

Abstract
The human-to-human transmitted respiratory illness in COVID-19 affected by the pathogenic Severe Acute Respiratory Syndrome Corona Virus 2 (SARS-CoV-2), which appeared in the last of December 2019 in Wuhan, China, and rapidly spread in many countries. Thereon, based on the urgent need for therapeutic molecules, we conducted in silico based docking and simulation molecular interaction studies on repurposing drugs, targeting SARS-CoV-2 spike protein. Further, the best binding energy of doxorubicin interacting with virus spike protein (PDB: 6VYB) was observed to be −6.38 kcal/mol and it was followed by exemestane and gatifloxacin. The molecular simulation dynamics analysis of doxorubicin, Reference Mean Square Deviation (RMSD), Root Mean Square fluctuation (RMSF), Radius of Gyration (Rg), and formation of hydrogen bonds plot interpretation suggested, a significant deviation and fluctuation of Doxorubicin-Spike RBD complex during the whole simulation period. The Rg analysis has stated that the Doxorubicin-Spike RBD complex was stable during 15,000–35,000 ps MDS. The results have suggested that doxorubicin could inhibit the virus spike protein and prevent the access of the SARS-CoV-2 to the host cell. Thus, in-vitro/in-vivo research on these drugs could be advantageous to evaluate significant molecules that control the COVID-19 disease.
Keywords: Antiviral drugs, Doxorubicin, Exemestane, Gatifloxacin, Pandemic, SARS-CoV-2, Spike protein
Abbreviations: kcal/mol, Kilocalories per mole; Hbond, Hydrogen bond; uM, Micro Molar; Å, Angstrom; nm, nanometer; ns, nanosecond; ps, picosecond
1. Introduction
Coronavirus Disease 2019 (COVID-19) is caused by a virus from an unknown source to a human of China and continuously spreading to many countries. As soon as possible the virus has been identified as Severe Acute Respiratory Syndrome, Corona Virus 2 (SARS-CoV-2), and more commonly the disease is known as COVID-19, and the virus is also known as the 2019 novel Corona Virus (2019-nCoV) (Zhou et al., 2020, Wang et al., 2020, Ali et al., 2021, Rehman et al., 2020 May 15). A large number of humans have affected soon and therefore the World Health Organization confirmed this disease as a pandemic. As of 09 April 2021, 133 146 550 confirmed cases of COVID-19, and 2 888 530 deaths have been reported by the World Health Organization (WHO) https://www.who.int/emergencies/diseases/novel-coronavirus-2019. It is thought that the virus is originated from the bat and its similarity with SARS has been reported well (Lu et al., 2020, Ansari et al., 2020a, Ansari et al., 2020b). The targets of virus-like protease, peptidase, and spike protein have been identified for drug discovery including vaccine development as well as to manage the disease. Many natural compounds against these targets have been screened by computational biologists and suggested for further in vitro and in vivo evaluation against the virus (WHO, 2020; Lu et al., 2020, Prajapat et al., 2020).
One of the targets of SARS-CoV-2 is spike protein belong to class I fusion glycoprotein. The spike protein helps the virus for attachment via the Angiotensin-converting enzyme 2 (ACE-2) receptor and fuses with the involvement of the S2 domain of protein facilitating the entry of the pathogen into the host cell (Khan et al., 2020). The ACE-2 receptor interacts with the S1 subunit of glycoprotein and assisted the virus attachment. Subsequent to the attachment protein goes under some conformational changes resulted in the fusion of membrane and entry of the virus into the host cell using the S2 domain of virus surface protein (Wan et al., 2020, Li et al., 2005). With the union of spike proteins, the virus pathogen seems as a trimeric form that provided a crown-like appearance, due to this it is commonly known as a coronavirus (Belouzard et al., 2012). It has been reported that the S1 domain of spike protein and dipeptidyl peptidase-4 (DPP4) for Middle East Respiratory Syndrome Corona Virus (MERS-CoV) and host receptor ACE-2 for SARS-CoV-2 involved in viral and host cell membrane fusion and facilitate the S2 domain-mediated entry to the host cell (Rehman et al., 2020b; Jamal et al., 2021). Moreover, the host cell produces an immunological response against the virus S protein. It is thought that spike protein is a potential target for drug discovery and research (Li, 2016). The hydroxychloroquine, chloroquine, and antiviral drugs like favipiravir, remdesivir, ritonavir, lopinavir, and oseltamivir have been studied by computational docking to screen out their potential against virus SARS-CoV-2 spike protein.
Vaccination therapy has been developed and used recently to treat COVID-19 disease. FDA- approved some repurposing drugs have been tested with nanoparticles (NPs) as adjuvants. NPs technological tested combination has been reported with some serious side effects and continuous efforts are going on in search of new molecules as well as a new therapy. The anticovid-19 therapeutic potential of some natural molecules like Berbamine, cepharanthine, fangchinoline, and tetrandrine), saponins and triterpenes (glycyrrhizin), glycyrrhizinic acid, anthraquinones (hypericin), saikosaponins, (rutin and quercetin like flavonoids and FDA approved some repurposing drugs have been described (Gomez et al., 2021). The significant interacting potential of doxorubicin against SARS-CoV-2 main protease through docking and simulation studies was also reported (Jamal et al., 2021). Urolithin, an active chemical of pomegranate has been reported to interact with virus protease and peptidase more significantly as compared to repurposing drugs (Ahmad et al., 2020).
