

Abstract
Compounds combining dual inhibitory action against FAAH and cyclooxygenase (COX) may be potentially useful analgesics. Here, we describe a novel flurbiprofen analogue, N-(3-bromopyridin-2-yl)-2-(2-fluoro-(1,1'-biphenyl)-4-yl)propanamide (Flu-AM4). The compound is a competitive, reversible inhibitor of FAAH with a Ki value of 13 nM and which inhibits COX activity in a substrate-selective manner. Molecular modelling suggested that Flu-AM4 optimally fits a hydrophobic pocket in the ACB region of FAAH, and binds to COX-2 similarly to flurbiprofen. In vivo studies indicated that at a dose of 10 mg/kg, Flu-AM4 was active in models of prolonged (formalin) and neuropathic (chronic constriction injury) pain and reduced the spinal expression of iNOS, COX-2, and NFκB in the neuropathic model. Thus, the present study identifies Flu-AM4 as a dual-action FAAH/substrate-selective COX inhibitor with anti-inflammatory and analgesic activity in animal pain models. These findings underscore the potential usefulness of such dual-action compounds.
Keywords: Flurbiprofen amides, FAAH inhibition, fatty acid amide hydrolase, endocannabinoid, cyclooxygenase, non-steroidal anti-inflammatory drugs, hyperalgesia, allodynia
Graphical Abstract

Introduction
The endogenous cannabinoid system is involved in a number of physiological functions, including the regulation of pain1. Anandamide (AEA) and 2-arachidonoylglycerol (2-AG) are the two best-characterised endocannabinoids identified to date. They are synthesised on demand from membrane lipids, after which they are released and bind and activate the cannabinoid receptors (CB) that mediate multiple intracellular signal transduction pathways. Endocannabinoids are tightly regulated not only with respect to their biosynthesis but also with respect to their degradation. The two major hydrolytic enzymes responsible for the catabolism of AEA and 2-AG in the brain are fatty acid amide hydrolase (FAAH)2, and monoacylglycerol lipase (MAGL)3, respectively.
FAAH is a dimeric enzyme with a complex catalytic site characterised by three main interaction sites: the membrane access channel (MAC) where two charged residues favour the entrance of the polar head groups of fatty acid substrates, an adjacent “acyl-chain binding” cavity which likely contributes to the accommodation of substrates during catalysis, and a solvent-exposed “cytosolic port” (CP) which represents access to the cytosolic compartment of the cell. Once the substrate is bound to FAAH, the catalytic reaction occurs in the core of its binding site where an unusual catalytic triad (Ser241 − Ser217 − Lys142) performs the hydrolysis of the AEA. This occurs in two steps: firstly, a deprotonated Lys142 activates Ser241 through a proton exchange chain involving Ser217 leading to the protonation of Lys142. Then, the activated Ser241 attacks the substrate carbonyl group allowing the protonation of the leaving group ethanolamine which exits the enzyme through the CP4. Finally, a reverse proton transfer between Lys142 and Ser217 occurs, followed by a third proton transfer that re-establishes the original protonation state of the FAAH catalytic triad and leads to the formation of arachidonic acid.
Given the role of FAAH in the AEA hydrolysis, several classes of FAAH selective inhibitors, have been described in the scientific and patent literature5. FAAH inhibitors show efficacy in preclinical models of pain6,7, nausea8, and anxiety9,10 without the occurrence of tolerance11,12. However, FAAH inhibitors did not exhibit significant efficacy in several phase II clinical trials of osteoarthritis13, painful diabetic neuropathy14, pelvic pain and bladder dysfunction15 despite being well tolerated with little to no adverse events reported in each of these studies. One compound that was not well tolerated in man, however, was the irreversible FAAH inhibitor BIA10-2474, which left one person dead and four others with severe neurological symptoms16. However, these have been related to off-target effects due to the compound binds to a number of other serine hydrolases not exhibited by other FAAH inhibitors evaluated clinically13 and interactions with proteins other than FAAH are likely to mediate these extreme adverse events17. These clinical failures highlight the lack of translation between animal models and humans at least regarding the treatment of pain associated with inflammation. The real causes of the failure are not yet been identified, but it is reasonable that in a complex signalling pathway such as the endocannabinoid system, other metabolic enzymes and/or endocannabinoid targets may negate the effects of the FAAH inhibition.
The search for innovative strategies for pain management remains of key importance. In the literature, there are many examples of new potential analgesic molecules, which can selectively interact with known or new targets. Most of them are COX inhibitors18, TRPV antagonists19, or even FAAH inhibitors. Nevertheless, the usual approach of “one target-one drug” may not be the most optimal approach, which opens the way to the concept that molecules interacting with more targets that are possibly relevant for the same disease may show higher therapeutic effects and a safer biological profile. Therefore, in the last years, several studies have explored the dual targeting of FAAH and other related targets involved on signalling pathways of fatty acids and their derivatives, such as FAAH/MAGL inhibitors20, FAAH/TRPV1-receptor blockers21,22, FAAH/PGF2α-EA-receptor blockers23, and FAAH/COX-2 inhibitors24–29. In particular, the two latter strategies are based on the discovery that AEA and 2-AG are also COX-2 substrates that are converted to biologically active prostamides (prostaglandin ethanolamides) and prostaglandin glyceryl esters. Indeed, the prostamide F2α, produced by the cyclooxygenation of AEA, induces inflammatory and pro-algesic effects30. Therefore, dual blockade of FAAH and COX-2 could reduce prostamide biosynthesis while at the same time increasing AEA concentrations. In support of this hypothesis, it was seen in a key study that there was a synergistic interaction between the FAAH inhibitor URB597 and the non-steroidal anti-inflammatory drug (NSAID) diclofenac with respect to analgesia. Moreover, the co-administration also improved safety, whereby the FAAH inhibition lowered the incidence of ulceration produced by the NSAID31.
The development of dual FAAH/COX inhibitors started in 1997 when one of us found that a series of NSAID compounds inhibited anandamide deamidation albeit with a modest potency32,33. These findings, in particular for ibuprofen and flurbiprofen (1), led to the development of amide derivatives with increased FAAH potency while retaining COX-inhibitory potency. Among these, compounds deriving from the condensation of ibuprofen and flurbiprofen with 2-amino-3-methylpyridine (Ibu-AM5 and Flu-AM1) were identified as the most promising dual inhibitors. In particular, Ibu-AM5 was 2-3 orders of magnitude more potent than ibuprofen against FAAH24,34,35 albeit a weaker COX-1 and COX-2 than ibuprofen. Flu-AM1 was about 60-fold more potent against FAAH than flurbiprofen, and was a substrate selective inhibitor against COX-2 in that it inhibited the cyclooxygenation of 2-AG more potently than the cyclooxygenation of arachidonic acid (AA) by either form of COX24,26,34. Using molecular docking, molecular dynamics and evaluation of free energy using a QM/MM approach, we found that Ibu-AM5 and Flu-AM1 showed very similar binding modes in the same site located between acyl binding channel (ACB) and the MAC35.
Although these FAAH/COX compounds look very promising in vitro, the only in vivo data so far presented were with Ibu-AM5, which showed efficacy in a model of visceral pain without producing ulcerations21,22. An alternative approach, to combine the elements of flurbiprofen and the irreversible FAAH inhibitor URB597, led to a FAAH/COX dual inhibitor that was active in models of inflammation and also did not produce ulcerations36, suggesting that this profile is a class effect of FAAH/COX dual inhibitors. Finally, a hydrogen sulphide releasing analogue of ketoprofen with enhanced FAAH inhibitory properties compared to ketoprofen has recently been shown to produce analgesic effects without gastrointestinal damage37.
With respect to improving the potency of our compounds, we found that for Ibu-AM5, the replacement of the 2-amino-3-methylpyridine moiety with a 2-amino-3-halolpyridine caused an increased potency towards FAAH and a selective inhibition of COX-2 when 2-AG is used as substrate38,39. In particular the N-(3-bromopyridin-2-yl)-2-(4-isobutylphenyl)propanamide (Ibu-AM68) and 2-(6-chloro-9H-carbazol-2-yl)-N-(3-chloropyridin-2-yl)propanamide (Carpro-AM6) showed the best dual FAAH/COX-2 inhibition activity. In the present paper we report the design, synthesis, and the in vitro and in vivo evaluation of the pharmacological activity of a new series of halogenated flurbiprofen amide derivatives, retaining the COX inhibition profile of the leads, but with an improved FAAH inhibition profile.
