

Summary
Utilizing T cells expressing chimeric antigen receptors (CARs) to identify and attack solid tumors has proven challenging, in large part because of the lack of tumor-specific targets to direct CAR binding. Tumor selectivity is crucial because on-target, off-tumor activation of CAR T cells can result in potentially lethal toxicities. This study presents a stringent hypoxia-sensing CAR T cell system that achieves selective expression of a pan-ErbB-targeted CAR within a solid tumor, a microenvironment characterized by inadequate oxygen supply. Using murine xenograft models, we demonstrate that, despite widespread expression of ErbB receptors in healthy organs, the approach provides anti-tumor efficacy without off-tumor toxicity. This dynamic on/off oxygen-sensing safety switch has the potential to facilitate unlimited expansion of the CAR T cell target repertoire for treating solid malignancies.
Keywords: chimeric antigen receptor, T cell, hypoxia, HIF1α, cytokine release syndrome, toxicity, cancer, immunotherapy, CAR T cells, HypoxiCAR
Graphical abstract
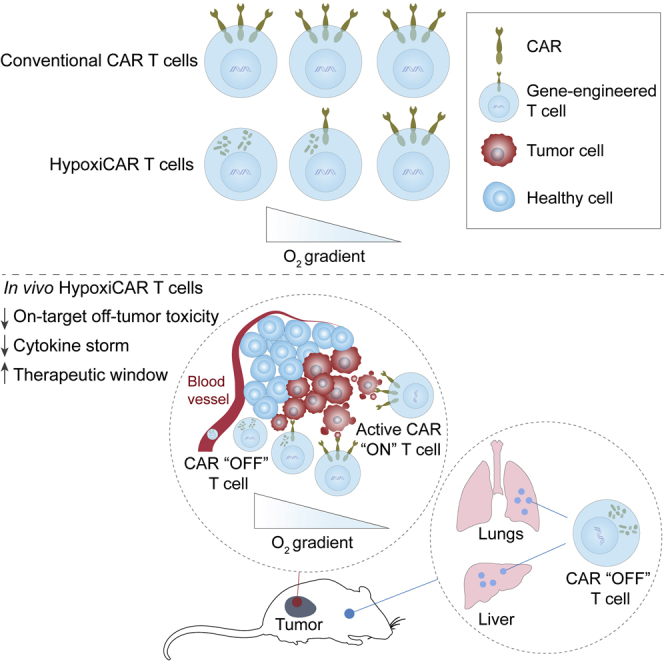
Highlights
A dual oxygen-sensing switch provides stringent hypoxia-dependent regulation of a CAR
HypoxiCAR T cells deliver tumor-selective CAR expression and anti-tumor efficacy
HypoxiCAR T cells prevent on-target, off-tumor activation and cytokine release syndrome
HypoxiCAR provides a strategy to expand the CAR repertoire for solid malignancies
Utilizing CAR T cells to attack solid tumors has proven challenging because of the lack of tumor-specific CAR targets. Kosti et al. demonstrate that re-engineering CARs to stringently and selectively express in response to tumor hypoxia prevents their off-tumor activation and toxicity while delivering robust anti-tumor efficacy.
Introduction
There has been significant interest in the prospects of chimeric antigen receptor (CAR) T cell therapy for solid malignancies, and multiple clinical trials are in progress.1 However, the scope of these trials has been restricted by the lack of availability of tumor-specific targets. Upon antigen binding, CARs initiate robust T cell activation and subsequent cytolytic killing of the target cell through an intracellular signaling domain that most commonly contains CD3ζ and co-stimulatory elements.2 However, the selectivity of CAR-mediated killing of tumor cells is currently dictated solely by the biodistribution of the CAR antigen. Tumor specificity is vital for the success and safe use of CAR therapy. Intravenously (i.v.) infused anti-ErbB2 CAR T cells resulted in lethal toxicity in an individual with metastatic colon cancer because of uncontrolled cytokine release syndrome (“cytokine storm”) following CAR T cell activation in the lungs.3 The ErbB family of receptors is widely expressed on normal epithelial cells. Nonetheless, ErbB receptors remain attractive tumor-associated targets because they are expressed, or overexpressed, in a wide range of cancers.4,5 Identifying approaches to circumvent off-tumor toxicity has the potential to unlock an entirely new repertoire of CAR antigen targets for carcinomas, which are currently limited. In this study, we investigate the opportunities for harnessing tumor hypoxia, a characteristic common to most solid tumors, as a physical cue for licensing CAR T cell activation specifically in the tumor microenvironment (TME). We find that expressing a CAR under control of a stringent hypoxia-sensing safety switch avoids on-target, off-tumor activation of CAR T cells while delivering efficient anti-tumor killing. This study provides an approach to overcome a major hurdle for utilization of CAR T cell therapy against solid malignancies.
Results
I.v. administration of pan-anti-ErbB CAR T cells results in lethal toxicity in mice
To investigate the issue of on-target, off-tumor CAR T cell activation, we utilized a second-generation pan-anti-ErbB CAR T1E28z, T4-CAR, which has specificity toward 8 of 9 of the possible ErbB receptor homo- and heterodimers and crosses the species barrier, binding human and mouse receptors equivalently.6 T4-CAR co-expresses a chimeric cytokine receptor (4αβ) that delivers an intracellular interleukin-2 (IL-2)/IL-15 signal upon binding of IL-4 to the extracellular domain, providing a means to selectively enrich CAR T cells during ex vivo expansion (Figures 1A and 1B)7, 8, 9 without affecting the CAR-dependent killing capabilities of these cells.7 T4-CAR has been proven to be safe for intra-tumoral (i.t.) delivery in individuals with head and neck squamous cell carcinoma (HNSCC),10 but i.v. infusion is required to treat individuals with metastatic disease. However, preclinically, i.v. infusion of human T4-CAR T cells into non-obese diabetic (NOD)-scid IL2Rgammanull (NSG) mice bearing HN3 tumors (Figures 1C, 1D, and S1A), which express ErbB1-4 (Figures S1B and S1C), resulted in lethal toxicity, evident by rapid weight loss in these animals (Figure 1D). As observed clinically,3 analysis of the blood of these mice revealed evidence of cytokine storm (Figure 1E). Using a sub-lethal dose of CAR T cells concurrently expressing the reporter luciferase (Luc), we evaluated the biodistribution of these cells acutely after i.v. infusion (Figures 1C and 1F). Imaging revealed that the majority of the infused CAR T cells accumulated in the lungs and liver, whereas only a minority reached the tumor despite expression of ErbB1-4 on tumor cells (Figures 1G, S1B, and S1C). Profiling of ErbB1-4 mRNA expression confirmed that all four receptors were expressed across all vital organs, including in the lungs and liver (Figures S1D–S1G). Hematoxylin and eosin (H&E)-stained tissue sections of the liver and lungs of T4-CAR T cell-infused mice revealed the presence of myeloid cell infiltrates, indicative of CAR-mediated inflammation in these tissues (Figures 1H and 1I). These data indicate that the liver and lungs are two key organs for off-tumor CAR T cell activation.
Figure 1.

I.v.-infused T4-CAR T cells cause inflammation in healthy organs
(A) Diagram depicting T4-CAR.
(B) Example histograms of surface CAR expression on live (7-aminoactinomycin D [7AAD]−) CD3+ T4-CAR or non-transduced human T cells, assessed using flow cytometry before and after expansion in IL-4.
(C–E) On day 13 after subcutaneous HN3 tumor cell inoculation, mice were infused i.v. with vehicle or 10 × 106 non-transduced or T4-CAR T cells (n = 5).
(C) Schematic depicting the experiment.
(D) Weight change of the mice. The arrow denotes T cell infusion, and the cross denotes the humane endpoint.
(E) Serum cytokines 24 h after infusion.
(F) Low-dose human ErbB-CAR/Luc T cells (4.5 × 106) were infused i.v. into SKOV3 tumor-bearing NSG mice, and 4 days later, bioluminescence imaging was performed on the whole body and dissected organs. LN, inguinal lymph node; SI, small intestine.
(G) Quantification of the photons per second per unit area as percentage of all organs (n = 6).
(H and I) H&E-stained sections (left) and quantitation of myeloid infiltration (right) in the lungs (H) and liver (I) 5 days after i.v. infusion of low-dose 4.5 × 106 T4-CAR, non-transduced T cells, or vehicle. Arrows indicate myeloid infiltrates.
(J and K) Immunohistochemistry (IHC) staining of tissue sections for reductively activated pimonidazole in tumor bearing NSG mice (J) and quantitation of the staining, scoring between 0–3, from no staining (0) to intense staining (3), as percent area of the tissue (K).
All experiments are representative of a biological repeat. In line charts, the dots mark the mean and error bars SEM. Bar charts show the mean and points individual mice. ∗p < 0.05, ∗∗p < 0.01.
