

Abstract
Methamphetamine (METH) is a major public health and safety problem worldwide. METH is psychostimulant that activates microglia via the toll-like receptor (TLR) 4/MD2 complex, modulating the abundant production of pro-inflammatory cytokines in the central nervous system (CNS). The TLR4/MD2 complex on the surface of microglia recognizes pathogen-associated molecular patterns such as lipopolysaccharide (LPS) resulting in brain tissue inflammation and neuronal damage. Since METH has been associated with microglia-induced neurotoxicity, we hypothesized that METH impairs the expression of TLR4 and activation of NF-κB in NR-9460 microglia-like cells after LPS challenge. We demonstrated that METH decreases the distribution and expression of TLR4 receptors on the surface of microglia-like cells after incubation with endotoxin. Moreover, METH impairs the TLR4/MD2 complex signaling pathways, compromises the activation of NF-κB, and reduces the production of pro-inflammatory mediators in microglia-like cells upon LPS stimulation. Interestingly, microglia-like cells treated with METH and challenged with LPS showed considerable cellular morphological changes including enlarged nuclei and ruffled surface. Our results suggest that METH may have a significant impact on microglial-induced neuroinflammation, neurotoxicity, and the CNS defense against infection. It also highlights the importance of studying the effects of METH on the molecular and cellular components of users’ CNS immunity. Finally, animal studies exploring the role of METH on the effectors functions of microglia after antigenic exposure are necessary to understand drug-related inflammation and neural damage in users.
Keywords: LPS, methamphetamine, microglia, minocycline, TLR4
1. Introduction
Methamphetamine (METH) is a strong psychostimulant globally abused. In 2017, METH was responsible for ~15% of all drug overdose deaths in the United States and its consumption has increased 7.5 times in the last 10 years (Hedegaard et al., 2018). The drug’s ability to rapidly release high levels of dopamine in reward areas of the brain strongly reinforces drug-taking behavior, making the user want to repeat the experience leading to its abuse and addiction. METH causes alterations of the users’ behavior, making them susceptible to engaging in unsafe activities like unprotected sex and sharing of contaminated needles, leading to the acquisition of infectious diseases (Ellis et al., 2003). METH contributes to increased transmission of AIDS (Ellis et al., 2003), hepatitis (Gonzales et al., 2006), tuberculosis (Mankatittham et al., 2009), herpes (Valencia et al., 2012), and other communicable diseases (Galindo et al., 2012). Although most of the current investigations and published literature have focused on the impact of METH on human psychology and behavior, little is known about the effects of METH on cells and molecules associated with immunity, particularly in the central nervous system (CNS).
In the brain, microglia, the resident surveillance cells of the CNS, act as its primary active immune defense, suggesting that they play an important role controlling infectious diseases (Kambugu et al., 2008). METH impairs immune cellular and molecular functions and is deadly to phagocytic cells including microglia (Aslanyan et al., 2019), which may result in increased susceptibility of users to acquire CNS-related infectious diseases (Cherner et al., 2005; Eugenin et al., 2013; Langford et al., 2003; Najera et al., 2016; Patel et al., 2013). In addition, microglia have been associated with METH-induced neurotoxicity and brain tissue degeneration (Sharikova et al., 2018; Xu et al., 2017), which is possibly attributed to a variety of mechanisms including: increase in neuronal firing rate, increased concentrations of intracellular Ca+2 and Na+ ions, dysregulation of mitochondrial function, neuronal energetic imbalance, and overproduction of reactive oxygen species (Shaerzadeh et al., 2018). Even though microglia are critical in controlling microbial CNS colonization (Koutsouras et al., 2017) and synapse homeostasis (Colonna and Butovsky, 2017), their responses to microbial antigens in the setting of METH remain understudied.
Toll-like receptor 4 (TLR4) plays important roles in innate immune and inflammatory responses, attracting considerable attention. TLR4 is a pattern recognition receptor, highly expressed in microglia (Wendeln et al., 2018). TLR4 is activated by LPS, whereas CD14 and MD2 act as accessory proteins for LPS/TLR4 binding (Park et al., 2009). Upon ligand binding, TLR4 dimerizes, and recruits downstream adaptor molecules such as MyD88/MAL and TRIF/TRAM to mount an inflammatory response (Yesudhas et al., 2014). The activated MyD88/MAL then activates IRAK4, TRAF6, TAK1, and IKK complexes, while TRIF/TRAM signals through RIP1 to TRAF6/TAK1 and IKK. After this, both these pathways converge at NF-κB. The cytoplasmic NF-κB complex is maintained in the inactive state by IκB, which is in turn degraded by proteasomes, resulting in the translocation of NF-κB into the nucleus. Besides activating NF-κB, TAK1 also phosphorylates MAPKs to further reinforce the inflammatory response. The TRIF/TRAM pathway not only activates NF-κB but also triggers IRF3 to mount an antiviral response. METH binds to MD2 and TLR4 activation leads to the production of pro-inflammatory cytokines (Wang et al., 2019). TLR4 and its downstream signaling pathways play pivotal roles in the initiation and progression of the neurodegenerative disease due to the persistent activation of the microglial cells, provoking an increase in pro-inflammatory mediators and reactive oxygen species, which lead to cellular macromolecular damage and apoptosis (Du et al., 2017). However, whether TLR4 is implicated in METH-induced microglial toxicity remains unknown.
In this study, we explored the impact of METH on NR-9460 microglia-like cell TLR4 signaling pathways and production of inflammatory mediators upon stimulation with LPS. We hypothesized that METH alters TLR4 expression, signaling, and cytokine secretion in microglia-like cells. We aimed to understand the impact of METH on TLR4 expression and effector functions of microglia, which may have important implications in neuroinflammation and/or defense of the CNS against infection. Targeting TLR4 activity may constitute a potential intervention strategy to minimize METH-mediated neuroinflammation and susceptibility to infectious diseases.
2. Materials and methods
2.1. NR-9460 microglial-like cells
The NR-9460 cells, a murine microglial line derived from wild type mouse brain tissue, were immortalized by being infected with the ecotropic transforming replication-deficient retrovirus J2 (BEI Resources, NIAID, National Institutes of Health; NIH). The cell line was cultured in Dulbecco modified eagle medium (DMEM) supplemented with 11% fetal bovine serum (FBS; Atlanta Biologicals), 2 μM of L-glutamine (Sigma), 1 μM of sodium pyruvate (Sigma) and incubated at 37°C and CO2. Characterization based on immunofluorescence and flow cytometry demonstrated that the NR-9460 cell line retains its morphological, functional, and surface expression properties.
2.2. Rationale for METH doses used in these studies
Controlled studies indicated that a single 260-mg dose peaks at a level of 7.5 μM (Melega et al., 2007). Thus, a single dose of 260 mg would be expected to produce 7.5 to 28.8 μM blood METH levels. Intravenous drug users tend to self-administer METH in binges, and as the drug exhibits a half-life of 11.4 to 12 h, this can lead to higher drug levels (Cho et al., 2001; Harris et al., 2003). Binge patterns of use in individuals have shown that the fourth administration of 260 mg during a single day produces blood levels of 17 μM and could reach 20 μM on the second day of such a binge (Melega et al., 2007). Thus, binge doses of 260 to 1,000 mg produce 17 to 80 μM blood METH levels and levels in the micromolar range of hundreds in organs, including the brain and the spleen (Harris et al., 2003). Therefore, we selected 25 μM METH to perform our experiments.