Materials and methods
General methods
Anandamide [ethanolamine-1-3H] (specific activity 2.22 TBq mmol-1) was purchased from American Radiolabeled Chemicals, Inc (St.Louis, MO). Ovine COX-1 (cat. no. 60100), human recombinant COX-2 (cat. no. 60122), COX-2 polyclonal antibody (rabbit antimouse, cat #: 160106), arachidonic acid, 2-AG, AEA and URB597 (cyclohexylcarbamic acid 30-carbamoylbiphenyl-3-yl ester) were purchased from the Cayman Chemical Co. (Ann Arbour, MI, USA). Substrates were dissolved in ethanol or DMSO as appropriate. Polyclonal goat anti-rabbit immunoglobulin/HRP was obtained from Dako (Glostrup, Denmark). Protease inhibitor cocktail set III was obtained from Merck Millipore (Darmstadt, Germany). All commercially available solvents and reagents were used without further purification and were purchased from Sigma-Aldrich (Milan, Italy). 1H-NMR spectra were recorded on an Inova 500 spectrometer (Varian, Palo Alto, CA), 13 C-NMR spectra were recorded on a Bruker Advance III HD 600. The chemical shifts (δ) are reported in part per million downfield from tetramethylsilane (TMS), which was used as internal standard, and the spectra were recorded in hexadeuteriodimethylsulphoxide (DMSO-d6). Infra-red spectra were recorded on a Vector 22 spectrometer (Bruker, Bremen, Germany). The main bands are given in cm−1. Positive-ion electrospray ionisation (ESI) mass spectra were recorded on a double-focusing MAT 95 instrument (Finnigan, Waltham, MA) with BE geometry. Melting points (mp) were determined on a SMP1 Melting Point apparatus (Stuart Scientific, Stone, UK) and are uncorrected. All products reported showed spectra in agreement with the assigned structures. The purity of the tested compounds was determined by combustion elemental analyses conducted by the Microanalytical Laboratory, Department of Chemical and Pharmaceutical Sciences of the University of Ferrara with a MT-5 CHN recorder elemental analyser (Yanagimoto, Kyoto, Japan) and the values found were within 0.4% of theoretical values.
General procedure for synthesis of flurbiprofen amides
The solution of Flurbiprofen (1) (0.24 g, 1 mmol), EDC (0.19 g, 1.1 mmol) and HOBt (0.13 g, 1 mmol) in anhydrous MeCN (10 ml) was stirred at r.t., after 30 min the opportune amine (1 mmol). The mixture was stirred at r.t. for 72 h and the solvent was then removed under vacuum. The residue was dissolved in AcOEt (20 ml) and washed sequentially with brine (2 × 5 ml), 10% citric acid (2 × 5 ml), saturated NaHCO3 aqueous solution (2 × 5 ml) and water (2 × 5 ml). The organic layer was dried over anhydrous Na2SO4 and evaporated under vacuum to give the title amides.
2–(2-Fluoro-(1,1'-biphenyl)-4-yl)-N-(3-(trifluoromethyl)pyridin-2-yl)propanamide (Flu-AM3)
Obtained following the general procedure by the condensation between flurbiprofen and 2-amino-3-(trifluoromethyl)pyridine. Yield 45%. m.p. 110–112 °C. 1H NMR (DMSO-d6) δ 1.45 (d, J = 7.0 Hz, 3H, CH3), 3.99 (m, 1H, CH), 7.28–8.73 (m, 11H, Ar), 10.39 (s, 1H, NH). IR (Nujol) 3267, 1673, 1583, 1515 cm−1. m/z 389 (M + H)+. Elemental analysis: calculated for C21H16F4N2O (388.37)% C 64.95; H 4.15; N 7.21; found % C 65.00; H 4.14; N 7.17.
N-(3-Bromopyridin-2-yl)-2-(2-fluoro-(1,1'-biphenyl)-4-yl)propanamide (Flu-AM4)
Obtained following the general procedure by the condensation between flurbiprofen and 2-amino-3-bromopyridine. Yield 60%. m.p. 89–90 °C. 1H NMR (DMSO-d6) δ 1.45 (d, J = 7.0 Hz, 3H, CH3), 3.99 (q, J = 7.0 Hz, 1H, CH), 7.23-8.42 (m, 11H, Ar), 10.35 (s, 1H, NH). 13 C NMR (DMSO-d6) δ 18.4, 44.7, 115.2, 115.3, 115.4, 117.6, 124.2 (2 C), 128.0 (2 C), 128.8, 128.9 (2 C), 142.2, 143.2, 143.3, 147.7, 158.3, 159.9, 175.2. IR (Nujol) 3265, 1667, 1511 cm−1. m/z 399, 401(M + H)+. Elemental analysis: calculated for C20H16BrFN2O (398.26)% C 60.17; H 4.04; N 7.02; found % C 60.25; H 4.06; N 6.99.
N-(3-Chloropyridin-2-yl)-2-(2-fluoro-(1,1'-biphenyl)-4-yl)propanamide (Flu-AM6)
Obtained following the general procedure by the condensation between flurbiprofen and 2-amino-3-chloropyridine. Yield 60%. m.p. 61–63 °C. 1H NMR (DMSO-d6) δ 1.48 (d, J = 7.0 Hz, 3H, CH3), 4.01 (q, J = 7.0 Hz, 1H, CH), 7.30–7.56 (m, 9H, Ar), 7.98 (d, J = 8.0 Hz, 1H, Ar), 8.40 (m, 1H, Ar), 10.41 (s, 1H, NH). 13 C NMR (DMSO-d6) δ 18.4, 44.6, 115.1, 115.3, 123.3, 124.2 (2 C), 127.9 (2 C), 128.7, 128.8 (2 C), 130.6, 130.7, 138.9, 147.0, 148.0, 158.1, 159.8, 171.9. IR (Nujol) 3280, 1671, 1578 cm−1. m/z 327, 329 (M + H)+. Elemental analysis: calculated for C20H16ClFN2O (326.75)% C 67.70; H 4.55; N 7.90; found % C 67.63; H 4.54; N 7.93.
Computational methods
QM optimisation and electrostatic potential
Quantum mechanics (QM) calculations have been used to evaluate the strength of interaction between the ligands (FluAM-1 and FluAM-4) and the rat FAAH (rFAAH) Phe381 sidechain, and to analyse the electrostatic potential surface (ESP) of the compounds. The starting conformation was retrieved from the published FluAM-1 binding mode35. The Phe381 and fluoro-biphenyl moieties of the ligand were fixed during QM optimisation to adjust the geometry of the complex ligand:Phe381 using Gaussian software40 at B3LYP/6–311 + G(d,p) level of theory. Interaction energy (ΔE) between a ligand (L) and the Phe381 (P) was calculated as:
where ELP, EL and EP are the energies of the ligand:Phe381 complex, the ligand alone and the Phe381 alone, respectively.
Electrostatic potential maps were generated on the optimised conformations of the compounds. ESP surfaces were represented in pymol 2.3.241 using 0.004 isodensity surface and consequently the surfaces were mapped by colour where the ESP varies from red (the most negative) to blue (the most positive).
FAAH/COXs receptors and ligands preparation
The crystal structure of rFAAH was downloaded from the Protein Data Bank (PDB ID: 3QK542). For the COXs isoforms, the ovine COX-1 PDB model 3N8Z43 and the mouse COX-2 PDB model 3RR344 were selected. The presence of the non-selective NSAID Flurbiprofen as co-crystallized ligand was used as main selection criterion of the two isoforms. However, the human COX-2 complexed with flurbiprofen was not found, thus the mouse COX-2 was chosen as isoform-2 target, given its 87% identity with human COX-2. Both monomers A and B of rFAAH and COXs were treated with the Protein Preparation Wizard tool implemented in Maestro ver. 11.1245, in order to add all the hydrogen atoms and assign the correct bond orders. Subsequently, the co-crystallized ligands (QK5 and Flurbiprofen) and water molecules were removed. Residue Lys142 of rFAAH was considered in its deprotonated form, according to the proposed catalytic mechanism46. The 3D structure of Flu-AM3-6 amides described above were built using the Graphical User Graphical User Interface (GUI) of Maestro ver. 11.12. Their protonation state at pH 7.4 in water has been calculated using the Epik module. Finally, each compound was then subjected to a minimisation protocol as described in our previous work38.