The interaction studies with spike protein, molecular dynamics simulation, and potential of drug candidates like doxorubicin yet not have been reported. Therefore, this study is the first study to find out the potentiality of the Food and Drug Administration (FDA) approved drugs to manage the COVID-19 disease more significantly.
2. Material and methods
2.1. Drug compounds preparation
The physicochemical and 2 Dimensional (2D) structural information of FDA approved drugs azithromycin (CID:447043), doxorubicin (CID:31703), exemestane (CID:60198), gatifloxacin (CID:5379), sulbactam (CID:130313), and tobramycin (CID:36294) (Table 1) were accessed and downloaded from PubChem Database provided by National Center for Biotechnology Information (NCBI) (https://pubchem.ncbi.nlm.nih.gov/). The 2D drugs chemical structure need to be further converted to Protein Data Bank (PDB) (https://www.rcsb.org/) (Berman et al., 2000) files, hence the 2D files were converted into three dimensional (3D) PDB files using Open Babel online platform (O'Boyle et al., 2011) and further, prepared 3D spike protein PDB file was energy minimized by choosing the CHARMm forcefield from the simulation module in Discovery Studio Visualizer 2020 (Brooks et al., 2009, Dassault Systèmes BIOVIA, 2020).
Table 1.
Details of selected drugs with their physicochemical information for the molecular interaction analysis.
| S.No. | Drug Name | Molecular Formula | Molecular Weight | Chemical Structure | SMILES IDs | PubChem IDs |
|---|---|---|---|---|---|---|
| Azithromycin | C38H72N2O12 | 749 g/mol |  |
CCC1C(C(C(N(CC(CC(C(C(C(C(C(=O)O1)C)OC2CC(C(C(O2)C)O)(C)OC)C)OC3C(C(CC(O3)C)N(C)C)O)(C)O)C)C)C)O)(C)O | CID:447043 | |
| Doxorubicin | C27H29NO11 | 543.5 g/mol |  |
CC1C(C(CC(O1)OC2CC(CC3 = C2C(=C4C(=C3O)C(=O)C5 = C(C4 = O)C(=CC = C5)OC)O)(C(=O)CO)O)N)O | CID:31703 | |
| Exemestane | C20H24O2 | 296.4 g/mol |  |
CC12CCC3C(C1CCC2 = O)CC(=C)C4 = CC(=O)C = CC34C | CID:60198 | |
| Gatifloxacin | C19H22FN3O4 | 375.4 g/mol |  |
CC1CN(CCN1)C2 = C(C = C3C(=C2OC)N(C = C(C3 = O)C(=O)O)C4CC4)F | CID:5379 | |
| Sulbactam | C8H11NO5S | 233.24 g/mol | 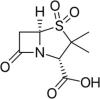 |
CC1(C(N2C(S1(=O) = O)CC2 = O)C(=O)O)C | CID:130313 | |
| Tobramycin | C18H37N5O9 | 467.5 g/mol | 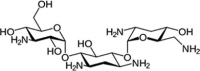 |
C1C(C(C(C(C1N)OC2C(C(C(C(O2)CO)O)N)O)O)OC3C(CC(C(O3)CN)O)N)N | CID:36294 |
2.2. Spike protein preparation as a receptor
The 3D structures of SARS-CoV-2 spike ectodomain (open state) (PDB: 6VYB with Resolution: 3.20 Å) (Fig. 1) (Walls et al., 2020) was accessed and downloaded from the PDB database (Berman et al., 2000). In order to prepare a downloaded 3D crystal structure for molecular docking-based interaction studies, it is required to remove the water molecules and HETATM from the native 3D structure. Also, active site of the pre-bonded ligand molecules was taken after visualizing the 3D structure and active site of the amino acid residues information found in order to utilized during docking of the drugs on the analyzed binding site. Further, all editing of 3D structure and energy minimization was done after implementing CHARMm forcefield by simulation protocol available in Discovery Studio Visualizer 2020 (Brooks et al., 2009, Dassault Systèmes BIOVIA, 2020).
Fig. 1.
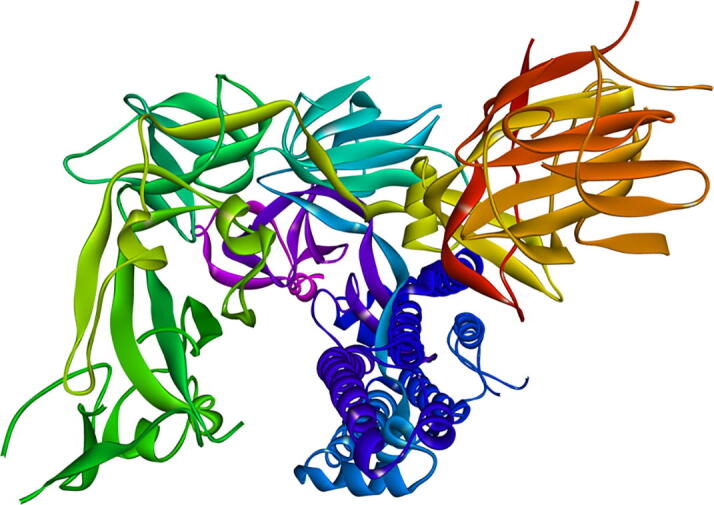
Showing the 3D crystal structure of SARS-CoV-2 spike ectodomain structure (open state) (PDB: 6VYB).
