

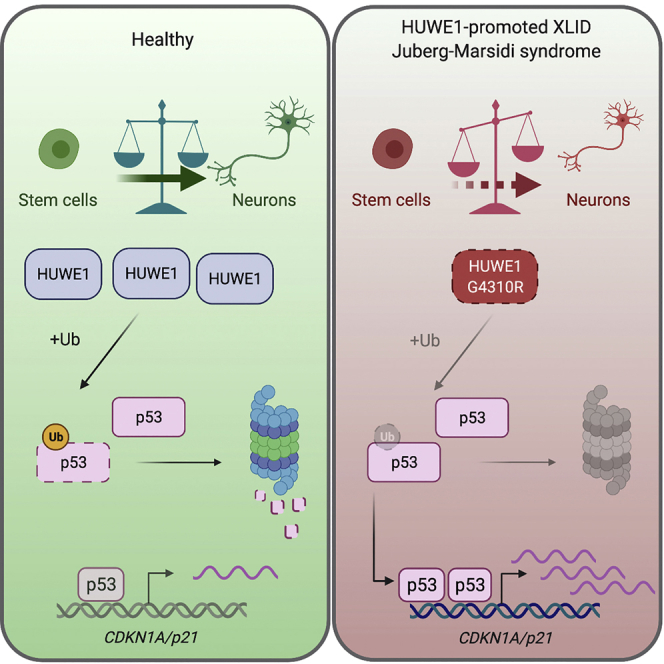
Summary
Essential E3 ubiquitin ligase HUWE1 (HECT, UBA, and WWE domain containing 1) regulates key factors, such as p53. Although mutations in HUWE1 cause heterogenous neurodevelopmental X-linked intellectual disabilities (XLIDs), the disease mechanisms common to these syndromes remain unknown. In this work, we identify p53 signaling as the central process altered in HUWE1-promoted XLID syndromes. By focusing on Juberg-Marsidi syndrome (JMS), one of the severest XLIDs, we show that increased p53 signaling results from p53 accumulation caused by HUWE1 p.G4310R destabilization. This further alters cell-cycle progression and proliferation in JMS cells. Modeling of JMS neurodevelopment reveals majorly impaired neural differentiation accompanied by increased p53 signaling. The neural differentiation defects can be successfully rescued by reducing p53 levels and restoring the expression of p53 target genes, in particular CDKN1A/p21. In summary, our findings suggest that increased p53 signaling underlies HUWE1-promoted syndromes and impairs XLID JMS neural differentiation.
Keywords: X-linked intellectual disability, E3 ubiquitin ligase, HUWE1, p53, neurodevelopment
Graphical abstract
Highlights
p53 signaling is hyperactivated in HUWE1-promoted intellectual disabilities
Neural differentiation is impaired in Juberg-Marsidi syndrome with HUWE1 p.G4310R
Juberg-Marsidi brain organoids have reduced size and aberrant cellular organization
Restored p53 signaling rescues neural development in Juberg-Marsidi syndrome
Using transcriptome analysis, Aprigliano et al. identify increased p53 signaling as the most significantly deregulated process in HUWE1-promoted XLIDs. Modeling of a severe HUWE1-promoted XLID, Juberg-Marsidi syndrome, reveals impaired neural differentiation capacity, which is successfully rescued by p53 knockdown and restored expression of p53 target genes, such as CDKN1A/p21.
Introduction
Neurodevelopment is a complex, finely regulated process in which ubiquitination plays a fundamental role by controlling key factors that act in cell proliferation, differentiation, death, and migration.1 The specificity of this posttranslational multistep mechanism is determined by E3 ubiquitin ligases that transfer ubiquitin to the substrate protein. Alterations in E3 ubiquitin ligases were associated with different neurological diseases. Mutations in E3 ligase HUWE1 (HECT, UBA, and WWE domain containing 1; also known as Mule, ARF-BP1, E3Histone, UREB1, HectH9, and LASU1) cause neurodevelopmental X-linked intellectual disability (XLID) syndromes.2, 3, 4, 5, 6, 7 HUWE1-promoted XLID syndromes are heterogenous, presenting with a spectrum of clinical findings that vary in severity and range from dysmorphic facial features and mild intellectual disability (ID) to extreme ID and early death. Juberg-Marsidi syndrome (JMS) is one of the severest forms of XLID, characterized by acute learning disability, generalized undergrowth, microcephaly, seizures, and reduced lifespan.5 We recently showed that XLID JMS is caused by p.G4310R HUWE1 mutation (Figure 1A) and is accompanied by p53 accumulation.4 If and to which extent p53 accumulation contributes to JMS and HUWE1-promoted XLID in general is unclear. Altogether, although different HUWE1 mutations clearly cause XLID syndromes, the underlying pathomechanisms through which HUWE1 mutations contribute to onset of XLIDs remain largely unknown.
Figure 1.

p53 signaling is hyperactivated in cells from XLID patients with mutated HUWE1
(A) Schematic representation of XLID-causative HUWE1 mutations analyzed in this study (p.R2981H, p.R4187C, and JMS-p.G4310R; HUWE1 duplication: 2×). Depicted HUWE1 domains: ARLD1/2, Armadillo repeat-like domains 1/2; UBA, ubiquitin-association domain; WWE, tryptophan-tryptophan-glutamate domain; BH3, Bcl-2 homology 3 domain; HECT, homologous to E6-AP carboxyl terminus domain.
(B) Top five most significant KEGG pathway terms as determined by gene set enrichment analysis (GSEA) of common differentially expressed genes in XLID patient-derived lymphoblastoid cells (LCs) compared with healthy individual cells (Benjamini corrected p < 0.05).
(C) Heatmap of common differentially expressed genes in XLID compared with healthy LCs belonging to the KEGG p53 signaling term.
(D–F) mRNA levels of p53 target genes CDKN1A/p21 (D), GADD45α (E), and BBC3/PUMA (F) determined by qRT-PCR analysis of four healthy (GM03798, GM07535, GM16113, and GM16119) and five XLID LCs with mutated HUWE1 (p.R2981H, p.R4187C, JMS-p.G4310R, and HUWE1 duplication).
All error bars indicate mean ± SEM (n = 3, biological replicates). Two-tailed unpaired t test; ∗∗p ≤ 0.01, ∗∗∗∗p ≤ 0.0001.
E3 ubiquitin ligase Huwe1 was demonstrated to have vital functions in neurodevelopment, and its loss leads to lethality in mice.8, 9, 10 Huwe1 affects laminar patterning by ensuring timely cell-cycle exit and differentiation of neural progenitor cells (NPCs) in the cerebral cortex and of neuronal and glia progenitors in the cerebellum.8,11,12 We suggested that similar to mouse progenitors, HUWE1 has an important role in human NPCs.13 HUWE1 mediates its vital roles by regulating activity and stability of key cellular factors, such as p53. In unstressed cells, HUWE1 was suggested to be one of the key E3 ligases responsible for p53 ubiquitination and degradation.14 Transcription factor p53 controls expression of genes involved in the cell cycle, apoptosis, and differentiation. Although p53 has been extensively studied as a tumor suppressor whose loss promotes cancer, several lines of evidence suggested that altered p53 activity contributes to developmental defects in different human genetic syndromes.15 By regulating the balance among apoptosis, proliferation, and differentiation, p53 is suggested to play an important role in brain organogenesis. Lack of p53 was shown to promote expansion of NPCs and alter their differentiation,16,17 whereas p53 increase in neurons triggered developmental programmed cell death.18, 19, 20 In addition, reduced p53 expression was suggested to impair cytoarchitecture in human brain organoids, resulting in a disorganized stem cell layer and reduced number of progenitor cells and neurons.21 Although both HUWE1 and p53 play an important role in regulation of proliferation and differentiation, the extent to which these two factors co-operate in ensuring unperturbed neurodevelopment remains largely unknown.
In this work, we showed that increased p53 signaling is the common feature of the HUWE1-promoted XLID syndromes. Comparison of transcriptomes from XLID and healthy age-matched control lymphoblastoid cells (LCs) identified a set of common differentially expressed genes (DEGs) belonging to the p53 signaling pathway. To explore the functional importance of deregulated p53 signaling, we focused on one of the severest XLID forms: JMS caused by HUWE1 p.G4310R. The p.G4310R mutation did not affect HUWE1 activity but instead reduced its stability, leading to p53 accumulation and consequently increased expression of p53 target genes. The increased p53 signaling in JMS patient-derived LCs resulted in impaired cell-cycle progression and significantly reduced proliferation. Interestingly, subsequent modeling of disease neurodevelopment, through human induced pluripotent stem cells (hiPSCs) derived from a JMS patient, revealed majorly impaired neural differentiation capacity. This was accompanied by increased p53 signaling in JMS neural cells. Defects in JMS neurodevelopment were further evident in altered cortical cytoarchitecture, resulting in a disorganized layer of proliferating NPCs and reduced JMS brain organoid size. The observed neural differentiation defects were directly caused by elevated p53 activity, because p53 knockdown and restored expression of p53 target genes, in particular of CDKN1A/p21, efficiently rescued neurodevelopmental potential of JMS hiPSCs. Altogether, our findings identify increased p53 signaling as the pathomechanism common to HUWE1-promoted XLIDs and suggest a crucial role for the HUWE1-p53 pathway during human neurodevelopment.
