

Abstract
Hepatocellular carcinoma (HCC) is the fastest growing cancer worldwide in part due to the obesity epidemic and fatty liver disease, particularly nonalcoholic steatohepatitis (NASH). Chronic inflammation with the release of cytokines and chemokines with activation of hepatic stellate cells results in changes of the liver extracellular matrix (ECM) that predisposes to the development of HCC. Blood levels of the gastrointestinal peptide cholecystokinin (CCK) are increased in humans and mice consuming a high-fat diet. We found that the CCK-B receptor (CCK-BR) expression increased in the livers of mice with NASH. Treatment of mice with a CCK-BR antagonist, proglumide, prevented NASH, lowered hepatic inflammatory cytokines and chemokines, reduced oxidative stress, decreased F4/80+ hepatic macrophages, and prevented HCC. CCK-AR and CCK-BR expression was increased in both murine and human HCC cell lines compared to that of normal liver, and CCK stimulated the growth of wildtype and CCK-A receptor knockout HCC cells in vitro, but not CCK-BR knockout cells suggesting that the CCK-BR mediates proliferation. Proglumide therapy significantly reduced growth by 70% and 73% in mice bearing Dt81Hepa1-6 or in RIL-75 HCC tumors, respectively. Immunohistochemistry of a human liver tissue array with a selective CCK-BR antibody revealed staining of human HCC and no staining in normal liver.
Keywords: cholecystokinin, tumor growth, proglumide, CCK-B receptor, Liver cancer
Introduction
Today, the fastest growing cause of cancer-related death is hepatocellular carcinoma (HCC) (1). Infection with chronic hepatitis B or C virus is currently the dominant risk factor worldwide accounting for more than 1.3 million deaths per year from HCC (2). Once HCC develops surgical resection or orthotopic transplantation offer the only hope for a cure. Transarterial chemoembolization (TACE) provides palliation, but this therapy is typically only a bridge to transplantation (3). Recently, treatment with immune checkpoint antibodies has shown promise in HCC, but response is only 25–40% (4). Tyrosine kinase inhibitors such as Sorafenib and Regorafenib may extend survival a few months but also are not curative (5). Although vaccination for hepatitis B and treatment of hepatitis C has helped to decrease the liver cancer incidence from viral etiologies, the new obesity pandemic associated with the rising prevalence of non-alcoholic steatohepatitis (NASH) has become a new risk factor for cirrhosis and HCC (6, 7).
With chronic inflammation and injury, the liver undergoes tissue repair and regeneration, a process which often leads to hepatic fibrosis (8). Most cases of HCC arise in damaged livers with fibrosis or cirrhosis (9). The components of a premalignant liver and the liver extracellular matrix (ECM) are now well recognized as important factors involved in the initiation of HCC (10). The hepatic stellate cell, (HSC) (8, 10) is an important cell in the hepatic ECM that plays an essential role in fibrogenesis and hepatic inflammation. Hepatic fibrosis results from the activation of tissue myofibroblasts or HSCs that proliferate, migrate, and produce ECM components, such as type I collagen, and express cytokines and chemokines(11). HSCs often respond to inflammatory stimuli (9) such as cytokines and chemokines from macrophages (12) and other immune cells resulting in HSC activation and migration.
Strategies to reduce hepatic inflammation and reverse fibrosis are under investigation. Although it was thought for years that cirrhosis and fibrosis were not reversible, studies have now shown that treatment of hepatitis C (13) and hepatitis B (14) actually can reverse cirrhosis. Anti-fibrotic strategies have been attempted with agents that inhibit the activation of HSCs, agents that block HSC function, anti-cytokine or anti-growth factor therapies, and of course, elimination of the insulting agent (such as alcohol or hepatitis). Some agents under investigation include peroxisome proliferation-activated receptor-γ ligands, angiotensin receptor blockers, angiotensin-converting enzyme inhibitors antioxidants, serine-protease inhibitors, and NADPH oxidase inhibitors (15). Unfortunately, since many of these compounds are not specific to the fibroblasts, they have side effects including possible deleterious effects on other organs. The HSCs also play an important role in the normal function of the liver and are thought to help with regeneration after injury and maintenance of cell function (16). Therefore, agents that eliminate or injury the HSCs would be counterproductive.
The evolution of inflammation in nonalcoholic fatty liver disease leading to NASH and subsequently increasing the risk for HCC has been hypothesized to involve the multiple parallel hits hypothesis (17). Numerous molecular mechanisms have been proposed in the etiology of NASH, including genetic predisposition, abnormal lipid metabolism, oxidative stress, mitochondrial dysfunction, altered production of cytokines and adipokines, gut dysbiosis and endoplasmic reticulum stress (18). In this investigation, we are taking a paradigm shift in the possible mechanisms involved in dietary fat and NASH-related hepatic carcinogenesis. Our focus in this investigation concerns the role of a mitogenic, pro-inflammatory gastrointestinal peptide; cholecystokinin (CCK), that we hypothesize is a key instigator of NASH-associated HCC. Since diets high in saturated fat raise blood CCK levels in humans and mice (19, 20); and because CCK is involved in insulin regulation, lipid metabolism, and inflammation, we studied the role of CCK and CCK receptors in NASH and HCC. In a study with immunocompetent C57BL/6 mice fed a saturated fat choline deficient ethionine (CDE) supplemented diet, we found that a CCK receptor antagonist proglumide not only prevented steatosis, inflammation, fibrosis, and HCC, but also was able to reverse the histologic and biochemical effects of NASH (21). Herein, we explore the possible mechanisms by which interruption of CCK signaling at its receptor may be protective for the ‘at risk’ liver thus preventing progression to HCC. Furthermore, we explore the role of CCK-receptor blockade in established HCC as a novel therapeutic approach to this aggressive malignancy.
Methods and Materials
HCC cell lines
Two murine HCC and one human HCC cell lines were used in this study. Dt81Hepa1-6 murine HCC cells (22) were a gift from Dr. Marc Bilodeau (Montreal, Canada) and are a highly metastatic cell line derived from the ATCC parent Hepa1–6 cells. The Dt81Hepa1-6 cells were tested by the MU Research Animal Diagnostic Laboratory using the IMPACT I PCR Profile that included testing for the following: Ectromelia, EDIM, Hantaan, K virus, LCMV, LDEV, MAD, mCMV, MHV, MNV, MPV, MTV, MVM, Mycoplasma sp., Polyoma, PVM, REO3, Sendai, TMEV GDVII. All of these tests were negative. RIL-175 murine HCC cells were derived by ex vivo genetic manipulation of embryonic liver progenitor cells that were manipulated with retroviral gene transfer of oncogenes or short hairpin RNAs targeting tumor suppressor genes to develop tumor that are p53–/–overexpressing c-myc (23). The RIL-175 cell line was characterized and donated by Dr. Tim Greten (NCI, Bethesda, MD). The RIL-175 cells were tested at the Animal Health Diagnostic laboratory, NCI Frederick, MD using the Molecular Testing of Biological Materials-Mouse/Rat (MTBM-M/R) Test, and all the tests were negative. The Dt81Hepa1-6 and HepG2 cells were maintained in DMEM standard growth media and the RIL-175 were maintained in RPMI-1640 Medium supplemented with 10% fetal bovine serum in humidified air with 5% CO2.
HepG2 human HCC cancer cell line was obtained from the ATCC through the Tissue and Cell culture repository of the Lombardi Comprehensive Cancer Center. This cell line that has been used extensively to study human HCC has been characterized as a hepatoblastoma-derived cell line (24). These cells were maintained in DMEM media supplemented with 10% fetal bovine serum in humidified air with 5% CO2.