2.3. Drugs-spike protein interaction analysis
MGL tool AutoDock 4.2 has been utilized for the calculation of binding affinity between drug compounds and spike protein. AutoDock molecular docking method follows Lamarckian Genetic Algorithm (LGA) as a scoring function. Molecular docking methods followed by searching the top conformation of drug compounds and receptor (i.e. protein or enzyme) complex by utilizing the following formula in order to calculate the final binding energy (ΔG).
Here, ΔG gauss: attractive term for dispersion of two gaussian functions, ΔGrepulsion: square of the distance if closer than a threshold value, ΔGhbond: ramp function - also used for interactions with metal ions, ΔGhydrophobic: ramp function, ΔGtors: proportional to the number of rotatable bonds (Morris et al., 1998, Jamal, 2020).
Furthermore, water molecules were deleted from the selected SARS-CoV-2 spike protein (PDB: 6VYB) native PDB file. Hydrogen atoms were added, Kollman united charges, and solvation parameters were also implemented to the spike protein. drug compounds were charged by Gasteiger charge. The Grid box was set to cover the maximum part of the spike protein and drug compounds. The values were set to 60 × 60 × 60° in X, Y, and Z-axis of a grid point. The values for XYZ coordinates were set as 175.032, 253.721, and 217.497, respectively. The default grid points, spacing was set as 0.375 Å. The default LGA parameters population size (ga_pop_size), energy evaluations (ga_num_generation), mutation rate, crossover rate and step size were set to 150, 2500000, 27000, 0.02, 0.8 and 0.2 Å, respectively. Finally, 10 LGA runs were executed (Morris et al., 2009, Haneef et al., 2014). After LGA execution, the spike protein and drug compounds complexes were prepared after extraction of the total binding energy and inhibition constants data from the AutoDock output files. 3D visualization analysis of the complexes and observation of different types of interactions was performed using Discovery Studio Visualizer 2020 (Dassault Systèmes BIOVIA, 2020, Jamal, 2020).
2.4. MDS experimentation
Based on the final intermolecular interaction energies output generated from the AutoDock tool, we have selected the Doxorubicin-Spike protein receptor-binding domain (RBD) complex needs to be further analyzed through MDS. Hence, the MDS has performed for 50 (ns) simulation using GROningen MAchine for Chemical Simulations (GROMACS) tool 2018 version developed by the University of Groningen, Netherlands (Van Der Spoel et al., 2005).
pdb2gmx module was used to generate the required Spike RBD (PDB:6VYB) topology file followed by CHARMM27 all-atom forcefield selection. Also, doxorubicin topology files were prepared and downloaded from the SwissParam server (Zoete et al., 2011).
A triclinic-shaped water molecules-filled solvation unit cell was built. Addition of Na+ and Cl− ions was done for stabilization of the system followed by energy minimization. Equilibrium setup of the system containing Doxorubicin-Spike RDB complex required and it was done followed by two-step ensembles NVT (constant number of particles, pressure, and temperature) and NPT (constant number of particles, pressure, temperature) (Jamal et al., 2021). All necessary ensembles deliver control over temperature, pressure coupling provides steadfastness and stabilization of the system during the whole simulation (Gupta et al., 2020). GORMACS have many inbuilt packages, for Doxorubicin-Spike RBD complex MDS analysis, gmx rms was utilized for Root Mean Square Deviation (RMSD) (Kufareva and Abagyan, 2012), gmx rmsf for Root Mean Square fluctuation (RMSF), gmx gyrate for the complete assessment of Radius of Gyration (Rg) (Kuzmanic and Zagrovic, 2010) and gmx hbond for the calculation of numbers of hydrogen-bond (Hbond) formed during the interaction. In the last, after 50 ns MDS, output trajectory files analyzed and graph plots were generated using Xmgrace program (Turner, 2005).
3. Results
Molecular interaction analysis needs pre-processing and editing of datasets downloaded from the structural database before providing them as an input for the AutoDock tool. Therefore, required preparation of drug compounds and native spike protein 3D structure (PDB: 6VYB) were edited and energy minimized by CHARMm forcefield by Discovery Studio Visualizer 2020 (Brooks et al., 2009, Dassault Systèmes BIOVIA, 2020).
Molecular docking results further analyzed on the basis of binding energy, inhibition constants, and Hbond formation while drug compounds interact with spike protein as a receptor (Ferreira et al., 2015). The molecular interaction results of repurposing drugs with virus spike protein, observed by docking experimentation are represented in Table 2. The repurposing drugs showed significant binding interaction efficiency with SARS-CoV-2 spike protein and could interfere with the virus entry into the host cell.
Table 2.