Results
Increased p53 signaling is a process commonly deregulated in XLID cells harboring different HUWE1 mutations
Mutations in HUWE1 cause heterogenous neurodevelopmental XLID syndromes. To determine the process that underlies XLIDs caused by different HUWE1 mutations, and because HUWE1 regulates multiple transcription factors important for neurodevelopment,10,22 we performed RNA sequencing analysis. The LC transcriptomes from five XLID patients harboring different HUWE1 mutations (Figure 1A) were compared with the transcriptomes from four control healthy age- and gender-matched LCs. This led to identification of 277 common DEGs in all XLID LCs, with a ≥1.5-fold change and a false discovery rate (FDR) ≤ 0.3. KEGG pathway analysis of the 277 DEGs revealed p53 signaling as the most significantly enriched pathway across the XLID LCs with different HUWE1 mutations (Figure 1B). Nearly all DEGs regulated by p53 were upregulated in XLID patient LCs (Figure 1C) with the exception of RRM2, a ribonucleotide reductase gene known to be suppressed by p53.23 To validate this, the expression of well-known p53 target genes BBC3/PUMA, GADD45α, and CDKN1A/p21, was analyzed by qRT-PCR. In line with the RNA sequencing results, these genes were significantly overexpressed in XLID LCs compared with healthy control cells (Figures 1D–1F). Altogether, our results suggest that increased p53 signaling and subsequent deregulation of p53 target genes are the common features of XLID cells with different HUWE1 mutations.
Deregulated expression of p53 target genes contributes to an altered cell cycle and reduced proliferation in HUWE1-promoted JMS
To explore the impact of increased p53 signaling (Figure 1) on cellular functioning, we focused on one of the severest XLID forms: JMS caused by mutated HUWE1 p.G4310R. We first confirmed increased p53 signaling by detecting significant overexpression of p53 targets BBC3/PUMA and GADD45a in LCs from two JMS patients compared with healthy controls (Figure S1). The subsequent immunoblot analysis of JMS LCs revealed a two-fold increase in p21 protein levels (Figure 2A), suggesting that increased p53 activity is directly reflected in the deregulated protein status of the p53 targets. In line with our previous work,4 HUWE1 p.G4310R levels were reduced, and p53 accumulated in JMS LCs (Figure 2A). To better understand how mutated HUWE1 p.G4310R causes p53 accumulation, we next compared the activities of wild-type (WT) and G4310R HUWE1 catalytic HECT domains. In vitro ubiquitination assays indicated that both HECT G4310R and HECT WT ubiquitinate p53 with similar efficiencies (Figure 2B), thus indicating that G4310R does not affect HUWE1 catalytic HECT domain activity. Because HUWE1 p.G4310R levels are reduced in JMS cells (Figure 2A),4 we next tested whether the mutation potentially affects HUWE1 stability by calculating change in the folding free energy (ΔΔG) between WT and G4310R proteins. Several tools (mCSM,24 SDM,25 DUET,26 iMutant2.0,27 and PoPMuSiC28) revealed negative ΔΔG, indicating that G4310R mutation destabilizes HUWE1 (Figure 2C). Altogether, these results suggest that JMS causing G4310R mutation negatively affects HUWE1 stability, which in turn promotes p53 accumulation and leads to increased signaling. On a functional level, increased p53 signaling was accompanied by impaired cell-cycle progression, with significant JMS LC accumulation in the G1 phase and reduction in the S phase of the cell cycle (Figure 2D). Besides cell-cycle impairments, a slight, although not significant, increase in the apoptosis rate measured by annexin V staining (Figure 2E) and significantly reduced proliferation (Figure 2F) were observed in the two JMS LCs. Altogether, these findings suggest that in differentiated JMS cells, such as LCs, increased p53 signaling results in impaired cell-cycle progression and reduced proliferation.
Figure 2.

p53 accumulation and activation, caused by HUWE1 p.G4310R, perturb the cell cycle and impair proliferation in JMS patient-derived cells
(A) Immunoblot analysis of the HUWE1, p53, and p21 protein levels in healthy control LCs and LCs from two JMS patients (JMS1 and JMS2). p53 protein levels relative to tubulin, serving as loading control, are indicated.
(B) In vitro ubiquitination of purified recombinant p53 with increasing amounts of wild-type (WT) and p.G4310R HECT proteins.
(C) In silico analysis of difference in the folding free energy change (ΔΔG) of WT and G4310R HUWE1 using the indicated prediction tools.
(D) Cell-cycle distribution determined by flow cytometry of healthy control, JMS1, and JMS2 LCs.
(E) Fraction of annexin V-positive apoptotic cells measured by flow cytometry.
(F) Proliferation rate of healthy control, JMS1, and JMS2 LCs.
All error bars indicate mean ± SEM (n ≥ 3, biological replicates). Statistical significance was determined by two-way ANOVA with Tukey post-test (D) and Bonferroni post-test (F); one-way ANOVA with Bonferroni post-test; ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001, n.s. ≥ 0.05. See also Figure S1.
Increased p53 signaling leads to impaired neural differentiation of JMS patient hiPSCs
To determine whether the elevated p53 signaling observed in Figure 1 affects JMS neurodevelopment, we modeled the disease in a set of neural induction experiments, using two independent JMS hiPSC clones (JMS-cl.1 and JMS-cl.2) generated by reprograming of fibroblasts from a JMS patient (III-2).4 Both JMS-cl.1 and JMS-cl.2 were characterized by unperturbed expression of pluripotency markers OCT4, SSEA, and SOX2 and embryoid body (EB) formation comparable to that of healthy control hiPSCs (WT-DYS0100 and WT-CRL(S)23) (Figures S2A–S2H). Furthermore, the germ layer markers GATA4 (endoderm), FOXC1 (mesoderm), and NES/NESTIN (ectoderm) were expressed at similar levels in JMS and healthy control EBs (Figures S2I–S2K). In summary, these results indicate the comparable pluripotent ability of JMS-cl.1, JMS-cl.2, and healthy control WT-DYS0100 and WT-CRL(S)23 hiPSCs. Further characterization of hiPSCs indicated that as in two JMS LC lines (Figure 2A), p53 accumulates in JMS hiPSC clones compared with controls (Figure S2L). Interestingly, despite increased p53 levels, no difference in cell-cycle progression was observed between control hiPSCs and JMS clones (Figure S2M).
To model JMS, we next performed neural induction experiments up to the stage of rosettes.29 Although both healthy control and JMS hiPSCs formed EBs with comparable capacities (Figure S2H), JMS-cl.1 and JMS-cl.2 hiPSCs failed to undergo neural differentiation (Figure 3A), forming significantly fewer neural rosettes compared with control WT-DYS0100 and WT-CRL(S)23 hiPSCs (Figure 3B). This observation was supported by the mRNA profiles, which upon neural induction revealed dramatically reduced expression of neural differentiation markers TUBB3/TUJ1 and DCX in JMS-cl.1 and JMS-cl.2 (Figures 3E and 3F). To a lesser extent, upon differentiation, the levels of proliferating NPC maker NES/NESTIN were also significantly reduced in the JMS background compared with healthy controls (Figure 3D). The levels of pluripotency marker OCT4 were comparable in JMS and healthy hiPSCs and as expected significantly reduced upon differentiation (Figure 3C). Similar to the changes on the mRNA level, TUJ1 staining was reduced and TUJ1-positive branches were dramatically altered in JMS-cl.1 and JMS-cl.2 rosettes compared with control samples (Figures 3G and 3H).
Figure 3.

Neural differentiation of JMS patient hiPSCs is impaired and accompanied by activation of p53 signaling
(A) Representative bright-field images of neural differentiation of healthy control hiPSCs (WT-DYS0100 and WT-CRL(S)23) and two hiPSC clones from a JMS patient expressing HUWE1 p.G4310R (JMS-cl.1 and JMS-cl.2) at day 11; neural rosette structures are visible.
(B) Relative number of rosettes formed in WT and JMS clones (n ≥ 3, biological replicates).
(C–F) qRT-PCR analysis of gene expression of OCT4 (C), NES/NESTIN (D), TUBB3/TUJ1 (E), and DCX (F) in WT and JMS hiPSCs and neural cells (collected at day 13) (n = 3, biological replicates).
(G) Immunofluorescence analysis of TUJ1 in WT and JMS hiPSCs at day 13 of neural differentiation.
(H) Relative intensity of the TUJ1 signal in WT and JMS clones from experiments like the one in (G) (n ≥ 2, biological replicates).
(I–L) qRT-PCR analysis of mRNA levels of the p53 target genes CDKN1A/p21 (I), GADD45α (J), BBC3/PUMA (K), and BAX (L) in WT and JMS hiPSCs and neural cells (collected at day 13) (n = 3, biological replicates).
All error bars indicate mean ± SEM; one-way ANOVA followed by Tukey post-test (B); two-way ANOVA followed by Tukey post-test (C–F and I–L); ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001, n.s. ≥ 0.05. Scale bar: 400 μm. See also Figures S2 and S3.
