

Abstract
Background
Severe acute respiratory syndrome-2 (SARS-CoV-2) exhibits a broad spectrum of clinical manifestations. Despite the fact that SARS-CoV-2 has slower evolutionary rate than other coronaviruses, different mutational hotspots have been identified along the SARS-CoV-2 genome.
Methods
We performed whole-genome high throughput sequencing on isolates from 50 Egyptian patients to see if the variation in clinical symptoms was related to mutations in the SARS-CoV-2 genome. Then, we investigated the relationship between the observed mutations and the clinical characteristics of the patients.
Results
Among the 36 most common mutations, we found two frameshift deletions linked to an increased risk of shortness of breath, a V6 deletion in the spike glycoprotein's signal peptide region linked to an increased risk of fever, longer fever duration and nasal congestion, and L3606-nsp6 deletion linked to a higher prevalence of cough and conjunctival congestion. S5398L nsp13-helicase was linked to an increased risk of fever duration and progression. The most common mutations (241, 3037, 14,408, and 23,403) were not linked to clinical variability. However, the E3909G-nsp7 variant was more common in children (2–13 years old) and was associated with a shorter duration of symptoms. The duration of fever was significantly reduced with E1363D-nsp3 and E3073A-nsp4.
Conclusions
The most common mutations, D614G/spike-glycoprotein and P4715L/RNA-dependent-RNA-polymerase, were linked to transmissibility regardless of symptom variability. E3909G-nsp7 could explain why children recover so quickly. Nsp6-L3606fs, spike-glycoprotein-V6fs, and nsp13-S5398L variants may be linked to clinical symptom worsening. These variations related to host-virus interactions might open new therapeutic avenues for symptom relief and disease containment.
Keywords: SARS-CoV-2; Mutation; Clinical symptoms; Spike glycoprotein signal peptide; nsp6, nsp13-helicase; nsp7
Graphical abstract
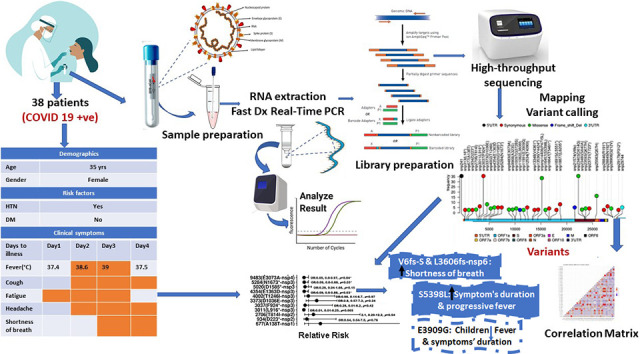
1. Background
The Corona virus disease 2019 (COVID 19) outbreak, initially emerged in late December 2019 in Wuhan, China, has subsequently spread to other countries around the world, posing a global public health threat [1]. Whole genome sequencing of the highly contagious virus revealed that the causative pathogen is closely related to a group of SARS-like coronaviruses (CoV) with 89.1% similarity, and it was eventually termed SARS-CoV-2 [2,3].
SARS-CoV-2 is a large, enveloped virus with a 29.9 kb single-stranded positive sense RNA genome. Two-thirds of the viral genome at the 5’terminus, according to the most recent annotation (MN908947.3), contains an open reading frame (ORF), ORF1a, which encodes 16 non-structural proteins (nsps). These nsps play a role in virus replication and, possibly, evasion of host immune surveillance. Alternatively, one-third of the 3'end contains genes that encode conserved structural proteins like spike glycoprotein (S), envelope protein (E), membrane protein (M), nucleocapsid protein (N), and several accessory proteins (3a, 6, 7a, 7b, 8 and 10) [4,5].
Coronavirus replication occurs in the host cell's cytoplasm, conveyed by the assembly of replication transcription complexes (RTCs) to the host cytoplasmic membranes. The hydrophobic transmembrane domains of nsp3, nsp4, and nsp6 mediate the rearrangement of RTC-anchored double membrane vesicles (DMVs) [6,7]. Interaction of nsp13-helicase with viral holoenzyme (RNA-dependent-RNA-polymerase (RdRp) in association with nsp7 and nsp8) is crucial for virus replication.
The sign and symptoms of SARS-CoV-2 infection can be categorized into four different classes, systemic, respiratory, gastrointestinal, and cardiovascular. COVID 19 presents a broad range of clinical features across a spectrum of disease severity ranging from asymptomatic/mild symptoms to severe conditions that can progress to acute respiratory distress and multiorgan dysfunction syndromes (MODs). The majority of infected people have mild to moderate symptoms within the first week of infection [8]. The most common mild symptoms of COVID 19 have been identified as fever, fatigue, and a dry cough. Other infected patients, on the other hand, may develop serious complications with varying degrees of severity, such as shortness of breath, MODs, severe pneumonia, and, eventually, death [8,9].
A combination of symptoms, particularly fever, myalgia or arthralgia, fatigue, and headache, indicates the presence of COVID 19 [10]. However, because the symptoms of COVID 19 mild cases overlap with those of influenza, SARS-CoV-2 may be misdiagnosed as an influenza-like illness [11]. Accordingly, COVID 19 diagnosis cannot be based on a specific symptomatic detection pattern. The majority of signs and symptoms had a high specificity but a low sensitivity for COVID 19 diagnosis. The most sensitive and specific diagnostic methods are currently computed tomography and positive detection of viral genome reverse transcription-polymerase chain reaction (RT-PCR) [12,13].
The variation in COVID 19 clinical manifestations across countries and within Egypt could be due to virus mutations or angiotensin-converting-enzyme-2 polymorphisms. In this study, we used a high throughput sequencing technique to look for mutation hotspots in Egyptian patients and identify dominant variants that could be linked to differences in clinical symptoms.
2. Materials and methods
2.1. Sample collection and virus genome extraction
The study was endorsed by the Ministry of Health and Populations' Ethics Committee. Training and Research sector with office of human research protections (OHRP): FWA00016183 23 March 2020, IORG0005704/ IRB0000687 8 November 2020, and each participant provided informed consent. Nasopharyngeal swabs were collected from patients at the National Cancer Institute and Army hospitals between March and April 2020 for molecular analysis (RT-PCR). Samples were curated for duplicates and acquired family infection to improve assessment of virus variability.