2.3. Determining TLR4 expression on the surface of NR-9460 cells using flow cytometry
Monolayers of 105 microglial-like cells were grown in a 24-well microtiter plate (Corning) in the absence or presence of METH (25 μM; Sigma) or Minocycline (MC; 1 μM; Sigma) for 2 h at 37°C and 5% CO2. MC inhibits microglial activation and was used as a positive control (Liu et al., 2013). Then, lipopolysaccharide (LPS; 10 μg/ml; Sigma), a component of the cell wall of Gram-negative bacteria and a potent immune modulator, was added to NR-9460 cells and incubated for 24 h at 37°C and 5% CO2. For flow cytometry analysis, microglia-like cells were detached with 0.05% trypsin (Corning) treatment for 1 min followed by centrifugation at 980 rpm for 5 min at room temperature (RT). Cells were washed 3X with phosphate buffer saline (PBS) and centrifuged as described above. A goat-anti mouse TLR4 Alexa Fluor® 488-conjugated monoclonal antibody (1:200 dilution; R&D Systems) was added on 1% bovine serum albumin (BSA; Thermo Fisher; TF) in PBS to the cells followed by an incubation at 37°C for 1 h. Samples were processed (10,000 events per sample) on a Gallios flow cytometer (Beckman Coulter) and TLR4 expression on microglia-like cells was analyzed using the Kaluza software (Beckman Coulter).
2.4. Fluorescent microscopy
For fluorescent microscopy, monolayers were fixed on a glass-bottom petri dish (TF) with 4% formaldehyde (TF) for 20 min at RT. The cells were washed 3X with 5% Tween 20 (TF) in PBS and blocked with 1% BSA in PBS for 1 h at RT. After blocking, the cells were again washed 3X with 5% Tween 20 in PBS and incubated with an anti-TLR4 conjugated to Alexa Fluor® 488 (green; 1:100 dilution; R&D Systems), in blocking solution in an orbital shaker (New Brunswick Galaxy 170S) at 150 rpm and 37°C for 1h. The samples were washed 3X with blocking buffer and incubated with 4′, 6-diamidino-2-phenylindole (DAPI; blue; TF) to stain nuclei for 1 h at 37°C. The slides were washed 3X with PBS, coverslips were affixed, and each sample was viewed to determine the TLR4 distribution on the surface of NR-9460 cells with a Zeiss LSM 700 Confocal Laser Scanning Microscope (Carl Zeiss) at a magnification of ×60. Images were collected using an AxioCam digital camera and analyzed using Zen Lite digital imaging software (Carl Zeiss). The cell volume (V = 4/3 πr³) of nine cells per group was measured with Adobe Photoshop CC2017 by tracing the radius of each cell at the equatorial plane.
2.5. Western blot analysis
To further understand the impact of METH on the TLR4/MD2 complex in NR-9460 microglia-like cells upon LPS stimulation, a density of 105 microglial-like cells were incubated in 24-well microtiter plate with PBS (untreated), 25 μM METH, or 1 μM MC for 2 h. Then, the cells were washed 3X with PBS and incubated in the presence of 10 μM LPS for 24h at 37°C and 5% CO2. Western blot analysis was conducted using cytoplasmic extracts made with an NE-PER nuclear and cytoplasmic extraction kit (TF). The mixture was centrifuged at 10,000 × g for 10 min at 4°C, and the resulting protein content of the supernatant was determined using the Bradford method, employing a Pierce™ BCA protein assay kit (Bio-Rad). Lysates were preserved in protease inhibitor cocktail (TF) and stored at −20°C until use. Extracts were diluted with 2x Laemmli sample buffer (Bio-Rad) and β-mercaptoethanol (Sigma). The mixture was heated to 90°C for 5 min. Twenty-three micrograms of protein were applied to each lane of a gradient gel (7.5%; Bio-Rad). Proteins were separated by electrophoresis at a constant 130 V/gel for 90 min and transferred to a nitrocellulose membrane on the Trans-Blot® Turbo™ Transfer System (Bio-Rad) at 25 V for 7 min. The membranes were blocked with 5% BSA in tris-buffered saline (TBST; 0.1% Tween 20) for 2 h at RT. Primary mouse monoclonal anti-TLR4 (1:100 dilution; Santa Cruz Biotech; SCB), anti-MyD88 (1:400 dilution; SCB), anti-IRAK1 (1:200 dilution; SCB), anti-TRAF6 (1:800 dilution; SCB), anti-IRF3 (1:800 dilution; SCB), anti-IKKi (1:800 dilution; SCB), and anti-NF-κB (1:200 dilution; SCB) antibodies were incubated overnight at 4°C with TBST (5% BSA). After washing the membranes 3X with TBST for 10 min, a goat anti-mouse IgG (H+L) conjugated to horseradish peroxidase (HRP) was used as a secondary antibody (1:1000; Southern Biotech) and incubated with TBST (5% BSA) for 1 h at RT. The membranes were washed as described above. Protein bands were measured using the iBright FL100 imaging system (Invitrogen) after staining each membrane with chemiluminescence detection reagents (TF). Quantitative measurements of individual band intensities in Western blot analyses for TLR4, MyD88, IRAK1, TRAF6, IRF3, IKKi, and NF-κB were performed using ImageJ software (NIH). Actin (dilution, 1:1,000; TF), a structural housekeeping protein and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; dilution, 1:1,000; BD), a cytoplasmic housekeeping protein, were used as a loading control to determine the relative intensity ratio.
2.6. Cytokines determinations
IFN-γ, MCP-1, TNF-α, IL-6, IL-10, and IL-12p70 were determined using a Cytometric Bead Array mouse inflammation kit (CBA; BD Biosciences). Supernatant samples were mixed to capture specific beads for each cytokine. Anti-IFN-γ, anti-MCP-1, anti-TNF-α, anti-IL-6, anti-IL-10, and anti-IL-12p70 antibodies were added, conjugated with phycoerythrin and incubated for 2 h at RT, protected from light. Tubes were then centrifuged (200G for 5 min) and the supernatant was carefully aspired and discarded. The pellets containing beads were re-suspended and the samples were analyzed on the Gallios flow cytometer (Beckman Coulter). The data obtained were analyzed by the Kaluza software.
2.7. Statistical analysis.
All data were subjected to statistical analysis using Prism 7.0 (GraphPad). P values for multiple comparisons were calculated by analysis of variance (ANOVA) and were adjusted by use of the Tukey’s multiple comparison analysis. P values for individual comparisons were calculated using student’s t-test analysis. P values of <0.05 were considered significant.
3. Results
3.1. METH reduces TLR4 distribution and expression on NR-9460 cells upon LPS stimulation.
Since TLR4 activation leads to inflammation, we investigated the impact of METH on the distribution of TLR4 on the surface of NR-9460 microglial-like cells after challenged with LPS using flow cytometry (Fig. 1). We found that all the conditions have increased TLR4 distribution on the surface of NR-9460 microglia-like cells compared to control naïve microglia (P<0.05) (Fig. 1B). LPS- and MC + LPS-treated microglia demonstrated the highest distribution of TLR4 (P<0.05) (Fig. 1B). Although METH showed no difference in the distribution of TLR4 on the surface of NR-9460 cells relative to the untreated microglia, METH significantly reduces the distribution of these innate immunity receptors upon activation after treatment with LPS (P<0.05) (Fig. 1B). Given that minocycline (MC) inhibits microglial activation, this antibiotic was used as a positive control. MC did not alter the distribution of TLR4 on LPS-activated NR-9460 cells. To further confirm the possibility that METH impairs the expression of TLR4 receptors after exposure to LPS, we performed western blot analysis comparing untreated and METH-treated glial cells after incubation with LPS (Fig. 1C). METH + LPS-treated NR-9460 cells demonstrated a significant decreased in the expression of TLR4 relative to LPS-treated microglia-like cells (P<0.05) (Fig. 1D). These results indicate that METH reduces TLR4 distribution and expression on microglial-like cells and may attenuate microglial activation upon interaction with microbial antigens.
Fig. 1. Methamphetamine (METH) reduces TLR4 distribution and expression on the surface of NR-9460 microglial-like cells upon lipopolysaccharide (LPS) stimulation.
