

Abstract
Background:
While growing evidence suggests a link between periodontal disease (PD) and cardiovascular disease (CVD), the independence of this association and the pathway remain unclear. Herein, we tested the hypotheses that: (1) inflammation of the periodontium (PDinflammation) predicts future CVD independently of disease risk factors shared between CVD and PD, and (2) the mechanism linking the two diseases involves heightened arterial inflammation.
Methods:
18F-fluorodeoxyglucose positron emission tomography/computed tomography (18F-FDG-PET/CT) imaging was performed in 304 individuals (median age 54 years; 42.4% male) largely for cancer screening; individuals without active cancer were included. PDinflammation and arterial inflammation were quantified using validated 18F-FDG-PET/CT methods. Additionally, we evaluated the relationship between PDinflammation and subsequent major adverse cardiovascular events (MACE) using Cox models and log-rank tests.
Results:
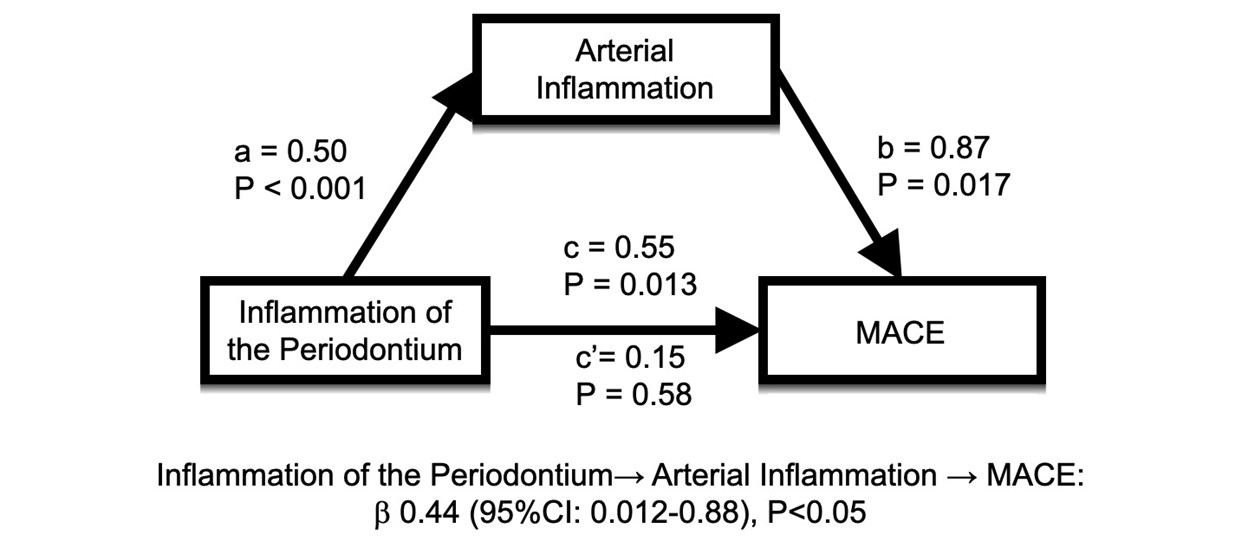
Thirteen individuals developed MACE during follow-up (median 4.1 years). PDinflammation associated with arterial inflammation, remaining significant after adjusting for PD and CVD risk factors (standardized β [95% CI]: 0.30 [0.20–0.40], P < 0.001). PDinflammation predicted subsequent MACE (standardized HR [95% CI]: 2.25 [1.47 to 3.44], P <0.001, remaining significant in multivariable models), while periodontal bone loss did not. Furthermore, mediation analysis suggested that arterial inflammation accounts for 80% of the relationship between PDinflammation and MACE (standardized log odds ratio [95% CI]: 0.438 [0.019–0.880], P = 0.022).
Conclusion:
PDinflammation is independently associated with MACE via a mechanism that may involve increased arterial inflammation. These findings provide important support for an independent relationship between PDinflammation and CVD.
Keywords: atherosclerosis, cardiovascular disease, periodontal disease, positron emission tomography
1 |. INTRODUCTION
Periodontal disease, an inflammatory condition impacting the supporting structures of teeth,1 afflicts 42% of dentate US adults aged ≥30 years, with 8% having severe periodontitis. Overall, 3% of all periodontally probed sites (9% of all teeth) had periodontal probing depth (PD) of ≥4 mm, and 19% of sites (37% of teeth) had clinical attachment loss of ≥3 mm. Severe periodontitis was most prevalent among adults aged ≥65 years, Mexican Americans, non-Hispanic Blacks, and smokers. A large body of epidemiological evidence links periodontal disease to cardiovascular disease (CVD).2–5 However, it remains unclear whether that relationship is causal, whereby the chronic inflammatory milieu elicited by periodontitis potentiates inflammatory conditions like CVD,6,7 or is non-causal, whereby periodontitis and CVD simply share risk factors like smoking, diabetes mellitus, age, sex, and genetic background.4,8,9
Standard quantitative assessment of periodontitis includes measures of attachment loss and pocket depth, which provide an index of accumulated damage to the periodontium. However, these traditional structural/anatomical measures of periodontitis might not accurately reflect the ongoing biological processes that drive periodontal disease progression. Alternatively, PDinflammation, which initially prompts bone loss and pocket formation,10 may be a more appropriate measure of ongoing periodontal disease activity. Indeed, inflammatory biomarkers in gingival crevicular fluid correlate well with periodontal disease progression (disease activity).11,12 Periodontal inflammation has repeatedly been shown to associate with arterial inflammation,13,14 which may be particularly important, since arterial inflammation associates with major adverse cardiovascular events (MACE) risk.15 However, it remains untested whether PDinflammation associates with MACE.
18F-fluorodeoxyglucose positron emission tomography/computed tomography (18F-FDG-PET/CT) is used clinically to identify metabolically active tissues non-invasively and is useful for the identification of inflamed or infected tissues as well as for measuring atherosclerotic inflammation.16–18 The uptake of 18F-FDG-PET/CT is a reliable marker for the measurement of tissue glycolysis and 18F-FDG-PET/CT imaging has furthermore been validated as an effective means of measuring active periodontal inflammation14,18–21 and evaluating anti-inflammatory therapies.14,22 Accordingly, using 18F-FDG-PET/CT as the measure of PDinflammation, we sought to: 1) test whether PDinflammation independently associates with the risk for subsequent MACE and 2) evaluate whether arterial inflammation may mediate the link between periodontitis and CVD.
