

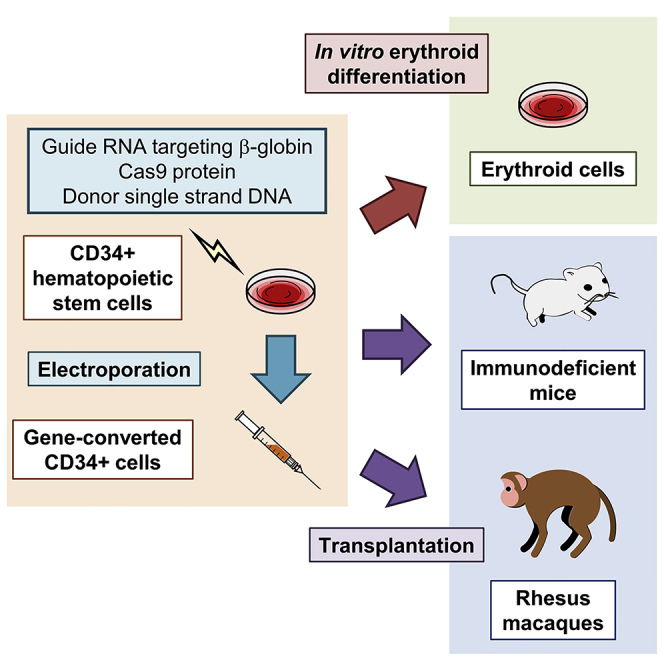
Summary
Sickle cell disease (SCD) is caused by a 20A > T mutation in the β-globin gene. Genome-editing technologies have the potential to correct the SCD mutation in hematopoietic stem cells (HSCs), producing adult hemoglobin while simultaneously eliminating sickle hemoglobin. Here, we developed high-efficiency viral vector-free non-footprint gene correction in SCD CD34+ cells with electroporation to deliver SCD mutation-targeting guide RNA, Cas9 endonuclease, and 100-mer single-strand donor DNA encoding intact β-globin sequence, achieving therapeutic-level gene correction at DNA (∼30%) and protein (∼80%) levels. Gene-edited SCD CD34+ cells contributed corrected cells 6 months post-xenograft mouse transplant without off-target δ-globin editing. We then developed a rhesus β-to-βs-globin gene conversion strategy to model HSC-targeted genome editing for SCD and demonstrate the engraftment of gene-edited CD34+ cells 10–12 months post-transplant in rhesus macaques. In summary, gene-corrected CD34+ HSCs are engraftable in xenograft mice and non-human primates. These findings are helpful in designing HSC-targeted gene correction trials.
Keywords: genome editing, CRISPR/Cas9, sickle cell disease, hematopoietic stem cell, transplantation, electroporation, gene correction, β-globin gene, non-human primate, large animal model
Graphical abstract
Highlights
Sickle cell disease (SCD) is a blood disease caused by a mutation in the β-globin gene
A viral vector-free non-footprint gene correction is developed for SCD CD34+ cells
Achieve therapeutic-level SCD gene correction of DNA (∼30%) and protein (∼80%)
Demonstrate engraftment of gene-edited CD34+ cells in xenografts and non-human primates
Sickle cell disease (SCD) is caused by a point mutation in the β-globin gene and can be cured by the replacement of hematopoietic stem cells (HSCs). Uchida et al. demonstrate a high-efficiency gene correction method for the SCD mutation and engraftment of gene-edited CD34+ HSCs in xenograft mice and non-human primates.
Introduction
Sickle cell disease (SCD) is the most common single-gene disorder in the world. It is caused by a 20A > T mutation in the β-globin gene, resulting in the sickling of red blood cells (RBCs), hemolytic anemia, pain crisis, organ damage, and early mortality.1 There are few treatment options for patients with SCD. The first US Food and Drug Administration (FDA)-approved medication is hydroxyurea and acts by fetal hemoglobin (HbF) induction, thereby reducing RBC sickling; however, lifelong treatment is needed and therapeutic effects vary among patients.2 A P-selectin antibody (crizanlizumab) treatment designed to reduce blood cell adhesion was recently approved by the FDA, and l-glutamine administration has been shown to reduce pain crises in patients with SCD.3,4 Sibling hematopoietic stem cell (HSC) transplantation allows for a one-time cure for SCD, but histocompatible sibling donors can be found in only ∼10% of patients.5,6 In current gene therapy trials, SCD can be cured by therapeutic β-globin gene addition into a patient’s autologous HSCs with lentiviral transduction; however, this method relies upon random vector integration, leaving the SCD mutation intact and potentially inducing insertional mutagenesis.7
State-of-the-art genome editing technologies have the potential to correct the SCD mutation without genome-wide integration of vector sequence, producing adult hemoglobin (HbA) by homology-directed repair (HDR) while simultaneously eliminating sickle hemoglobin (HbS) by either the HDR conversion to HbA or incapacitating the HbS allele by non-homologous end joining (NHEJ) small insertion or deletion (indel) formation.8,9 Engineered endonucleases, including zinc finger nucleases (ZFNs) and CRISPR/Cas9 (including guide RNA and Cas9 endonuclease), allow for site-specific DNA breakage, enhancing HDR-based gene correction with donor DNA encoding an intact gene sequence.10 Prior HSC gene correction research in SCD used mainly an adeno-associated virus (AAV) vector encoding large-sized donor DNA as well as occasionally electroporation-mediated delivery of small-sized donor single-stranded DNA (ssDNA) including silent mutations to escape from guide RNA targeting (Table 1), but data regarding engraftment of gene-corrected HSCs was limited.8,11, 12, 13, 14, 15 Here, we investigated CRISPR/Cas9-mediated gene correction for patient-derived SCD CD34+ cells with non-viral vector delivery using an approach that results in seamless repair of the SCD mutation. This work was complemented by using the converse approach to seamlessly introduce the SCD mutation into non-human primate-derived CD34+ cells. We demonstrated high-efficiency gene correction in patient-derived SCD CD34+ cells and gene conversion to SCD in non-human primate CD34+ cells at both DNA and protein levels, resulting in intact β-globin sequences without any editing footprint (no silent mutation in the guide RNA target site). Gene-edited CD34+ cells with HDR engrafted and remained detectable during follow-up at 6 months post-transplant in xenograft mice and at 10–12 months post-transplant in non-human primates.
Table 1.
Summary of SCD gene correction in CD34+ cells with CRISPR/Cas9 system
| Target site in β-globin gene | Donor DNA size (base) | Silent mutation in donor DNA | Delivery method | Pre-stimulation for CD34+ cells | HDR/indel in vitro | Protein conversion in vitro | Engraftment in xenograft mice | HDR/indel in xenograft mice | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Exon 1 | 960 (AAV6) | yes (GFP insertion, β-to-βs-globin conversion) | AAV6 (donor DNA) and Lonza Nucleofector 2b | 1 day in StemSpan with SCF, FL, TPO, IL-6, and SR1 | 50%/~40% | NA | 7.0% in BM (16W) | 3.5%/NA in BM (16W) | 8 |
| 138–195 (ssDNA) | yes (β-to-βs-globin conversion) | Lonza 4D Nucleofector | 1 day in StemSpan with CC110 cocktail | up to 25%/~60% | up to 29.3% | 37% in BM (16W) | 4%/~40% in BM (16W) | 11 | |
| 168 (ssDNA) | yes (β-to-βs-globin conversion) | BTX ECM 830 Electroporator | 2 days in X-VIVO15 with SCF, FL, and TPO | ~20%/~40% | NA | ~10% in BM (5M) | ~2%/~10% in BM (5M) | 12 | |
| Exon 1, including SCD mutation site | 168 (ssDNA) | yes (β-to-βs-globin conversion) | Lonza 4D Nucleofector | 2 days in SCGM with SCF, FL, TPO, and IL-6 | 12%/17% | 20% | ~80% in BM (12–14W) | ~4%/~4% in BM (12–14W) | 13 |
| no (β-to-βs-globin conversion) | 17%/20% | 19% | NA | NA | |||||
| 120 (ssDNA) | yes | Lonza 4D Nucleofector | 2 days in SCGM with SCF, FL, TPO, and IL-3 | 25%/42% | ~40% | 30% in BM (19W) | 10%/33% in BM (19W) | 14 | |
| 100 (ssDNA) | no | MaxCyte GT System | 2 days in StemSpan with SCF, FL, and TPO | ~30%/~50% | ~80% | 8%–29% in PB (24W) | 10%–38%/13%–36% in PB (24W) | current data |
AAV, adeno-associated virus; BM, bone marrow; FL, FMS-related tyrosine kinase 3 ligand; IL-3, interleukin-3; IL-6, interleukin-6; M, months; NA, not applicable; PB, peripheral blood; SCF, stem cell factor; SCGM, stem cell growth medium; SR1, StemRegenin1; ssDNA, single-stranded DNA; TPO, thrombopoietin; W, weeks.
Results
Therapeutic-level gene correction of the SCD mutation in patient CD34+ cells with electroporation of editing tools
To develop a clinically relevant βs-to-β-globin gene correction strategy in SCD, we designed SCD mutation-targeting guide RNA and donor ssDNA encoding a normal β-globin gene (Figure 1A), which were validated and optimized in a SCD B cell line model we created (Figures S1A and S1B). The SCD mutation-targeting guide RNA allowed more specific editing for the βs-globin sequence, but less for the normal β-globin sequence, evaluated by a lentiviral delivery system.16 For these early development studies, we used β-globin donor DNA that also included synonymous base pair changes that introduced a new HindIII enzyme site (6 bases). We detected ∼30% HDR-mediated HindIII site insertion in the βs-globin gene as evaluated by both enzyme digestion of polymerase chain reaction (PCR) products (Figure S1C) and targeted deep sequencing (Figure S1D). The use of normal β-globin donor DNA without a HindIII site for seamless conversion to the normal wild-type sequence resulted in a similar efficiency of gene correction (HDR), compared to DNA containing the HindIII site insertion. In addition, we detected similar HDR efficiencies in healthy donor CD34+ cells for HDR-mediated HindIII site insertion as well as reverse β-to-βs-globin gene conversion using a normal β-globin gene-targeting guide RNA, which targets the same site as the SCD mutation in the normal β-globin gene, and donor DNA encoding the βs-globin sequence (Figures S1C and S1D).