Evaluation of CCK receptor mRNA expression in murine liver tissues and cells
RNA was extracted (Qiagen) from log-phase murine liver cancer cells (Dt81Hepa1-6 and RIL-175) and also from normal and CDE-fed murine liver from C57BL/6 mice to determine mRNA expression of CCK-A receptors (CCK-AR) and CCK-B receptors (CCK-BR). cDNA was generated and subjected to real-time PCR (qRT-PCR) using SYBR® Green (Life Technologies) in an Applied Biosystems 7300 thermal cycler with the following conditions: initial incubation for 10 minutes at 95°C followed by 40 cycles of 95°C × 30sec, 60°C × 1 minutes, and 72°C for 30 sec. CCK-AR cDNA validated primers included: 5’CTTTTCTGCCTGGATCAACCT3’ (forward); 5’ACCGTGATAACCAGCGTGTTC3’ (reverse). CCK-BR primers included: 5’GATGGCTGCTACGTGCAACT3’ (forward) and 5’CGCACCACC- CGCTTCTTAG3’ (reverse). HPRT was used as a reference gene and HPRT murine cDNA validated primers included: 5’TCAGTCAACGGGG-GACATAAA 3’ (forward); and 5’GGGGCTGTACTGCTTAACCAG3’ (reverse).
CCK-BR protein expression by flow cytometry (Dt81Hepa1-6)
Flow cytometry was performed to measure surface Ab staining of mouse liver epithelial cells. One million viable Dt81Hepa1-6 cells per 2 mls of DMEM complete media were placed in each flow cytometry tube (Falcon Ref #352058, Bedford, MA). Volumes were equalized with PBS, and cells were pelleted by centrifugation at 1000 rpm for 5 minutes Then the CCK analogue, cerulein, (100 ng/ml) (Sigma, SRP4492–20μg, dissolved in dH2O) was added to the cells resulting in a final concentration of 1 μM. Brefeldin A solution, 1 μl/ml, (Biolegend, Cat: 420601) was added to each well for 4 hours. Cells were washed and then blocked by adding 5 μl Purified Rat Anti-Mouse CD16/CD32 (Mouse BD Fc Block™ brand reagent; BD Biosciences, San Jose, CA) and then 1 μl of CCK-BR (Abcam, ab7707) conjugated flow antibody with Dylight 488 (SA5–10078) was added. Cells were fixed with 300 μl IC Fixation buffer (Invitrogen, cat#00–8222-49) and incubated at room temperature for 20 minutes followed by the addition of Permeabilization Buffer (Invitrogen, cat#00–8333-56). Flow cytometry was performed using a FACSARIATM IIu brand cell sorter (BD Biosciences).
Gene editing by CRISPR Cas9 knockout experiments
Dt81Hepa1-6 liver cancer cells underwent gene editing with CRISPR-Cas9 technology to selectively knockout the CCK-AR or the CCK-BR. The Dt81Hepa1-6 liver cancer cell line was transfected with CRISPR vector pSpCas9 BB-2A-GFP PX485 to knockout the CCK-AR with CRISPR Cas9 technology using Lipofectamine 2000 (Life Technologies). Similarly, the CCK-BR in the Dt81Hepa1-6 cells was selectively knocked out with CCK-BR CRISPR Guide RNA or crRNA1 (GeneScript). To confirm successful knockout of the CCK-AR and CCK-BR, qRT-PCR was done as above of the clonal cells and compared to wild-type cells.
Cell proliferation assay
In vitro growth studies were performed with Dt81Hepa1-6 wild type cells with exogenously administered CCK (0.1, 1.0 of 10 nM) versus PBS (controls) (N=6 each). Cells were plated 25,000 cells into each well of 12-well plates in media containing 10% fetal bovine serum (FBS). After 24 hours, the media was changed to 1% FBS and the cells were treated with PBS (control) or CCK (0.1, 1.0 of 10 nM). Wild type, CCK-AR-KO or CCK-BR-KO Dt81Hepa1-6 cells (40,000 cells in each well) were plated in the 12-well plate (N=6 wells/ treatment). After allowing 24 hours for adherence to the plate, the cells were treated with PBS or CCK 1.0 nM. In each in vitro growth study, the media was changed daily and appropriate concentration of CCK peptide re-added or PBS to each appropriate well. After 4-days of growth, the cells were harvested with trypsin containing 0.025% EDTA. Viable cells were counted either manually or by mechanical methods using the trypan blue dye method. Growth of RIL-175 cells was evaluated using the MTT [3-(4, 5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide] colorimetric assay. Cells (5,000) were plated into each well of a 96-well plate and treated with media alone (Control), CCK (1 nM), the CCK-AR antagonist L364,718 (1 nM, Tocris), the CCK-BR antagonist L365,260 (1 nM, Tocris), or a combination of CCK with each antagonist. After 24 hours the media was reacted with the MTT Cell Proliferation Assay reagent (ATCC, catalog #30–1010K, Manassas, VA) and read in a plate reader at 540 nm.
Animal studies
NASH-inducing diet:
All mouse studies were performed in an ethical fashion and approved by the Institutional Animal Care and Use Committee (IACUC) at Georgetown University. Sixty-five female C57BL/6 mice (Charles Rivers, Germantown, MD), were used in a study that involved both a prevention and a NASH-reversal study as previously described (21). Mice (N=65) were fed either a modified 75% choline deficient ethionine supplemented (CDE) saturated fat diet (MP Biomedicals, LLC; Solon, Ohio) or a control diet with the same fat (lard), carbohydrate, and protein content as the CDE diet but with choline and without ethionine for 12 or 18 weeks. Some cohorts of mice were treated with the CCK receptor antagonist proglumide in the drinking water (Tocris Bioscience, Bristol, UK) at a concentration of 0.1 mg/ml.
HCC studies:
Two separate HCC studies were conducted to evaluate the role of proglumide therapy in decreasing growth of established HCC in murine models. In the first study, 6-week old male athymic nude mice (N=20) were injected subcutaneously in each flank with a 0.1 ml suspension of (106) Dt81Hepa1-6 murine HCC cells. Nude mice were used since we had attempted to grow the Dt81Hepa1-6 cells in C57BL/6, but did not achieve adequate tumor take. All mice were placed on a high-fat (58Y1, blue; TestDiet, St. Louis, MO) composed of 34.9% fat from lard in order to raise endogenous CCK blood levels.
In the second study, twenty C57BL/6 mice were inoculated over one flank subcutaneously with 300,000 murine syngeneic RIL-175 murine HCC cells and mice received standard chow. In both animal HCC growth studies, half of the mice received drinking water treated with proglumide (0.1 mg/ml) and the remaining mice (control mice) received untreated drinking water. Tumors were measured weekly with calipers and volumes calculated using L × (W)2 × 0.5. After approximately four weeks when control tumors reached a diameter of approximately 20 mm, the mice were ethically euthanized, tumors dissected and weighed.
Hepatic cytokine and chemokine PCR array from livers of NASH mice
RNA was isolated from 18-week old CDE-fed mouse livers and RNA integrity confirmed with RIN values. Synthesis of cDNA was performed using the RT2 First Strand Kit (Cat # 330401 Qiagen). PCR was performed using the mouse cytokines & chemokines RT2 Profiler PCR Array that evaluates profiles the gene expression of 84 key secreted cytokines, chemokines, and other proteins central to the immune response (Qiagen; Catalog #PAMM-150Z) with an ABI StepOne Real time PCR machine. Analysis of the array results and comparison between groups was performed using the array software provide on the Qiagen website.
Hepatic glutathione assay
Mouse NASH liver tissue samples from the CDE-fed mice were homogenized and deproteinization carried out with ice cold perchloric acid (4 M), according to the Glutathione Assay Kit protocol (Abcam, ab65322). This fluorometric, dye-based assay provides a simple in vitro assay for detection of total glutathione changes from tissue homogenates. The assay uses the dye monochlorobimane (MCB), to form an adduct with glutathione in a reaction catalyzed by glutathione-S-Transferase (GST). After 1 hour incubation at 37°C, fluorescence was measured in a plate reader at Ex/Em = 360 ± 20 nm / 460 ± 20 nm.
Hepatic expression of heat shock proteins
RNA isolated from 18-week old CDE-fed mouse livers cDNA was subjected to qRT-PCR using SYBR® Green (Life Technologies) in a 7300 ABI thermal cycler. PCR was performed in a 20 ml volume using 40 cycles and an annealing temperature of 60°C. Primers are shown in Supplementary Table 1.
