

Abstract
Although RNA sequencing (RNA-seq) has become the most advanced technology for transcriptome analysis, it also confronts various challenges. As we all know, the workflow of RNA-seq is extremely complicated and it is easy to produce bias. This may damage the quality of RNA-seq dataset and lead to an incorrect interpretation for sequencing result. Thus, our detailed understanding of the source and nature of these biases is essential for the interpretation of RNA-seq data, finding methods to improve the quality of RNA-seq experimental, or development bioinformatics tools to compensate for these biases. Here, we discuss the sources of experimental bias in RNA-seq. And for each type of bias, we discussed the method for improvement, in order to provide some useful suggestions for researcher in RNA-seq experimental.
1. Introduction
With the development of massive parallel sequencing, high-throughput sequencing (NGS) of RNA (RNA-seq) has become a very common tool in molecular biology. It almost affects our understanding for the function of genomic [1] and provides valuable resources for other scientific disciplines. However, RNA-seq is a process of extremely intricate, including RNA extraction and purification, library construction, sequencing, and bioinformatics analysis. These processes can inevitably introduce some deviations (Table 1), which influence the quality of RNA-seq datasets and result in their erroneous interpretation. Therefore, understanding these biases is critical to avoiding erroneous interpretation of the data and to realize the full potential of this powerful technology.
Table 1.
Sources of main bias in RNA-seq.
| Bias sources |
|---|
| Sample preservation (1) Degradation of RNA: such as tissue autolysis; nucleic acid degradation and cross-linking during the preparation of formalin-fixed; formalin-fixed paraffin-embedded (FFPE) [6] (2) RNA extraction: such as using TRIzol [12] (3) Alien sequence contamination [73] (4) Low-quality and/or low-quantity RNA [23] |
| Library preparation (1) mRNA enrichment bias: such as 3′-end capture bias [74] (2) RNA fragmentation bias [31] (3) Primer bias: such as random hexamer bias; mispriming; nonspecific binding [75] (4) Adapter ligation bias: such as adaptor contamination [41] (5) Reverse transcription bias [76] (6) PCR amplification bias [77] (7) Machine failure; for example, incorrect PCR cycling temperatures [17] |
| Sequencing and imaging (1) Experimenter bias: such as cluster crosstalk caused by overloading the flowcell [78] (2) Sequencing platform bias [65] (3) Sequence context: such as AT/GC enrichment [79] (4) Machine failure: such as failure of laser, hard drive, software, and fluidics |
Generally, the representative workflow of RNA-seq analysis includes the extraction and purification of RNA from cell or tissue, the preparation of sequencing library, including fragmentation, linear or PCR amplification, RNA sequencing, and the processing and analysis of sequencing data (Figure 1). Commonly used NGS platforms, including Illumina and Pacific Biosciences, need PCR amplification during library construction to increase the number of cDNA molecules to meet the needs of sequencing. Nevertheless, the most problematic step in sample preparation procedures is amplification. It is due to the fact that PCR amplification stochastically introduces biases, which can propagate to later cycles [2]. In addition, PCR also amplifies different molecules with unequal probabilities, leading to the uneven amplification of cDNA molecules [3, 4]. Recently, researchers have proposed several different methods in order to reduce PCR amplification, such as PCR-free protocols and isothermal amplification. Nevertheless, these methods are not perfect and still present some artifacts and biases of sequencing. Consequently, understanding these biases is critical to get reliable data and will provide some useful advice to the researcher.
Figure 1.
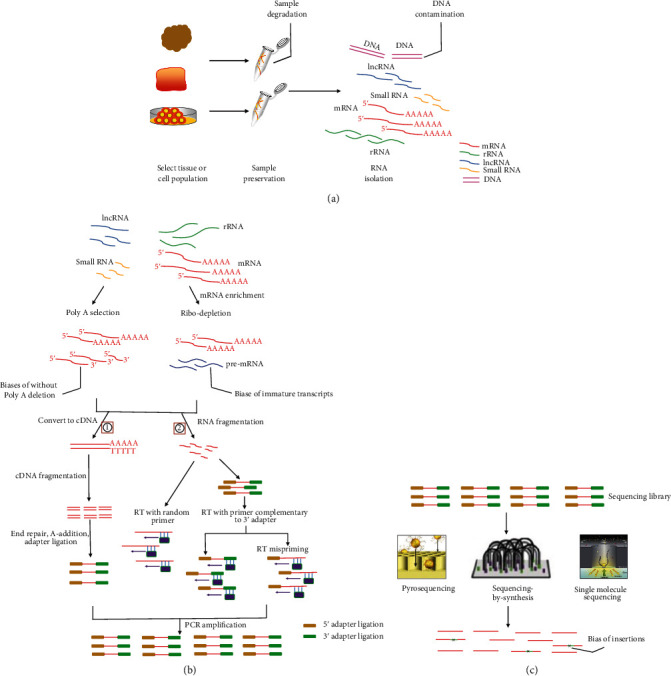
Simplified protocol of RNA-seq experiment and sources of bias. (a) Sample preservation and isolation. These biases can include sample degradation, DNA contamination. (b) Strategies for cDNA library construction. ①: the RNA directly converts to cDNA; then, cDNA was fragmented and library preparation. ②: classical a protocol. One method involves reverse transcription (RT) using random primers first, subsequently adapter ligations and sequencing (left). The other method is to first sequentially ligate 3′ and 5′ adapters, followed by performing cDNA synthesis with a primer complementary to the adapter (RT-primer), subsequently sequencing (right). On using the RT primer with a specific sequence, mispriming could occur due to annealing of the RT-primer to transcript sequences with some complementarity (RT mispriming). (c) RNA-seq platform (including Pyrosequencing, sequencing-by-synthesis, and single-molecule sequencing). These biases can be introduced by insertions and deletions, raw single-pass data, etc.
In this perspective article, we summarize the current situation and solutions on biases and discuss the source of bias in RNA-seq. The key point will be on solutions to reduce bias and improve the quality of library sequencing platform. Furthermore, we highlight the bias sources of different methods of amplification and how can amplification bias be reduced.
2. Sample Preservation and Isolation
Despite many studies have shown that RNA-seq has many advantages, it is still a rapidly developing biotechnology and faces several challenges. Among them, one often overlooked aspect is the sample preparation process, which may also bring potential variations and deviations on RNA-seq experiment, including RNA isolation, sample processing, library storage time, RNA input level (such as the difference in the number of start-up RNA), and sample cryopreservation (such as fresh or frozen preservation). Generally, good preservation of sample that may be used for transcriptome studies is more important, because many transcriptome protocols require high-quantity and high-quality nucleic acids [5]. Therefore, we will discuss the bias of sample in different preservation and isolation methods. A sum up and improvement suggestions are shown in Table 2.