Molecular docking
To investigate in detail the recognition of the Flurbiprofen amide halogenated derivatives Flu-AM3-6 to rFAAH and both COXs isoforms, docking simulations were carried out with the Glide software package, using the Standard Precision (SP) algorithm of the GlideScore function and the OPLS 2005 force field47. The docking binding site has been defined by means of a grid box of 29 × 29 × 29 Å centred in the ligand-binding cavity of rFAAH, and of 15 × 15 × 15 Å centred to cover the cyclooxygenase site of both COX-1 and COX-2. In addition, given the presence of halogen atoms in the designed ligands that could act both as halogen bond donors (between the σ-hole and a Lewis-base acceptor in the receptor) and hydrogen bond acceptors (between the lone pairs and an H-atom donor in the receptor), three different Glide protocols were adopted in the grid generation: i) default Glide parameters; ii) halogens treated as halogen bond donors; ii) halogens treated as hydrogen bond acceptors. A total amount of 200 ligand poses was generated and the conformational sampling of the ligand was enhanced by two times, as reported by the default setting of Glide. Docking conformations of Flu-AM3-6 were then clusterized based on their RMSD cut-off of 2 Å.
Molecular dynamics (MD)
The poses 1 and 2 of the most interesting compound Flu-AM4 retrieved by the docking procedure were submitted to 500 ns of MD simulation. The same docking pose of Flu-AM4 obtained in monomer A was loaded also in monomer B, then each monomer has been treated separately for the MD analysis. MD was performed with the AMBER16 software48. Each complex was immersed in a pre-equilibrated octahedral box of TIP3P water molecules, and then the system was neutralised. The final systems contained about 106,000 atoms. All simulations were performed with the Amber ff14SB force field49 for the protein, while ligand charges were obtained by computing the ESP by means of the Gaussian09 package40 using a B3LYP/6–31G* DFT level of theory, and then the restrained electrostatic potential (RESP) charges50 were obtained by a two-stage fitting procedure using Antechamber51 and the General Amber Force Field (GAFF) force field52. The geometry of the two systems was minimised using convergence criterion for the energy gradient set to 0.01 kcal (mol.Å)−1 in three steps, which include: (i) minimisation of hydrogen atoms in the system (5000 steps of steepest descent and 10000 steps of conjugate gradient); (ii) minimisation of hydrogen atoms, water molecules and counterions (2000 steps of steepest descent and 18,000 steps of conjugate gradient); finally (iii) the whole system (2000 steps of steepest descent and 18,000 steps of conjugate gradient). Thermal and pressure equilibrations of the systems were performed in three steps: 200 ps increasing the temperature from 0 to 298 K restraining the protein backbone and ligand; then, 800 ps in which the restraining force was gradually decreased and, finally, 200 ps were performed in order to equilibrate the system density at constant pressure (1 bar), constant temperature (298 K). Production runs of 500 ns were then performed under NPT conditions and a time step of 2fs. Trajectories and data were processed and analysed using the cpptraj module53 and the Visual Molecular Dynamics (VMD) graphics54. Combined clusterization analysis was carried out with the DBSCAN algorithm55. All figures were rendered using PyMOL (http://www.pymol.org).
Pharmacology
FAAH assay
Ethical permission for the in vitro animal experiments was obtained from the Umeå Ethical Committee for Animal Research, Umeå, Sweden (n° A67-09). Brains (minus cerebella) from adult Wistar or Sprague-Dawley rats (killed by decapitation) or from male B6CBAF1/J mice (killed by cervical dislocation), stored at −80 °C, were thawed, weighed and homogenised in cold 20 mM HEPES, 1 mM MgCl2, pH 7.0 and thereafter centrifuged at 35000 x g for 20 min at 4 °C. Homogenates were washed (by centrifugation followed by resuspension in the buffer) twice and incubated at 37 °C for 15 min in order to hydrolyse all endogenous FAAH substrates. After a further centrifugation, pellets were resuspended in 50 mM Tris-HCl buffer, pH 7.4, containing 1 mM EDTA and 3 mM MgCl2, and frozen at −80 °C in aliquots until used for the assay. For the FAAH assay56 test compounds, homogenates (usually 0.5–0.8 µg protein per assay, diluted with 10 mM Tris-HCl, 1 mM EDTA pH 7.4) and 25 µL of [3H]AEA in 10 mM Tris- HCl, 1 mM EDTA, pH 7.4, containing 1% w/v fatty acid-free bovine serum albumin, final substrate concentration of 0.5 µM) were incubated for 10 min at 37 °C (final assay volume 200 µL). Reactions were stopped by placing the tubes on ice. Final assay concentrations of the solvents used for the compounds (ethanol or DMSO) were in the range 1–5%. Activated charcoal (80 µL + 320 µL 0.5 M HCl) was added and the samples were mixed and left at room temperature for about 30 min. Following centrifugation at 2000 g for 10 min, aliquots (200 µL) of the supernatants were analysed for tritium content by liquid scintillation spectroscopy with quench correction. Blank values were obtained by the use of buffer rather than homogenate. In general, FAAH assays upon three homogenates were undertaken using separate inhibitor dilution series (from a stock solution), with 6 concentrations of inhibitor in half-log concentrations (i.e. 1, 3, 10 µM etc) ranging from 0.3–100 µM. Data were expressed as % of vehicle control and analysed using the algorithm log(inhibitor) vs. response - variable slope (four parameters) built into the GraphPad Prism computer program for the Macintosh (GraphPad Software Inc., San Diego, CA). Two different curve fits were chosen: one where the top (uninhibited) value was set to 100 and the bottom (maximum inhibition) was set to 0, and one where the top was set to 100 and the bottom allowed to float. The best model (when the bottom value returned >0) was chosen by Akaike’s informative criteria. For the compounds here, the simpler model was always returned. Since the program uses log10 inhibitor concentrations, the IC50 values for the inhibitable fraction are derived from the corresponding -log10(IC50) (pI50) values. Hence the SE values are for the pI50 values rather than the IC50 values. In consequence, we report both pI50 and IC50 values to indicate the SE values. With this assay, the selective FAAH Inhibitor URB597 completely blocked the hydrolysis of 0.5 µM [3H]AEA by rat brain homogenates at a concentration of 100 nM and caused 98% inhibition at 10 nM following a preincubation of 60 min between inhibitor and substrate, indicating that the assay as used here is specific for FAAH57. For the kinetic experiments, the data were analysed using algorithms for competitive, mixed-type or non-competitive inhibition in the GraphPad Prism program, and the best fit determined using Akaike's Informative Criteria.
COX-1 and 2 inhibition experiments
The assay was performed according to Meade et al.58 with minor modifications27. An oxygen electrode chamber with integral stirring (Oxygraph System, Hansatech Instruments, King’s Lynn, U.K.) was calibrated daily to ambient temperature and air pressure. The assay buffer contained 100 mM Tris-HCl buffer pH 7.4, 1 µM haematin, 2 mM phenol, 5 mM EDTA, 10 µM substrate (arachidonic acid or 2-AG) (final assay volume was 2 ml). After addition of Flu-AM4 dissolved in ethanol (final assay concentration 1%), a baseline was established for 5 min before initiation of reaction by addition of 200 units of either ovine COX-1 or human recombinant COX-2. The change in oxygen consumption as a measurement of enzyme activity was monitored for approximately 5 min.
In vivo pharmacological activity
Animals
The behavioural experiments were performed on CD1 male mice (25–30 g Charles Rivers-Italy). They were housed in the animal care facility of the Department Experimental of Pharmacology, University of Naples. The animals were acclimated to their environment for 1 week and had ad libitum access to standard rodent chow pellets VRF1 (purchased from Special Diet Service-SDS). All behavioural tests were performed between 9:00 AM and 13:00 PM, and the animals were only used once. Procedures involving animals and their care were conducted in conformity with international and national law and policies (EU Directive 2010/63/EU for animal experiments). The procedures reported here were approved by the Institutional Committee on the Ethics of Animal Experiments (CSV) of the University of Naples “Federico II” and by the “Ministero della Salute”, protocol no. 2014–0084607. At the end of all experiments for all pain models, the animals were euthanized by CO2 overdose. As suggested by the animal welfare protocol, all efforts were made to minimise animal suffering and to use only the number of animals necessary to produce reliable scientific data.
Drug treatments
During this study, Flu-AM4 was administered systemically (intraperitoneally) and locally (intraplantar 100 microg/paw). For systemic administration, the compound was dissolved in PEG400 20% v/v, TWEEN 80 5% (v/v) and sterile saline and used at different doses (1–3–10 mg/kg). Vehicle group was only treated with PEG, TWEEN and sterile saline. For intraplantar administration Flu-AM4 was dissolved in PEG400 10% v/v, TWEEN 80 5% (v/v) and formalin 5% in sterile saline.