2 |. MATERIALS AND METHODS
2.1 |. Study population
Participants (N = 304) were selected from all individuals who underwent clinical 18F-FDG-PET/CT imaging, largely for cancer screening, at the Massachusetts General Hospital from 2005 through 2008. All individuals with adequate periodontal tissue imaging who were included in a prior study evaluating the relationship between arterial inflammation and MACE23 were included in the current study (Fig. 1). Pre-defined inclusion criteria for study participants were: 1) absence of prior cancer or remission from cancer for at least 1 year before imaging and throughout the follow-up period, 2) absence of clinical CVD at the time of imaging, 3) absence of documented acute or chronic inflammatory or autoimmune disease or use of chronic anti-inflammatory treatment at the time of imaging or during the follow-up period, and 4) age >30 years. Those included had at least three clinical visit notes over at least 1 year for the assessment of baseline and follow-up status. The study protocol was approved by the Partners Human Research Committee.
FIGURE 1.
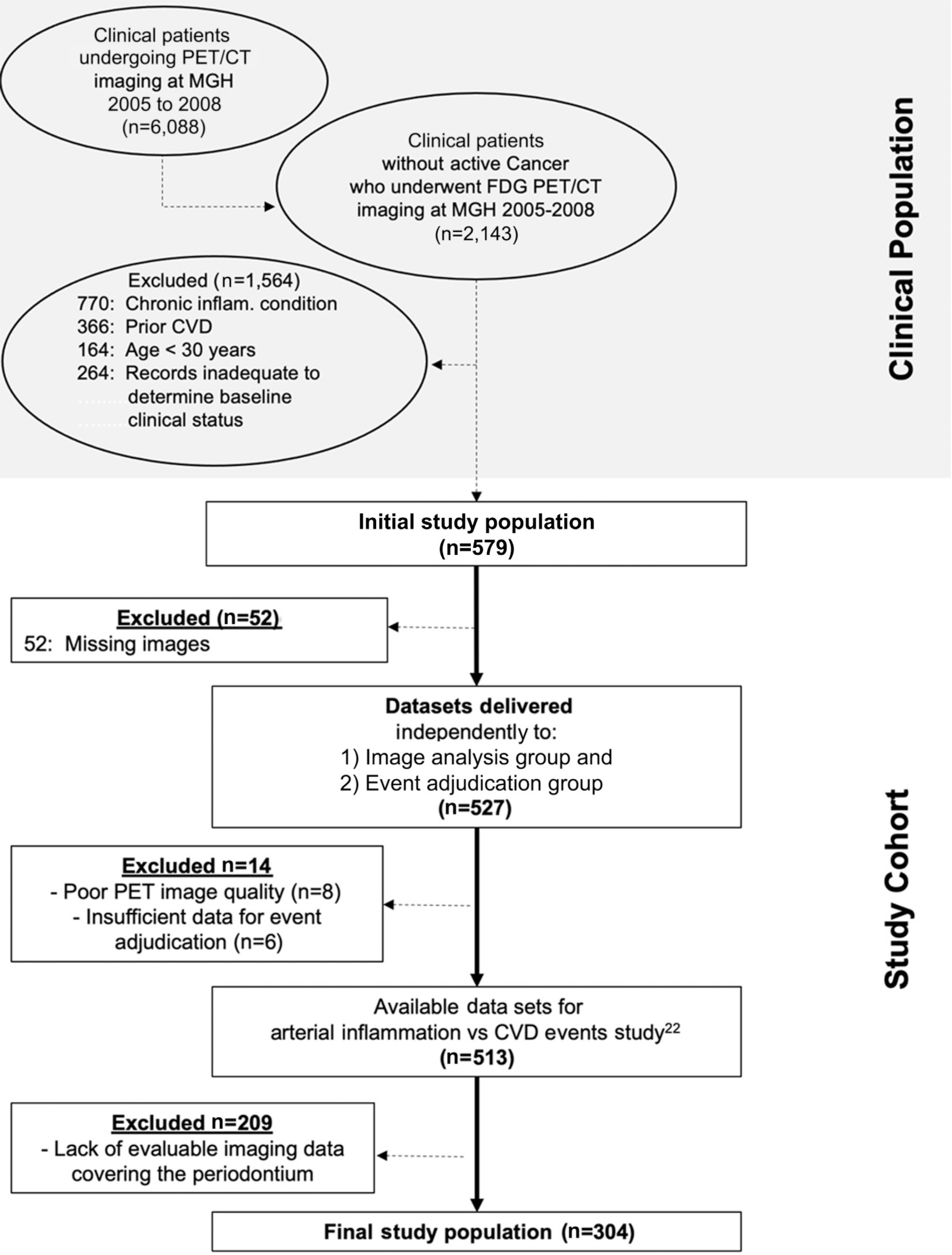
Flowchart of study participant selection. CVD, cardiovascular disease; CT, computed tomography; FDG, fluorodeoxyglucose; MGH, Massachusetts General Hospital; PET, positron emission tomography
2.2 |. 18F-FDG-PET/CT protocol
18F-FDG-PET/CT imaging was performed on a Biograph 64 (Siemens, Erlangen, Germany) or a similar system using previously validated approaches.22 18F-FDG at a dose of 370 MBq was injected intravenously, and PET images were acquired in three-dimensional mode ≈60 minutes after 18F-FDG administration. Low-dose, non-enhanced CT (120 KVp, ~50 mAs) was performed for attenuation correction before the PET scan. The images were co-registered at a workstation (Leonardo–TrueD, Siemens Medical Solutions, Forchheim, Germany)24 to facilitate image analysis.
2.3. 18F-FDG uptake measurement in the periodontal tissue
Images were blinded to patient identifiers and were analyzed using Leonardo TrueD software (Siemens, Forchheim, Germany). PDinflammation was measured as the periodontal accumulation of18F-FDG, according to previously published methods.13,14 The identification of the periodontal tissues was aided by use of the CT images; 18F-FDG uptake was evaluated on the co-registered images by manually drawing a rectangular region of interest (ROI) around the teeth from the mesial surface of the first premolar to the distal surface of the second molar carefully avoiding any spill-over from the surrounding tissues (e.g., pharyngeal and buccal mucosa and tongue). Four ROIs were drawn for each participant for the right and left mandibular and maxillary periodontal tissue, and the maximum standardized uptake values (SUVs) of 18F-FDG were registered for each quadrant. The SUV values for the periodontal quadrants were averaged to yield an average of the maximum periodontal SUVs.
2.4. 18F-FDG uptake measurement in the arterial wall
Arterial inflammation was assessed as a ratio of aortic wall to background venous blood metabolic activity. Circular ROIs were placed in the axial plane around the vessel wall every 3 mm starting 1 cm above the aortic valve annulus and continuing to the bottom of the aortic arch. Maximum SUV values were registered for each segment, and the mean of the SUV values was calculated for the entire ascending aorta. The background SUV was obtained by placing regions of interest in the superior vena cava in an area devoid of significant spillover activity from the surrounding tissues. Thereafter, a target-to-background-ratio was calculated by dividing the aortic SUV by the background venous SUV (Fig. 2).