Figure 1.

Efficient gene correction of the sickle cell disease (SCD) mutation in CD34+ cells in vitro
(A) Schematic design of guide RNA targeting the SCD mutation in β-globin gene and donor single-stranded DNA (ssDNA) encoding normal β-globin gene.
(B) A βs-to-β-globin gene correction model in SCD CD34+ cells by electroporation-mediated delivery of guide RNA, Cas9 endonuclease, and donor ssDNA, followed by in vitro erythroid differentiation.
(C) CD34+ cell viability 1 and 2 days after electroporation, evaluated by trypan blue stain. ∗p < 0.05, ∗∗p < 0.01, evaluated by Dunnett’s test, compared with no electroporation (n = 1–6). Standard error of the mean (SEM) shown as error bars.
(D) βs-to-β-globin gene correction (homology-directed repair [HDR]) and indels (small insertions and deletions), evaluated by targeted deep sequencing in cultured edited cells 2–5 days post-electroporation. ∗∗p < 0.01, evaluated by Dunnett’s test, compared with no electroporation (n = 1–6). SEM shown as error bars.
(E) Biallelic correction (HDR/HDR) and monoallelic correction (HDR/indel and HDR/sickle) in colony-forming units (CFUs), evaluated by quantitative polymerase chain reaction (qPCR) single-nucleotide polymorphism (SNP) genotyping (n = 1).
(F–H) Normal β-globin protein production in SCD CD34+ cell-derived erythroid cells following gene correction, evaluated by hemoglobin electrophoresis (F), and reversed-phase high-performance liquid chromatography (RP-HPLC) (G and H). ∗∗p < 0.01, evaluated by Dunnett’s test, compared with no electroporation (n = 1–6). SEM shown as error bars.
We then electroporated patient-derived plerixafor-mobilized SCD CD34+ cells to deliver SCD mutation-targeting guide RNA, Cas9 mRNA or protein, and correct-sequence β-globin donor 100-mer ssDNA without any additional mutations (80, 120, or 200 μg/mL) (Figure 1B). Cell viability was reduced to 76%–87% and 71%–81% 1 and 2 days after electroporation (p < 0.01, except 80 μg/mL of donor DNA), compared to 90% and 94% in a no-electroporation control, respectively (Figure 1C). Following erythroid differentiation, high-efficiency genome editing at the DNA level was observed in both Cas9 mRNA (29%–33% gene correction and 50%–58% indels with donor DNA concentration dependence) and Cas9 protein (37% correction and 44% indels) (Figure 1D). Importantly, the gene-corrected DNA sequence was completely identical to the intact β-globin gene. A total of 15%–23% biallelic and 17%–26% monoallelic gene corrections were detected at the clonal level by colony assay (Figure 1E). High-level normal β-globin protein production was observed by hemoglobin electrophoresis (Figure 1F) and reversed-phase high-performance liquid chromatography (RP-HPLC) (Figures 1G and 1H) using Cas9 mRNA (82%–85%) as well as Cas9 protein (81%), while βs-globin amounts were markedly reduced under both conditions (15%–19%). Interestingly, a similar efficiency of gene correction was observed before and after erythroid differentiation (Figure S1E). These data demonstrate that this viral vector-free non-footprint gene editing method allows for efficient gene correction in SCD CD34+ cells, exceeding the therapeutic threshold of 20% in SCD.17
We evaluated off-target effects on the highly homologous δ-globin gene, previously reported to be a major off-target site in β-globin gene editing,18 and no increase in off-target events (0.6%–1.3% indels) was observed, as compared to controls (1.2% indels) (Figure S1F). Interestingly, the 9T > C polymorphism on the β-globin gene (11 bp upstream of SCD mutation) was found in SCD CD34+ cells (Figure S1G), and C-to-T gene conversion (13%–14%) occurred along with SCD gene correction (43%–54%) at 1 bp upstream of the DNA cleavage site, suggesting that gene conversion more efficiently occurs at nucleotides nearer to the DNA cleavage site.
To investigate whether indels in the β-globin gene can affect globin expression, we also evaluated βs-globin gene editing without donor ssDNA in SCD CD34+ cells (Figure S1H). In this donor DNA-free editing, 64%–71% indels (p < 0.01) were generated at the DNA level (Figure S1I), and mainly βs-globin was detected at the protein level, with minimal γ-globin and undetectable normal β-globin (Figures S1J and S1K). We also observed no increase in γ-globin protein production in gene-corrected SCD CD34+ cells, compared to no electroporation control (Figure S2A). These data demonstrate that minimal HbF expression is induced by indels in β-globin gene editing.
In addition, we evaluated genome editing among subpopulations of CD34+ cells from 3 healthy donors under the same conditions but using reagents that would achieve the converse effect of β-to-βs-globin gene conversion (Figure 2A). We observed similar editing efficiencies (HDR and indels) among more immature (CD34+CD133+CD90+) and relatively differentiated populations (CD34+CD133+CD90−, CD34+CD133−, and CD34−) (Figure 2B), as well as among cells at different phases of the cell cycle (G0/G1, S, and G2/M) (Figure 2C), suggesting that similar gene correction efficiencies are obtained in all CD34+ cell populations, potentially including the likely long-term repopulating HSC population.
Figure 2.

Similar-level β-to-βs-globin gene conversion among subpopulations of CD34+ cells
(A) A β-to-βs-globin gene conversion model to evaluate HDR and indels among sorted subpopulations of edited CD34+ cells 2 days post-electroporation.
(B) HDR and indels among subpopulations (CD34+CD133+CD90+, CD34+CD133+CD90−, CD34+CD133−, and CD34−) of edited CD34+ cells. Not significant, evaluated by Dunnett’s test, compared with bulk cells (n = 3). SEM shown as error bars.
(C) HDR and indels among cell cycles (G0/G1, S, and G2/M) in edited CD34+ cells. Not significant, evaluated by Dunnett’s test, compared with bulk cells (n = 3). SEM shown as error bars.
Engraftment of gene-edited cells with the SCD mutation in xenograft mouse and rhesus transplantation models
To evaluate the engraftment of gene-corrected CD34+ cells, gene-edited SCD CD34+ cells from 3 patient donors (4–5 × 105 cells per mouse) were transplanted into immunodeficient NOD/B6/SCID/IL-2rγ−/−/KitW41/W41 (NBSGW) mice (Figure 3A; Table S1). Efficient gene correction in vitro was observed in SCD CD34+ cells from all 3 donors at both DNA and protein levels (Figures 3B, 3C, and S2B). During the first 24 weeks after xenograft transplantation of gene-edited CD34+ cells, 8%–29% human cell engraftment was observed (Figure 3D); 34%–59% gene correction was detected at the early time point, but these levels decreased to 10%–38% over a 24-week follow-up (Figure 3E). The indel ratios were relatively similar between 4 weeks (28%–33%) and 24 weeks (13%–36%) post-transplant. We also observed a negative correlation between human cell engraftment and gene correction efficiency (p < 0.01), but not between engraftment and indels (Figures S3A and S3B), suggesting that gene correction in engrafting CD34+ cells is less efficient as compared to indels or that undergoing HDR is detrimental to engraftment. Off-target δ-globin gene editing was not detectable (<1%) 24 weeks post-transplant (Figure S3C). In addition, we performed a second set of xenograft mouse transplantation with gene-corrected SCD CD34+ cells, and 2%–7% gene correction and 23%–52% indels (p < 0.05) were observed in bone marrow cells 24 weeks post-transplant (Figures S3D and S3E), similar to peripheral blood data in the first set of transplantation (Figure 3E). We observed more efficient gene editing with Cas9 protein (HDR 2%–5%, indel 44%–52%) in xenograft bone marrow cells, compared to Cas9 mRNA (HDR 2%–7%, indel ∼23%); therefore, we used Cas9 protein for rhesus transplantation rather than Cas9 mRNA.
Figure 3.

Engraftment of gene-corrected SCD CD34+ cells in xenograft mice
(A) Xenograft mouse transplantation for SCD CD34+ cells from 3 donors following βs-to-β-globin gene correction.
(B) Gene correction (HDR) and indels in edited SCD CD34+ cells in vitro, evaluated by targeted deep sequencing (n = 1).
(C) Normal β-globin protein production in erythroid cells in vitro differentiated from edited SCD CD34+ cells, evaluated by RP-HPLC (n = 1).
(D) Engraftment of edited SCD CD34+ cells 4–24 weeks post-xenograft transplantation, evaluated by human CD45 expression in flow cytometry. ∗p < 0.05, ∗∗p < 0.01, evaluated by Dunnett’s test, compared with no electroporation (n = 2–5). SEM shown as error bars.
(E) Gene correction (HDR) and indels in peripheral blood cells 4–24 weeks post-xenograft transplantation, evaluated by targeted deep sequencing. ∗p < 0.05, ∗∗p < 0.01, evaluated by Dunnett’s test, compared with no electroporation (n = 1–5). SEM shown as error bars.
Given that there is no SCD non-human primate model, we decided to model SCD gene correction in rhesus macaques using a reverse β-to-βs globin conversion model of our design (Figure S4A). Mobilized rhesus CD34+ cells from 2 animals were electroporated to deliver rhesus β-globin-targeting guide RNA (the same target site as the SCD mutation-targeting guide RNA), Cas9 protein, and donor ssDNA including the SCD mutation (20A > T). We also added an adjuvant (i53 mRNA) to improve gene conversion (HDR) efficiencies.19,20 We observed high-efficiency genome editing without the adjuvant (29% ± 0% gene conversion and 63% ± 1% indels), and further enhanced genome editing with the adjuvant (56% ± 2% gene conversion and 37% ± 1% indels) (Figure S4B). Erythroid differentiation of edited CD34+ cells resulted in the production of βs-globin protein (∼100%) and undetectable production of normal β-globin in gene-edited cells, analyzed by hemoglobin electrophoresis (Figure S4C) and RP-HPLC (Figures S4D and S4E). High-level γ-globin expression was observed as a background of rhesus erythroid differentiation (Figure S2C), perhaps reducing a sensitivity of β-globin production.