Formalin-evoked hind-paw licking
Mice received intraplantar injections of formalin in saline (10%,vol/vol; 10 µl/paw). Paw licking was monitored by an observer blind to the experimental treatment for periods of 0–15 min (early phase) and 15–60 min (late phase) immediately after formalin administration. Flu-AM4 was administrated both by local injection (100 µg) and systemic administration (10 mg/kg).
Carrageenan-induced paw eodema
Paw edoema was induced by a subplantar injection of 0.1 ml of sterile saline containing 1% λ-carrageenan (Sigma Aldrich) into the right hind paw. Paw volumes were measured by a plethysmometer (Ugo Basile, Italy) at different times after the paw injection. The increase in paw volume was evaluated as the difference between the paw volume measured (Vm) at each time point (1, 2, 4, 6 and 48 h post carrageenan) and the basal paw volume (Vb) measured immediately before carrageenan injection (Δ volume = Vm – Vb).
Mechanical hyperalgesia
Paw withdrawal threshold (PWT) to mechanical pressure was measured with a Randall-Selitto Analgesy-Meter for mice (Ugo Basile, Italy). PWT was measured on the ispsilateral paw at 1, 2, 4, 6 and 48 h post carrageenan injection. Each paw was tested twice per session. Cut-off force was set at 100 g.
Chronic constriction injury (CCI) model of neuropathic pain
Neuropathic pain was induced by ligation of the sciatic nerve as described previously59. Briefly, mice (n = 6 each group) were first anaesthetised with xylazine (10 mg/kg i.p.) and ketamine (100 mg/kg i.p.), and the left thigh was shaved and scrubbed with betadine, then a small incision in the middle left thigh (2 cm in length) was performed to expose the sciatic nerve. The nerve was loosely ligated at two distinct sites (spaced at a 2-mm interval) around the entire diameter of the nerve using silk sutures (7–0). The surgical area was closed and finally scrubbed with betadine. At day 7 and 14 Mechanical Allodynia (von Frey test) and Mechanical hyperalgesia (Randall-Selitto test) were assessed. In sham-operated animals, the nerve was exposed but not ligated.
Mechanical allodynia
To assess for changes in sensation or in the development of mechanical allodynia, sensitivity to tactile stimulation was measured using the Dynamic Plantar Aesthesiometer (DPA, Ugo Basile, Italy). Ligated animals were placed in a chamber with a mesh metal floor covered by a plastic dome that enabled the animal to walk freely, but not to jump. The mechanical stimulus was then delivered in the mid-plantar skin of the hind paw. The cut-off was fixed at 5 g, while the increasing force rate (ramp duration) was settled at 20 s. The DPA automatically records the force at which the foot is withdrawn and the withdrawal latency. Each paw was tested twice per session. This test did not require any special pre-training, just an acclimation period to the environment and testing procedure. Testing was performed on the ispsilateral (ligated) paw at day 7 and 14 after ligation, 1 h post dose.
Statistical analysis
In vivo data are presented as mean ± SEM. In CCI experiments, DPA data are presented as paw withdrawal threshold (g), cold allodynia data are presented as number of paw withdrawal. The significance of differences between groups was determined by one-way repeated measurements ANOVA followed by post hoc Bonferroni's test.
Results and discussion
Design
On the basis of our previous work on Flu-AM1, we have designed a novel series of Flu-AM1 derivatives where the pyridine methyl is substituted by halogen atoms. The most stable binding mode of Flu-AM1 obtained in the QM/MM calculation showed the methyl group of the methyl-pyridine moiety located into a hydrophobic pocket defined by the Met436, Leu404 and Phe381. Taking advantage of these finding, we computationally validated our design exploring the possibility of enhancing this interaction by substituting the methyl group with halogens, in order to exploit halogen bonding of the σ-hole of the halogen with the aromatic ring of Phe381. To evaluate this possibility, we transformed Flu-AM1 into Flu-AM4 and performed an DFT geometry optimisation with the program Gaussian at the B3LYP/6–311 + G(d,p) level of the conformation of the ligand. To this aim, we defined a simplified system containing only the ligand and Phe381 and performed a DFT optimisation to evaluate energetic differences in the CH-π interaction when transforming methyl to bromine. The results (Supporting information Figure S1 and Table S1) clearly showed the greater tendency of bromine-derivative than methyl derivative to interact with Phe381.
Synthesis
The target amides of flurbiprofen with halogenated pyridinamines were synthesised according to the Scheme 1. The amides Flu-AM 3, 4, 6 were obtained by coupling the flurbiprofen (1) with 2-amino-3-halopyridines in the presence of 1–(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) and 1-hydroxybenzotriazole hydrate (HOBt) in dry acetonitrile (MeCN) solution. This synthetic pathway was found to be clean and high yielding.
Scheme 1.

Synthesis of flurbiprofen amides: (i) EDCI, HOBt, MeCN, r.t. 72 h.
FAAH inhibition
The obtained halogenated amides (Flu-AM3, 4 and 6), along with the reference compound Flu-AM1 were evaluated for their ability to inhibit FAAH. The inhibition assays were performed using 0.5 µM [3H]AEA as substrate and rat or mouse brain homogenates as the enzyme source. The results of these primary assays are shown in Table 1 and Figure 1(A). The results revealed that the introduction of a halogenated moiety at 3-position of pyridine ring resulted in an increased ability to inhibit FAAH as compared to the reference Flu-AM1. All the new amides showed enhanced activity as compared with the reference. The replacement of the methyl on the pyridine ring of Flu-AM1 with a trifluoromethyl group led to about 4-fold increase in activity (Flu-AM3 IC50 0.11 µM). The introduction in the same position of chlorine or bromine atom produced further increase in activity being amides Flu-AM6 and Flu-AM4 with nanomolar potency (IC50 19 and 21 nM respectively, i.e. more than three orders of magnitude greater than flurbiprofen) and approximately 20-fold more potent than Flu-AM1.
Table 1.
pI50 (-logIC50) and the derived IC50 values for inhibition of rat brain AEA hydrolysis by flurbiprofen amides. In all cases, the maximum inhibition was 100%.
| Compound | pI50 | IC50 (µM) | |
|---|---|---|---|
| Flurbiprofena | 4.53 ± 0.02 | 29 | |
| Flu-AM1a | 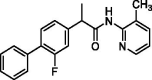 |
6.36 ± 0.02 | 0.44 |
| Flu-AM3 | 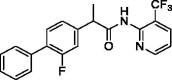 |
6.96 ± 0.12 | 0.11 |
| Flu-AM4 | 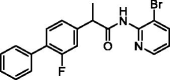 |
7.67 ± 0.10 | 0.021 |
| Flu-AM6 | 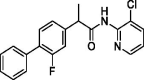 |
7.72 ± 0.07 | 0.019 |
aThe data for flurbiprofen and Flu-AM1, shown for comparative purposes, are from Ref. 26. Data are means ± s.e.m., n = 3–4.
Figure 1.

Mode of inhibition of FAAH by Flu-AM4 showing a reversible and competitive mode of FAAH inhibition. Inhibition of rat (unless otherwise shown) brain [3H]AEA hydrolysis by Flu-AM4. Panel A shows the inhibition curves for both rat and mouse brain homogenates from which the IC50 values were derived. In Panel B, rat brain homogenates were preincubated for the times shown prior to addition of substrate and incubation for a further 10 min. Panel C shows the kinetics of the inhibition of rat brain [3H]AEA hydrolysis by Flu-AM4. The data were better fitted by a model assuming competitive inhibition (Ki value 0.013 µM) than by a mixed model inhibition. All data are means ± s.e.m. (unless hidden by the symbols) n = 3–4. In Panel D, rat brain homogenates (at 20-fold normal strength) were preincubated with either vehicle, 0.1, 0.2 or 0.3 µM Flu-AM4 for 60 min. Aliquots were then diluted 20-fold and assayed for FAAH activity. These are shown as 0.1 → 0.005, 0.2 → 0.01 and 0.3 → 0.015. Concomitantly, Flu-AM4 was added to vehicle-preincubated aliquots to give concentrations of 0.005, 0.01 and 0.015 µM (representing free concentrations after a 20-fold dilution), 0.1, 0.2 and 0.3 µM final concentrations. The panel shows the data as % of corresponding control. For a fully reversible inhibitor, the preincubated + diluted samples should match the corresponding “diluted” rather than “concentrated” concentrations added after the preincubation phase (i.e. the 0.1 → 0.005 samples should match the 0.005 and not the 0.1 µM samples). Data are means ± s.e.m. (unless hidden within the symbols), n = 3–4.