FIGURE 2.
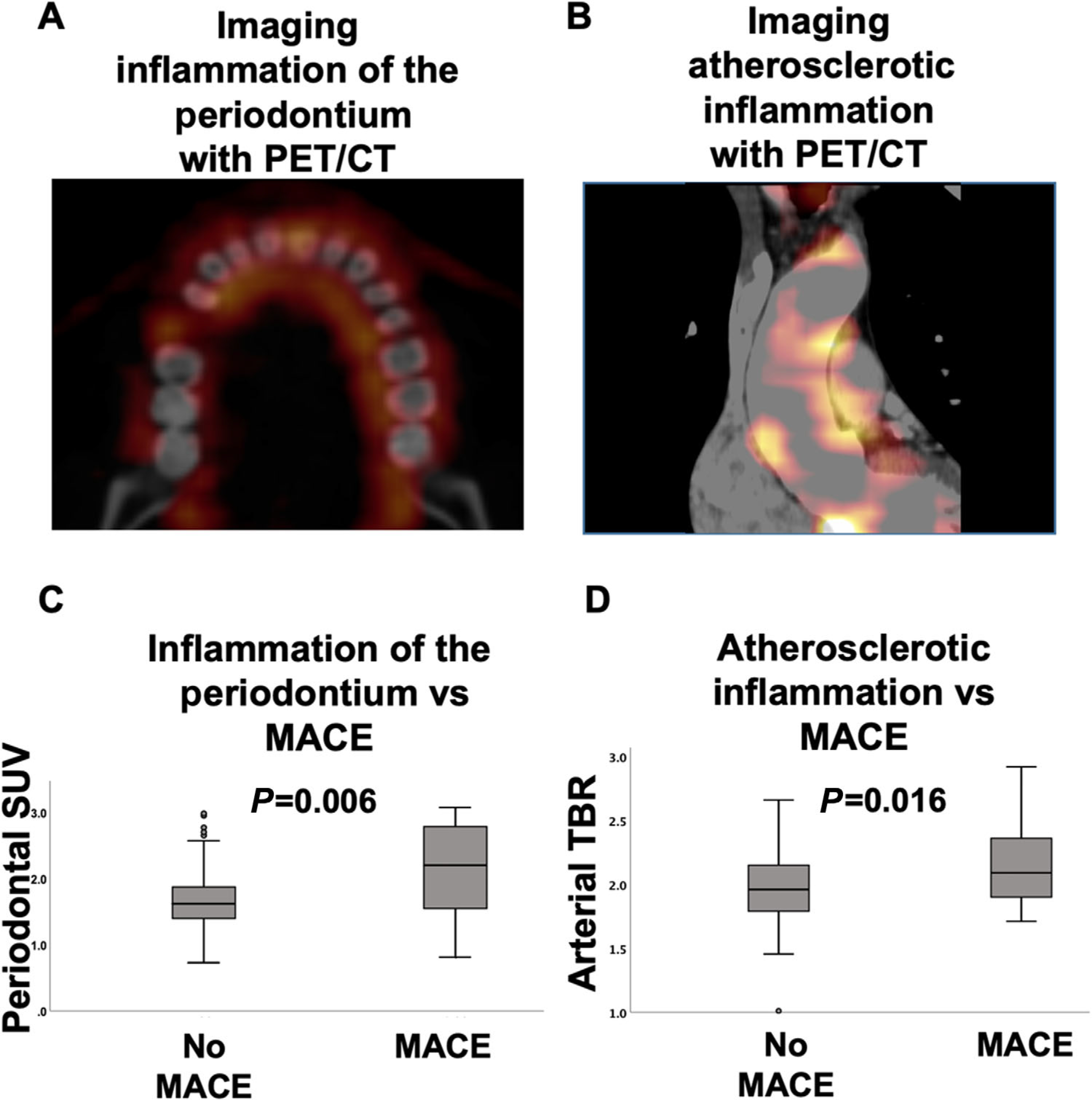
Periodontal and arterial 18F-FDG uptake. A) shows axial images at the level of periodontium from CT and 18F-FDG-PET-CT. 1 B) shows coronal images of the ascending aorta. C and D) demonstrate box plots of periodontal inflammation and of atherosclerotic inflammation among individuals with versus without subsequent MACE. CT, computed tomography; 18F-FDG, 18F-fluorodeoxyglucose; PET, positron emission tomography
2.5 |. Periodontal bone loss assessment
Periodontal bone loss was measured using the attenuation correction CT images acquired before the PET scan. Evaluation of the periodontal CT images was performed by an experienced periodontist masked to all PET imaging and clinical data using previously validated methods.14 Alveolar bone resorption was assessed semi-quantitatively in each of the four dental quadrants and was graded as follows: edentulous, none, mild (limited to coronal one-third of the root), mild-to moderate, moderate (including the middle one-third of the root), moderate-to-severe, or severe (including the apical one-third of the root) bone loss, respectively. Classification of each individual’s disease severity was based on each individual’s most severely diseased quadrant. Periodontium of anterior teeth (incisors and canines) and third molars were not evaluated for bone resorption scoring.
2.6 |. Coronary calcium assessment
Coronary artery calcium (CAC) score was quantitatively assessed by a separate investigator who was blinded to clinical and PET data, using standardized methods.25 A Hounsfield Unit (HU) threshold was pre-specified for this analysis: range of > 130 HU in more than three continuous voxels on non-gated CT images obtained from a hybrid PET/CT scanner. Since it is known that non-gated CT scans can underestimate the presence of CAC.26–28 Accordingly, the lowest CAC scores (0 to 10) were combined into one group,29 such that CAC was categorized into three groups: 0 to 10,11 to 99, and >100.
2.7 |. MACE adjudication
Two cardiologists masked to imaging data adjudicated MACE using the available clinical records. MACE was defined as a composite of CVD death, myocardial infarction, unstable angina, cerebrovascular accident, heart failure, or coronary or peripheral artery revascularization.30
2.8 |. Statistical analysis
All analyses were performed using SPSS (IBM, Version 25.0, Armonk, NY). Data are presented as mean ± SD for continuous parametric variables, median (P25-P75) for continuous non-parametric variables, and frequency (%) for categorical variables. Pearson correlation coefficients were used to assess associations for normally distributed variables, and Spearman correlations were used for variables that are not normally distributed. Kaplan–Meier curves of MACE-free survival were generated, and log-rank tests were performed to determine statistical significance between groups. For these analyses, two approaches were employed to categorize PDinflammation values as “higher” versus “lower”. The first approach identified “higher” values based on data distribution, as those PDinflammation SUV values that are > 1 SD above the mean. The second approach used an ROC approach to define higher values as those that are more predictive of MACE, using the Youden method. The Youden index to identify the PDinflammation value that best predicts MACE, based on the ROC analysis. This index gives equal weight to sensitivity and specificity and identifies the PDinflammation SUV threshold that provides the greatest sum of sensitivity + (1-specificity). Univariable and multivariable Cox proportional hazards regression analysis were used (employing a continuous variable for PDinflammation) to calculate hazard ratios (HRs) and 95% confidence intervals (CIs). Continuous variables were standardized. When >1 covariable was entered in the model, a backward stepwise (conditional) method was used.