Table 2.
Sources of bias in RNA-seq sample preservation and suggestions for improvement.
| Description | Suggestion for improvement |
|---|---|
| Sample preservation | |
| FFPE methods: causes modifications of biomolecules, such as cross-linkage of nucleic acids with proteins | Use of non-cross-linking organic fixatives and methacarn solution [6] |
| RNA extraction | |
| Using TRIzol: small RNA loss at low concentrations | Use high concentrations of RNA samples or avoid TRIzol extraction altogether [80] Use alternative protocols such as the mirVana miRNA isolation kit [14] |
2.1. The Storage and Preservation Methods of RNA
Studies have been demonstrated that RNA degradation is closely related to sample preservation or fixation method. At the present, as far as we know, the standard storage of tissues for RNA-seq has been in liquid nitrogen or freeze stored at -80°C. Unfortunately, frozen specimens are not widely available because they are costly to collect and maintain. Therefore, in diagnostic pathology archives, most tissue samples rely on the formalin-fixed and paraffin-embedded (FFPE) method for preservation [6]. Nevertheless, nucleic acids are more difficult to extract from FFPE tissue, because of the need to remove paraffin and counteract the covalent protein DNA interaction during the fixation process [7, 8]. Additionally, fixation delay, fixation process, tissue preparation, paraffin embedding, and archival preservation may lead to fragmentation, cross-linking, and chemical modification of FFPE tissue-derived nucleic acids, resulting in poor sequencing libraries [9]. Recently, the researcher proposed an optimization scheme [9]; the main problem to be considered with this method is as follows: (1) by minimizing the sample processing and freezing and thawing cycles, ensure that RNA is preserved as best as possible after extraction; (2) for degradation samples, it is best to use high sample input; (3) in the reverse transcription step, use random priming instead of oligo-dT or specific sequence as primers. These suggestions might help to mitigate some sequencing biases or errors to some extent, so that we can make full use of the FFPE sample to obtain reliable results.
2.2. The Isolation and Extraction of RNA
High-quality RNA purification is the premise of RNA-seq. However, due to the widespread existence of RNA degrading enzymes (RNases) [10–12], successful isolation of high-quality RNA remains challenging. At the present, the RNA extraction method can be divided into two types, including TRIzol (phenol: chloroform extraction) and Qiagen (silica-gel-based column procedures). These methods were mainly developed to extract long mRNAs and have been based on the assumption that all RNAs are equally purified, when these methods are applied to noncoding RNAs, which may be resulted in RNA degradation [13]. Currently, the mirVana kit was reported to be the best tool for producing high-yield and high-quality RNA [14].
3. Library Construction
After RNA isolation and extraction, the next step is library construction of transcriptome sequencing. Library construction usually begins with the depletion of ribosomal RNA (rRNA) or the enrichment of mRNA enrichment, because most of the total RNA of cellular or tissue is rRNA. For eukaryotic transcriptome, polyadenylated mRNAs are usually extracted by oligo-dT beads, or rRNAs are selectively depleted. Unlikely, prokaryote mRNAs are not stably polyadenylated. Hence, oligo d(T)-mediated messenger enrichment is not suitable; there is only the second option. Then, RNA is usually fragmented to a certain size range by physical or chemical method. The subsequent steps differ among experimental design and NGS platforms. However, studies indicated that most of the protocols currently used for library construction may introduce serious deviations. For example, RNA fragmentation can introduce length biases or errors, subsequently propagating to later cycles. Furthermore, library amplification may also be affected by primer bias, such as primer bias in multiple displacement amplification (MDA) [15], primer mismatch in PCR amplification [16, 17]. As a consequence, it may introduce nonlinear effects and inevitably compromise the quality of RNA-seq dataset, leading to the result of erroneous interpretation. Consequently, in the next section, we will describe and summarize the bias sources of library preparation, including mRNA enrichment, fragmentation, primer bias, adapter ligation, reverse transcription, and especially PCR. A sum up suggestions for improvement is presented in Table 3.
Table 3.
Sources of bias in RNA-seq library preparation and suggestions for improvement.
| Description | Suggestion for improvement |
|---|---|
| mRNA enrichment | |
| 3′-end capture bias that is introduced during poly (A) enrichment in RNA sequencing | Use rRNA depletion [81]. |
| Fragmentation | |
| RNA fragmentation by RNase III: not completely random, leading to reduced complexity | Use chemical treatment (e.g., zinc) rather than RNase III for RNA fragmentation [31] Intact RNAs can be reverse transcribed to cDNA by reverse transcriptase, then which was fragmented by mechanical or enzymatic methods [82] |
| Priming bias | |
| Random hexamer priming bias | RNA is not converted to dscDNA using random priming, instead of sequencing adapters that are ligated directly onto RNA fragments [39] A read count reweighing scheme was proposed that adjusts for the bias and makes the distribution of reads more uniform [40] |
| Adapter ligation | |
| Adapter ligation bias: due to substrate preferences of T4 RNA ligases | Use adapters with random nucleotides at the extremities to be ligated [42] |
| PCR | |
| (1) Bias due to preferential amplification of with neutral GC% (2) The number of cycles of high PCR amplification |
Use Kapa HiFi rather than Phusion polymerase [49] For extremely AT/GC-rich genomes, use the PCR additive TMAC or betaine, or lower extension temperatures and extended denaturation times [17, 48] Reduce the number of cycles of amplification [55] For the amplification of minute quantities of genomic DNA (single cell), use MDA rather than PCR [83] A large number of starting material, use amplification-free PCR [47] |
3.1. Input RNA
Notwithstanding, RNA-seq can be used to measure transcripts of any sample in principle; it has been a challenge to apply standard protocols to samples with either very low quantity or low quality (partially degraded) input RNA. It is due to the fact that the bias associated with low amounts of input RNA has strong and harmful effects on downstream analysis. If not noticed, this may have significant impact on the subsequent biological interpretation. Recently, the researcher has proposed several different methods to overcome the challenges of low-quality or low-quantity RNA sample, including RNase H (also known as SDRNA) [18], Ribo-Zero [19], SMART4 [20], Ovation RNA-seq System (NuGEN) [21], and Duplex-Specific Nuclease (DSN) light normalization [22]. Adiconis et al. [23] compared the relative merits of each method with a standard high-input and high-quality sample group, determining its applicability for a specific project. This result showed that RNase H was the best method for detecting low-quality RNA and even could effectively replace the standard RNA-seq method based on oligo (dT). For low-quantity RNA, the SMART and NuGEN approaches had lower duplication rates and significantly decreased the necessary amount of starting material compared to other methods.