We then evaluated the engraftment of gene-edited rhesus CD34+ cells with β-to-βs-globin conversion (n = 2, 13U005 and 12U011) to model gene correction in humans. Mobilized rhesus CD34+ cells (3.4 × 107 [5.9 × 106/kg] and 3.8 × 107 [6.8e × 106/kg], respectively) were electroporated to deliver editing tools and cryopreserved after electroporation (Figure 4A). Small aliquots of edited cells (before and after cryopreservation) were differentiated into erythroid cells in vitro, resulting in 17%–26% gene conversion and 57%–72% indels at the DNA level (Figure 4B) and 42%–100% of βs-globin production at the protein level (Figures 4C and S2D), with no difference observed between aliquots taken before and after cryopreservation. Lower amounts of βs-globin protein production was observed in 2nd-mobilization CD34+ cells in 13U005, likely due to slightly less efficient genome editing at the DNA level. Following myeloablative total body irradiation (9.5 Gy), the frozen edited CD34+ cells (2.2 × 107 [3.8e × 1066/kg] and 1.6 × 107 [2.9 × 106/kg], respectively) were injected into autologous macaques.
Figure 4.

Engraftment of gene-edited CD34+ cells with β-to-βs-globin conversion in rhesus macaques
(A) Rhesus transplantation of autologous CD34+ cells following β-to-βs-globin gene conversion in 2 animals (13U005 and 12U011).
(B) Gene conversion (HDR) and indels in edited rhesus CD34+ cells in in vitro culture, evaluated by targeted deep sequencing.
(C) βs-globin protein production in erythroid cells in vitro differentiated from edited rhesus CD34+ cells, evaluated by RP-HPLC.
(D) Gene correction (HDR) and indels in peripheral blood cells post-transplant, evaluated by targeted deep sequencing.
(E and F) βs-globin protein production in red blood cells post-transplant, evaluated by hemoglobin electrophoresis.
We observed a robust recovery of blood counts in 13U005 (without transfusion), while peripheral blood recovery was delayed in 12U011, which was supported by whole-blood transfusion at days 28 and 39 and platelet-rich plasma transfusion at days 23, 30, and 33 (Figures S4F and S4G). In 13U005 and 12U011, 7%–11% and 1%–2% of β-globin gene conversion were observed in both granulocytes and lymphocytes at the early time point (3 weeks) post-transplant, respectively, and the gene conversion ratios plateaued at ∼1% in both animals 10–16 weeks post-transplant (Figure 4D). At the early time point, 44%–54% and 45%–51% of indels were detected, but these levels plateaued to 8%–10% and 7%–8%, respectively. HbS production in RBCs was ∼17% and ∼8% at the early time point in 13U005 and 12U011 (Figure 4E), plateauing at ∼10% and ∼3% 10–12 months post-transplant, respectively (Figure 4F). Interestingly, 10%–30% of HbF production was observed at early time points post-transplant in 12U011, but was undetectable in 13U005, likely due to stress hematopoiesis accompanying the delayed recovery (Figure S2E). E7V amino acid replacement (resulting from β-to-βs-globin conversion) and E7 deletion (resulting from indel-mediating GAG deletion in the SCD mutation site) were detected in β-globin protein from both animals 5–6 months post-transplant (Figures S4H and S4I). These data demonstrate that gene-edited CD34+ cells with gene conversion and indels are engraftable in xenograft mice to 6 months and in rhesus macaques to 10–12 months.
Discussion
We developed an efficient gene correction strategy for the SCD mutation in the β-globin gene with electroporation-mediated delivery of editing tools into SCD CD34+ cells, achieving therapeutic-level gene correction at the DNA level (∼30%) and at the protein level (∼80%) (Figure 1). This virus-free gene correction method using SCD mutation-targeting guide RNA, Cas9 mRNA/protein, and donor ssDNA encoding the normal β-globin sequence (without silent mutations in the guide RNA targeting sites) results in seamless correction to the normal wild-type β-globin sequence without any editing footprint. The gene-edited SCD CD34+ cells with correction to normal β-globin gene sequence were engraftable for 6 months as measured in xenografts in immunodeficient mice (Figure 3), and engraftment of gene-edited CD34+ cells with conversion from normal to βs-globin gene sequence was observed for 10–12 months post-transplant in a rhesus HSC gene correction model (Figure 4).
SCD is the most common single-gene disorder; therefore, development of a curative strategy for SCD is a primary goal in the field of gene therapy research.19,21 In the present gene therapy approach, a therapeutic β-globin gene is added to CD34+ HSCs with a lentiviral vector, allowing for therapeutic-level gene marking and phenotypic correction in SCD.7 However, the lentiviral vector backbone is integrated throughout the HSC genome, potentially inducing insertional mutagenesis. Unlike lentiviral gene therapies, gene correction methods can repair the SCD mutation without genome-wide vector integration. Initial research into SCD gene correction used AAV6 vector delivery of large-size donor DNA, which encoded a β-globin gene with silent mutations (Table 1).8 This method allows the donor DNA to escape from guide RNA targeting; however, the corrected β-globin gene included these silent mutations as footprints of genome editing, which should not change the resulting amino acid sequence but may affect its transcriptional activity, since codon optimization using silent mutations was demonstrated to change gene expression levels,22,23 and various transcriptional factor binding sites exist around the β-globin gene.24 In addition, while recombinant AAV vectors generally persist episomally, the random integration of AAV vector genomes was reported in a canine gene therapy model.25 Recently, donor ssDNA was used for β-globin gene editing (Table 1); however, gene correction data in SCD CD34+ cells was limited, and almost all donor DNAs included silent mutation(s) to escape from guide RNA targeting. AAV-based delivery of large-sized donor DNA permits the increase of gene correction in CD34+ cells in vitro, compared to electroporation-mediated donor ssDNA delivery; however, gene correction for engrafting cells in xenograft mice has not yet achieved the therapeutic threshold (∼20%) in SCD. In this study, we designed SCD mutation-targeting guide RNA that can specifically target the mutation in the βs-globin gene and not the normal β-globin sequence, allowing us to use normal wild-type β-globin sequence donor 100-mer ssDNA without introducing silent mutations in the target site. We also used viral vector-free electroporation-mediated delivery of editing tools, allowing for gene correction in the SCD mutation without potential integration of vector backbone or the potential adverse cellular responses that can occur with the AAV vector for delivery of donor sequence. This viral vector-free non-footprint gene correction method should result in transcriptional activity that is indistinguishable from that of normal and an improved safety profile that would be beneficial for clinical translation.
Previous phenotypic analysis in SCD patients suggests that 20% HbF (or potentially HbA) per RBC is a therapeutic threshold to reduce clinical events,26 and in allogeneic HSC transplantation following reduced intensity conditioning, 20% donor cell chimerism (mostly HbAS donor) can overcome the SCD phenotype.17 Based on this rationale, we expect that ≥20% β-globin correction per cell at the protein level (allowed by both biallelic and monoallelic correction) in ≥20% RBCs derived from gene-corrected HSCs is needed to produce long-term therapeutic effects. In this study, our viral vector-free non-footprint method allowed for ∼30% of gene correction (∼20% biallelic and ∼20% monoallelic correction) at the DNA level and ∼80% of normal β-globin production at the protein level in SCD CD34+ cells in vitro (Figure 1), which should be sufficient for initiating animal experiments to evaluate the engraftment of gene-edited CD34+ cells. Indels (∼50%) in the β-globin gene lead to a frameshift mutation (except when indel base pairs are divisible by 3); therefore, indels should strongly reduce RBC generation similar to β-thalassemia. Based on this observation, ∼30% of gene correction and ∼10% of remaining SCD mutation results in ∼80% of normal β-globin protein production (Figure 1). Similarly, β-to-βs-globin gene conversion ratios at the protein level (8%–17%) in rhesus RBCs were higher than gene conversion ratios at the DNA level (1%–11%) in granulocytes and lymphocytes, likely due to indel (44%–54%)-mediated inadequate erythropoiesis (Figure 4). If high-level indels (near 100%) were obtained in the β-globin gene, it may induce severe anemia such as β-thalassemia major; however, in the present transplanted macaques, hemoglobin concentration and RBC counts were recovered to the normal range (Figures S4F and S4G), probably due to insufficient indel levels for the thalassemia-major phenotype. Interestingly, E7 deletion (indel-mediating GAG deletion) was detected at both DNA (3%–4%) and protein levels in transplanted rhesus macaques (Figures S4H and S4I), and the E7 deletion was previously described as Hb Leiden with similar oxygen affinity and anti-sickling effects as normal HbA.27 The E7 deletion (GAG) may be enhanced up to detectable levels by microhomology in the rhesus β-globin gene (GAGGAG). Indels in the βs-globin gene can reduce pathogenic HbS production, allowing one to prevent the sickling of RBCs;28 thus, they should contribute positively to SCD gene correction therapy. This high-efficiency gene editing method would allow for therapeutic-level gene correction in SCD CD34+ cells assessed in vitro.