Showing docking results of selected drugs with receptor SARS-CoV-2 spike protein (PDB: 6VYB). Where vdW = Van der Waals, Hbond = Hydrogen bond, and desolv Energy = desolvation energy and in Hbond name column UNK1 = selected drug compounds.
| S. No. | Drug Name | Final Intermolecular Energy (Kcal/mol) | vdW + Hbond + desolv Energy (Kcal/mol) | Electrostatic Energy (Kcal/mol) | Inhibition Constant | Hbond name | Hbond length (Angstrom) | Residues involved in hydrophobic interaction |
|---|---|---|---|---|---|---|---|---|
| 1. | Azithromycin | −4.60 | −4.83 | +0.23 | 832.15 uM | A:ARG214:HH21 - :UNK1:O20 | 1.72 | Ala27,Tyr28,Thr29,Asn30,Trp64,Arg214,Asp215 |
| :UNK1:H - A:TYR28:O | 1.67 | |||||||
| :UNK1:C31 - A:ASP215:OD1 | 3.11 | |||||||
| :UNK1:C31 - A:ASP215:OD2 | 2.98 | |||||||
| 2. | Doxorubicin | −6.38 | −5.35 | −1.04 | 98.42 uM | :UNK1:H66 - A:ASP215:OD2 | 1.83 | Tyr28,Thr29,Asn30,Phe32,Phe59,Trp64,Arg214,Asp215,Gln218 |
| :UNK1:H67 - A:ASP215:OD1 | 2.31 | |||||||
| :UNK1:H65 - A:TYR28:O | 1.80 | |||||||
| :UNK1:C6 - A:ASP215:OD2 | 3.30 | |||||||
| :UNK1:C2 - A:ASP215:OD1 | 3.42 | |||||||
| 3. | Exemestane | −5.49 | −5.37 | −0.12 | 94.22 uM | A:ARG214:HH11 - :UNK1:O11 | 2.02 | Ala27,Trp64,Phe65,His66,Ile68,Arg214 |
| 4. | Gatifloxacin | −5.54 | −3.62 | −1.92 | 476.05 uM | :UNK1:H38 - A:ASP215:OD2 | 1.74 | Ala27, Tyr28,Thr29,Asn30,Trp64,Arg214,Asp215 |
| A:TYR28:HN - :UNK1 | 2.66 | |||||||
| 5. | Sulbactam | −5.47 | −3.05 | −2.42 | 112.85 uM | A:ALA27:HN2 - :UNK1:O13 | 1.96 | Ala27,Thr63,Trp64,Phe65,His66 |
| A:ALA27:HN3 - :UNK1:O14 | 1.75 | |||||||
| A:HIS66:HN - :UNK1:O7 | 2.02 | |||||||
| :UNK1:H23 - A:TRP64:O | 2.01 | |||||||
| 6. | Tobramycin | −5.25 | −4.80 | −0.46 | 52.94 mM | A:ASN30:HN - :UNK1:O19 | 2.05 | Tyr28,Thr29,Asn30,Phe32,Trp64,Leu21,Arg214,Asp215,Leu216,Pro217, Gln218 |
| A:ARG214:HH21 - :UNK1:O14 | 2.82 | |||||||
| :UNK1:H49 - A:TYR28:O | 2.10 | |||||||
| :UNK1:H50 - A:TYR28:O | 2.95 | |||||||
| :UNK1:H53 - A:ASP215:OD2 | 1.90 | |||||||
| :UNK1:H66 - A:ASP215:OD1 | 2.35 | |||||||
| :UNK1:H66 - A:LEU216:O | 2.21 | |||||||
| :UNK1:H67 - A:ASP215:OD1 | 2.08 | |||||||
| :UNK1:C4 - A:ASP215:OD2 | 3.75 | |||||||
| :UNK1:C12 - A:TYR28:O | 3.05 | |||||||
Azithromycin interacted to spike protein (PDB: 6VYB) with total intermolecular energy −4.60 kcal/mol, inhibition constant 832.15 uM, 4 Hbonds (A:ARG214:HH21 - :UNK1:O20, :UNK1:H - A:TYR28:O, :UNK1:C31 - A:ASP215:OD1, :UNK1:C31 - A:ASP215:OD2) formed with lengths of 1.72, 1.67, 3.11 and 2.98 Angstrom (Å), respectively. Ala27, Tyr28, Thr29, Asn30, Trp64, Arg214, and Asp215 amino acid residues are involved in hydrophobic interaction (Table 2). The interacting amino acid could be involved in the formation of a binding pocket for azithromycin. Moreover, azithromycin has also been reported to interact with ACE-2 receptor as hydroxychloroquine significantly (Oldenburg and Doan, 2020).