Mediation (path) analysis was performed using the SPSS PROCESS macro, as previously described.30 We examined the pre-specified single mediator path: (1) PDinflammation ⟶ arterial inflammation ⟶ MACE. A two-sided P value <0.05 was considered statistically significant for all analyses.
3 |. RESULTS
3.1 |. Study participant characteristics
A total of 304 individuals were included with a median [P25 to P75] follow-up of 4.0 [3.0 to 5.1] years. The median age was 54.0 [44.0 to 63.8] years and 129 (41.6%) were males. Additional characteristics are listed in Table 1 and in Tables S1A and S1B in the online Journal of Periodontology.
TABLE 1.
Study participant characteristics
| Characteristics | Full cohort (N = 304) |
|---|---|
| Age (years) | 54.0 (44.0 to 63.8) |
| Male (%) | 129 (42.4%) |
| Body mass index (kg/m2)a | 26.4 (23.3 to 30.9) |
| Current smoker (%) | 25 (8.3%) |
| Diabetes mellitus (%) | 20 (6.6%) |
| Dyslipidemia (%) | 78 (25.7%) |
| Hypertension (%) | 95 (31.3%) |
| Statin use (%) | 54 (17.8%) |
| Periodontal bone loss (% of populationa) | Edentulous (1.5%) None (0%) Mild (37.4%) Mild-to-moderate (1.1%) Moderate (30.5%) Moderate-to-severe (4.2%) Severe (25.2%) |
| Subsequent cardiovascular event (%) | 13 (4.3%) |
| Framingham Risk Scoreb | |
| <10 | 114 (82.0%) |
| 10–19.9 | 21 (15.1%) |
| ≥20 | 4 (2.9%) |
Values are median (P25 to P75), or n (%).
Available in 295 subjects
Available in 139 subjects.
3.2. Periodontal disease activity independently associates with arterial inflammation
We previously reported a significant association between PDinflammation and arterial inflammation in this cohort31. Herein, we additionally observe that the relationship between PDinflammation and arterial inflammation remained significant after adjusting for CVD risk factors (age, sex, hypertension, diabetes, smoking, and dyslipidemia) (standardized β [95% CI]: (0.30 [0.20 to 0.40], P <0.001) or after additionally adjusting for statin use (0.30 [0.20 to 0.40], P <0.001). Additionally, there was a non-significant trend towards an association between PDinflammation and CAC (r = 0.12, P = 0.06).
3.3. Periodontal disease activity independently associates with subsequent MACE
During follow-up, 13 participants in the outcomes study (4.2%) developed MACE, including four myocardial infarctions, one coronary revascularization, five strokes, one cardiovascular death, and two peripheral arterial disease events. PDinflammation and arterial inflammation were higher among individuals with subsequent MACE (Fig. 2). Moreover, PDinflammation predicted the risk of subsequent MACE (Table 2 and Fig. 3), yielding a 2.25-fold increased risk per SD increase in periodontal SUV in univariable analysis (standardized HR [95% CI]: 2.25 [1.47 to 3.44], P <0.001). Furthermore, the relationship between PDinflammation and MACE remained significant after adjustments for: 1) risk factors shared by periodontal and CVD (i.e., age, sex, smoking, hypertension, and diabetes mellitus), 2) Framingham risk score, 3) preexisting atherosclerotic disease burden (i.e., CAC score), and 4) periodontal bone loss at baseline (Table 2). Further, PDinflammation remained predictive of CVD after adjustment for history of prior cancer (standardized HR [95% CI]: 2.31 [1.49 to 3.58], P <0.0001).
TABLE 2.
Uni- and multivariable analysis of periodontal activity versus MACE
| Model Predictor | Model Covariablesa | Model outcome |
|
|---|---|---|---|
| MACE | |||
| Standardized HR (95% CI) | P | ||
| Inflammation of the periodontium (PDinflammation) a | None | 2.25 (1.47 to 3.44) | <0.001 |
| Age and sex | 1.94 (1.31 to 2.87) | 0.001 | |
| Framingham Risk Scoreb | 1.79 (1.16 to 2.75) | 0.008 | |
| Age, sex, smoking, HTN, and DM | 1.93 (1.31 to 2.84) | 0.001 | |
| Statin use | 2.09 (1.36 to 3.23) | 0.001 | |
| CAC | 2.19 (1.41 to 3.39) | <0.001 | |
| Periodontal bone lossc | 3.16 (1.60 to 6.22) | 0.001 | |
| Arterial inflammationd | 1.80 (1.12 to 2.89) | 0.016 | |
Continuous variables were standardized; a backward (conditional) model was used when more than one covariable was entered into the model.
The predictor, Inflammation of the periodontium, was entered as a continuous variable.
Framingham Risk Score data for these sub-analyses are available in 106 individuals.
Periodontal bone loss was evaluated on a five-point scale; the analysis excluded individuals who were edentulous.
Arterial Inflammation is measured as target-to-background on positron emission tomography.
DM, diabetes mellitus; HTN, arterial hypertension; CAC, coronary artery calcium; HR, hazard ratio, CI, confidence interval.
FIGURE 3.