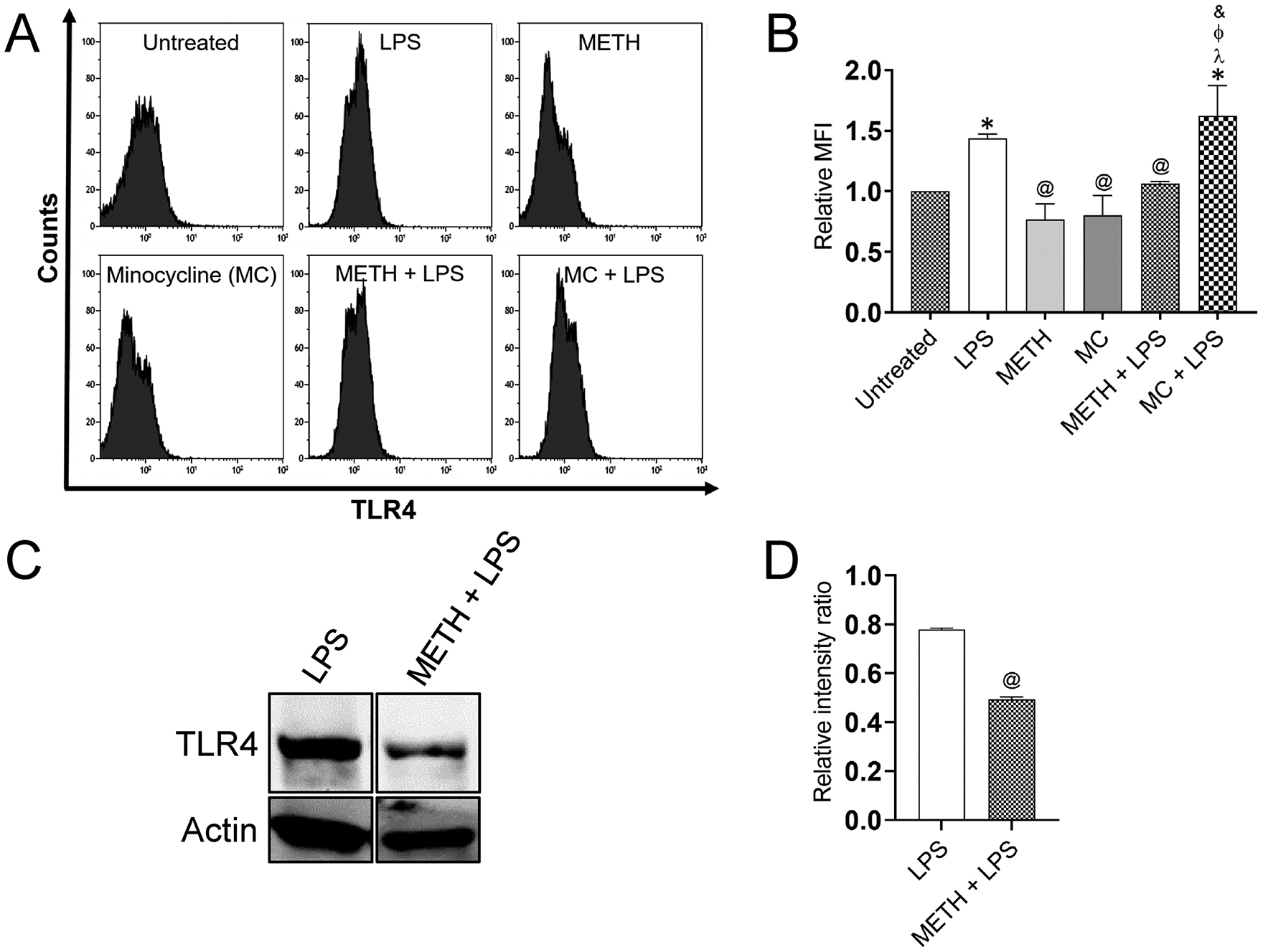
(A) The mean fluorescence intensity (MFI) of TLR4 molecules on NR-9460 cells was analyzed by flow cytometry. A density of 105 NR-9460 cells were treated with 25 μM METH or 1 μM minocycline (MC) for 2 h followed by an incubation with 10 μM LPS for 24 h. Representative histograms of untreated, LPS, METH, MC, METH + LPS, and MC + LPS treated microglia like-cells are shown. Each plot was generated after 10,000 events were analyzed. This experiment was performed 4X and similar results were obtained each time. (B) The relative MFI of TLR4 receptor was analyzed. Bars represent the mean of 4 independent experiments (n=4) and error bars indicate standard deviations. Symbols (*, @, λ, Φ, and &) indicate P value significance (P < 0.05) calculated using ANOVA and adjusted by use of the Tukey’s multiple comparison analysis. *, λ, and Φ indicate significantly higher relative MFI than in microglia from the untreated, METH, MC, and METH + LPS-treated groups, respectively. @ indicate significantly lower protein expression than in microglia from the LPS-treated group, respectively. (C) The expression of TLR4 in NR-9460 cells was determined by Western blot analysis. Microglia-like cells were incubated without or with 25 μM METH for 2 h, followed by an incubation with 10 μM LPS for 24 h. Actin was used as a housekeeping gene control. (D) The levels of expression of TLR4 were measured by determining the relative intensity ratios. Individual band intensities from the Western blot in panel C were quantified using ImageJ software. The actin gene was used as a reference to determine the relative intensity ratios shown in panel D. Bars represent the mean of 3 independent experiments (n=3) and error bars indicate standard deviations. @ indicates P value significance (P < 0.05) calculated using student’s t-test analysis. This experiment was performed 3X and similar results were obtained each time.
3.2. METH affects the morphology of NR-9460 cells and alters surface TLR4 receptors.
We used fluorescent microscopy to determine the impact of METH on microglial cell TLR4 distribution (Fig. 2A) and morphology (Fig. 2B). Untreated NR-9460 cells displayed a typical rounded morphology and large nuclei. LPS- and MC-treated cells demonstrated a slightly larger morphology, characterized by larger cytoplasm. METH significantly reduced microglia-like cell volume compared to all the other conditions. METH-treated NR-9460 cells were considerably small, rounded, and with minimal cytoplasmic region. Combination of METH and LPS treatments resulted in significantly larger cells compared to all the other conditions. METH- and LPS-treated microglia were characterized by large nuclei and the presence of visible filopodia extending from the cell cytoplasmic lamellopodium. Microglia treated with MC and LPS showed larger volume relative to untreated, LPS, METH, and MC. Our findings suggest that METH has profound effects in microglial morphology and TLR4 surface distribution.
Fig. 2. METH alters the distribution of TLR4 receptor and causes morphological changes in NR-9460 microglia-like cells.