Doxorubicin showed a significant binding affinity with −6.38 kcal/mol, inhibition constant 98.42 uM, and 5 Hbonds (:UNK1:H66 - A:ASP215:OD2, :UNK1:H67 - A:ASP215:OD1, :UNK1:H65 - A:TYR28:O, :UNK1:C6 - A:ASP215:OD2, :UNK1:C2 - A:ASP215:OD1) formed with the lengths of 1.83, 2.31, 1.80, 3.30 and 3.42 Å, respectively. Tyr28, Thr29, Asn30, Phe32, Phe59, Trp64, Arg214, Asp215, Gln218 amino acids were involved in hydrophobic interaction (Fig. 2a, b, Table 2). Also, other interactions were observed. Thr29 was forming Amide Pi-stacked. Asn30 was involved in Pi-Sigma bond and Phe32 and Phe59 were involved in Pi-Alkyl contacts (Fig. 2b). Exemestane has shown a binding affinity −5.49 kcal/mol, inhibition constant 94.22 uM, single Hbond (A:ARG214:HH11 - :UNK1:O11) formed with the length of 2.02 Å. Phe140, Ala27, Trp64, Phe65, His66, Ile68, Arg214 amino acids were involved in hydrophobic interaction (Fig. 2c, d, Table 2). After analyzing complex interaction it was also found that Ala27, Trp64, His66, Ile68 were forming Alkyl and Pi-Alkyl bonds (Fig. 2d). Gatifloxacin interacted with −5.54 kcal/mol, inhibition constant 476.05 uM, 2 Hbonds (:UNK1:H38 - A:ASP215:OD2 and A:TYR28:HN - :UNK1) with the length of 1.74 and 2.66 Å. Ala27, Tyr28, Thr29, Asn30, Trp64, Arg214, Asp215 amino acids were involved in hydrophobic interaction (Fig. 2e, f, Table 2). Apart from traditional conventional bonds other types of interactions were also observed. Ala27 was forming the Pi-Alkyl bond. Tyr28 was also involved in Pi-Lone pairing and Arg214 was forming a halogen-fluorine interaction (Fig. 2f).
Fig. 2.

Showing the 3D interaction of SARS-CoV-2 spike protein (in rainbow color with ribbon pattern) with a (Doxorubicin), c (Exemestane) and e (Gatifloxacin). b (Doxorubicin), d (Exemestane), and f (Gatifloxacin) showing 2D graphical representation amino acid residues involved in hydrophobic interaction (shown by different color in a sphere) and different color dotted line showing different types of bonding including hydrogen bonds formation during drug and spike protein interaction. All drug compounds shown by grey color in the center with stick pattern. 3D and 2D graphics were generated by Discovery Studio Visualizer 2020.
Sulbactam showed binding energy −5.47 kcal/mol, inhibition constant 112.85 uM, and 4 Hbonds (A:ALA27:HN2 - :UNK1:O13, A:ALA27:HN3 - :UNK1:O14, A:HIS66:HN - :UNK1:O7, :UNK1:H23 - A:TRP64:O) formed with the lengths of 1.96, 1.75, 2.02 and 2.01 Å, respectively. Ala27, Thr63, Trp64, Phe65, His66 amino acids were involved in hydrophobic interaction (Table 2).
Binding energy has also been observed during Tobramycin and spike protein interaction. It was −5.25 kcal/mol, inhibition constant 52.94 mM and 10 Hbonds (A:ASN30:HN - :UNK1:O19, A:ARG214:HH21 - :UNK1:O14, :UNK1:H49 - A:TYR28:O, :UNK1:H50 - A:TYR28:O, :UNK1:H53 - A:ASP215:OD2, :UNK1:H66 - A:ASP215:OD1, :UNK1:H66 - A:LEU216:O, :UNK1:H67 - A:ASP215:OD1, :UNK1:C4 - A:ASP215:OD2, :UNK1:C12 - A:TYR28:O) formed with the length of 2.05, 2.82, 2.10, 2.95, 1.90, 2.35, 2.21, 2.08, 3.75 and 3.05 Å, respectively. Tyr28, Thr29, Asn30, Phe32, Trp64, Leu212, Arg214, Asp215, Leu216, Pro217, Gln218 amino acids were involved in hydrophobic interaction (Table 2). It was also observed that Leu212 and Pro217 were involved in Alkyl interaction.
Besides the ionic interactions as positively and negatively charged amino acids are observed to interact with spike protein. Moreover, it was also facilitated by the hydrophobic interaction due to Trp64, involved in Pi-Alkyl interaction. Thus, the interacting bonds are believed to form the stronger interaction to stabilize the drug-spike protein complex and could beneficial to stop the entry of the virus into the host cellular system using these drugs.
At the next level, 50 ns MDS output shown that RMSD values were ranged between 2.5 and 3.0 nm (Fig. 3a). RMSF calculation per residues has shown an average value between 0.5 and 1.5 nm. Few fluctuations were observed at 300–350 and the highest fluctuation was observed around 460–480 amino acid residues regions (Fig. 3b). The hydrogen bond plot showed the formation of 1–4 hydrogen bonds during the 20,000–45,000 ps period (Fig. 3c). The radius of gyration analysis provides the details of the assessment of the compactness and stability of spike protein structure during the whole simulation period due to the availability of drug compounds. The observed value of Rg was between 3.75 and 5.50 nm. The Rg plot showed that the most of time compactness was maintained with the value near 5.0 nm except sudden decrease at the time of 30,000–40,000 ps (Fig. 3d) after that regain stability until 50,000 ps.
Fig. 3.

Graphical representation showing a RMSD plot of Doxorubicin-Spike RBD complex deviation during 50 ns period; b RMSF plot with fluctuation per residue; c Hydrogen bond plot showing formation of hydrogen bonds during 50000 ps period; d Radius of gyration (Rg) plot showing compactness of Spike RBD molecule during 50 ns simulation. Where nm = nanometer; ns = nanoseconds; ps = picoseconds.