Murine hepatic macrophage quantification by immunohistochemistry in the NASH liver
Five μm cuts of paraffin-embedded liver sections from mice fed CDE diet with untreated water (N= 15) and mice fed CDE diet with proglumide-treated water (N=15) were mounted on slides. Slides were reacted with an F4/80 Ab (1:40) at 4°C overnight; (eBioscience; Cat # 14–4801-85) followed by incubation with a secondary M.O.M antibody (ImmPRESS; Cat# MP-2400) for 30 minutes at room temperature. After counterstaining with hematoxylin (1:9) the number of immunoreactive cells per slide, were counted with ImageJ computer software.
Effects of CCK receptor blockade on fibrosis
Sections of livers (5 μm) were cut and mounted from the mice fed the CDE diet to induce NASH and were treated with either the CCK receptor antagonist proglumide or untreated water. Fibrosis was evaluated by Masson’s Trichrome stain and images were captured using a 20X objective lens on an Olympus BX61 microscope with a DP73 camera (N=10 per group). Analysis of fibrosis was done using software by ImageJ.
Using a 380 miRNA qPCR-based array (Qiagen) we conducted differential miRNA expression analysis of pancreas tissue from KRAS mice treated for 4 months with proglumide and pancreas tissue from KRAS mice receiving no treatment. Histology confirmed that proglumide reversed inflammation and fibrosis in this model.
Translational studies with normal human liver tissue and liver cancer
Next we examined whether the CCK-A and CCK-B receptors are also present in normal human liver tissues and human HCC cancer cells. First we studied the mRNA expression for these receptors and next we examined the CCK-BR protein expression by immunohistochemistry using a liver tissue array. MRNA expression by qRT-PCR was done using human HepG2 HCC cells compared to normal human liver tissue. Five samples of flash frozen tissues were obtained from surgical resections of human subjects with benign liver conditions shown in Supplementary Table 1. RNA was extracted from the normal human tissues and subjected to qRT-PCR. CCK receptor expression was evaluated in the normal human liver tissues and the human HepG2 HCC cells using the following primers: CCK-BR: 5’ TACACCACC ATCAGCACACT3’ (forward); and 5’ TCAGTTGTGGTTTGTGTTTTCTAT3’ (reverse); and the CCK-AR: 5’ CAGGCAAGGATGGAT-GTGGT3’ (forward); and 5’ GCATCCGCTTGTTCCGAATC3’ (reverse). GAPDH was used as a reference gene.
In order to determine if the CCK-BR was the receptor responsible for the proliferative effects in cancer, we examined the effects of exogenous gastrin (a peptide that binds only to the CCK-BR and not the CCK-AR; (25, 26) treatment with or without the CCK receptor antagonist proglumide in an in vitro clonogenic assay. HepG2 cells were seeded at 2,500 cells/well in a 12-well plate. Once attached, the cells were treated in serum-free medium with medium alone (control), gastrin 0.4 nM, proglumide 40 nM, or a combination of gastrin and proglumide for 5 days to visualize colony formation. Cells were then stained with methylene blue (10 mg/ml, in 1:1 methanol: water) for 10 minutes, then washed with deionized water. Plates were scanned and images were analyzed on ImageJ.
In order to study the CCK-BR protein expression in human liver tissues and human HCC, a tissue array (Cat# BC03117) was obtained from US Biomax, Rockville, MD. This array contained 48 cases of hepatocellular carcinoma and five tissues from normal human liver. After deparaffination and antigen retrieval procedures, the array was reacted with a goat polyclonal CCK-BR primary antibody (Abcam, ab7707) at 1:200 overnight at 4°C. The next day after washing x3, the array was incubated with 1–3 drops of Biotinylated secondary antibody (R&D systems catalog # CTS008) for 60 minutes. The slide was rewashed with1x PBS three times, for 2 minutes each, and reacted with DAB Chromogen (R&D systems catalog # CTS008) for 3 minutes. After washing in 1x PBS three times; 2 minutes each, the slide was counterstained with Hematoxylin (Fisher, Harris Modified Hematoxylin), blued in 1% ammonium hydroxide, dehydrated, and mounted with Paramount. Images were captured using an Olympus BX61 microscope with a DP73 camera.
Statistical analysis
Mean values for each treatment group were compared to controls or baseline groups using computer software by PRISM GraphPad 7 and / or Minitab version 19. Student t-tests were used to evaluate statistical significance with a p < 0.05 considered to be statistically significant when two groups were compared. Where multiple comparisons were made to a control the Bonferroni adjustment was made, and if the data was skewed, a nonparametric analysis with Mann Whitney was performed. RT-PCR assays were performed in triplicate and results were expressed as pairwise Student t-tests on the normalized mean ΔCT (the difference between the cycle counts of the gene of interest minus the count of an endogenous control) values for each group.
Results
Expression of CCK receptors on hepatic cells and tissues
Both CCK-AR and CCK-BR mRNA expression were only minimally detected by qRT-PCR in control murine liver. The CCK-BR expression was significantly increased (Fig. 1A) in NASH livers of mice fed a CDE saturated fat diet (P=0.008) compared to that of control mouse liver. In contrast, CCK-AR mRNA levels were significantly decreased in mice fed the saturated fat CDE diet (Fig. 1A) compared to control mouse liver. CCK-AR and CCK-BR expression in murine RIL-175 murine HCC cells and murine Dt81Hepa1-6 HCC cells were both significantly increased compared to the expression of these receptors in normal mouse liver (Fig. 1B). To confirm the CCK-BR mRNA receptor was translated into receptor protein, Dt81Hepa1-6 cells were subjected to flow cytometry using a Dylight-Alexa488 labeled CCK-BR antibody. Flow cytometry confirmed that over 90% of the Dt81Hepa1-6 cells were immunoreactive and expressed fluorescence consistent with the CCK-BR (Fig. 1C).
Figure 1.

Expression of CCK receptors in the murine liver and HCC cells and response to CCK. A, CCK-BRs are significantly increase in the NASH livers of mice on the CDE diet compared to livers of control mice (P=0.008). CCK-ARs are downregulated compared to control livers in mice fed a high saturated fat CDE diet (P=0.0014). B, Compared to normal mouse liver, CCK-BR and CC-ARs are significantly increased in mouse RIL-175 and Dt81Hepa1-6 HCC cells. C, CCK-BR immunofluorescence is detected by flow cytometry in Dt81Hepa1-6 HCC cells. †reproduced with permission from Dig Dis Sci (21). D, CCK peptide stimulates growth of Dt81Hepa1-6 HCC murine cells in cell culture with the greatest effect with 1nM, a concentration equal to the physiologic binding affinity at the CCK-BR. E, Means ± SD of Dt81Hepa1-6 wild-type, CCK-AR-KO or CCK-BR-KO cells after four days of exposure to PBS or CCK (1 nM). CCK peptide stimulated growth of wild-type cells and CCK-AR-KO cells but not CCK-BR-KO cells. F, CCK (1 nM) stimulated growth of RIL-175 murine HCC cells compared to controls. The CCK-increased growth was blocked only by the CCK-BR antagonist, L365,260, and not the CCK-AR antagonist, L364,718. G, In vivo, Dt81Hepa1-6 tumor size was significantly less in mice treated with proglumide compared to controls. H, In vivo, weekly tumor volumes of RIL-175 murine HCC tumors were significantly less in mice that received drinking water supplemented with proglumide. Significantly different than controls: *P<0.05, **P<0.01, ***P<0.001.
CCK mediates growth of HCC by the CCK-BR in vitro and in vivo
In vitro growth studies performed with Dt81Hepa1-6 HCC cells (Fig. 1D) in the presence of CCK (0.1, 1, or 10 nM) or PBS (0) demonstrated the characteristic bell-shaped curve with the greatest proliferative effect found at the CCK 1 nM dose, a dose consistent with the physiologic binding affinity (Kd) of CCK at its receptor. With the intention to determine whether the CCK-AR or the CCK-BR was involved in promoting HCC tumor growth, we selectively performed gene editing for each receptor. Confirmation of successful knockout using CRISPR Cas9 technology confirmed that the established clones had either selective CCK-AR (Fig. S1–A) or CCK-BR knockout (Fig. S1–B). Using this optimal concentration of CCK, growth of wild-type HCC cells was compared to growth of CCK-AR-KO or CCK-BR-KO cells in the presence or absence of CCK peptide. CCK stimulated growth of wild-type and CCK-AR-KO HCC cells, but not CCK-BR-KO cells (Fig. 1E). This finding suggests that the CCK-proliferative effects are mediated by the CCK-BR and not the CCK-AR. Mouse RIL-175 cells were also stimulated in cell culture by CCK and this effect was blocked only by the CCK-BR antagonist, L365,260, not by the CCK-AR antagonist, L364,718 (Fig. 1F), again supporting that the proliferative effects of CCK on HCC cells are mediated by the CCK-BR and not the CCK-AR.