Fifty nasopharyngeal swabs from SARS-CoV-2 positive patients were chosen for virus sequencing using an ion-torrent sequencing method. Samples with a high viral load and a Ct value of <25 met the inclusion criteria. The samples were kept at −80 °C until they were analysed.
Swabs were immersed in viral transfer media (600 L). The QIAamp®DSP Virus Kit was used to extract total viral RNA (Qiagen, Hilden, Germany). The quality of the isolated RNA was evaluated on an Agilent 2100 Bioanalyzer using the Agilent RNA 6000 Nano Kit. TaqPathTM 1-Step RT-qPCR Master Mix (ThermoFisher Scientific, Applied BiosystemsTM, USA) was used to quantitatively amplify the extracted SARS-CoV-2 RNA on an Applied BiosystemsTM 7500 Fast Dx Real-Time PCR Instrument (Thermo Fisher Scientific) according to the manufacturer's instructions.
2.2. Library preparation
Following viral RNA extraction, the DNA library was prepared for complete sequencing using the Ion AmpliSeq SARS-CoV-2 Research Panel (ThermoFisher Scientific, USA) according to the manufacturer's instructions. This panel describes a targeted amplicon sequencing approach that includes 237 amplicons specific to SARS-CoV-2 with amplicon sizes ranging from 125 to 275 bp. Preparing the library included the following steps: 1) RNA reverse transcription using the ThermoFisher Scientific SuperSrcriptTM VILO cDNA Synthesis Kit (ThermoFisher Scientific, USA), 2) target amplification for 15–20 cycles, 3) partial amplicon digestion, 4) adapter ligation 5) library purification using Agencourt™ AMPure™ XP reagent at room temperature, and 6) quantification of the amplified library by Qubit™ Fluorometer kit. Barcoded libraries were loaded to the Ion Chef™ Instrument (Thermo Fisher Scientific) for emulsion PCR, enrichment, and loading onto the Ion Chip followed by high-throughput sequencing reactions run on Ion GeneStudio S5, ion torrent sequencer (ThermoFisher Scientific).
2.3. Bioinformatic analysis
Consensus sequences were generated using a double-assembly method that included denovo assembly and multiple rounds of mapping to the reference genome. Prior to analysis, paired end (PE) raw reads were subjected to a quality control using FastQC to eliminate low quality error prone reads. Trimmomatic was used to improve the read quality by removing adapters, barcodes, and low quality read edges. PE reads were rapidly mapped to an indexed reference genome (MN908947.3) using Burrows Wheeler aligner and samtools program. After that, the aligned reads were sorted by position on the reference genome, and PCR duplicates were removed. The flagstat samtools count the total number of reads, the percentage of reads mapped to the reference genome, and the percentage of reads-pair mapped. The number of reads covering each position of the reference genome was determined using QualiMap, which indicates the number of reads covering each position of the reference genome to ensure that each data feature is real and not a result of sequencing error. To call variants, samtools mpileup and bcftools were used to generate a variant call file (VCF) and variant sites, respectively.
2.4. Statistical analysis
All statistical tests were done using R studio software (version 3.6). Normality of data was detected by Shapiro-Wilk test. Lollipop plot represented the position of mutation hotspots along the virus genome. Association of mutation hot spot with patients' demographic and risk factors was done using Fischer exact-test and displayed as Mosaic bar plot. Log-binomial regression was used to assess the risk ratio of each variant for prediction of clinical symptoms. The pwr package of R studio was used to calculate the power of the test for the given sample size (n = 50) taking in consideration the level of significance (0.05) and the effect size (0.5–0.8). Independent t-test was used to compare the mean difference of age, duration of symptoms and fever duration between reference and altered alleles. Correlation matrix represented the Pearson correlations between mutational hotspots frequencies as well as between risks factors and clinical symptoms. Statistical significance was two-tailed at p-value <0.05. All p-values were adjusted for multiple comparisons according to Benjamini and Hochberg [14].
3. Results
3.1. Patients' demographic, risk factors and clinical symptoms
The clinical characteristics of the patients and their COVID 19 symptoms are summarized in Table 1, Table 2 . The mean age of patients was 34.76 ± 18.04 yrs. (range, 2–84 yrs.). The study examined the association of age with different mutation sites in three age groups (2–13, 20–45, and > 45 years). There were 29 males and 21 females in our sample. Exposure to infected individuals (21/50, 42.0%) and hypertension (17/50, 34.0%) were the most frequently occurring risk factors. The mean duration of symptoms was 19.03 ± 19.0 days. The most frequently encountered clinical symptom (60.0%) was fever, followed by severe headache (46.0%). The mean duration of fever was 2.38 ± 2.71 days. 60.0% of patients who developed fever experienced a regressive fever, 23.3% experienced a stationary fever, and 16.7% experienced a progressive fever. Only 32.0% of cases progressed to the point of experiencing shortness of breath.
Table 1.
Patients' demographic and risk factors.
| Characteristic | N (%) Total N = 50 |
|---|---|
| Demographic | |
| Age (mean ± SD) | 34.76 ± 18.04 |
| Age(yrs.) | |
| 2–13 | 9(18.0) |
| 20–45 | 30(60.0) |
| >45 | 11(22.0) |
| Gender | |
| Female | 21(42.0) |
| Male | 29(58.0) |
| Risk factors | |
| Asthma | |
| No | 41(82.0) |
| Yes | 9(18.0) |
| History of COPD | |
| No | 44(88.0) |
| Yes | 6(12.0) |
| Diabetes mellitus | |
| No | 43(86.0) |
| Yes | 7(14.0) |
| Chronic liver disease | |
| No | 44(88.0) |
| Yes | 6(12.0) |
| Rheumatic heart disease | |
| No | 46(92.0) |
| Yes | 4(8.0) |
| Immunodeficiency | |
| No | 46(92.0) |
| Yes | 4(4.0) |
| Hypertension | |
| No | 33(66.0) |
| Yes | 17(34.0) |
| Smoking | |
| No | 40(80.0) |
| Yes | 10(20.0) |
| Exposed to cases | |
| No | 29(58.0) |
| Yes | 21(42.0) |
COPD; chronic obstructive pulmonary disease.
Table 2.