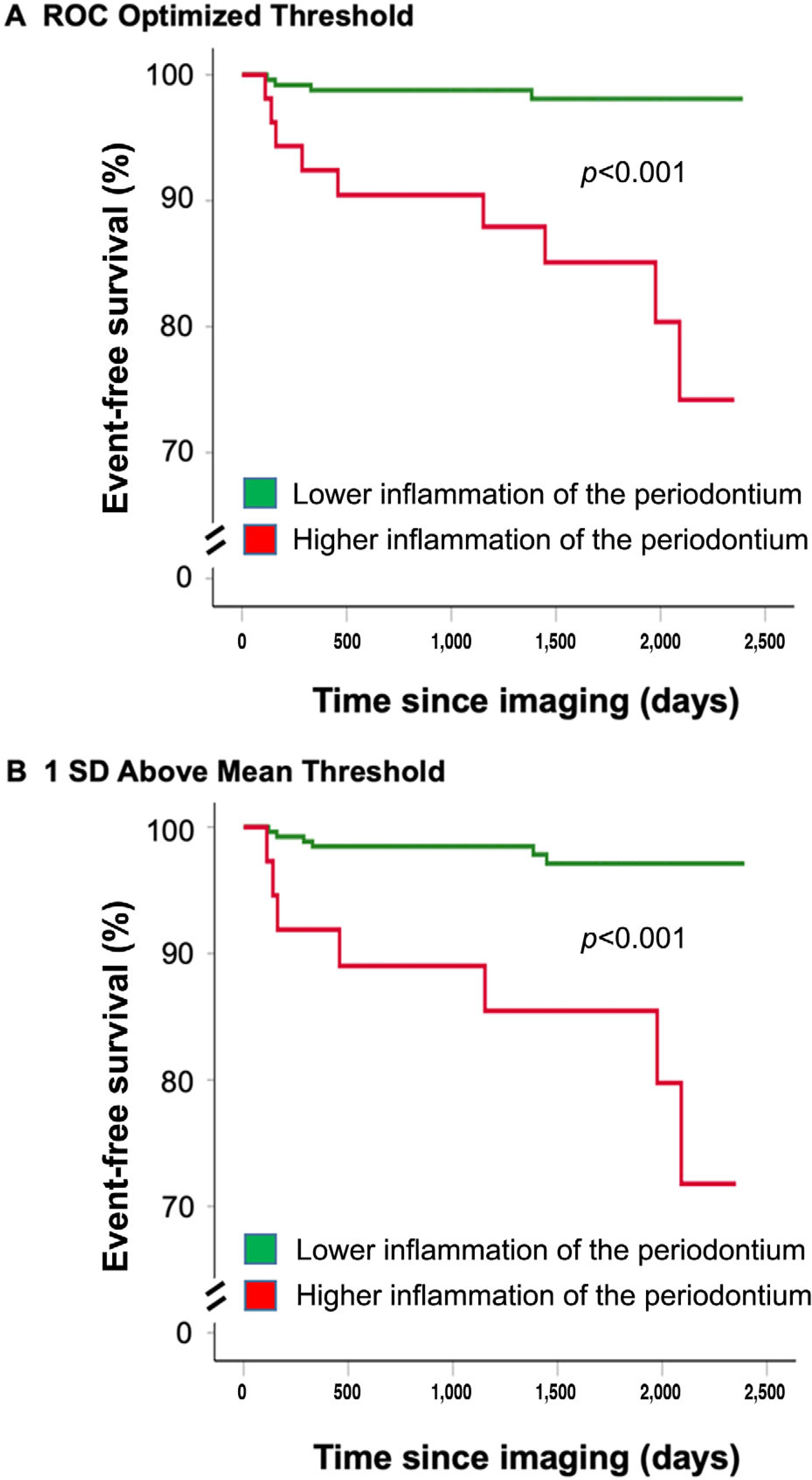
Kaplan-Meier survival curves showing MACE-free survival by periodontal activity. Survival curves relating PDinflammation and MACE are shown. PDinflammation was dichotomously characterized as “high” or “low” based on two distinct approaches: A) The first approach dichotomized PDinflammation values by identifying which PDinflammation threshold most accurately predicted future MACE events using receiver-operator curve (ROC) analyses. B) The second approach dichotomized PDinflammation based on data distribution with a defined threshold SUV value ≥1 SD above the mean periodontal SUV. MACE, major adverse cardiovascular event; PDinflammatlon, periodontal disease inflammation; ROC, receiver operating curve; SUV, standardized uptake value
Unlike the findings with PDinflammation, periodontal bone loss did not associate with MACE, either in univariable analyses (1.24 [0.74 to 2.08], P = 0.405), or after adjustments for age, sex, smoking, hypertension, and diabetes (1.09 [0.66 to 1.82], P = 0.731) or for statin use (1.22 [0.72 to 2.02], P = 0.488).
3.4. Association between PDinflammation and MACE events is mediated by arterial inflammation
We performed mediation (pathway) analyses to test the hypothesis that the relationship between PDinflammation and MACE was significantly mediated by increased arterial inflammation. Arterial inflammation accounted for most of the relationship between PDinflammation and MACE (standardized logs odds ratio, [95% CI]: 0.438 [0.019 to 0.880], P = 0.022), or 80% of the total effect (see Figure S1 in online Journal of Periodontology). This relationship (path) remained significant in analyses that adjusted for age and sex (P = 0.032), periodontal and CVD risk factors (i.e., age, sex, smoking, diabetes, arterial hypertension, dyslipidemia and family history of coronary artery disease; P = 0.036), as well as CAC and periodontal bone loss (P = 0.043). While unable to establish causality, this analysis delineates a significant pathway (PDinflammation ⟶ arterial inflammation ⟶ MACE). On the other hand, the path that excludes arterial inflammation becomes non-significant (P = 0.579).
4 |. DISCUSSION
To our knowledge, this is the first advanced imaging study to link periodontal inflammation to subsequent major adverse cardiovascular events, showing that inflammation of the periodontium predicts MACE after correction for potential confounders and shared risk factors. Further, we observe that objectively measured inflammation of the periodontium (i.e., PDinflammation) may represent a better predictor of MACE than measures of periodontal bone loss. Additionally, the current study suggests that the mechanism mediating the link between PDinflammation and CVD may involve increased arterial inflammation.
While the relationship between periodontal disease and CVD has been previously examined,5,8 important questions remained unanswered. Perhaps the most critical question pertains to the identification of the mechanistic pathway linking periodontal disease to CVD.2 We previously observed an independent association between PDinflammation and atherosclerotic inflammation that remained significant after robust adjustments for disease risk factors that are shared between PD and atherosclerotic diseases.13,14 We posited that the association between PD and arterial inflammation may be particularly important, since arterial inflammation robustly associates with MACE risk.15 We hypothesized that PD associates with CVD events through a mechanism that involves increased arterial inflammation (Fig. 4). In support of this hypothesis, the findings of the current study demonstrate that: 1) PDinflammation independently associates with both increased arterial inflammation as well as CVD after adjusting for risk factors for the diseases, and 2) mediation (pathway) analysis demonstrates that arterial inflammation significantly mediates the link between PDinflammation and CVD, accounting for 80% of the total effect.
FIGURE 4.