3.2. rRNA Depletion
rRNAs are very abundant, often constituting 80% to 90% of total RNA. Due to the fact that rRNA sequence rarely arouse people's interest in RNA-seq experiments, it is necessary to remove rRNA from sample before library construction. The aim is in order to prevent most of the library and the majority of sequencing reads from being rRNA. A standard solution is to enrich for the polyadenylated (poly (A)) RNA transcripts with oligo (dT) primers. For eukaryotes, studies have been shown that oligo (dT) provides technical convenience for enriching mRNA from sample; it is due to the fact that most mRNA and many long noncoding RNAs (lncRNAs) have poly (A) tail [24]. Nevertheless, in addition to rRNA, this method also removes all non-poly (A) RNAs, such as replication-dependent histones, various lncRNAs, and bacterial mRNA [25, 26]. Moreover, oligo (dT) is difficult to capture incomplete mRNA molecules (such as mRNAs lacking intact poly (A) tails). Therefore, if the starting materials are from the FFPE sample, it is not the best method, because the RNAs of FFPE were degraded to a small average size. On the other hand, the non-poly (A) tailed mRNA enrichment method can be used to isolate RNA from any eukaryotic organism [27].
The second rRNA depletion approach takes the opposite method, targeting rRNA molecules and removing them. This rRNA depletion method can be used for subsequent sequencing of all non-rRNA molecules and is not limited to complete mRNA molecules. Unlike the oligo (dT), rRNA depletion relies on the exact sequence content of the ribosomal RNA, so commercially available or each given kits will only be effective for a specific group of species whose rRNA sequences complement the probes in the kit [28]. However, because rRNA depletion depends on sequence-specific hybridization of probe, there is a risk of nonspecific cross-hybridization and transcript removal, leading to biased representation of that transcript in the sequencing data. The researcher gave two suggestions to the selection of kit, oligonucleotide probes and an antibody specific for RNA: DNA hybrids, to minimize the effects of the oligonucleotide mishybridization [28]. On the other hand, rRNA depletion may capture more immature transcripts, leading to a complexity increase of sequencing data [29]. However, neither method can enrich poly (A) transcripts, such as poly (A)-histone mRNAs, including histone H1 variants [27]. Moreover, the rRNA depletion method is remarkably more expensive than mRNA isolation.
3.3. RNA Fragmentation
Currently, RNA is usually fragmented due to read length restriction (<600 bp) of sequencing technologies and the sensitivity of amplification to long cDNA molecules. There are two major approaches of RNA fragmentation: chemical (using metal ions) and enzymatic (using RNase III) [30]. Commonly, RNA is fragmented using metal ions such as Mg++ and Zn++ in high temperatures and alkaline conditions. This method yields more accurate transcript identification than RNase III digestion [31]. This result was also confirmed in Wery et al. [31]. Furthermore, intact RNAs can be reverse transcribed (RT) to cDNA by reverse transcriptase, subsequently was fragmented. Then, the cDNA was fragmented using the enzymatic or physical method. Examples of the enzymatic method include DNase I digestion, nonspecific endonuclease (like NEBNext dsDNA Fragmentase from New England Biolabs), and transposase-mediated DNA fragmentation (Illumina Nextera XT). However, the Tn5 transposase method showed sequence-specific bias [32], which is the preferred method when only small quantities of cDNA are available, since the cDNA fragmentation and adapter ligation are connected in one step [33]. Studies have shown that nonspecific restriction endonucleases indicate less sequence bias and have been shown to perform similarly to the physical methods with respect to cleavage-site sequence bias and coverage uniformity of target DNA [34, 35]. Another advantage of the enzymatic method is that they are easy to automate [36]. The physical method includes acoustic shearing, sonication, and hydrodynamic [17, 37, 38], which also can present nonrandom DNA fragmentation bias [35]. However, the physical cDNA fragmentation method is less amenable to automation than RNA fragmentation. Therefore, the physical method will be replaced by commercially available kits and the enzymatic method.
3.4. Primer Bias
Commonly, after mRNA is fragmented, which can be reverse transcribed into cDNA by random hexamers. However, studies have been indicated that random hexamer primer can lead to the deviation of nucleotide content of RNA sequencing reads, which also affects the consistency of the locations of reads along expressed transcripts. This may result in low complexity of RNA sequencing data. Given this bias, Mamanova et al. [39] proposed an alternative to RNA-seq using the Illumina Genome Analyzer. The reverse transcription takes place directly on the flowcells which yield stranded reads and avoids the amplification of polymerase chain reaction. RNA is not transformed into dscDNA using random priming but directly connected to RNA fragment by sequencing adapters. Then, the ligated RNA library is reverse transcribed on the flowcells [40]. Thus, the deviation is avoided due to primer. In addition, the researcher proposed using a bioinformatics tool, via reweighing scheme to adjust for the bias and make the distribution of the reads more uniform.
3.5. Adapter Ligation
Generally, as for the deep sequencing of RNA library preparation, a critical step is the ligation of adapter sequences. The selection of T4 RNA ligase (Rnl1 or Rnl2) or other RNA ligase is very important. Subsequently, the ligation products were amplified by PCR. Or, nucleotide homopolymer sequences were added by poly (A) polymerase [41] or terminal deoxyribonucleotidyl transferase [41] but prevent the unambiguous determination of the termini of the input RNAs. This method has also been widely used in the construction of small RNA library. Recently, studies have shown that adapter ligation introduces a significant but widely overlooked bias in the results of NGS small RNA sequencing. Hence, in order to alleviate this bias, the new Bio Scientific NEXTflex V2 protocol uses a set of random nucleotide adapters at the ligation boundary. And the study indicated that this protocol can reliably detect several Illumina-based methods to evade the capture of miRNAs. Although these results did not show a clear standard for small RNA library preparation, the data of the NEXTflex protocol had the best correlation with RT-qPCR [42].
3.6. Reverse Transcription
Currently, the strategies of transcriptome analysis are still to convert RNA to cDNA before sequencing. A known feature of reverse transcriptases is that they tend to produce false second strand cDNA through DNA-dependent DNA polymerase. This may not be able to distinguish the sense and antisense transcript and create difficulties for the data analysis. The researcher proposed several modifications. Among them, the deoxyuridine triphosphate (dUTP) method, one of the leading cDNA-based strategies, can be specifically removed by enzymatic digestion [43]. It can provide excellent library complexity, chain specificity, coverage uniformity, consistency with known annotation, and accuracy for expression analysis [44]. However, the effectiveness of antisense transcription near highly expressed genes should be carefully measured, since a small number of reads (about 1%) have been observed on the opposite chain [45]. Another method is to synthesize the first strand of cDNA using labeled random hexamer primer and SSS using DNA-RNA template-switching primer. Nevertheless, the two methods are laborious [46]. Additionally, for the SSS method, it requires a nonstandard sequencing data analysis scheme, and as part of the genome, complexity is lost in the process of converting four bases into three bases; about 30% of the unique matching sequencing readings are lost. Furthermore, the combination of random primers and template switching may lead to uneven gene coverage.