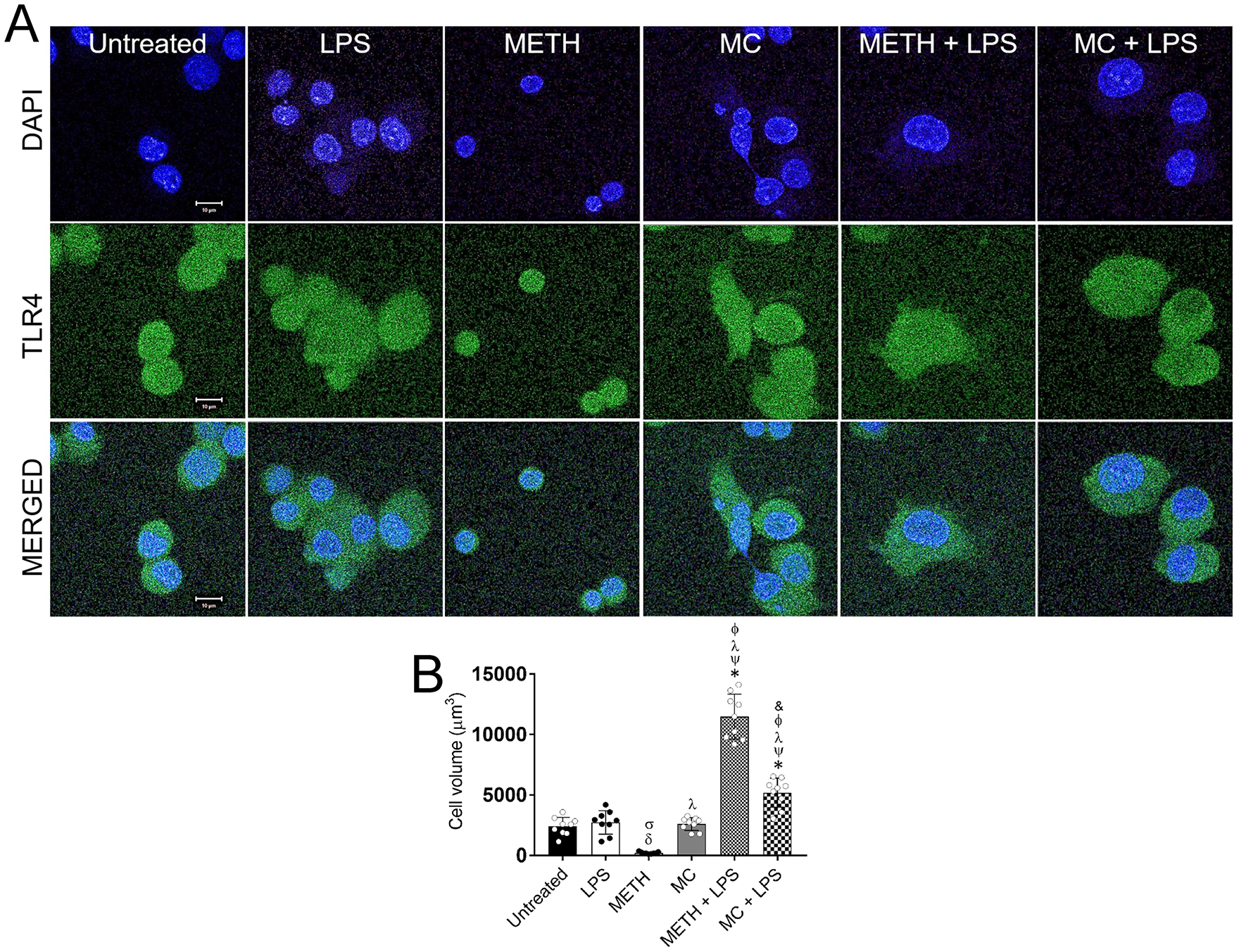
(A) Immunofluorescent images show the distribution of TLR4 (green; anti-TLR4 conjugated to Alexa Fluor 488) on the surface of untreated and treated NR-9460 cells with LPS, METH, MC, METH + LPS, and MC + LPS. Nuclei of microglia were stained in blue with DAPI. Scale bar, 10 μm. (B) The cell volume (V = 4/3 πr³) of untreated and treated microglia-like cells with LPS, METH, MC, METH + LPS, and MC + LPS was determined. Bars represent the mean of nine cell measurements (n=9; each symbol represents 1 microglia) and error bars indicate standard deviations. Symbols (δ, σ, *, ψ, λ, Φ, and &) indicate P value significance (P < 0.05) calculated using analysis of variance (ANOVA) and adjusted by use of the Tukey’s multiple comparison analysis. δ, σ, and & indicate significantly lower cell volume than in microglia from the untreated, LPS− and METH + LPS-treated groups, respectively. *, ψ, λ, and Φ indicate significantly higher cell volume than in microglia from the untreated, LPS, METH, and MC-treated groups, respectively. The experiments in A and B were performed twice and similar results were obtained each time.
3.3. METH alters the expression of downstream effector proteins in the TLR4 signaling pathway.
Given that METH modifies NR-9460 cell TLR4 expression and distribution, we used western blot analysis (Fig. 3A) to evaluate the effect of this substance of abuse on the expression of proteins associated with the TLR4/MD2 complex, which is responsible of activation of MyD88 dependent and independent pathways. The MyD88-dependent pathway is triggered by MyD88 recruitment of TRAF6 and IRAKs, for TAK1 activation. TAK1 phosphorylates IKKβ, leading to the activation of IκBa and NF-κB (Diamond et al., 2015) or MAPKs to further reinforce the inflammatory response via phosphorylation of p38 or JNK and activation of the transcription factors CREB or AP-1, respectively (Yesudhas et al., 2014). Therefore, we first determined the expression of MyD88 in NR-9460 microglia-like cells (Fig. 3B). LPS-treated microglia demonstrated the lowest expression of MyD88 in comparison to all the other conditions (P<0.05). Microglia treated with METH, MC, METH + LPS, and METH + MC showed significantly higher expression of MyD88 than untreated cells (P<0.05). METH + LPS treated cells had higher MyD88 expression compared to METH, MC, and MC + LPS microglia (P<0.05). IRAK1 was significantly and similarly expressed in microglia treated with MC, METH + LPS, and MC + LPS (P<0.05) (Fig. 3C). METH + LPS treated NR-9460 cells evinced significantly higher IRAK1 expression than LPS-treated cells (P<0.05). Untreated microglia showed significantly lower expression of TRAF6 compared to all the other conditions (P<0.05) (Fig. 3D). LPS-treated microglia displayed significantly higher expression of TRAF6 relative to METH-treated cells (P<0.05). Microglia treated with METH + LPS or METH + MC had the highest expression of TRAF6 (P<0.05). IKK expression was the highest in microglia treated with MC, METH + LPS, and MC + LPS (P<0.05) (Fig. 3E). LPS-treated NR-9460 cells showed the lowest expression of NF-κB (P<0.05) (Fig. 3F). Microglia treated with METH or MC evinced significantly higher NF-κB expression when compared to untreated, METH + LPS, and MC + LPS cells (P<0.05). p38 activates CREB, a transcription factor that supports the NF-κB activated immune response. We examined p38 expression in absence or presence of LPS, METH, or MC alone or combination of METH + LPS or MC + LPS (Fig. 3G). All the groups of microglial cells treated with LPS had reduced p38 expression compared to untreated-, METH-, or MC-treated cells (P<0.05) (Fig. 3G). The MyD88-independent pathway relies on TRIF recruitment of RIP1 or TRAF3. TRAF3 activates IRF3 through TBK1, inducing transcription of type I interferons (IFNs) and IFN-inducible genes (Diamond et al., 2015). We found that LPS-treated NR-9460 cells had lower IRF3 expression than all the other treatments (P<0.05) (Fig. 3H). Similarly, METH, MC, METH + LPS, and MC + LPS treated microglia showed significantly high IRF3 expression compared to untreated microglia (P<0.05). MC-treated microglia demonstrated a significantly higher expression of IRF3 when compared to METH, METH + LPS, and MC + LPS (P<0.05). Similarly, MC + LPS showed higher expression of IRF3 than METH + LPS (P<0.05). Taken together, METH reduces the expression of downstream proteins (e.g., NF-κB, p38, IRF3) in microglia, which are responsible for cytokine production upon stimulation with LPS.
Fig. 3. METH alters the expression of the TLR4/MD2 protein complex in microglial-like cells.