To determine the extent to which impairments in neural differentiation correlate with p53 signaling, the expression of several important p53 targets was analyzed. Although no differences were observed in the expression of CDKN1A/p21, GADD45α, BBC3/PUMA, and BAX in the hiPSCs, levels of all tested p53 targets were increased in JMS compared with healthy control cells (Figures 3I–3L), thereby indicating elevated p53 signaling upon neural induction of JMS hiPSCs. The most prominent and significantly increased p53 target gene was CDKN1A/p21 (Figure 3I). In summary, our results suggest that capacity to undergo neural differentiation is significantly reduced in the analyzed JMS patient hiPSCs and accompanied by elevated p53 signaling, whereas the stem cell identity remains unaltered.
JMS patient-specific cerebral organoids fail to fully develop
To assess the extent to which observed neural differentiation impairments influence neurogenesis, cerebral organoids were developed using WT-DYS0100 and JMS-cl.1 hiPSCs. Although similar to rosette development (Figure 3), no difference was observed at the stage of EB formation and growth (day 4), and major developmental failure occurred in JMS patient-specific cerebral organoids upon formation of neuroepithelial buds (day 14) (Figures S3A and S3B). Most JMS organoids failed to develop further, and only a few (1–2 of initial 96 in several independent experiments) passed day 35 of development. The JMS organoids that did develop were significantly smaller than the control organoids during days 14–60 (Figures S3A and S3B). Furthermore, the regions of cortical layering were dramatically reduced in the JMS patient-specific organoids and exhibited reduced cellularity and increased ventricle-like spaces (Figure S3C). The reduced cellularity was accompanied by visibly decreased and disorganized Ki67 staining, a marker of proliferation, in JMS organoids (Figures S3D and S3E), thus corresponding to the altered cortical layering observed in Figure S3C. Altogether, the reduced cellularity, altered layering, and decreased size observed in JMS patient-specific organoids closely recapitulate symptoms described in JMS patients, including microcephaly, enlarged ventricles, and reduced lifespan.4,5
p53 downregulation rescues neurodevelopmental defects in JMS patient hiPSCs
As shown in Figure 3, the impaired neural differentiation in JMS patient hiPSCs was accompanied by elevated p53 signaling. However, the extent to which elevated p53 levels directly cause JMS neurodevelopmental impairments remained unclear. To address this and test whether neurodevelopmental defects are a consequence of increased p53, we infected JMS-cl.1 and JMS-cl.2 hiPSCs with lentivirus encoding scrambled control short hairpin RNA (shRNA [shCtrl]), or three shRNAs targeting p53 (shp53a, shp53b, and shp53c) and confirmed knockdown efficiencies (Figures S4A and S5A). Importantly, p53 knockdown resulted in JMS neural rosette formation comparable to healthy controls, thus efficiently rescuing the impaired neural differentiation observed in JMS-cl.1, JMS-cl.1 shCtrl, JMS-cl.2, and JMS-cl.2 shCtrl (Figures 4A and S5B). Rescue of neural differentiation in JMS shp53 rosettes was accompanied by increased expression of maturation markers TUBB3/TUJ1 and DCX to a level significantly higher than in JMS and JMS shCtrl samples (Figures 4C, 4D, S5D, and S5E). In line with mRNA expression of maturation markers (Figures 4C and S5D), the TUJ1 signal and positive branches were restored in JMS-cl.1 and JMS-cl.2 cells upon p53 knockdown (Figures 4E, S4B, and S5I). In addition to the changes in maturation markers, p53 knockdown resulted in significantly reduced expression of p53 target CDKN1A/p21 (Figures 4F and S5F). Levels of GADD45α and BAX (Figures 4G and 4H) were also reduced; however, the reduction was not significant across JMS-cl.1 and JMS-cl.2. In summary, p53 knockdown in JMS-cl.1 and JMS-cl.2 hiPSCs resulted in significant rescue of the capacity to produce maturing neurons, thus suggesting that elevated p53 signaling is likely a predominant cause of impaired JMS neural differentiation.
Figure 4.

p53 downregulation rescues neurodevelopmental defects in XLID JMS patient hiPSCs
(A) Relative number of rosettes upon neural differentiation of WT-DYS0100, JMS-cl.1, and JMS-cl.1-expressing shRNA control (shCtrl) or shRNA targeting p53 (shP53a and shP53b) hiPSCs (n ≥ 3, biological replicates). JMS cells harbor HUWE1 p.G4310R.
(B–D) mRNA expression levels of NES/NESTIN (B), TUBB3/TUJ1 (C), and DCX (D) analyzed by qRT-PCR in WT and JMS neural cells (collected at day 13) addressed by qRT-PCR.
(E) Immunofluorescence analysis of the TUJ1 signal in WT-DYS0100, JMS-cl.1, JMS-cl.1 shCtrl, and JMS-cl.1 shP53a and shP53b at day 13 of neural differentiation.
(F–H) qRT-PCR analysis of CDKN1A/p21 (F), GADD45α (G), and BAX (H) expression in WT and JMS neural cells (collected at day 13).
All error bars indicate mean ± SEM (n = 3, biological replicates); one-way ANOVA followed by Dunnett’s post-test (A); one-tailed t test (B–D and F–H); ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, ∗∗∗∗p ≤ 0.0001, n.s. ≥ 0.05. See also Figures S4 and S5.
Discussion
HUWE1 is an essential E3 ubiquitin ligase that plays a decisive role in early stages of neurodevelopment.8,30 In humans, HUWE1 mutations cause XLID syndromes; however, the underlying pathomechanisms are still poorly understood. In this work, we demonstrate that p53 signaling is the process commonly altered in XLID cells with different HUWE1 mutations. Specifically, RNA sequencing performed on cells from five independent XLID patients with mutated HUWE1 identified p53 signaling as the most significantly altered pathway compared with the four healthy control cell lines (Figure 1). Analysis of expression changes in individual p53 target genes revealed increased p53 signaling in XLID cells. These findings support the hypothesis that p53 activation might be an important link among different genetic developmental disorders.15 Although augmented p53 signaling was indicated as the common feature of HUWE1-promoted XLIDs (Figure 1), to explore the importance of increased p53 signaling, we focused on JMS, one of the severest XLID forms. In addition to increased p53 signaling, JMS was characterized by reduced HUWE1 p.G4310R levels and p53 accumulation (Figure 2A).4 The p53 accumulation was likely a result not of altered HUWE1 p.G4310R activity (Figure 2B) but instead of reduced stability (Figure 2C). Besides p53 protein accumulation, specific patterns of posttranslational modifications, such as phosphorylation and acetylation, promote p53 signaling and the expression of p53 target genes.31 Although we did not detect a significant increase in p53 Serine 15 phosphorylation in JMS cells (data not shown), it will be interesting in the follow-up work to determine modifications and sites on p53 at which they occur to further explore p53 activity and regulation in JMS. By modeling JMS neurodevelopment with JMS patient-derived JMS-cl.1 and JMS-cl.2 hiPSCs, we show that increased p53 signaling significantly reduced the neural differentiation capacity of JMS stem cells, whereas the ability to form EBs remained unaffected (Figures 3 and S2). Interestingly, work focusing on an XLID-causing mutation in another E3 ubiquitin ligase, RNF12, indicated that similar to HUWE1 p.G4310R, loss of RNF12 function results in altered neural differentiation.32 The reduced neural differentiation capacity observed in the HUWE1-promoted JMS stem cells (Figure 3) is supported by studies in mice, which indicated the crucial importance of Huwe1 in the cell-cycle exit of NPCs and their subsequent neurogenic differentiation in both cerebral cortex11 and cerebellum.12 The impaired JMS neurogenic capacity was accompanied by increased levels of the p53 target genes CDKN1A/p21, GADD45α, BBC3/PUMA, and BAX at the final stages of neural differentiation, of which CDKN1A/p21 levels were most significantly affected (Figures 3I–3L). The impaired neural differentiation of JMS hiPSCs was reflected in majorly perturbed brain organogenesis, as demonstrated through the development of patient-specific JMS cerebral organoids (Figure S3). Only a few of the JMS organoids that developed had decreased size, which is in line with the microcephaly observed in JMS patients,4 and were characterized by reduced cellularity and lack of laminar patterning, features reported previously in Huwe1 mouse models.11,12 The reduced cellularity in JMS organoids (Figure S3) coincides with the apparent lowered DAPI intensity and reduced cell numbers in JMS samples undergoing neural differentiation (Figures 3 and 4). The observation that elevated p53 activity accompanies neurodevelopmental impairments in JMS is supported by studies showing that patients with germline TP53 mutations present with microcephaly,33 as well as that microcephaly and neurodevelopmental defects in several human neurodevelopmental disorders are p53 dependent.15 This supports the idea that deregulation of p53 activity could have central role in the onset of various neurological conditions. p53 knockdown in the hiPSCs from a JMS patient, and consequently restored expression of p53 targets, in particular of CDKN1A/p21, resulted in significant rescue of JMS capacity to form rosettes and the expression of neural maturation markers (Figures 4 and S5). Recent work similarly showed that p53 knockdown increases the pace of healthy neuroepithelial stem cell differentiation into neurons, which was accompanied by reduced CDKN1A/p21 expression.21 Furthermore, Huwe1 depletion in Drosophila results in impaired development, characterized by aberrant cell-cycle phasing and failure to enter the S phase; similar to our observations, all Huwe1 loss-of-function phenotypes were successfully suppressed by p53 knockdown.34 In mouse iPSCs, p53 accumulation was shown to cause impaired neural differentiation, which was successfully overcome by p53 inactivation.35 In summary, our findings suggest that augmented p53 signaling is the common process underlying HUWE1-promoted XLID syndromes. Our previous work showed that depending on the type of HUWE1 mutation, its stability and activity are differentially affected, which in turn can have specific impacts on p53 levels.4,13 In addition, because p53 regulation of target genes is strongly influenced by chromatin status and cell type36, 37, 38 and different HUWE1-promoted XLID syndromes present with unique symptoms, subsequent studies are needed to clarify how increased p53 signaling contributes to the different phenotypes.