3.7. PCR Amplification
PCR is a basic tool widely in molecular biology laboratories. In particular, the combination of PCR and NGS sequencing promoted the explosive development of RNA sequence acquisition. However, PCR amplification has been proved to be the main source of artifacts and base composition bias in the process of library construction, which may lead to misleading or inaccurate conclusions in data analysis. Therefore, it is essential to avoid PCR bias, and great efforts have been expended on trying to control and mitigate bias in current. In the next section, we will discuss the sources of bias in PCR amplification and suggestions for improvement.
3.8. The Sources of PCR Amplification Biases and Improvement Methods
3.8.1. Extremely AT/GC-Rich
Studies have been indicated that fragments of GC-neutral can be amplified more than GC-rich or AT-rich fragments. Therefore, the fragments with high AT or very high GC content often have little or no amplification at all [47, 48]. These unfavorable features result in difficulties in genome sequencing of extremely AT-rich, such as human malaria parasite [48], or high GC (Bordetella pertussis) genomes (average GC content, about 75%). Bearing this in mind, Aird et al. [17] present a protocol to reduces the introduction of GC bias in the PCR library preparation stage by switching the polymerases, prolonging the denaturation step, and reducing the annealing and extension temperature. On the other hand, researchers developed a without amplification library construction approach [39]. Through the use of custom adapters, the samples without amplification and ligation can be hybridized directly with the oligonucleotides on the flowcell surface, thus avoiding the biases and duplicates of PCR. However, the amplification-free method requires high sample input, which limits its widely used.
Besides, the Phusion polymerase method is commonly used in PCR amplification at present, compared with other polymerase methods, which have processivity and fidelity [49]. However, the amplification efficiency of extremely (G+C)-rich or (A+T)-rich fragments was lower efficiency than (G+C)-neutral fragments. Several laboratories have compared different PCR polymerases and conditions to minimize amplification bias. For example, Quail et al. [49] estimated a large series of polymerases. This result showed that the best total enzyme was Kapa HiFi (Kapa Biosystems), because the genome coverage with Kapa HiFi was more uniform than Phusion, which was very close to the result without PCR.
In addition, in order to overcome the amplification bias of AT/GC-rich, it is necessary to the addition of substances for enhancement PCR specificity and/or yield. The most effective PCR enhancing additives currently used are betaine [50]. It is an amino acid mimic that acts to balance the differential Tm between AT and GC base pairs and has been effectively used to improve the coverage of GC-rich templates [17]. Furthermore, Oyola et al. [48] tested various PCR amplification conditions by testing a series of polymerases and their tolerance to AT-rich templates in the absence or presence of tetramethylammonium chloride (TMAC). Their result showed that the TMAC can remarkably increase the amplification of AT-rich regions in Kapa HiFi in the presence. Additionally, a number of additives have been reported to play an important role in reducing the bias of PCR amplification, including small amides such as formamide, small sulfoxides such as dimethyl sulfoxide (DMSO), or reducing compounds such as β-mercaptoethanol or dithiothreitol (DTT) [50].
3.8.2. PCR Cycle
As we all know, PCR can exponentially amplify DNA/cDNA templates, thus leading to a significant increase of amplification bias with the number of PCR cycles [51]. Therefore, it is recommended that PCR be performed using as few cycle numbers as possible to mitigation bias [52, 53]. At the present, several laboratories have compared different PCR cycle number to reduce amplification bias. Wu et al. [54] performed a comprehensive analysis. The results of the study indicate that comparing with the lower cycle number, the higher cycle number can produce significant biases or artifacts in standard amplifications of mixed templates. In addition, Sze and Schloss's [55] study indicated that reducing the number of cycles of amplification can also decrease PCR biases and artifacts using a mock community and human stool samples.
3.9. Alternative Methods for Library Amplification
Although PCR amplification has many advantages, it also has some disadvantages. For example, time-consuming thermal cycling is needed to obtain the target sequence amplification at different temperatures, which led to the development of alternative amplification methods [56, 57]. Among them, isothermal amplification does not need any thermal cycle, which is easier to operate than PCR, and requires less energy. These characteristics greatly simplify the realization of isothermal amplification of diagnostic equipment in medical points. In 2006, Piepenburg and colleagues proposed new methods of recombinase polymerase amplification (RPA), which characteristic of the reaction is a constant temperature by a strand-displacement polymerase [58]. And the sensitivity of RPA is similar compared to PCR. Another method is linear amplification for deep sequencing (LADS), which connects the two different sequencing adapters to the blunt end repair and A-tailed fragments; then, one of them is extended with the sequence of T7 RNA polymerase promoter to form a linearly amplified library [59]. Compared to standard PCR-amplified libraries, T7-amplified libraries can remarkably reduce the bias of AT- or GC-rich, yet strand information is not maintained. Accordingly, if directionality needs to be maintained, it must either be introduced before adapter ligation or it requires modification of the LADS protocol. For instance, during the synthesis of the first strand of cDNA, the “barcode” can be incorporated on the antistrand and double-stranded cDNA can be generated to start LADS.
At present, the preparation of genomic from clinical samples is still a bottleneck in sequencing analysis and frequently limits by the amount of specimen available. Therefore, the amplification of samples is indispensable to get sufficient sample yield in sequencing. Researchers proposed a whole-genome amplification (WGA) method, which can generate a large amount of DNA/cDNA directly from small cell samples. Subsequently, the entire genome fragments were amplified by multiple displacement amplification (MDA) in 30°C condition, which uses φ29 DNA polymerase and random exonuclease-resistant primers [60]. In addition, Dean et al.'s [60] study showed that MDA could generate large quantities and high-quality cDNA directly from the starting material. For this reason, MDA became an optimal choice for WGA from single cells.
Additionally, what is worth mentioning here is that an amplification-free RNA-seq protocol has been reported [39]. In the method, the ligating adapters contain primers annealing and attachment to the flowcell surface, subsequently the amplification step of a standard cluster. Hence, the PCR amplification step is avoided during library preparation and efficiently tackles this problem of PCR amplification bias. Nevertheless, when only a limited amount of starting material is available, the method is inappropriate, because the amplification-free method needs several hundred nanograms of input sample for library preparation [47].