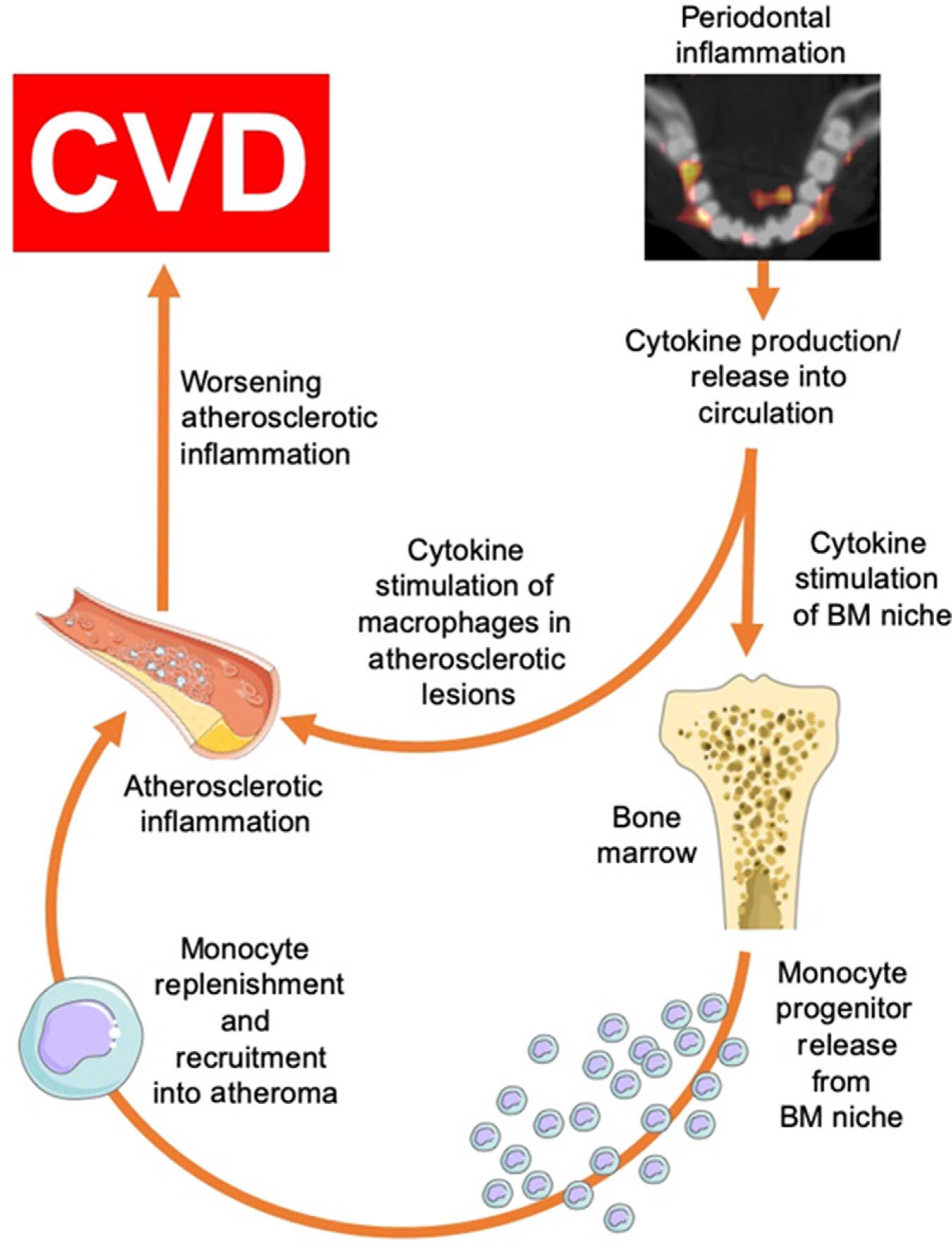
Hypothesized pathway linking periodontal disease to CVD
The major cause of MACE is rupture of the lipid-rich atherosclerotic plaque that causes blockage of a major artery, resulting in an acute coronary syndrome.32 Established risk factors, including dyslipidemia, hypertension, diabetes mellitus, and smoking are associated with MACE. Nevertheless, >40% of all heart attacks and strokes occur in patients with “normal” cholesterol levels.33 In humans, plaques at high-risk for rupture present distinct features, including intense inflammation and oxidative stress, large areas of necrosis composed of apoptotic cells with thinning, and inflammation of the protective fibrous cap.34,35 There are significant data to suggest that local inflammatory foci, particularly in the oral cavity,14,36,37 can impact the systemic inflammatory burden38,39 and may contribute to the risk for MACE. Animal studies strongly suggest that local periodontal inflammation is associated with an increase in the incidence of atherothrombosis.40–42 Furthermore, we previously reported, in this cohort, an association between inflammation of the periodontium and upregulated bone marrow leukopoiesis (an important source of inflammatory cells that traffic to the atherosclerotic milieu).31 Additionally, untreated, bacterially-induced periodontal inflammation may contribute to atherosclerotic plaque vulnerability and thrombosis by activating macrophages that elaborate enzymes including matrix metalloproteinases that degrade all components of the arterial extracellular matrix.43,44 Inflammation can also increase fibrinogen-stimulated coagulation and accumulation of activated platelets at the site of thin fibrous cap or fissure.45,46 Even low-grade inflammation may influence platelet activation potentially leading to platelet aggregation.47 Platelet activity and aggregation has been associated with periodontitis in previous reports. Perumal et al.48 demonstrated increase P-selectin surface expression on platelets and increased aggregation in periodontitis, as have others.49,50 Moreover, treatment of periodontitis seems to reverse the propensity for platelet activation.51,52
4.1 |. Limitations
The retrospective nature of this study limits the opportunity to compare the imaging findings with blood biomarkers, as these factors were variably assessed in the study population. Further, of the original cohort of imaged patients, 216 individuals either lacked PET coverage or had insufficient data quality to assess periodontal uptake of FDG (thus constraining the final sample size to 304 individuals). As a result, the number of observed clinical events was modest; only thirteen study participants developed MACE during the follow-up period. Notably, the current study participants were identified from a database of patients who had undergone 18F-FDG-PET/CT for clinical indications, mainly cancer screening. Further studies are needed to determine the generalizability of our findings to broader patient populations, especially since many risk factors are shared by malignancy and periodontal disease. We conscientiously included only patients that had documented cancer remission for at least 1 year. Additionally, the association between PDinflammation and CVD events remained robust after adjusting for history of cancer, suggesting that this potential confounder is not responsible for our findings. Nonetheless, it will be important to replicate these findings in future, larger studies. Nevertheless, these limitations are counterbalanced by significant strengths, including the unique, simultaneous, multi-modality assessment of disparate tissues and their association with MACE in humans.
4.2 |. Future directions
This study should prompt further evaluation of the mechanisms linking PDinflammation to CVD. These results underscore the intriguing hypothesis that improving oral health and aggressively treating PDinflammation might reduce arterial inflammation and builds upon the observation that treatment of periodontal inflammation reduces systemic inflammation 53 and improves endothelial function.54 As such, these findings should stimulate studies to test whether treating PDinflammation ameliorates arterial inflammation and reduces the risk of CVD events. Should future studies show such a benefit, PDinflammation could ultimately be recognized as a modifiable atherosclerotic risk factor, and more aggressive screening and therapy for PDinflammation could be pursued for individuals at higher risk for atherosclerotic diseases.