Patients' clinical symptoms.
| COVID 19 symptoms | N (%) |
|---|---|
| Symptoms duration (days) (mean ± SD) | 19.3 ± 19.0 |
| Fever | |
| No | 20(40.0) |
| Yes | 30(60.0) |
| Fever progression | |
| Regressive | 18(60.0) |
| Stationary | 7(23.3) |
| Progressive | 5(16.7) |
| Fever duration (mean ± SD) | 2.38 ± 2.71 |
| Myalgia/arthralgia | |
| No | 41(82.0) |
| Yes | 9(18.0) |
| Conjunctival congestion | |
| No | 41(82.0) |
| Yes | 9(18.0) |
| Nasal congestion | |
| No | 35(70.0) |
| Yes | 15(30.0) |
| Headache | |
| No | 27(54.0) |
| Yes | 13(46.0) |
| Cough | |
| No | 40(80.0) |
| Yes | 10(20.0) |
| Sore throat | |
| No | 40(80.0) |
| Yes | 10(20.0) |
| Sputum production | |
| No | 39(78.0) |
| Yes | 11(22.0) |
| Fatigue | |
| No | 40(80.0) |
| Yes | 10(20.0) |
| Shortness of breath | |
| No | 34(68.0) |
| Yes | 16(32.0) |
COVID; corona virus disease.
3.2. Mutation sites of SARS-CoV-2
Sequence reads were mapped to the SARS-CoV-2 genome Refseq (MN908947.3), revealing a total of 126 mutations. We focused on single nucleotide substitutions detected in >5% of the genome for detailed analysis of their association with clinical symptoms.
As shown in Table 3 , 36 single nucleotide substitutions occurred in >5% of SARS-CoV-2 genomes. The frequency and position of these nucleotide substitutions in the affected protein product, as well as variants in each sample and their classification, are depicted in Fig. 1, Fig. 2 . This included 15 synonymous mutations, 17 missense mutations, two frameshift mutations, and two non-coding region substitutions in the 5′ and 3’UTRs. The maximum number of mutations accumulated at ORF1a encoding RdRp, nsp3 and nsp7, at the S gene encoding the spike protein, and at ORF3a encoding Coronavirus accessory protein 3a (APA-viroporin). The recurrent mutations in ORF1a encoding RdRp were 6 synonymous at 13,536 (Y4424*) and 44 missense at 14,408 (P4715L), whereas those encoding nsp3 were synonymous mutations in 3011 (L916*, n = 9), 3037 (F924*, n = 45), 5020 (D1585*, n = 8), and 5284 (N1673*, n = 8). In the S gene encoding the spike glycoprotein, we observed missense mutations at 23,403 (D614G, n = 47), 23,480 (S640A, n = 6), and 23,593 (Q677H, n = 6), 8 frameshift deletions at 21,574 (V6fs), and 7 synonymous mutations at 23,731 (T723*). Additionally, 21 missense mutations in ORF3a were identified, resulting in APA-viroporin Q57H variants. Furthermore, ORF1a has accumulated numerous other mutations at various positions, including 3 synonymous at 313 (L16*) and 3 missense at 677 (A138T) encoding for nsp1, 4 synonymous at 934 (D223*) and 5 missense at 2706 (T814I) encoding for nsp2, 6 missense at 9483 (E3073A) and 9968 (A323S) encoding for nsp4, 6 missense at 10,097 (G3728S) encoding for 3C-like proteinase, 5 frameshifts at 11,082 encoding nsp6 (L3606fs), 5 missenses at 11,991 encoding nsp7-replicase (E3909G), 9 missenses at 12,534 encoding nsp8-replicase (T4040I), 3 synonymous at 13,348 encoding nsp10 (V4361*), 3 missense at 16,457 (S5398L), 7 synonymous at 16,647 (T5461*) and 4 synonymous at 16,915 (L5551*) encoding for nsp13-helicase, and 13 synonymous at 18877 encoding for nsp-11 3’exonuclease (L6205*). Out of the 25 mutations accumulating in ORF9/N gene encoding the nucleocapsid protein, we observed synonymous mutations at 28,846 (R191*, n = 5), 28,849 (N192*, n = 9) and 291,719 (P312*, n = 4) while, 7 missense at 28,908 (G212V). Additionally, the non-coding region of the 5’UTR at position 241 harbored 46 mutations, while the 3’UTR at position 29,744 harbored 9 mutations.
Table 3.
Single nucleotide substitution in SARS-CoV2 genome from Egyptian Patients at frequency > 5%.
| Refseq position | Ref | Alt | Gene | Product | Amino acid substitution | Type of mutation | Frequency of substitution |
|---|---|---|---|---|---|---|---|
| 241 | C | T | 5’UTR | – | 46 (92.0%) | ||
| 313 | C | T | ORF1a | nsp1 | L > L | Synonymous | 3 (6.0%) |
| 677 | G | A | T > I | Missense | 3 (6.0%) | ||
| 934 | C | T | nsp2 | D > D | Synonymous | 4(8.0%) | |
| 2706 | C | T | T > I | Missense | 5 (10.0%) | ||
| 3011 | T | C | nsp3 | L > L | Synonymous | 9 (18.0%) | |
| 3037 | C | T | P > P | Synonymous | 45 (90.0%) | ||
| 3373 | C | A | D > E | Missense | 12 (24.0%) | ||
| 4002 | G | T | T > I | Missense | 7 (14.0%) | ||
| 4354 | G | T | E > D | Missense | 8 (16.0%) | ||
| 5020 | C | T | D > D | Synonymous | 8 (16.0%) | ||
| 5284 | C | T | N > N | Synonymous | 8 (16.0%) | ||
| 9483 | A | C | nsp4 | E > A | Missense | 6 (12.0%) | |
| 9968 | G | T | A > S | Missense | 6 (12.0%) | ||
| 10,097 | G | A | 3C-like proteinase | G > S | Missense | 6 (12.0%) | |
| 11,082 | G | – | nsp6 | L > fs | Frameshift | 5 (10.0%) | |
| 11,991 | A | G | nsp7 (replicase) | E > G | Missense | 5 (10.0%) | |
| 12,534 | C | T | nsp8 (replicase) | T > I | Missense | 9 (18.0%) | |
| 13,348 | G | T | nsp10 | V > V | Synonymous | 3 (6.0%) | |
| 13,536 | C | T | nsp12 (RdRp) | Y > Y | Synonymous | 6 (12.0%) | |
| 14,408 | C | T | P > L | Missense | 44 (88.0%) | ||
| 16,457 | C | T | nsp13 (helicase) | S > L | Missense | 3 (6.0%) | |
| 16,647 | G | T | T > T | Synonymous | 7 (14.0%) | ||
| 16,915 | C | T | L > L | Synonymous | 4 (8.0%) | ||
| 18,877 | C | T | nsp-11 3’exonuclease | L > L | Synonymous | 13 (26.0%) | |
| 21,574 | C | – | S | Spike glycoprotein | V > fs | Frameshift | 8 (16.0%) |
| 23,403 | A | G | D > G | Missense | 47 (94.0%) | ||
| 23,480 | T | G | S > A | Missense | 6 (12.0%) | ||
| 23,593 | G | T | Q > H | Missense | 6 (12.0%) | ||
| 23,731 | C | T | T > T | Synonymous | 7 (14.0%) | ||
| 25,563 | G | T | ORF3a | APA3-viroporin (accessory protein) | Q > H | Missense | 21 (42.0%) |
| 28,846 | C | T | ORF9/N | Coronavirus nucleocapsid | R > R | Synonymous | 5 (10.0%) |
| 28,849 | C | T | N > N | Synonymous | 9 (18.0%) | ||
| 28,908 | G | T | G > V | Missense | 7 (14.0%) | ||
| 29,179 | G | T | P > P | Synonymous | 4 (8.0%) | ||
| 29,744 | G | A | 3’UTR | – | – | – | 9(18.0%) |
ORF; open reading frame, UTR; untranslated region, nsp; non-structural protein, RdRp; RNA dependent RNA polymerase.