Altogether, the findings presented in this study indicate that HUWE1 plays a vital role in regulation of p53 signaling during human neurodevelopment and suggest an important contribution of the HUWE1-p53 pathway in stem cell differentiation, an imbalance of which has the capacity to cause the onset of neurodevelopmental XLIDs.
Limitations of study
A frequent limitation in rare disease studies such as this one is a low number of participants and limited availability of samples from donors.39 This is a particular challenge in hiPSCs-based research, in which multiple patient samples are ideally reprogrammed and compared in parallel.39,40 Primarily, inter-individual heterogeneity across the genome, even in the individuals that carry identical pathogenic mutations, was reported to cause a certain level of hiPSC heterogeneity.40,41 Because the number of individuals with identical pathogenic mutations is frequently limited in rare disease studies, one important factor is the analysis of hiPSCs from patients exhibiting representative clinical phenotypes.40 The JMS analyzed in this work represents severe ID and dysmorphic features that are frequently observed across HUWE1-promoted XLIDs.5 However, because of limited material availability, all hiPSC clones analyzed in this study were from a single donor (Figures 3, 4, and S2–S5). Although analysis of the independent clones is of key importance to ensure the significance of impairments in neural differentiation capacities of hiPSCs from a JMS patient with HUWE1 p.G4310R (Figures 3, 4, and S2–S5), it does not account for the potential heterogeneity in differentiation of hiPSCs from different donors with the same genetic background. Parallel reprogramming of cells from different donors with HUWE1-promoted XLIDs would allow further delineation of the degree of effects caused by mutated HUWE1. An additional possible approach could be based on the recent advancements in genome-editing approaches and their increasing efficiencies to generate corrected, as well as isogenic, hiPSCs from well-characterized, preexisting hiPSC lines.
In summary, although the degree of heterogeneity in neural differentiation capacities of hiPSCs from different JMS individuals remains to be determined, the results of this work identified specific neurodevelopmental defects in cells with HUWE1 p.G4310R mutations and as such recapitulate important phenotypes observed in JMS patients.4
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| HUWE1 | Bethyl Laboratories | A300-486A; RRID:AB_2264590 |
| p53 | ThermoFisher Scientific | MA5-12571; RRID:AB_10986581 |
| p21 | ThermoFisher Scientific | 33-7000; RRID:AB_2533135 |
| Tubulin | Sigma-Aldrich | T9026; RRID:AB_477593 |
| β-Actin | Sigma-Aldrich | A1978; RRID:AB_476692 |
| Dye-conjugated secondary antibodies | Li-COR Bioscienecs | N/A |
| OCT4 | Cell signaling | C30A3; RRID:AB_2167691 |
| SSEA4 | Cell signaling | MC813; RRID:AB_1264259 |
| NESTIN | Abcam | ab22035; RRID:AB_446723 |
| TUJ1 | Covance | MMS-435P; RRID:AB_2313773 |
| Ki67 | Abcam | ab15580; RRID:AB_443209 |
| Donkey anti-Mouse, Alexa Fluor 488 | Invitrogen | A21202; RRID:AB_141607 |
| Goat anti-Rabbit, Alexa Fluor 594 | Invitrogen | A11037; RRID:AB_2534095 |
| Bacterial and virus strains | ||
| E.coli BL21-CodonPlus (DE) RP | Agilent | 230255 |
| Chemicals, peptides, and recombinant proteins | ||
| Halt Protease and Phosphatase Inhibitors Cocktail | Thermo Scientific | 78445 |
| Accutase | STEMCELL Technologies | 07922 |
| mTesR.1 medium | STEMCELL Technologies | 85850 |
| E8 medium | Thermo Scientific | A1517001 |
| DMEM/F12 medium | GIBCO | 11330057 |
| NEUROBASAL-A medium | GIBCO | 10888022 |
| Knock-out serum replacement (KSOR) | GIBCO | 10828-028 |
| Glutamax | Invitrogen | 35050-038 |
| MEM non-essential amino acids | Sigma | M7145 |
| Y27632 (Rock inhibitor) | Merck | 688000-100MG |
| Noggin | R & D Systems | 1967-NG-025 |
| 2-mercaptoethanol | GIBCO | 31350-010 |
| B27 supplement, 50x | GIBCO | 17504-044 |
| B27 supplement (50x), minus vitamin A | GIBCO | 12587-010 |
| N2 supplement (100x) | GIBCO | 175020-01 |
| Ascorbic acid | Merck | A8960-5G |
| BDNF | R&D Systems | 248-BDB-050 |
| P53 | Milipore | 23-034 |
| GDNF | R&D Systems | 212-GD-050 |
| dibutyryl-cAMP | Sigma | D0627-250MG |
| Critical commercial assays | ||
| SENSE mRNA-Seq library prep kit | Lexogen GmbH, Vienna, Austria | 001.96 |
| Mag-Bind RXNPure Plus beads | Omega Bio-tec, GA, USA | M1386-01 |
| KAPA Library Quantification Kit | Roche | 7960204001 |
| Agilent High Sensitivity DNA Kit on a Bioanalyzer | Agilent Technologies, CA, USA | 5067-4626 |
| cBot Cluster Generation System on HiSeq4000 flowcells | Illumina Inc., CA, USA | SY-301-2002 |
| Dead Cell Apoptosis Kit | Molecular Probes | V13241 |
| hPSC Genetic Analysis Kit | Stem Cell Technologies | 07550 |
| Deposited data | ||
| RNA sequencing of healthy control and patient LCs | GEO | GEO: GSE130551 |
| Experimental models: Cell lines | ||
| Healthy individual LCLs (GM03798, GM07535, GM16113, GM16119) | Coriell Cell Repository (Coriell Institute for Medical Research, USA) | N/A |
| XLID LCLs with HUWE1 duplication, or HUWE1 p.R2981H and p.R4187C mutations | Bosshard et al.13 | N/A |
| JMS1 and JMS2 and WT LCLs with mutation HUWE1 p.G4310R | Friez et al.4 | N/A |
| JMS1 patient-derived human induced pluripotent stem cells (hiPSCs) | Applied StemCell (California, USA) | N/A |
| Healthy control hiPSCs | ATCC, Manassas, USA CRL(S)23—in Siller et al.42 |
ATCC-DYS0100 |
| Oligonucleotides | ||
| Listed in Table S1. | N/A | |
| Recombinant DNA | ||
| shControl; pLKO.1 puro | Addgene | 8453 |
| shp53 pLKO.1 puro shRNA | Addgene | 19119 |
| shp53 pLKO.1 puro shRNA-427 | Addgene | 25636 |
| shp53 pLKO.1 puro shRNA-941 | Addgene | 25637 |
| pET28-HECT | Parsons et al.43 | N/A |
| Software and algorithms | ||
| bcl2fastq 2.20.0.422 | Illumina, Inc., CA, USA | N/A |
| STAR aligner | Dobin et al.44 | v2.4.0 |
| htseq-count | Anders et al.45 | v0.6.0 |
| limma voom | Ritchie et al.46 | N/A |
| DAVID (Database for Annotation, Visualization, and Integrated Discovery, v6.8) | Huang et al.47 | N/A |
| FlowJo™ | LLC software | v. 10.6.1 |
| Fiji ImageJ | National Institute of Health | v. 1.53c |
| iMaris | Bitplane | v. 8.2.1 |
| Other | ||
| RNeasy mini kit | QIAGEN | 74106 |
| MultiScribe Reverse Transcriptase | ThermoFisher Scientific | 4311235 |
| Power SYBR Green PCR Master Mix | Applied Biosystems | 43-676-59 |
| Illumina HiSeq4000 instrument | Illumina, Inc., CA, USA | N/A |
| BD FACSAria II | BD Biosciences | N/A |
| U-bottom ultra-low attachment 96-well plates | Corning | CLS7007 |
| Costar 24-well plates, flat bottom, ultra-low attachment | Corning | CLS3473-24EA |
| Matrigel | BD Biosciences | 356234 |
| Zeiss LSM 510 Meta live Confocal system | Zeiss | N/A |
| EVOS FL Auto Cell Imaging System | ThermoFisher Scientific | AMF5000 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Barbara van Loon (barbara.v.loon@ntnu.no).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
The data that support the findings of present study are available from the corresponding author on reasonable request. The RNA sequencing data reported in this paper are available in GEO under accession GEO: GSE130551. The study did not generate any new codes.