4. Discussion
The interaction energies analyzed from the in silico study of SARS-CoV-2 spike protein (PDB: 6VYB) with repurposing drugs were found high (7–8 (<4–5.5 kcal/mol) as compared to previously reported antiviral drugs Ritonavir, Oseltamivir, Remdesivir, Ribavirin, Chloroquine, Hydroxychloroquine, Favipiravir (<4–5.5 kcal/mol) (Narkhede et al., 2020).
The virus spike protein, a glycoprotein identified, binds with ACE-2 receptors and facilitates the entry of SARS-CoV-2 into the host cell. Then, bonded complex, spike protein-ACE-2 undergone some conformational changes to perform fusions supported by spike surface protease, TM protease serine 2 (TMPRSS2) and help the virus to activate the spike protein which facilitates the access of SARS-CoV-2 into the host cell (Pandey et al., 2020). The spike protein has been recognized with S1 and S2 domains which facilitate the binding and fusion respectively. (Huang et al., 2020).
This study signified that doxorubicin, exemestane, and gatifloxacin significantly interacting with the binding as well as the fusion domain of spike protein. Doxorubicin has been observed with maximum binding interaction with S protein. Recently, Jamal et al. (2021) also described the virus protease and peptidase inhibitory potential of doxorubicin.
The docking results indicated that the inhibitory potential of screened drugs could be due to hydrogen bonding and the involvement of many amino acids that facilitate the covalent, ionic as well as hydrophobic interaction. Angiotensin convertase enzyme 2 and its receptor work as a cellular doorway for SARS-CoV-2 entrance in the host cell through facilitating binding of the spike protein. The protein modeling of the RBD of the virus spike protein described that spike protein has interacted significantly with humanoid ACE-2. Moreover, ACE-2 protein expressed HeLa cells were reported more susceptible to COVID-19 infection while HeLa cells lacking ACE-2 protein expression they were found infection free with the virus (Xu et al., 2020).
The drugs have been found to interact with the receptor-binding domain of the S1 unit but the exact interacting amino acid between ACE-2 and S1 needs to be explored (Chen et al., 2020). The docking analysis was found that the doxorubicin drug has the highest docking interaction to counter COVID-19 through spike protein, followed by exemestane and gatifloxacin. Doxorubicin has also represented potential interaction that could be involved to interact with many macromolecules, and inhibit macromolecular biosynthesis required for activation and fusion. Doxorubicin, exemestane, and gatifloxacin could interact strongly with the spike S1 domain of the virus to offer greater stability to the complex that could facilitate to inhibit the attachment and entry of SARS-CoV-2 into the host cell. Recently, spike protein’s RBD model was tested to neutralize the SARS-CoV-2 with repurposing drugs like Azithromycin (Macrolides), Doxycycline, antiviral drugs like Tenofovir, Oseltamivir, Telbivudine, Zanamivir, Stavudine, Zalcitabine, Remdesivir -ATPase analog), synthetic peptide Colistin and Emblicanin A, natural compounds (Toor et al., 2021).
The docking studies with spike protein and hydroxychloroquine, chloroquine have also been reported with significant interaction to interact with spike protein. Since these drugs have been approved by FDA, it is suggested that studied drugs could be used against COVID-19 alone or in combination with other drugs (Al-Bari, 2017, Yao et al., 2020, Gautret et al., 2020). Previously conducted in vitro and clinical trial studies on COVID-19 patients explored the significant efficacy of combination drugs Azithromycin and Hydroxychloroquine against COVID-19 disease. The recently reported docking interaction of Hydroxychloroquine also supported the observed results of this study.
Furthermore, 50 ns MDS experimentation observed parameters like RMSD, RMSF, Rg, and formation of hydrogen bonds plot interpretation, revealed deviation and fluctuation of the Doxorubicin-Spike RBD complex during the whole simulation period. Observed RMSD values were between 2.5 and 3.0 nm after 5 ns complex gained stability until 25 ns after that small fluctuation observed between 25 and 30 ns but again around 35 ns until 50 ns stability regained by Doxorubicin-Spike RBD complex. Overall Rg analysis suggested that the Doxorubicin-Spike RBD complex was stable during 15,000–35,000 ps MDS with some fluctuations.
5. Conclusion
The interaction of doxorubicin was much high with the spike protein as compared to the other analyzed drugs. The computational studies provided possible types of bonds involved in the doxorubicin-spike complex that favors the stable bindings and promotes pathogen entry and multiplication. The drug doxorubicin has the highest binding energy (binding affinity) with COVID-19 spike protein which was validated with RMSD, RMSF, Rg, and Hbond plots data generated by MDS analysis. Thus, further research on doxorubicin could be beneficial to manage disease by inhibiting the spike protein RBD mediated viral entry to the host cell. So, we suggest conducting in vitro and in vivo antiviral potential of these drugs against the virus could be effective in the prevention of COVID-19.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgments
The authors are thankful to Qassim University, Saudi Arabia for providing necessary facilities to conduct the study.
Funding
Deanship of Scientific Research, Imam Abdulrahman Bin Faisal University, Dammam, Saudi Arabia, Grant number-Covid19-2020-002-IRMC.
Footnotes
Peer review under responsibility of King Saud University.
Contributor Information
Qazi Mohammad Sajid Jamal, Email: m.quazi@qu.edu.sa.