Fig. 1.

Mutation hotspots of SARS-CoV-2 genome. a) Lollipop plot showing mutations at different positions along SARS-CoV-2 genome according to Ensembl COVID-19 reference genome (MN908947.3). b) Bar plot of the percentage frequency amino acid change per amino acid unit length of SARS-CoV-2 proteins.
Fig. 2.

Summary of SARS-CoV-2 mutational variants. a) The number of variants in each sample as stacked barplot, summary of variant types and summary variant classification as boxplots. b) Oncoplots displaying the frequency of each mutation site in the samples under consideration.
3.3. Mutations sites in association to patients' characteristics
The correlation between patient demographics and the frequency of SARS-COV-2 mutations revealed that the mean age of patients with mutations at positions 11,991, 16,915, 28,849, and 27,944 was significantly lower than that of patients without mutations (p < 0.001, p = 0.038, and p = 0.023, respectively) by 76.08%, 75.68%, 47.05%, and 53.54%, respectively. On the other hand, the mean age of patients with mutations at 16,457 was 62.13% higher than that of patients without mutations (p = 0.034) (Supplementary Table 1). The age groupings were as follows: 2–13, 20–45, and > 45 yrs. As illustrated in Fig. 3 , the 2–13 yrs. group had a significantly higher mutation frequency at positions 5284 (62.5%), 11,991 (100.0%), and 16,915 (100.0%) when compared to the 20–45 yrs. group (25.0%, 0.0%, and 0.0%, respectively) and the >45 yrs. group (12.5%, 0.0%, and 0.0%, respectively) (p = 0.002, p < 0.001, and p < 0.001, respectively). However, mutations at 16,457 were significantly more prevalent in the over-45 age group (100.0%) than in the other two younger age groups (0.0%, p = 0.0035). There was no significant correlation between SARS-CoV-2 mutation sites and gender. Potential risk factors (asthma, family history of chronic obstructive pulmonary disease, diabetes mellitus, chronic liver disease, hypertension, smoking, and exposure to infected cases) were not significantly different between patients with and without SARS-CoV-2 variants.
Fig. 3.

Mosaic plot of patients' demographic and risk factors in association with frequent point mutations along SARS-CoV-2 genome. Rectangular tiles indicate residuals or deviation from a particular test model. Blue tiles mean positive residuals in which the observed frequency is greater than the expected (under independence). Red tiles mean negative residuals in which the observed frequency is less than the expected. The intensity of color reflects the magnitude of the residual. Residuals greater than 2 or less than −2 represent significant deviation from independence. *Significance at p < 0.05. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The mean duration of clinical symptoms was significantly shorter in patients with mutations at 677, 11991, 16,915, and 29,744 (p < 0.001, for all) than in those without mutations, by 90.20%, 94.37%, 95.21%, and 84.36%, respectively (Supplementary Table).
The mean duration of fever was significantly shorter in patients with mutations at 3011, 4354, 5284, 9483, and 29,744 (p < 0.001, for all) than in patients without mutations by 80.22%, 91.04%, 95.37%, 100.0%, and 92.28%, respectively. On the other hand, patients with mutations at 16457 and 21,574 had a significantly longer duration of fever (p = 0.013 and p = 0.017, respectively) by 359.2% and 100.98%, respectively, compared to those without mutations (Supplementary Table). Using Fischer exact test, the cooncoplots display the SARS-CoV-2 variants significantly associated with clinical symptoms ( Fig. 4 ). Development of fever in patients with mutations at 9483, 5284, 4345, 3011 and 29,744 (30%, 35%, 35%, 35% and 35%, respectively) was significantly lower than those without mutations (0%, 3%, 3%, 7% and 7%, respectively, p < 0.05). However, mutation at 21,574 was significantly associated with the development of fever, with 27% of cases with fever having the mutation, while 0% of those without fever had the mutation (p < 0.001). Progressive fever was significantly associated with missense mutation at 16,457, with 25% of cases with progressive fever having the mutation, compared to 0% of those with regressive and stable fever had the mutation (p < 0.001). Forty percent of patients who developed cough had the frameshift deletion at 11,082, while only 2% of those who did not develop cough had the mutation (p < 0.001). Conjunctival congestion was significantly associated with the frameshift deletion at 11,082, with 56% of individuals who had conjunctival congestion harbored the mutation, while 0% of those who did not develop conjunctival congestion had the mutation (p < 0.001). Forty one percent of patients who experienced nasal congestion had the frameshift deletion at 21,574, while only 0% of those who did not experience nasal congestion had the mutation (p < 0.001). Myalgia/arthralgia was significantly linked to synonymous mutation at 934, with 44% of patients who experience myalgia/arthralgia had the mutation, while 0% of those without myalgia/arthralgia had the mutation (p < 0.001). Forty percent of patients who experienced fatigue had mutation at 28,846, compared to 2% of those who did not experience fatigue had the mutation. Missense mutation at 28,908 and synonymous mutation at 28,846 were significantly associated with developing sore throat symptoms, with 40% and 50% of cases experiencing sore throat having mutation at 28,908 and 28,846, respectively, compared to 8% and 0% of those without experiencing sore throat having the respective mutations. Shortness of breath was significantly linked to mutations at 21,574, 11,082 and 3373, with 44%, 25% and 44% of cases who developed shortness of breath had mutations at 21,574, 11,082 and 3373, respectively, compared to 3%, 3% and 15% of those who did not develop shortness of breath had the respective mutations (p < 0.001, <0.001 and < 0.05, respectively). Nevertheless, 26%, 24% and 24% of patients not evolving shortness of breath had mutations at 29,744, 5384 and 4354, respectively, while 0%, 0% and 0% of those not evolving shortness of breath had the respective mutations (p < 0.05).