The effect of proglumide was evaluated in mice bearing subcutaneous murine HCC tumors. Proglumide therapy significantly decreased reduced growth of Dt81-Hepa1–6 tumors compared to controls (Fig. 1G). Proglumide also slowed the growth rate of RIL-175 tumors in C57BL/6 mice compared to controls (Fig. 1H). No significant differences (P=0.374) were identified in final animal body weights between untreated water and proglumide-treated water mice. In the RIL-175 study, the mean ±SEM of animal body weights were 16.3 ± 0.66 g (PBS) and 17.4 ± 0.68 g (Proglumide). Nor were any differences (P = 0.369) in the animals’ final body weight in the mice bearing Dt81-Hepa1–6 tumors: 30.2 ± 0.9 g (high fat diet/ untreated water) and 28.0 ± 2.0 g (high fat diet /proglumide-treated water)
Proglumide therapy alters the hepatic extracellular matrix making it less pro-carcinogenic
Inflammation is one of the hallmarks of chronic liver disease and the hepatic ECM and hepatic parenchyma is infiltrated with immune cells and their key effectors such as cytokines and chemokines (12). Cytokines play an active role in the development and the progression of NASH through stimulation of hepatic inflammation, cell necrosis and apoptosis, and induction of fibrosis (27). We compared the gene expression of hepatic cytokines and chemokines in livers of mice with confirmed NASH secondary to the CDE saturated fat diet to mice on the exact same diet but that received proglumide supplemented drinking water. The livers from CDE fed mice exhibited increased inflammation histologically (21) compared to control-diet mice with a marked increase in gene expression of several cytokines and chemokines (Fig. 2A). Most of these cytokines and chemokines are involved in macrophage and lymphocyte recruitment. One chemokine in particular, CCL20 (also called Macrophage Inflammatory Protein-3), was increased in the livers of CDE-fed mice and this level was reduced greater than 20-fold with proglumide (Fig. 2B). CCL20 is the only known ligand for the receptor CCR6, which in turn is the only known receptor for CCL20 (28). The CCR6 and CCL20 interaction is associated with chronic hepatic inflammation (29). Another cytokine, CCL2, that has been shown to play an important role in the regulation of HCC tumor growth, metastasis and host immune response (30), was also significantly reversed with proglumide in CDE-fed mice (Fig. 2B).
Figure 2.
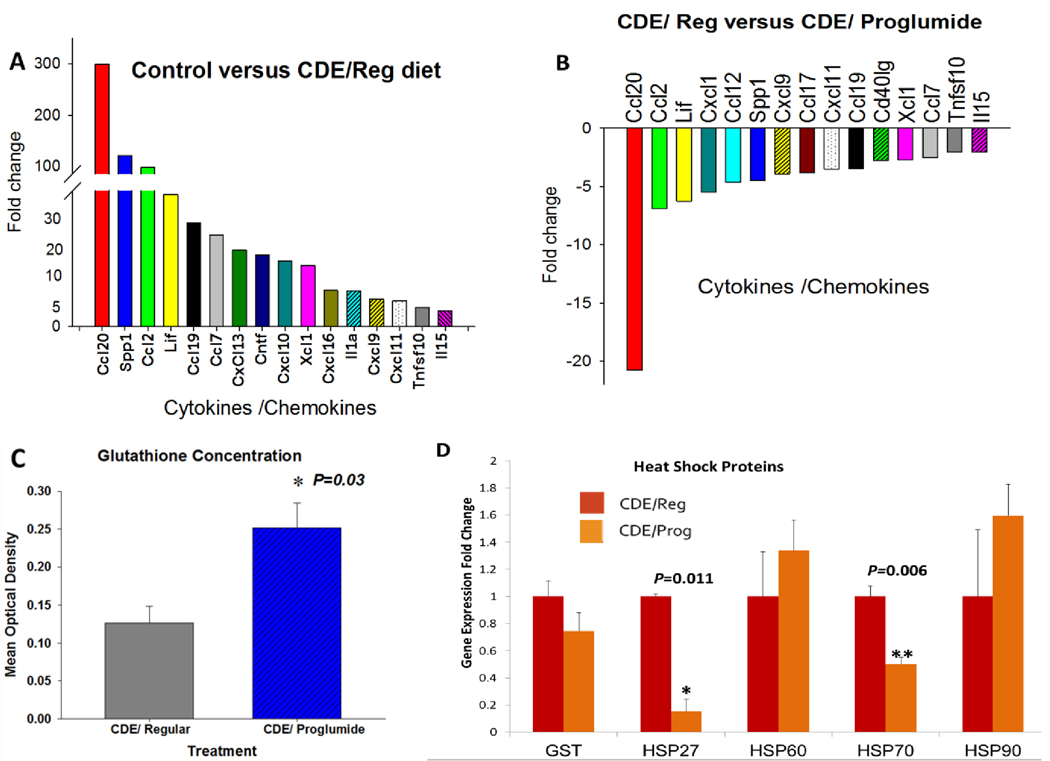
Proglumide therapy alters the hepatic extracellular matrix rendering it less pro-carcinogenic. A, Gene expression of cytokines and chemokines are increased in NASH livers of mice on the CDE diet with regular, untreated water compared to mice on the control saturated fat diet. B, Proglumide reverses gene expression of activated cytokines and chemokines in NASH livers of CDE fed mice compared to livers of the mice fed the CDE diet with untreated (Regular) drinking water. C, Liver glutathione levels are increased in CDE-fed mice treated with proglumide compared to CDE fed mice with regular (untreated water). D, Gene expression of selective heat shock proteins associated with hepatic inflammation and NASH demonstrate that HSP27 and HSP70 are significantly reduced in the CDE livers of mice treated with proglumide. Significantly different than controls: *P<0.05, **P<0.01.
Oxidative stress has been associated with the risk of liver cirrhosis. Glutathione is synthesized in the liver with its dependent antioxidant enzymes and plays an important role in the antioxidant defense system (31). CDE-fed mice that received proglumide had increased liver concentration of glutathione (Fig. 2C), suggesting that proglumide therapy is protective to hepatocellular injury and oxidative stress.
Heat shock proteins are often increased in tissues undergoing stress and have been identified as playing an important role in NASH and the development of HCC (32). Although the heat shock proteins we evaluated are ubiquitous and have numerous roles in tissues, we found that the hepatic expression of both HSP27 and HSP70 were significantly reduced when mice were treated concomitantly with proglumide (Fig. 2D). HSP27 was reported to be elevated in human subjects with known HCC compared to controls and was thought to serve as a potential biomarker for HCC (33). HSP70 is also elevated in the serum of patients with HCC and has been correlated with prognosis (34). Therefore, since proglumide decreases both of these heat shock proteins, we believe this property is most likely related to the anti-carcinogenic properties of this compound.
Infiltrating hepatic macrophages are decreased with proglumide
Macrophages release cytokines /chemokines that activate stellate cells. Activation and recruitment of infiltrating macrophages is involved with the progression of NASH (35). Livers from CDE-fed mice were stained with F4/80 to examine the number of hepatic macrophages compared to that of livers of CDE-fed mice that were treated with proglumide. Hepatic macrophages stained with F4/80 were abundant in the NASH livers of mice on the CDE diet (Fig. 3A). An area of the representative immunohistochemical slide, magnified to demonstrate macrophage staining in the mouse CDE NASH liver is presented (Fig. 3C). The number of immunoreactive macrophages is reduced in the livers of CDE-fed mice that were concomitantly treated with proglumide in the drinking water (Fig. 3B). The area within the box is magnified (Fig. 3D showing the paucity of macrophages in proglumide treated mice. The mean number of hepatic macrophages was analyzed by densitometry using ImageJ and was significantly decreased (P<0.0001) in the proglumide-treated mice (Fig. 3E).