HDR-mediated gene correction is thought to be enhanced by cell proliferation; however, cell proliferation may reduce the engraftment of gene-edited CD34+ HSCs.29,30 To investigate this hypothesis, we evaluated gene correction efficiencies in subpopulations and cell cycles in edited CD34+ cells 2 days after electroporation, since cell viability is generally reduced 1–2 days post-electroporation. In addition, the CD34+CD133+CD90+ fraction should contain engrafting cells at both day 2 (for electroporation) and day 4 (for cell sorting), and the ratios of G0/G1, S, and G2/M are similar between days 2 and 4 during CD34+ cell culture.31,32 HDR is thought to be regulated during the cell cycle;30 however, similar editing ratios were observed among subpopulations as well as cell cycles (Figure 2), and we also demonstrated similar editing efficiencies before and after erythroid differentiation in edited SCD CD34+ cells (Figure S1E). Similar editing ratios among subpopulations were previously described in healthy donor CD34+ cells with another target.33 These data demonstrate that gene editing efficiency does not vary between the subpopulations of CD34+ cells that we evaluated under the present conditions. To further investigate this hypothesis, we evaluated gene editing efficiency in repopulating blood cells derived from gene-edited CD34+ HSCs in xenograft mouse and non-human primate transplantation models. We observed the engraftment of gene-corrected human CD34+ cells from 3 SCD patient donors up to 6 months post-transplant in xenograft mice (Figure 3), and the converse β-to-βs-globin gene-converted CD34+ cells exhibited engraftment 10–12 months after rhesus transplantation (Figure 4), demonstrating that β-globin gene-edited HDR+ cells engraft in both xenograft mice and rhesus macaques. Multi-lineage engraftment of gene-modified CD34+ cells was previously demonstrated in xenograft mice.34 We also observed multi-lineage engraftment of gene-edited rhesus CD34+ cells in the present study among granulocytes and lymphocytes evaluated by DNA analysis (Figure 4), as well as RBCs evaluated by protein analysis (Figure 4). We did, however, observe a reduction in editing ratios during the first 3 months post-transplant, and editing levels plateaued over longer follow-up. Those trends can be explained by the fact that blood cells are still being reconstituted from hematopoietic progenitors during the first 3 months post-transplant, followed by long-term reconstitution derived from HSCs.35,36 This decrease in HDR+ cells is reminiscent of early studies by us and others examining retroviral gene transfer. Despite high efficiencies predicted by mouse models and various in vitro surrogate HSC assays, a marked decrease in engraftment was noted in both rhesus and human settings.37, 38, 39 Overall, these data demonstrate that gene-edited HDR+ cells are engraftable, as evaluated by xenograft mouse and rhesus HSC transplantation models. Long-term analysis following transplantation is thus essential to evaluate gene editing at the HSC level.
Xenograft mouse transplantation models are used to evaluate the blood reconstitution of human CD34+ cells; however, severe immunodeficiency in recipient mice is required for human cell engraftment.40 In our previous research evaluating lentiviral gene marking in HSCs, the engraftment of gene-modified CD34+ cells in xenograft mice appears to be overestimated compared to more relevant non-human primate transplantation, perhaps due to the unique immunological state that is required for xenograft transplantation.21,41, 42, 43, 44, 45 Because results from xenograft mouse transplantation are not necessarily predictive of human cell engraftment, non-human primate models are meaningful and desirable for translational research. Once we can reliably achieve therapeutic levels of gene modification without severe toxicity in rhesus macaques, we can move to clinical translation. In this study, the engraftment of gene-edited HDR+ cells was detected in peripheral blood cells from transplanted rhesus macaques at both DNA and protein levels; however, HDR ratios (∼1%) were not yet at the therapeutic threshold (∼20%) after gene-editing levels plateaued 3–4 months post-transplant (Figure 4). The adjuvant of i53 mRNA was used for both small-scale rhesus CD34+ cell editing in in vitro culture (Figures S4A–S4E) and large-scale rhesus CD34+ cell editing for transplantation (Figure 4). Gene conversion was less efficient in the large-scale editing, compared to the small scale, however. Further optimization of large-scale gene conversion in rhesus CD34+ cells should improve editing efficiency up to similar levels as the small scale, possibly allowing for long-term engraftment of gene-edited CD34+ cells at the therapeutic level sufficient for clinical translation.
In the analysis of βs-to-β-globin gene correction by targeted deep sequencing, false positive in no electroporation controls was detected up to 1%–5% in in vitro samples and >10% in xenograft samples. In rhesus β-to-βs-globin gene conversion, ∼0.1% HDR and ∼3% indels were detected in no electroporation controls. The background levels in β-globin gene correction are higher than the general error rates (∼0.1% per nucleotide), usually caused by PCR for preparing a DNA library and linear amplification for the sequencing process.46 However, the β-globin gene has strong homology to other β-globin series (ε-, γ1-, γ2-, and δ-globin genes) in the human genome as well as all β-globin series in the mouse genome, and these similar DNA sequences can induce skipping PCR,47 possibly resulting in false positives of β-globin gene correction as well as false negatives of β-to-βs-globin gene conversion. The false positive editing in β-globin gene (∼2%) was also reported by others.48 Therefore, a no-editing control should be important for targeted deep sequencing in the β-globin gene to evaluate a background level of gene editing rates.
HbF induction is an alternative genome editing strategy to potentially cure SCD. Potential treatment modalities include editing the BCL11A erythroid-specific enhancer or the γ-globin promoter (BCL11A-binding site editing), or alternatively creating a larger deletion in the δ-globin gene as seen in the hereditary persistence of fetal hemoglobin (HPFH).49, 50, 51 HbF induction can result from DNA breakage (indels) within these regions; thus, HDR is not needed for this strategy. Using the available genome editing technologies, this indel-only editing for HbF induction should be more efficient at the DNA level than HDR-mediated SCD gene correction. However, indel-mediated HbF induction could be less efficient at the protein level compared to HDR-mediated normal HbA production in SCD, since indel-mediated BCL11A interference cannot achieve 100% of HbF induction, but correction of the β-globin gene produces normal HbA only, with indels primarily leading to disrupted βs-globin production. In addition, BCL11A erythroid-specific enhancer editing may reduce erythroid output, as evaluated in xenograft mice.52 Indel ratios in BCL11A erythroid-specific enhancer editing decreased significantly over long-term follow-up in non-human primate transplantation models.53,54 The long-term persistence of HbF induction should be considered in clinical trials for BCL11A interference.
In summary, we developed a high-efficiency viral vector-free non-footprint gene correction method in SCD CD34+ cells, resulting in ∼30% gene correction at the DNA level and ∼80% normal β-globin production at the protein level. Gene-edited SCD CD34+ cells were engraftable 6 months post-xenograft transplantation. We also developed a rhesus β-to-βs-globin conversion model with HSC-targeted genome editing, resulting in the engraftment of gene-edited cells 10–12 months post-transplant in rhesus macaques. Our β-to-βs-globin gene conversion large-animal model demonstrates the persistence of HDR at significant levels, yet below the therapeutic threshold. It should allow us to comprehensively test strategies to improve the levels to the therapeutic threshold, permitting us to proceed to clinical application only after such levels are established. These findings are helpful in designing future HSC-targeted gene correction trials.
Limitations of study
While high levels of gene correction among human cells in vitro and in xenografted mice are now feasible, results in the rhesus HSC transplantation model demonstrated that the predictions made by these in vitro and in vivo assays overestimate the levels that are achievable in large animals and, likely, humans. These results demonstrate the limitations of these assays. Furthermore, longer follow-up of engrafted gene-edited CD34+ cells and evaluation of off-target effects are preferable in rhesus HSC transplantation, following further optimization to improve the engraftment of gene-edited rhesus CD34+ cells to the levels needed for therapeutic benefit in humans.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD34 antibody | BD Biosciences | Clone 581; RRID: AB_10563908 |
| CD90 antibody | BD Biosciences | Clone 5E10; RRID: AB_395970 |
| CD133 antibody | BD Biosciences | Clone W6B3C1; RRID: AB_2744280 |
| Human CD45 antibody | BD Biosciences | Clone HI30; RRID: AB_2033960 |
| Biological samples | ||
| SCD CD34+ cells | NHLBI, NIH | This paper (Donor information was shown in Table S2.) |
| Healthy donor CD34+ cells | NHLBI, NIH | This paper (Donor information was shown in Table S2.) |
| Chemicals, peptides, and recombinant proteins | ||
| Cas9 protein | MacroLab Facility | University of California, Berkeley, CA, USA |
| Stem cell factor | R&D Systems | 255-SC |
| FMS-related tyrosine kinase 3 ligand | R&D Systems | 308-FK |
| Thrombopoietin | R&D Systems | 288-TP |
| Erythropoietin | AMGEN | EPOGEN ® |
| Interleukin 3 | R&D Systems | 203-IL |
| Dexamethasone | VETone | 501012 |
| Estradiol | Pfizer | DEPO®-ESTRADIOL |
| Bovine serum albumin | Roche | 10738328103 |
| Busulfan | PDL BioPharma | BUSULFEX® |
| Experimental models: Cell lines | ||
| SCD B cell line | This paper | Not applicable |
| Experimental models: Organisms/strains | ||
| NBSGW mice | The Jackson Laboratory | 026622 |
| Rhesus macaque | NIH Animal Center | Not applicable |
| Oligonucleotides | ||
| SCD mutation-targeting guide RNA | Synthego Corporation | 5′-GUA ACG GCA GAC UUC UCC AC-3′ |
| Normal β-globin gene-targeting guide RNA | Synthego Corporation | 5′-GUA ACG GCA GAC UUC UCC UC-3′ |
| Rhesus β-globin-targeting guide RNA | Synthego Corporation | 5′-GUG ACG GCA UUC UUC UCC UC-3′ |
| Normal β-globin donor DNA | Integrated DNA Technologies, Inc. | 5′-TTC ATC CAC GTT CAC CTT GCC CCA CAG GGC AGT AAC GGC AGA CTT CTC CTC AGG AGT CAG ATG CAC CAT GGT GTC TGT TTG AGG TTG CTA GTG AAC ACA G-3′ |
| βs-globin donor DNA | Integrated DNA Technologies, Inc. | 5′-TTC ATC CAC GTT CAC CTT GCC CCA CAG GGC AGT AAC GGC AGA CTT CTC CAC AGG AGT CAG ATG CAC CAT GGT GTC TGT TTG AGG TTG CTA GTG AAC ACA G-3′ |
| Rhesus βs-globin donor DNA | Integrated DNA Technologies, Inc. | 5′-TTC ATC CAC GTT CAC CTT GCC CCA CAG GGT GGT GAC GGC ATT CTT CTC CAC AGG AGT CAG ATG CAC CAT GGT GTC TGT TTG AGG TTG CTC GTG AAC ACA G-3′ |
| Primers and probes | This paper | Table S3 |
| Software and algorithms | ||
| CRISPResso2 | Pinello Lab, Massachusetts General Hospital / Harvard Medical School, Boston, MA, USA | https://crispresso.pinellolab.partners.org/ |
| FlowJo v10 | BD Biosciences | https://www.flowjo.com/ |
| JMP 14 | SAS Institute Inc | https://www.jmp.com/en_us/home.geo.html |
| Other | ||
| StemSpan SFEM media | StemCell Technologies | 09650 |
| Iscove’s Modified Dulbecco’s Medium | Corning | 10-016-CV |
| KnockOut Serum Replacement | Thermo Fisher Scientific | 10828028 |
| MaxCyte GT System | MaxCyte, Inc. | https://maxcyte.com/gtx/ |
| Hemoglobin electrophoresis | Helena Laboratories | 4063 |
| QuantStudio 6 Flex Real-Time PCR System | Thermo Fisher Scientific | 4485689 |
| FACSAria II | BD Biosciences | 643180 |
| FACSCanto | BD Biosciences | 338960 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Naoya Uchida (uchidan@nhlbi.nih.gov).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate or analyze datasets.