Fig. 4.

Co-oncoplots displaying the mutation site significantly associated with clinical symptoms, a) fever, b) fever progression, c) cough, d) conjunctival congestion, e) nasal congestion, f) fatigue, h) myalgia/arthralgia, i) sore throat and j) shortness of breath, based on Fischer exact test (all p < 0.05).
After adjustment p-values for multiple testing, the log regression analysis revealed the SARS-CoV2 mutational hotspots linked to the relative risk of developing clinical symptoms as shown in Fig. 5 . Patients with the 21,574 (V6fs) mutation had a 90% increased risk of developing fever [RR: 1.90, 95% CI: 1.43–2.55, p.adjust < 0.001] compared to those without the mutation. A missense mutation at 16457 (S5398L) was associated with an increased risk of progressive fever [RR: 23.5, 95% CI: 6.06–30.0, p.adjust < 0.001]. Patients harboring a synonymous mutation at position 934 encoding nsp2 (D233*) had an 820% increased risk of developing myalgia/arthralgia [RR: 9.20, 95% CI: 4.02–21.05, p.adjust < 0.001]. Additionally, patients with a synonymous mutation in the 28,846 gene encoding the R191* N protein had a significantly increased risk of developing sore throat [RR: 9.00, 95% CI: 3.94–20.57, p.adjust < 0.001] and fatigue [RR: 6.0, 95% CI: 2.53–14.2, p.adjust < 0.001] compared to patients without the mutation by 800% and 500%, respectively. Missense mutation at 28,908 was also significantly associated with 310% increased risk of sore throat [RR: 4.10, 95% CI: 1.54–10.92, p.adjust < 0.05] compared to those without mutation. Frameshift deletion in 11,082 encoding L3606fs-nsp6 was significantly associated with increased prevalence of cough [RR: 6.00, 95%CI: 2.53–14.24, p.adjust < 0.001], conjunctival congestion [RR: 11.25, 95%CI: 4.40–28.0, p.adjust < 0.001] and shortness of breath [RR: 3.00, 95% CI: 1.56–5.77, p.adjust = 0.001] by 500%, 1025% and 200%, respectively as compared to the reference. Furthermore, patients with a frameshift deletion at the V6fs gene had a 359% and 308% increased prevalence of nasal congestion [RR: 4.59, 95% CI: 2.33–9.00, p.adjust < 0.001] and shortness of breath [RR: 4.08, 95% CI: 2.16–7.70, p.adjust < 0.001], respectively, when compared to those without the mutation.
Fig. 5.

Forest plot of clinicopathological feature distribution in association with frequent point mutations along SARS-CoV-2 genome. a) Fever, b) Fever progression, c) Headache, d) Cough, e) Conjunctival congestion, f) Nasal congestion, g) Fatigue, h) Myalgia/arthralgia, i) Sore throat, j) Sputum production and k) Shortness of breath. Significance of adjusted p-value, ***p < 0.001, *p < 0.01. Relative risk of clinical symptoms in relation to mutational variants was estimated using log-binomial regression and p-value adjusted by Benjamini and Hochberg.
3.4. Correlation between mutations in different SARS-CoV-2 genomic regions
Multiple co-occurring mutations were detected in various SARS-CoV-2 genomic positions ( Fig. 6 a ). The purpose of this study was to establish correlations between viral genome variants associated with clinical relevance. Significant co-occurring variations were observed in 11,991 encoding E3909G-nsp7/replicase and 16,915 encoding L5551*-nsp-13/helicase (r = 0.9, p < 0.001), 18,877 encoding L6205*-nsp-11/3’exonuclease (r = 0.6, p < 0.001) and 29,744–3’UTR (r = 0.4, p = 0.009). There was a strong positive correlation between mutations in ORF1a, including 9483 encoding E3073A-nsp4, and nsp3 mutations at 4345 (E1363) (r = 0.7, p < 0.0001) and 3011 (L916*) (r = 0.6, p < 0.001). The mutation at 21,574 encoding V6fs-spike glycoprotein was moderately correlated with the mutations at 2706 encoding T814I-nsp2, 3373 encoding D1036E-nsp3, and 16,647 encoding T5461*-nsp13 helicase (r = 0.5, p < 0.001), but was only weakly correlated with the mutations at 677 encoding A138T-nsp1 (r = 0.3, p = 0.01) and 9968 encoding for A323S-nsp4 (r = 0.3, p = 0.01). Alternatively, the most prevalent mutation at 14,408 encoding nsp12 RdRp was unrelated to mutations in other viral genomic regions.
Fig. 6.

Correlation matrix representing Pearson correlation coefficient a) between different mutation sites across SARS-CoV-2 genome and b) between patients' risk factors and clinical symptoms. Conj_Cong.; conjunctival congestion, Nasal_Cong; nasal congestion, DM; Diabetes mellitus, CHD; chronic heart disease, CLD; chronic liver disease; RHD, rheumatic heart disease, HTN; hypertension.