Figure 3.
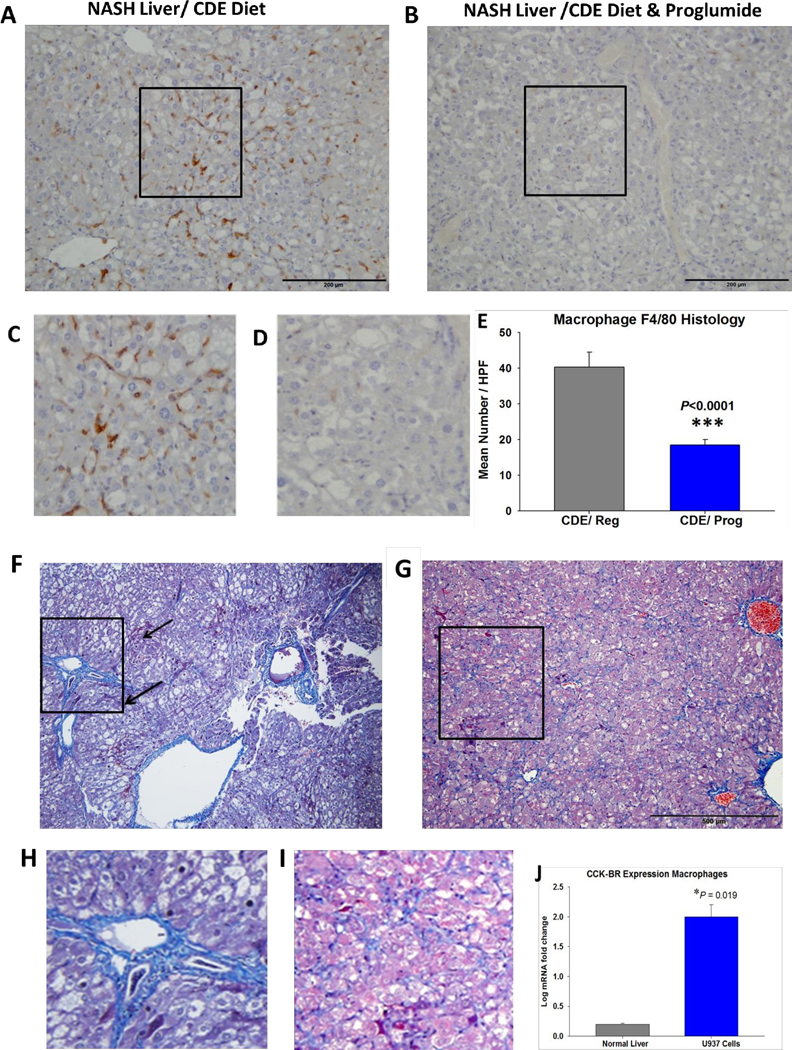
Proglumide treatment decreases influx of macrophages and reduces hepatic fibrosis. A, F4/80 positive hepatic macrophages are abundant in the NASH liver of a CDE fed mouse. Box in the center is magnified in C. B, Fewer macrophages are found in the hepatic parenchyma from a CDE-fed mouse that was treated with proglumide. Box in the center is magnified below in D. E, analysis of number of F4/80 positive macrophages is significantly reduced in the livers of CDE fed mice on proglumide (N=10) compared to that of livers of CDE-fed mice on regular untreated water (N=10). F, Masson’s trichrome stain shows marked fibrosis in a representative NASH liver section from a mouse fed the CDE diet for 18-weeks. Histologically there is macro-steatosis, balloon degeneration, inflammation, and dysplastic nodules (arrows). Box is magnified in H showing marked peri-portal fibrosis. G, Masson’s trichrome stain of a representative liver from a mouse fed the CDE diet for 18-weeks and treated with proglumide. The box is magnified in I demonstrating less fibrosis. J, Quantitative analysis of fibrosis in hepatic sections comparing fibrosis in livers of CDE fed mice receiving regular or proglumide-treated water. Significantly greater than controls: ** P<0.01; ****P<0.0001.
Hepatic fibrosis is decreased with proglumide therapy
Fibrosis was markedly increased in the livers of mice fed the CDE diet and regular water for 18-weeks (Fig. 3F). Many of these livers had early dysplastic nodules or early HCC as well as other hallmarks consistent with NASH including inflammation and balloon degeneration. A magnified insert of a fibrotic portal trial is shown (Fig. 3H). In contrast, there was less fibrosis in the CDE-fed mice at week 18 that were treated with proglumide (Fig. 3G) and magnified in Fig. 3I. When compared to CDE-fed mice on regular water analytically using ImageJ, there was significantly less fibrosis in the livers of CDE-fed mice that received proglumide (Fig. 3F). We previously performed Western blot analysis to measure changes in selective collagenous proteins in the extracellular matrix in the livers of CDE mice and found that proglumide therapy significantly reduced collagen IV and fibroblast activation protein (FAP) (21).
CCK- receptors are increased in human HCC compared to normal human liver
Both CCK-AR and CCK-BR mRNA expression was very low in all five of the normal liver tissue samples. Expression of the CCK-receptors was 3–4-fold greater in the human HepG2 liver cancer cells (Fig. 4A) compared to the mean expression in the normal human livers. A clonogenic assay was performed to evaluate whether the CCK-BR on HepG2 cells could be stimulated by, gastrin which binds to the CCK-BR with equal affinity to that of CCK without binding to the CCK-AR. Similar to the murine HCC cells, human HepG2 cells formed significantly more proliferative colonies in culture when treated with gastrin than controls (Fig. 4B). These results are the third piece of evidence supporting that the CCK-BR mediates the proliferative effects of ligands CCK and /or gastrin. Proglumide, reduced the colony formation by > 50%, yet this value did not quite reach statistical significance (P=0.06). Since proglumide is a weak antagonist, perhaps longer treatment or a higher concentration of proglumide would have had a more profound effect. We previously showed in mice bearing pancreatic cancer tumors, that when proglumide was administered in combination with an immune checkpoint antibody (36) or when administered with gemcitabine chemotherapy (AACR abstract 2020), that proglumide exhibited a significant additive anti-tumor effect compared to when given as monotherapy. Therefore, combination treatment using proglumide with other anti-tumor reagents in HCC may enhance the response.
Figure 4.
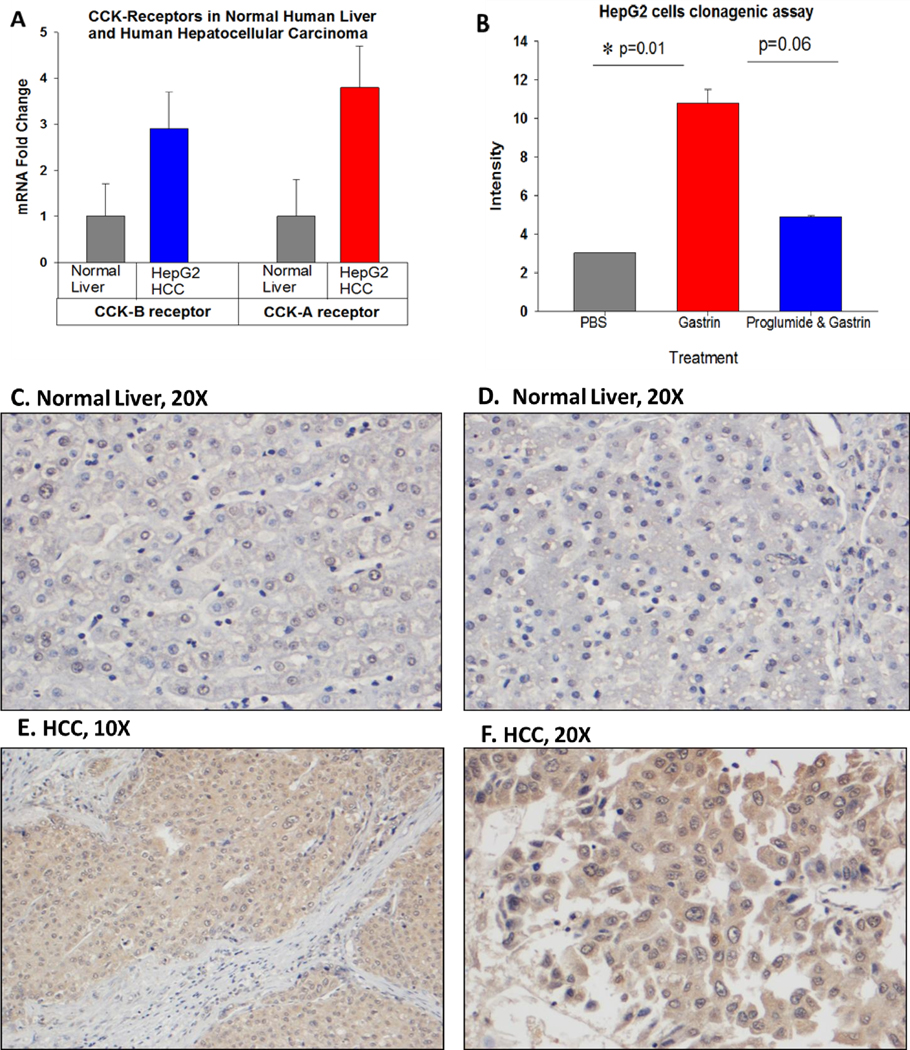
CCK receptors in human HCC compared to normal human liver. A, Mean qRT-PCR mRNA receptor expression is shown from the 5 normal human liver tissues for both the CCK-AR and CCK-BR. The expression of both the CCK-AR and CCK-BR is increase 3–4 fold in human HepG2 HCC cells. B, Colony formation was significantly increased by treatment of human HepG2 HCC cells with gastrin (0.4 nM) compared to PBS controls and the gastrin-accelerated growth was blocked by proglumide. C, D, Representative images from normal human liver from the Biomax tissue microarray reacted with the CCK-BR antibody does not show immunoreactivity. E, F, Representative images of human HCC stained with the anti-CCK-BR antibody from the human tissue microarray shows increased immunoreactivity.