Experimental model and subject details
Human donors
Granulocyte-colony stimulating factor-mobilized CD34+ cells from healthy donors and plerixafor-mobilized CD34+ cells from SCD patients were collected under studies (08-H-0156, 17-H-0124, and 03-H-0015) that were approved by the Institutional Review Board of the National Heart, Lung, and Blood Institute (NHLBI) (Table S2).55,56 Specific information on the donor demographics (genotype, age, and gender) are outlined in Table S2. CD34+ cells were cultured in serum free StemSpan SFEM media (StemCell Technologies, Vancouver, BC, Canada) containing 100ng/ml each of stem cell factor (SCF, R&D Systems, Minneapolis, MN, USA), FMS-related tyrosine kinase 3 ligand (FL, R&D Systems), and thrombopoietin (TPO, R&D Systems).
Non-human primates
Rhesus macaques were maintained, following the guidelines set out by the Public Health Service Policy on Humane Care and Use of Laboratory Animals under a protocol (H-0136) approved by the Animal Care and Use Committee of the NHLBI (information on genotype, age, and sex can be found in Table S2).35,36,57 Granulocyte colony-stimulating factor (Amgen, Thousand Oaks, CA, USA) and plerixafor (Amgen)-mobilized rhesus CD34+ cells were pre-stimulated in StemSpan containing 100ng/ml each of SCF, FL, and TPO for 2 days and electroporated to deliver editing tools.19 The gene-edited CD34+ cells were frozen several hours post-electroporation, and the frozen CD34+ cells were thawed and injected into autologous macaques (13U005: 5.9 years, female, and 12U011: 7.0 years, female) following total body irradiation (2x4.75Gy).
Mice
NOD/B6/SCID/IL2rγ−/−/KitW41/W41 mice (Jackson Laboratory, Bar Harbor, ME, USA) were maintained in a pathogen-free facility, following an animal care and use protocol (H-0228) approved by the Animal Care and Use Committee of the NHLBI (information on genotype, age, and sex can be found in Table S2). SCD CD34+ cells were pre-stimulated in StemSpam media containing the same cytokines (SCF, FL, and TPO) for 2 days and electroporated to deliver editing tools. The next day, gene-edited SCD CD34+ cells were injected into 7-week-old male mice.42,58,59 These mice received sublethal busulfan conditioning (25mg/kg, Busulfex, PDL BioPharma, Redwood City, CA, USA) 2 days before transplantation.
Cell line
Steady-state peripheral blood mononuclear cells (PBMCs) from SCD patients were collected under studies (08-H-0156, 17-H-0124, and 03-H-0015) that were approved by the Institutional Review Board of the NHLBI (Table S2). A SCD B cell line was established from SCD PBMCs with Epstein-Barr virus, as previously described.33 The SCD B cell line was cultured in Roswell Park Memorial Institute 1640 (RPMI, Corning, Tewksbury, MA, USA) including 20% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA).
Method details
Electroporation of human CD34+ cells to deliver guide RNA, Cas9 endonuclease, and donor DNA
Granulocyte-colony stimulating factor-mobilized CD34+ cells from healthy donors, and plerixafor-mobilized CD34+ cells and steady-state PBMCs from SCD patients were collected under studies (08-H-0156, 17-H-0124, and 03-H-0015) that were approved by the Institutional Review Board of the NHLBI.55,56 All individuals gave written informed consent for the sample donation and consent documents are maintained in the donor’s medical records. The consent document was approved by the Institutional Review Board prior to study initiation and is reviewed and updated annually. A SCD B cell line was established from SCD PBMCs with Epstein-Barr virus.33
SCD CD34+ cells were pre-stimulated for 2 days in serum free StemSpan SFEM media (StemCell Technologies, Vancouver, BC, Canada) containing 100ng/ml each of SCF (R&D Systems), FL (R&D Systems), and TPO (R&D Systems). The CD34+ cells were electroporated with a MaxCyte GT System (HSPC34-3 protocol, MaxCyte, Gaithersburg, MD, USA) to deliver 50 μg/ml modified guide RNA or 200 μg/ml unmodified guide RNA, 200 μg/ml Cas9 mRNA or 200 μg/ml Cas9 protein (MacroLab, University of California, Berkeley, CA, USA), and 80-200 μg/ml donor ssDNA. SCD mutation-targeting guide RNA (5′-GUA ACG GCA GAC UUC UCC AC-3′) and normal β-globin donor DNA (5′-TTC ATC CAC GTT CAC CTT GCC CCA CAG GGC AGT AAC GGC AGA CTT CTC CTC AGG AGT CAG ATG CAC CAT GGT GTC TGT TTG AGG TTG CTA GTG AAC ACA G-3′) were used for human βs-to-β-globin gene correction, while in human β-to-βs-globin gene conversion, normal β-globin gene-targeting guide RNA (5′-GUA ACG GCA GAC UUC UCC UC-3′) and βs-globin donor DNA (5′-TTC ATC CAC GTT CAC CTT GCC CCA CAG GGC AGT AAC GGC AGA CTT CTC CAC AGG AGT CAG ATG CAC CAT GGT GTC TGT TTG AGG TTG CTA GTG AAC ACA G-3′) were used. HindIII site insertion donor DNA (5′- TC ATC CAC GTT CAC CTT GCC CCA CAG GGC AGT AAC GGC AGA CTT CTC AAG CTT CAC AGG AGT CAG ATG CAC CAT GGT GTC TGT TTG AGG TTG CTA GTG AA-3′) was used for HindIII site insertion experiments. Cell viability was evaluated by trypan blue staining 1 day and 2 days post-electroporation.
One day post-electroporation, electroporated cells were differentiated into erythroid cells using differentiation media for 5 days with Iscove’s Modified Dulbecco’s Medium (IMDM, Corning) containing 20% KnockOut Serum Replacement (KSR, Thermo Fisher Scientific), 2U/ml erythropoietin (EPO, Amgen), 10ng/ml SCF, 1.0ng/ml interleukin 3 (R&D systems), 1.0 μM dexamethasone (VETone, Boise, ID, USA), 1.0 μM estradiol (Pfizer, NY, NY, USA) followed by maturation media for 11-13 days with IMDM containing 2U/ml EPO, 10ng/ml insulin (Lilly, Indianapolis, IN, USA), 0.5mg/ml holo-transferrin (Sigma Aldrich, Saint Louis, MO, USA), and 2% bovine serum albumin (Roche, Indianapolis, IN, USA).60
Efficiency of HDR (βs-to-β-globin gene correction) and indels was evaluated at DNA levels by targeted deep sequencing or quantitative PCR (qPCR)-based single nucleotide polymorphism (SNP) genotyping. Normal β-globin and βs-globin protein production was evaluated by hemoglobin electrophoresis (Helena Laboratories, Beaumont, TX, USA) and RP-HPLC.21,61
Targeted deep sequencing to evaluate β-globin gene editing
Efficiency of HDR (βs-to-β-globin gene correction) and indels was evaluated in gene-edited cells by targeted deep sequencing. We amplified both human β-globin gene and δ-globin gene using universal primers to detect both genes: BDG targeted seq F1 (5′-GTG TTC ACT AGC AAC CTC AAA-3′) and BDG targeted seq R1 (5′-ACC CAA GAG TCT TCT CTG TC-3′) and added P5 and P7 adapters using several forward and reverse primer sets, respectively: BDG targeted seq F2a (5′-AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT CTT TCC CTA CAC GAC GCT CTT CCG ATC TAC TAG CAA CCT CAA ACA GAC ACC-3′) and BDG targeted seq R2a (5′-CAA GCA GAA GAC GGC ATA CGA GAT GTC GTG ATG TGA CTG GAG TTC AGA CGT GTG CTC TTC CGA TCT AAC CTT GAT ACC AAC CTG CCC-3′), BDG targeted seq F2b (5′-AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT CTT TCC CTA CAC GAC GCT CTT CCG ATC TCA CTA GCA ACC TCA AAC AGA CAC C-3′) and BDG targeted seq R2b (5′-CAA GCA GAA GAC GGC ATA CGA GAT GTC GTG ATG TGA CTG GAG TTC AGA CGT GTG CTC TTC CGA TCA ACC TTG ATA CCA ACC TGC CC-3′), BDG targeted seq F2c (5′-AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT CTT TCC CTA CAC GAC GCT CTT CCG ATC TTT CAC TAG CAA CCT CAA ACA GAC ACC-3′) and BDG targeted seq R2c (5′-CAA GCA GAA GAC GGC ATA CGA GAT GTC GTG ATG TGA CTG GAG TTC AGA CGT GTG CTC TTC CGA TCC TTG ATA CCA ACC TGC CCA-3′), and BDG targeted seq F2d (5′-AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT CTT TCC CTA CAC GAC GCT CTT CCG ATC TGT TCA CTA GCA ACC TCA AAC AGA CAC C-3′) and BDG targeted seq R2d (5′-CAA GCA GAA GAC GGC ATA CGA GAT GTC GTG ATG TGA CTG GAG TTC AGA CGT GTG CTC TTC CGA TCT TGA TAC CAA CCT GCC CA-3′). HDR and indels in β-globin or δ-globin gene were detected by DNA sequencing. Deep sequencing and analysis were performed by the Cancer Research Technology Program, Leidos Biomedical Research, Inc., Frederick National Laboratory for Cancer Research.