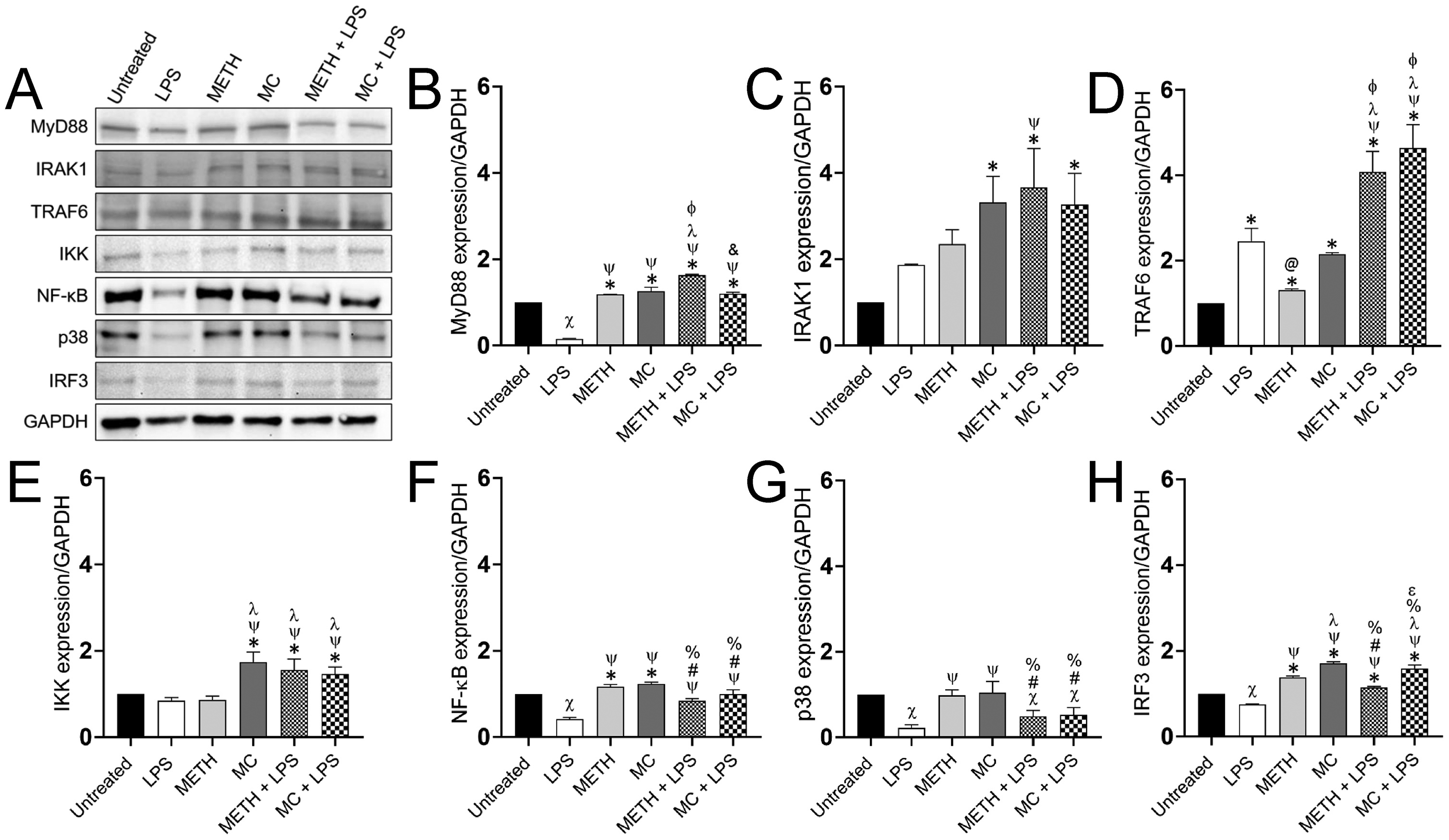
(A) The expression of MyD88, IRAK1, TRAF6, IKK, NF-kB, p38, and IRF3 in microglia-like cells grown in absence (untreated) or presence of LPS, METH, MC, METH + LPS, and MC + LPS was determined by a Western blot analysis and a representative gel is shown. A density of 105 NR-9460 cells was treated with 25 μM METH or 1 μM MC for 2 h followed by an incubation with 10 μM LPS for 24 h. The housekeeping gene Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a control. The levels of expression of (B) MyD88, (C) IRAK1, (D) TRAF6, (E) IKK, (F) NF-kB, (G) p38, and (H) IRF3 were measured. Individual band intensities from the Western blot in panel A were quantified using ImageJ software. GAPDH was used to determine the relative intensity ratios shown in graphs B-G. Symbols (*, X, @, #, ψ, λ, Φ, &, %, and ε) indicate P value significance (P < 0.05) calculated using ANOVA and adjusted by use of the Tukey’s multiple comparison analysis. *, ψ, λ, Φ, and ε indicate significantly higher protein expression than in microglia from the untreated, LPS−, METH−, MC−, and METH + LPS-treated groups, respectively. X, @, #, %, and & indicate significantly lower protein expression than in microglia from the untreated, LPS−, METH−, MC− and METH + LPS-treated groups, respectively. Each gel was run 3X and similar results were obtained each time.
3.4. METH reduces cytokine production by NR-9460 microglia-like cells upon activation with LPS.
TLR-induced inflammation is a well-established phenomenon and is perpetuated by several cytokines, ILs, and TNF-α, all of which are known to substantially regulate immune cells and inflammatory responses against infectious microorganisms (Swann et al., 2008; Yesudhas et al., 2014). Hence, we examined the impact of METH on microglia production of inflammatory cytokines upon activation with LPS (Fig. 4). We found that IFN-γ production by microglia was significantly higher in LPS, METH + LPS, and METH + MC than in untreated cells (P<0.05) (Fig. 4A). LPS-treated NR-9460 cells produced higher IFN-γ levels compared to METH, MC, METH + LPS, and METH + MC treated cells (P<0.05). MC-treated microglia alone or stimulated with LPS showed significantly lower levels of IFN-γ than METH-treated cells alone or challenge with LPS (P<0.05). MCP-1 and TNF-α was more elevated in NR-9460 cells treated with LPS, METH + LPS, and MC + LPS compared to the other groups (P<0.05) (Fig. 4B–C). METH-treated microglia evinced higher MCP-1 and TNF-α production than MC-treated cells (P<0.05). There was no difference observed in the production of MCP-1 in microglia-like cells treated with LPS, METH + LPS, and MC + LPS. METH + LPS-treated microglia demonstrated highest levels of TNF-α (Fig. 4C) whereas MC + LPS-treated cells had higher levels of TNF-α relative to LPS-treated cells. All the LPS-treated microglia groups secreted the highest levels of IL-6 (Fig. 4D), IL-10 (Fig. 4E), IL-12p70 (Fig. 4F) with LPS > METH + LPS > MC + LPS. There was no difference observed in the production of IL-6 (Fig. 4D) and IL-10 (Fig. 4E) in untreated or METH and MC-treated microglia-like cells. METH and MC-treated groups displayed higher IL-12p70 levels than untreated microglia (Fig. 4E). We found that METH alters microglia-like cells ability to secrete cytokines upon LPS activation and may interfere with these cells’ responses to microbial pathogens and antigens.
Fig. 4. METH compromises the production of pro-inflammatory mediators by microglia upon activation with LPS.