Representative images from the human liver tissue microarray are shown. Normal liver tissues reacted with the CCK-BR antibody lack staining for the CCK-BR (Figs. 4C, D). In contrast, representative images from human HCC demonstrated specific immunoreactivity to the CCK-BR (Figs 4 E, F). Immunoreactivity to CCK-BR in various human HCC samples intensified as the liver tissue progressed to higher stage and grade (Fig. S2)
miRNA Gene Expression Array
Using a 380 miRNA qPCR-based array (Qiagen) we conducted differential miRNA expression analysis of pancreas tissue from KRAS mice treated for 4 months with proglumide and pancreas tissue from KRAS mice receiving no treatment. Histology confirmed that proglumide reversed inflammation and fibrosis in this model (37). Differential expression of multiple miRNAs between the proglumide treated and untreated vehicle control mice were identified (Heat map, Fig. S3).
Discussion
The novel discovery in this investigation is that CCK receptors play an important role and become over-expressed in NASH and in hepatocellular carcinoma. We showed that the CCK-BR becomes upregulated in livers of mice on a NASH-inducing diet and treatment with a CCK-receptor antagonist, proglumide, can reverse the histologic features of the liver. CCK-receptor blockade decreased hepatic fibrosis, inflammatory cytokines and chemokines, hepatic macrophages, and increased glutathione all rendering the liver extracellular matrix less carcinogenic. Proglumide therapy remarkably, prevented the development of hepatic dysplastic nodules and HCC in spite of maintaining the CDE-saturated fat diet. Another finding from our investigation was that proglumide therapy decreased growth rates of murine HCC tumors suggesting that this receptor could potentially be a new target for therapeutics. Our translational studies confirmed that compared to normal human liver tissues, the CCK-Rs are also up-regulated and proglumide therapy decreased gastrin-stimulated growth in vitro.
CCK receptors have been reported in a number of gastrointestinal cancers, medullary thyroid cancer and small cell lung cancer (38); however, there are very few reports that have studied CCK receptors in normal or malignant liver. Early publications concerning CCK receptor mRNA expression in liver or liver cancer were inconsistent, but most investigations did not detect CCK receptor RNA in the liver particularly in animal models (39). Weinberg studied the mRNA expression of CCK-ARs and CCK-BRs in normal and malignant human tissues with end-point RT-PCR (40) and reported that normal human livers had detectable CCK-BR mRNA but not CCK-A receptors. His investigation did not include human liver cancers. In a study of 59 histologically confirmed human HCC tissue samples, Reubi et al (41) found no evidence of detectable CCK-ARs or CCK-BRs using receptor autoradiography. More recently, Roy and colleagues (42) reported immunoreactivity in 42 of 43 HCC human tissues using a custom humanized mouse monoclonal anti-human CCK-BR antibody. Unfortunately, in this same study (42), normal tissues from the esophagus, liver, lung, pancreas, spleen, stomach and thyroid also stained positive with this antibody and no controls were presented. Although the cell of origin for HCC is uncertain, many theories believe this cancer may arise from liver progenitor cells or stem cells (43). We detected very high CCK receptor expression by qRT-PCR or flow cytometry in HCC cells and two of the cell lines were studied, HepG2 and RIL-175, were derived from embryonic stem cells. Similarly, CCK receptor expression has been reported within the neck and isthmus region of the gastric gland, which is thought to host the putative stem cell compartment of the stomach (44). These data would suggest that perhaps a CCK-BR positive hepatic stem cell gives rise to HCC.
In this study, we showed that CCK-BR expression is very low in the normal whole mouse liver but is increased in mice fed the saturated fat CDE-diet. In contrast, CCK-AR expression is down-regulated by consumption of a high saturated fat diet. We previously demonstrated that in cultured murine HCC epithelial cells that CCK-BR expression was inversely and epigenetically regulated by miR148a (21). CCK-BR expression has been shown to be regulated by miR148. The 3’-UTR of CCK-BR has two sites in conserved and poorly conserved regions that can bind with the seed region of miR-148a and miR-148b (45), miR148a and miR148b are dysregulated in obese individuals (46). Dietary fat has been shown to induce hypermethylation of miR-148a (47), and decreased expression of miR-148a has been shown to increase CCK-BR expression (21). Based upon our results, we speculate an initiating factor in the NASH to HCC pathway is that consumption of a diet high in saturated fat leads to miR148a hypermethylation and down regulation of miR148, thus inversely activating the expression of CCK-BRs in liver epithelial cells, stellate cells, and perhaps immune cells in the hepatic ECM.
Another important finding in our investigation was that both CCK and gastrin stimulated growth of HCC in vitro. The proliferative effect was greatest at a concentration (0.4– 1.0 nM) consistent with the physiologic binding affinity (Kd) for the CCK-BR and these ligands. Although CCK binds to both CCK-ARs and CCK-BRs, using gene editing and selective CCK-AR and CCK-BR antagonists, this research revealed that CCK-stimulated growth is mediated by the CCK-BR and not the CCK-AR. Within the animal or human GI tract, the CCK peptide is produced and released from the I-cells of the duodenum in response to dietary fat and protein, and saturated fatty acids with a chain length >C12 are effective releasers of this GI peptide (48). CCK blood levels are elevated in human subjects (19) and in mice consuming a high saturated fat diet (20). Together with the increased CCK-BR expression in livers of mice on a high-fat diet, elevated CCK blood levels can stimulate the CCK receptor and downstream signaling to promote cellular proliferation and perhaps increased risk for HCC. In addition to blocking the proliferative effects of CCK and gastrin in HCC cells in vitro, we found that CCK-BR antagonism could decrease growth of HCC tumor growth in two mouse models. New agents that can add to the landscape of therapeutic options for HCC are desperately needed.