4. Sequencing and Imaging
It is very important for the selection of sequencing platform in RNA-seq experiment. Currently, commercially available NGS platforms include Illumina/Solexa Genome Analyser, Life Technologies/ABI SOLiD System, and Roche/454 Genome Sequencer FLX [61]. These platforms use a sequencing-by-synthesis approach to sort tens of millions of sequence clusters in parallel. Generally, the NGS platform can be classified as either ensemble-based (sequencing multiple identical copies of a DNA molecule) or monomolecular (sequencing a single DNA molecule). Nevertheless, studies have found that sequencing technologies often have systematic defects. For example, when the wrong bases are introduced in the process of template cloning and amplification, substitution bias may appear in platforms such as Illumina and SOLiD®, which limits the utility of data. In addition, studies pointed out that sequence-specific bias may be caused by single-strand DNA folding or sequence-specific changes in enzyme preference [62]. Pacific Biosciences SMRT platform produces long single molecular sequences that are vulnerable to misinsertion from nonfluorescent nucleotides [63, 64]. Besides, the sequencing platform can produce representative biases, that is, some base composition regions (especially those with very high or very low GC composition) are not fully represented, thus leading to bias in the results [65]. Consequently, we will briefly discuss the bias of sequencing platforms, mainly including the Illumina and single-molecule-based platforms. A sum up of suggestions for improvement is presented in Table 4.
Table 4.
The bias sources of major sequencing platforms.
| Company | Platforms | Sequencing | Dominant bias type | Suggestion for improvement |
|---|---|---|---|---|
| Roche/454 Life Sciences | GS FLX Titanium XL+ | Pyrosequencing | The bias of sequencing was introduced by PCR amplification prior to sequencing. | Reduction of the number of PCR cycles and use of DNA polymerases with even higher fidelity [84] |
| GS FLX Titanium XLR70 | ||||
| GS Junior | ||||
| HiSeq 2000 | ||||
|
| ||||
| Illumina | Genome Analyzer IIx | Sequencing-by-synthesis with reversible terminator | Substitution type miscalls are the major source of bias. | Quality trimming (sickle) combined with error correction (BayesHammer) followed by read overlapping (PANDAseq) as the most suitable approach, reducing substitution biases [85] |
| MiSeq | ||||
| SOLiD™ 4 system | ||||
| Ion PGM™ sequencer (318 chip) | ||||
|
| ||||
| Helicos BioSciences | HeliScope™ single molecule sequencer | Single-molecule sequencing | Biases were introduced by insertions and deletions. | If a low sequencing bias is needed, Illumina or SOLiD are often the best choices [86, 87] |
|
| ||||
| Pacific Biosciences | PacBio RS | Single-molecule sequencing | High bias of raw single-pass data | |
Currently, the Illumina HiSeq platform is the most widely used next-generation RNA sequencing technology and has become the standard of NGS sequencing. The platform has two flowcells, each of which provides eight separate channels for sequencing reaction. The sequencing reaction takes 1.5 to 12 days to complete, depending on the total read length of the library. Minoche et al.'s [66] study discovered that the HiSeq platform exists error types of GC content bias. In addition, Illumina released the MiSeq, which integrates NGS instruments and provides end-to-end sequencing solutions using reversible terminator sequencing-by-synthesis technology. The MiSeq instrument is a desktop classifier with low throughput but faster turnaround (generating about 30 million paired-end reads in 24 h). Simultaneously, it can perform on-board cluster generation, amplification, and data analysis in a single run, including base calls, alignment, and variant calling. At the present, MiSeq has become a dominant platform for gene amplification and sequencing in microbial ecology. Nevertheless, various technical problems still remain, such as reproducibility, hence hampered harnessing its true potential to sequence. Furthermore, Fadrosh et al.'s [67] study found that MiSeq 16S rRNA gene amplicon sequencing may arise “low sequence diversity” problems in the first several cycles.
Furthermore, the emergence of single-molecule sequencing platforms such as PacBio makes single-molecule real-time (SMRT) sequencing possible [68]. In this method, DNA polymerase and fluorescent-labeled nucleoside were used for uninterrupted template-directed synthesis. One advantage of SMRT is that it does not include the PCR amplification step, as a consequence avoiding amplification bias. At the same time, this sequencing approach can produce extraordinarily long reads with average lengths of 4200 to 8500 bp, which greatly improves the detection of new transcriptional structures [69, 70], in addition, due to the relatively low cost per run of PacBio's, which can reduce the cost of RNA-seq. However, PacBio can usually introduce high error rates (∼5%) compared to Illumina and 454 sequencing platform [71]. Due to the fact that it is difficult to the matching erroneous reads to the reference genome, thus the high error rate may be lead to misalignment and loss of sequencing reads. Furthermore, Fichot and Norman's [72] study showed that PacBio's sequencing platform can shun enrichment bias of extremely GC/AT.
5. Discussion and Conclusion
At the present, RNA-seq has been widely used in biological, medical, clinical, and pharmaceutical research. However, all these sequencing studies are limited by the accuracy of underlying sequencing experiments, because RNA-seq technology may introduce various errors and biases in sample preparation, library construction, sequencing and imaging, etc.
It is well known that RNA is extremely labile and degradable. Therefore, if the sample cannot be separated immediately after collection, which can be kept in intermediary solution. At the present, RNAlater (Thermo Fisher Scientific and Qiagen) and RNAstable (Sigma-Aldrich) are commonly used stabilizers, which can prevent RNA degradation and maintain RNA integrity. Additionally, the extraction and isolation of RNA have been proved as one of the most bias sources. Currently, TRIzol is a frequently used method. Furthermore, some protocols of RNA extraction and isolation may carry over some gDNA into total RNA samples, which can be removed by DNA enzyme treatment to prevention gDNA contamination (false positive signal).
Additionally, library construction methods are frequently biased, which is a main concern for RNA-seq data quality. Among them, PCR amplification is the major source of bias. A previous study showed that GC content has a virtual influence on PCR amplification efficiency. Therefore, we suggest that Kapa HiFi (Kapa Biosystems, Wilmington, MA) or AccuPrime Taq DNA Polymerase High Fidelity (Life Technologies) can be selected to PCR amplification. It has been shown that these enzymes can minimize the amplification bias caused by the extreme GC content. Furthermore, Aird et al. [17] found that, for extremely high GC samples, the amplification bias can remarkably be reduced by modification of the denaturation time and subsequent PCR melt cycles. Moreover, the reduction number of amplification cycles can also improve PCR bias. Therefore, the methods of no-PCR amplification were developed, but these require a lot of input material [39], resulting in a limitation for a low input sample. Therefore, when the amount of input material is limited, amplification is indispensable.
In summary, the major goal of constructing the sequencing library is to minimize the bias. The bias was frequently defined as the systematic distortion of data due to the experimental protocols. Therefore, it is impossible to eliminate all sources of experimental bias. The best strategies are as follows: (i) to understand how the bias is generated and to take measures to minimize it; (ii) to pay attention to the experimental design and minimize the influence of irreducible bias on the final analysis.
Acknowledgments
This work was supported by the grant from the National Natural Science Foundation of China (81827901 and 61801108) and the Natural Science Foundation of Jiangsu Province (BK20201148).