A dual oxygen-sensing system provides stringent hypoxia-regulated expression of a CAR
Hypoxia is a characteristic of most solid tumors, where proliferative and high metabolic demands of the tumor cells, alongside inefficient tumor vasculature, result in a state of inadequate oxygen supply (<2% O2) compared with healthy organs/tissues (5%–10% O2).11 Clinically, hypoxia is associated with a poor prognosis12 and resistance to chemotherapy13 and radiotherapy.14 Because hypoxia differentiates the TME from healthy, normoxic tissue, it is a desirable marker for induction of CAR T cell expression (Figures 1J and 1K). Although hypoxia has been linked to immune suppression,15 the killing capacity of T4-CAR T cells was not affected negatively by even extreme levels of hypoxia (0.1% O2) (Figure S2A). Cells have evolved an elegant biological machinery to detect and rapidly respond to hypoxia through the constitutively expressed transcription factor hypoxia-inducible-1 factor alpha (HIF1α).16 A previous study has investigated CARs fused with an oxygen-dependent degradation domain (ODD) of HIF1α.17 Under conditions of normoxia, the ODD becomes ubiquitinated, targeting the protein/CAR for proteasomal degradation.18,19 Although a CAR-ODD endowed CAR T cells with an improved ability to kill tumor cells under hypoxic conditions, the authors observed residual tumor cell killing under normoxic conditions.17 In an attempt to create a stringent hypoxia-regulated CAR expression system, we developed a dual oxygen-sensing approach for T4-CAR (Figure 2A). This was achieved by appending a C-terminal 203-amino-acid ODD20 onto the CAR while modifying the CAR’s promoter in the long terminal repeat (LTR) enhancer region of the vector to contain a series of 9 consecutive hypoxia-responsive elements (HREs),21 which permitted HIF1α-mediated transcription of the CAR. This CAR, called HypoxiCAR, demonstrated stringent hypoxia-specific presentation of CAR molecules on the cell surface of human T cells in vitro (Figure 2B). HypoxiCAR T cells expressed CAR molecules per cell equivalent to constitutive T4-CAR T cells when activated under hypoxic (0.1% O2) conditions (Figure S2B). The dual hypoxia-sensing system incorporated into HypoxiCAR proved to be superior to single hypoxia-sensing modules of the 9xHRE cassette or ODD, which displayed leakiness in CAR expression (Figures S2C and S2D) and tumor cell killing (Figure S2E) under conditions of normoxia. HypoxiCAR’s expression of the CAR, utilizing the dual hypoxia-sensing system, was restricted stringently to hypoxic environments and also highly dynamic, representing a switch that could be turned on and off in an O2-dependent manner (Figures 2B, 2E, and S2D). Because an ODD was not appended to the 4αβ receptor of T4-CAR, the leaky expression of the 9xHRE promoter under conditions of normoxia (Figure S2D) was sufficient to allow IL-4-mediated in vitro expansion of HypoxiCAR T cells under culture conditions of normoxia (Figure 2A). In further in vitro characterization, the exquisite O2 sensitivity of HypoxiCAR was confirmed; CAR expression was absent under O2 concentrations consistent with healthy organs (≥5%) but became detectable on the cell surface at O2 concentrations equivalent to those found in the TME (≤1%) (Figure 2F).
Figure 2.

HypoxiCAR T cell CAR surface expression and effector function are restricted stringently to hypoxic environments
(A) Diagram depicting HypoxiCAR in normoxia and hypoxia.
(B) Example histograms of surface CAR expression on live (7AAD−) CD3+ T4-CAR, HypoxiCAR, and non-transduced human T cells in normoxic or 18-h hypoxic (0.1% O2) conditions, assessed using flow cytometry.
(C) Genomic DNA from T4-CAR, HypoxiCAR, and non-transduced T cell preparations subjected to qPCR for T2A copy number relative to that of Tbp in the genomic DNA.
(D) The relative prevalence of CD4+/CD8+ T cells among CD3+ T cells, assessed using flow cytometry in the T cell, T4-CAR, and HypoxiCAR preparations (n = 6).
(E) Surface CAR expression on HypoxiCAR T cells at the indicated times under hypoxia (0.1% O2) and re-exposure to normoxia, assessed normalized to 18-h hypoxia (n = 6).
(F) Surface CAR expression on HypoxiCAR T cells after 18-h exposure to 20%, 5%, 1%, and 0.1% O2 (n = 6)’ values are normalized to 0.1% O2.
(G–I) In vitro SKOV3 tumor cell killing by T4-CAR, HypoxiCAR, CD3ζ-truncated HypoxiCAR (CD3ζ-; to prevent intracellular signaling), and non-transduced T cells (effector to target cell ratio 1:1) under normoxic and 0.1% O2 hypoxic conditions.
(H and I) Quantification of IL-2 (H) and IFN-γ (I) released into the medium from the respective T cells after 24-h and 48-h exposure to SKOV3 cells, respectively, under normoxic and 0.1% O2 hypoxic conditions.
All experiments are representative of a biological repeat. Bars on charts show the mean and points an individual healthy donor. In line charts, the dots mark the mean and error bars SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
HypoxiCAR T cells provide hypoxia-restricted tumor cell killing in vitro
Having validated HypoxiCAR’s ability to sense hypoxia, we sought to investigate its ability to elicit hypoxia-dependent killing of tumor target cells. SKOV3 ovarian cancer cells were seeded onto culture plates and co-incubated with T4-CAR or HypoxiCAR T cells under normoxic and hypoxic (0.1% O2) conditions. Despite equivalent transduction efficiencies and CD4+:CD8+ T cells ratios (Figures 2C and 2D), HypoxiCAR T cells displayed efficient hypoxia-dependent killing of SKOV3 cells, almost equivalent to T4-CAR T cells, with no significant killing observed under normoxic conditions (Figure 2G). Target cell destruction was strictly CAR dependent because, when the intracellular tail of HypoxiCAR was truncated to prevent CD3ζ signaling, killing was abrogated (Figure 2G). In addition, HypoxiCAR T cells exhibited stringent hypoxia-restricted secretion of IL-2 (Figure 2H) and IFN-γ (Figure 2I), two cytokines that play an important role in the T cell response.22,23
HypoxiCAR T cells express CAR selectively in the TME in vivo
To evaluate whether hypoxia could restrict HypoxiCAR expression to the TME while remaining switched off in healthy organs in vivo, human HypoxiCAR T cells were injected concurrently i.v. and i.t. in NSG mice bearing hypoxic HN3 tumors with an approximate volume of 500 mm3 (Figures 3A, 1J, and 1K). Three days after HypoxiCAR T cell infusion, tissue-resident T cells were assessed for CAR expression ex vivo using flow cytometry. As predicted by the in vitro analyses (Figure 2), HypoxiCAR T cells had no detectable surface CAR molecules when recovered from the blood, lungs, or liver of mice but did express surface CAR molecules in the hypoxic TME (Figures 3B, 3C, and S2F). A similar observation was made in NSG mice bearing SKOV3 tumors (Figures 3D and 3E). To establish whether the Hypoxi construct elements would remain active at different stages of tumor growth, a Hypoxi-Luc reporter was developed in which the HRE promoter was used to drive expression of a Luc-ODD. This reporter was stably transduced into the SKOV3 and HN3 cell lines (Figures S3A and S3B). Luc-ODD, despite not being detectable in tumor cells under normoxic conditions (Figure S3C), was detected in vivo at all stages of tumor growth, even prior to the tumor becoming palpable, in SKOV3 (Figure S3D) and HN3 tumors (Figure S3E). This highlights the potential for HypoxiCAR T cells to target tumors from an early stage of development.
Figure 3.

HypoxiCAR T cells express CAR on their surface specifically in tumors
(A) Schematic depicting the experiment.
(B–E) Subcutaneous tumor-bearing NSG mice were injected concurrently i.v. and i.t. with human HypoxiCAR T cells (2.5 × 105 i.t. and 7.5 × 105 i.v.) 72 h prior to sacrifice. Representative histograms showing surface CAR expression on live nucleated (7AAD−, Ter119−) CD45+ CD3+ HypoxiCAR T cells in the indicated enzyme-dispersed tissues and blood (B) and frequency of CAR expression (C) in HN3 tumor-bearing mice (n = 9). (D) and (E) represent the same respective analysis in SKOV3 tumor-bearing NSG mice (n = 8).
All experiments are representative of a biological repeat. Bar charts shows the mean and each point an individual mouse. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
HypoxiCAR T cells circumvent treatment-limiting toxicities and provide anti-tumor efficacy in vivo
To test the anti-tumor efficacy of HypoxiCAR, high-dose T4-CAR, HypoxiCAR, or non-transduced human T cells were infused into mice on day 16 after injection of HN3 tumor cells, just prior to tumors becoming palpable (Figures 4A and S3E). In keeping with the absence of CAR expression on T cells in normoxic tissues (Figure 3), HypoxiCAR circumvented the treatment-limiting toxicity seen following i.v. infusion of high-dose T4-CAR T cells. Mice infused with HypoxiCAR T cells displayed no acute drop in weight after infusion (Figure 4B), no evidence of cytokine storm in the systemic circulation (Figure 4C), and no signs of tissue damage in the lungs or liver (Figures 4D and 4E). Importantly, although mice infused i.v. with human T4-CAR T cells all reached their humane endpoints at 28 h (Figure 4B), those infused with HypoxiCAR T cells displayed no signs of toxicity while tumor growth was prevented effectively (Figure 4F). To directly confirm that HypoxiCAR T cells accumulated at the site of disease when tumor control was observed, HypoxiCAR T cells were co-transduced to express constitutively expressed Renilla (r)Luc and injected into mice bearing established SKOV3 tumors (Figure 4G). HypoxiCAR T cells suppressed tumor growth in mice bearing palpable SKOV3 tumors (Figure 4H) without obvious toxicity (Figure S4A). Tracking the biodistribution of reporter HypoxiCAR T cells in vivo confirmed their infiltration into the TME and persistence in these animals as well as presence in the tumor for at least 26 days after infusion (Figures 4I, 4J, and S4B). However, there was an observable drop in the prevalence of HypoxiCAR T cells 11 days after infusion which preceded the loss of tumor control (Figure S4B). Therefore, HypoxiCAR overcomes a major hurdle that currently precludes systemic administration of CAR T cells targeting antigens that are expressed in normal tissues throughout the body.3
Figure 4.

HypoxiCAR T cells provide anti-tumor efficacy without systemic toxicity
(A-C) Sixteen days after subcutaneous HN3 tumor cell inoculation, mice were infused i.v. with vehicle or 10 × 106 T4-CAR, HypoxiCAR, or non-transduced human T cells (control) (n = 4 mice).
(A) Schematic depicting the experiment.
(B) Weight change of the mice.
(C) Serum cytokines 24 h after infusion.
(D and E) Low-dose (4.5 × 106) T4-CAR or HypoxiCAR T cells were infused i.v. into NSG mice, and five days later, the indicated tissues were excised, and myeloid infiltration was scored in the lungs (D) and liver (E) (n = 5–6).
(F) HN3 tumor growth curves from (A)–(C), with an arrow marking the point of CAR T cell infusion.
(G–J) Schematic depicting the experiment in which HypoxiCAR T cells or T cells were transduced to express a constitutive rLuc/EGFP reporter to allow in vivo tracking (G). Mice bearing established SKOV3 tumors were infused i.v. with vehicle (n = 6) or 10 × 106 reporter HypoxiCAR (n = 7) or reporter T cells (n = 5) (H). Bioluminescence imaging was performed on the whole body of mice to track the biodistribution of the infused HypoxiCAR T cells on day 26 after infusion. The red box marks the SKOV3 tumor (I). Also shown is quantification of the percent photon flux (photons per second per unit area) signal detected specifically in the tumor out of total photon flux across the whole body (J).