Mode of inhibition of FAAH by Flu-AM4
The mode of inhibition of FAAH by Flu-AM4 is shown in Figure 1. The compound acted as a reversible inhibitor of rat brain FAAH (no time-dependent inhibition, loss of inhibition following dilution of the enzyme-inhibitor complex prior to addition of substrate). With respect to kinetics of inhibition, the Flu-AM4 inhibition data were better fitted by a model assuming competitive inhibition (Ki value 13 nM) than by a mixed-model inhibition. The compound was, however, an order of magnitude more potent towards rat brain FAAH than mouse brain FAAH (pI50 6.37 ± 0.10, IC50 value 430 nM). This is consistent with our previous work with Ibu-AM5 and Flu-AM135 and is presumably related to species differences of the amino acid compositions in the key binding sites of the enzymes. Species differences have also been reported for other types of FAAH inhibitor60,61. Nonetheless, Flu-AM1 has sub-micromolar potency in the mouse, which is of relevance for in vivo studies.
COX inhibitory activity of Flu-AM4
Given the ability of Ibu-AM5 and Flu-AM1 to inhibit COX24,26, we investigated the ability of Flu-AM4 to inhibit COX-1 and COX-2 (Figure 2). Two substrates were investigated, arachidonic acid (AA, for both COX isoforms) and 2-AG (for COX-2; COX-1 does not catalyse the cyclooxygenation of this compound). At a concentration of 10 µM, Flu-AM4 completely inhibited the cyclooxygenation of 2-AG by COX-2, while only partially inhibiting the cyclooxygenation of AA by COX-1 (2-AG is not a substrate of COX-1). This pattern of substrate selectivity is similar to that seen with flurbiprofen and Flu-AM123.
Figure 2.

Inhibition of COX-1 and COX-2 by Flu-AM4. Inhibition of the activities of ovine COX-1 (towards arachidonic acid, AA) and human recombinant COX-2 (towards either arachidonic acid or 2-AG) by Flu-AM4. Shown are means ± s.e.m., n = 3 for the change in oxygen tension following addition of enzyme in the presence of the concentrations of the compounds (µM) shown on the right of each panel. The enzyme isoform and substrate (10 μM concentration) used are shown above each panel. “F” in Panel A refers to 10 µM flurbiprofen, used as a positive control for COX-1.
On the basis of enzymatic assays, Flu-AM4 was considered the most interesting derivative and investigated in vivo.
Molecular modelling: FAAH inhibition
To study the effect of the substitution of the methyl group of Flu-AM1 with halogens we performed a molecular docking investigation of the compounds Flu-AM3-6 in monomer A of the crystal structure of rat FAAH (rFAAH) (PDB ID: 3QK5), with the Standard Precision (SP) algorithm of the Glide program (Schrödinger Inc.) by using three different protocols: i) default parameters ii) halogens treated as electron acceptors (halogen bond) and iii) halogens treated as hydrogen bond acceptors. The results of all the protocols, ranked together in both terms of Glide SP score and cluster population, clearly indicated as favoured poses those showing the biphenyl moiety pointing towards the catalytic triad and the pyridine ring entering the MAC (Supporting information Text S1.1, Table S2 and Figure S2), in a very similar orientation as found for Flu-AM1 obtained after Molecular Dynamics (MD) refinement35.
Considering Flu-AM4 as the most promising compound of the series, we refined the two best docking poses of Flu-AM4 through 500 ns of classical MD simulations. The MD simulations were carried out on the rFAAH homodimer by loading the same docking pose of Flu-AM4 obtained in both monomers A. MD analysis was performed considering the simulations of the two monomers as independent of each other, according to previous reports62. Therefore, four independent MD trajectories were analysed, two for each pose, for a total MD simulation time of 2 µs. The RMSD analysis of Flu-AM4 revealed stable trajectories with small fluctuation within 1 Å of the ligand after an initial adjustment within the hydrophobic ACB channel (Supplementary Figure S3). The combined clusterization of the four trajectories, performed with the Density-Based Spatial Clustering of Applications with Noise (DBSCAN) algorithm55 showed that both docking poses converged to the same binding mode, represented by a main cluster accounting for 70% of the overall population (Table 2).
Table 2.
Combined cluster analysis of the MD simulation of pose 1 and pose 2 in monomers A and B.
| Cluster | Population% | Population(%) Pose1_MonoA | Population(%) Pose1_MonoB | Population(%) Pose2_MonoA | Population(%) Pose2_MonoB |
|---|---|---|---|---|---|
| c0 | 70% | 66% | 19% | 100% | 95% |
| c1 | 16% | 0% | 64% | 0% | 0% |
| c2 | 12% | 31% | 15% | 0% | 0% |
The minimised representative structure of the best MD cluster (Figure 3(A)) showed an L-shaped conformation of the ligand and a binding mode mainly based on hydrophobic and aromatic interactions. The terminal phenyl ring fitted a hydrophobic pocket lined by Ile238, Val276, Ala377, Leu380, and Val495, with the central fluorine-substituted phenyl ring resulted stacked between two T-shaped π-π interactions, with Phe194 and Phe381, and flanked by Leu192 and Ile491. The bromo-pyridine ring showed hydrophobic contacts with Leu404, Leu429, Phe432, Leu433, Met436, Thr488 and a T-shaped π–π interaction with Trp531. Moreover, the bromine atom pointed towards Phe381, at a distance of 3.8 Å, thus indicating the possibility of a direct halogen bond interaction. Finally, a discontinuous hydrogen bond interaction (H-bond) was observed between the carbonyl of the ligand and the hydroxyl group of Thr488, with an occupancy percentage of 38% over the MD run. Taken together, the MD results suggested that the introduction of a bromine instead of the methyl on the pyridine ring induced a slightly different conformational behaviour, leading to a more compact structure that perfectly fits the hydrophobic environment of the ACB. The comparison of the proposed Flu-AM4 binding mode to that previously published for Flu-AM1 (Supporting information Figure S4) revealed how the different binding conformations of the two close compounds, likely driven by different internal interaction of the bromine or the methyl with the fluorine-substituted phenyl ring, determined a different ability to exploit the hydrophobic residue that characterises the ACB channel. In particular, the biphenyl moiety of Flu-AM4 optimally fit a hydrophobic pocket establishing apolar contacts with 6 residues, while Flu-AM1 only with 3 residues (Phe194, Ile238 and Ile491). Therefore, we can conclude that the introduction of the bromine enhanced the affinity for the FAAH, by means of the optimisation of hydrophobic interactions within the ACB channel.
Figure 3.

(A) The Flu-AM4 L-shaped conformation (magenta stick) of the best cluster obtained from the DBSCAN analysis and minimised considering the explicit water molecules. (B) General view of the rFAAH enzyme (PDB ID: 3QK5) complexed with Flu-AM4 in the same conformation (magenta spheres). rFAAH is displayed as wheat cartoon with the catalytic triad residues Lys142, Ser217 and Ser241 highlighted as magenta sticks. Amino acids involved in ligand/protein interactions with Flu-AM4 are shown as yellow lines. Hydrogen bond interaction between the carbonyl of Flu-AM4 and the hydroxyl group of Thr488 is shown as dashed black line.
Molecular modelling: COX-2 inhibition
To study the molecular basis of COXs inhibition by Flu-AM4, docking experiments were carried out on COX-1 and COX-2 isoforms (PDB IDs: 3N8Z and 3RR3 respectively). Docking results of Flu-AM4 in the COX-2 showed a clear preference for a single pose, strictly resembling the binding mode of the complexed flurbiprofen (1, Figure 4(B)), with the amide carbonyl oxygen and the pyridine nitrogen mimicking the interaction of the carboxylic acid group of 1, directly engaging hydrogen bond interactions with the key residue Arg120. Accordingly, the analysis of the electrostatic potential surface of compounds Flu-AM1 and Flu-AM4 clearly showed a negative electrostatic potential localised between the pyridine nitrogen atom and the carbonyl group, supporting the hypothesis that the capability to retain COX-2 inhibitory properties is due to the peculiar behaviour of the amide-pyridine group to mimic the carboxylic group in NSAIDs. Interestingly, within the COX-1 Flu-AM4 was not able to fit into the known active site occupied by 1, lacking the key interactions with Arg120 (Figure 4(C)). In accordance with these observations, the resulting Glide SP scores are consistently higher in the case of COX-1 isoform (Supporting Information, Table S3). NSAIDs selectivity towards COX-2 has been achieved exploiting the different volume of the orthosteric binding site, mainly due to the presence of Ile523 in the catalytic domain of COX-1 (Val523 in COX-2), which reduces the volume of the cyclooxygenase site by closing the hydrophilic side pocket located above the Arg-120/Tyr-355 residues63. In our case, docking results of Flu-AM4 against COX-1 highlighted also the role of Leu357 (Phe357 in COX-2) that further reduces the catalytic pocket, thus hampering the bromine-pyridine ring to adopt the same binding mode observed in COX-2 (Figure 4(C)).