Conflicts of Interest
There are no conflicts of interest to declare.
Authors' Contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- 1.Wang Z., Gerstein M., Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dohm J. C., Lottaz C., Borodina T., Himmelbauer H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Research. 2008;36(16, article e105) doi: 10.1093/nar/gkn425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li S., Tighe S. W., Nicolet C. M., et al. Multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study. Nature Biotechnology. 2014;32(9):915–925. doi: 10.1038/nbt.2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Dijk E. L., Jaszczyszyn Y., Thermes C. Library preparation methods for next-generation sequencing: tone down the bias. Experimental Cell Research. 2014;322(1):12–20. doi: 10.1016/j.yexcr.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 5.Camacho-Sanchez M., Burraco P., Gomez-Mestre I., Leonard J. A. Preservation of RNA and DNA from mammal samples under field conditions. Molecular Ecology Resources. 2013;13(4):663–673. doi: 10.1111/1755-0998.12108. [DOI] [PubMed] [Google Scholar]
- 6.Groelz D., Sobin L., Branton P., Compton C., Wyrich R., Rainen L. Non-formalin fixative versus formalin-fixed tissue: a comparison of histology and RNA quality. Experimental and Molecular Pathology. 2013;94(1):188–194. doi: 10.1016/j.yexmp.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 7.Hedegaard J., Thorsen K., Lund M. K., et al. Next-generation sequencing of RNA and DNA isolated from paired fresh-frozen and formalin-fixed paraffin-embedded samples of human cancer and normal tissue. PLoS One. 2014;9(5) doi: 10.1371/journal.pone.0098187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li P., Conley A., Zhang H., Kim H. L. Whole-Transcriptome profiling of formalin-fixed, paraffin-embedded renal cell carcinoma by RNA-seq. BMC Genomics. 2014;15(1, article 1087) doi: 10.1186/1471-2164-15-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levin Y., Talsania K., Tran B., Shetty J., Zhao Y. M., Mehta M. Optimization for sequencing and analysis of degraded FFPE-RNA samples. Jove-Journal of Visualized Experiments. 2020;e61060(160) doi: 10.3791/61060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gayral P., Weinert L., Chiari Y., Tsagkogeorga G., Ballenghien M., Galtier N. Next-generation sequencing of transcriptomes: a guide to RNA isolation in nonmodel animals. Molecular Ecology Resources. 2011;11(4):650–661. doi: 10.1111/j.1755-0998.2011.03010.x. [DOI] [PubMed] [Google Scholar]
- 11.Hillyard G., Clark M. S. RNA preservation of Antarctic marine invertebrates. Polar Biology. 2012;35(4):633–636. doi: 10.1007/s00300-011-1088-9. [DOI] [Google Scholar]
- 12.Riesgo A., Pérez-Porro A. R., Carmona S., Leys S. P., Giribet G. Optimization of preservation and storage time of sponge tissues to obtain quality mRNA for next-generation sequencing. Molecular Ecology Resources. 2012;12(2):312–322. doi: 10.1111/j.1755-0998.2011.03097.x. [DOI] [PubMed] [Google Scholar]
- 13.Wang W.-X., Wilfred B. R., Baldwin D. A., et al. Focus on RNA isolation: obtaining RNA for microRNA (miRNA) expression profiling analyses of neural tissue. Biochimica et Biophysica Acta. 2008;1779(11):749–757. doi: 10.1016/j.bbagrm.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown R. A., Epis M. R., Horsham J. L., Kabir T. D., Richardson K. L., Leedman P. J. Total RNA extraction from tissues for microRNA and target gene expression analysis: not all kits are created equal. BMC Biotechnology. 2018;18(1):p. 16. doi: 10.1186/s12896-018-0421-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hutchison C. A., Smith H. O., Pfannkoch C., Venter J. C. Cell-free cloning using φ29 DNA polymerase. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(48):17332–17336. doi: 10.1073/pnas.0508809102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hodges E., Xuan Z., Balija V., et al. Genome-wide _in situ_ exon capture for selective resequencing. Nature Genetics. 2007;39(12):1522–1527. doi: 10.1038/ng.2007.42. [DOI] [PubMed] [Google Scholar]
- 17.Aird D., Ross M. G., Chen W. S., et al. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biology. 2011;12(2, article R18) doi: 10.1186/gb-2011-12-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morlan J. D., Qu K., Sinicropi D. V. Selective depletion of rRNA enables whole transcriptome profiling of archival fixed tissue. PLoS One. 2012;7(8, article e42882) doi: 10.1371/journal.pone.0042882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang R., Jaritz M., Guenzl P., et al. An RNA-Seq strategy to detect the complete coding and non-coding transcriptome including full-length imprinted macro ncRNAs. PLoS One. 2011;6(11, article e27288) doi: 10.1371/journal.pone.0027288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramskold D., Luo S., Wang Y.-C., et al. Author Correction: Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nature Biotechnology. 2020;38(3):374–374. doi: 10.1038/s41587-020-0427-1. [DOI] [PubMed] [Google Scholar]
- 21.Tariq M. A., Kim H. J., Jejelowo O., Pourmand N. Whole-transcriptome RNAseq analysis from minute amount of total RNA. Nucleic Acids Research. 2011;39(18, article e120) doi: 10.1093/nar/gkr547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yi H., Cho Y.-J., Won S., et al. Duplex-specific nuclease efficiently removes rRNA for prokaryotic RNA-seq. Nucleic Acids Research. 2011;39(20, article e140) doi: 10.1093/nar/gkr617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adiconis X., Borges-Rivera D., Satija R., et al. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nature Methods. 2013;10(7):623–629. doi: 10.1038/nmeth.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sultan M., Amstislavskiy V., Risch T., et al. Influence of RNA extraction methods and library selection schemes on RNA-seq data. BMC Genomics. 2014;15(1):675–675. doi: 10.1186/1471-2164-15-675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Conesa A., Madrigal P., Tarazona S., et al. A survey of best practices for RNA-seq data analysis. Genome Biology. 2016;17(13) doi: 10.1186/s13059-016-0881-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang X.-O., Yin Q.-F., Chen L.-L., Yang L. Gene expression profiling of non-polyadenylated RNA-seq across species. Genomics Data. 2014;2:237–241. doi: 10.1016/j.gdata.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Munafó D. B., Langhorst B. W., Chater C. L., Sumner C. J., Davis T. B. Selective Depletion of Abundant RNAs to Enable Transcriptome Analysis of Low-Input and Highly Degraded Human RNA. John Wiley & Sons, Inc.; 2016. [DOI] [PubMed] [Google Scholar]
- 28.O'Neil D., Glowatz H., Schlumpberge M. Ribosomal RNA depletion for efficient use of RNA-seq capacity. Current Protocols in Molecular Biology. 2013;103(1):4.19.1–4.19.8. doi: 10.1002/0471142727.mb0419s103. [DOI] [PubMed] [Google Scholar]