Experimental model and subject details
Cell lines
The XLID LCs with HUWE1 duplication, HUWE1 p.R2981H or HUWE1 p.R4187C mutation were used as described previously.3 The JMS1 and JMS2 LCs with mutated HUWE1 p.G4310R were obtained from the individuals III-2 and IV-4, and WT LCs from healthy relative IV-1, from the original family reported by Juberg and Marsidi48 and described by Friez et al.4 Informed consent was obtained from participants enrolled in the study of X-linked Intellectual Disability according to the regulations of the Institutional Review Board of Self Regional Healthcare and Greenwood Genetic Centre, South Carolina, USA. In addition to the five XLID patient LCs, four healthy gender- and age- matched LCs (GM03798, GM07535, GM16113, GM16119) were obtained from Coriell Cell Repository (Coriell Institute for Medical Research, USA). All LCs were maintained in RPMI 1640 (Corning) with 15% FBS (ThermoFisher Scientific), 1% penicillin/streptomycin (Sigma), in a humidified 5% CO2 atmosphere at 37°C. The cell lines were tested for mycoplasma.
Patient-derived hiPSCs were generated by Applied StemCell (California, USA) by viral-free episomal reprogramming of JMS individual III-2 fibroblasts through electroporation with three reprogramming plasmids containing human sequences for OCT4, SOX2, KLF4, LIN28, L-MYC. Two independent clones from the same reprograming experiment (JMS cl.1 and cl.2) were used further. Using similar approach were generated healthy control CRL(S)23 cells (WT-CRL(S)23).42 The JMS hiPSC clones (cl.1 and cl.2), WT-CRL(S)23, as well as additional healthy control WT-DYS0100 (ATCC, Manassas, USA) were grown in mTeSR1 (STEMCELL Technologies) and E8 medium (ThermoFisher Scientific), on CellMatrix Basement Membrane Gel (ATCC) coated dishes. All cell lines were grown in a humidified 5% CO2 atmosphere at 37°C, and regularly tested for mycoplasma. The hiPSC cell lines were verified for the expression of pluripotency genes by immunofluorescence and RT-qPCR (Figure S2), karyotyped using hPSC Genetic Analysis Kit (Stem Cell Technologies).
Samples were allocated to the healthy and XLID experimental groups based on the HUWE1 status, which was confirmed by sequencing. The sample size was determined in line with our previous work,4,13 as well as tested by G∗Power 3 analysis49 to reach statistical power of 0.8.
Method details
Number of biological replicates are indicated in the Figure Legends. Sample size was determined as described above. No data was excluded from the experiments. In all experiments random cultures of healthy and XLID cells were used. The RNA-sequencing, gene expression analysis in LCs and neural differentiation experiments, germ layer marker analysis and Hematoxylin Eosin Saffron staining of organoids were performed by investigators blinded to the outcome data.
RNA isolation and quantitative PCR
RNA isolation was performed using RNeasy mini kit (QIAGEN) and cDNA subsequently generated with MultiScribe Reverse Transcriptase (ThermoFisher Scientific) according to the manufacturer’s instructions. Quantitative PCR (qPCR) was performed with Power SYBR Green PCR Master Mix (Applied Biosystems) on a StepOnePlus Real-Time PCR System. qPCR was performed with primer pairs (Sigma) listed in Table S1.
RNA sequencing
RNA sequencing libraries were generated using SENSE mRNA-Seq library prep kit according to manufacturer’s instructions (Lexogen GmbH, Vienna, Austria). In brief, 300 ng of total RNA was prepared and incubated with magnetic beads coated with oligo-dT, then all other RNAs except mRNA were removed by washing. Library preparation was then initiated by random hybridization of starter/stopper heterodimers to the poly(A) RNA still bound to the magnetic beads. These starter/stopper heterodimers contain Illumina-compatible linker sequences. A single-tube reverse transcription and ligation reaction extends the starter to the next hybridized heterodimer, where the newly synthesized cDNA insert was ligated to the stopper. Second-strand synthesis was performed to release the library from the beads. The resulting double-stranded library was purified and amplified (13 PCR cycles) prior to adding the adaptors and indexes. Finally, libraries were purified using the Mag-Bind RXNPure Plus beads (Omega Bio-tec, GA, USA), quantitated by qPCR using KAPA Library Quantification Kit (Roche, Kapa Biosystems, Inc., MA, USA) and validated using Agilent High Sensitivity DNA Kit on a Bioanalyzer (Agilent Technologies, CA, USA). The size range of the DNA fragments were measured to be in the range of app. 200-450 bp and peaked around 270 bp.
Libraries were normalized and pooled to 2.4 nM and subject to clustering (by a cBot Cluster Generation System on HiSeq4000 flowcells (Illumina Inc., CA, USA), according to manufacturer’s instructions. The sequencing (75 cycles single end reads) was performed on an Illumina HiSeq4000 instrument, in accordance with the manufacturer’s instructions (Illumina, Inc., CA, USA). FASTQ files were created with bcl2fastq 2.20.0.422 (Illumina, Inc., CA, USA).
Bioinformatic analysis
RNA-seq analysis
Raw reads were demultiplexed and mapped to the human genome (Ensembl GRCh38.84) using STAR aligner v2.4.0.44 Read counts per gene were calculated with htseq-count45 v0.6.0, with features being counted at the exon level. Normalization and differential expression were analyzed with limma voom.46 Read counts were filtered to remove genes with less than an average of 1 read per sample post-normalization. Fold changes > 1 and FDR (false discovery rate) < 0.3 were considered significant.
Functional annotation
Pathway analysis of differentially expressed genes (DEGs) with fold change > 1 and FDR < 0.3 was performed using the functional annotation tool DAVID (Database for Annotation, Visualization, and Integrated Discovery, v6.8).47 Significance of KEGG_PATHWAY terms was evaluated by hypergeometric testing (as a function in DAVID), using p values with Benjamini correction for multiple hypothesis testing. Pathways were considered significantly enriched with a Benjamini corrected p value < 0.05.
Heatmap
Log-CPM (copies per million) normalized expression values of DEGs that appeared on the KEGG p53 signaling pathway list were used as input to the R package ‘pheatmap’ v1.0.12 (R version 3.4.1) to create a heatmap. Log-CPM values were scaled across rows (genes) to generate Z-scores, which were then used for coloring the heatmap.
Whole cell extracts
LCs were collected and washed twice with ice cold PBS, followed by flash freezing in liquid N2. Whole cell extracts (WCEs) were obtained similarly as described previously.50 Briefly, cell pellets were resuspended in lysis buffer I (10 mM Tris-HCl pH 7.8, 200 mM KCl, 0.1 mM MG-132, 1 μM PMSF, halt protease and phosphatase inhibitor cocktail (ThermoScientific)); followed by addition of lysis buffer II (10 mM Tris-HCl pH 7.8, 600 mM KCl, 2 mM EDTA, 40% glycerol, 0.2% NP-40, 0.1 mM MG-132, 1 μM PMSF, halt protease and phosphatase inhibitor cocktail); rotated 30 min at 4 °C, sonicated and centrifuged. The supernatants representing WCEs were collected. hiPSCs WCEs were prepared as described by van Loon and Samson.51 Briefly, hiPSCs were resuspended in two packed cell volumes (PCV) hypotonic lysis buffer (20 mM HEPES at pH 7.9, 2 mM MgCl2, 0.2 mM EGTA, 10% (v/v) glycerol, 0.1 mM PMSF, 2 mM DTT, complemented with Halt Protease and Phosphatase Inhibitors Cocktail) and incubated 5 min at 4°C, followed by three freeze/thaw cycles. NaCl and Nonidet-P40 were added to final concentration 0.5 M and 0.5% (v/v), respectively, and samples incubated 20 min at 4°C, sonicated and centrifuged. The supernatants were diluted with eight PCV of lysis buffer containing 50 mM NaCl and used for subsequent analysis.
Immunoblot analysis
WCE proteins were separated on 4%–12% Bis–Tris polyacrylamide gels (Invitrogen) followed by transfer to Amersham Hybond® P western blotting membranes, PVDF (Sigma). Primary antibodies HUWE1 (A300-486A, Bethyl Laboratories), p53 (MA5-12571, ThermoFisher Scientific), p21 (33-7000, ThermoFisher Scientific), Tubulin (T9026, Sigma-Aldrich), β-Actin (A1978, Sigma-Aldrich)) were detected using infrared (IR) Dye-conjugated secondary antibodies (Li-COR Bioscienecs) and the signal visualized by Odyssey Scanner, LI-COR Biosciences.
Plasmids for the expression of HUWE1 HECT WT and G4310R
Site-directed mutagenesis was performed on pET28a-HECT WT plasmid43 to obtain the plasmid encoding introduce HUWE1 G4310R mutantion by using Phusion® High-Fidelity DNA Polymerase (ThermoFisher Scientific), according to the manufacturer’s instructions using HECT_JMS Fw and Rev primer pairs (Table S1). The insertion of the mutation was confirmed by sequencing. PCR conditions: denaturation 98°C for 2 min; amplification (25 cycles) 98°C for 30 s, 72°C for 1 min, 72°C for 3 min; extension 72°C for 5 min.
