual Property: I was a co-inventor of the DLBCL cell of origin patent recently filed by the NIH
Kanwal P. S. Raghav
Consulting or Advisory Role: AstraZeneca, Bayer, Eisai, Daiichi Sankyo
Funda Meric-Bernstam
Employment: MD Anderson Cancer Center
Honoraria: Mayo Clinic, Rutgers Cancer Institute of New Jersey
Consulting or Advisory Role: Genentech, Inflection Biosciences, Samsung Bioepis, Spectrum Pharmaceuticals, Aduro Biotech, OrigiMed, Xencor, Debiopharm Group, Mersana, Seattle Genetics, Silverback Therapeutics, Immunomedics, IBM, Roche, PACT Pharma, eFFECTOR Therapeutics, Jackson Laboratory for Genomic Medicine, Kolon Life Sciences, Parexel International, Pfizer, Tyra Biosciences, Zymeworks, Puma Biotechnology, Zentalis, Alkermes
Speakers' Bureau: Chugai Pharma
Research Funding: Novartis, AstraZeneca, Taiho Pharmaceutical, Genentech, Calithera Biosciences, Debiopharm Group, Bayer, Aileron Therapeutics, Puma Biotechnology, CytomX Therapeutics, Jounce Therapeutics, Zymeworks, Curis, Pfizer, eFFECTOR Therapeutics, Abbvie, Boehringer Ingelheim, Guardant Health, Daiichi Sankyo, GlaxoSmithKline, Seattle Genetics, Millennium
Travel, Accommodations, Expenses: Taiho Pharmaceutical, Beth Israel Deaconess Medical Center
Stephen Leong
Employment: Merck Sharp & Dohme
Stock and Other Ownership Interests: Antares Pharmaceuticals, Spectrum Pharmaceuticals
Consulting or Advisory Role: Bristol Myers Squibb
Research Funding: Deciphera, Karyopharm Therapeutics, Bristol Myers Squibb, Lilly
Travel, Accommodations, Expenses: Genentech/Roche
Saiama Waqar
Research Funding: Spectrum Pharmaceuticals, Lilly, Pfizer, Genentech/Roche, Daiichi Sankyo, Newlink Genetics, EMD Serono, Puma Biotechnology, Novartis, Xcovery, Synermore Biologics, Celgene, Vertex, Bristol Myers Squibb, Stemcentrx, Hengrui Therapeutics, Checkpoint Therapeutics, Ignyta, AstraZeneca, ARIAD, Roche, Merck
Biswajit Das
Research Funding: Illumina
Chris Karlovich
Stock and Other Ownership Interests: Clovis Oncology
Research Funding: Illumina
Travel, Accommodations, Expenses: Illumina
Chih-Jian Lih
Employment: Exelixis
Stock and Other Ownership Interests: Exelixis
Eric Polley
Research Funding: GRAIL
Richard Simon
Consulting or Advisory Role: AbbVie, Amgen, Janssen, Bristol Myers Squibb, Pfizer, Onco-Nano
Travel, Accommodations, Expenses: Amgen
No other potential conflicts of interest were reported.