Figure 4.

(A) General view of the COX-1 (dark-green cartoon) and COX-2 isoforms (orange cartoon) complexed, in the cyclooxygenase site, with the best docking results of Flu-AM4 (dark-green and orange spheres, respectively in COX-1 and COX-2). (B) Best docking pose of Flu-AM4 (orange stick) in the COX-2 isoform, superimposed with the co-crystallographic binding mode of flurbiprofen (violet stick) (PDB ID: 3RR3). (C) Best docking pose of Flu-AM4 (dark-green stick) in the COX-1 isoform, superimposed with the binding mode of Flu-AM4 against COX-2 (orange dots) to highlight the steric hindrance between the bromine atom and the Leu357, which hampers a full accommodation of Flu-AM4 within the catalytic domain of COX-1. Key enzymes residues Arg120 and Tyr355 in COX-1 and COX-2 are shown as dark-green and orange lines, respectively. Amino acid residues influencing the binding mode of Flu-AM4 between COX-1 and COX-2 are shown as spheres. Hydrogen bond interactions are shown as dashed black lines.
Flu-AM4 activity in acute and chronic pain mice models
The efficacy of FAAH inhibitors in several animal models of pain and inflammation have been described6,63,64. Most data are from irreversible inhibitors, but the reversible FAAH inhibitor OL-135 also reduces mechanical allodynia in a rat spinal nerve ligation model65. On the basis of enzymatic assays, Flu-AM4 was considered the most interesting derivative and investigated in vivo in order to evaluate its anti-inflammatory and analgesic proprieties. In particular, we choose acute pain/inflammation animal models, such as carrageenan and formalin tests, to assess the ability of our compound to reduce oedema and inflammatory pain. Moreover, using the CCI model we have determinate the capability of Flu-AM4 in a chronic neuropathic pain model.
The nocifensive response to formalin injection into the paw is characterised by two phases, an early sensorial phase (phase I) lasting the first 15 min, and a late inflammatory phase lasting from 20 to 60 min after the injection. This can be evaluated by determining the amount of time that the animals spend licking or biting the injected paw (Figure 5 white bars). Systemic Flu-AM4 administration (10 mg/kg) was able to reduce the behavioural responses in both phases (Figure 5(A) black bars, ***p < 0.001). Moreover, we also evaluated the activity of Flu-AM4 after local injection. Co-administration of formalin 5% and Flu-AM4 (100 µg) reduced the paw licking time in both phases compared to formalin alone, suggesting that peripheral effects of the compound can contribute to its efficacy in this model (Figure 5(B) black bars, ***p < 0.001).
Figure 5.

Effect of vehicle and Flu-AM4 after A, systemic administration and B, local application in formalin-induced paw licking. In Panel A, 1 h before formalin injection, mice were treated with vehicle (white column) and Flu-AM4 (10 mg/kg ip; black columns). In Panel B, mice were treated with vehicle + formalin in paw (white column) and Flu-AM4 (100 µg) + formalin in paw (black columns). The cumulative licking time were counted for 15 min (phase I) and 50 min (phase II). Data are shown as mean ± SEM of 6 animals per group. ** p < 0.01 and *** p < 0.001 vs. vehicle, unpaired t-test.
The anti-inflammatory properties of Flu-AM4 were investigated further in the carrageenan paw oedema model. This is a classic inflammatory model in which injection of carrageenan induced plasma extravasation (oedema) and prolonged hyperalgesia until 72 h after injection. Systemic administration of Flu-AM4 (10 mg/kg) reduced paw oedema at 2 and 4 h after carrageenan injection (Figure 6(A), *p < 0.05), moreover, the compound was able to reduce hyperalgesia seen 2 h after carrageenan (Figure 6(B), *p < 0.001).
Figure 6.
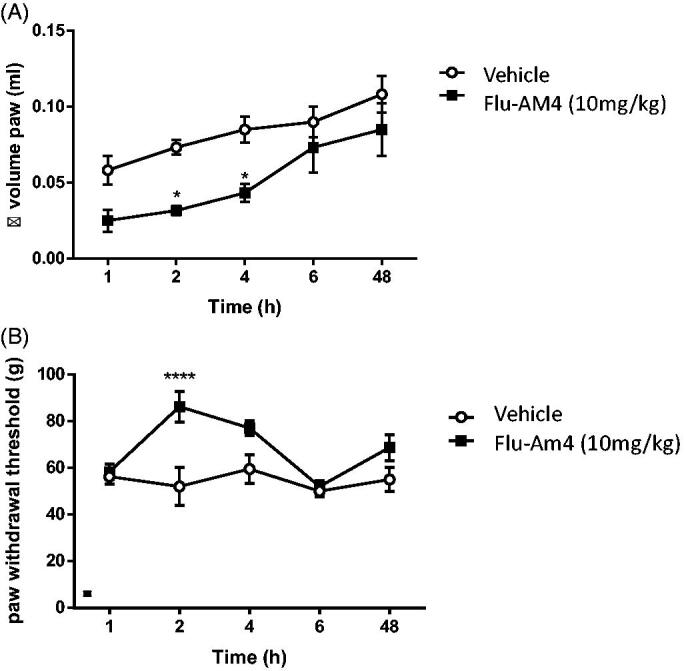
Effect of vehicle and Flu-AM4 upon carrageenan-induced paw oedema (A) and paw hyperalgesia (B). Five min after carrageenan injection, mice were treated with vehicle (white circle) or Flu-AM4 (10 mg/kg ip; black square). Data are shown as mean ± SEM of 6 animals per group. * p < 0.05, *** p < 0.001 vs. vehicle, unpaired t-test.
Finally, we evaluated Flu-AM4 effect at different doses with respect to the mechanical allodynia and hyperalgesia produced in a neuropathic pain model, sciatic nerve ligation (CCI). We observed that 7- and 14-days vehicle CCI-treated mice (ctr) showed a significant reduction of the paw withdrawal threshold in both Von Frey and Randall-Selitto tests respect to sham not-ligated mice (Figure 7; p < 0.001 vs. sham). Systemic Flu-AM4 treatment (1 h before test) significantly decreased the mechanical allodynia and hyperalgesia. In particular, the high dose, 10 mg/kg increased paw withdrawal latency in both test and in all experimental time (day 7 and 14), the dose of 3 mg/kg resulted efficacy only at 14 days in both tests and on both days 7 and 14. The dose of 3 mg/kg showed efficacy only at the 14-day time point in both tests, and the 1 mg/kg dose did not show any effect.
Figure 7.

Effect of vehicle and Flu-AM4 in CCI-induced mechanical (A) and hyperalgesia (B) allodynia on day 7 and 14. Mice 1 h before test were treated with vehicle (grey bars) and Flu-AM4 (1, 3 or 10 mg/kg ip). Data are shown as mean ± SEM of 6 animals per group. *p < 0.05 and p < 0.01 vs. vehicle, 2-way repeated-measures ANOVA with post-hoc Bonferroni tests.
We evaluated the expression of key factors involved in the development and maintenance of the inflammatory and pain state, such as COX-2, iNOS and NFκB in spinal cord of neuropathic-mice. Results clearly showed that sciatic nerve ligation induced an increase of iNOS (A), COX-2 (B) and NFκB-p65 (C) expression in the spinal cord. Acute Flu-AM4 treatment significantly reduced this expression (Figure 8).
Figure 8.

iNOS (A), COX-2 (B) and NF-κB-p65 (C) expression in the spinal cord of CCI-mice. Mice were treated 1 h before with vehicle (black bars) and Flu-AM4 (10 mg/kg ip, grey bars). Sham mice represent animal without sciatic nerve ligation. Data are shown as mean ± SEM of 6 animals per group; representative blot are shown. *p < 0.05 vs. sham; #p < 0.05 and ##p < 0.01 vs. vehicle, one-way repeated-measures ANOVA with post-hoc Bonferroni tests.