(K–M) Example IHC-stained human HNSCC section for HIF1α (red) and CD3 (brown) (K). The abundance of inter-epithelial T cells (IETs) (L) (example marked by a black arrow in K), low/absent n = 40 and high n = 52, was assessed against the HIF1α stabilization score of the tumor (L). For tumors where IETs were high, tumor-infiltrating lymphocytes (TILs) directly infiltrating HIF-1α stabilized regions of the tumor (H-TILs; examples marked by white arrows in K) were scored as absent (n = 6 of 52 tumors) or present (n = 46 of 52 tumors) and plotted against the HIF1α stabilization score of the tumor (M).
(N) Immunofluorescence images from a human oral tongue carcinoma stained with DAPI (nuclei, blue) and antibodies against CD3 (green) and HIF1α (red); white denotes CD3 and HIF1α co-localization.
All experiments are representative of a biological repeat. Bar charts shows the mean and each point an individual mouse. In line charts, the dots mark the mean and error bars SEM. Boxplots show median and upper/lower quartiles, and whiskers show the highest and lowest value.∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
T cells efficiently infiltrate and stabilize HIF1α in hypoxic tumor islands in human cancer
Hypoxia has been extensively studied in HNSCC.12,24,25 To assess which individuals might be most appropriate for HypoxiCAR T cell immunotherapy, we first generated an HRE-regulated gene signature using tumor sample transcriptomic data that included PGK1, SLC2A1, CA9, ALDOA, and VEGFA (Figure S5A). Expression of this 5-gene signature, although expressed across all tumors, did increase with tumor size (suggesting that larger tumors were more hypoxic; Figure S5B), but did not differentiate between HNSCC subtypes (Figure S5C) and was prognostic of poorer survival in stage 3 and 4 cancers (Figure S5D). Other groups have also demonstrated hypoxia gene signatures to be predictive of adverse prognosis in HNSCC,26, 27, 28 and such signatures could be utilized to guide selection of individuals for HypoxiCAR therapy.
Immunohistochemistry staining of HNSCC tumor sections for stabilized HIF1α, the master transcription factor for HypoxiCAR expression, revealed large regions of the tumors where HIF1α had become stabilized (Figure 4K). Although several factors can stabilize HIF1α, hypoxia is the most probable explanation for this observation.29 Heterogeneity in HIF1α stabilization and i.t. T cell infiltration was seen between individuals; however, tumors with the highest prevalence and/or intensity of HIF1α stabilization did not exclude T cells from entering the inter-epithelial space (Figure 4L), nor from entering HIF1α-stabilized regions of the tumor (Figure 4M). Using immunofluorescence, we also confirmed that CD3+ T cells infiltrating HIF1α-stabilized tumor regions also stabilized HIF1α themselves (Figure 4N and S5E), suggesting that, in these environments, HypoxiCAR T cells would become activated. These observations suggest that HypoxiCAR could find clinical application in hypoxic tumor types such as HNSCC, where gene expression (Figure S5), staining of biopsy samples for HIF1α/CD3 (Figure 4K), and imaging techniques such as positron emission tomography (PET)/computed tomography (CT) using a hypoxia radiotracer such as copper-64-diacetyl-bis(N4-methylthiosemicarbazone) (64Cu-ATSM)25 might provide biomarkers to confirm the presence of a hypoxic TME and guide selection of affected individuals.30
Discussion
Approaches to improve the tumor specificity of CAR T cells have been developed, such as T cell receptor-mimetic CARs with specificity for human leukocyte antigen (HLA)-presented antigens,31 combined targeting of tumor antigens,32, 33, 34 or tuning of CAR affinity to preferentially target high density antigens.35 This study demonstrates an alternative approach to achieve cancer-selective immunotherapy, exploiting one of the most innate characteristics of the TME. The dual hypoxia-sensing system described here achieves compelling anti-tumor efficacy while abrogating off-tumor toxicity of a CAR that recognizes multiple targets in normal tissues.
T cells that have infiltrated the TME can egress,36 highlighting a potential safety concern if hypoxia-experienced HypoxiCAR T cells expressing CAR were to re-enter healthy normoxic tissue. However, CD8+ T cell migration has been demonstrated to cease in regions where it encounters tumor cells expressing its cognate antigen,37 suggesting that, when HypoxiCAR T cells have expressed their CAR and engaged with tumor cells expressing ErbB receptors, their ability to egress could be limited. Furthermore, based on our in vitro observations (Figure 2E), CAR expression is reduced rapidly upon potential exit from the tumor, substantially limiting the risk of unwanted off-tumor activation of the infused CAR T cells.
The hypoxic TME is not conducive to efficient immune reactions,15 where hypoxia can activate immune-suppressive programs in stromal cells such as macrophages,38 regulate the expression of immune checkpoint molecules,39 and promote a more aggressive tumor cell phenotype.40 T cell activation/killing can also result in increased hypoxia in the TME.41 Encouragingly, however, and in agreement with the findings by others,42 we found that hypoxia did not negatively affect T cell effector function in vitro (Figures 2G–2I and S2A). HypoxiCAR T cells were also able to prevent growth of hypoxic tumors (Figures 4F and 4H), suggesting that, in the tested models, the TME was not an absolute barrier to HypoxiCAR’s effector function. Although the HypoxiCAR T cell dose used in this study did not entirely eradicate SKOV3 tumors (Figure 4H), the loss of tumor control coincided with an observable drop in the number of HypoxiCAR T cells resident in the mice (Figures S4B), which could account for this observation. Also, SKOV3 tumors induced lower overall expression of CAR compared with HN3 tumors (Figure 3), which agrees with SKOV3 tumors being relatively less hypoxic (Figures S3D and S3E). These observations highlight important considerations for translation and appropriately selecting the dose and individual receiving HypoxiCAR T cells to achieve efficient anti-tumor control. Importantly, because we also demonstrated that T cells are not excluded from HIF1α stabilized regions of human tumors (Figures 4K–4N), it is likely that HypoxiCAR T cells should be able to access the appropriate TMEs to activate CAR expression. Although we did not observe evidence of treatment-limiting toxicity in mice infused with high therapeutic doses of HypoxiCAR T cells (Figures 4B–4E), there are microenvironments in healthy tissues, such as the intestinal mucosa, where “physiologic hypoxia” has been observed.43 Such tissues might be sites where off-tumor activation of HypoxiCAR T cells could take place. However, an additional suicide switch44 could be incorporated into HypoxiCAR to provide an additional level of safety for clinical testing.
HypoxiCAR provides a stringent and broadly applicable strategy to overcome the paucity of safe targets available for the treatment of solid malignancies.
Limitations of study
The current study utilizes a single CAR in xenograft tumor models with immunocompromised mice. Utilizing the HypoxiCAR approach in other CARs and tumor models, potentially including syngeneic mouse CAR T cells in immunocompetent preclinical models of cancer, would provide further insight into the effectiveness and utility of the approach.
The current study also does not establish a maximal tolerated dose of HypoxiCAR T cells compared with constitutive CAR T cells or the effectiveness of multiple dose regimens, which would provide important further insight for translation. Given that HypoxiCAR T cells do not express CARs on their surface during their in vitro expansion phase, it would also be interesting to study whether, in the absence of “tonic” signaling, HypoxiCAR T cells have a superior phenotype for adoptive cell therapy.