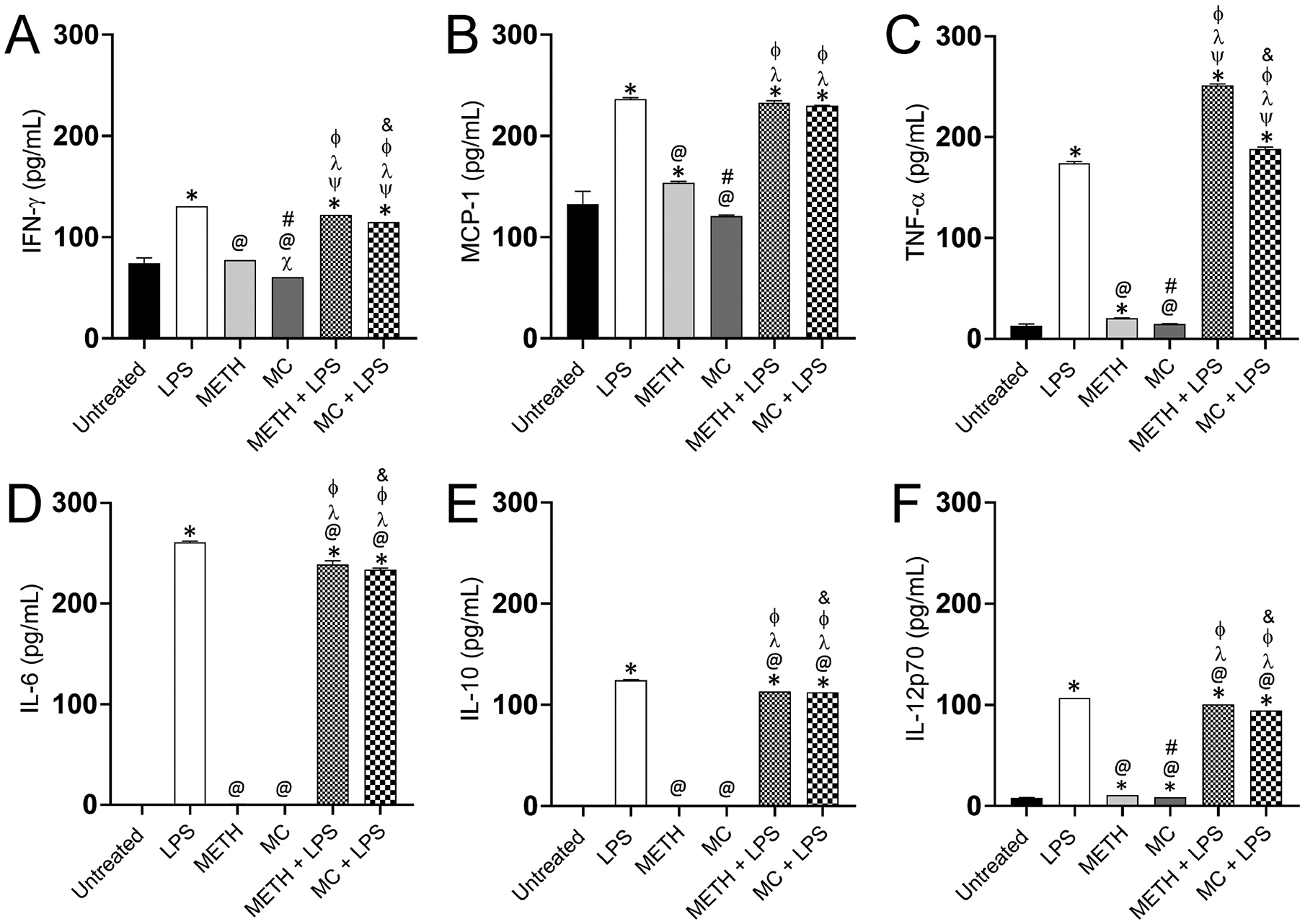
The supernatants (n = 3 supernatants per group) from untreated, LPS, METH, MC, METH + LPS, and MC + LPS treated microglia like-cells grown for 24 h were processed and analyzed for (A) IFN-γ, (B) MCP-1, (C) TNF-α, (D) IL-6, (E) IL-10, and (F) IL-12p70 levels with the BD cytometric bead array mouse inflammation kit. Bars represent the mean values; error bars indicate standard deviations. Symbols (*, X, @, #, ψ, λ, Φ, and &) indicate P value significance (P < 0.05) calculated using ANOVA and adjusted by use of the Tukey’s multiple comparison analysis. *, ψ, λ, and Φ indicate significantly higher cytokine levels than in microglia from the untreated, LPS−, METH−, and MC-treated groups, respectively. X, @, # and & indicate significantly lower cytokine levels than in microglia from the untreated, LPS−, METH−, and METH + LPS-treated groups, respectively. Cytokine quantification was performed twice with similar results obtained.
4. Discussion
METH is a powerful CNS stimulant that causes microglia-induced neuroinflammation resulting in neurotoxicity in users. The mechanisms underlying METH-induced microglial activation remain poorly understood. Recent evidence indicates that METH exacerbates neuroinflammation, at least partly, through the activation of TLR4 after binding to its co-receptor MD2 (Park et al., 2009; Wang et al., 2019). Given the direct interaction of METH and TLR4, we investigated the effect of METH on TLR4 signaling pathways and its upregulation of pro-inflammatory cytokines after LPS-stimulation. Our results demonstrate that LPS alone enhances the distribution of TLR4 on microglia-like cells compared to naïve cells. However, METH-treated microglia challenged with LPS evinced a reduction in the distribution and expression of TLR4 compared to glial cells only exposed to LPS. It is plausible that a reduction in microglial-TLR4 expression after METH treatment impairs the response of these phagocytic cells to CNS Gram-negative bacterial infections (e.g., Neisseria meningitides). METH use is a risk factor for the acquisition of meningococcal disease among men who have sex with men (Ridpath et al., 2015). In addition, LPS has been abundantly identified and quantified (e.g., ~100,000 pg/mL) in the cardiovascular system (e.g., heart and veins) of deceased METH-users (Zhu et al., 2005). METH increases the blood brain barrier (BBB) injury biomarker levels in blood, permeability, and penetration (Huang et al., 2013), suggesting that the acquisition of a CNS bacterial infection in the setting of METH use is possible.
Combination of METH and LPS alter the morphology of microglia. Microglia treated with METH and challenged with LPS showed larger cellular size compared to those cells in the other groups. Our results are in agreement with previous studies demonstrating that chronic exposure of cardiomyocytes to METH facilitate the development of cellular hypertrophy (Maeno et al., 2000a; Maeno et al., 2000b). METH increases the cross surface area of cardiomyocytes and enhances the formation of actin bundles and microtubular structures (Maeno et al., 2000a), which are associated with cardiac functional disorder (Maeno et al., 2000b). It is probable that similar activation of microglia occurs after exposure to METH and LPS resulting in neuroinflammation and neurotoxicity. This hypothesis is supported by the observation that microglia exposed to METH and LPS produce high levels of TNF-α, a cytokine significantly released during activation of phagocytic cells and inflammation. Even though microglia show similar trend in the production of IL-10, an anti-inflammatory cytokine, we have previously demonstrated in mice that these anti-inflammatory mediators might not be sufficient to prevent neuroinflammation and neurotoxicity (Martinez et al., 2009). MC-treated microglia challenged with LPS also showed cellular hypertrophy and high levels of TNF-α, which is indicative that microglial size increase or activation is driven by exposure to the drug and endotoxin. Microglia treated with METH alone displayed small cells with large nuclei and a limited cytoplasmic region. It is likely that METH inhibits actin polymerization in microglia, which may impair TLR4 distribution on the cell surface. We have previously shown that METH immobilize MAC-1 receptors in macrophages (Martinez et al., 2009). Moreover, METH reduces the expression of Fcγ receptors on the surface of macrophages and microglia (Aslanyan et al., 2019), which are involved in the recognition of opsonized antigen and antibody-mediated phagocytosis. METH modulates actin cytoskeletal dynamics and causes internalization of surface molecules such as the tight junction molecule occludin (Park et al., 2013). Likewise, we demonstrated that METH alters BBB integrity and modifies the expression of tight junction and adhesion molecules using a murine model of METH administration, we demonstrated that METH alters BBB integrity and modifies the expression of tight junction and adhesion molecules (Eugenin et al., 2013). Furthermore, pharmacological levels of METH in human blood and organs are cytotoxic and cause apoptosis in phagocytes (Aslanyan et al., 2019).
The TLR4/MD2 complex determines the initiation of the NF-κB activation signaling cascade resulting in the production of pro-inflammatory cytokines, which are important in the amplification of the immune response (Chen et al., 2012). Surprisingly, we found that with the exception of TRAF6, LPS inhibited the proteins involved in the NF-κB signaling pathway in microglia, although LPS-treated cells in absence or presence of LPS produced elevated levels of pro-inflammatory cytokines, TNF-α and IL-6. It is conceivable that LPS-induced neuroinflammation and activation of microglia is mediated by microRNAs (miR) (Karthikeyan et al., 2016). For instance, miR-155 is produced upon LPS challenge targeting Socs-1, a negative regulator of inflammation in microglia, which increases the production of TNF-α and IL-6 (Cardoso et al., 2012). We recently demonstrated that genes for TNF-α and IL-6 along with IL-1α, IL-β, KC, and MIP-2 were highly expressed or evinced similar increasing trend in untreated and METH-treated murine brain tissue after LPS challenge (DiCaro et al., 2019).