Rhesus β-globin gene was amplified by a primer set containing partial P5 and P7 adaptor sequences: rBG TargSeq F1 GW2 (5′-ACA CTC TTT CCC TAC ACG ACG CTC TTC CGA TCT AGT CAG GGC AGA GCC ATC TAT TG-3′) and rBG TargSeq R1 GW2 (5′-GAC TGG AGT TCA GAC GTG TGC TCT TCC GAT CTT GCC TCC TTA AGC ATG TCT TGT AAC C-3′). Deep sequencing was performed by GENEWIZ (South Plainfield, NJ, USA), and DNA sequences were analyzed by the CRISPResso2 online software.58
Quantitative polymerase chain reaction (qPCR)-based single nucleotide polymorphism (SNP) genotyping
In CFUs,36 the SCD mutation and normal β-globin sequences were separately detected by qPCR-based SNP genotyping (QuantStudio 6 Flex Real-Time PCR System, Thermo Fisher Scientific) using Sickle FWD4 primer: 5′-GG CAG AGC CAT CTA TTG CTT AC-3′, Sickle REV2 primer: 5′-CCA ACT TCA TCC ACG TTC ACC-3′, Sickle FAM probe (SCD mutation): 5′-FAM-CTG ACT CCT GTG GAG AA-3′, Sickle VIC probe (normal β-globin): 5′-VIC-CTG ACT CCT GAG GAG AA-3′, and Sickle Reference probe (for all β-globin sequences): 5′-Cy5-CCT CAA ACA GAC ACC AT-3′.41 The SCD mutation and normal β-globin signals were standardized by the sickle reference, and the percentages of SCD mutation and normal β-globin were measured by comparison to a βs-globin control (100% of SCD mutation) and an intact β-globin control (100% of intact β-globin), respectively. The indel percentages were calculated by “indel (%) = reference (100%) - SCD mutation (%) - normal β-globin (%).”
Cell sorting in flow cytometry
Healthy donor CD34+ cells were pre-stimulated for 2 days and electroporated for β-to-βs-globin conversion. Two days post-electroporation, gene-edited CD34+ cells were stained with CD34 (clone 581, BD Biosciences, East Rutherford, NJ, USA), CD90 (clone 5E10, BD Biosciences), and CD133 (clone W6B3C1, BD Biosciences) antibodies or propidium iodide (PI/RNase Staining Buffer, BD Biosciences) and sorted by subpopulation (CD34+CD133+CD90+, CD34+CD133+CD90-, CD34+CD133-, and CD34-) or stage in the cell cycle (G0/G1, S, and G2/M) by flow cytometry (FACSAria II, BD Biosciences), respectively (Figures S1L and S1M).9,33,62 Each fraction was separately analyzed by targeted deep sequencing.
Xenograft transplantation of gene-edited SCD CD34+ cells
SCD CD34+ cells from three donors were pre-stimulated in StemSpam media containing the same cytokines (SCF, FL, and TPO) for 2 days and electroporated to deliver SCD mutation-targeting guide RNA, Cas9 mRNA, and normal β-globin donor DNA. The next day, gene-edited SCD CD34+ cells were injected into 7-week-old male NOD/B6/SCID/IL2rγ−/−/KitW41/W41 mice (NOD.Cg-KitW-41J Tyr + Prkdcscid Il2rgtm1Wjl/ThomJ, Jackson Laboratory).42,58,59 These mice received sublethal busulfan conditioning (25mg/kg, Busulfex, PDL BioPharma) 2 days before transplantation. The use of mice in transplantation experiment (H-0228) was approved by the Animal Care and Use Committee of the NHLBI. A small aliquot of gene-edited CD34+ cells were cultured to evaluate in vitro βs-to-β-globin gene correction at DNA and protein levels. After transplantation, human cell engraftment was evaluated by human CD45 antibody (clone HI30; BD Biosciences) in flow cytometry (FACSCanto, BD Biosciences) during the first 6 months post-transplant.
Autologous gene-edited CD34+ cell transplantation in rhesus macaques
We previously developed an autologous HSC transplantation model with lentiviral gene modification in rhesus macaques, following the guidelines set out by the Public Health Service Policy on Humane Care and Use of Laboratory Animals under a protocol (H-0136) approved by the Animal Care and Use Committee of the NHLBI.35,36,57 Granulocyte colony-stimulating factor (Amgen) and plerixafor (Amgen)-mobilized rhesus CD34+ cells were pre-stimulated in StemSpan containing 100ng/ml each of SCF, FL, and TPO for 2 days and electroporated to deliver rhesus β-globin-targeting guide RNA (5′-GUG ACG GCA UUC UUC UCC UC-3′), Cas9 protein, βs-globin donor DNA including a SCD mutation (5′-TTC ATC CAC GTT CAC CTT GCC CCA CAG GGT GGT GAC GGC ATT CTT CTC CAC AGG AGT CAG ATG CAC CAT GGT GTC TGT TTG AGG TTG CTC GTG AAC ACA G-3′), and an adjuvant to enhance HDR (100 μg/ml i53 mRNA, CELLSCRIPT, Madison, WI, USA).19 The gene-edited CD34+ cells were frozen several hours post-electroporation, and the frozen CD34+ cells were thawed and injected into autologous macaques following total body irradiation (2x4.75Gy). A small aliquot of gene-edited CD34+ cells were cultured to evaluate in vitro β-to-βs-globin gene conversion at DNA and protein levels.
After engraftment of gene-edited cells, β-to-βs-globin gene conversion (HDR) and indels were evaluated in granulocytes and lymphocytes by targeted deep sequencing. βs-globin protein production in red blood cells was evaluated by hemoglobin electrophoresis and mass spectrometry,21 to confirm the rhesus β- and βs-globin peaks in RP-HPLC.
Quantification and statistical analysis
Statistical analyses were performed using the JMP 14 software (SAS Institute Inc., Cary, NC, USA). Two averages were evaluated with Student’s t test. The averages in various conditions were evaluated by Dunnett’s test (one-way ANOVA for a control). A p value of < 0.01 or 0.05 was deemed significant. Standard error of the mean was shown as error bars in all figures.
Acknowledgments
This work was supported by the intramural research program of the National Heart, Lung, and Blood Institute (NHLBI) and the National Institute of Diabetes, Digestive and Kidney Diseases (NIDDK) at NIH. The contributions of authors H.M. and S.D. were supported by the intramural research program of the National Institute of Allergy and Infectious Diseases (NIAID) at NIH. The contributions of L.L., C.A., and S.S.D.R. were supported by Maryland Stem Cell Research Fund (MSCRF) grant 2015-MSCRFP-1752. We thank the animal care staff and technicians for their care and handling of animals. We thank Dr. Duck-Yeon Lee from the NHLBI Biochemistry Core for RP-HPLC analysis help. We thank Keyvan Keyvanfar at the NHLBI for help with flow cytometry and cell sorting. We thank Drs. Marjan Gucek and Yanling Yang from the NHLBI Proteomics Core for mass spectrometry analysis help. We thank Dr. Xiaolin Wu from the Cancer Research Technology Program, Leidos Biomedical Research, Frederick National Laboratory for Cancer Research, for targeted deep sequencing analysis help. We thank CELLSCRIPT (Madison, WI, USA) for help with i53 mRNA production.
Author contributions
N.U. designed the research, performed the experiments, analyzed the results, created the figures, and wrote the paper; L.L. designed the research, performed the experiments, and analyzed the results; T.N. performed the experiments; C.M.D. performed the experiments and wrote the paper; M.Y. performed the experiments; J.G. performed the experiments; J.J.H.-M. performed the experiments; S.D. performed the experiments; A.C.B. performed the experiments; A.E.K. performed the experiments; N.S.L. performed the experiments; C.A. designed the research; M.V.P. designed the research; S.S.D.R. designed the research; R.E.D. designed the research; H.L.M. designed the research; and J.F.T. designed the research and wrote the paper.
Declaration of interests
L.L., C.A., and M.V.P. were employees at MaxCyte during the period of this work.
Published: April 20, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100247.