REFERENCES
- 1.Hyman DM, Taylor BS, Baselga J.Implementing genome-driven oncology Cell 168584–5992017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Le Tourneau C, Borcoman E, Kamal M.Molecular profiling in precision medicine oncology Nat Med 25711–7122019 [DOI] [PubMed] [Google Scholar]
- 3.Senft D, Leiserson MDM, Ruppin E, et al. Precision oncology: The road ahead Trends Mol Med 23874–8982017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tate JG, Bamford S, Jubb HC, et al. COSMIC: The catalogue of somatic mutations in cancer Nucleic Acids Res 47D941–D9472019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lih C-J, Sims DJ, Harrington RD, et al. Analytical validation and application of a targeted next-generation sequencing mutation-detection assay for use in treatment assignment in the NCI-MPACT trial J Mol Diagn 1851–672016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao Y, Polley EC, Li M-C, et al. GeneMed: An informatics hub for the coordination of next-generation sequencing studies that support precision oncology clinical trials Cancer Inform 1445–552015. suppl 2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur J Cancer 45228–2472009 [DOI] [PubMed] [Google Scholar]
- 8.Ottmann OG, Druker BJ, Sawyers CL, et al. A phase 2 study of imatinib in patients with relapsed or refractory Philadelphia chromosome-positive acute lymphoid leukemias Blood 1001965–19712002 [DOI] [PubMed] [Google Scholar]
- 9.Sawyers CL, Hochhaus A, Feldman E, et al. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: Results of a phase II study Blood 993530–35392002 [DOI] [PubMed] [Google Scholar]
- 10.O'Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia N Engl J Med 348994–10042003 [DOI] [PubMed] [Google Scholar]
- 11.Perez EA, Romond EH, Suman VJ, et al. Four-year follow-up of trastuzumab plus adjuvant chemotherapy for operable human epidermal growth factor receptor 2-positive breast cancer: Joint analysis of data from NCCTG N9831 and NSABP B-31 J Clin Oncol 293366–33732011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perez EA, Romond EH, Suman VJ, et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2-positive breast cancer: Planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831 J Clin Oncol 323744–37522014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fukuoka M, Wu YL, Thongprasert S, et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS) J Clin Oncol 292866–28742011 [DOI] [PubMed] [Google Scholar]
- 14.Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial Lancet Oncol 13239–2462012 [DOI] [PubMed] [Google Scholar]
- 15.Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial Lancet 380358–3652012 [DOI] [PubMed] [Google Scholar]
- 16.Hyman DM, Puzanov I, Subbiah V, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations N Engl J Med 373726–7362015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haslem DS, Van Norman SB, Fulde G, et al. A retrospective analysis of precision medicine outcomes in patients with advanced cancer reveals improved progression-free survival without increased health care costs J Oncol Pract 13E108–E1192017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Massard C, Michiels S, Ferte C, et al. High-throughput genomics and clinical outcome in hard-to-treat advanced cancers: Results of the MOSCATO 01 trial Cancer Discov 7586–5952017 [DOI] [PubMed] [Google Scholar]
- 19.Hainsworth JD, Meric-Bernstam F, Swanton C, et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: Results from MyPathway, an open-label, phase IIa multiple basket study J Clin Oncol 36536–5422018 [DOI] [PubMed] [Google Scholar]
- 20.Prasad V, Fojo T, Brada M.Precision oncology: Origins, optimism, and potential Lancet Oncol 17e81–e862016 [DOI] [PubMed] [Google Scholar]
- 21.Saad ED, Paoletti X, Burzykowski T, et al. Precision medicine needs randomized clinical trials Nat Rev Clin Oncol 14317–3232017 [DOI] [PubMed] [Google Scholar]
- 22.Moscow JA, Fojo T, Schilsky RL.The evidence framework for precision cancer medicine Nat Rev Clin Oncol 15183–1922018 [DOI] [PubMed] [Google Scholar]
- 23.Sicklick JK, Kato S, Okamura R, et al. Molecular profiling of cancer patients enables personalized combination therapy: The I-PREDICT study Nat Med 25744–7502019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Freidlin B, Allegra CJ, Korn EL.Moving molecular profiling to routine clinical practice: A way forward? J Natl Cancer Inst 112773–7782019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le Tourneau C, Delord J-P, Gonçalves A, et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial Lancet Oncol 161324–13342015 [DOI] [PubMed] [Google Scholar]