Mohammad Azam Ansari, Email: maansari@iau.edu.sa.
Suriya Rehman, Email: surrehman@iau.edu.sa.
References
- Ahmad V. A Molecular docking study against COVID-19 protease with a pomegranate phyto-constituents 'urolithin' and other repurposing drugs: from a supplement to ailment. J. Pharm. Res. Int. 2020;51–62 doi: 10.9734/jpri/2020/v32i1130545. [DOI] [Google Scholar]
- Al-Bari, M.A.A., 2017 Targeting endosomal acidification by chloroquine analogs as a promising strategy for the treatment of emerging viral diseases. Pharmacol Res Perspect 5(1), e00293. Published 2017 Jan 23. doi:10.1002/prp2.293. [DOI] [PMC free article] [PubMed]
- Ali S.G., Ansari M.A., Alzohairy M.A., Almatroudi A., Alomary M.N., Alghamdi S., Rehman S., Khan H.M. Natural products and nutrients against different viral diseases: prospects in prevention and treatment of SARS-CoV-2. Medicina. 2021;57(2):169. doi: 10.3390/medicina57020169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari M.A., Almatroudi A., Alzohairy M.A., AlYahya S., Alomary M.N., Al-Dossary H.A., Alghamdi S. Lipid-based nano delivery of Tat-peptide conjugated drug or vaccine–promising therapeutic strategy for SARS-CoV-2 treatment. Expert Opin. Drug Deliv. 2020;17(12):1671–1674. doi: 10.1080/17425247.2020.1813712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari M.A., Jamal Q.M., Rehman S., Almatroudi A., Alzohairy M.A., Alomary M.N., Tripathi T., Alharbi A.H., Adil S.F., Khan M., Malik M.S. TAT-peptide conjugated repurposing drug against SARS-CoV-2 main protease (3CLpro): potential therapeutic intervention to combat COVID-19. Arabian J. Chem. 2020;13(11):8069–8079. doi: 10.1016/j.arabjc.2020.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belouzard S., Millet J.K., Licitra B.N., Whittaker G.R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses. 2012;4:1011–1033. doi: 10.3390/v4061011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H.M., Westbrook J., Feng Z. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B.R., Brooks C.L., 3rd, Mackerell A.D., Jr CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30(10):1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Guo Y., Pan Y., Zhao Z.J. Structure analysis of the receptor binding of 2019-nCoV [published online ahead of print, 2020 Feb 17] Biochem. Biophys. Res. Commun. 2020;525(1):135–140. doi: 10.1016/j.bbrc.2020.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronavirus disease 2019. https://www.who.int/emergencies/diseases/novel-coronavirus-2019 Accessed: 2021-04-09.
- Dassault Systèmes BIOVIA . Dassault Systèmes; San Diego: 2020. Discovery Studio Visualizer], [Software version 2020. [Google Scholar]
- Ferreira, L.G., Dos Santos, R.N., Oliva, G., Andricopulo, A.D., 2015. Molecular docking and structure-based drug design strategies. Molecules. 2015;20(7):13384‐13421. Published Jul 22. doi:10.3390/molecules200713384. [DOI] [PMC free article] [PubMed]
- Gautret P., Lagier J.C., Parola P. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial [published online ahead of print, 2020 Mar 20] Int. J. Antimicrob. Agents. 2020;105949 doi: 10.1016/j.ijantimicag.2020.105949. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Gomez C., Espinoza I., Faruque F., Hasan M.M., Rahman K., Walker L., Muhammad I. Therapeutic intervention of COVID-19 by natural products: a population-specific survey directed approach. Molecules. 2021;26:1191. doi: 10.3390/molecules26041191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S., Tiwari N., Verma J., Waseem M., Subbarao N., Munde M. Estimation of a stronger heparin binding locus in fibronectin domain III14 using thermodynamics and molecular dynamics. RSC Adv. 2020;10:20288–20301. doi: 10.1039/D0RA01773F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haneef M., Lohani M., Dhasmana A., Jamal Q.M.S., Shahid S.M.A., Firdaus S. Molecular Docking of Known Carcinogen 4-(Methyl-nitrosamino)-1-(3-pyridyl)-1-butanone (NNK) with Cyclin Dependent Kinases towards Its Potential Role in Cell Cycle Perturbation. Bioinformation. 2014;10:526–532. doi: 10.6026/97320630010526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Y., Yang, C., Xu, X.-feng, Xu, W., Liu, S.-wen, 2020. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacologica Sinica 41, 1141–1149. doi:10.1038/s41401-020-0485-4. [DOI] [PMC free article] [PubMed]
- Jamal QMS. (2020). Structural Recognition and Binding Pattern Analysis of Human Topoisomerase II Alpha with Steroidal Drugs: In Silico Study to Switchover the Cancer Treatment. Asian Pacific J. Cancer Prevent.: APJCP, 21(5), 1349–1355. 10.31557/APJCP.2020.21.5.1349. [DOI] [PMC free article] [PubMed]
- Jamal Q.M.S., Alharbi A.H., Ahmad V. Identification of doxorubicin as a potential therapeutic against SARS-CoV-2 (COVID-19) protease: a molecular docking and dynamics simulation studies. J. Biomol. Struct. Dyn. 2021;1–15 doi: 10.1080/07391102.2021.1905551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamal Qazi Mohammad Sajid, Khan Saif, Khan Mahvish, Ansai Awais Abrar, Ashraf Jalaluddin Mohammad, Habibullah Mahmoud, Farasani Abdullah, Madkhali Aymen Mohammed, Lohani Mohtashim. Smoking May Increase the Risk of COVID-19 Infection: Evidence from In Silico Analysis. J. Pharmaceutical Res.Int. 2021;33(22B):12–21. doi: 10.9734/jpri/2021/v33i22B31394. [DOI] [Google Scholar]