Supplemental information
References
- 1.Steinberg M.H. Management of sickle cell disease. N. Engl. J. Med. 1999;340:1021–1030. doi: 10.1056/NEJM199904013401307. [DOI] [PubMed] [Google Scholar]
- 2.Bridges K.R., Barabino G.D., Brugnara C., Cho M.R., Christoph G.W., Dover G., Ewenstein B.M., Golan D.E., Guttmann C.R., Hofrichter J. A multiparameter analysis of sickle erythrocytes in patients undergoing hydroxyurea therapy. Blood. 1996;88:4701–4710. [PubMed] [Google Scholar]
- 3.Ataga K.I., Kutlar A., Kanter J., Liles D., Cancado R., Friedrisch J., Guthrie T.H., Knight-Madden J., Alvarez O.A., Gordeuk V.R. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N. Engl. J. Med. 2017;376:429–439. doi: 10.1056/NEJMoa1611770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Niihara Y., Miller S.T., Kanter J., Lanzkron S., Smith W.R., Hsu L.L., Gordeuk V.R., Viswanathan K., Sarnaik S., Osunkwo I., Investigators of the Phase 3 Trial of l-Glutamine in Sickle Cell Disease A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N. Engl. J. Med. 2018;379:226–235. doi: 10.1056/NEJMoa1715971. [DOI] [PubMed] [Google Scholar]
- 5.Hsieh M.M., Fitzhugh C.D., Weitzel R.P., Link M.E., Coles W.A., Zhao X., Rodgers G.P., Powell J.D., Tisdale J.F. Nonmyeloablative HLA-matched sibling allogeneic hematopoietic stem cell transplantation for severe sickle cell phenotype. JAMA. 2014;312:48–56. doi: 10.1001/jama.2014.7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hsieh M.M., Kang E.M., Fitzhugh C.D., Link M.B., Bolan C.D., Kurlander R., Childs R.W., Rodgers G.P., Powell J.D., Tisdale J.F. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N. Engl. J. Med. 2009;361:2309–2317. doi: 10.1056/NEJMoa0904971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ribeil J.A., Hacein-Bey-Abina S., Payen E., Magnani A., Semeraro M., Magrin E., Caccavelli L., Neven B., Bourget P., El Nemer W. Gene Therapy in a Patient with Sickle Cell Disease. N. Engl. J. Med. 2017;376:848–855. doi: 10.1056/NEJMoa1609677. [DOI] [PubMed] [Google Scholar]
- 8.Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haro-Mora J.J., Uchida N., Demirci S., Wang Q., Zou J., Tisdale J.F. Biallelic correction of sickle cell disease-derived induced pluripotent stem cells (iPSCs) confirmed at the protein level through serum-free iPS-sac/erythroid differentiation. Stem Cells Transl. Med. 2020;9:590–602. [Google Scholar]
- 10.Kim H., Kim J.S. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014;15:321–334. doi: 10.1038/nrg3686. [DOI] [PubMed] [Google Scholar]
- 11.DeWitt M.A., Magis W., Bray N.L., Wang T., Berman J.R., Urbinati F., Heo S.J., Mitros T., Muñoz D.P., Boffelli D. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci. Transl. Med. 2016;8:360ra134. doi: 10.1126/scitranslmed.aaf9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Romero Z., Lomova A., Said S., Miggelbrink A., Kuo C.Y., Campo-Fernandez B., Hoban M.D., Masiuk K.E., Clark D.N., Long J. Editing the Sickle Cell Disease Mutation in Human Hematopoietic Stem Cells: Comparison of Endonucleases and Homologous Donor Templates. Mol. Ther. 2019;27:1389–1406. doi: 10.1016/j.ymthe.2019.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pattabhi S., Lotti S.N., Berger M.P., Singh S., Lux C.T., Jacoby K., Lee C., Negre O., Scharenberg A.M., Rawlings D.J. In Vivo Outcome of Homology-Directed Repair at the HBB Gene in HSC Using Alternative Donor Template Delivery Methods. Mol. Ther. Nucleic Acids. 2019;17:277–288. doi: 10.1016/j.omtn.2019.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park S.H., Lee C.M., Dever D.P., Davis T.H., Camarena J., Srifa W., Zhang Y., Paikari A., Chang A.K., Porteus M.H. Highly efficient editing of the β-globin gene in patient-derived hematopoietic stem and progenitor cells to treat sickle cell disease. Nucleic Acids Res. 2019;47:7955–7972. doi: 10.1093/nar/gkz475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin R.M., Ikeda K., Cromer M.K., Uchida N., Nishimura T., Romano R., Tong A.J., Lemgart V.T., Camarena J., Pavel-Dinu M. Highly Efficient and Marker-free Genome Editing of Human Pluripotent Stem Cells by CRISPR-Cas9 RNP and AAV6 Donor-Mediated Homologous Recombination. Cell Stem Cell. 2019;24:821–828.e5. doi: 10.1016/j.stem.2019.04.001. [DOI] [PubMed] [Google Scholar]
- 16.Uchida N., Drysdale C.M., Nassehi T., Gamer J., Yapundich M., DiNicola J. Cas9 protein delivery non-integrating lentiviral vectors for gene correction in sickle cell disease. Mol. Ther. Methods Clin. Dev. 2021;21:121–132. doi: 10.1016/j.omtm.2021.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fitzhugh C.D., Cordes S., Taylor T., Coles W., Roskom K., Link M., Hsieh M.M., Tisdale J.F. At least 20% donor myeloid chimerism is necessary to reverse the sickle phenotype after allogeneic HSCT. Blood. 2017;130:1946–1948. doi: 10.1182/blood-2017-03-772392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Long J., Hoban M.D., Cooper A.R., Kaufman M.L., Kuo C.Y., Campo-Fernandez B., Lumaquin D., Hollis R.P., Wang X., Kohn D.B., Romero Z. Characterization of Gene Alterations following Editing of the β-Globin Gene Locus in Hematopoietic Stem/Progenitor Cells. Mol. Ther. 2018;26:468–479. doi: 10.1016/j.ymthe.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Canny M.D., Moatti N., Wan L.C.K., Fradet-Turcotte A., Krasner D., Mateos-Gomez P.A., Zimmermann M., Orthwein A., Juang Y.C., Zhang W. Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR-Cas9 genome-editing efficiency. Nat. Biotechnol. 2018;36:95–102. doi: 10.1038/nbt.4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jayavaradhan R., Pillis D.M., Goodman M., Zhang F., Zhang Y., Andreassen P.R., Malik P. CRISPR-Cas9 fusion to dominant-negative 53BP1 enhances HDR and inhibits NHEJ specifically at Cas9 target sites. Nat. Commun. 2019;10:2866. doi: 10.1038/s41467-019-10735-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Uchida N., Hsieh M.M., Raines L., Haro-Mora J.J., Demirci S., Bonifacino A.C., Krouse A.E., Metzger M.E., Donahue R.E., Tisdale J.F. Development of a forward-oriented therapeutic lentiviral vector for hemoglobin disorders. Nat. Commun. 2019;10:4479. doi: 10.1038/s41467-019-12456-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanson G., Coller J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 2018;19:20–30. doi: 10.1038/nrm.2017.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ward N.J., Buckley S.M., Waddington S.N., Vandendriessche T., Chuah M.K., Nathwani A.C., McIntosh J., Tuddenham E.G., Kinnon C., Thrasher A.J., McVey J.H. Codon optimization of human factor VIII cDNAs leads to high-level expression. Blood. 2011;117:798–807. doi: 10.1182/blood-2010-05-282707. [DOI] [PubMed] [Google Scholar]
- 24.Forget B.G., Hardison R.C. The Normal Structure and Regulation of Human Globin Gene Clusters. In: Forget B.G., Weatherall D.J., Higgs D.R., Steinberg M.H., editors. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management. Cambridge University Press; 2009. pp. 46–61. [Google Scholar]
- 25.Nguyen G.N., Everett J.K., Raymond H., Kafle S., Merricks E.P., Kazazian H.H., Nichols T.C., Bushman F.D., Sabatino D.E. Long-Term AAV-Mediated Factor VIII Expression in Nine Hemophilia A Dogs: A 10 Year Follow-up Analysis on Durability, Safety and Vector Integration. Blood. 2019;134:611. [Google Scholar]
- 26.Powars D.R., Weiss J.N., Chan L.S., Schroeder W.A. Is there a threshold level of fetal hemoglobin that ameliorates morbidity in sickle cell anemia? Blood. 1984;63:921–926. [PubMed] [Google Scholar]
- 27.Nagel R.L., Rieder R.F., Bookchin R.M., James G.W., 3rd Some functional properties of hemoglobin Leiden. Biochem. Biophys. Res. Commun. 1973;53:1240–1245. doi: 10.1016/0006-291x(73)90598-6. [DOI] [PubMed] [Google Scholar]
- 28.Henry E.R., Cellmer T., Dunkelberger E.B., Metaferia B., Hofrichter J., Li Q., Ostrowski D., Ghirlando R., Louis J.M., Moutereau S. Allosteric control of hemoglobin S fiber formation by oxygen and its relation to the pathophysiology of sickle cell disease. Proc. Natl. Acad. Sci. USA. 2020;117:15018–15027. doi: 10.1073/pnas.1922004117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uchida N., Weitzel R.P., Evans M.E., Green R., Bonifacino A.C., Krouse A.E., Metzger M.E., Hsieh M.M., Donahue R.E., Tisdale J.F. Evaluation of engraftment and immunological tolerance after reduced intensity conditioning in a rhesus hematopoietic stem cell gene therapy model. Gene Ther. 2014;21:148–157. doi: 10.1038/gt.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hustedt N., Durocher D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2016;19:1–9. doi: 10.1038/ncb3452. [DOI] [PubMed] [Google Scholar]
- 31.Shin J., Wyman S.K., Dewitt M.A., Bray N.L., Vu J., Corn J.E. Controlled cycling and quiescence enables homology directed repair in engraftment-enriched adult hematopoietic stem and progenitor cells. bioRxiv. 2018:301176. doi: 10.1016/j.celrep.2020.108093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu M.H., Smith S.L., Danet G.H., Lin A.M., Williams S.F., Liebowitz D.N., Dolan M.E. Optimization of culture conditions to enhance transfection of human CD34+ cells by electroporation. Bone Marrow Transplant. 2001;27:1201–1209. doi: 10.1038/sj.bmt.1703054. [DOI] [PubMed] [Google Scholar]
- 33.De Ravin S.S., Li L., Wu X., Choi U., Allen C., Koontz S., Lee J., Theobald-Whiting N., Chu J., Garofalo M. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci. Transl. Med. 2017;9:eaah3480. doi: 10.1126/scitranslmed.aah3480. [DOI] [PubMed] [Google Scholar]