3.5. Correlation between co-occurring clinical symptoms
As illustrated in Fig. 6 b, hypertension was moderately correlated with the duration of symptoms (r = 0.5, p < 0.001), the duration of fever (r = 0.4, p = 0.001), the fever itself (r = 0.4, p = 0.002), and the progression of the fever (r = 0.5, p < 0.001), but was only weakly correlated with shortness of breath (r = 0.3, p = 0.02). Asthma, rheumatic heart disease, chronic liver disease, and immunodeficiency were all associated with the development of sore throat (r = 0.4, 0.4, 0.6, and 0.6, p = 0.002, 0.003, 0.001, and 0.001, respectively) and fatigue symptoms (r = 0.4, 0.4, 0.6, and 0.6, p = 0.002, 0.003, 0.001, and 0.001, respectively). Additionally, smoking and exposure to infected cases were associated with fever duration (r = 0.4, p = 0.003& 0.005, respectively), fever (r = 0.3, p = 0.03& 0.04, respectively), and fever progression (r = 0.3, p = 0.016& 0.014, respectively).
We identified a positive correlation between the duration of clinical symptoms and progressive disease, including progressive fever (r = 0.4, p = 0.01) and shortness of breath (r = 0.3, p = 0.02). Shortness of breath was moderately correlated with fever duration (r = 0.5, p < 0.001), fever (r = 0.5, p < 0.001), and progression of fever (r = 0.4, p = 0.001), but was only weakly correlated with nasal congestion (r = 0.3, p = 0.03) and conjunctival congestion (r = 0.3, p = 0.01). Furthermore, in our cohort of patients, fatigue was significantly associated with the development of sore throat (r = 0.7, p < 0.001), cough (r = 0.4, p = 0.007), and nasal congestion (r = 0.3, p = 0.02).
4. Discussion
Due to the slow evolutionary rate of SARS-CoV-2, the occurrence of even a single mutation with a single amino acid substitution that can alter the virus's phenotype is noteworthy. Other coronaviruses, such as MERSE-CoV and SARS-CoV-1, have been associated with resistance to neutralizing antibodies, which may result in an increase in disease severity and mortality risk [15]. Until now, no study has reported changes in clinical symptoms associated with SARS-CoV-2 mutations. COVID 19 has a broad clinical spectrum, ranging from asymptomatic to mild symptoms to life-threatening disease. A recent study by Yao, Lu [16] has reported SARS-CoV-2 variants had an impact on viral infectivity and blood clotting functions using in vitro viral cell line model. However, no apparent changes in the patient's clinical symptoms could be linked to the observed virus mutations in their study.
Our study revealed that severe clinical symptoms were weakly to moderately correlated to patients' risk factors including hypertension, exposure to infected cases, smoking, chronic liver disease, rheumatic heart diseases, immunodeficiency, and asthma. Moreover, there was a positive significant correlation between shortness of breath and fever progression along with duration longer fever and symptoms duration. There were no associations between risk factors and any of clinical symptoms observed.
To investigate whether the variation of clinical symptoms might be associated with evolution of the virus over the last few months, we performed a full-length genome high-throughput sequencing. Then, we compared the sequence reads with SARS-CoV-2 reference genome to detect important mutation positions across the viral genome associated with various SARS-CoV-2-specific symptoms across Egyptian patients. We identified 36 frequent point mutations in >5% of the viral genome. Four highly recurrent mutations (>85%) were observed in S-gene at 23,403 encoding for D614G-spike glycoprotein, in 241–5’UTR and in ORF1a at 3037 (F924*-nsp3) and 14,408 (P4715L-RdRp). This type of mutation is known to have higher transmissibility in Europe and is similar to those seen elsewhere [17]. However, different mutations were detected in North America and none of these mutations were observed in Asia [18].
The most common amino acid variations among SARS-CoV-2 proteins are spike glycoproteins, followed by ORF1a polyproteins. According to reports, these variants cause epitope loss, which has been linked to increased viral pathogenesis and transmissibility regardless of the severity of symptoms [19]. In the current study, we found that these recurrent mutation hot spots had no effect on the risk of developing clinical symptoms. The missense mutation in 23,403 (A to G) encoding for the spike glycoprotein resulted in an amino acid substitution D614G with a significantly different isoelectric point. Korber, Fischer [20] found that D614G status was significantly associated with higher viral loads without being significantly related to disease severity as indicated by hospitalization. In contrast to previous findings in Europe that showed the co-occurrence of mutations in 14,408 encoding for RNA dependent RNA polymerase (RdRp) and G614 variants, no significant correlation was found between genomes isolated from our Egyptian patient's cohort harboring mutations in 14,408 (P4715L-RdRp) and 23,403 (D614G-spike glycoprotein). The P4715L RdRPp variant was caused by a missense mutation in 14,408. When compared to proline, leucine has similar physiochemical properties and an isoelectric point. This could explain the lack of an association between variants with P4715L RdRp mutations and clinical manifestation variability.
In the current study, we detected two rare frameshift deletions: one in the ORF1a at 11,082 encoding for the L3606-nsp6 variant, which affected 10% of the sequenced genomes, and one in the low complexity region signal peptide sequence of S gene (21,574) encoding for the V6 spike glycoprotein variant, which affected 16% of the sequenced virus genomes.
Our principal finding was the detection of significant association of L3606fs-nsp6 and V6fs-spike glycoprotein variants with increased risk of shortness of breath. We identified that patients harboring frameshift mutation in 11,082 leading to leucine deletion at amino acidic region 3606 (corresponding to the amino acid residue 37 of nsp6) experienced a significant increase in risk of developing cough, conjunctival congestion, and shortness of breath by 500%, 1025% and 200%, respectively as compared to those without mutations. There has been debate over whether nsp6 variants promote viral replication and allow viruses to evade immune surveillance or the reverse.
SARS-nsp6 CoV-2's protein, like that of other coronaviruses, has seven transmembrane helices that mediate autophagosome formation. It has previously been reported that the phenylalanine residue-rich region at the C-terminus of nsp6's outer membrane mediates its stable binding to the ER membrane of the virus-infected cell, resulting in the formation of small autophagosomes such as DMVs [21,22]. This binding limits autophagosome degradation in lysosomes which enhances coronavirus infection. According to Benvenuto, Angeletti [23], a leucine to phenylalanine substitution of the 37th amino acid residue in the phenylalanine rich region of nsp6 decreased protein structure stability. This discovery was made solely on the basis of amino acid stability analysis, without taking into account transmembrane positions or other protein interactions. Mutations in these viral regions should be closely monitored because it could cause considerable changes in host-virus interaction, potentially affecting SARS-CoV-2 pathogenicity on host cells. Furthermore, we found that deletion of V6 in the signal peptide region of spike protein was associated with a 90%, 359%, and 308% increase in the risk of fever, nasal congestion, and shortness of breath, respectively, when compared to the reference. Furthermore, the V6fs-S glycoprotein variant was 100.98% associated with a longer duration of fever when compared to the reference. Valine deletion, by affecting the signal peptide of spike protein, may aid in protein folding and virus secretion. Further studies on host-virus interactions are needed to prove the exact relevance of this variation.