Although the HypoxiCAR approach is a strategy to circumvent off-tumor activation, there are other hurdles that are known to hinder the effectiveness of CAR immunotherapy for treating solid malignancies. These include a lack of cell trafficking and entry to the tumor and the immune-suppressive TME, which may require further consideration to achieve the maximal efficacy of the approach for translation. However, some of these hurdles may be addressed by evaluating HypoxiCAR T cells with combination therapy strategies, such as alongside immune checkpoint blockade or even radiotherapy, where the latter could potentially preferentially target more oxygenated regions of the tumor. The long-term effect of HypoxiCAR T cells on healthy organs where regions of HIF-1α stabilization have been described, and also pathophysiological conditions such as ischemia, which would represent environments where off-tumor activation may be possible, also require careful investigation for translation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-human CD3ε Brilliant Violet 421 (clone SK7) | Biolegend | Cat# 344834; RRID:AB_2565675 |
| Anti-human CD8α Alexa Fluor 488 (clone RPA-T8) | Biolegend | Cat# 301021; RRID:AB_2561281 |
| Anti-human CD4 PE (clone RPA-T4) | eBioscience | Cat# 12-0049-42, RRID:AB_1582249 |
| Anti-human CD45 Brilliant Violet 510 (clone HI30) | Biolegend | Cat# 304036; RRID:AB_2561940 |
| Anti-mouse CD16/32 (clone 2.4G2) | Biolegend | Cat# 101301; RRID:AB_312800 |
| Anti-human/primate EGF biotinylated | R&D Systems | Cat# BAF236; RRID:AB_356307 |
| Streptavidin APC | Biolegend | Cat# 405207 |
| Anti-human ErbB1 (ICR62) | Institute of Cancer Research | N/A |
| Anti-human ErbB2 (ICR12) | Institute of Cancer Research | N/A |
| Anti-human ErbB3/HER-3 PE (clone 1B4C3) | Biolegend | Cat# 324706; RRID:AB_2099569 |
| Anti-human ErbB4/Her4 (clone H4.77.16 (Ab77)) | Novus | Cat# NB120-3104; RRID:AB_789269 |
| Goat anti-mouse IgG (H+L) highly cross-adsorbed secondary Alexa Fluor Plus 488 | Thermo Fisher Scientific | Cat# A32723; RRID:AB_2633275 |
| Goat anti-rat IgG (minimal x-reactivity) APC | Biolegend | Cat# 405407; RRID:AB_315018 |
| 7-amino actinomycin D | Cayman Chemical Company | Cat# 11397 |
| Anti-mouse TER-119 PerCP-Cyanine5.5 (clone TER-119) | Thermo Fisher Scientific | Cat# 45-5921-82; RRID:AB_925765 |
| Anti-Hypoxyprobe antibody, PAb2627 | Hypoxyprobe | Cat# HP PAb2627 |
| Anti-human CD3 (clone F7.2.38) | Dako | Cat# M7254 |
| Rabbit anti-HIF1α monoclonal (clone EP1215Y) | Abcam | Cat# ab51608; RRID:AB_880418 |
| Donkey anti-mouse IgG (H+L) Alexa Fluor Plus 488 | Thermo Fisher Scientific | Cat# A32766; RRID:AB_2762823 |
| Donkey anti-rabbit IgG (H+L) Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A10042; RRID:AB_2534017 |
| Anti-mouse CD4 FITC (clone RM4-5) | eBioscience | Cat# 11-0042-82; RRID:AB_464896 |
| Anti-mouse CD8α eFluor 450 (clone 53-6.7) | eBioscience | Cat# 48-0081-82; RRID:AB_1272198 |
| Anti-mouse CD3ε PE (clone 145-2C11) | eBioscience | Cat# 12-0031-82; RRID:AB_465496 |
| Bacterial and virus strains | ||
| Stbl3 E. coli | Thermo Fisher Scientific | Cat# C737303 |
| Biological samples | ||
| Human blood/T cells | Healthy volunteers | Guy’s and St Thomas’ Research Ethics Committee (REC reference 09/H0804/92) |
| Chemicals, peptides, and recombinant proteins | ||
| Ficoll-Paque PLUS | GE Healthcare | Cat# GE17-1440-02 |
| T4 DNA ligase | Thermo Fisher Scientific | Cat# EL0011 |
| RetroNectin recombinant human fibronectin fragment | Takara | Cat# T100B |
| Dynabeads human T-activator CD3/CD28 | Thermo Fisher Scientific | Cat# 11131D |
| Restriction endonucleases | New England Biolabs | Various, as per this paper |
| FuGENE HD transfection reagent | Promega | Cat# E2311 |
| Recombinant human IL-4 | PeproTech | Cat# 200-04 |
| Proleukin (aldesleukin), human recombinant IL-2 | Clinigen Group | N/A |
| Polybrene | Santa Cruz Biotechnology | Cat# NC9840454 |
| KiCqStart SYBR green qPCR ReadyMix, with ROX | Sigma-Aldrich | Cat# KCQS02 |
| TRIzol reagent | Thermo Fisher Scientific | Cat# 15596026 |
| Cobalt(II) chloride | Sigma-Aldrich | Cat# 60818 |
| XenoLight D-Luciferin - K+ salt bioluminescent substrate | PerkinElmer | Cat# 122799 |
| RediJect coelenterazine h bioluminescent substrate | PerkinElmer | Cat# 760506 |
| Collagenase I from Clostridium Histolyticum | Sigma-Aldrich | Cat# C0130 |
| DNase I | AppliChem | Cat# A3778 |
| Hypoxyprobe 1 Kit Hpi | Hypoxyprobe | Cat# HP1-200Kit |
| Dako EnVision + system-HRP (DAB) | Agilent Technologies | Cat# K4010 |
| DAPI (4’,6-diamidino-2-phenylindole, dihydrochloride) | Thermo Fisher Scientific | Cat# D1306 |
| Critical commercial assays | ||
| TaqMan gene expression assay | Thermo Fisher Scientific | Cat# 4331182 |
| QIAGEN Plasmid Mini, Midi and Maxi Kits | QIAGEN | Cat# 12125, 12145, 12163 |
| QIAGEN DNeasy Blood & Tissue Kit | QIAGEN | Cat# 69506 |
| Pan T Cell Isolation Kit, human | Miltenyi Biotec | Cat# 130-096-535 |
| EXPRESS One-Step Superscript qRT-PCR Kit | Thermo Fisher Scientific | Cat# 11781200 |
| Human IL-2 ELISA Ready-SET-Go! Kit, 2. Generation | eBioscience | Cat# 15590997 |
| Human IFN-Gamma DuoSet ELISA | R&D Systems | Cat# DY285B |
| UltraView Universal DAB Detection Kit | Roche | Cat# 760-500; RRID:AB_2753116 |
| UltraView Universal Alkaline Phosphatase Red Detection Kit | Roche | Cat# 760-501 |
| Experimental models: cell lines | ||
| SKOV3 | ATCC | Cat# HTB-77; RRID:CVCL_0532 |
| HN3 | Ludwig Institute for Cancer Research, London | RRID:CVCL_8126 |
| HEK293T | ATCC | Cat# CRL-3216; RRID:CVCL_0063 |
| Experimental models: organisms/strains | ||
| NOD-scid IL2Rγnull (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) | Charles River | RRID:BCBC_4142 |
| Oligonucleotides | ||
| gBlocks gene fragments | Integrated DNA Technologies | Various, as per this paper |
| GCCTACCAAGAACAACTGGAC | Integrated DNA Technologies | Fwd binds upstream AgeI |
| GGCCCTGCCCCCTCGCGCCCCAGCCGCTGGA | Integrated DNA Technologies | Fwd to fuse CD3ζ-ODD |
| TCCAGCGGCTGGGGCGCGAGGGGGCAGGGCC | Integrated DNA Technologies | Rev to fuse CD3ζ-ODD |
| GACTAATCCGGATCCTCGAGTGGCTGTTACTGGA ATACTGTAACTGTGCTTTGAGG |
Integrated DNA Technologies | Rev to ODD stop XhoI |
| CCATGGTGAAGCGTGAGAAAAATG | Integrated DNA Technologies | Fwd to amplify reporter |
| CTCGAGTTACTTGTACAGCTCGTCCATGC | Integrated DNA Technologies | Rev to amplify reporter |
| GAGAAGGCCGGCGGTGCCCCAGCCGCTGGA | Integrated DNA Technologies | Fwd to fuse Luc-ODD |
| TCCAGCGGCTGGGGCACCGCCGGCCTTCTC | Integrated DNA Technologies | Rev to fuse Luc-ODD |
| CCTCAAAGCACAGTTACAGTATTCCAGGGAAGCGG AGCTACTAACTTCAG |
Integrated DNA Technologies | Fwd to fuse Luc-ODD-2A |
| CTGAAGTTAGTAGCTCCGCTTCCCTGGAATACTGTA ACTGTGCTTTGAGG |
Integrated DNA Technologies | Rev to fuse Luc-ODD-2A |
| GCGGGCCTCTTCGCTATTA | Integrated DNA Technologies | Rev clone 9HRE in SFG |
| ATCCGCCACAACATCGAG | Integrated DNA Technologies | Fwd clone 9HRE in SFG |
| Recombinant DNA | ||
| SFG CBG99Luc-P2A-EGFP | This study | P1 |
| SFG HRE9 CBG99luc-ODD401-603-P2A-GFP | This study | P20; HypoxiLuc reporter |
| SFG 4αβ-2A-T1E-CD28-CD3z-ODD | This study | P22 |
| SFG HRE9 4αβ-2A-T1E-CD28-CD3z | This study | P26 |
| SFG HRE9 4αβ-2A-T1E-CD28-CD3z-ODD | This study | P23; HypoxiCAR |
| Software and algorithms | ||
| FlowJo v.10 Software | TreeStar Inc. | https://www.flowjo.com/ |
| Prism 6 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Snapgene | GSL Biotech | https://www.snapgene.com/ |
| NIS-Elements Imaging Software | Nikon | https://www.microscope.healthcare.nikon.com/products/software |
| R version 3.5.1 | The R Foundation | https://www.r-project.org/ |
| Other | ||
| Hypoxia incubator chamber | STEMCELL Technologies | Cat# 27310 |
| Gas cylinders | BOC | Custom (at indicated gas mixes) |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. James Arnold (james.n.arnold@kcl.ac.uk).
Materials availability
Constructs and other reagents generated in this study will be made available from the lead contact for academic/non-commercial research purposes on request without restriction under a Material Transfer Agreement. Commercial use of the constructs generated or derivatives would be subject to a licensing agreement as intellectual property rights are in place.
Data and code availability
The datasets supporting the current study have not been deposited in a public repository but are available from the corresponding author upon request.
Experimental model and subject details
Mice
NSG (NOD-scid IL2Rgammanull) mice were purchased from Charles River. Male NSG mice were used for studies involving HN3 (a human male cell line) and female NSG mice were used for studies involving SKOV3 (a human female cell line). All mice used for ectopic tumor studies were adults of 6-8 weeks of age and approximately 22 g in weight. Mice were maintained in individually ventilated cages in the King’s College London Biological Services Unit. The use of animals for this study was approved by the Ethical Review Committee at King’s College London and the Home Office, UK. Experiments using animals were performed under Home Office License P95C5B41D.
Cell lines
SKOV3 human (female) ovarian adenocarcinoma cells45 were originally purchased from ATCC and were re-authenticated for this study by ATCC. HN3 human (male) head and neck adenocarcinoma cells6,8 were acquired from Ludwig Institute for Cancer Research, London and grown in D10 medium, Dulbecco’s modified Eagle’s medium (DMEM; GIBCO) supplemented with 10% Fetal calf serum (FCS; Thermo Fisher Scientific) and GlutaMAX (Thermo Fisher Scientific). Cell lines were confirmed to be free of mycoplasma for this study using the MycoAlert® Mycoplasma Detection Kit (Lonza).
Primary human cells
Primary T cells were isolated from peripheral blood mononuclear cells (PBMCs) from fresh blood obtained as described below from healthy volunteers and cultured in RPMI 1640 supplemented with 5% human serum (Sigma-Aldrich) and 1X penicillin/streptomycin and the indicated cytokines. Donors were between 20-40 years of age with an equal ratio of male:female donors used. No age/gender-specific differences were observed. Blood was obtained from healthy volunteers under approval of the Guy’s and St Thomas’ Research Ethics Committee (REC reference 09/H0804/92).