5 |. CONCLUSIONS
This study demonstrates that periodontal disease inflammation predicts the risk of MACE independently of risk factors for atherosclerosis or PD. The study may also shed light on the mechanism mediating that association, by suggesting that increased arterial inflammation links PDinflammation to CVD. Accordingly, the current study suggests that PDinflammation might represent an independent and potentially modifiable risk factor for atherosclerotic diseases and provides novel insights into the mechanism mediating that association.
Supplementary Material
{kind=link}
ACKNOWLEDGMENTS
The following NIH grants provided support for this study: R01HL122177 (AT), R01HL137913 (AT), R01HL129856 (PH), T32HL076136 (MTO), R01DE02520 (TVD) and KL2TR002542 (MTO). Harvard Catalyst provided statistical support. Some of the findings described were presented at the American Heart Association Conference in November 2016 in New Orleans, Louisiana.
Dr. Hsue reports personal fees from Gilead and Merck that are unrelated to the submitted work. Dr. Osborne reports consulting fees from Intrinsic Imaging for unrelated work. Dr. Tawakol reports grants from Genentech and personal fees from Actelion and Esperion during this study for research outside the submitted work. The remaining authors report no conflicts of interest related to this study.
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
REFERENCES
- 1.Cekici A, Kantarci A, Hasturk H, Van Dyke TE. Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontology 2000. 2014;64:57–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tonetti MS, Van Dyke TE. working group 1 of the joint EFPAAPw. Periodontitis and atherosclerotic cardiovascular disease: consensus report of the Joint EFP/AAP workshop on periodontitis and systemic diseases. J Periodontol. 2013;84:S24–29. [DOI] [PubMed] [Google Scholar]
- 3.Beck J, Garcia R, Heiss G, Vokonas PS, Offenbacher S. Periodontal disease and cardiovascular disease. J Periodontol. 1996;67:1123–1137. [DOI] [PubMed] [Google Scholar]
- 4.Lockhart PB, Bolger AF, Papapanou PN, et al. Periodontal disease and atherosclerotic vascular disease: does the evidence support an independent association?: a scientific statement from the American Heart Association. Circulation. 2012;125:2520–2544. [DOI] [PubMed] [Google Scholar]
- 5.Yu YH, Chasman DI, Buring JE, Rose L, Ridker PM. Cardiovascular risks associated with incident and prevalent periodontal disease. J Clin Periodontal. 2015;42:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iacopino AM. Periodontitis and diabetes interrelationships: role of inflammation. Ann Periodontal Am Acad Periodontal. 2001;6:125–137. [DOI] [PubMed] [Google Scholar]
- 7.Kholy KE, Genco RJ, Van Dyke TE. Oral infections and cardiovascular disease. Trends Endocrinol Metabolism: TEM. 2015;26:315–321. [DOI] [PubMed] [Google Scholar]
- 8.Gomes MS, Hugo FN, Hilgert JB, et al. Apical periodontitis and incident cardiovascular events in the Baltimore longitudinal study of ageing. Int Endodon J. 2016;49:334–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schaefer AS, Richter GM, Groessner-Schreiber B, et al. Identification of a shared genetic susceptibility locus for coronary heart disease and periodontitis. PLoS Genetics. 2009;5:e1000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dietrich T, Sharma P, Walter C, Weston P, Beck J. The epidemiological evidence behind the association between periodontitis and incident atherosclerotic cardiovascular disease. J Periodontal. 2013;84(Suppl 4):S70–84. [DOI] [PubMed] [Google Scholar]
- 11.Taba M Jr., Kinney J, Kim AS, Giannobile WV. Diagnostic biomarkers for oral and periodontal diseases. Dent Clin North Am. 2005;49:551–571.vi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gupta S, Chhina S, Arora SA. A systematic review of biomarkers of gingival crevicular fluid: their predictive role in diagnosis of periodontal disease status. J Oral Biol Craniofac Res. 2018;8:98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fifer KM, Qadir S, Subramanian S, et al. Positron emission tomography measurement of periodontal (18)f-fluorodeoxyglucose uptake is associated with histologically determined carotid plaque inflammation. J Am Coll Cardiol. 2011;57:971–976. [DOI] [PubMed] [Google Scholar]
- 14.Subramanian S, Emami H, Vucic E, et al. High-dose atorvastatin reduces periodontal inflammation: a novel pleiotropic effect of statins. J Am Coll Cardiol. 2013;62:2382–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marnane M, Merwick A, Sheehan OC, et al. Carotid plaque inflammation on 18F-fluorodeoxyglucose positron emission tomography predicts early stroke recurrence. Ann Neurology. 2012;71:709–718. [DOI] [PubMed] [Google Scholar]
- 16.Blankstein R, Osborne M, Naya M, et al. Cardiac positron emission tomography enhances prognostic assessments of patients with suspected cardiac sarcoidosis. J Am College Cardiol. 2014;63:329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saby L, Laas O, Habib G, et al. Positron emission tomography/computed tomography for diagnosis of prosthetic valve endocarditis: increased valvular 18F-fluorodeoxyglucose uptake as a novel major criterion. J Am Coll Cardiol. 2013;61:2374–2382. [DOI] [PubMed] [Google Scholar]
- 18.Tawakol A, Migrino RQ, Bashian GG, et al. In vivo 18F-fluorodeoxyglucose positron emission tomography imaging provides a noninvasive measure of carotid plaque inflammation in patients. J Am Coll Cardiol. 2006;48:1818–1824. [DOI] [PubMed] [Google Scholar]
- 19.Fifer KM, Qadir S, Subramanian S, et al. Positron emission tomography measurement of periodontal 18F-fluorodeoxyglucose uptake is associated with histologically determined carotid plaque inflammation. J Am College Cardiol. 2011;57:971–976. [DOI] [PubMed] [Google Scholar]
- 20.Shimamoto H, Tatsumi M, Kakimoto N, et al. (18)F-FDG accumulation in the oral cavity is associated with periodontal disease and apical periodontitis: an initial demonstration on PET/CT. Ann Nuclear Med. 2008;22:587–593. [DOI] [PubMed] [Google Scholar]
- 21.Kito S, Koga H, Kodama M, et al. Reflection of (1)(8)F-FDG accumulation in the evaluation of the extent of periapical or periodontal inflammation. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;114:e62–69. [DOI] [PubMed] [Google Scholar]
- 22.Tawakol A, Fayad ZA, Mogg R, et al. Intensification of statin therapy results in a rapid reduction in atherosclerotic inflammation: results of a multicenter fluorodeoxyglucose-positron emission tomography/computed tomography feasibility study. J Am Coll Cardiol. 2013;62:909–917. [DOI] [PubMed] [Google Scholar]
- 23.Figueroa AL, Abdelbaky A, Truong QA, et al. Measurement of arterial activity on routine FDG PET/CT images improves prediction of risk of future CV events. JACC Cardiovascular Imaging. 2013;6:1250–1259. [DOI] [PubMed] [Google Scholar]