Purification of His-tagged-HUWE1 HECT WT and G4310R
Expression of the His-tagged HUWE1 HECT WT and G4310R proteins was carried out in the E. coli strain BL21-CodonPlus (DE) RP competent cells. Cells were transformed with the respective pET28a-HECT WT or pET28a-HECT G4310R vectors and grown overnight in LB medium at 37°C. The next day the overnight culture was further incubated in a rotatory shaker at 37°C, until the absorbance at 595 nm reached 0.6. The culture was induced by 0.5 mM IPTG (Sigma) for 3 h at 37°C. Cells were collected by centrifugation for 40 min at 4000 rpm, 4°C, and washed with cold PBS. Total cell extracts were obtained by sonication (4 cycles, 20 s ON + 20 s OFF; tubes were put on ice for 1 min between each sonication cycle) of the cells in Sonication buffer (20 mM Tris HCl pH 8, 500 mM NaCl, 10% Glycerol, 1 mM PMSF, 1X proteases inhibitors). The lysates were then cleared by centrifugation at 4°C, 18000 rcf., for 15 min, to separate insoluble proteins from the soluble extract. Protein expression and solubility was analyzed by gel electrophoresis and visualized by Comassie-Blue staining. The proteins were purified by incubating the lysates for 2 h, at 4°C, with rotation with TALON metal affinity resins pre-equilibrated with Sonication buffer. After extensive washing, His-tagged-HUWE1 HECT WT and G4310R were eluted in Sonication buffer containing increasing concentration of imidazole (8 × 1 mL sonication buffer with (1 × 100 mM imidazole; 2 × 200 mM imidazole; 3 × 300 mM imidazole; 1 × 400mM imidazole; 1 × 500 mM imidazole)). The fractions were dialyzed against 20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 20% glycerol, and 1 mM DDT and stored at −80°C until use. Protein purity was assessed by SDS/PAGE and visualized by Comassie-Blue staining.
In vitro ubiquitination assay
Ubiquitin, E1 ubiquitin-activating enzyme and the UbcH7 E2 ubiquitin-conjugating enzymes (U-Boston Biochem) were pre-mixed with recombinant p53 (Milipore) and 2,5x ubiquitin buffer (125 mM Tris-HCl pH 7.5, 12,5 mM MgCl2, 5 mM DTT, 5mM ATP). To this pre-mix, increasing amounts of His-tagged recombinant HECT (WT or G4310R) were added. The in vitro ubiquitination was performed at 30 °C for 90 min, stopped with SDS-PAGE loading dye and separated on a NuPAGE® Novex® 4%–12% Bis-Tris Protein Gels (Life Technologies) and immunoblotted as described above.
Change of folding free energy upon missense mutation (ΔΔG)
To predict missense mutations’ effect the protein folding stability (ΔΔG) was calculated. The webservers used in this study include mCSM,24 SDM,25 DUET,26 iMutant2.0,27 and PoPMuSiC.28
Cell cycle analysis
Cells were fixed with 70% ethanol, stained with propidium iodide (Sigma Aldrich) at a final concentration of 20 μg/ml in PBS with the addition of 100 μg/ml RNase A (37°C, 30 min in the dark). Flow cytometry analysis was performed with the BD FACS CANTO SYSTEM (BD Biosciences). Data were analyzed in FlowJo LLC software (USA).
Analysis of apoptosis
The apoptotic WT, JMS1 and JMS2 LCs were identified using Dead Cell Apoptosis Kit (Molecular Probes). WT LCs treated with 250 μM H2O2, for 2 h, at 37 °C served as a positive control. LCs were washed with cold PBS and diluted to 1 × 106 cells/mL in 1X annexin-binding buffer; 100 μL of cell suspension was labeled with Annexin V Alexa Fluor® 488 and Propidium Iodide (Sigma), incubated for 15 min at room temperature and analyzed on a BD FACSAria II (BD Biosciences). The apoptotic cell fraction was determined by FlowJo, LLC software (USA).
Cell proliferation
LCs (1.5 × 104 per well) were seeded in 96 well plates and growth rate monitored by counting every 22h (Countess II Automated cell counter, ThermoFisher Scientific).
EB formation for germ layer marker analysis
WT-DYS0100, WT-CRL(S)23 and JMS-cl.1 and cl.2 hiPSCs were dissociated with Accutase (STEMCELL Technologies), neutralized with DMEM/F12 and pelleted. Next, cells were resuspended with EB Medium (DMEM/F12, 20% Knock-Out Serum Replacement (KSOR), 1% Glutamax, 1% nonessential amino acids (NEAA)) containing 10 μM Rock inhibitor (only for D1). For 1 well of ultra-low attachment 6-well plate, 400000 cells per ml were seeded. The EBs were incubated on an orbital shaker (71 rpm) at 37°C. Medium was changed every other day and EBs were collected at D4 for RT-qPCR analysis of germ layer markers (Table S1).
Neural differentiation
Neural differentiation was performed according to modified protocol.29 WT-DYS0100, WT-CRL(S)23 and JMS-cl.1-2 hiPSC colonies were pre-incubated with 10 μM ROCK inhibitor (Y-27632, Merck) for 1 h, and dissociated with Accutase (STEMCELL Technologies). Cells were re-aggregated (10000 cells per well) in U-bottom ultra-low attachment 96-well plates (Corning) and cultured in “EB-medium” (DMEM/F12-GLUTAMAX medium, 2% B27 supplement without vitamin A (GIBCO), 1% N2 supplement (GIBCO), 50 μM 2-mercaptoethanol (2-ME), 5 μM Y-27632 and 100 ng/ml recombinant mouse Noggin (R&D Systems, 1967-NG-025)). After 2 days half of the culture medium was replaced with “EB-medium” with vitamin A. After 2 additional days free-floating EBs were transferred on Matrigel (BD Biosciences) coated plates and cultured in neural medium (DMEM/F12-GlutaMAX containing 2% B27 with Vitamin A, 1% N2, 50 uM 2-mercaptoethanol) for 2 days. Terminal differentiation was induced using a NEUROBASAL-A medium (ThermoFisher Scientific) with 1% N2, 2% B27 with vitamin A, 1% Glutamax, 1% NEAA and 50 μM 2-Mercaptoethanol, 200 nM ascorbic acid, 10 ng/ml BDNF (R&D Systems), 10 ng/ml GDNF (R&D Systems) and 1 mM dibutyryl-cAMP (Sigma), and medium exchanged every 48h. After 6 days of terminal differentiation the neural cells were collected for subsequent immunostaning or RT-qPCR analysis.
shRNA knockdown
JMS-cl.1 and JMS-cl.2 hiPSCs were transduced with lentiviruses encloding for the non-specific control short-hairpin RNA (shControl; pLKO.1 puro (Addgene ID: 8453)) or the p53 targeting shRNAs; shP53a (shp53 pLKO.1 puro shRNA (Addgene ID: 19119)52), shP53b (shp53 pLKO.1 puro shRNA-427 (Addgene ID: 25636)53) and shP53c (shp53 pLKO.1 puro shRNA-941(Addgene ID: 25637)53). 72 h after infection, puromycin selection (1 μg/ml) was started and carried out for 48 h. Stable transduced colonies were expanded and p53 downregulation assessed by RT-qPCR analysis.
Cerebral organoids
hiPSCs have been differentiated to form cerebellar organoids following protocol described in Lancaster and Knoblich.54 Briefly, healthy control WT-DYS0100 and JMS cl.1 hiPSCs were dissociated and induced to form EBs, which after six days were transferred in neural induction medium allowing neuroectoderm formation. Neural ectoderm induced EBs were embedded in Matrigel (BD Biosciences, 356234) and, upon outgrowth of neuroepithelaial buds, transferred to spinning bioreactor. 60 days after the initiation of differentiation cerebral organoids were collected and prepared for cryosectioning.