METH and MC similarly increased the expression of MyD88, IRAK1, TRAF6, and NF-κB in microglia-like cells relative to naïve cells. METH binds to MD2, the co-receptor of TLR4, activating the NF-κB signaling pathway (Wang et al., 2019). The interaction of METH with the TLR4/MD2 complex promotes the release of pro-inflammatory mediators, which amplify the reward changes in neuronal activity (Wang et al., 2019). Although we observed an increased in microglial-NF-κB activation after exposure to METH, we had low levels of pro-inflammatory cytokines compared to all the LPS-treated conditions. This discrepancy with recent findings by Wang and colleagues (Wang et al., 2019) can be explained based on the fact that we used a different microglial cell line (NR-9460 vs. BV-2 cells) and a physiological METH dose (25 vs. 200 μM; 7 -fold difference) in our studies. MC-treated microglia evinced high NF-κB activity but similar to METH, low pro-inflammatory cytokines production. MC inhibits neuroinflammation by microglia via suppression of adenosine A 2A (Tao et al., 2017) or P2X4 receptors (Long et al., 2018). Interestingly, microglia incubated with METH or MC and challenged with LPS demonstrated a progressive upregulation of MyD88, IRAK1, and TRAF6, the early proteins involved in the activation of NF-κB. However, this trend was not sustained and was considerably reduced after the TRAF6 intersection indicating that the TLR4/MD2 complex signaling diverge to NF-κB, MAPks, and IRF3. In this regard, the TLR4/MD2 complex activates members of the MAPKs to activate an alternative closely related pathway that phosphorylates p38 and c-Jun N-terminal Kinase (JNK). p38 was substantially reduced in microglia incubated with METH or MC and sensitized with LPS, supporting the notion that miR may be involved in the modulation of pro-inflammatory cytokines in these CNS resident immune cells. In response to various TLR ligands, reduced activity of NF-κB, JNK, and p38 was observed in B cells and embryonic fibroblasts derived from TAK1-deficient mice (Sato et al., 2005). Alternatively, TRIF recruits TRAF3, TBK1, IKK, and IRF3 resulting in the production of the type I interferon (IFN). METH, MC, or MC + LPS enhance IRF3 expression in microglia-like cells. Nevertheless, METH impairs IRF3 expression in microglia upon LPS exposure, although IFN-γ and IL-12 production was stimulated by the endotoxin. IL-12 is indispensable for viral clearance and regulates the production of IFN-γ and other cytokines (Guo et al., 2019). MiR-9 regulates microglial-associated inflammation by downregulating the expression of the target protein, monocyte chemotactic protein-induced protein 1 (MCPIP1) (Yao et al., 2014). It is possible that miR-9 is modulating the production of IFN-γ in microglia independently of IRF3. Furthermore, this notion is supported by the comparable levels of MCP-1 and IFN-γ produced by microglia-like cells.
In conclusion, we demonstrated that METH decreases TLR4 distribution and expression on the surface of microglia-like cells upon LPS exposure. METH also causes cellular morphological changes, impairs the TLR4/MD2 complex signaling pathways, compromises the activation of NF-κB, and reduces the microglial production of pro-inflammatory mediators after LPS challenge. Our findings highlight the importance of investigating the impact of METH on the CNS molecular and cellular immunity of users. Finally, future in vivo studies testing the effects of METH on microglial function upon antigenic challenge are warranted to better understand the role of these CNS resident cells in neuroinflammation and neurotoxicity.
Highlights.
METH-induced microglia neuroinflammation might be important in neural toxicity.
METH causes morphological changes in NR-9460 microglia-like cells after LPS challenge.
METH decreases TLR4 distribution and expression on the surface of microglia-like cells upon LPS stimulation.
METH impairs the TLR4/MD2 complex signaling pathways and interferes NF-κB activation.
METH reduces the microglial production of pro-inflammatory cytokines.
Acknowledgements
A.M.R. and L.R.M. were supported by the National Institute of Allergy and Infectious Diseases of the NIH under award number R01AI145559. D.E.R.R. was supported by the National Institute of General Medical Sciences (NIGMS) of the NIH under the UPR-Ponce Research Initiative for Scientific Enhancement (RISE) training grant R25GM096955. L.R.M. was supported by the NIGMS of the NIH under award number 1R15GM117501. L.R.M. was supported by the Border Biomedical Research Center (BBRC; National Institute on Minority Health and Health Disparities (NIMHHD) award number 5U54MD007592), UTEP’s Research Centers in Minority Institutions (RCMI) Program. We are grateful to the personnel of the DNA Sequencing and Gene Expression Analysis (Mrs. Ana Betancourt) and the Cytometry, Screening, and Imaging (Dr. Armando Varela-Ramirez and Mrs. Gladys Almodovar) core facilities at UTEP. These facilities are part of the BBRC and supported by NIMHHD award number 5U54MD007592.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no conflict of interest.
References
- Aslanyan L, Lee HH, Ekhar VV, Ramos RL, Martinez LR, 2019. Methamphetamine Impairs IgG1-Mediated Phagocytosis and Killing of Cryptococcus neoformans by J774.16 Macrophage- and NR-9640 Microglia-Like Cells. Infect Immun 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso AL, Guedes JR, Pereira de Almeida L, Pedroso de Lima MC, 2012. miR-155 modulates microglia-mediated immune response by down-regulating SOCS-1 and promoting cytokine and nitric oxide production. Immunology 135, 73–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Jalabi W, Shpargel KB, Farabaugh KT, Dutta R, Yin X, Kidd GJ, Bergmann CC, Stohlman SA, Trapp BD, 2012. Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J Neurosci 32, 11706–11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherner M, Letendre S, Heaton RK, Durelle J, Marquie-Beck J, Gragg B, Grant I, Group HIVNRC, 2005. Hepatitis C augments cognitive deficits associated with HIV infection and methamphetamine. Neurology 64, 1343–1347. [DOI] [PubMed] [Google Scholar]
- Cho AK, Melega WP, Kuczenski R, Segal DS, 2001. Relevance of pharmacokinetic parameters in animal models of methamphetamine abuse. Synapse 39, 161–166. [DOI] [PubMed] [Google Scholar]
- Colonna M, Butovsky O, 2017. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol 35, 441–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond CE, Khameneh HJ, Brough D, Mortellaro A, 2015. Novel perspectives on non-canonical inflammasome activation. Immunotargets Ther 4, 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCaro D, Lee HH, Belisario C, Ramos RL, Martinez LR, 2019. Combination of acute intravenous methamphetamine injection and LPS challenge facilitate leukocyte infiltration into the central nervous system of C57BL/6 mice. Int Immunopharmacol 75, 105751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du SH, Qiao DF, Chen CX, Chen S, Liu C, Lin Z, Wang H, Xie WB, 2017. Toll-Like Receptor 4 Mediates Methamphetamine-Induced Neuroinflammation through Caspase-11 Signaling Pathway in Astrocytes. Front Mol Neurosci 10, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis RJ, Childers ME, Cherner M, Lazzaretto D, Letendre S, Grant I, Group HIVNRC, 2003. Increased human immunodeficiency virus loads in active methamphetamine users are explained by reduced effectiveness of antiretroviral therapy. J Infect Dis 188, 1820–1826. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, Greco JM, Frases S, Nosanchuk JD, Martinez LR, 2013. Methamphetamine alters blood brain barrier protein expression in mice, facilitating central nervous system infection by neurotropic Cryptococcus neoformans. J Infect Dis 208, 699–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galindo GR, Casey AJ, Yeung A, Weiss D, Marx MA, 2012. Community associated methicillin resistant Staphylococcus aureus among New York City men who have sex with men: qualitative research findings and implications for public health practice. J Community Health 37, 458–467. [DOI] [PubMed] [Google Scholar]