Human tumor tissue
Human FFPE HNSCC tumor tissue was obtained with informed consent under ethical approval from the King’s Health Partners Head and Neck Cancer Biobank (REC reference 12/EE/0493) and stained for immunohistochemistry (IHC) and immunofluorescence analyses as described below. The patient demographic of HNSCC tissue included in this study was; total n = 92 / gender: female (F) n = 34, male (M) n = 58 / ages (years): 66 ± 11 (s.d.) / T-stage (T): T1 n = 18, T2 n = 16, T3 n = 17, T4 n = 41). Which was composed of the following patient demographic for the indicated tumor subtypes; hypopharynx (total n = 3 / gender: F n = 2, M n = 1 / ages (years): 59, 74,78 / T4 n = 3), larynx (total n = 20 / gender: F n = 0, M n = 20 / average age (years): 63 ± 10 (s.d.) / T1 n = 1, T3 n = 5, T4 n = 14), oral cavity (total n = 67 / gender: F n = 32, M n = 35 / average age (years): 67+/−11 (s.d.) / T1 n = 16, T2 n = 15, T3 = 12, T4 = 24), tonsil (total n = 2 / gender: F n = 1, M n = 1 / ages (years): 65,39 / T1 n = 1, T2 n = 1). No subtype-specific differences were observed.
Microbes
One Shot Stbl3 chemically competent E. coli (Thermo Fisher Scientific) were grown in Luria Bertani (LB) or LB Agar (Sigma-Aldrich) plates containing 100 μg/ml ampicillin (Santa Cruz Biotechnology) in a 37°C incubator.
Method details
CAR/Reporter construct cloning
Human T1E28z CAR (pan-ErbB-targeting CAR)-containing SFG retroviral vector6,8,46 was modified to generate the constructs utilized in this study. The full-length ODD cDNA encoding for amino acids 401-603 from human HIF1α was synthesized as a gBlock® (Integrated DNA Technologies) and was appended onto the C terminus of CD3ζ within the T1E28z CAR through overlap PCR using Platinum Pfx DNA polymerase (Thermo Fisher Scientific) according to the manufacturer’s instructions. The forward primer 5′-GCCTACCAAGAACAACTGGAC-3′ and reverse primer 5′-TCCAGCGGCTGGGGCGCGAGGGGGCAGGGCC-3′ were used to amplify 4αβ-T2A-T1E28z CAR while introducing ODD-compatible sticky ends. The ODD was amplified using forward primer 5′-GGCCCTGCCCCCTCGCGCCCCAGCCGCTGGA-3′ and reverse primer 5′-GACTAATCCGGATCCTCGAGTGGCTGTTACTGGAATACTGTAACTGTGCTTTGAGG-3′ which also introduced a CD3ζ-compatible sticky end. The PCR fragments were then fused using the forward primer 5′-GCCTACCAAGAACAACTGGAC-3′ and the reverse primer 5′-GACTAATCCGGATCCTCGAGTGGCTGTTACTGGAATACTGTAACTGTGCTTTGAGG-3′. PCR products were run on 1.2% Agarose (Sigma-Aldrich) gels and product size was estimated against a 1kb Plus DNA ladder (Thermo Fisher Scientific). Fragments of the expected size were excised and purified using the QIAquick® Gel Extraction kit. The 4αβ T2A T1E28z CAR-ODD sequence which contained flanking AgeI and XhoI cleavage sites was cloned into the SFG vector to replace the wild-type T1E28z CAR with the T1E28z ODD CAR. AgeI and XhoI restriction endonucleases (New England Biolabs) were used to cleave AgeI and XhoI restriction enzyme cleavage sites in the SFG plasmid to remove the existing 4αβ T2A CAR from the vector backbone. AgeI and XhoI restriction endonucleases were also used to cleave the AgeI and XhoI restriction enzyme sites which were built to flank the 4αβ T2A T1E28z CAR-ODD cDNA. Vector and constructs that had been restriction endonuclease-digested were agarose gel-purified using the QIAquick® Gel Extraction kit (QIAGEN) and ligated using T4 ligase (Thermo Fisher Scientific). The final ODD modified 4αβ T2A T1E28z CAR-ODD construct was 3212bp in length (including the 609bp ODD) from the start to the stop codon. Plasmids were transformed into One Shot Stbl3 chemically competent E. coli (Thermo Fisher Scientific). Transformed E. coli were selected using ampicillin (100μg/ml; Santa Cruz Biotechnology) containing Luria Bertani (LB) Agar (Sigma-Aldrich) plates. Transformed colonies were then grown up in LB broth (Sigma-Aldrich) with 100 μg/ml ampicillin and purified using either QIAGEN Plasmid Midi or Maxi kits. Final constructs were sequence verified (Source BioScience). The constitutively expressed reporter construct has previously been described47 and contained a Click Beetle Luciferase (Luc) and eGFP separated by a viral P2A sequence.48 The reporter construct was PCR amplified using Platinum Pfx DNA polymerase (Thermo Fisher Scientific) according to the manufacturer’s protocol with the forward primer 5′- CCATGGTGAAGCGTGAGAAAAATG-3′ and the reverse primer 5′- CTCGAGTTACTTGTACAGCTCGTCCATGC-3′. The amplified product was digested with NcoI and XhoI restriction endonuclease (New England Biolabs) and cloned into the SFG vector using the NcoI and XhoI restriction sites and T4 DNA ligase (Thermo Fisher Scientific). A full length ODD (as described above) was also appended onto the C terminus of Luc from the reporter construct by overlap PCR using the primers: forward 5′-GAGAAGGCCGGCGGTGCCCCAGCCGCTGGA-3′ and reverse 5′-CCTCAAAGCACAGTTACAGTATTCCAGGGAAGCGGAGCTACTAACTTCAG-3′ to amplify the ODD flanked with complementary overhangs. Subsequently, overlapping fusion PCR using primers: forward 5′-CCATGGTGAAGCGTGAGAAAAATG-3′ and reverse 5′-CTCGAGTTACTTGTACAGCTCGTCCATGC-3′ was performed to generate a fragment encoding Luciferase-ODD-P2A-eGFP flanked by NcoI and XhoI restriction sites, which were used to insert Luciferase-ODD-P2A-eGFP into the SFG vector. The HRE modification was targeted in the 3′ LTR of the SFG retroviral vector, as the 3′ LTR region is copied to the 5′ LTR upon integration.49 DNA containing 9 tandem 5′-GGCCCTACGTGCTGTCTCACACAGCCTGTCTGAC-3′ HRE motifs (total length 306bp) derived from the human EPO gene, containing both HIF-binding and ancillary sites, was synthesized as a gBlock® (Integrated DNA Technologies) and sub-cloned into the 3′ LTR of the SFG vector, to replace an equivalent sized fragment within the enhancer region, between the NheI and XbaI restriction endonuclease sites upstream of the native murine leukemia virus promoter. As NheI and XbaI were not unique restriction sites in the vector, to achieve specific modification of the 3′ LTR, we generated using overlapping fusion PCR a larger fragment flanked by XhoI/EcoRI unique restriction endonuclease sites. This fragment was identical to the vector DNA XhoI/EcoRI region except that the NheI/XbaI region was replaced by the 9 HRE gBlock®. The T1E28z CAR ODD CD3- truncated control construct (for CD3-truncated HypoxiCAR) was synthesized as gBlock® (Integrated DNA Technologies) with flanking SbfI and XhoI restriction sites and sub-cloned into the HRE-modified SFG vector using SbfI and XhoI restriction endonucleases (New England Biolabs). To generate the bicistronic Luciferase-T2A-CAR construct for in vivo tracking CAR T cells, a gBlock® (Integrated DNA Technologies), which was designed to include Luciferase-T2A-T1E peptide binder flanked with AgeI and NotI restriction sites, was inserted into the T1E28z CAR construct.
Human T cell isolation
Blood obtained from healthy volunteers, under approval of the Guy’s and St Thomas’ Research Ethics Committee (REC reference 09/H0804/92), was collected into Falcon tubes containing anti-coagulant (10% citrate), mixed at 1:1 with RPMI 1640 and layered over Ficoll-Paque Plus (GE Healthcare). Samples were centrifuged at 750 g for 30 mins at 20°C (acceleration and brake set to 0) to separate the PBMC cell fraction. The interface between the plasma and the Ficoll layer, which contained the PBMCs, was harvested using a sterile Pasteur pipette and washed in RPMI 1640. T cells were purified from the PBMC fraction using human Pan T cell isolation kit (Miltenyi Biotec) and isolated using a MidiMACs separator and LS columns (Miltenyi Biotec) according to the manufacturer’s protocol. Purified human T cells were activated using CD3/CD28 Human T-Activator Dynabeads (GIBCO) at a 1:1 cell to bead ratio and seeded in tissue culture plates at 3x106/ml in RPMI 1640 supplemented with 5% human serum (Sigma-Aldrich) and 1X penicillin/streptomycin. The following day, 100 IU/ml recombinant human IL-2 (PROLEUKIN) was added to the cultures.
Retroviral transduction
To produce retrovirus with tropism for human cells, RD114 pseudotyped retroviral particles were generated by triple transfection, using Peq-Pam plasmid (Moloney GagPol; a gift from Dr Martin Pule, UCL), RDF plasmid (RD114 envelope; a gift from Prof. Mary Collins, UCL) and the SFG plasmid of interest, using FuGENE HD transfection reagent (Promega), of HEK293T cells as previously described.41 For the in vivo experiments evaluating the therapeutic efficacy of CAR T cells in which the SKOV3 tumor cells expressed Click Beetle luciferase, T cells were tracked by co-transduction with a construct containing Renilla luciferase. Co-transduction was conducted using 1:1 ratios of virus containing a red-shifted Renilla reniformis luciferase 8.6-535 variant (rluc; Genscript),50 which contained the Renilla luciferase separated from eGFP by a furin-T2A sequence (rluc/eGFP),51 alongside the respective retroviral particles containing the indicated CAR constructs. Supernatants containing viral particles were harvested and incubated with the cells of interest for at least 48 h to allow their transduction. T cells were transduced in non-tissue culture treated plates that were pre-coated with 4 μg/cm2 RetroNectin (Takara Bio) overnight at 4°C. Prior to the retroviral transduction of human T cells, CD3/CD28 Human T-Activator Dynabeads (GIBCO) were removed and fresh IL-2 was added as stated in the T cell isolation section. In the case of T cell transduction with the bicistronic 4αβ-T2A-CAR construct, following T cell transduction, human IL-4 (Peprotech) at 30 ng/ml final concentration was added to the culture every 2-3 days to selectively enrich the transduced T cell population. Adherent cell lines, including SKOV3 and HN3, were transduced with retrovirus, produced as indicated before, in media solution containing Polybrene (Santa Cruz Biotechnology Inc) at 4μg/ml final concentration to increase infection efficiency. Cells modified to express Luc/eGFP were purified by cell sorting using BD FACSAria III (BD Biosciences) based on their eGFP fluorescence.