- 24.Emami H, Singh P, MacNabb M, et al. Splenic metabolic activity predicts risk of future cardiovascular events: demonstration of a cardiosplenic axis in humans. JACC Cardiovascular Imaging. 2015;8:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blaha MJ, Budoff MJ, DeFilippis AP, et al. Associations between C-reactive protein, coronary artery calcium, and cardiovascular events: implications for the JUPITER population from MESA, a population-based cohort study. Lancet. 2011;378:684–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Einstein AJ, Johnson LL, Bokhari S, et al. Agreement of visual estimation of coronary artery calcium from low-dose CT attenuation correction scans in hybrid PET/CT and SPECT/CT with standard Agatston score. J Am Coll Cardiol. 2010;56:1914–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Budoff MJ, Nasir K, Kinney GL, et al. Coronary artery and thoracic calcium on noncontrast thoracic CT scans: comparison of ungated and gated examinations in patients from the COPD Gene cohort. J Cardiovasc Comput Tomogr. 2011;5:113–n8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirsch J, Buitrago I, Mohammed TL, Gao T, Asher CR, Novaro GM. Detection of coronary calcium during standard chest computed tomography correlates with multi-detector computed tomography coronary artery calcium score. Int J of Cardiovasc Imaging. 2012;28:1249–1256. [DOI] [PubMed] [Google Scholar]
- 29.Shaw LJ, Raggi P, Schisterman E, Berman DS, Callister TQ. Prognostic value of cardiac risk factors and coronary artery calcium screening for all-cause mortality. Radiology. 2003;228:826–833. [DOI] [PubMed] [Google Scholar]
- 30.Tawakol A, Ishai A, Takx RA, et al. Relation between resting amygdalar activity and cardiovascular events: a longitudinal and cohort study. Lancet. 2017;389:834–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ishai A, Osborne MT, El Kholy K, et al. Periodontal disease associates with arterial inflammation via potentiation of a hematopoietic-arterial axis. JACC Cardiovascular Imaging. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crea F, Libby P. Acute coronary syndromes: the way forward from mechanisms to precision treatment. Circulation. 2017;136:1155–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Erne P, Gutzwiller F, Urban P, et al. Characteristics and outcome in acute coronary syndrome patients with and without established modifiable cardiovascular risk factors: insights from the Nationwide AMIS Plus Registry 1997–2010. Cardiology. 2012;121:228–236. [DOI] [PubMed] [Google Scholar]
- 34.Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47:C13–18. [DOI] [PubMed] [Google Scholar]
- 35.Tabas I Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10:36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grau AJ, Becher H, Ziegler CM, et al. Periodontal disease as a risk factor for ischemic stroke. Stroke. 2004;35:496–501. [DOI] [PubMed] [Google Scholar]
- 37.Virtanen E, Nurmi T, Soder PO, Airila-Mansson S, Soder B, Meurman JH. Apical periodontitis associates with cardiovascular diseases: a cross-sectional study from Sweden. BMC Oral Health. 2017;17:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tawakol A, Grinspoon SK. Imaging atherosclerotic burden and inflammation: insights into the spectrum of atherosclerotic disease in HIV. J Nuclear Cardiol: official publication of the Am Society of Nuclear Cardiol. 2015;22:381–384. [DOI] [PubMed] [Google Scholar]
- 39.Hajishengallis G Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol. 2015;15:30–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hasturk H, Abdallah R, Kantarci A, et al. Resolvin E1 (RvE1) attenuates atherosclerotic plaque formation in diet and inflammation-induced atherogenesis. Arterioscler, Thromb, Vasc Biol. 2015;35:1123–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gibson FC 3rd, Yumoto H, Takahashi Y, Chou HH, Genco CA. Innate immune signaling and Porphyromonas gingivalis-accelerated atherosclerosis. J Dent Res. 2006;85:106–121. [DOI] [PubMed] [Google Scholar]
- 42.Ekuni D, Tomofuji T, Sanbe T, et al. Periodontitis-induced lipid peroxidation in rat descending aorta is involved in the initiation of atherosclerosis. J Periodontal Res. 2009;44:434–442. [DOI] [PubMed] [Google Scholar]
- 43.Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. 2016;118:653–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shalhoub J, Falck-Hansen MA, Davies AH, Monaco C. Innate immunity and monocyte-macrophage activation in atherosclerosis. J Inflammation. 2011;8:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tatsumi K, Mackman N. Tissue factor and atherothrombosis. J Atheroscler Thromb. 2015;22:543–549. [DOI] [PubMed] [Google Scholar]
- 46.Westrick R, Fredman G, Early Career C. Platelets: context-dependent vascular protectors or mediators of disease. Arterioscler Thrombosis Vascular Biol. 2015;35:e25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Larsen SB, Grove EL, Wurtz M, Neergaard-Petersen S, Hvas AM, Kristensen SD. The influence of low-grade inflammation on platelets in patients with stable coronary artery disease. Thromb Haemost. 2015;114:519–529. [DOI] [PubMed] [Google Scholar]
- 48.Perumal R, Rajendran M, Krishnamurthy M, Ganji KK, Pendor SD. Modulation of P-selection and platelet aggregation in chronic periodontitis: a clinical study. J Indian Societ Periodontal. 2014;18:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhan Y, Lu R, Meng H, Wang X, Hou J. Platelet activation and platelet-leukocyte interaction in generalized aggressive periodontitis. J Leukocyte Biol. 2016;100:1155–1166. [DOI] [PubMed] [Google Scholar]
- 50.Papapanagiotou D, Nicu EA, Bizzarro S, et al. Periodontitis is associated with platelet activation. Atherosclerosis. 2009;202:605–611. [DOI] [PubMed] [Google Scholar]
- 51.Arvanitidis E, Bizzarro S, Alvarez Rodriguez E, Loos BG, Nicu EA. Reduced platelet hyper-reactivity and platelet-leukocyte aggregation after periodontal therapy Thromb J. 2017;15:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laky M, Anscheringer I, Wolschner L, et al. Periodontal treatment limits platelet activation in patients with periodontitis-a controlled-randomized intervention trial. J Clin Periodontal. 2018;45:1090–1097. [DOI] [PubMed] [Google Scholar]
- 53.Offenbacher S, Beck JD, Moss K, et al. Results from the periodontitis and vascular events (PAVE) Study: a pilot multicentered, randomized, controlled trial to study effects of periodontal therapy in a secondary prevention model of cardiovascular disease. J Periodontal. 2009;80:190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tonetti MS, D’Aiuto F, Nibali L, et al. Treatment of periodontitis and endothelial function. New England J Med. 2007;356:911–920. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.