- Gonzales R, Marinelli-Casey P, Shoptaw S, Ang A, Rawson RA, 2006. Hepatitis C virus infection among methamphetamine-dependent individuals in outpatient treatment. J Subst Abuse Treat 31, 195–202. [DOI] [PubMed] [Google Scholar]
- Guo Y, Cao W, Zhu Y, 2019. Immunoregulatory Functions of the IL-12 Family of Cytokines in Antiviral Systems. Viruses 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris DS, Boxenbaum H, Everhart ET, Sequeira G, Mendelson JE, Jones RT, 2003. The bioavailability of intranasal and smoked methamphetamine. Clin Pharmacol Ther 74, 475–486. [DOI] [PubMed] [Google Scholar]
- Hedegaard H, Bastian BA, Trinidad JP, Spencer M, Warner M, 2018. Drugs Most Frequently Involved in Drug Overdose Deaths: United States, 2011–2016. Natl Vital Stat Rep 67, 1–14. [PubMed] [Google Scholar]
- Huang SH, Wang L, Chi F, Wu CH, Cao H, Zhang A, Jong A, 2013. Circulating brain microvascular endothelial cells (cBMECs) as potential biomarkers of the blood-brain barrier disorders caused by microbial and non-microbial factors. PLoS One 8, e62164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambugu A, Meya DB, Rhein J, O’Brien M, Janoff EN, Ronald AR, Kamya MR, Mayanja-Kizza H, Sande MA, Bohjanen PR, Boulware DR, 2008. Outcomes of cryptococcal meningitis in Uganda before and after the availability of highly active antiretroviral therapy. Clin Infect Dis 46, 1694–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karthikeyan A, Patnala R, Jadhav SP, Eng-Ang L, Dheen ST, 2016. MicroRNAs: Key Players in Microglia and Astrocyte Mediated Inflammation in CNS Pathologies. Curr Med Chem 23, 3528–3546. [DOI] [PubMed] [Google Scholar]
- Koutsouras GW, Ramos RL, Martinez LR, 2017. Role of microglia in fungal infections of the central nervous system. Virulence 8, 705–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langford D, Adame A, Grigorian A, Grant I, McCutchan JA, Ellis RJ, Marcotte TD, Masliah E, Group HIVNRC, 2003. Patterns of selective neuronal damage in methamphetamine-user AIDS patients. J Acquir Immune Defic Syndr 34, 467–474. [DOI] [PubMed] [Google Scholar]
- Liu X, Su H, Chu TH, Guo A, Wu W, 2013. Minocycline inhibited the pro-apoptotic effect of microglia on neural progenitor cells and protected their neuronal differentiation in vitro. Neurosci Lett 542, 30–36. [DOI] [PubMed] [Google Scholar]
- Long T, He W, Pan Q, Zhang S, Zhang Y, Liu C, Liu Q, Qin G, Chen L, Zhou J, 2018. Microglia P2X4 receptor contributes to central sensitization following recurrent nitroglycerin stimulation. J Neuroinflammation 15, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeno Y, Iwasa M, Inoue H, Koyama H, Matoba R, 2000a. Methamphetamine induces an increase in cell size and reorganization of myofibrils in cultured adult rat cardiomyocytes. Int J Legal Med 113, 201–207. [DOI] [PubMed] [Google Scholar]
- Maeno Y, Iwasa M, Inoue H, Koyama H, Matoba R, Nagao M, 2000b. Direct effects of methamphetamine on hypertrophy and microtubules in cultured adult rat ventricular myocytes. Forensic Sci Int 113, 239–243. [DOI] [PubMed] [Google Scholar]
- Mankatittham W, Likanonsakul S, Thawornwan U, Kongsanan P, Kittikraisak W, Burapat C, Akksilp S, Sattayawuthipong W, Srinak C, Nateniyom S, Tasaneeyapan T, Varma JK, 2009. Characteristics of HIV-infected tuberculosis patients in Thailand. Southeast Asian J Trop Med Public Health 40, 93–103. [PubMed] [Google Scholar]
- Martinez LR, Mihu MR, Gacser A, Santambrogio L, Nosanchuk JD, 2009. Methamphetamine enhances histoplasmosis by immunosuppression of the host. J Infect Dis 200, 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melega WP, Cho AK, Harvey D, Lacan G, 2007. Methamphetamine blood concentrations in human abusers: application to pharmacokinetic modeling. Synapse 61, 216–220. [DOI] [PubMed] [Google Scholar]
- Najera JA, Bustamante EA, Bortell N, Morsey B, Fox HS, Ravasi T, Marcondes MC, 2016. Methamphetamine abuse affects gene expression in brain-derived microglia of SIV-infected macaques to enhance inflammation and promote virus targets. BMC Immunol 17, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO, 2009. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458, 1191–1195. [DOI] [PubMed] [Google Scholar]
- Park M, Kim HJ, Lim B, Wylegala A, Toborek M, 2013. Methamphetamine-induced occludin endocytosis is mediated by the Arp2/3 complex-regulated actin rearrangement. J Biol Chem 288, 33324–33334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D, Desai GM, Frases S, Cordero RJ, DeLeon-Rodriguez CM, Eugenin EA, Nosanchuk JD, Martinez LR, 2013. Methamphetamine enhances Cryptococcus neoformans pulmonary infection and dissemination to the brain. MBio 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridpath A, Greene SK, Robinson BF, Weiss D, Meningococcal Investigation T, 2015. Risk Factors for Serogroup C Meningococcal Disease during Outbreak among Men who Have Sex with Men, New York City, New York, USA. Emerg Infect Dis 21, 1458–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Matsumoto K, Takeuchi O, Akira S, 2005. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol 6, 1087–1095. [DOI] [PubMed] [Google Scholar]
- Shaerzadeh F, Streit WJ, Heysieattalab S, Khoshbouei H, 2018. Methamphetamine neurotoxicity, microglia, and neuroinflammation. J Neuroinflammation 15, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharikova AV, Quaye E, Park JY, Maloney MC, Desta H, Thiyagarajan R, Seldeen KL, Parikh NU, Sandhu P, Khmaladze A, Troen BR, Schwartz SA, Mahajan SD, 2018. Methamphetamine Induces Apoptosis of Microglia via the Intrinsic Mitochondrial-Dependent Pathway. J Neuroimmune Pharmacol 13, 396–411. [DOI] [PubMed] [Google Scholar]
- Swann JB, Vesely MD, Silva A, Sharkey J, Akira S, Schreiber RD, Smyth MJ, 2008. Demonstration of inflammation-induced cancer and cancer immunoediting during primary tumorigenesis. Proc Natl Acad Sci U S A 105, 652–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao T, Fu J, Liu YG, Li ZX, Li XG, 2017. [Minocycline Prevent Microglial Activation via Suppression of Adenosine A 2A Receptor in a Rat Stroke Ischemia/Reperfusion Model]. Sichuan Da Xue Xue Bao Yi Xue Ban 48, 221–224. [PubMed] [Google Scholar]
- Valencia F, Bubar MJ, Milligan G, Cunningham KA, Bourne N, 2012. Influence of methamphetamine on genital herpes simplex virus type 2 infection in a mouse model. Sex Transm Dis 39, 720–725. [DOI] [PubMed] [Google Scholar]
- Wang X, Northcutt AL, Cochran TA, Zhang X, Fabisiak TJ, Haas ME, Amat J, Li H, Rice KC, Maier SF, Bachtell RK, Hutchinson MR, Watkins LR, 2019. Methamphetamine Activates Toll-Like Receptor 4 to Induce Central Immune Signaling within the Ventral Tegmental Area and Contributes to Extracellular Dopamine Increase in the Nucleus Accumbens Shell. ACS Chem Neurosci 10, 3622–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, Wagner J, Hasler LM, Wild K, Skodras A, Blank T, Staszewski O, Datta M, Centeno TP, Capece V, Islam MR, Kerimoglu C, Staufenbiel M, Schultze JL, Beyer M, Prinz M, Jucker M, Fischer A, Neher JJ, 2018. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556, 332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu E, Liu J, Liu H, Wang X, Xiong H, 2017. Role of microglia in methamphetamine-induced neurotoxicity. Int J Physiol Pathophysiol Pharmacol 9, 84–100. [PMC free article] [PubMed] [Google Scholar]
- Yao H, Ma R, Yang L, Hu G, Chen X, Duan M, Kook Y, Niu F, Liao K, Fu M, Hu G, Kolattukudy P, Buch S, 2014. MiR-9 promotes microglial activation by targeting MCPIP1. Nat Commun 5, 4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yesudhas D, Gosu V, Anwar MA, Choi S, 2014. Multiple roles of toll-like receptor 4 in colorectal cancer. Front Immunol 5, 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu BL, Ishikawa T, Michiue T, Quan L, Maeda H, 2005. Postmortem serum endotoxin level in relation to the causes of death. Leg Med (Tokyo) 7, 103–109. [DOI] [PubMed] [Google Scholar]