Quantitative PCR
Genomic DNA was extracted from cells using a DNeasy Blood & Tissue Kit (QIAGEN) according to manufacturer’s protocol and quantitative PCR was performed using KiCqStart SYBR Green qPCR ReadyMix with ROX (Sigma-Aldrich) according to the manufacturer’s protocol using custom designed primers to generate amplicons from Tbp, Luc or T2A sequences in the genome. The primers used were: human TBP 5′- TTTGGTGTTTGCTTCAGTCAG-3′ and 5′-ATACCTAGAAAACAGGAGTTGCTCA-3′, T2A 5′-CGGAGAAAGCGCAGC-3′ and 5′- GGGTCCGGGGTTCTCTT-3′. Amplification of the genes of interest was detected on an ABI 7900HT Fast Real Time PCR instrument (Thermo Fisher Scientific).
Quantitative reverse transcriptase PCR
mRNA was extracted using TRIzol (Thermo Fisher Scientific) method and quantitative reverse transcription (qRT) PCR was performed as previously described52 using the EXPRESS one-step Superscript RT PCR kit and the following primers/probes purchased from Thermo Fisher Scientific: Erbb1 Mm01187858_m1, Erbb2 Mm00658541_m1, Erbb3 Mm01159999_m1, Erbb4 Mm01256793_m1 and Tbp Mm01277045_m1. Expression of all genes is represented relative to the house-keeping gene Tata-binding protein (Tbp) for both human and murine experiments. Assays were performed using an ABI 7900HT Fast Real Time PCR instrument (Thermo Fisher Scientific).
In vitro studies
In vitro hypoxia was achieved using a hypoxia incubator chamber (StemCell Technologies, Inc.) purged at 25L/min for 4 mins with gas containing either; 0.1, 1 or 5% O2, 5% CO2 and nitrogen as a balance (BOC), after which the chamber was sealed. This process was repeated again after 1 h. Hypoxia-mediated HIF1α stabilization was, in some cases, mimicked by using the chemical CoCl2 (Sigma-Aldrich), which inhibits HIF1α hydroxylation, at 100μM final concentration, unless otherwise stated. In in vitro cytotoxicity assays 1x104 Luc/eGFP-expressing SKOV3 cells were seeded in 96-well tissue culture plates and transduced or non-transduced T cells were added in the well at the indicated effector to target ratios. Co-cultures were incubated for 24, 48 and 72 h time points in normoxia or experimental hypoxia as indicated and target cell viability was determined by luciferase quantification following the addition of 1μl of 15mg/ml XenoLight D-luciferin (PerkinElmer) in PBS per 100μl of media. Luminescence was quantified using a FLUOstar Omega plate reader (BMG Labtech). At the 24 and 48 h co-culture time points a sample of media was taken from the co-culture and subsequently used for IL-2 and IFN-γ quantification, respectively. IL-2 was quantified using Human IL-2 ELISA Ready-SET-Go! Kit, 2nd Generation (eBioscience) as per manufacturer’s protocol. IFN-γ was quantified using Human IFN-γ DuoSet ELISA kit (Bio-Techne) as per manufacturer’s protocol. In both ELISAs, cytokine concentration was determined by absorbance measurements at 450 nm on a Fusion alpha-FP spectrophotometer (Perkin-Elmer).
In vivo studies
Tumor cell lines (2.5x105 cells in PBS) were inoculated by subcutaneous (s.c.) injection into female (for SKOV3) and male (for HN3) mice that were six to eight weeks of age. Once tumors were palpable, digital caliper measurements of the long (L) and short (S) dimensions of the tumor were performed every 2 or 3 days. Tumor volume was established using the following equation: Volume = (S2xL)/2. The indicated doses of CAR T cells were injected in 200μl PBS through the tail vein using a 26 G needle. Where i.t. injection was used, cells were injected directly into the tumor in 50μl PBS. Blood samples were taken from mice in EDTA-coated Microvette tubes (Sarstedt) and plasma was extracted by centrifugation of these samples at 2,000 g for 5 mins. Cytokine concentrations were blindly measured externally by Abcam using the FirePlex Human Th1/Th2/Th17 and FirePlex Mouse Inflammation Immunoassay panels. After mice had been humanely sacrificed at the end of a study period, where tissues were excised they were immersed in excess formalin solution (10%) neutral buffered (Sigma-Aldrich), paraffin embedded, sectioned and stained with H&E using standard protocols. Blinded analysis of histopathology was performed by a FRCPath-qualified specialist in veterinary pathology. Scores were assigned for evidence of inflammation using a non-linear semiquantitative grading system from 0 to 5 where 0 = no significant change and 5 = whole organ or tissue affected for each observation. Tumor tissue, and other organs, for flow cytometry analyses were enzyme-digested to release single cells as previously described.41 In brief, tissues were minced using scalpels, and then single cells were liberated by incubation for 60 mins at 37°C with 1 mg/ml Collagenase I from Clostridium Histolyticum (Sigma-Aldrich) and 0.1 mg/ml Deoxyribonuclease I (AppliChem) in RPMI (GIBCO). Released cells were then passed through a 70 μm cell strainer prior to staining for flow cytometry analyses. Viable cells were numerated using a hemocytometer with trypan blue (Sigma-Aldrich) exclusion.
Bioluminescence Imaging
For assessing Luc bio-distribution in vivo mice were injected intraperitoneally (i.p.) with 200μl (15mg/ml) XenoLight D-luciferin (PerkinElmer) in sterile PBS 10 mins prior to imaging to detect Click Beetle Luciferase. Renilla reniformis Luciferase was detected using 100μl (150μg/ml) RediJect Coelenterazine Bioluminescent Substrate (PerkinElmer) immediately prior to imaging. Animals were anesthetized for imaging and emitted light was detected using the In vivo Imaging System (IVIS®) Lumina Series III (PerkinElmer) and data analyzed using the Living Image software (PerkinElmer). Light was quantified in photons/second/unit area.
Immunohistochemistry
To measure hypoxia within tissues, mice were injected i.p. with 60mg/kg pimonidazole HCl (Hypoxyprobe, HPI, Inc) dissolved in PBS, 2 h before sacrifice. Tissues were transferred into formalin solution (10%) neutral buffered (Sigma-Aldrich) for at least 24 h prior to paraffin embedding. Paraffin-embedded tissues were cut into 5 μm sections and mounted onto glass microscope slides (VWR). Sections were dewaxed in Histo-Clear (National Diagnostics) for 6 mins, prior to rehydration through 100%, 95%, 70% ethanol and tap water for 3 min each at room temperature (RT). Antigens were retrieved using Access Revelation (Biocore LLC) at 95°C for 20 mins. Sections were washed three times in 100 mM Tris, 140 mM NaCl, 0.1% Tween 20 pH7.4 (TBST) wash buffer for 10 mins prior to applying a wax circle. Tissue peroxidases were quenched using 0.3% H2O2 (Sigma-Aldrich) for 10 mins at RT. Sections were washed again in TBST prior to blocking with 10% goat serum (Sigma-Aldrich), 0.1% Triton X-100 in TBST for 1 h at RT. The stable protein adducts formed with the reductively activated pimonidazole in hypoxic tissue were detected using rabbit anti-pimonidazole antisera (1:100 Pab2627, Hypoxyprobe, HPI, Inc) O.N. at 4°C. Sections were washed with TBST as above and bound rabbit IgG was detected using Dako EnVision+ System-HRP (DAB) (K4010, Agilent Technologies) according to the manufacturer’s instructions. Tissue sections were counterstained with hematoxylin and washed clear with tap water prior to dehydration through 70%, 95%, 100% ethanol, and Histo-Clear (National Diagnostics) for 3 mins each prior to mounting with a coverslip using DePex (SERVA).
FFPE human HNSCC tumor tissues sections were deparaffinized and dual-antibody stained using a BenchMark ULTRA IHC/ISH system (Roche). Deparaffinized sections were pretreated with Cell Conditioning 1 (CC1) buffer (Roche) for 36 minutes and then incubated with mouse anti-human CD3ε (1:50 F7.2.38 Dako) for 32 mins at 37°C followed by amplification and detection using ultraView Universal DAB Detection Kit (Roche). Subsequently, tissues were incubated with Rabbit anti-human HIF1α (1:1000 EP1215Y, Abcam) for 32 mins at 37°C followed by amplification and detection using ultraView Universal Alkaline Phosphatase Red Detection Kit (Roche). Tissues were counterstained with hematoxylin and bluing reagent (Roche), dehydrated and mounted with a coverslip.
Images were acquired using a NanoZoomer Digital Slide Scanner (Hamamatsu) and IHC staining for stable protein adducts of hypoxyprobe and HIF1α were quantified using an H-score which represented the sum of the respective stain intensities from 0-3 (3 being highest), multiplied by the percent area that each intensity occupied across the tumor. Intra-epithelial T cells (IETs) were scored as low/absent if CD3+ cells were sparse or absent and ‘high’ if prevalent in the stromal regions surrounding the tumor. Tumor infiltrating lymphocytes (TILs) were scored as ‘present’ if there was > 3 areas across the section where CD3+ cells could be found within the tumor tissue.
Immunofluorescence
Sections from formalin fixed paraffin embedded (FFPE) human head and neck cancer, principally oral cavity (tongue) and tonsil, were de-paraffinized and antigen retrieved using a Ventana® BenchMark ULTRA (Roche Tissue Diagnostics). Immunofluorescence was performed as previously described.47 The following antibodies were used at 1:100 dilutions, mouse anti-CD3ε (F7.2.38, Dako) and Rabbit anti-HIF1α (EP12151, Abcam). Primary antibodies were detected using donkey IgG antibodies purchased from Thermo Fisher Scientific at 1:100: AlexaFluor® 488 anti-mouse IgG and AlexaFluor® 568 anti-rabbit IgG. Nuclei were stained using 1.25 μg/ml 4’,6-diamidino-2-phenylindole dihydrochloride (DAPI) (Thermo Fisher Scientific). Images were acquired using a Nikon Eclipse Ti-E Inverted Spinning Disk confocal microscope system and associated NIS Elements software. Co-localization of staining was evaluated using thresholding on the NIS Elements Software.