Another intriguing finding in the current study was the significant association between a missense mutation at 16457 encoding for nsp13/helicase-S5398L and an increase in fever duration and mean age of infected patients. This variant was also found to be significantly related to the prevalence of progressive fever. This substitution corresponds to the 74th amino acid residue of the SARS-CoV-2 nsp13/helicase protein, which is located on zinc-finger3 (ZF3) motif of the zinc-binding domain (ZBD) of the protein. Nsp13/helicase of SARS-CoV-2 plays a pivotal role in the unwinding of DNA and RNA duplexes by coordinating its five main domains, ZBD, stalk domain, and three core domains (1B, 1A and 2A). The ZBD is required for helicase activity due to its interaction with the stalk domain [24,25]. This stalk domain represents a link that transfer the signals coming from ZBD down to the helicase core, indicating the coordination of the five domains required for helicase activity. Furthermore, it has been reported that nsp12-RdRp can bind to nsp13-helicase on the ZF3 motif of ZBD, and that this bound nsp13-helicase increased its unwinding activity by 2-fold [25].
We hypothesize that the association of S5398L-nsp13 with longer fever duration and progressive fever observed in this study might be due to the fact that this variant is in a region that could enhance the replication transcription complex (RTC) to unwind more double-stranded nucleic acid, eventually leading to more virus self-reproduction.
In the current study, we observed a link between two synonymous mutations, 28,846 encoding R191* N-protein and 934 encoding D223*-nsp2, and clinical manifestations. The mutation at 28,846 was associated with higher risk developing sore throat and fatigue, whereas the mutation at 934 was linked to the development of myalgia/arthralgia. Since these are synonymous mutations, they are unrelated to amino acid variation. These observed variations in clinical symptoms, however, could be related to a C to T transition, which could affect codon optimization, mRNA structure and function, and thus might influence fitness.
Previous studies have revealed that SARS-CoV-2 RNA RdRp forms a hollow cylinder supercomplex (holo-RdRp) with other viral cofactors including nsp7/helicase, which activates and confers RdRp processivity and proofreading capability [18,26]. Moreover, it has been reported that mutant SARS-COV RNA genomes with altered position of nsp7/nsp8 replicase coding sequences result in viral viability loss [27]. In the present study, we found that E3909G nsp7replicase variant encoded by ORF1a at 11,991 was significantly more common among children (2–13 yrs.), affecting 100% of this age group while being absent in other age groups and being significantly associated with a reduction in duration of COVID 19 symptoms. These findings suggest that this mutant variant may contribute to hindering of nsp7/replicase binding to RdRp and proofreading capability that eventually lead to viral viability loss and improvement of clinical symptoms, which may explain the fast recovery observed in children.
Coronaviruses, like other positive stranded RNA viruses, induce the formation of membranous structures in host infected cells, including double membrane vesicles (DMVs) and convoluted membranes (CMs), which serve as a platform for the virus RTC. Furthermore, co-expression of nsp3 with nsp4 and nsp6 has been shown to play an important role in the formation of replicative structures and inducing membrane rearrangements in virus host cells [6,28].
We found that a missense mutation in the 9483 encoding nsp4 resulted in an E3073A substitution within the amino terminal transmembrane domain of nsp4. The E3073A-nsp4 variant was related to a significant reduction in fever duration. A charged amino acid to alanine substitution within the amino terminal of nsp4 has been attributed to a reduction in mutant hepatitis viral RNA synthesis [29]. Furthermore, it has been reported that charged amino acid to alanine variations of viral proteins produced a temperature sensitive and attenuated mutant virus phenotype [30]. Thus, substitution of the charged glutamate amino acid with alanine within the amino terminal of nsp4 of SARS-CoV-2 genome observed in our study could result in a temperature sensitive phenotype with a significant reduction in fever duration. In line with this finding, we observed a missense mutation in 4354 of nsp3 resulting in amino acid change from glutamate to aspartate (E1363D). Despite the fact that aspartate and glutamate have similar isoelectric points and physiochemical properties, the D1363 variant is associated with significantly shorter fever duration. The significant strong positive correlation discovered between nsp4 A3073 and nsp3 D1363 variants could explain this change in clinical symptoms (r = 0.7, p < 0.0001).
In conclusion, we found that the most recurrent P4715L-RdRp and D614G-spike-glycoprotein amino acid alterations were not significantly associated with variations in clinical symptoms, indicating that these mutations are linked to viral spread and persistence within the host regardless of clinical symptoms. Certain mutation hot spots were associated with specific age groups, with 11,991 and 16,915 being significantly prevalent among children while, 16,457 being significantly associated with older age groups (>45 yrs.).
Three mutations were found to be significantly related to clinical symptom worsening. Shortness of breath was significantly associated with frameshift deletions affecting spike glycoprotein (V6fs) and nsp6 (L3606fs). Missense mutation leading to S5398L helicase was significantly associated with progressive fever. Other mutation sites, on the other hand, were linked to better outcomes. E3909G-nsp7 was associated a shorter duration of symptoms, and E3073G-nsp4 along with E1363D-nsp3 were linked to a significant decrease in fever duration. Further large-scale research is needed to explore the mechanistic insights underlying these intriguing findings.
The following are the supplementary data related to this article.
Mean age, duration of symptoms and duration among mutational hotspot.
Funding
This study was supported by Science and Technology Development Fund (STDF) Egypt, Grant ID grg (ACSE41907).
Intellectual property
The authors confirm that our institutions' intellectual property regulations have been strictly followed.
Research ethics
The study was endorsed by the Ministry of Health and Populations' Ethics Committee. Training and Research sector with office of human research protections (OHRP): FWA00016183 23 March 2020, IORG0005704/ IRB0000687 8 November 2020, and each participant provided informed consent.