- 29.Bush S. J., Mcculloch M. E. B., Summers K. M., Hume D. A., Clark E. L. Integration of quantitated expression estimates from polyA-selected and rRNA-depleted RNA-seq libraries. BMC Bioinformatics. 2017;18(1):p. 301. doi: 10.1186/s12859-017-1714-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ares M. Fragmentation of whole-transcriptome RNA using E. coli RNase III. Cold Spring Harbor Protocols. 2013;2013(5):479–481. doi: 10.1101/pdb.prot074369. [DOI] [PubMed] [Google Scholar]
- 31.Wery M., Descrimes M., Thermes C., Gautheret D., Morillon A. Zinc-mediated RNA fragmentation allows robust transcript reassembly upon whole transcriptome RNA-Seq. Methods. 2013;63(1):25–31. doi: 10.1016/j.ymeth.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 32.Parkinson N. J., Maslau S., Ferneyhough B., et al. Preparation of high-quality next-generation sequencing libraries from picogram quantities of target DNA. Genome Research. 2012;22(1):125–133. doi: 10.1101/gr.124016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Caruccio N. Preparation of next-generation sequencing libraries using Nextera™ technology: simultaneous DNA fragmentation and adaptor tagging by in vitro transposition. Methods in Molecular Biology. 2011;733:p. 241. doi: 10.1007/978-1-61779-089-8_17. [DOI] [PubMed] [Google Scholar]
- 34.Knierim E., Lucke B., Schwarz J. M., Schuelke M., Seelow D., Gilbert M. T. P. Systematic comparison of three methods for fragmentation of long-range PCR products for next generation sequencing. PLoS One. 2011;6(11, article e28240) doi: 10.1371/journal.pone.0028240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poptsova M. S., IlIcheva I. A., Nechipurenko D. Y., et al. Non-random DNA fragmentation in next-generation sequencing. Scientific Reports. 2014;4 doi: 10.1038/srep04532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar R., Ichihashi Y., Kimura S., et al. A high-throughput method for Illumina RNA-Seq library preparation. Frontiers in Plant Science. 2012;3:p. 202. doi: 10.3389/fpls.2012.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adey A., Morrison H. G., Asan, et al. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biology. 2010;11:1–17. doi: 10.1186/gb-2010-11-12-r119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perkins T., Kingsley R. A., Fookes M., et al. A strand-specific RNA-seq analysis of the transcriptome of the typhoid bacillus Salmonella typhi. PLoS Genetics. 2009;5:1–13. doi: 10.1371/journal.pgen.1000569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mamanova L., Andrews R. M., James K. D., et al. FRT-seq: amplification-free, strand-specific transcriptome sequencing. Nature Methods. 2010;7(2):130–132. doi: 10.1038/nmeth.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hansen K. D., Brenner S. E., Dudoit S. Biases in Illumina transcriptome sequencing caused by random hexamer priming. Nucleic Acids Research. 2010;38(12, article e131) doi: 10.1093/nar/gkq224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharma S. V., Lee D. Y., Li B., et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141(1):69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baran-Gale J., Kurtz C. L., Erdos M. R., et al. Addressing bias in small RNA library preparation for sequencing: a new protocol recovers micro RNAs that evade capture by current methods. Frontiers in Genetics. 2015;6:p. 352. doi: 10.3389/fgene.2015.00352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parkhomchuk D., Borodina T., Amstislavskiy V., et al. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Research. 2009;37(18, article e123) doi: 10.1093/nar/gkp596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levin J. Z., Yassour M., Adiconis X., et al. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nature Methods. 2010;7(9):709–715. doi: 10.1038/nmeth.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zeng W., Mortazavi A. Technical considerations for functional sequencing assays. Nature Immunology. 2012;13(9):802–807. doi: 10.1038/ni.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Z., Theurkauf W. E., Weng Z., Zamore P. D. Strand-specific libraries for high throughput RNA sequencing (RNA-Seq) prepared without poly (A) selection. Silence. 2012;3(1):p. 9. doi: 10.1186/1758-907X-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kozarewa I., Ning Z., Quail M. A., Sanders M. J., Berriman M., Turner D. J. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G+C)-biased genomes. Nature Methods. 2009;6(4):291–295. doi: 10.1038/nmeth.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oyola S. O., Otto T. D., Gu Y., et al. Optimizing Illumina next-generation sequencing library preparation for extremely AT-biased genomes. BMC Genomics. 2012;13(1):p. 1. doi: 10.1186/1471-2164-13-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quail M. A., Otto T. D., Gu Y., et al. Optimal enzymes for amplifying sequencing libraries. Nature Methods. 2012;9(1):10–11. doi: 10.1038/nmeth.1814. [DOI] [PubMed] [Google Scholar]
- 50.Henke W., Herdel K., Jung K., Schnorr D., Loening S. A. Betaine improves the PCR amplification of GC-rich DNA sequences. Nucleic Acids Research. 1997;25(19):3957–3958. doi: 10.1093/nar/25.19.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qiu X., Wu L., Huang H., et al. Evaluation of PCR-generated chimeras, mutations, and heteroduplexes with 16S rRNA gene-based cloning. Applied and Environmental Microbiology. 2001;67(2):880–887. doi: 10.1128/AEM.67.2.880-887.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Acinas S. G., Sarma-Rupavtarm R., Klepac-Ceraj V., Polz M. F. PCR-induced sequence artifacts and bias: insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Applied and Environmental Microbiology. 2005;71(12):8966–8969. doi: 10.1128/AEM.71.12.8966-8969.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sipos R., Székely A. J., Palatinszky M., Révész S., Márialigeti K., Nikolausz M. Effect of primer mismatch, annealing temperature and PCR cycle number on 16S rRNA gene-targetting bacterial community analysis. FEMS Microbiology Ecology. 2007;60(2):341–350. doi: 10.1111/j.1574-6941.2007.00283.x. [DOI] [PubMed] [Google Scholar]
- 54.Wu J.-Y., Jiang X.-T., Jiang Y.-X., Lu S.-Y., Zou F., Zhou H.-W. Effects of polymerase, template dilution and cycle number on PCR based 16 S rRNA diversity analysis using the deep sequencing method. BMC Microbiology. 2010;10(1):p. 255. doi: 10.1186/1471-2180-10-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sze M. A., Schloss P. D. The impact of DNA polymerase and number of rounds of amplification in PCR on 16S rRNA gene sequence data. mSphere. 2019;4 doi: 10.1128/msphere.00163-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Asiello P. J., Baeumner A. J. Miniaturized isothermal nucleic acid amplification, a review. Lab on a Chip. 2011;11(8):1420–1430. doi: 10.1039/c0lc00666a. [DOI] [PubMed] [Google Scholar]
- 57.Kim J., Easley C. J. Isothermal DNA amplification in bioanalysis: strategies and applications. Bioanalysis. 2011;3(2):227–239. doi: 10.4155/bio.10.172. [DOI] [PubMed] [Google Scholar]