Flow cytometry
Flow cytometry was performed as previously described.53 The following antibodies were purchased from suppliers indicated in the Key Resources Table and were used at 1 μg/ml unless stated otherwise: anti-human CD3ε Brilliant Violet 421 (SK7; Biolegend®), anti-human CD8α Alexa Fluor 488 (RPA-T8), anti-human CD4 PE (RPA-T4), anti-human CD45 Brilliant Violet 510 (HI30 Biolegend®), anti-mouse CD4 FITC (RM4-5), anti-mouse CD8α eFluor®450 (53-6.7), anti-mouse CD3ε PE (145-2C11), neutralizing anti-mouse CD16/CD32 (2.4G2). Background fluorescence was established using fluorescence minus one staining. The T1E28z CAR was stained with a biotinylated anti-human EGF antibody (Bio-Techne: BAF236) and detected using Streptavidin APC. Human ErbB family members were detected using anti-ErbB1 (ICR62), anti-ErbB2 (ICR12; both ICR antibodies were gifts of Professor Suzanne Eccles, Institute of Cancer Research, Sutton), anti-ErbB3 PE (BioLegend®, 1B4C3), anti-ErbB4 (NOVUS Biologicals, H4.77.16). Antibodies that there not conjugated to a fluorochrome were detected using goat anti-Mouse IgG (H+L) highly cross-adsorbed secondary conjugated to Alexa Fluor Plus 488 (Thermo Fisher Scientific) or goat anti-rat IgG APC (BioLegend®) as appropriate. eGFP was detected by its native fluorescence. Dead cells and red blood cells were excluded using 1 μg/ml 7-amino actinomycin D (Cayman Chemical Company) alongside anti-Ter-119 PerCP-Cy5.5 (Ter-119; Thermo Fisher Scientific). Data were collected on a BD FACS Canto II (BD Biosciences). Data was analyzed using FlowJo software (Freestar Inc.).
Quantification and statistical analysis
Computational analysis of cancer patient data
RSEM normalized expression datasets from the Cancer Genome Atlas (TCGA) were downloaded from the Broad Institute Firehose resource (https://gdac.broadinstitute.org/). The HRE-regulated gene expression signature was generated by taking the mean normalized log2-transformed expression value of the component signature genes. The HRE-regulated gene signature was comprised of HRE-regulated genes for which a positive correlation was observed between all genes. As VEGFA and ENO1 did not correlate with each other (Figure S5A), we elected to include VEGFA over ENO1 in the signature as, by comparison, VEGFA provided a greater sensitivity for tumor T-stage and patient prognosis. This final signature included genes associated with glucose metabolism (ALDOA, PGK1, SLC2A1), pH regulation (CA9) and angiogenesis (VEGFA). All TCGA data was analyzed using R version 3.5.1 (https://www.r-project.org/). Overall survival analyses were generated by partitioning all HNSCC patients into quartiles based on hypoxia score and taking the top and bottom quartile expression ranked hypoxia score values. Kaplan-Meier survival curves were plotted using GraphPad Prism (GraphPad).
Statistics
Normality and homogeneity of variance were determined using a Shapiro-Wilk normality test and an F-test, respectively. Statistical significance was then determined using a two-sided unpaired Students t test for parametric or Mann-Whitney test for nonparametric data using GraphPad Prism 6 software. When comparing paired data, a paired ratio Students t test was performed. A Welch’s correction was applied when comparing groups with unequal variances. Statistical analysis of in vitro killing curves was performed using repeated-measures ANOVA followed by Tukey’s post hoc tests. Homoscedasticity of residual variance and normality assumptions were met. Statistical analysis of tumor growth curves was performed as described.54 Correlation analyses were performed using Pearson correlation. The pairwise Wilcoxon Rank Sum Test with Benjamini-Hochberg correction was used to measure statistical differences between clinical groups and hypoxia score in cancer patient data from the TCGA. The log-rank (Mantel-Cox) test was used to determine statistical significance for overall survival in cancer patient data from TCGA. No outliers were excluded from any data presented.
Acknowledgments
The authors thank Dr. Yasmin Haque, King’s College London, for cell sorting and flow cytometry assistance; Dr. Adam Ajina (KCL) for taking blood samples; Miss Dominika Sosnowska for help with tail vein injections; and Dr. Gilbert Fruhwirth for helpful discussions. This work was funded by the European Research Council (335326) and King’s Commercialisation Institute. P.K. and J.W.O. are supported by the UK Medical Research Council (MR/N013700/1) and are KCL members of the MRC Doctoral Training Partnership in Biomedical Sciences. K.I.L.-M. is supported by the Consejo Nacional de Ciencia y Tecnología (CONACyT) and Leucid Bio. The research was supported by the Cancer Research UK King’s Health Partners Centre and Experimental Cancer Medicine Centre at King’s College London and the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy's and St Thomas' NHS Foundation Trust and King’s College London. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health.
Author contributions
P.K., J.M., and J.N.A. conceived the project, designed the approach, interpreted the data, and wrote the manuscript. P.K., J.W.O., K.I.L.-M., R.H.-S., C.L.S., M.O., M.Y.M.T., T.M., and D.L.-Y. performed experiments and interpreted the data. D.M.D., N.W., C.E.G., and S.T. provided key expertise and interpretation.
Declarations of interest
J.M. is co-founder and chief scientific officer, T.M. is an employee, and D.M.D. and D.L.-Y. are consultants of Leucid Bio, a spinout company focused on development of cellular therapeutic agents. J.N.A., J.M., and P.K. are named inventors on a patent submitted in relation to this work.
Published: April 9, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100227.
Supplemental information
References
- 1.Johnson L.A., June C.H. Driving gene-engineered T cell immunotherapy of cancer. Cell Res. 2017;27:38–58. doi: 10.1038/cr.2016.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kosti P., Maher J., Arnold J.N. Perspectives on Chimeric Antigen Receptor T-Cell Immunotherapy for Solid Tumors. Front. Immunol. 2018;9:1104. doi: 10.3389/fimmu.2018.01104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morgan R.A., Yang J.C., Kitano M., Dudley M.E., Laurencot C.M., Rosenberg S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holbro T., Hynes N.E. ErbB receptors: directing key signaling networks throughout life. Annu. Rev. Pharmacol. Toxicol. 2004;44:195–217. doi: 10.1146/annurev.pharmtox.44.101802.121440. [DOI] [PubMed] [Google Scholar]
- 5.Karachaliou N., Lazzari C., Verlicchi A., Sosa A.E., Rosell R. HER3 as a Therapeutic Target in Cancer. BioDrugs. 2017;31:63–73. doi: 10.1007/s40259-016-0205-2. [DOI] [PubMed] [Google Scholar]
- 6.Davies D.M., Foster J., Van Der Stegen S.J., Parente-Pereira A.C., Chiapero-Stanke L., Delinassios G.J., Burbridge S.E., Kao V., Liu Z., Bosshard-Carter L. Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Mol. Med. 2012;18:565–576. doi: 10.2119/molmed.2011.00493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilkie S., Burbridge S.E., Chiapero-Stanke L., Pereira A.C., Cleary S., van der Stegen S.J., Spicer J.F., Davies D.M., Maher J. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J. Biol. Chem. 2010;285:25538–25544. doi: 10.1074/jbc.M110.127951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Stegen S.J., Davies D.M., Wilkie S., Foster J., Sosabowski J.K., Burnet J., Whilding L.M., Petrovic R.M., Ghaem-Maghami S., Mather S. Preclinical in vivo modeling of cytokine release syndrome induced by ErbB-retargeted human T cells: identifying a window of therapeutic opportunity? J. Immunol. 2013;191:4589–4598. doi: 10.4049/jimmunol.1301523. [DOI] [PubMed] [Google Scholar]
- 9.Klampatsa A., Achkova D.Y., Davies D.M., Parente-Pereira A.C., Woodman N., Rosekilly J., Osborne G., Thayaparan T., Bille A., Sheaf M. Intracavitary ‘T4 immunotherapy’ of malignant mesothelioma using pan-ErbB re-targeted CAR T-cells. Cancer Lett. 2017;393:52–59. doi: 10.1016/j.canlet.2017.02.015. [DOI] [PubMed] [Google Scholar]
- 10.Papa S., Adami A., Metoudi M., Achkova D., van Schalkwyk M., Parente Pereira A., Bosshard-Carter L., Whilding L., van der Stegen S., Davies D. A phase I trial of T4 CAR T-cell immunotherapy in head and neck squamous cancer (HNSCC) J. Clin. Oncol. 2018;36 3046–3046. [Google Scholar]
- 11.Muz B., de la Puente P., Azab F., Azab A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl.) 2015;3:83–92. doi: 10.2147/HP.S93413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nordsmark M., Bentzen S.M., Rudat V., Brizel D., Lartigau E., Stadler P., Becker A., Adam M., Molls M., Dunst J. Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. An international multi-center study. Radiother. Oncol. 2005;77:18–24. doi: 10.1016/j.radonc.2005.06.038. [DOI] [PubMed] [Google Scholar]
- 13.Guo Q., Lu L., Liao Y., Wang X., Zhang Y., Liu Y., Huang S., Sun H., Li Z., Zhao L. Influence of c-Src on hypoxic resistance to paclitaxel in human ovarian cancer cells and reversal of FV-429. Cell Death Dis. 2018;8:e3178. doi: 10.1038/cddis.2017.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Overgaard J. Hypoxic modification of radiotherapy in squamous cell carcinoma of the head and neck--a systematic review and meta-analysis. Radiother. Oncol. 2011;100:22–32. doi: 10.1016/j.radonc.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 15.Barsoum I.B., Koti M., Siemens D.R., Graham C.H. Mechanisms of hypoxia-mediated immune escape in cancer. Cancer Res. 2014;74:7185–7190. doi: 10.1158/0008-5472.CAN-14-2598. [DOI] [PubMed] [Google Scholar]