One of the hallmarks of NASH that distinguishes this condition from simple hepatic steatosis is hepatocyte injury and death manifested histologically by balloon degeneration. Injured hepatocytes release factors that promote the recruitment and accumulation of immune cells that produce hepatotoxic substances and provoke further injury and inflammation. Liver inflammation is regulated by cytokines and chemokines, which regulate the migration and activities of cells in the hepatic ECM including hepatocytes, macrophages, stellate cells, and circulating immune cells (49). Chemokines mediate their effects through interface and downstream signaling through G-protein coupled receptors (GPCR). In our study, proglumide significantly reduced inflammation, inflammatory cytokines, and chemokines. The mechanism by which blockade of the CCK-BR decreases cytokines and chemokines is unknown; however, CCK receptors and chemokine receptors are both G-protein coupled receptors (GPCRs). GPCRs are known to “cross-talk” or influence the action of the other GPCRs, either by sensitizing or desensitizing the intracellular signaling or downstream pathways or by regulating the actions by forming heterodimers (50). We postulate that one of the mechanisms through which proglumide may have decreased inflammation is by blocking the cross-talk interaction between the CCK-BRs and chemokine receptors. We also demonstrated that cells from the hepatic ECM that are known producers of cytokines and chemokines, in particular macrophages and stellate cells, also express CCK-BR; therefore, proglumide could have a direct effect on these inflammatory cell mediators by direct antagonism at the CCK-BR.
Fibrosis is a complex area of research and in the liver; it is thought to be the greatest risk factor for the development of HCC. Hepatic fibrosis is derived from activated hepatic stellate cells (HSC) and from the recruitment of tissue myofibroblasts. CCK receptors have also been identified on tissue fibroblasts (51) and pancreatic stellate (52) cells, and when these receptors are stimulated, the cells become activated to produce desmoplastic stroma characteristic of the microenvironment. Most likely, the mechanism for the reduction in fibrosis in these livers was due to CCK-BR antagonism on the stellate cells or diminished chemokines in the hepatic cellular matrix.
Tissues from mice treated with proglumide compared to controls exhibited significant epigenetic changes if miRNAs involved with hepatic inflammation and fibrosis. These data also suggest that miRNAs might be involved in regulation of the CCK-BR downstream signaling pathway. Of the many miRNAs affected by proglumide, we draw your attention to a number of key miRNAs. Those miRNAs upregulated by proglumide in the pancreas have been shown to inhibit fibrosis, inflammation or both, including miR-185-5p, miR-346-5p, miR-378a-3p, miR708-5p, and miR-302d-3p. And, a significant portion of miRNAs that were down-regulated by proglumide in the pancreas have been shown to promote fibrosis, inflammation or both (e.g.miR-21, miR-103-3p, miR-142-5p, and miR-126, miR-135b-5p). Several of the miRNAs down regulated by proglumide have been reported for their oncogenic potential (e.g. miR-27a-3p, miR-21a-5p, miR-19b-3p, and miR-30c, miR-338-3p, miR-9-5p, miR-32-5p).
NASH is a very common condition that increases the risk for cirrhosis and HCC (53). If we can understand the mechanisms involved in the precancerous stage, strategies to intervene can be developed. Many have approached NASH by targeting one cell type such as macrophages (54) or chemokine receptors (55). Proglumide is an older drug that was originally developed for peptic ulcer disease. Since the development of proton pump inhibitors, the commercialization of proglumide was halted, but it was found to have an excellent safety profile when tested in human subjects. Targeting the CCK-BR with an orally bioavailable agent that can alter the hepatic extracellular matrix making it less carcinogenic could represent an alternative novel therapy for HCC prevention. Furthermore, the anti-proliferative effects of proglumide on growth of cancer cells and tumors found in this study may lead to increased opportunities for new therapies in treating subjects with HCC.
Supplementary Material
Significance:
This investigation demonstrates the proliferative role of CCK in HCC. CCK-BR blockade reverses the premalignant state of the hepatic extracellular matrix (ECM), rendering it less susceptible to the development of HCC. CCK-BR blockade may provide a novel approach for the prevention or treatment of HCC.
Acknowledgments:
We thank the staff from the Lombardi Comprehensive Cancer Center Histology Core facility and flow cytometry Core for their assistance in experimental procedures and preparation of tissues. We also thank the staff in the Division of Comparative Medicine for their care and oversight of the animal experiments.
Funding: This work was supported by a 2019 AACR-Bayer Innovation and Discovery Grant, Grant Number 19-80-44-SMIT to JPS, and also supported by the Ruesch Center for the Cure of Gastrointestinal Cancers, Georgetown University Medical Center to JPS and AK. AK acknowledges funding support from the National Institute of Allergy and Infectious Diseases (R01AI132389; R21AI130800). Post-doctoral support was provided by a NIH training grant to Martha Gay TL1TR001431. These studies were conducted in part at the Lombardi Comprehensive Cancer Center Shared resource facilities NIH CA051008.
Footnotes
Conflict of interest: Georgetown University has intellectual property for the use of proglumide in liver disease.
References
- 1.Mittal S, El-Serag HB, Sada YH, Kanwal F, Duan Z, Temple S, et al. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans is Associated With Nonalcoholic Fatty Liver Disease. Clin Gastroenterol Hepatol 2016;14:124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McGlynn KA, Petrick JL, London WT. Global epidemiology of hepatocellular carcinoma: an emphasis on demographic and regional variability. Clin Liver Dis 2015;19:223–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruix J, Reig M, Sherman M. Evidence-Based Diagnosis, Staging, and Treatment of Patients With Hepatocellular Carcinoma. Gastroenterology 2016;150:835–53. [DOI] [PubMed] [Google Scholar]
- 4.Kudo M. Immuno-Oncology in Hepatocellular Carcinoma: 2017 Update. Oncology 2017;93 Suppl 1:147–59. [DOI] [PubMed] [Google Scholar]
- 5.Boland P, Wu J. Systemic therapy for hepatocellular carcinoma: beyond sorafenib. Chin Clin Oncol 2018;7:50. [DOI] [PubMed] [Google Scholar]
- 6.Dyson J, Jaques B, Chattopadyhay D, Lochan R, Graham J, Das D, et al. Hepatocellular cancer: the impact of obesity, type 2 diabetes and a multidisciplinary team. J Hepatol 2014;60:110–7. [DOI] [PubMed] [Google Scholar]
- 7.Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet 2018;391:1301–14. [DOI] [PubMed] [Google Scholar]
- 8.Baglieri J, Brenner DA, Kisseleva T. The Role of Fibrosis and Liver-Associated Fibroblasts in the Pathogenesis of Hepatocellular Carcinoma. Int J Mol Sci 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 2005;115:209–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim BM, Abdelfattah AM, Vasan R, Fuchs BC, Choi MY. Hepatic stellate cells secrete Ccl5 to induce hepatocyte steatosis. Sci Rep 2018;8:7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trautwein C, Friedman SL, Schuppan D, Pinzani M. Hepatic fibrosis: Concept to treatment. J Hepatol 2015;62:S15–S24. [DOI] [PubMed] [Google Scholar]
- 12.Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology 2015;61:1066–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arthur MJ. Reversibility of liver fibrosis and cirrhosis following treatment for hepatitis C. Gastroenterology 2002;122:1525–8. [DOI] [PubMed] [Google Scholar]
- 14.Bedossa P. Reversibility of hepatitis B virus cirrhosis after therapy: who and why? Liver Int 2015;35 Suppl 1:78–81. [DOI] [PubMed] [Google Scholar]
- 15.Sun M, Kisseleva T. Reversibility of liver fibrosis. Clin Res Hepatol Gastroenterol 2015;39 Suppl 1:S60–S63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bansal MB. Hepatic stellate cells: fibrogenic, regenerative or both? Heterogeneity and context are key. Hepatol Int 2016;10:902–8. [DOI] [PubMed] [Google Scholar]
- 17.Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 2010;52:1836–46. [DOI] [PubMed] [Google Scholar]