- 34.Hayakawa J., Hsieh M.M., Anderson D.E., Phang O., Uchida N., Washington K., Tisdale J.F. The assessment of human erythroid output in NOD/SCID mice reconstituted with human hematopoietic stem cells. Cell Transplant. 2010;19:1465–1473. doi: 10.3727/096368910X314161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uchida N., Hargrove P.W., Lap C.J., Evans M.E., Phang O., Bonifacino A.C., Krouse A.E., Metzger M.E., Nguyen A.D., Hsieh M.M. High-efficiency transduction of rhesus hematopoietic repopulating cells by a modified HIV1-based lentiviral vector. Mol. Ther. 2012;20:1882–1892. doi: 10.1038/mt.2012.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uchida N., Washington K.N., Hayakawa J., Hsieh M.M., Bonifacino A.C., Krouse A.E., Metzger M.E., Donahue R.E., Tisdale J.F. Development of a human immunodeficiency virus type 1-based lentiviral vector that allows efficient transduction of both human and rhesus blood cells. J. Virol. 2009;83:9854–9862. doi: 10.1128/JVI.00357-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dunbar C.E., Cottler-Fox M., O’Shaughnessy J.A., Doren S., Carter C., Berenson R., Brown S., Moen R.C., Greenblatt J., Stewart F.M. Retrovirally marked CD34-enriched peripheral blood and bone marrow cells contribute to long-term engraftment after autologous transplantation. Blood. 1995;85:3048–3057. [PubMed] [Google Scholar]
- 38.Tisdale J.F., Hanazono Y., Sellers S.E., Agricola B.A., Metzger M.E., Donahue R.E., Dunbar C.E. Ex vivo expansion of genetically marked rhesus peripheral blood progenitor cells results in diminished long-term repopulating ability. Blood. 1998;92:1131–1141. [PubMed] [Google Scholar]
- 39.Stewart A.K., Sutherland D.R., Nanji S., Zhao Y., Lutzko C., Nayar R., Peck B., Ruedy C., McGarrity G., Tisdale J., Dubé I.D. Engraftment of gene-marked hematopoietic progenitors in myeloma patients after transplant of autologous long-term marrow cultures. Hum. Gene Ther. 1999;10:1953–1964. doi: 10.1089/10430349950017310. [DOI] [PubMed] [Google Scholar]
- 40.Shultz L.D., Ishikawa F., Greiner D.L. Humanized mice in translational biomedical research. Nat. Rev. Immunol. 2007;7:118–130. doi: 10.1038/nri2017. [DOI] [PubMed] [Google Scholar]
- 41.Uchida N., Evans M.E., Hsieh M.M., Bonifacino A.C., Krouse A.E., Metzger M.E., Sellers S.E., Dunbar C.E., Donahue R.E., Tisdale J.F. Integration-specific In Vitro Evaluation of Lentivirally Transduced Rhesus CD34(+) Cells Correlates With In Vivo Vector Copy Number. Mol. Ther. Nucleic Acids. 2013;2:e122. doi: 10.1038/mtna.2013.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uchida N., Hsieh M.M., Hayakawa J., Madison C., Washington K.N., Tisdale J.F. Optimal conditions for lentiviral transduction of engrafting human CD34+ cells. Gene Ther. 2011;18:1078–1086. doi: 10.1038/gt.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uchida N., Nassehi T., Drysdale C.M., Gamer J., Yapundich M., Bonifacino A.C., Krouse A.E., Linde N., Hsieh M.M., Donahue R.E. Busulfan Combined with Immunosuppression Allows Efficient Engraftment of Gene-Modified Cells in a Rhesus Macaque Model. Mol. Ther. 2019;27:1586–1596. doi: 10.1016/j.ymthe.2019.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uchida N., Nassehi T., Drysdale C.M., Gamer J., Yapundich M., Demirci S., Haro-Mora J.J., Leonard A., Hsieh M.M., Tisdale J.F. High-Efficiency Lentiviral Transduction of Human CD34+ Cells in High-Density Culture with Poloxamer and Prostaglandin E2. Mol. Ther. Methods Clin. Dev. 2019;13:187–196. doi: 10.1016/j.omtm.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uchida N., Weitzel R.P., Shvygin A., Skala L.P., Raines L., Bonifacino A.C., Krouse A.E., Metzger M.E., Donahue R.E., Tisdale J.F. Total body irradiation must be delivered at high dose for efficient engraftment and tolerance in a rhesus stem cell gene therapy model. Mol. Ther. Methods Clin. Dev. 2016;3:16059. doi: 10.1038/mtm.2016.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pfeiffer F., Gröber C., Blank M., Händler K., Beyer M., Schultze J.L., Mayer G. Systematic evaluation of error rates and causes in short samples in next-generation sequencing. Sci. Rep. 2018;8:10950. doi: 10.1038/s41598-018-29325-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Potapov V., Ong J.L. Examining Sources of Error in PCR by Single-Molecule Sequencing. PLoS ONE. 2017;12:e0169774. doi: 10.1371/journal.pone.0169774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoban M.D., Cost G.J., Mendel M.C., Romero Z., Kaufman M.L., Joglekar A.V., Ho M., Lumaquin D., Gray D., Lill G.R. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 2015;125:2597–2604. doi: 10.1182/blood-2014-12-615948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Traxler E.A., Yao Y., Wang Y.D., Woodard K.J., Kurita R., Nakamura Y., Hughes J.R., Hardison R.C., Blobel G.A., Li C., Weiss M.J. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat. Med. 2016;22:987–990. doi: 10.1038/nm.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bauer D.E., Kamran S.C., Lessard S., Xu J., Fujiwara Y., Lin C., Shao Z., Canver M.C., Smith E.C., Pinello L. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342:253–257. [Google Scholar]
- 51.Sankaran V.G., Xu J., Byron R., Greisman H.A., Fisher C., Weatherall D.J., Sabath D.E., Groudine M., Orkin S.H., Premawardhena A., Bender M.A. A functional element necessary for fetal hemoglobin silencing. N. Engl. J. Med. 2011;365:807–814. doi: 10.1056/NEJMoa1103070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chang K.-H., Sanchez M., Heath J., deDreuzy E., Haskett S., Vogelaar A., Gogi K., Da Silva J., Wang T., Sadowski A. Comparative Studies Reveal Robust HbF Induction By Editing of HBG1/2 Promoters or BCL11A Erythroid-Enhancer in Human CD34+ Cells but That BCL11A Erythroid-Enhancer Editing Is Associated with Selective Reduction in Erythroid Lineage Reconstitution in a Xenotransplantation Model. Blood. 2018;132:409. [Google Scholar]
- 53.Humbert O., Radtke S., Samuelson C., Carrillo R.R., Perez A.M., Reddy S.S., Lux C., Pattabhi S., Schefter L.E., Negre O. Therapeutically relevant engraftment of a CRISPR-Cas9-edited HSC-enriched population with HbF reactivation in nonhuman primates. Sci. Transl. Med. 2019;11:eaaw3768. doi: 10.1126/scitranslmed.aaw3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Demirci S., Zeng J., Wu Y., Uchida N., Gamer J., Yapundich M., Drysdale C., Bonifacino A.C., Krouse A.E., Linde N.S. Durable and Robust Fetal Globin Induction without Anemia in Rhesus Monkeys Following Autologous Hematopoietic Stem Cell Transplant with BCL11A Erythroid Enhancer Editing. Blood. 2019;134:4632. [Google Scholar]
- 55.Uchida N., Leonard A., Stroncek D., Panch S.R., West K., Molloy E., Hughes T.E., Hauffe S., Taylor T., Fitzhugh C. Safe and efficient peripheral blood stem cell collection in patients with sickle cell disease using plerixafor. Haematologica. 2020;105:e497. doi: 10.3324/haematol.2019.236182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Uchida N., Fujita A., Hsieh M.M., Bonifacino A.C., Krouse A.E., Metzger M.E., Donahue R.E., Tisdale J.F. Bone Marrow as a Hematopoietic Stem Cell Source for Gene Therapy in Sickle Cell Disease: Evidence from Rhesus and SCD Patients. Hum. Gene Ther. Clin. Dev. 2017;28:136–144. doi: 10.1089/humc.2017.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uchida N., Bonifacino A., Krouse A.E., Metzger M.E., Csako G., Lee-Stroka A., Fasano R.M., Leitman S.F., Mattapallil J.J., Hsieh M.M. Accelerated lymphocyte reconstitution and long-term recovery after transplantation of lentiviral-transduced rhesus CD34+ cells mobilized by G-CSF and plerixafor. Exp. Hematol. 2011;39:795–805. doi: 10.1016/j.exphem.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Clement K., Rees H., Canver M.C., Gehrke J.M., Farouni R., Hsu J.Y., Cole M.A., Liu D.R., Joung J.K., Bauer D.E., Pinello L. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019;37:224–226. doi: 10.1038/s41587-019-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Leonard A., Yapundich M., Nassehi T., Gamer J., Drysdale C.M., Haro-Mora J.J., Demirci S., Hsieh M.M., Uchida N., Tisdale J.F. Low-Dose Busulfan Reduces Human CD34+ Cell Doses Required for Engraftment in c-kit Mutant Immunodeficient Mice. Mol. Ther. Methods Clin. Dev. 2019;15:430–437. doi: 10.1016/j.omtm.2019.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Uchida N., Demirci S., Haro-Mora J.J., Fujita A., Raines L.N., Hsieh M.M., Tisdale J.F. Serum-free Erythroid Differentiation for Efficient Genetic Modification and High-Level Adult Hemoglobin Production. Mol. Ther. Methods Clin. Dev. 2018;9:247–256. doi: 10.1016/j.omtm.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Uchida N., Haro-Mora J.J., Fujita A., Lee D.Y., Winkler T., Hsieh M.M., Tisdale J.F. Efficient Generation of β-Globin-Expressing Erythroid Cells Using Stromal Cell-Derived Induced Pluripotent Stem Cells from Patients with Sickle Cell Disease. Stem Cells. 2017;35:586–596. doi: 10.1002/stem.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kumkhaek C., Aerbajinai W., Liu W., Zhu J., Uchida N., Kurlander R., Hsieh M.M., Tisdale J.F., Rodgers G.P. MASL1 induces erythroid differentiation in human erythropoietin-dependent CD34+ cells through the Raf/MEK/ERK pathway. Blood. 2013;121:3216–3227. doi: 10.1182/blood-2011-10-385252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate or analyze datasets.