- 16.Semenza G.L., Wang G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992;12:5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Juillerat A., Marechal A., Filhol J.M., Valogne Y., Valton J., Duclert A., Duchateau P., Poirot L. An oxygen sensitive self-decision making engineered CAR T-cell. Sci. Rep. 2017;7:39833. doi: 10.1038/srep39833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang L.E., Gu J., Schau M., Bunn H.F. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaakkola P., Mole D.R., Tian Y.M., Wilson M.I., Gielbert J., Gaskell S.J., von Kriegsheim A., Hebestreit H.F., Mukherji M., Schofield C.J. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 20.Harada H., Hiraoka M., Kizaka-Kondoh S. Antitumor effect of TAT-oxygen-dependent degradation-caspase-3 fusion protein specifically stabilized and activated in hypoxic tumor cells. Cancer Res. 2002;62:2013–2018. [PubMed] [Google Scholar]
- 21.Liu Y., Cox S.R., Morita T., Kourembanas S. Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells. Identification of a 5′ enhancer. Circ. Res. 1995;77:638–643. doi: 10.1161/01.res.77.3.638. [DOI] [PubMed] [Google Scholar]
- 22.Bhat P., Leggatt G., Waterhouse N., Frazer I.H. Interferon-γ derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis. 2017;8:e2836. doi: 10.1038/cddis.2017.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rollings C.M., Sinclair L.V., Brady H.J.M., Cantrell D.A., Ross S.H. Interleukin-2 shapes the cytotoxic T cell proteome and immune environment-sensing programs. Sci. Signal. 2018;11:eaap8112. doi: 10.1126/scisignal.aap8112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bredell M.G., Ernst J., El-Kochairi I., Dahlem Y., Ikenberg K., Schumann D.M. Current relevance of hypoxia in head and neck cancer. Oncotarget. 2016;7:50781–50804. doi: 10.18632/oncotarget.9549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suh Y.E., Lawler K., Henley-Smith R., Pike L., Leek R., Barrington S., Odell E.W., Ng T., Pezzella F., Guerrero-Urbano T., Tavassoli M. Association between hypoxic volume and underlying hypoxia-induced gene expression in oropharyngeal squamous cell carcinoma. Br. J. Cancer. 2017;116:1057–1064. doi: 10.1038/bjc.2017.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toustrup K., Sørensen B.S., Nordsmark M., Busk M., Wiuf C., Alsner J., Overgaard J. Development of a hypoxia gene expression classifier with predictive impact for hypoxic modification of radiotherapy in head and neck cancer. Cancer Res. 2011;71:5923–5931. doi: 10.1158/0008-5472.CAN-11-1182. [DOI] [PubMed] [Google Scholar]
- 27.Buffa F.M., Harris A.L., West C.M., Miller C.J. Large meta-analysis of multiple cancers reveals a common, compact and highly prognostic hypoxia metagene. Br. J. Cancer. 2010;102:428–435. doi: 10.1038/sj.bjc.6605450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaupel P., Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. 2007;26:225–239. doi: 10.1007/s10555-007-9055-1. [DOI] [PubMed] [Google Scholar]
- 29.Majmundar A.J., Wong W.J., Simon M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Evans S.M., Koch C.J. Prognostic significance of tumor oxygenation in humans. Cancer Lett. 2003;195:1–16. doi: 10.1016/s0304-3835(03)00012-0. [DOI] [PubMed] [Google Scholar]
- 31.Rafiq S., Purdon T.J., Daniyan A.F., Koneru M., Dao T., Liu C., Scheinberg D.A., Brentjens R.J. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms Tumor 1 antigen. Leukemia. 2017;31:1788–1797. doi: 10.1038/leu.2016.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roybal K.T., Rupp L.J., Morsut L., Walker W.J., McNally K.A., Park J.S., Lim W.A. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell. 2016;164:770–779. doi: 10.1016/j.cell.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilkie S., van Schalkwyk M.C., Hobbs S., Davies D.M., van der Stegen S.J., Pereira A.C., Burbridge S.E., Box C., Eccles S.A., Maher J. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J. Clin. Immunol. 2012;32:1059–1070. doi: 10.1007/s10875-012-9689-9. [DOI] [PubMed] [Google Scholar]
- 34.Grada Z., Hegde M., Byrd T., Shaffer D.R., Ghazi A., Brawley V.S., Corder A., Schönfeld K., Koch J., Dotti G. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol. Ther. Nucleic Acids. 2013;2:e105. doi: 10.1038/mtna.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caruso H.G., Hurton L.V., Najjar A., Rushworth D., Ang S., Olivares S., Mi T., Switzer K., Singh H., Huls H. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res. 2015;75:3505–3518. doi: 10.1158/0008-5472.CAN-15-0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Torcellan T., Hampton H.R., Bailey J., Tomura M., Brink R., Chtanova T. In vivo photolabeling of tumor-infiltrating cells reveals highly regulated egress of T-cell subsets from tumors. Proc. Natl. Acad. Sci. USA. 2017;114:5677–5682. doi: 10.1073/pnas.1618446114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boissonnas A., Fetler L., Zeelenberg I.S., Hugues S., Amigorena S. In vivo imaging of cytotoxic T cell infiltration and elimination of a solid tumor. J. Exp. Med. 2007;204:345–356. doi: 10.1084/jem.20061890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henze A.T., Mazzone M. The impact of hypoxia on tumor-associated macrophages. J. Clin. Invest. 2016;126:3672–3679. doi: 10.1172/JCI84427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noman M.Z., Desantis G., Janji B., Hasmim M., Karray S., Dessen P., Bronte V., Chouaib S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014;211:781–790. doi: 10.1084/jem.20131916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim M.C., Hwang S.H., Kim N.Y., Lee H.S., Ji S., Yang Y., Kim Y. Hypoxia promotes acquisition of aggressive phenotypes in human malignant mesothelioma. BMC Cancer. 2018;18:819. doi: 10.1186/s12885-018-4720-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kraman M., Bambrough P.J., Arnold J.N., Roberts E.W., Magiera L., Jones J.O., Gopinathan A., Tuveson D.A., Fearon D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330:827–830. doi: 10.1126/science.1195300. [DOI] [PubMed] [Google Scholar]
- 42.Nakagawa Y., Negishi Y., Shimizu M., Takahashi M., Ichikawa M., Takahashi H. Effects of extracellular pH and hypoxia on the function and development of antigen-specific cytotoxic T lymphocytes. Immunol. Lett. 2015;167:72–86. doi: 10.1016/j.imlet.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 43.Zheng L., Kelly C.J., Colgan S.P. Physiologic hypoxia and oxygen homeostasis in the healthy intestine. A Review in the Theme: Cellular Responses to Hypoxia. Am. J. Physiol. Cell Physiol. 2015;309:C350–C360. doi: 10.1152/ajpcell.00191.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Di Stasi A., Tey S.K., Dotti G., Fujita Y., Kennedy-Nasser A., Martinez C., Straathof K., Liu E., Durett A.G., Grilley B. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morimoto H., Safrit J.T., Bonavida B. Synergistic effect of tumor necrosis factor-alpha- and diphtheria toxin-mediated cytotoxicity in sensitive and resistant human ovarian tumor cell lines. J. Immunol. 1991;147:2609–2616. [PubMed] [Google Scholar]
- 46.Maher J., Brentjens R.J., Gunset G., Rivière I., Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta /CD28 receptor. Nat. Biotechnol. 2002;20:70–75. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 47.Muliaditan T., Caron J., Okesola M., Opzoomer J.W., Kosti P., Georgouli M., Gordon P., Lall S., Kuzeva D.M., Pedro L. Macrophages are exploited from an innate wound healing response to facilitate cancer metastasis. Nat. Commun. 2018;9:2951. doi: 10.1038/s41467-018-05346-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tiffen J.C., Bailey C.G., Ng C., Rasko J.E., Holst J. Luciferase expression and bioluminescence does not affect tumor cell growth in vitro or in vivo. Mol. Cancer. 2010;9:299. doi: 10.1186/1476-4598-9-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Levin J.G., Mitra M., Mascarenhas A., Musier-Forsyth K. Role of HIV-1 nucleocapsid protein in HIV-1 reverse transcription. RNA Biol. 2010;7:754–774. doi: 10.4161/rna.7.6.14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loening A.M., Wu A.M., Gambhir S.S. Red-shifted Renilla reniformis luciferase variants for imaging in living subjects. Nat. Methods. 2007;4:641–643. doi: 10.1038/nmeth1070. [DOI] [PubMed] [Google Scholar]
- 51.Whilding L.M., Parente-Pereira A.C., Zabinski T., Davies D.M., Petrovic R.M.G., Kao Y.V., Saxena S.A., Romain A., Costa-Guerra J.A., Violette S. Targeting of Aberrant alphavbeta6 Integrin Expression in Solid Tumors Using Chimeric Antigen Receptor-Engineered T Cells. Mol. Ther. 2017;25:2427. doi: 10.1016/j.ymthe.2017.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arnold J.N., Magiera L., Kraman M., Fearon D.T. Tumoral immune suppression by macrophages expressing fibroblast activation protein-α and heme oxygenase-1. Cancer Immunol. Res. 2014;2:121–126. doi: 10.1158/2326-6066.CIR-13-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muliaditan T., Opzoomer J.W., Caron J., Okesola M., Kosti P., Lall S., Van Hemelrijck M., Dazzi F., Tutt A., Grigoriadis A. Repurposing Tin Mesoporphyrin as an Immune Checkpoint Inhibitor Shows Therapeutic Efficacy in Preclinical Models of Cancer. Clin. Cancer Res. 2018;24:1617–1628. doi: 10.1158/1078-0432.CCR-17-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Laajala T.D., Corander J., Saarinen N.M., Makela K., Savolainen S., Suominen M.I., Alhoniemi E., Makela S., Poutanen M., Aittokallio T. Improved statistical modeling of tumor growth and treatment effect in preclinical animal studies with highly heterogeneous responses in vivo. Clin. Cancer Res. 2012;18:4385–4396. doi: 10.1158/1078-0432.CCR-11-3215. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the current study have not been deposited in a public repository but are available from the corresponding author upon request.