Immunostaining
WT-DYS0100, WT-CRL(S)23, JMS-cl.1 and JMS-cl.2 neural cells at day 13 (Figures 3G, 4E, and S5), or hiPSCs WT-DYS0100, WT-CRL(S)23, JMS-cl.1 and JMS-cl.2 (Figure S2) were fixed for 15 min with 4% paraformaldehyde or ice-cold methanol, respectively. Upon permeabilized at RT with 0.1% Triton-X in 1X PBS for 15 min, samples were incubated with 5% BSA, 5% goat gut serum, 0.1% Triton-X in 1X PBS for 45 min. Primary antibodies (OCT4 (diluted 1:200, Cell signaling C30A3); SSEA4 (diluted 1:200, Cell signaling MC813), NESTIN (diluted 1:400, Abcam ab22035), TUJ1 (diluted 1:750, Covance MMS-435P), Ki67 (diluted 1:200, Abcam ab15580) diluted in 0.5% BSA, 0.5% goat gut serum, 0.1% Tween-20 in 1X PBS (PBS-T), were added to samples and incubated over-night at 4°C. After washing with PBS-T, samples were incubated with fluorophore-conjugated secondary antibody from Invitrogen (Donkey anti-Mouse, Alexa Fluor 488, Invitrogen, A21202; Goat anti-Rabbit, Alexa Fluor 594, Invitrogen, A11037) diluted 1:400 in 1X PBS-T or 0.5% BSA, 0.5% goat gut serum, 0.1% Tween-20 in 1X PBS at RT for 1 h. Upon washing with PBS-T, cells were stained with DAPI for 10 min at RT and washed 3 times with PBS. The 18-μm thick cerebral organoid cryosections were immunostained following previously described procedure13 and primary and secondary antibodies indicated above. Images of WT and JMS cerebral organoids were captured with the Zeiss LSM 510 Meta live Confocal system, using ZEN 2009 software, UV laser (405 nm) and Argon laser (488 nm). The images of cerebral organoids are a compilation of confocal Z stacks comprising of up to 33 optical spices (0.31 um intervals) into 2D using maximum intensity projection. The images of neural induction experiments were captured with the EVOS FL Auto Cell Imaging System and are a compilation of 4 to 7 Z stacks comprised of 7.112 μm intervals. Images of WT-DYS0100, WT-CRL(S)23, JMS-cl.1 and JMS-cl.2 hiPSCs (Figure S2) were captured with the EVOS FL Auto Cell Imaging System for stemness markers analysis.
Quantification and statistical analysis
Quantification of gene expression levels
The relative expression levels were determined by normalization of qPCR signals for gene targets to GAPDH or ACTB/β-Actin signal, as indicated in the figure legends.
Protein level analysis
Signals obtained by immunoblot analysis were quantified using Fiji ImageJ (National Institutes of Health). Signal of each analyzed protein target was normalized to tubulin (Figure 2A) or β-actin (Figure S2L), which served as loading controls.
Analysis of immunofluorescence images
To automatically count the number of rosette structures in the DAPI channel, images resulting from immunofluorescence analysis were preprocessed and analyzed using the Analyze Particles function in Fiji ImageJ (National Institutes of Health, v. 1.53c). At least one well from three or more biologically independent neural induction experiments has been analyzed. Signal intensities of TUJ1 (Figures 3, S4, and S5) were quantified using the Cells and Surfaces functions in iMaris (Bitplane, v. 8.2.1) in one or more wells per each neural rosette experiment, as indicated in the figure legends. To quantify signal intensities of OCT4 in stem cells (Figure S2) the same approach was applied to at least two wells from two biologically independent experiments.
Statistical analysis
To determine significance of observed changes Student t test, one-way or two-way ANOVA were used in Prism 9, as indicated in the figure legends. p < 0.05 was considered to be significant.
Acknowledgments
This work was supported by the Central Norway Regional Health Authority (90270800 to B.v.L.) and the Liaison Committee between the Central Norway Regional Health Authority and the Norwegian University of Science and Technology (46055600-91 to B.v.L.). It was supported in part by an Onsager Fellowship (to B.v.L.), a NINDS grant (1R01NS07385A to C.E.S.), and a grant from the South Carolina Department of Disabilities and Special Needs (SCDDSN). The RNA library preparation and sequencing were performed at the Genomics Core Facility (GCF), and microscopy was performed at the Cellular and Molecular Imaging Core Facility (CMIC), Norwegian University of Science and Technology (NTNU). GCF and CMIC are funded by the Faculty of Medicine and Health Sciences at NTNU and Central Norway Regional Health Authority. We thank Siren Berland, Ingvild Aukrust, Marte Gjøl Haug, Ulrich Hübscher, and Giovanni Maga for input and discussions on the project.
Author contributions
B.v.L. led the study. R.A. performed most experiments. M.E.A. contributed to the stem cell profiling, generation of organoids, and together with B.M. and W.W., the neural induction and the immunofluorescence analysis. S.B. contributed to the RNA sequencing, cell culturing, and together with M.E.A., B.M., K.R., N.P.M., and D.L.B., the qRT-PCR and immunoblot analysis.; S.L.F.M., the RNA sequencing analysis; K.M.G., immunofluorescence quantifications; B.v.L., E.A., and Y.P., the protein analysis; and N.-B.L., FACS analysis. M. Bosshard, G.J.S., and C.S. contributed reagents. B.v.L., R.A., W.W., and M. Bjørås interpreted the experiments. B.v.L., R.A., and B.M. wrote the manuscript and generated figures. B.v.L. and C.E.S. had the original idea. All authors approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: April 8, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100240.
Supplemental information
References
- 1.Upadhyay A., Joshi V., Amanullah A., Mishra R., Arora N., Prasad A., Mishra A. E3 Ubiquitin Ligases Neurobiological Mechanisms: Development to Degeneration. Front. Mol. Neurosci. 2017;10:151. doi: 10.3389/fnmol.2017.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Froyen G., Belet S., Martinez F., Santos-Rebouças C.B., Declercq M., Verbeeck J., Donckers L., Berland S., Mayo S., Rosello M. Copy-number gains of HUWE1 due to replication- and recombination-based rearrangements. Am. J. Hum. Genet. 2012;91:252–264. doi: 10.1016/j.ajhg.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Froyen G., Corbett M., Vandewalle J., Jarvela I., Lawrence O., Meldrum C., Bauters M., Govaerts K., Vandeleur L., Van Esch H. Submicroscopic duplications of the hydroxysteroid dehydrogenase HSD17B10 and the E3 ubiquitin ligase HUWE1 are associated with mental retardation. Am. J. Hum. Genet. 2008;82:432–443. doi: 10.1016/j.ajhg.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friez M.J., Brooks S.S., Stevenson R.E., Field M., Basehore M.J., Adès L.C., Sebold C., McGee S., Saxon S., Skinner C. HUWE1 mutations in Juberg-Marsidi and Brooks syndromes: the results of an X-chromosome exome sequencing study. BMJ Open. 2016;6:e009537. doi: 10.1136/bmjopen-2015-009537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moortgat S., Berland S., Aukrust I., Maystadt I., Baker L., Benoit V., Caro-Llopis A., Cooper N.S., Debray F.G., Faivre L. HUWE1 variants cause dominant X-linked intellectual disability: a clinical study of 21 patients. Eur. J. Hum. Genet. 2018;26:64–74. doi: 10.1038/s41431-017-0038-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muthusamy B., Nguyen T.T., Bandari A.K., Basheer S., Selvan L.D.N., Chandel D., Manoj J., Gayen S., Seshagiri S., Chandra Girimaji S., Pandey A. Exome sequencing reveals a novel splice site variant in HUWE1 gene in patients with suspected Say-Meyer syndrome. Eur. J. Med. Genet. 2020;63:103635. doi: 10.1016/j.ejmg.2019.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ibarluzea N., Hoz A.B., Villate O., Llano I., Ocio I., Martí I., Guitart M., Gabau E., Andrade F., Gener B., Tejada M.I. Targeted Next-Generation Sequencing in Patients with Suggestive X-Linked Intellectual Disability. Genes (Basel) 2020;11:51. doi: 10.3390/genes11010051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao X., Heng J.I., Guardavaccaro D., Jiang R., Pagano M., Guillemot F., Iavarone A., Lasorella A. The HECT-domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N-Myc oncoprotein. Nat. Cell Biol. 2008;10:643–653. doi: 10.1038/ncb1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kon N., Zhong J., Qiang L., Accili D., Gu W. Inactivation of arf-bp1 induces p53 activation and diabetic phenotypes in mice. J. Biol. Chem. 2012;287:5102–5111. doi: 10.1074/jbc.M111.322867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giles A.C., Grill B. Roles of the HUWE1 ubiquitin ligase in nervous system development, function and disease. Neural Dev. 2020;15:6. doi: 10.1186/s13064-020-00143-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao X., D’Arca D., Lim W.K., Brahmachary M., Carro M.S., Ludwig T., Cardo C.C., Guillemot F., Aldape K., Califano A. The N-Myc-DLL3 cascade is suppressed by the ubiquitin ligase Huwe1 to inhibit proliferation and promote neurogenesis in the developing brain. Dev. Cell. 2009;17:210–221. doi: 10.1016/j.devcel.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D’Arca D., Zhao X., Xu W., Ramirez-Martinez N.C., Iavarone A., Lasorella A. Huwe1 ubiquitin ligase is essential to synchronize neuronal and glial differentiation in the developing cerebellum. Proc. Natl. Acad. Sci. USA. 2010;107:5875–5880. doi: 10.1073/pnas.0912874107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bosshard M., Aprigliano R., Gattiker C., Palibrk V., Markkanen E., Backe P.H., Pellegrino S., Raymond F.L., Froyen G., Altmeyer M. Impaired oxidative stress response characterizes HUWE1-promoted X-linked intellectual disability. Sci. Rep. 2017;7:15050. doi: 10.1038/s41598-017-15380-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brooks C.L., Gu W. p53 ubiquitination: Mdm2 and beyond. Mol. Cell. 2006;21:307–315. doi: 10.1016/j.molcel.2006.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bowen M.E., Attardi L.D. The role of p53 in developmental syndromes. J. Mol. Cell Biol. 2019;11:200–211. doi: 10.1093/jmcb/mjy087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Armesilla-Diaz A., Bragado P., Del Valle I., Cuevas E., Lazaro I., Martin C., Cigudosa J.C., Silva A. p53 regulates the self-renewal and differentiation of neural precursors. Neuroscience. 2009;158:1378–1389. doi: 10.1016/j.neuroscience.2008.10.052. [DOI] [PubMed] [Google Scholar]