- 58.Piepenburg O., Williams C. H., Stemple D. L., Armes N. A. DNA detection using recombination proteins. PLoS Biology. 2006;4(7, article e204) doi: 10.1371/journal.pbio.0040204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoeijmakers W. A., Bártfai R., Françoijs K.-J., Stunnenberg H. G. Linear amplification for deep sequencing. Nature Protocols. 2011;6(7):1026–1036. doi: 10.1038/nprot.2011.345. [DOI] [PubMed] [Google Scholar]
- 60.Dean F. B., Hosono S., Fang L., et al. Comprehensive human genome amplification using multiple displacement amplification. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(8):5261–5266. doi: 10.1073/pnas.082089499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Metzker M. L. Sequencing technologies -- the next generation. Nature Reviews. Genetics. 2010;11(1):31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 62.Nakamura K., Oshima T., Morimoto T., et al. Sequence-specific error profile of Illumina sequencers. Nucleic Acids Research. 2011;39(13, article e90) doi: 10.1093/nar/gkr344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fuller C. W., Middendorf L. R., Benner S. A., et al. The challenges of sequencing by synthesis. Nature Biotechnology. 2009;27(11):1013–1023. doi: 10.1038/nbt.1585. [DOI] [PubMed] [Google Scholar]
- 64.Roberts R. J., Carneiro M. O., Schatz M. C. The advantages of SMRT sequencing. Genome Biology. 2013;14(6):p. 405. doi: 10.1186/gb-2013-14-6-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Robasky K., Lewis N. E., Church G. M. The role of replicates for error mitigation in next-generation sequencing. Nature Reviews. Genetics. 2014;15(1):56–62. doi: 10.1038/nrg3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Minoche A. E., Dohm J. C., Himmelbauer H. Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and genome analyzer systems. Genome Biology. 2011;12(11, article R112) doi: 10.1186/gb-2011-12-11-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fadrosh D. W., Ma B., Gajer P., et al. An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome. 2014;2(1):p. 6. doi: 10.1186/2049-2618-2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eid J., Fehr A., Gray J., et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323(5910):133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 69.Au K. F., Sebastiano V., Afshar P. T., et al. Characterization of the human ESC transcriptome by hybrid sequencing. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(50):E4821–E4830. doi: 10.1073/pnas.1320101110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sharon D., Tilgner H., Grubert F., Snyder M. A single-molecule long-read survey of the human transcriptome. Nature Biotechnology. 2013;31(11):1009–1014. doi: 10.1038/nbt.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Carneiro M. O., Russ C., Ross M. G., Gabriel S. B., Nusbaum C., DePristo M. A. Pacific biosciences sequencing technology for genotyping and variation discovery in human data. BMC Genomics. 2012;13(1):p. 375. doi: 10.1186/1471-2164-13-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fichot E. B., Norman R. S. Microbial phylogenetic profiling with the Pacific Biosciences sequencing platform. Microbiome. 2013;1(1):p. 10. doi: 10.1186/2049-2618-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lin M. T., Tseng L. H., Kamiyama H., et al. Quantifying the relative amount of mouse and human DNA in cancer xenografts using species-specific variation in gene length. BioTechniques. 2010;48:351–355. doi: 10.2144/000113363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Innis M. A., Gelfand D. H., Sninsky J. J., White T. J. PCR Protocols: A Guide to Methods and Applications. Academic press; 2012. [Google Scholar]
- 75.Nagalakshmi U., Wang Z., Waern K., et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320(5881):1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Perocchi F., Xu Z., Clauder-Münster S., Steinmetz L. M. Antisense artifacts in transcriptome microarray experiments are resolved by actinomycin D. Nucleic Acids Research. 2007;35(19, article e128) doi: 10.1093/nar/gkm683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Akbari M., Hansen M. D., Halgunset J., Skorpen F., Krokan H. E. Low copy number DNA template can render polymerase chain reaction error prone in a sequence-dependent manner. The Journal of Molecular Diagnostics. 2005;7(1):36–39. doi: 10.1016/S1525-1578(10)60006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Whiteford N., Skelly T., Curtis C., et al. Swift: primary data analysis for the Illumina Solexa sequencing platform. Bioinformatics. 2009;25(17):2194–2199. doi: 10.1093/bioinformatics/btp383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Loman N. J., Misra R. V., Dallman T. J., et al. Performance comparison of benchtop high-throughput sequencing platforms. Nature Biotechnology. 2012;30(5):434–439. doi: 10.1038/nbt.2198. [DOI] [PubMed] [Google Scholar]
- 80.Kim Y.-K., Yeo J., Kim B., Ha M., Kim V. N. Short structured RNAs with low GC content are selectively lost during extraction from a small number of cells. Molecular Cell. 2012;46(6):893–895. doi: 10.1016/j.molcel.2012.05.036. [DOI] [PubMed] [Google Scholar]
- 81.Schuierer S., Carbone W., Knehr J., et al. A comprehensive assessment of RNA-seq protocols for degraded and low-quantity samples. BMC Genomics. 2017;18(1):p. 442. doi: 10.1186/s12864-017-3827-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marine R., Polson S. W., Ravel J., et al. Evaluation of a transposase protocol for rapid generation of shotgun high-throughput sequencing libraries from nanogram quantities of DNA. Applied and Environmental Microbiology. 2011;77(22):8071–8079. doi: 10.1128/AEM.05610-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dabney J., Meyer M. Length and GC-biases during sequencing library amplification: a comparison of various polymerase-buffer systems with ancient and modern DNA sequencing libraries. BioTechniques. 2012;52(2):87–94. doi: 10.2144/000113809. [DOI] [PubMed] [Google Scholar]
- 84.Brodin J., Mild M., Hedskog C., et al. PCR-induced transitions are the major source of error in cleaned ultra-deep pyrosequencing data. PLoS One. 2013;8(7, article e70388) doi: 10.1371/journal.pone.0070388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schirmer M., Ijaz U. Z., D'Amore R., Hall N., Sloan W. T., Quince C. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Research. 2015;43(6, article e37) doi: 10.1093/nar/gku1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chu Y., Corey D. R. RNA sequencing: platform selection, experimental design, and data interpretation. Nucleic Acid Therapeutics. 2012;22(4):271–274. doi: 10.1089/nat.2012.0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rhoads A., Au K. F. PacBio sequencing and its applications. Genomics, Proteomics & Bioinformatics. 2015;13(5):278–289. doi: 10.1016/j.gpb.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]