- 18.Caligiuri A, Gentilini A, Marra F. Molecular Pathogenesis of NASH. Int J Mol Sci 2016;17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gibbons C, Finlayson G, Caudwell P, Webb DL, Hellstrom PM, Naslund E, et al. Postprandial profiles of CCK after high fat and high carbohydrate meals and the relationship to satiety in humans. Peptides 2016;77:3–8. [DOI] [PubMed] [Google Scholar]
- 20.Nadella S, Burks J, Al-Sabban A, Inyang G, Wang J, Tucker RD, et al. Dietary fat stimulates pancreatic cancer growth and promotes fibrosis of the tumor microenvironment through the cholecystokinin receptor. Am J Physiol Gastrointest Liver Physiol 2018;315:G699–G712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tucker RD, Ciofoaia V, Nadella S, Gay MD, Cao H, Huber M, et al. A Cholecystokinin Receptor Antagonist Halts Nonalcoholic Steatohepatitis and Prevents Hepatocellular Carcinoma. Dig Dis Sci 2020;65:189–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lacoste B, Raymond VA, Cassim S, Lapierre P, Bilodeau M. Highly tumorigenic hepatocellular carcinoma cell line with cancer stem cell-like properties. PLoS One 2017;12:e0171215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zender L, Xue W, Cordon-Cardo C, Hannon GJ, Lucito R, Powers S, et al. Generation and analysis of genetically defined liver carcinomas derived from bipotential liver progenitors.Cold Spring Harb Symp Quant Biol 2005;70:251–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lopez-Terrada D, Cheung SW, Finegold MJ, Knowles BB. Hep G2 is a hepatoblastoma-derived cell line. Hum Pathol 2009;40:1512–5. [DOI] [PubMed] [Google Scholar]
- 25.Gardner JD, Walker MD, Martinez J, Priestly GP, Natarajan S, Bodanszky M. The importance of the amino acid in position 27 of cholecystokinin in determining its biological activity on pancreatic acini. Biochim Biophys Acta 1980;630:323–9. [DOI] [PubMed] [Google Scholar]
- 26.Jensen SL, Holst JJ, Nielsen OV, Rehfeld JF. Effect of sulfation of CCK-8 on its stimulation of the endocrine and exocrine secretion from the isolated perfused porcine pancreas. Digestion 1981;22:305–9. [DOI] [PubMed] [Google Scholar]
- 27.Braunersreuther V, Viviani GL, Mach F, Montecucco F. Role of cytokines and chemokines in non-alcoholic fatty liver disease. World J Gastroenterol 2012;18:727–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nandi B, Pai C, Huang Q, Prabhala RH, Munshi NC, Gold JS. CCR6, the sole receptor for the chemokine CCL20, promotes spontaneous intestinal tumorigenesis. PLoS One 2014;9:e97566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chu X, Jin Q, Chen H, Wood GC, Petrick A, Strodel W, et al. CCL20 is up-regulated in non-alcoholic fatty liver disease fibrosis and is produced by hepatic stellate cells in response to fatty acid loading. J Transl Med 2018;16:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhuang H, Cao G, Kou C, Liu T. CCL2/CCR2 axis induces hepatocellular carcinoma invasion and epithelial-mesenchymal transition in vitro through activation of the Hedgehog pathway. Oncol Rep 2018;39:21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Andrade KQ, Moura FA, dos Santos JM, de Araujo OR, de Farias Santos JC, Goulart MO. Oxidative Stress and Inflammation in Hepatic Diseases: Therapeutic Possibilities of N-Acetylcysteine. Int J Mol Sci 2015;16:30269–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang C, Zhang Y, Guo K, Wang N, Jin H, Liu Y, et al. Heat shock proteins in hepatocellular carcinoma: Molecular mechanism and therapeutic potential. Int J Cancer 2016;138:1824–34. [DOI] [PubMed] [Google Scholar]
- 33.Feng JT, Liu YK, Song HY, Dai Z, Qin LX, Almofti MR, et al. Heat-shock protein 27: a potential biomarker for hepatocellular carcinoma identified by serum proteome analysis. Proteomics 2005;5:4581–8. [DOI] [PubMed] [Google Scholar]
- 34.Qu B, Jia Y, Liu Y, Wang H, Ren G, Wang H. The detection and role of heat shock protein 70 in various nondisease conditions and disease conditions: a literature review. Cell Stress Chaperones 2015;20:885–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kazankov K, Jorgensen SMD, Thomsen KL, Moller HJ, Vilstrup H, George J, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol 2019;16:145–59. [DOI] [PubMed] [Google Scholar]
- 36.Smith JP, Wang S, Nadella S, Jablonski SA, Weiner LM. Cholecystokinin receptor antagonist alters pancreatic cancer microenvironment and increases efficacy of immune checkpoint antibody therapy in mice. Cancer Immunol Immunother 2018;67:195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith JP, Cooper TK, McGovern CO, Gilius EL, Zhong Q, Liao J, et al. Cholecystokinin receptor antagonist halts progression of pancreatic cancer precursor lesions and fibrosis in mice. Pancreas 2014;43:1050–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith JP, Fonkoua LK, Moody TW. The Role of Gastrin and CCK Receptors in Pancreatic Cancer and other Malignancies. Int J Biol Sci 2016;12:283–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lay JM, Jenkins C, Friis-Hansen L, Samuelson LC. Structure and developmental expression of the mouse CCK-B receptor gene. Biochem Biophys Res Commun 2000;272:837–42. [DOI] [PubMed] [Google Scholar]
- 40.Weinberg DS, Ruggeri B, Barber MT, Biswas S, Miknyocki S, Waldman SA. Cholecystokinin A and B receptors are differentially expressed in normal pancreas and pancreatic adenocarcinoma. J Clin Invest 1997;100:597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reubi JC, Zimmermann A, Jonas S, Waser B, Neuhaus P, Laderach U, et al. Regulatory peptide receptors in human hepatocellular carcinomas. Gut 1999;45:766–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roy J, Putt KS, Coppola D, Leon ME, Khalil FK, Centeno BA, et al. Assessment of cholecystokinin 2 receptor (CCK2R) in neoplastic tissue. Oncotarget 2016;7:14605–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sia D, Villanueva A, Friedman SL, Llovet JM. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017;152:745–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmitz F, Goke MN, Otte JM, Schrader H, Reimann B, Kruse ML, et al. Cellular expression of CCK-A and CCK-B/gastrin receptors in human gastric mucosa. Regul Pept 2001;102:101–10. [DOI] [PubMed] [Google Scholar]
- 45.Song Y, Xu Y, Wang Z, Chen Y, Yue Z, Gao P, et al. MicroRNA-148b suppresses cell growth by targeting cholecystokinin-2 receptor in colorectal cancer. Int J Cancer 2012;131:1042–51. [DOI] [PubMed] [Google Scholar]
- 46.Shi C, Zhang M, Tong M, Yang L, Pang L, Chen L, et al. miR-148a is Associated with Obesity and Modulates Adipocyte Differentiation of Mesenchymal Stem Cells through Wnt Signaling. Sci Rep 2015;5:9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He XX, Kuang SZ, Liao JZ, Xu CR, Chang Y, Wu YL, et al. The regulation of microRNA expression by DNA methylation in hepatocellular carcinoma. Mol Biosyst 2015;11:532–9. [DOI] [PubMed] [Google Scholar]
- 48.McLaughlin J, Grazia LM, Jones MN, D’Amato M, Dockray GJ, Thompson DG. Fatty acid chain length determines cholecystokinin secretion and effect on human gastric motility. Gastroenterology 1999;116:46–53. [DOI] [PubMed] [Google Scholar]
- 49.Marra F, Tacke F. Roles for chemokines in liver disease. Gastroenterology 2014;147:577–94. [DOI] [PubMed] [Google Scholar]
- 50.Werry TD, Wilkinson GF, Willars GB. Mechanisms of cross-talk between G-protein-coupled receptors resulting in enhanced release of intracellular Ca2+. Biochem J 2003;374:281–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Singh P, Owlia A, Espeijo R, Dai B. Novel gastrin receptors mediate mitogenic effects of gastrin and processing intermediates of gastrin on Swiss 3T3 fibroblasts. Absence of detectable cholecystokinin (CCK)-A and CCK-B receptors. J Biol Chem 1995;270:8429–38. [DOI] [PubMed] [Google Scholar]
- 52.Berna MJ, Seiz O, Nast JF, Benten D, Blaker M, Koch J, et al. CCK1 and CCK2 receptors are expressed on pancreatic stellate cells and induce collagen production. J Biol Chem 2010;285:38905–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Diehl AM, Day C. Nonalcoholic Steatohepatitis. N Engl J Med 2018;378:781. [DOI] [PubMed] [Google Scholar]
- 54.Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol 2017;66:1300–12. [DOI] [PubMed] [Google Scholar]
- 55.Sumida Y, Yoneda M. Current and future pharmacological therapies for NAFLD/NASH. J Gastroenterol 2018;53:362–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.