Conclusion
A growing body of evidence suggesting that dual inhibition of COX and FAAH enzymes offers a potential therapeutic strategy to attenuate inflammatory and neuropathic pain states24,31,36. The development of dual FAAH-COX inhibitors would be of great potential value as a therapeutic strategy to treat different types of pain. Indeed, efforts are underway to synthesise such compounds26,29,36. In this paper we have described the design, the synthesis, the molecular mechanism of action and the pharmacological characterisation of a series of pyridine amide derivatives of flurbiprofen. In particular, enzymatic assays identified Flu-AM4 the most promising compound within the series, docking and molecular dynamics studies suggested the molecular basis of dual action of pyridine-amide flurbiprofen derivatives. Moreover, our in vivo studies clearly indicated that the administration of a dual compound reduced inflammation and pain. Flu-AM4, indeed, was able to modulate the inflammatory process developed by carrageenan and formalin injection restoring the expression of important enzymes such as COX-2 and iNOS in the spinal cord of inflamed-mice. Moreover, Flu-AM4 reduced also neuropathic pain by sciatic nerve ligation suggesting that its activity can influence much more elaborate and chronic pathways. Our results are in agreement with other studies where co-administration of the FAAH inhibitor URB597 and COX-1/2 inhibitor diclofenac produced synergistic antinociceptive effects in the acetic acid abdominal stretching assay31. Likewise, combination of the peripherally restricted FAAH inhibitor URB937 and the COX-1/2 inhibitor indomethacin significantly reduced nociceptive behaviour in the carrageenan and CCI models in a synergistic fashion64.
In conclusion, we have identified Flu-AM4 as a dual-action reversible FAAH/substrate-selective COX inhibitor anti-inflammatory and analgesic activity in several animal pain models. These findings underscore the potential usefulness of such dual-action compounds and suggest that Flu-AM4 is an ideal molecule with which to explore further this exciting possibility.
Supplementary Material
Acknowledgements
CF would like to thank the Research Funds of the Medical Faculty, Umeå University, for financial support.
Funding Statement
VO would like to thank the University of Cagliari (grant FIR 2018–19). FM was supported by a fellowship of Regione Campania (n. B61C17000070007 – SATIN). BC gratefully acknowledges the support of NVIDIA Corporation with the donation of the Titan Xp GPU used for this research, and the SCoPE datacenter of the University Federico II for the access to HPC infrastructures.
Author contributions
AD, JK, FM, MS, CC, CMM performed the experiments and analysed the data. CJF, RR, BC and VO conceived the experiments, supervised, and analysed the data. CJF, RR, BC and VO have written the manuscript with contributions from all authors. All authors have given approval to the final version of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary data
These data include: docking studies on Flu-AM3-6 in the rFAAH; QM/MM energetic and structural values, and molecular electrostatic potential surface calculated for the interaction of Flu-AM1 and Flu-AM4 with Phe381; comparison of Flu-AM1 and Flu-AM4 binding modes (PDF); Molecular model of Flu-AM4 binding mode against FAAH (pdb); Molecular model of Flu-AM4 binding mode against COX-2 (pdb).
References
- 1.Woodhams SG, Chapman V, Finn DP, et al. The cannabinoid system and pain. Neuropharmacology 2017;124:105–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deutsch DG, Chin SA.. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem Pharmacol 1993;46:791–6. [DOI] [PubMed] [Google Scholar]
- 3.Dinh TP, Carpenter D, Leslie FM, et al. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci U S A 2002;99:10819–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKinney MK, Cravatt BF.. Structure and function of fatty acid amide hydrolase. Annu Rev Biochem 2005;74:411–32. [DOI] [PubMed] [Google Scholar]
- 5.Tripathi RKP. A perspective review on fatty acid amide hydrolase (FAAH) inhibitors as potential therapeutic agents. Eur J Med Chem 2020;188:111953. [DOI] [PubMed] [Google Scholar]
- 6.Guindon J, Lai Y, Takacs SM, et al. Alterations in endocannabinoid tone following chemotherapy-induced peripheral neuropathy: effects of endocannabinoid deactivation inhibitors targeting fatty-acid amide hydrolase and monoacylglycerol lipase in comparison to reference analgesics following cisplatin treatment. Pharmacol Res 2013;67:94–109. doi: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khasabova IA, Khasabov S, Paz J, et al. Cannabinoid type-1 receptor reduces pain and neurotoxicity produced by chemotherapy. J Neurosci 2012;32:7091–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parker LA, Limebeer CL, Rock EM, et al. A comparison of novel, selective fatty acid amide hydrolase (FAAH), monoacyglycerol lipase (MAGL) or dual FAAH/MAGL inhibitors to suppress acute and anticipatory nausea in rat models. Psychopharmacology (Berl) 2016;233:2265–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hill MN, Gorzalka BB.. The endocannabinoid system and the treatment of mood and anxiety disorders. CNS Neurol Disord Drug Targets 2009;8:451–8. [DOI] [PubMed] [Google Scholar]
- 10.Kathuria S, Gaetani S, Fegley D, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med 2003;9:76–81. [DOI] [PubMed] [Google Scholar]
- 11.Schlosburg JE, Carlson BLA, Ramesh D, et al. Inhibitors of endocannabinoid-metabolizing enzymes reduce precipitated withdrawal responses in THC-dependent mice. Aaps J 2009;11:342–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schlosburg JE, Blankman JL, Long JZ, et al. Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci 2010;13:1113–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huggins JP, Smart TS, Langman S, et al. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-04457845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain 2012;153:1837–46. [DOI] [PubMed] [Google Scholar]
- 14.Bradford D, Stirling A, Ernault E, et al. The MOBILE Study-A Phase IIa Enriched Enrollment randomized withdrawal trial to assess the analgesic efficacy and safety of ASP8477, a fatty acid amide hydrolase inhibitor, in patients with peripheral neuropathic pain. Pain Med 2017;18:2388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wagenlehner FME, van Till JWO, Houbiers JGA, et al. Fatty acid amide hydrolase inhibitor treatment in men with chronic prostatitis/chronic pelvic pain syndrome: an adaptive double-blind, randomized controlled trial. Urology 2017;103:191–7. [DOI] [PubMed] [Google Scholar]
- 16.Moore N. Lessons from the fatal French study BIA-10-2474. BMJ 2016;353:i2727. [DOI] [PubMed] [Google Scholar]
- 17.van Esbroeck ACM, Janssen APA, Cognetta AB, et al. Activity-based protein profiling reveals off-target proteins of the FAAH inhibitor BIA 10-2474. Science 2017;356:1084–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharma V, Bhatia P, Alam O, et al. Recent advancement in the discovery and development of COX-2 inhibitors: Insight into biological activities and SAR studies (2008-2019). Bioorg Chem 2019;89:103007. [DOI] [PubMed] [Google Scholar]
- 19.Brito R, Sheth S, Mukherjea D, et al. TRPV1: a potential drug target for treating various diseases. Cells 2014;3:517–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakin YS, Dogrul A, Ilkaya F, et al. The effect of FAAH, MAGL, and Dual FAAH/MAGL inhibition on inflammatory and colorectal distension-induced visceral pain models in Rodents. Neurogastroenterol Motil 2015;27:936–44. [DOI] [PubMed] [Google Scholar]
- 21.de Novellis V, Vita D, Gatta L, et al. The blockade of the transient receptor potential vanilloid type 1 and fatty acid amide hydrolase decreases symptoms and central sequelae in the medial prefrontal cortex of neuropathic rats. Mol Pain 2011; 7:71744-8069-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Malek N, Mrugala M, Makuch W, et al. A multi-target approach for pain treatment: dual inhibition of fatty acid amide hydrolase and TRPV1 in a rat model of osteoarthritis. Pain 2015;156:890–903. [DOI] [PubMed] [Google Scholar]