CRediT authorship contribution statement
Abdel-Rahman N. Zekri: Hypothesis, Experimental Design, Revising the manuscript. Marwa Mohanad: Data analysis, Bioinformatics analysis, Interpretation of data, Writing of manuscript. Mohammed M Hafez: Experimental analysis, Revising the manuscript. Hany K. Soliman: Experimental analysis. Zainab K. Hassan: Experimental design. Mohamed Abouelhoda: Bioinformatics analysis. Khaled E. Amer: Collection of patient's clinical data. Mohamed G. Seadawy: Collection of patient's clinical data, Experimental analysis. Ola S Ahmed: Experimental analysis.
Declaration of competing interest
The authors declare that there are no conflicts of interest, and that they have equally contributed to every part of this work.
Acknowledgments
The authors would like to express their gratitude to Prof. Dr. Mohamed Othman Elkhosht, President of Cairo University, for his assistance with this work.
References
- 1.Shigemura J., et al. Public responses to the novel 2019 coronavirus (2019-nCoV) in Japan: mental health consequences and target populations. Psychiatry Clin. Neurosci. 2020;74(4):281–282. doi: 10.1111/pcn.12988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu F., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gorbalenya, A.E., et al., Severe acute respiratory syndrome-related coronavirus: the species and its viruses – a statement of the Coronavirus Study Group. bioRxiv, 2020: p. 2020.02.07.937862.
- 4.Robson F., et al. Coronavirus RNA proofreading: molecular basis and therapeutic targeting. Mol. Cell. 2020;79(5):710–727. doi: 10.1016/j.molcel.2020.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hagemeijer M.C., et al. Dynamics of coronavirus replication-transcription complexes. J. Virol. 2010;84(4):2134–2149. doi: 10.1128/JVI.01716-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Angelini M.M., et al. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. MBio. 2013;4(4) doi: 10.1128/mBio.00524-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Snijder E.J., et al. A unifying structural and functional model of the coronavirus replication organelle: tracking down RNA synthesis. PLoS Biol. 2020;18(6) doi: 10.1371/journal.pbio.3000715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang C., et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie Y., et al. Epidemiologic, clinical, and laboratory findings of the COVID-19 in the current pandemic: systematic review and meta-analysis. BMC Infect. Dis. 2020;20(1):640. doi: 10.1186/s12879-020-05371-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Struyf T., et al. Signs and symptoms to determine if a patient presenting in primary care or hospital outpatient settings has COVID-19 disease. Cochrane Database Syst. Rev. 2020;(7) doi: 10.1002/14651858.CD013665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zipfel C.M., Bansal S. Assessing the interactions between COVID-19 and influenza in the United States. medRxiv: The Preprint Server for health Sciences. 2020;2020.03.30.20047993 [Google Scholar]
- 12.Bernheim A., et al. Chest CT findings in coronavirus disease-19 (COVID-19): relationship to duration of infection. Radiology. 2020;295(3):685–691. doi: 10.1148/radiol.2020200463. 200463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ai T., et al. Correlation of chest CT and RT-PCR testing for coronavirus disease 2019 (COVID-19) in China: a report of 1014 cases. Radiology. 2020;296(2):E32–e40. doi: 10.1148/radiol.2020200642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995;57(1):289–300. [Google Scholar]
- 15.Tang X.-C., et al. Identification of human neutralizing antibodies against MERS-CoV and their role in virus adaptive evolution. Proc. Natl. Acad. Sci. 2014;111(19):E2018–E2026. doi: 10.1073/pnas.1402074111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yao H., et al. Patient-derived SARS-CoV-2 mutations impact viral replication dynamics and infectivity in vitro and with clinical implications in vivo. Cell Discov. 2020;6(1):76. doi: 10.1038/s41421-020-00226-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yin C. Genotyping coronavirus SARS-CoV-2: methods and implications. Genomics. 2020;112(5):3588–3596. doi: 10.1016/j.ygeno.2020.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pachetti M., et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020;18(1):179. doi: 10.1186/s12967-020-02344-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupta A.M., Chakrabarti J., Mandal S. Non-synonymous mutations of SARS-CoV-2 leads epitope loss and segregates its variants. Microbes Infect. 2020;22(10):598–607. doi: 10.1016/j.micinf.2020.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korber, B., et al., Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell, 2020. 182(4): p. 812–827.e19. [DOI] [PMC free article] [PubMed]
- 21.Cottam E.M., et al. Coronavirus nsp6 proteins generate autophagosomes from the endoplasmic reticulum via an omegasome intermediate. Autophagy. 2011;7(11):1335–1347. doi: 10.4161/auto.7.11.16642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cottam E.M., Whelband M.C., Wileman T. Coronavirus NSP6 restricts autophagosome expansion. Autophagy. 2014;10(8):1426–1441. doi: 10.4161/auto.29309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benvenuto D., et al. Evolutionary analysis of SARS-CoV-2: how mutation of non-structural protein 6 (NSP6) could affect viral autophagy. J. Inf. Secur. 2020;81(1):e24–e27. doi: 10.1016/j.jinf.2020.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singleton M.R., Dillingham M.S., Wigley D.B. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007;76(1):23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 25.Jia Z., et al. Delicate structural coordination of the severe acute respiratory syndrome coronavirus Nsp13 upon ATP hydrolysis. Nucleic Acids Res. 2019;47(12):6538–6550. doi: 10.1093/nar/gkz409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao Y., et al. Structure of RNA-dependent RNA polymerase from 2019-nCoV, a major antiviral drug target. bioRxiv. 2020;2020.03.16.993386 [Google Scholar]
- 27.Deming D.J., et al. Processing of open reading frame 1a replicase proteins nsp7 to nsp10 in murine hepatitis virus strain A59 replication. J. Virol. 2007;81(19):10280–10291. doi: 10.1128/JVI.00017-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hagemeijer M.C., et al. Membrane rearrangements mediated by coronavirus nonstructural proteins 3 and 4. Virology. 2014;458:125–135. doi: 10.1016/j.virol.2014.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sparks J.S., Lu X., Denison M.R. Genetic analysis of murine hepatitis virus nsp4 in virus replication. J. Virol. 2007;81(22):12554–12563. doi: 10.1128/JVI.01257-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang R.S., et al. Clustered charge-to-alanine mutagenesis of human respiratory syncytial virus L polymerase generates temperature-sensitive viruses. Virology. 2002;302(1):207–216. doi: 10.1006/viro.2002.1596. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mean age, duration of symptoms and duration among mutational hotspot.