- 17.Gil-Perotin S., Marin-Husstege M., Li J., Soriano-Navarro M., Zindy F., Roussel M.F., Garcia-Verdugo J.M., Casaccia-Bonnefil P. Loss of p53 induces changes in the behavior of subventricular zone cells: implication for the genesis of glial tumors. J. Neurosci. 2006;26:1107–1116. doi: 10.1523/JNEUROSCI.3970-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aloyz R.S., Bamji S.X., Pozniak C.D., Toma J.G., Atwal J., Kaplan D.R., Miller F.D. p53 is essential for developmental neuron death as regulated by the TrkA and p75 neurotrophin receptors. J. Cell Biol. 1998;143:1691–1703. doi: 10.1083/jcb.143.6.1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacobs W.B., Kaplan D.R., Miller F.D. The p53 family in nervous system development and disease. J. Neurochem. 2006;97:1571–1584. doi: 10.1111/j.1471-4159.2006.03980.x. [DOI] [PubMed] [Google Scholar]
- 20.Kaplan D.R., Miller F.D. Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- 21.Marin Navarro A., Pronk R.J., van der Geest A.T., Oliynyk G., Nordgren A., Arsenian-Henriksson M., Falk A., Wilhelm M. p53 controls genomic stability and temporal differentiation of human neural stem cells and affects neural organization in human brain organoids. Cell Death Dis. 2020;11:52. doi: 10.1038/s41419-019-2208-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y., Argiles-Castillo D., Kane E.I., Zhou A., Spratt D.E. HECT E3 ubiquitin ligases—emerging insights into their biological roles and disease relevance. J. Cell Sci. 2020;133:jcs228072. doi: 10.1242/jcs.228072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He Z., Hu X., Liu W., Dorrance A., Garzon R., Houghton P.J., Shen C. P53 suppresses ribonucleotide reductase via inhibiting mTORC1. Oncotarget. 2017;8:41422–41431. doi: 10.18632/oncotarget.17440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pires D.E., Ascher D.B., Blundell T.L. mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics. 2014;30:335–342. doi: 10.1093/bioinformatics/btt691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Worth C.L., Preissner R., Blundell T.L. SDM—a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011;39:W215–W222. doi: 10.1093/nar/gkr363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pires D.E., Ascher D.B., Blundell T.L. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014;42:W314–W319. doi: 10.1093/nar/gku411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Capriotti E., Fariselli P., Casadio R. I-Mutant2.0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005;33:W306–W310. doi: 10.1093/nar/gki375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilis D., Rooman M. PoPMuSiC, an algorithm for predicting protein mutant stability changes: application to prion proteins. Protein Eng. 2000;13:849–856. doi: 10.1093/protein/13.12.849. [DOI] [PubMed] [Google Scholar]
- 29.Mariani J., Coppola G., Zhang P., Abyzov A., Provini L., Tomasini L., Amenduni M., Szekely A., Palejev D., Wilson M. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell. 2015;162:375–390. doi: 10.1016/j.cell.2015.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hao Z., Duncan G.S., Su Y.W., Li W.Y., Silvester J., Hong C., You H., Brenner D., Gorrini C., Haight J. The E3 ubiquitin ligase Mule acts through the ATM-p53 axis to maintain B lymphocyte homeostasis. J. Exp. Med. 2012;209:173–186. doi: 10.1084/jem.20111363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brooks C.L., Gu W. New insights into p53 activation. Cell Res. 2010;20:614–621. doi: 10.1038/cr.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bustos F., Segarra-Fas A., Chaugule V.K., Brandenburg L., Branigan E., Toth R., Macartney T., Knebel A., Hay R.T., Walden H., Findlay G.M. RNF12 X-Linked Intellectual Disability Mutations Disrupt E3 Ligase Activity and Neural Differentiation. Cell Rep. 2018;23:1599–1611. doi: 10.1016/j.celrep.2018.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toki T., Yoshida K., Wang R., Nakamura S., Maekawa T., Goi K., Katoh M.C., Mizuno S., Sugiyama F., Kanezaki R. De Novo Mutations Activating Germline TP53 in an Inherited Bone-Marrow-Failure Syndrome. Am. J. Hum. Genet. 2018;103:440–447. doi: 10.1016/j.ajhg.2018.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yanku Y., Bitman-Lotan E., Zohar Y., Kurant E., Zilke N., Eilers M., Orian A. Drosophila HUWE1 Ubiquitin Ligase Regulates Endoreplication and Antagonizes JNK Signaling During Salivary Gland Development. Cells. 2018;7:151. doi: 10.3390/cells7100151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Z., Zhang C., Skamagki M., Khodadadi-Jamayran A., Zhang W., Kong D., Chang C.W., Feng J., Han X., Townes T.M. Elevated p53 Activities Restrict Differentiation Potential of MicroRNA-Deficient Pluripotent Stem Cells. Stem Cell Reports. 2017;9:1604–1617. doi: 10.1016/j.stemcr.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sammons M.A., Zhu J., Drake A.M., Berger S.L. TP53 engagement with the genome occurs in distinct local chromatin environments via pioneer factor activity. Genome Res. 2015;25:179–188. doi: 10.1101/gr.181883.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karsli Uzunbas G., Ahmed F., Sammons M.A. Control of p53-dependent transcription and enhancer activity by the p53 family member p63. J. Biol. Chem. 2019;294:10720–10736. doi: 10.1074/jbc.RA119.007965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hafner A., Kublo L., Tsabar M., Lahav G., Stewart-Ornstein J. Identification of universal and cell-type specific p53 DNA binding. BMC Mol. Cell Biol. 2020;21:5. doi: 10.1186/s12860-020-00251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Freel B.A., Sheets J.N., Francis K.R. iPSC modeling of rare pediatric disorders. J. Neurosci. Methods. 2020;332:108533. doi: 10.1016/j.jneumeth.2019.108533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anderson R.H., Francis K.R. Modeling rare diseases with induced pluripotent stem cell technology. Mol. Cell. Probes. 2018;40:52–59. doi: 10.1016/j.mcp.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu B.Y., Weick J.P., Yu J., Ma L.X., Zhang X.Q., Thomson J.A., Zhang S.C. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc. Natl. Acad. Sci. USA. 2010;107:4335–4340. doi: 10.1073/pnas.0910012107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siller R., Naumovska E., Mathapati S., Lycke M., Greenhough S., Sullivan G.J. Development of a rapid screen for the endodermal differentiation potential of human pluripotent stem cell lines. Sci. Rep. 2016;6:37178. doi: 10.1038/srep37178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parsons J.L., Tait P.S., Finch D., Dianova I.I., Edelmann M.J., Khoronenkova S.V., Kessler B.M., Sharma R.A., McKenna W.G., Dianov G.L. Ubiquitin ligase ARF-BP1/Mule modulates base excision repair. EMBO J. 2009;28:3207–3215. doi: 10.1038/emboj.2009.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anders S., Pyl P.T., Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Juberg R.C., Marsidi I. A new form of X-linked mental retardation with growth retardation, deafness, and microgenitalism. Am. J. Hum. Genet. 1980;32:714–722. [PMC free article] [PubMed] [Google Scholar]
- 49.Faul F., Erdfelder E., Lang A.G., Buchner A. G∗Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods. 2007;39:175–191. doi: 10.3758/bf03193146. [DOI] [PubMed] [Google Scholar]
- 50.Tanaka M., Lai J.S., Herr W. Promoter-selective activation domains in Oct-1 and Oct-2 direct differential activation of an snRNA and mRNA promoter. Cell. 1992;68:755–767. doi: 10.1016/0092-8674(92)90150-b. [DOI] [PubMed] [Google Scholar]
- 51.van Loon B., Samson L.D. Alkyladenine DNA glycosylase (AAG) localizes to mitochondria and interacts with mitochondrial single-stranded binding protein (mtSSB) DNA Repair (Amst.) 2013;12:177–187. doi: 10.1016/j.dnarep.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Godar S., Ince T.A., Bell G.W., Feldser D., Donaher J.L., Bergh J., Liu A., Miu K., Watnick R.S., Reinhardt F. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62–73. doi: 10.1016/j.cell.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim J.S., Lee C., Bonifant C.L., Ressom H., Waldman T. Activation of p53-dependent growth suppression in human cells by mutations in PTEN or PIK3CA. Mol. Cell. Biol. 2007;27:662–677. doi: 10.1128/MCB.00537-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lancaster M.A., Knoblich J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014;9:2329–2340. doi: 10.1038/nprot.2014.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of present study are available from the corresponding author on reasonable request. The RNA sequencing data reported in this paper are available in GEO under accession GEO: GSE130551. The study did not generate any new codes.