- 23.Ligresti A, Martos J, Wang J, et al. Prostamide F2a receptor antagonism combined with inhibition of FAAH may block the pro-inflammatory mediators formed following selective FAAH inhibition. Br J Pharmacol 2014;171:1408–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fowler CJ, Björklund E, Lichtman AH, et al. Inhibitory properties of ibuprofen and its amide analogues towards the hydrolysis and cyclooxygenation of the endocannabinoid anandamide. J Enzyme Inhib Med Chem 2013;28:172–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cocco MT, Congiu C, Onnis V, et al. Synthesis of ibuprofen heterocyclic amides and investigation of their analgesic and toxicological properties. Eur J Med Chem 2003;38:513–8. [DOI] [PubMed] [Google Scholar]
- 26.Cipriano M, Björklund E, Wilson AA, et al. Inhibition of fatty acid amide hydrolase and cyclooxygenase by the N-(3-methylpyridin-2-yl)amide derivatives of flurbiprofen and naproxen. Eur J Pharmacol 2013;720:383–90. [DOI] [PubMed] [Google Scholar]
- 27.Onnis V, Congiu C, Björklund E, et al. Synthesis and evaluation of paracetamol esters as novel fatty acid amide hydrolase inhibitors. J Med Chem 2010;53:2286–98. [DOI] [PubMed] [Google Scholar]
- 28.Goodman MC, Xu S, Rouzer CA, et al. Dual cyclooxygenase-fatty acid amide hydrolase inhibitor exploits novel binding interactions in the cyclooxygenase active site. J Biol Chem 2018;293:3028–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Favia AD, Habrant D, Scarpelli R, et al. Identification and characterization of carprofen as a multitarget fatty acid amide hydrolase/cyclooxygenase inhibitor. J Med Chem 2012;55:8809–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alhouayek M, Muccioli GG.. COX-2-derived endocannabinoid metabolites as novel inflammatory mediators. Trends Pharmacol Sci 2014;35:284–92. [DOI] [PubMed] [Google Scholar]
- 31.Naidu PS, Booker L, Cravatt BF, Lichtman AH.. Synergy between enzyme inhibitors of fatty acid amide hydrolase and cyclooxygenase in visceral nociception. J Pharmacol Exp Ther 2009;329:48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fowler CJ, Stenström A, Tiger G.. Ibuprofen inhibits the metabolism of the endogenous cannabimimetic agent anandamide. Pharmacol Toxicol 1997;80:103–7. [DOI] [PubMed] [Google Scholar]
- 33.Fowler CJ, Janson U, Johnson RM, et al. Inhibition of Anandamide Hydrolysis by the Enantiomers of Ibuprofen, Ketorolac, and Flurbiprofen. Arch Biochem Biophys 1999;362:191–6. [DOI] [PubMed] [Google Scholar]
- 34.Holt S, Paylor B, Boldrup L, et al. Inhibition of fatty acid amide hydrolase, a key endocannabinoid metabolizing enzyme, by analogues of ibuprofen and indomethacin. Eur J Pharmacol 2007;565:26–36. [DOI] [PubMed] [Google Scholar]
- 35.Karlsson J, Morgillo CM, Deplano A, et al. Interaction of the N-(3-Methylpyridin-2-yl)amide derivatives of flurbiprofen and ibuprofen with FAAH: enantiomeric selectivity and binding mode. PLoS One 2015;10:e0142711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sasso O, Migliore M, Habrant D, et al. Multitarget Fatty acid amide hydrolase/cyclooxygenase blockade suppresses intestinal inflammation and protects against nonsteroidal anti-inflammatory drug-dependent gastrointestinal damage. Faseb J 2015;29:2616–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Costa SKPF, Muscara MN, Allain T, et al. Enhanced analgesic effects and gastrointestinal safety of a novel, hydrogen sulfide-releasing anti-inflammatory drug (ATB-352): a role for endogenous cannabinoids. Antioxid Redox Signal 2020;33(14):1003–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deplano A, Cipriano M, Moraca F, et al. Benzylamides and piperazinoarylamides of ibuprofen as fatty acid amide hydrolase inhibitors. J Enzyme Inhib Med Chem 2019;34:562–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deplano A, Karlsson J, Fowler CJ, Onnis V.. The fatty acid amide hydrolase and cyclooxygenase-inhibitory properties of novel amide derivatives of carprofen. Bioorg Chem 2020;101:104034. [DOI] [PubMed] [Google Scholar]
- 40.Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09. 2009.
- 41.DeLano WL. The PyMOL molecular graphics system. Schrödinger LLC. 2014. http://www.pymol.org.
- 42.Gustin DJ, Ma Z, Min X, et al. Identification of potent, noncovalent fatty acid amide hydrolase (FAAH) inhibitors. Bioorg Med Chem Lett 2011;21:2492–6. [DOI] [PubMed] [Google Scholar]
- 43.Sidhu RS, Lee JY, Yuan C, Smith WL.. Comparison of cyclooxygenase-1 crystal structures: cross-talk between monomers comprising cyclooxygenase-1 homodimers. Biochemistry 2010;49:7069–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duggan KC, Hermanson DJ, Musee J, et al. (R)-Profens are substrate-selective inhibitors of endocannabinoid oxygenation by COX-2. Nat Chem Biol 2011;7:803–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schroedinger . Maestro Release 2019-1. 2019.
- 46.Bracey MH, Hanson MA, Masuda KR, et al. Structural adaptations in a membrane enzyme that terminates endocannabinoid signaling. Science (80-) 2002;298:1793–6. [DOI] [PubMed] [Google Scholar]
- 47.Jorgensen WL, Maxwell DS, Tirado-Rives J.. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc 1996;118:11225–36. [Google Scholar]
- 48.Case DA, Cerutti DS, Cheatham TEI, et al. Univ California, San Fr. 2017:AMBER 2017, University of California, San Francisc; 2017. doi:citeulike-article-id:2734527
- 49.Maier JA, Martinez C, Kasavajhala K, et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 2015;11:3696–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bayly CI, Cieplak P, Cornell W, Kollman PA.. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J Phys Chem 1993;97:10269–80. [Google Scholar]
- 51.Wang J, Wang W, Kollman PA, Case DA.. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model 2006;25:247–60. [DOI] [PubMed] [Google Scholar]
- 52.Wang J, Wolf RM, Caldwell JW, Kollman PA, et al. Development and testing of a general Amber force field. J Comput Chem 2004;25:1157–74. [DOI] [PubMed] [Google Scholar]
- 53.Roe DR, Cheatham TE.. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J Chem Theory Comput 2013;9:3084–95. [DOI] [PubMed] [Google Scholar]
- 54.Shim J, Humphreys GI, Venkatesan BM, et al. Detection and quantification of methylation in DNA using solid-state nanopores. Sci Rep 2013;3:1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ester M, Kriegel H-P, Sander J, Xu X. A density-based algorithm for discovering clusters in large spatial databases with noise. In: KDD’96: Proceedings of the Second International Conference on Knowledge Discovery and Data Mining. 1996, p. 226–231. [Google Scholar]
- 56.Boldrup L, Wilson SJ, Barbier AJ, Fowler CJ.. A simple stopped assay for fatty acid amide hydrolase avoiding the use of a chloroform extraction phase. J Biochem Biophys Methods 2004;60:171–7. [DOI] [PubMed] [Google Scholar]
- 57.A. Wilson A, W. Hicks J, Sadovski O, et al. Radiosynthesis and evaluation of [11C-Carbonyl]-labeled carbamates as fatty acid amide hydrolase radiotracers for positron emission tomography. J Med Chem 2013;56:201–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Meade EA, Smith WL, DeWitt DL.. Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other non-steroidal anti-inflammatory drugs. J Biol Chem 1993;268:6610–4. [PubMed] [Google Scholar]
- 59.Russo R, De Caro C, Avagliano C, et al. Sodium butyrate and its synthetic amide derivative modulate nociceptive behaviors in mice. Pharmacol Res 2016;103:279–91. [DOI] [PubMed] [Google Scholar]
- 60.Mileni M, Johnson DS, Wang Z, et al. Structure-guided inhibitor design for human FAAH by interspecies active site conversion. Proc Natl Acad Sci USA 2008;105:12820–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Estiarte MA, Johnson RJ, Kaub CJ, et al. 2-Amino-5-arylbenzoxazole derivatives as potent inhibitors of fatty acid amide hydrolase (FAAH). Med Chem Commun 2012;3:611–9. [Google Scholar]
- 62.Patel JS, Berteotti A, Ronsisvalle S, et al. Steered molecular dynamics simulations for studying protein-ligand interaction in cyclin-dependent kinase 5. J Chem Inf Model 2014;54:470–80. [DOI] [PubMed] [Google Scholar]
- 63.Ahn K, Johnson DS, Mileni M, et al. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem Biol 2009;16:411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sasso O, Bertorelli R, Bandiera T, et al. Peripheral FAAH inhibition causes profound antinociception and protects against indomethacin-induced gastric lesions. Pharmacol Res 2012;65:553–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chang L, Luo L, Palmer JA, et al. Inhibition of fatty acid amide hydrolase produces analgesia by multiple mechanisms. Br J Pharmacol 2006;148:102–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.