- 26.Herbst RS, Gandara DR, Hirsch FR, et al. Lung master protocol (lung-MAP)-A biomarker-driven protocol for accelerating development of therapies for squamous cell lung cancer: SWOG S1400 Clin Cancer Res 211514–15242015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goncalves A, Bachelot T, Lusque A, et al. High-throughput genome analysis and therapeutic decision for patients with HER2-negative metastatic breast cancer: First feasibility and molecular results of the randomized phase II study SAFIR02 BREAST (UCBG-0105/1304) Cancer Res. 2017;77 (abstr PD1-08) [Google Scholar]
- 28.Kaplan R, Maughan T, Crook A, et al. Evaluating many treatments and biomarkers in oncology: A new design J Clin Oncol 314562–45682013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodon J, Soria JC, Berger R, et al. Genomic and transcriptomic profiling expands precision cancer medicine: The WINTHER trial Nat Med 25751–7582019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tuxen IV, Rohrberg KS, Oestrup O, et al. Copenhagen prospective personalized oncology (CoPPO)—Clinical utility of using molecular profiling to select patients to phase I trials Clin Cancer Res 251239–12472019 [DOI] [PubMed] [Google Scholar]
- 31.Chakravarty D, Gao JJ, Phillips S, et al. OncoKB: A precision oncology knowledge base. JCO Precis Oncol. 10.1200/PO.17.00011 [epub ahead of print on May 16, 2017] [DOI] [PMC free article] [PubMed]
- 32.Chen AP, Eljanne M, Harris L, et al. National Cancer Institute basket/umbrella clinical trials: MATCH, LungMAP, and beyond Cancer J 25272–2812019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Griffith M, Spies NC, Krysiak K, et al. CIViC is a community knowledgebase for expert crowdsourcing the clinical interpretation of variants in cancer Nat Genet 49170–1742017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li MM, Datto M, Duncavage EJ, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists J Mol Diagn 194–232017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mangat PK, Halabi S, Bruinooge SS, et al. Rationale and design of the targeted agent and profiling utilization registry (TAPUR) study. JCO Precis Oncol. 10.1200/PO.18.00122 [epub ahead of print on July 11, 2018] [DOI] [PMC free article] [PubMed]
- 36.Rugo HS, Olopade OI, DeMichele A, et al. Adaptive randomization of veliparib–carboplatin treatment in breast cancer N Engl J Med 37523–342016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients Nat Med 23703–7132017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sabapathy K, Lane DP.Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others Nat Rev Clin Oncol 1513–302018 [DOI] [PubMed] [Google Scholar]
- 39.Dummer R, Schadendorf D, Ascierto PA, et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial Lancet Oncol 18435–4452017 [DOI] [PubMed] [Google Scholar]
- 40.Dai X, Xia H, Zhou S, et al. Zoledronic acid enhances the efficacy of the MEK inhibitor trametinib in KRAS mutant cancers Cancer Lett 442202–2122019 [DOI] [PubMed] [Google Scholar]
- 41.Hatzivassiliou G, Haling JR, Chen H, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers Nature 501232–2362013 [DOI] [PubMed] [Google Scholar]
- 42.Sun C, Hobor S, Bertotti A, et al. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3 Cell Rep 786–932014 [DOI] [PubMed] [Google Scholar]
- 43.Tannock IF, Hickman JA.Limits to personalized cancer medicine N Engl J Med 3751289–12942016 [DOI] [PubMed] [Google Scholar]
- 44.McGranahan N, Swanton C.Biological and therapeutic impact of intratumor heterogeneity in cancer evolution Cancer Cell 2715–262015 [DOI] [PubMed] [Google Scholar]
- 45.Brady SW, McQuerry JA, Qiao Y, et al. Combating subclonal evolution of resistant cancer phenotypes. Nat Commun. 2017;8:1231. doi: 10.1038/s41467-017-01174-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Landau DA, Carter SL, Stojanov P, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia Cell 152714–7262013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rossi D, Khiabanian H, Spina V, et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia Blood 1232139–21472014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hong MKH, Macintyre G, Wedge DC, et al. Tracking the origins and drivers of subclonal metastatic expansion in prostate cancer. Nat Commun. 2015;6:6605. doi: 10.1038/ncomms7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferry-Galow KV, Datta V, Makhlouf HR, et al. What can be done to improve research biopsy quality in oncology clinical trials? J Oncol Pract 14e722–e7282018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmitt MW, Loeb LA, Salk JJ.The influence of subclonal resistance mutations on targeted cancer therapy Nat Rev Clin Oncol 13335–3472016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lipinski KA, Barber LJ, Davies MN, et al. Cancer evolution and the limits of predictability in precision cancer medicine Trends Cancer 249–632016 [DOI] [PMC free article] [PubMed] [Google Scholar]