- Khan S., Tombuloglu H., Hassanein S.E., Rehman S., Bozkurt A., Cevik E., Abdel-Ghany S., Nabi G., Ali A., Sabit H. Coronavirus diseases 2019: current biological situation and potential therapeutic perspective. Eur. J. Pharmacol. 2020 Nov;5(886):173447. doi: 10.1016/j.ejphar.2020.173447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufareva I., Abagyan R. Methods of protein structure comparison. Methods Mol. Biol. 2012;857:231–257. doi: 10.1007/978-1-61779-588-6_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmanic A., Zagrovic B. Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys. J. 2010;98(5):861–871. doi: 10.1016/j.bpj.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 2016;3:237–261. doi: 10.1146/annurev-virology-110615-042301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F., Li W., Farzan M., Harrison S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- Lu R., Zhao X., Li J. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet (London, England) 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G.M., Goodsell D.S., Halliday R.S., Huey R., Hart W.E., Belew R.K. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998;19:1639–1662. [Google Scholar]
- Morris G.M., Huey R., Lindstrom W. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkhede R.R., Cheke R.S., Ambhore J.P., Shinde S.D. The molecular docking study of potential drug candidates showing anti-COVID-19 activity by exploring of therapeutic targets of SARS-CoV-2. Eurasian J. Med. Oncol. 2020;4(3):185–195. doi: 10.14744/ejmo.2020.31503. [DOI] [Google Scholar]
- O'Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. Open Babel: an open chemical toolbox. J. Cheminf. 2011;3 doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenburg C.E., Doan T. Azithromycin for severe COVID-19. Lancet. 2020;396(10256):936–937. doi: 10.1016/S0140-6736(20)31863-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey P., Rane J.S., Chatterjee A., Kumar A., Khan R., Prakash A., Ray S. Targeting SARS-CoV-2 spike protein of COVID-19 with naturally occurring phytochemicals: anin silicostudy for drug development. J. Biomol. Struct. Dyn. 2020;1–11 doi: 10.1080/07391102.2020.1796811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prajapat M., Sarma P., Shekhar N. Drug targets for corona virus: a systematic review. Indian J Pharmacol. 2020;52:56–65. doi: 10.4103/ijp.IJP_115_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman S., Majeed T., Ansari M.A., Ali U., Sabit H., Al-Suhaimi E.A. Current scenario of COVID-19 in pediatric age group and physiology of immune and thymus response. Saudi J. Biol. Sci. 2020 May 15;27(10):2567–2573. doi: 10.1016/j.sjbs.2020.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman S., Majeed T., Ansari M.A., Al-Suhaimi E.A. Syndrome resembling Kawasaki disease in COVID-19 asymptomatic children. J. Infect. Public Health. 2020 Aug 20 doi: 10.1016/j.jiph.2020.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toor H.G., Banerjee D.I., Lipsa Rath S., Darji S.A. Computational drug re-purposing targeting the spike glycoprotein of SARS-CoV-2 as an effective strategy to neutralize COVID-19. Eur. J. Pharmacol. 2021;890:173720. doi: 10.1016/j.ejphar.2020.173720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, P., 2005. XMGRACE, Version 5.1. 19. Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology, Beaverton, OR.
- Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A.E., Berendsen H.J. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Walls A.C., Park Y.J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020;181(2):281–292.e6. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y., Shang J., Graham R., Baric R.S., Li F. Receptor recognition by the novel coronavirus from wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol. 2020;94(7):e00127–e220. doi: 10.1128/JVI.00127-20. Published 2020 Mar 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C., Horby, P.W., Hayden, F.G., Gao, G.F., 2020. A novel coronavirus outbreak of global health concern [published correction appears in Lancet. 2020 Jan 29;:].Lancet 395(10223):470‐473. doi:10.1016/S0140-6736(20)30185-9. [DOI] [PMC free article] [PubMed]
- Xu X., Chen P., Wang J., Feng J., Zhou H., Li X., Zhong W., Hao P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020;63:457–460. doi: 10.1007/s11427-020-1637-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, X., Ye, F., Zhang, M., et al., 2020. In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) [published online ahead of print, 2020 Mar 9]. Clin Infect Dis ciaa237. doi:10.1093/cid/ciaa237. [DOI] [PMC free article] [PubMed]
- Zhou P., Yang X.L., Wang X.G. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoete V., Cuendet M.A., Grosdidier A., Michielin O. SwissParam: a fast force field generation tool for small organic molecules. J. Comput. Chem. 2011;32(11):2359–2368. doi: 10.1002/jcc.21816. [DOI] [PubMed] [Google Scholar]